Abstract
Cellular senescence is an irreversible growth arrest that occurs as a result of different damaging stimuli, including DNA damage, telomere shortening and dysfunction or oncogenic stress. Senescent cells exert a pleotropic effect on development, tissue aging and regeneration, inflammation, wound healing and tumor suppression. Strategies to remove senescent cells from aging tissues or preneoplastic lesions can delay tissue dysfunction and lead to increased healthspan. However, a significant hurdle in the aging field has been the identification of a universal biomarker that facilitates the unequivocal detection and quantification of senescent cell types in vitro and in vivo. Mammalian skin is the largest organ of the human body and consists of different cell types and compartments. Skin provides a physical barrier against harmful microbes, toxins, and protects us from ultraviolet radiation. Increasing evidence suggests that senescent cells accumulate in chronologically aged and photoaged skin; and may contribute to age-related skin changes and pathologies. Here, we highlight current biomarkers to detect senescent cells and review their utility in the context of skin aging. In particular, we discuss the efficacy of biomarkers to detect senescence within different skin compartments and cell types, and how they may contribute to myriad manifestations of skin aging and age-related skin pathologies.
Cellular Senescence and Organismal Aging
Aging is the result of a gradual functional decline at the cellular, and ultimately, organismal level, resulting in the development of myriad chronic illnesses including heart disease, stroke and diabetes. On a cellular level, aging was first described by Hayflick and Moorhead (1961), who demonstrated that somatic mammalian cells have a finite propensity for cell division, after which they enter an irreversible growth arrest termed cellular senescence. Senescent cells are characterized by their inability to proliferate, resistance to apoptosis, and secretion of factors that promote inflammation and tissue deterioration (Campisi and d’Adda di Fagagna, 2007; Kuilman et al., 2010; He and Sharpless, 2017). The molecular basis underlying Hayflick’s observations came to light only decades later. In the early 1970s, Watson and Olovnikov proposed that during semi-conservative DNA replication of linear chromosomes, the removal of the terminal RNA primer, required to initiate lagging strand synthesis, would result in a gap that can no longer be filled by conventional DNA polymerases. As a result of this “end replication problem", chromosome terminal sequences, termed telomeres (Blackburn and Challoner, 1984) shorten during each replication cycle (Hayflick and Moorhead, 1961; Watson, 1972; Olovnikov, 1973; Allsopp et al., 1992). Critically shortened telomeres trigger a persistent activation of DNA damage response (DDR) pathways, thereby resulting in cellular senescence (D’Adda Di Fagagna et al., 2003). It is now clear that several types of cellular stressors can trigger senescence. These include telomere shortening and dysfunction (D’Adda Di Fagagna et al., 2003; Takai et al., 2003), inadvertent activation of oncogenes, termed oncogene-induced senescence (OIS) (Serrano et al., 1997; Michaloglou et al., 2005; Suram et al., 2012), changes in chromatin structure and epigenetic stress (Bakkenist and Kastan, 2003; Munro et al., 2004), oxidative stress, mitochondrial dysfunction (Wiley et al., 2016), or persistent activation of DNA damage checkpoints (D’Adda Di Fagagna et al., 2003; Rodier et al., 2009). There is increasing evidence suggesting that senescent cells accumulate in aging tissues and organs, thereby impairing physiological processes, including regeneration, and contributing to organismal aging (Dimri et al., 1995; Krishnamurthy et al., 2004; Jeyapalan et al., 2007).
Senescence Biomarkers
The identification of unique markers that unequivocally detect and quantify senescent cells, particularly in vivo, remains challenging (Sharpless and Sherr, 2015). Senescent cells display an enlarged and flattened cell shape (Hayflick, 1965; Chen et al., 2000, 2008), and elevated senescence-associated β-galactosidase (SA-β-gal) activity, which remains the gold standard to identify senescent cells in culture and tissue samples (Dimri et al., 1995; Debacq-Chainiaux et al., 2009). However, this assay requires active enzymatic SA-β-gal activity, which is retained in fresh tissue samples but often lost in fixed or cryopreserved tissues (Severino et al., 2000; Lee et al., 2006). In addition, non-specific SA-β-gal activity can be detected in early passage adult melanocytes proliferating in culture, in hair follicles, sebaceous glands, and eccrine glands and ducts in both young and old human skin (Dimri et al., 1995), as well as in proliferating surface luminal cells of the duodenum (Going et al., 2002). Importantly, it remains difficult to multiplex the conventional detection method for SA-β-gal with other senescence-associated or cell-type specific markers, hence limiting the ability to identify senescent cell types or populations within a complex tissue or organ. Toward this goal, a modified flow cytometry-based method of multiplexing SA-β-gal with other senescence markers has been reported (Biran et al., 2017). However, such multiplexing strategies involving SA-β-gal in tissue biopsies in situ remains laborious. Despite being a hallmark of senescent cells, SA-β-gal activity does not play a mechanistic role in triggering senescence: cells from patients with autosomal recessive G(M1)-gangliosidosis lack lysosomal β-gal activity, but retain their ability to senesce (Lee et al., 2006).
As senescent cells are terminally growth arrested, cell cycle regulators such as p16INK4a, p21CIP1 and p53 (Collado and Serrano, 2010) are commonly employed to detect senescent cells. p16INK4a, p21CIP1 and p53 were simultaneously upregulated in human skin fibroblasts post-ultraviolet (UV) radiation (Chen et al., 2008). p16INK4a plays a key role in cell cycle control upstream of the retinoblastoma tumor suppressor protein, and p16INK4a-positive senescent cells accumulate in an age-dependent manner in multiple tissues including the skin (Zindy et al., 1997; Krishnamurthy et al., 2004; Michaloglou et al., 2005; Herbig et al., 2006; Ressler et al., 2006; Coppé et al., 2011; Waaijer et al., 2012). p16INK4a-positive cells accumulate in preneoplastic lesions, including melanocyte-rich benign human nevi, caused by activating mutations in N-RAS or its downstream target BRAF (Michaloglou et al., 2005; Ivanov et al., 2013). Considering the role of p16INK4a in mediating senescence, it is not surprising that this locus is frequently mutated in a variety of human cancers, including skin epithelial tumors (Soufir et al., 1999; De Snoo et al., 2008).
Although less well understood, senescence is also characterized by widespread chromatin remodeling. Normal cellular aging is associated with global heterochromatin loss, characterized by markers H3K9me3 and H3K27me3 (Tsurumi and Li, 2012). In agreement with these findings, cells from patients with the accelerated aging syndrome Hutchinson–Gilford progeria syndrome (HGPS) exhibit a profound loss of heterochromatin (Misteli and Scaffidi, 2005; Shumaker et al., 2006; Chojnowski et al., 2015). Predominantly during OIS in vitro, heterochromatin is redistributed into 30–50 punctate DNA-dense senescence-associated heterochromatin foci (SAHF). SAHF are silent domains that co-localize with H3K9me3 and heterochromatin protein 1 (HP1) and may lock cells in a senescent state by transcriptionally repressing genes involved in cell proliferation (Narita et al., 2003; Sadaie et al., 2013; Shah et al., 2013). In contrast, SAHF formation was not observed in HGPS patient cells or preneoplastic lesions that express other senescence markers (Scaffidi and Misteli, 2006; Shumaker et al., 2006; Kosar et al., 2011; Ivanov et al., 2013). Long-term monitoring of senescent cells in vitro revealed progressive proteolysis of Histones 3 and 4 without DNA loss. Reduced histone content was also observed in nevus melanocytes, as compared to neighboring non-senescent melanocytes and keratinocytes in vivo (Ivanov et al., 2013). These studies confirm the dramatic structural changes of chromatin in senescent cells. In addition, the same authors also noted the presence of cytoplasmic chromatin fragments (CCFs) in ∼20% of cells undergoing replicative senescence (RS) or OIS in vitro. CCFs were positive for H3K9me3 and γ-H2A-X, but negative for 53BP1 and lamin A/C, and targeted for degradation by autophagy. The accumulation of such chromatin fragments within the cytoplasm suggests that the integrity of the nuclear envelope becomes compromised in senescent cells (Ivanov et al., 2013).
Indeed, the nuclear lamina, a proteinaceous network that lies beneath the inner nuclear membrane, undergoes dramatic remodeling during cellular senescence (Shimi et al., 2011; Freund et al., 2012; Dreesen et al., 2013a). Lamin B1, an intermediate filament protein expressed in all somatic cells (Stewart and Burke, 1987; Hoger et al., 1988), is downregulated in cells undergoing RS, OIS and UV-induced senescence in vitro (Shimi et al., 2011; Freund et al., 2012; Dreesen et al., 2013a; Ivanov et al., 2013; Sadaie et al., 2013; Shah et al., 2013; Wang et al., 2017). Lamin B1 levels also decline during chronological aging of human skin in vivo (Dreesen et al., 2013a,b), in senescent melanocytes within human nevi (Ivanov et al., 2013), in UV-exposed mouse skin epidermis (Wang et al., 2017), irradiated mouse liver (Freund et al., 2012) and in kidneys of a premature aging mouse model (Baar et al., 2017). Importantly, by co-staining with cell-type specific markers, lamin B1 staining facilitated the identification and quantification of senescent melanocytes within nevi as compared to neighboring keratinocytes within the epidermis (Ivanov et al., 2013). Similarly, co-staining lamin B1 with a keratinocyte differentiation marker followed by single cell analysis enabled us to quantify the accumulation and clearance of senescent cells in different epidermal compartments after UV exposure and upon regeneration, respectively (Wang et al., 2017). In addition to lamin B1, the inner nuclear membrane protein lamin B receptor (LBR) and the lamina-associated polypeptide-α (LAP2α) are also downregulated in senescent cells (Dreesen et al., 2013a; Ivanov et al., 2013; Lukášová et al., 2017). However, LBR levels vary within the different epidermal layers in mouse skin, indicating that its expression may be altered during keratinocyte differentiation (Solovei et al., 2013). Hence, this may limit the usage of LBR as an adequate senescence marker in human skin. Moreover, loss of LAP2α is not specific to senescent cells and also occurs in quiescent cells (Pekovic et al., 2007; Dreesen et al., 2013a). Thus, co-staining of lamin B1 and LAP2α distinguishes senescent from quiescent cells (Dreesen et al., 2013a). The mechanism of senescence-induced lamin B1 downregulation has been elucidated in detail by several groups. LMNB1 transcription is decreased (Shimi et al., 2011; Freund et al., 2012; Dreesen et al., 2013a) and LMNB1 mRNA is destabilized, possibly via targeted degradation by miR-23a (Lin and Fu, 2009; Dreesen et al., 2013a). Additionally, lamin B1 protein is shuttled to the cytoplasm and degraded via autophagy (Dou et al., 2015). It should be noted that LMNB1 mRNA levels alone are not a reliable indicator of senescence. For instance, LMNB1 mRNA levels decrease in contact-inhibited quiescent fibroblasts, whereas lamin B1 protein levels remain stable as cells do not turn over (Dreesen et al., 2013a). Taken together, although the mechanistic role of lamin B1 during cellular senescence is not fully understood, loss of lamin B1 is emerging as a potentially important biomarker to identify and quantify senescent cells in vitro and in vivo.
Concomitant with senescence-associated loss in chromatin organization, overall DNA methylation and expression of the DNA methyl-transferase DNMT1 decrease during cellular aging. These altered DNA methylation signatures can distinguish cells based on their developmental potential and may be used to predict chronological and biological age (Horvath, 2013; Raddatz et al., 2013). Such distinct methylation patterns are also associated with chronologically aged and photoaged skin (Grönniger et al., 2010).
Persistent DNA damage is a critical trigger of cellular senescence and can be identified by the presence of γ-H2A-X and 53BP1 foci (D’Adda Di Fagagna et al., 2003) and activated ataxia-telangiectasia mutated (ATM) kinase (Zou, 2007). However, while DNA damage per se, is not a marker for cellular senescence, the occurrence of telomere-associated DNA damage foci (TAF) has been used to detect senescent cells and quantify tissue aging (Herbig et al., 2006; Jeyapalan et al., 2007; Hewitt et al., 2012; Waaijer et al., 2018). Replicative senescent baboon skin fibroblasts and skin biopsies from aged baboons displayed high incidence of TAF as indicated by co-localization of 53BP1 and γ-H2A-X on telomeric DNA (Herbig et al., 2006; Jeyapalan et al., 2007). In agreement with these findings, shortened telomeres have been detected in skin from aged individuals, in sun-exposed skin and premalignant skin lesions including actinic keratosis (AK) (Sugimoto et al., 2006; Ikeda et al., 2014). Other age-related genomic changes occur in mitochondrial DNA (mtDNA): age-dependent increase of mtDNA mutations and a common 4977 bp mtDNA deletion have been reported in several human tissues and cell types (Passos et al., 2007; Wiley et al., 2016), as well as in photoaged human skin (Berneburg et al., 1997; Koch et al., 2001). Lastly, a histone variant H2A.J accumulates in an age-dependent manner in mouse and human skin, and in irradiated mouse skin. Both H2A.J and p16INK4a levels were elevated in carcinogen-induced preneoplastic lesions but decreased in more advanced papillomas in mouse skin. H2A.J upregulation promotes expression of inflammatory genes, further exacerbating the senescence-induced hyperinflammation (Contrepois et al., 2017).
For long, senescent cells were considered passive bystanders that ceased to proliferate. Research in the past decade revealed that senescence can dramatically change cell function. For instance, senescent cells modulate their environment by secreting inflammatory cytokines, chemokines, matrix metalloproteinases (MMPs) and growth factors, collectively known as the senescence-associated secretory phenotype (SASP) (Acosta et al., 2008; Coppé et al., 2008b; Kuilman et al., 2008; Rodier et al., 2009) or senescence-messaging secretome (SMS) (Kuilman and Peeper, 2009). As such, the presence of MMPs (e.g., MMP3 and MMP9), chemokines (e.g., CXCR2), cytokines (e.g., IL-6 and IL-8) (Acosta et al., 2008; Kuilman et al., 2008) and insulin-like growth factor binding protein 7 (IGFBP7) (Wajapeyee et al., 2008) has been used as markers for senescent dermal fibroblasts and melanocytes in vitro. In vivo, elevated IL-6 has been detected in nevi melanocytes (Ghosh and Capell, 2016) while MMPs are detected in chronologically aged and photoaged skin, and are responsible for breakdown of the extracellular matrix (ECM) (Quan et al., 2009; Quan and Fisher, 2015). In addition, the plasminogen activator inhibitor type 1 (PAI-1), an inhibitor of serine proteases, is elevated in dermal fibroblasts derived from patients with the premature aging syndromes HGPS and Werner (Murano et al., 1991; Goldstein et al., 1994; Chojnowski et al., 2015), chronologically aged donors (Goldstein et al., 1994), and in tissues of premature aging BubR1 mutant mice (Baker et al., 2008, 2011). PAI-1 plays a role in triggering senescence downstream of p53 (Kortlever et al., 2006) and is both a mediator and marker of senescence. Interestingly, most studies focused on fibroblasts in vitro and it remains to be determined whether the secretome of other senescent cell types differs – and how this may affect the function of neighboring cells. It is noteworthy that expression of p53 or p16INK4a alone may not be sufficient to trigger a SASP as a persistent DDR is mandatory (Rodier et al., 2009; Coppé et al., 2011).
High mobility group box-1 (HMGB1) belongs to the family of alarmins, which is an inflammatory mediator important in tissue damage signaling (Bianchi, 2006; Yamada and Maruyama, 2007). In senescent cells, HMGB1 translocates from the nucleus to the cytoplasm and extracellular space, facilitating the release of SASP factors including IL-1β, IL-6 and MMP3 (Davalos et al., 2013; Biran et al., 2017). UVB-exposure stimulated the release of HMGB1, IL-1 and IL-6 in human keratinocytes in vitro, and reduced nuclear Hmgb1 expression in mouse skin epidermis in vivo (Johnson et al., 2013). Although HMGB1 is used as a marker to detect senescent cells in various contexts (Davalos et al., 2013; Wiley et al., 2016; Biran et al., 2017), its utility to detect senescent cells in human skin remains to be investigated.
Taken together, various senescence markers are currently being used individually and in combination, in different biological contexts in vitro and in vivo (Figure 1 and Table 1). Nevertheless, further characterization of current markers and the identification of additional markers are indispensable to reliably detect and quantify senescent cells in vitro and in vivo. In particular, it is essential to develop better methods to multiplex senescence markers with other cell-type specific markers to identify and quantify senescent cell types in multi-compartment organs such as the skin and its appendages including sebaceous glands and hair follicles. Here, we will focus on what is known about the accumulation of senescent cells in skin, how they may affect skin function and contribute to age-related skin pathologies.
FIGURE 1
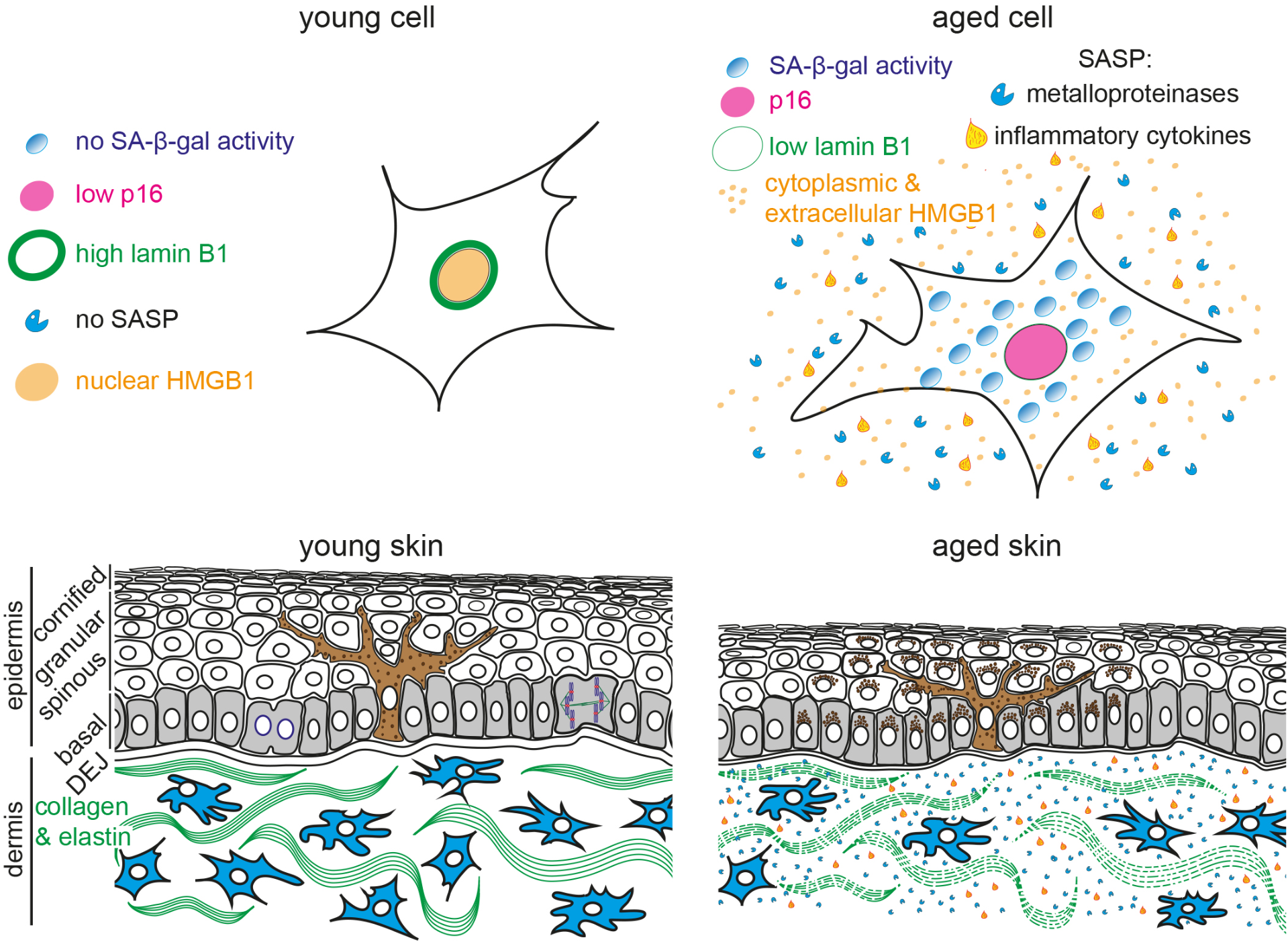
Schematic representation of young and aged human skin. (Top) Illustration of young (Left) and aged senescent cells (Right). Senescent cells are characterized by (1) enlarged and flattened cell morphology, (2) increased senescence-associated β-galactosidase (SA-β-gal) activity, (3) p16 upregulation, (4) reduced lamin B1 expression, (5) translocation of nuclear HMGB1 into the cytoplasm and extracellular space and (6) secretion of senescence-associated secretory phenotype (SASP) factors, including inflammatory cytokines and metalloproteinases. (Bottom) The epidermis consists of keratinocytes, arranged into basal (gray), spinous, granular and cornified layers. Keratinocytes progressively flatten as they move apically and lose their nuclei. Melanocytes (brown) reside within the basal layer; their dendrites branch to neighboring keratinocytes to facilitate pigment transfer. The dermal-epidermal junction (DEJ) separates the epidermis from the underlying dermis. Dermal fibroblasts (blue) reside among collagen and elastin fibers (green fibers) in the dermis. Aged skin (Right) becomes atrophic due to reduced proliferation, exhibits abnormal pigmentation, increased inflammation and breakdown of collagen fibers.
Table 1
| Biomarkers | In vitro | In vivo | Reference |
|---|---|---|---|
| SA-β-gal | RS fibroblasts, keratinocytes, melanocytes (Hu), UVI keratinocytes and fibroblasts (Hu) | CA epidermis (Hu) UVI epidermis (Ms) | Dimri et al., 1995Chen et al., 2008; Wang et al., 2017 |
| Enlarged, flattened cell morphology | RS fibroblasts (Hu, Bb), UVI keratinocytes and fibroblasts (Hu) | Not well documented | Hayflick and Moorhead, 1961; Chen et al., 2000, 2008; Jeyapalan et al., 2007; Wang et al., 2017 |
| p16INK4a | OIS melanocytes (Hu), RS fibroblasts (Bb) and ROT-treated fibroblasts (Hu) | Nevi melanocytes (Hu), premalignant papillomas (Ms), CA skin (Bb), CA epidermis, dermis and hair follicles (Hu) | Collado et al., 2005; Michaloglou et al., 2005; Herbig et al., 2006; Ressler et al., 2006; Jeyapalan et al., 2007; Waaijer et al., 2012, 2018; Ivanov et al., 2013 |
| Lamin B1 | UVI keratinocytes (Hu) and OIS melanocytes (Hu) | CA epidermis (Hu), nevi melanocytes (Hu) and UVI epidermis (Ms) | Dreesen et al., 2013a,b; Ivanov et al., 2013; Wang et al., 2017 |
| SASP (cytokines, MMPs, growth factors) | OIS melanocytes (IL-1, IL-6, IL8, IGFBP7; Hu), CA fibroblasts (IL-6, IL-8; Hu), UVI keratinocytes (IL-1, IL-6, HMGB1; Hu), HGPS and WS fibroblasts (PAI-1; Hu) | CA and UVI epidermis (MMPs, Hu), UVI skin (Hmgb1; Ms), nevi melanocytes (IL-6; Hu), and DNA damaged fibroblasts (IL-6, IL-8, Mmp1, Mmp3; Ms) | Murano et al., 1991; Goldstein et al., 1994; Coppé et al., 2008b; Kuilman et al., 2008; Wajapeyee et al., 2008; Jun and Lau, 2010; Johnson et al., 2013; Chojnowski et al., 2015; Quan and Fisher, 2015; Ghosh and Capell, 2016 |
| Telomere-associated foci (TAFs) | RS fibroblasts (Bb) and ROT-treated fibroblasts (Hu) | CA skin (Bb) | Herbig et al., 2006; Jeyapalan et al., 2007; Waaijer et al., 2018 |
| mtDNA modification | UVI keratinocytes (Hu) | UVI skin (Hu) | Berneburg et al., 1997; Koch et al., 2001 |
| Chromatin modification | HGPS fibroblasts (H3K27me3; Hu) and OIS melanocytes (CCFs, H3, H4; Hu) | Nevi melanocytes (H3; Hu), CA skin (H2A.J; Ms, Hu), UVI and premalignant lesions (H2A.J; Ms) | Ivanov et al., 2013; Chojnowski et al., 2015; Contrepois et al., 2017 |
Biomarkers previously described in skin aging.
Hu, human; Ms, mouse; Bb, baboon; SA-β-gal, senescence-associated β-galactosidase; UVI, ultraviolet irradiated; RS, replicative senescent; OIS, oncogene-induced senescent; ROT, rotenone; CA, chronologically aged; SASP, senescence-associated secretory phenotype; MMPs, matrix metalloproteinases; mtDNA, mitochondria DNA; CCFs, cytoplasmic chromatin fragments; H3, histone 3; H4, histone 4.
Skin Aging
Skin is the largest organ of the human body: it provides a barrier against harmful organisms and substances, protects against UV radiation and regulates water loss and body temperature (Blanpain and Fuchs, 2006). The skin is a complex organ, consisting of several compartments with different functions. The outermost epidermis is stratified into four sublayers (basal, spinous, granular and cornified layer) with keratinocytes being the predominant cell type. Pigment-producing melanocytes reside within the basal layer of the epidermis, determine skin color and possess photo-protecting properties (Figure 1). The dermal-epidermal junction (DEJ) connects the epidermis to the underlying dermis, which harbors dermal fibroblasts and appendages such as hair follicles, sebaceous glands, and sweat glands.
Skin aging is a multi-factorial process that affects nearly every aspect of its biology and function; it is driven by both intrinsic (e.g., time, genetic factors, hormones) and extrinsic (e.g., UV exposure, pollution) factors. Characteristics of intrinsic or chronological aging include visible signs such as thin and dry skin, fine wrinkles, decreased elasticity, aberrant pigmentation, hair graying and hair loss. Epidermal thinning is, in part, caused by decreased proliferation and renewal capacity of basal keratinocytes and reduced epidermal stem cell number (Kligman, 1979; Montagna and Carlisle, 1979; Mimeault and Batra, 2010; Dreesen et al., 2013a; López-Otín et al., 2013). Besides the epidermis, both the DEJ and dermis also become thinner (Lavker et al., 1986). Fibroblasts residing within the dermis generate an ECM that provides the skin with structural integrity and elasticity. During aging, the ECM undergoes structural alterations and degradation (Figure 1), thought to result in dermal thinning, increased wrinkling and loss of elasticity (Shuster et al., 1975). Although the number of senescent cells increases during chronological aging of human skin (Dimri et al., 1995; Ressler et al., 2006; Waaijer et al., 2012; Dreesen et al., 2013a; Ghosh and Capell, 2016), a more detailed characterization as to which cell types or skin compartment undergoes senescence is necessary.
UV and Skin Aging
In addition to chronological aging, extrinsic factors including exposure to sunlight, pollutants and cigarette smoke can accelerate skin aging. While most UVC is blocked by the ozone layer, UVA and UVB rays reach the earth’s surface and contribute to skin aging and cancer development. UVA is a weak mutagen, yet penetrates into the dermis contributing to oxidative stress and tissue inflammation. UVB is more mutagenic as it directly interacts with DNA to generate dipyrimidine photoproducts, resulting in DNA damage during DNA replication (Ravanat et al., 2001; You et al., 2001). Acute UV radiation results in sunburns, aberrant pigmentation and immune suppression, whilst chronic exposure is associated with premature skin aging and a predisposition to develop malignancies (de Gruijl, 1999; Ichihashi et al., 2003; Quan et al., 2009; Behrens et al., 2014; Rittié and Fisher, 2015). Sun-exposed skin appears thick and rough, marked with coarse wrinkles, visible appearance of blood vessels under the skin surface (telangiectasia) and aberrant pigmentation (Ma et al., 2001; Wlaschek et al., 2001). A key histological hallmark of photodamaged skin is the accumulation of amorphous elastic fibers (solar elastosis), accompanied by fragmented and disorganized collagen in the dermis (Varani et al., 2004, 2006; Quan et al., 2009; Watson et al., 2014). Solar elastosis may be a consequence of impaired elastic and fibrillin production, elevated breakdown by MMPs secreted by senescent cells, or a direct consequence of UV exposure (Sherratt, 2013; Quan and Fisher, 2015; Pittayapruek et al., 2016). In vitro, UVB-exposed skin cell types (fibroblasts, keratinocytes) exhibit DNA damage, cell cycle arrest and express senescence biomarkers such as increased SA-β-gal activity, p16INK4a, p21CIP1, p53 activation and lamin B1 downregulation (Chainiaux et al., 2002; Debacq-Chainiaux et al., 2005; Lewis et al., 2008; McCart et al., 2017; Wang et al., 2017). In vivo, chronic low dose exposure to UVB resulted in accumulation of DNA damage and lamin B1-low senescent cells within the mouse epidermis, but not the dermis (Wang et al., 2017), a reduction in stem cell numbers in the hair follicle, and p21CIP1 accumulation in epidermis and hair follicle stem cell region (McCart et al., 2017).
Although the number of melanocytes in our skin decreases and skin color in sun-protected areas lightens with age (Gilchrest et al., 1979; Ortonne, 1990), aberrant pigmentation (hyper- and hypopigmentation; i.e., sun spots and freckles) occurs at chronically sun-exposed body parts (Ortonne, 1990; Chung, 2003). Interestingly, in a 3D organotypic skin model comprised of keratinocytes, melanocytes and fibroblasts, the presence of aged fibroblasts resulted in increased melanogenic gene transcription, increased epidermal melanin and hyperpigmentation (Duval et al., 2014). Diverse pigmentary changes occur in aged skin and future studies will investigate whether senescent cell types accumulate at these lesions and how they may affect pigmentation. We anticipate that multiplexing senescence markers with cell-type specific markers will shed light as to how chronological aging and/or UV exposure modulate the complex interplay between pigment-producing melanocytes and their neighboring epidermal keratinocytes and dermal fibroblasts.
Pollution and Skin Aging
More recently, it has been shown that air pollution contributes to various aspects of skin aging. Residents living near highly polluted areas and exposed to high levels of traffic-related particles and nitrogen dioxide developed pigmentary changes and increased coarse wrinkles (Vierkötter et al., 2010; Hüls et al., 2016). Similarly, exposure to tobacco smoke accelerates skin aging, resulting in increased wrinkling, elastosis, telangiectasia and laxity in the skin (Schroeder et al., 2006; Morita, 2007; Vierkötter et al., 2010). Application of tobacco extracts to skin and oral fibroblasts in vitro triggered several hallmarks of senescence including premature cell cycle arrest, oxidative DNA damage, secretion of inflammatory cytokines and MMPs, and downregulation of cell junction proteins E-cadherin and ZO-1 (Coppé et al., 2008a). The ability to detect and quantify senescent cells in aged skin is indispensable to further elucidate how pollution may accelerate senescence and how this in turn alters skin appearance and function.
Whilst aging results in dry, itchy and more irritable skin, the underlying cause is likely multifactorial and involves changes in skin barrier function, androgen levels and sebaceous gland function (Kligman, 1979; Zouboulis and Boschnakow, 2001). Sebaceous glands contribute to skin oiliness and moisture by secreting a complex mix of lipids called sebum. While the number of sebaceous glands does not change during aging, its activity drastically decreases in postmenopausal females and males over 80 years old. Somewhat counterintuitive, contrasting its declining function and sebum production, the size of the sebaceous gland increases with age (Plewig and Kligman, 1978; Zouboulis and Boschnakow, 2001). Chronic UV exposure has also been implicated in sebaceous hyperplasia and carcinoma (Lesnik et al., 1992; Harwood et al., 2001). The specific involvement of cellular senescence in aging of the pigmentary units and sebaceous glands remains to be explored in greater detail.
Genetic Diseases and Skin Aging
Multiple genetic syndromes that result in persistent DNA damage or telomere dysfunction (telomeropathies) and cellular senescence, predispose the skin to accelerated aging and/or carcinogenesis (Harada et al., 1999; Davis et al., 2007; Opresko and Shay, 2017). HGPS is a rare early-onset premature aging syndrome caused by a mutation in lamin A, resulting in a truncated mutant protein termed progerin. HGPS patients exhibit signs of accelerated aging at 12–18 months after birth and die in their mid-teens as a result of cardiovascular complications. Early clinical signs include alopecia, sclerotic skin, loss of subcutaneous fat and aberrant skin pigmentation (Merideth et al., 2008). At the cellular level, progerin expression results in atypical nuclear architecture, heterochromatin loss, DNA damage and premature senescence (Dreesen and Stewart, 2011; Vidak and Foisner, 2016; Kubben and Misteli, 2017). The primary driver mechanisms of these complex cellular phenotypes remain unclear. Progerin sequestration of NRF2, a major factor in a cell’s response to oxidative stress, has been suggested to play a role in progerin-dependent heterochromatin loss and DNA damage (Kubben et al., 2016). Other studies linked progerin-induced DNA damage to replication defects (Hilton et al., 2017; Wheaton et al., 2017) and telomere dysfunction (Kudlow et al., 2008; Decker et al., 2009; Benson et al., 2010; Chojnowski et al., 2015; Wood et al., 2015).
Mandibuloacral dysplasia (MAD) and restrictive dermopathy (RD) are also caused by nuclear lamina aberrations. Similar to HGPS, MAD is caused by a lamin A mutation and patients exhibit growth retardation, skeletal abnormalities, skin atrophy and hyperpigmentation (Novelli et al., 2002). RD is a rare neonatal lethal disease in which newborns exhibit intrauterine growth retardation, tight, erosive skin with hyperkeratosis, and sparse eyebrows and hair. RD is caused by loss of ZMPSTE24, a zinc metalloproteinase critical for the correct processing of lamin A (Navarro et al., 2005). While RD does not reflect skin aging phenotypes per se, the occurrence of these skin-specific laminopathies highlights the importance of nuclear lamina functions in the development and homeostasis of skin and hair.
Werner syndrome (WS), also referred to as adult-onset progeria, is regarded as a milder form of progeria. WS patients start to exhibit clinical features including hair loss and graying, scleroderma-like skin, ulcers and osteoporosis in their 30’s and die in their 50’s due to myocardial infarction and cancer. WS has been linked to mutations in WRN, a member of the RecQ helicase family (Yu et al., 1996) that lead to defects in DNA replication, repair, and premature cellular senescence (Oshima et al., 2017). Loss of function mutations in WRN result in defective telomere lagging strand synthesis, telomere loss and genomic instability (Crabbe et al., 2004, 2007).
Bloom syndrome is another disorder caused by loss of function mutations in the RecQ DNA helicase and affects DNA replication and repair (Bachrati and Hickson, 2003). Its clinical symptoms include the development of skin rash in sun-exposed areas, accelerated skin aging, alopecia, poikiloderma (irregular pigmentation, telangiectasia, and atrophy) and a predisposition to cancer (Goto, 2001; German and Sanz, 2013).
Xeroderma pigmentosum (XP) is a rare autosomal recessive syndrome characterized by extreme sensitivity to sunlight, resulting in sunburn, aberrant pigmentation, dry skin, premature skin aging, and increased risk to develop skin malignancies, including squamous cell carcinoma (SCC), basal cell carcinoma (BCC) and melanoma (Bukowska and Karwowski, 2018). The median age of death for XP patients is 29–32 years and most frequently caused by metastatic skin cancers and neurodegeneration (Bradford et al., 2011; DiGiovanna and Kraemer, 2012). The etiology of XP is characterized by defects in nucleotide excision repair (NER), a key pathway required to repair UVB-induced dipyrimidine photoproducts (Cleaver, 1968). Defects in NER lead to the accumulation of DNA damage, genomic instability, impaired cell growth, premature senescence and/or cell death (Niedernhofer et al., 2011; Black, 2016). Similarly, two other disorders caused by NER defects are Cockayne syndrome and trichothiodystrophy (TTD); both diseases are characterized by premature aging, neurological abnormalities and skin photosensitivity. Additionally, TTD patients display rough skin and brittle hair (Itin et al., 2001; Tan et al., 2005).
Mutations in ATM, an important molecule involved in activating checkpoint signaling in response to DNA double strand breaks, cause Ataxia-telangiectasia, another rare early-onset neurodegenerative disease in which patients display skin abnormalities including vitiligo, hair graying and telangiectasia in sun-exposed skin (Zou, 2007; Derheimer and Kastan, 2010; Rothblum-Oviatt et al., 2016).
Dyskeratosis congenita (DKC) is multi-systemic premature aging syndrome resulting in some skin defects. Initial clinical features appear during childhood and include abnormal skin pigmentation and nail dystrophy. During the second and third decade, patients develop bone marrow failure, ultimately resulting in their death. In addition, some patients exhibit an increased risk to develop premalignant and malignant lesions, including AK and SCC (Powell et al., 2014). DKC is caused by mutations in various components of telomerase (TCAP1, DKC1, TERC, TERT, NHP2, NOP10) or shelterin (TINF2), a protein complex involved in telomerase recruitment and telomere protection. These mutations result in impaired telomere maintenance, presumably in adult stem cell compartments, and premature telomere shortening (Mitchell et al., 1999; Batista et al., 2011; Dokal, 2011).
A common feature of the abovementioned genetic syndromes is the accumulation of persistent DNA damage, either by impaired DNA replication, repair, or signaling, as well as by DNA damage at unrepairable loci such as telomeres, resulting in premature cellular senescence and accelerated skin aging (Carrero et al., 2016). These phenotypes can be recapitulated in patient-derived fibroblasts, cell- or iPSC-based disease models and mouse models (Crabbe et al., 2004, 2007; Lombard et al., 2005; Batista et al., 2011; Zhang et al., 2011, 2015; Chojnowski et al., 2015). However, the rarity of these genetic diseases coupled with limited access to tissue biopsies have hampered efforts to test whether senescent cells accumulate within patient tissues in vivo. How are these accelerated aging syndromes relevant to normal aging? During chronological aging, at least the accumulation of genomic alterations correlates with the onset of age-related phenotypes and diseases (Chen et al., 2007; Vijg and Suh, 2013). Furthermore, an increase susceptibility to DNA damage (Xu et al., 2000) and decline in DNA repair capacity (Moriwaki et al., 1996; Yamada et al., 2006) have also been described in aging skin. Chronological aging can be accelerated by extrinsic factors that result in DNA damage, including UV radiation, particles from traffic pollution and cigarette smoke. In conclusion, increased DNA damage, impaired repair or signaling play a key conserved feature of premature aging syndromes and normal aging.
Role of Cellular Senescence in Wounds and Cancers
Aging results in a progressive decline of skin function, leading to fragility, impaired barrier function, increased susceptibility to physical insults, infection, accumulation of age-related pathologies such as impaired wound healing and an increased risk of cancer (Lewis et al., 2011; Maru et al., 2014).
Skin wound healing occurs in four stages: hemostasis, inflammation, proliferation and remodeling. Wound closure takes place during the proliferation phase and depends on the formation of granulation tissue and the generation of contractile myofibroblasts. A ground breaking study by Demaria et al. (2014) demonstrated that senescent cells transiently accumulate during the proliferative stage and secrete platelet-derived growth factor AA (PDGF-AA) to induce myofibroblasts differentiation and maturation, essential for wound closure. Elimination of senescent cells reduced the number of myofibroblasts, delayed wound closure and increased fibrosis, all of which could be prevented by ectopic administration of PDGF-AA (Jun and Lau, 2010; Demaria et al., 2014). Similarly, senescent cells prevent fibrosis during liver regeneration (Krizhanovsky et al., 2008; Kim et al., 2013). These results demonstrate that senescent cells play an important role in tissue repair via cell-autonomous mechanisms: in the skin through the secretion of PDGF-AA; in the regenerating liver by secreting Mmp9, Mmp13 and IL-6. Thus, senescent cells may limit tissue regeneration in order to protect against unrestricted cell proliferation. On the flip side, a prolonged presence of senescent cells in the elderly prevents re-epithelialization and wound closure resulting in chronic wounds (Holt et al., 1992; Mendez et al., 1998; Vande Berg and Robson, 2003; Telgenhoff and Shroot, 2005; Vande Berg et al., 2005). Taken together, cellular senescence plays a complex role during normal wound healing as well as in chronic wounds.
Senescence and Skin Cancer
Cellular senescence is a double-edged sword: on the one hand, the accumulation of senescent cells may contribute to various age-related complications; on the other hand, RS and OIS suppress uncontrolled proliferation of cells that are at risk for preneoplastic transformation, thereby limiting tumorigenesis. As a result, cells from premalignant clinical specimens of breast, colon, lung and bladder tumors exhibited persistent DNA damage checkpoint activation and expressed some senescence markers (Bartkova et al., 2005, 2006; Gorgoulis et al., 2005; Nuciforo et al., 2007; Suram et al., 2012). Besides promoting non-cell-autonomous growth inhibition, the production of SASP is also critical for the recruitment of immune cells that create an anti-tumorigenic microenvironment important for the clearance of senescent cells (Coppé et al., 2010).
In the skin, the vast majority of benign nevi are caused either by activating mutations in N-RAS or BRAF, and are unlikely to progress toward melanoma. Melanocytes within these preneoplastic lesions are positive for multiple senescence markers, including SA-β-gal, p16INK4a and loss of lamin B1 (Michaloglou et al., 2005; Gray-Schopfer et al., 2006; Suram et al., 2012; Ivanov et al., 2013), which are absent in more advanced melanoma (Michaloglou et al., 2005; Suram et al., 2012). Kras mutant mice also expressed increased senescence markers in premalignant lung and pancreatic adenomas, and in chemically induced skin papillomas (Collado et al., 2005).
Skin cancer represents the most common human cancer type and its incidence increases with age and UV exposure (Narayanan et al., 2010). Long-term exposure to sunlight results in persistent DNA damage, increased cellular senescence and formation of premalignant lesions such as AK. AK affects approximately 60% of individuals above the age of 40 with a history of sun exposure. If left untreated, a percentage of AK lesions will progress toward invasive SCCs (Berman, 2012; Siegel et al., 2017). Clinically, 97% of SCCs are contiguous with AK (Hurwitz and Monger, 1995) yet the risk of progression or regression remains difficult to assess. Although cells within AK lesions exhibit genomic instability and telomere shortening (Rehman et al., 1994; Ikeda et al., 2014), it is unclear whether senescent cells accumulate within AK lesions. As senescent cells generally accumulate in premalignant, but not malignant lesions, the identification and quantification of senescent cells within AK lesions may serve as a valuable tool to diagnose and distinguish AK from SCC and predict the course of disease progression.
Senescent Cells: Therapeutic Opportunities for Cancer and Aging
Numerous studies have aimed at preventing the accumulation of senescent cells within aging tissues. However, strategies that involve blocking p16INK4a and p53, or activating telomerase to extend the proliferative capacity of cells inevitably lead to an increased cancer risk. Thus, a safer approach might be to either selectively eliminate senescent cells from tissues or modulate their function (particularly the SASP). The use of a progeroid mouse model expressing a mutated form of the mitotic checkpoint BubR1 has proven instrumental in demonstrating that p16INK4a-positive senescent cells drive age-related pathologies, and that selective elimination of these cells can prevent or delay age-related tissue deterioration (Baker et al., 2008, 2011). Similarly, clearance of p16INK4a-positive cells in normal-aging mice extended their median lifespan, delayed tumorigenesis, improved healthspan indices including heart and kidney function, and delayed age-related decline in exploratory behavior (Baker et al., 2016). Multiple recent studies confirmed that clearing senescent cells improved tissue function in various age-related pathologies including degenerative joint disease, hepatic fat accumulation and liver chemotoxicity (Xu et al., 2015; Baar et al., 2017; Farr et al., 2017; Jeon et al., 2017; Ogrodnik et al., 2017). These studies established senescent cells as potential therapeutic targets in the treatment of aging and age-related diseases. As a result, compounds that selectively eliminate senescent cells (senolytics) are under investigation to treat and prevent various age-related diseases (Baar et al., 2017; Kirkland and Tchkonia, 2017). While several molecular pathways that trigger senescence are under consideration for the development of senolytics, the majority are directed at the pathways that confer apoptosis-resistance in senescent cells (in particular the anti-apoptotic BCL protein family) (Chang et al., 2016; Yosef et al., 2016; Zhu et al., 2016). However, few studies have probed how treatment with senolytics may affect skin function and homeostasis. In one study involving a skin-specific keratin 5-driven p14ARF transgenic mouse model, senescent epidermal cells were efficiently eliminated upon delivery of ABT-737, a BCL-specific senolytic compound (Tokarsky-Amiel et al., 2013; Yosef et al., 2016). Although elimination of senescent cells from the epidermis led to increased proliferation within hair follicles, the underlying mechanism or other consequences on skin function were not investigated. Thus, it remains to be determined whether senolytics offer novel therapeutic approaches to treat chronic wounds, skin inflammation, age-related pigmentation disorders and ECM breakdown, or whether their clearance would impair wound healing or other skin functions in younger individuals.
A prerequisite for any such studies is a better understanding as to which skin compartment or cell type becomes senescent in the myriad age-related skin pathologies. Multiplexing senescence biomarkers with cell-type specific markers will pave the way to address this question. In addition, it is important to better understand how senescence affects or modulates the function of various skin cell types. To date, most SASP studies have focused on dermal fibroblasts and little is known about the SASP of other skin resident cell types, including melanocytes, keratinocytes or cell types from skin appendages, such as sebocytes. This is particularly relevant in skin as the interplay between these different cell types is essential to regulate skin functions including pigmentation, barrier function, sebum production, wound healing and cancer progression.
Conclusion
The recognition of the role of cellular senescence in tissue aging has progressed tremendously in the past decade and this is in part attributed to better characterization of senescence biomarkers which allows qualitative and quantitative detection of senescent cells in vitro and in situ. Recent studies provided evidence that senescent cells accumulate in aged skin and may be driving the functional deterioration that characterizes aging skin and age-related skin diseases. Improved methodologies to detect senescent cells and a better understanding how senescence affects cellular function will enable us to determine how they contribute to various age-related skin changes and pathologies, including impaired wound healing and tumorigenesis. Moreover, the careful evaluation of senolytics in animal models and 3D reconstructed skin organotypics will facilitate the successful translation of senolytics as a therapeutic intervention for skin aging. The accessibility of human skin offers advantages to test these hypotheses.
Statements
Author contributions
OD and AW wrote the manuscript and prepared the figures.
Funding
This work was supported by the Singapore Biomedical Research Council (A∗STAR, Singapore).
Acknowledgments
We thank Philipp Kaldis, Steven Thng Tien Guan, and the members of the Dreesen lab for discussions and careful reading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AcostaJ. C.O’LoghlenA.BanitoA.GuijarroM. V.AugertA.RaguzS.et al (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence.Cell1331006–1018. 10.1016/j.cell.2008.03.038
2
AllsoppR. C.VaziriH.PattersonC.GoldsteinS.YounglaiE. V.FutcherA. B.et al (1992). Telomere length predicts replicative capacity of human fibroblasts.Proc. Natl. Acad. Sci. U.S.A.8910114–10118. 10.1073/pnas.89.21.10114
3
BaarM. P.BrandtR. M. C.PutavetD. A.KleinJ. D. D.DerksK. W. J.BourgeoisB. R. M.et al (2017). Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging.Cell169132–147.e16. 10.1016/j.cell.2017.02.031
4
BachratiC. Z.HicksonI. D. (2003). RecQ helicases: suppressors of tumorigenesis and premature aging.Biochem. J.374577–606. 10.1042/bj20030491
5
BakerD. J.ChildsB. G.DurikM.WijersM. E.SiebenC. J.ZhongJ.et al (2016). Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan.Nature530184–189. 10.1038/nature16932
6
BakerD. J.Perez-TerzicC.JinF.PitelK.NiederländerN. J.JeganathanK.et al (2008). Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency.Nat. Cell Biol.10825–836. 10.1038/ncb1744
7
BakerD. J.WijshakeT.TchkoniaT.LebrasseurN. K.ChildsB. G.Van De SluisB.et al (2011). Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders.Nature479232–236. 10.1038/nature10600
8
BakkenistC. J.KastanM. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation.Nature421499–506. 10.1038/nature01368
9
BartkovaJ.HořejšíZ.KoedK.KrämerA.TortF.ZlegerK.et al (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis.Nature434864–870. 10.1038/nature03482
10
BartkovaJ.RezaeiN.LiontosM.KarakaidosP.KletsasD.IssaevaN.et al (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints.Nature444633–637. 10.1038/nature05268
11
BatistaL. F.PechM. F.ZhongF. L.NguyenH. N.XieK. T.ZaugA. J.et al (2011). Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells.Nature474399–402. 10.1038/nature10084
12
BehrensA.Van DeursenJ. M.RudolphK. L.SchumacherB. (2014). Impact of genomic damage and ageing on stem cell function.Nat. Cell Biol.16201–207. 10.1038/ncb2928
13
BensonE. K.LeeS. W.AaronsonS. A. (2010). Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence.J. Cell Sci.1232605–2612. 10.1242/jcs.067306
14
BermanB. (2012). New developments in the treatment of actinic keratosis: focus on ingenol mebutate gel.Clin. Cosmet. Investig. Dermatol.5111–122. 10.2147/CCID.S28905
15
BerneburgM.GattermannN.StegeH.GreweM.VogelsangK.RuzickaT.et al (1997). Chronically ultraviolet-exposed human skin shows a higher mutation frequency of mitochondrial DNA as compared to unexposed skin and the hematopoietic system.Photochem. Photobiol.66271–275. 10.1111/j.1751-1097.1997.tb08654.x
16
BianchiM. E. (2006). DAMPs, PAMPs and alarmins: all we need to know about danger.J. Leukoc. Biol.811–5. 10.1189/jlb.0306164
17
BiranA.ZadaL.Abou KaramP.VadaiE.RoitmanL.OvadyaY.et al (2017). Quantitative identification of senescent cells in aging and disease.Aging Cell16661–671. 10.1111/acel.12592
18
BlackJ. O. (2016). Xeroderma pigmentosum.Head Neck Pathol.10139–144. 10.1007/s12105-016-0707-8
19
BlackburnE. H.ChallonerP. B. (1984). Identification of a telomeric DNA sequence in Trypanosoma brucei.Cell36447–457. 10.1016/0092-8674(84)90238-1
20
BlanpainC.FuchsE. (2006). Epidermal stem cells of the skin.Annu. Rev. Cell Dev. Biol.22339–373. 10.1146/annurev.cellbio.22.010305.104357
21
BradfordP. T.GoldsteinA. M.TamuraD.KhanS. G.UedaT.BoyleJ.et al (2011). Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair.J. Med. Genet.48168–176. 10.1136/jmg.2010.083022
22
BukowskaB.KarwowskiB. T. (2018). Actual state of knowledge in the field of diseases related with defective nucleotide excision repair.Life Sci.1956–18. 10.1016/j.lfs.2017.12.035
23
CampisiJ.d’Adda di FagagnaF. (2007). Cellular senescence: when bad things happen to good cells.Nat. Rev. Mol. Cell Biol.8729–740. 10.1038/nrm2233
24
CarreroD.Soria-VallesC.López-OtínC. (2016). Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells.Dis. Model. Mech.9719–735. 10.1242/dmm.024711
25
ChainiauxF.MagalhaesJ.-P.EliaersF.RemacleJ.ToussaintO. (2002). UVB-induced premature senescence of human diploid skin fibroblasts.Int. J. Biochem. Cell Biol.341331–1339. 10.1016/S1357-2725(02)00022-5
26
ChangJ.WangY.ShaoL.LabergeR. M.DemariaM.CampisiJ.et al (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice.Nat. Med.2278–83. 10.1038/nm.4010
27
ChenJ. H.HalesC. N.OzanneS. E. (2007). DNA damage, cellular senescence and organismal ageing: causal or correlative?Nucleic Acids Res.357417–7428. 10.1093/nar/gkm681
28
ChenQ. M.TuV. C.CataniaJ.BurtonM.ToussaintO.DilleyT. (2000). Involvement of Rb family proteins, focal adhesion proteins and protein synthesis in senescent morphogenesis induced by hydrogen peroxide.J. Cell Sci.1134087–4097.
29
ChenW.KangJ.XiaJ.LiY.YangB.ChenB.et al (2008). p53-related apoptosis resistance and tumor suppression activity in UVB-induced premature senescent human skin fibroblasts.Int. J. Mol. Med.21645–653. 10.3892/ijmm.21.5.645
30
ChojnowskiA.OngP. F.WongE. S. M.LimJ. S. Y.MutalifR. A.NavasankariR.et al (2015). Progerin reduces LAP2a-telomere association in Hutchinson-Gilford progeria.eLife41–21. 10.7554/eLife.07759
31
ChungJ. H. (2003). Photoaging in Asians.Photodermatol. Photoimmunol. Photomed.19109–121. 10.1034/j.1600-0781.2003.00027.x
32
CleaverJ. E. (1968). Defective repair replication of DNA in xeroderma pigmentosum.Nature218652–656. 10.1038/218652a0
33
ColladoM.GilJ.EfeyanA.GuerraC.SchuhmacherA. J.BarradasM.et al (2005). Tumour biology: senescence in premalignant tumours.Nature436:642. 10.1038/436642a
34
ColladoM.SerranoM. (2010). Senescence in tumours: evidence from mice and humans.Nat. Rev. Cancer1051–57. 10.1038/nrc2772
35
ContrepoisK.CoudereauC.BenayounB. A.SchulerN.RouxP. F.BischofO.et al (2017). Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression.Nat. Commun.8:14995. 10.1038/ncomms14995
36
CoppéJ.-P.BoysenM.SunC. H.WongB. J. F.KangM. K.ParkN.-H.et al (2008a). A role for fibroblasts in mediating the effects of tobacco-induced epithelial cell growth and invasion.Mol. Cancer Res.61085–1098. 10.1158/1541-7786.MCR-08-0062
37
CoppéJ.-P.PatilC. K.RodierF.SunY.MuñozD. P.GoldsteinJ.et al (2008b). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor.PLoS Biol.6:e301. 10.1371/journal.pbio.0060301
38
CoppéJ.-P.DesprezP.-Y.KrtolicaA.CampisiJ. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression.Annu. Rev. Pathol. Mech. Dis.599–118. 10.1146/annurev-pathol-121808-102144
39
CoppéJ. P.RodierF.PatilC. K.FreundA.DesprezP. Y.CampisiJ. (2011). Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype.J. Biol. Chem.28636396–36403. 10.1074/jbc.M111.257071
40
CrabbeL.JauchA.NaegerC. M.Holtgreve-GrezH.KarlsederJ. (2007). Telomere dysfunction as a cause of genomic instability in Werner syndrome.Proc. Natl. Acad. Sci. U.S.A.1042205–2210. 10.1073/pnas.0609410104
41
CrabbeL.VerdunR. E.HaggblomC. I.KarlsederJ. (2004). Defective telomere lagging strand synthesis in cells lacking WRN helicase activity.Science3061951–1953. 10.1126/science.1103619
42
D’Adda Di FagagnaF.ReaperP. M.Clay-FarraceL.FieglerH.CarrP.Von ZglinickiT.et al (2003). A DNA damage checkpoint response in telomere-initiated senescence.Nature426194–198. 10.1038/nature02118
43
DavalosA. R.KawaharaM.MalhotraG. K.SchaumN.HuangJ.VedU.et al (2013). p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes.J. Cell Biol.201613–629. 10.1083/jcb.201206006
44
DavisT.WyllieF. S.RokickiM. J.BagleyM. C.KiplingD. (2007). The role of cellular senescence in Werner syndrome: toward therapeutic intervention in human premature aging.Ann. N. Y. Acad. Sci.1100455–469. 10.1196/annals.1395.051
45
de GruijlF. R. (1999). Skin cancer and solar UV radiation.Eur. J. Cancer352003–2009. 10.1016/S0959-8049(99)00283-X
46
De SnooF. A.BishopD. T.BergmanW.Van LeeuwenI.Van Der DriftC.Van NieuwpoortF. A.et al (2008). Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families.Clin. Cancer Res.147151–7157. 10.1158/1078-0432.CCR-08-0403
47
Debacq-ChainiauxF.BorlonC.PascalT.RoyerV.EliaersF.NinaneN.et al (2005). Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-beta1 signaling pathway.J. Cell Sci.118743–758. 10.1242/jcs.01651
48
Debacq-ChainiauxF.ErusalimskyJ. D.CampisiJ.ToussaintO. (2009). Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo.Nat. Protoc.41798–1806. 10.1038/nprot.2009.191
49
DeckerM. L.ChavezE.VultoI.LansdorpP. M. (2009). Telomere length in hutchinson-gilford progeria syndrome.Mech. Ageing Dev.130377–383. 10.1016/j.mad.2009.03.001
50
DemariaM.OhtaniN.YoussefS. A.RodierF.ToussaintW.MitchellJ. R.et al (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA.Dev. Cell31722–733. 10.1016/j.devcel.2014.11.012
51
DerheimerF. A.KastanM. B. (2010). Multiple roles of ATM in monitoring and maintaining DNA integrity.FEBS Lett.5843675–3681. 10.1016/j.febslet.2010.05.031
52
DiGiovannaJ. J.KraemerK. H. (2012). Shining a light on xeroderma pigmentosum.J. Invest. Dermatol.132785–796. 10.1038/jid.2011.426
53
DimriG. P.LeeX.BasileG.AcostaM.ScottG.RoskelleyC.et al (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo.Proc. Natl. Acad. Sci. U.S.A.929363–9367. 10.1073/pnas.92.20.9363
54
DokalI. (2011). Dyskeratosis congenita.Hematol. Am. Soc. Hematol. Educ. Progr.2011480–486. 10.1182/asheducation-2011.1.480
55
DouZ.XuC.DonahueG.ShimiT.PanJ.-A.ZhuJ.et al (2015). Autophagy mediates degradation of nuclear lamina.Nature527105–109. 10.1038/nature15548
56
DreesenO.ChojnowskiA.OngP. F.ZhaoT. Y.CommonJ. E.LunnyD.et al (2013a). Lamin B1 fluctuations have differential effects on cellular proliferation and senescence.J. Cell Biol.200605–617. 10.1083/jcb.201206121
57
DreesenO.OngP. F.ChojnowskiA.ColmanA. (2013b). The contrasting roles of lamin B1 in cellular aging and human disease.Nucleus4283–290. 10.4161/nucl.25808
58
DreesenO.StewartC. L. (2011). Accelerated aging syndromes, are they relevant to normal human aging?Aging3889–895. 10.18632/aging.100383
59
DuvalC.CohenC.ChagnoleauC.FlouretV.BourreauE.BernerdF. (2014). Key regulatory role of dermal fibroblasts in pigmentation as demonstrated using a reconstructed skin model: impact of photo-aging.PLoS One9:e114182. 10.1371/journal.pone.0114182
60
FarrJ. N.XuM.WeivodaM. M.MonroeD. G.FraserD. G.OnkenJ. L.et al (2017). Targeting cellular senescence prevents age-related bone loss in mice.Nat. Med.231072–1079. 10.1038/nm.4385
61
FreundA.LabergeR.-M.DemariaM.CampisiJ. (2012). Lamin B1 loss is a senescence-associated biomarker.Mol. Biol. Cell232066–2075. 10.1091/mbc.E11-10-0884
62
GermanJ.SanzM. (eds). (2013). “Bloom’s syndrome,” inBrenner’s Encyclopedia of Genetics, (New York, NY: Elsevier), 353–355. 10.1016/B978-0-12-374984-0.00160-1
63
GhoshK.CapellB. C. (2016). The senescence-associated secretory phenotype: critical effector in skin cancer and aging.J. Invest. Dermatol.1362133–2139. 10.1016/j.jid.2016.06.621
64
GilchrestB. A.BlogF. B.SzaboG. (1979). Effects of aging and chronic sun exposure on melanocytes in human skin.J. Invest. Dermatol.73141–143. 10.1111/1523-1747.ep12581580
65
GoingJ. J.StuartR. C.DownieM.Fletcher-MonaghanA. J.KeithW. N. (2002). “Senescence-associated” beta-galactosidase activity in the upper gastrointestinal tract.J. Pathol.196394–400. 10.1002/path.1059
66
GoldsteinS.MoermanE. J.FujiiS.SobelB. E. (1994). Overexpression of plasminogen activator inhibitor type-1 in senescent fibroblasts from normal subjects and those with Werner syndrome.J. Cell. Physiol.161571–579. 10.1002/jcp.1041610321
67
GorgoulisV. G.VassiliouL.-V. F.KarakaidosP.ZacharatosP.KotsinasA.LiloglouT.et al (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions.Nature434907–913. 10.1038/nature03485
68
GotoM. (2001). Clinical characteristics of Werner syndrome and other premature aging syndromes: pattern of aging in progeroid syndromes.Gann Monogr. Cancer Res.4927–39.
69
Gray-SchopferV. C.CheongS. C.ChongH.ChowJ.MossT.Abdel-MalekZ. A.et al (2006). Cellular senescence in naevi and immortalisation in melanoma: a role for p16?Br. J. Cancer95496–505. 10.1038/sj.bjc.6603283
70
GrönnigerE.WeberB.HeilO.PetersN.StäbF.WenckH.et al (2010). Aging and chronic sun exposure cause distinct epigenetic changes in human skin.PLoS Genet.6:e1000971. 10.1371/journal.pgen.1000971
71
HaradaY. N.ShiomiN.KoikeM.IkawaM.OkabeM.HirotaS.et al (1999). Postnatal growth failure, short life span, and early onset of cellular senescence and subsequent immortalization in mice lacking the xeroderma pigmentosum group G gene.Mol. Cell. Biol.192366–2372. 10.1128/MCB.19.3.2366
72
HarwoodC. A.SwaleV. J.BatailleV. A.QuinnA. G.GhaliL.PatelS. V.et al (2001). An association between sebaceous carcinoma and microsatellite instability in immunosuppressed organ transplant recipients.J. Invest. Dermatol.116246–253. 10.1046/j.1523-1747.2001.01233.x
73
HayflickL. (1965). The limited in vitro lifetime of human diploid cell strains.Exp. Cell Res.37614–636. 10.1016/0014-4827(65)90211-9
74
HayflickL.MoorheadP. S. (1961). The serial cultivation of human diploid cell strains.Exp. Cell Res.25585–621. 10.1016/0014-4827(61)90192-6
75
HeS.SharplessN. E. (2017). Senescence in health and disease.Cell1691000–1011. 10.1016/j.cell.2017.05.015
76
HerbigU.FerreiraM.CondelL.CareyD.SedivyJ. M. (2006). Cellular senescence in aging primates.Science311:1257. 10.1126/science.1122446
77
HewittG.JurkD.MarquesF. D. M.Correia-MeloC.HardyT.GackowskaA.et al (2012). Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence.Nat. Commun.3:708. 10.1038/ncomms1708
78
HiltonB. A.LiuJ.CartwrightB. M.LiuY.BreitmanM.WangY.et al (2017). Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes.FASEB J.313882–3893. 10.1096/fj.201700014R
79
HogerT. H.KrohneG.FrankeW. W. (1988). Amino acid sequence and molecular characterization of murine lamin B as deduced from cDNA clones.Eur. J. Cell Biol.47283–290.
80
HoltD. R.KirkS. J.ReganM. C.HursonM.LindbladW. J.BarbulA. (1992). Effect of age on wound healing in healthy human beings.Surgery112293–297; discussion 297–298. 10.5555/URI:PII:003960609290223M
81
HorvathS. (2013). DNA methylation age of human tissues and cell types.Genome Biol.14:3156. 10.1186/gb-2013-14-10-r115
82
HülsA.VierkötterA.GaoW.KrämerU.YangY.DingA.et al (2016). Traffic-related air pollution contributes to development of facial lentigines: further epidemiological evidence from Caucasians and Asians.J. Invest. Dermatol.1361053–1056. 10.1016/j.jid.2015.12.045
83
HurwitzR. M.MongerL. E. (1995). Solar Keratosis: an evolving squamous cell carcinoma. benign or malignant?Dermatol. Surg.21184–184. 10.1111/j.1524-4725.1995.tb00141.x
84
IchihashiM.UedaM.BudiyantoA.BitoT.OkaM.FukunagaM.et al (2003). UV-induced skin damage.Toxicology18921–39. 10.1016/S0300-483X(03)00150-1
85
IkedaH.AidaJ.HatamochiA.HamasakiY.Izumiyama-ShimomuraN.NakamuraK. I.et al (2014). Quantitative fluorescence in situ hybridization measurement of telomere length in skin with/without sun exposure or actinic keratosis.Hum. Pathol.45473–480. 10.1016/j.humpath.2013.10.009
86
ItinP. H.SarasinA.PittelkowM. R. (2001). Trichothiodystrophy: update on the sulfur deficient brittle hair syndromes.J. Am. Acad. Dermatol.44891–920. 10.1067/mjd.2001.114294
87
IvanovA.PawlikowskiJ.ManoharanI.TuynJ.Van NelsonD. M.Singh RaiT.et al (2013). Lysosome-mediated processing of chromatin in senescence.J. Cell Biol.202129–143. 10.1083/jcb.201212110
88
JeonO. H.KimC.LabergeR. M.DemariaM.RathodS.VasserotA. P.et al (2017). Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment.Nat. Med.23775–781. 10.1038/nm.4324
89
JeyapalanJ. C.FerreiraM.SedivyJ. M.HerbigU. (2007). Accumulation of senescent cells in mitotic tissue of aging primates.Mech. Ageing Dev.12836–44. 10.1016/j.mad.2006.11.008
90
JohnsonK. E.WulffB. C.OberyszynT. M.WilgusT. A. (2013). Ultraviolet light exposure stimulates HMGB1 release by keratinocytes.Arch. Dermatol. Res.305805–815. 10.1007/s00403-013-1401-2
91
JunJ.LauL. F. (2010). The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing.Nat. Cell Biol.12676–685. 10.1038/ncb2070
92
KimK.-H.ChenC.-C.MonzonR. I.LauL. F. (2013). Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts.Mol. Cell. Biol.332078–2090. 10.1128/MCB.00049-13
93
KirklandJ. L.TchkoniaT. (2017). Cellular senescence: a translational perspective.eBiomedicine2121–28. 10.1016/j.ebiom.2017.04.013
94
KligmanA. M. (1979). Perspectives and problems in cutaneous gerontology.J. Invest. Dermatol.7339–46. 10.1111/1523-1747.ep12532758
95
KochH.WitternK. P.BergemannJ. (2001). In human keratinocytes the common deletion reflects donor variabilities rather than chronologic aging and can be induced by ultraviolet A irradiation.J. Invest. Dermatol.117892–897. 10.1046/j.0022-202X.2001.01513.x
96
KortleverR. M.HigginsP. J.BernardsR. (2006). Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence.Nat. Cell Biol.8877–884. 10.1038/ncb1448
97
KosarM.BartkovaJ.HubackovaS.HodnyZ.LukasJ.BartekJ. (2011). Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner, and follow expression of p16ink4a.Cell Cycle10457–468. 10.4161/cc.10.3.14707
98
KrishnamurthyJ.TorriceC.RamseyM. R.KovalevG. I.Al-RegaieyK.SuL.et al (2004). Ink4a/Arf expression is a biomarker of aging.J. Clin. Invest.1141299–1307. 10.1172/JCI200422475
99
KrizhanovskyV.YonM.DickinsR. A.HearnS.SimonJ.MiethingC.et al (2008). Senescence of activated stellate cells limits liver fibrosis.Cell134657–667. 10.1016/j.cell.2008.06.049
100
KubbenN.MisteliT. (2017). Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases.Nat. Rev. Mol. Cell Biol.18595–609. 10.1038/nrm.2017.68
101
KubbenN.ZhangW.WangL.VossT. C.YangJ.QuJ.et al (2016). Repression of the antioxidant NRF2 pathway in premature aging.Cell1651361–1374. 10.1016/j.cell.2016.05.017
102
KudlowB. A.StanfelM. N.BurtnerC. R.JohnstonE. D.KennedyB. K. (2008). Suppression of Proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53.Mol. Biol. Cell195238–5248. 10.1091/mbc.E08-05-0492
103
KuilmanT.MichaloglouC.MooiW. J.PeeperD. S. (2010). The essence of senescence.Genes Dev.242463–2479. 10.1101/gad.1971610
104
KuilmanT.MichaloglouC.VredeveldL. C. W.DoumaS.van DoornR.DesmetC. J.et al (2008). Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network.Cell1331019–1031. 10.1016/j.cell.2008.03.039
105
KuilmanT.PeeperD. S. (2009). Senescence-messaging secretome: SMS-ing cellular stress.Nat. Rev. Cancer981–94. 10.1038/nrc2560
106
LavkerR. M.ZhengP. S.DongG. (1986). Morphology of aged skin.Dermatol. Clin.4379–389.
107
LeeB. Y.HanJ. A.ImJ. S.MorroneA.JohungK.GoodwinE. C.et al (2006). Senescence-associated beta-galactosidase is lysosomal beta-galactosidase.Aging Cell5187–195. 10.1111/j.1474-9726.2006.00199.x
108
LesnikR. H.KligmanL. H.KligmanA. M. (1992). Agents that cause enlargement of sebaceous glands in hairless mice. I. Topical substances.Arch. Dermatol. Res.284100–105. 10.1007/BF00373378
109
LewisD. A.TraversJ. B.MachadoC.SomaniA. K.SpandauD. F. (2011). Reversing the aging stromal phenotype prevents carcinoma initiation.Aging3407–416.
110
LewisD. A.YiQ.TraversJ. B.SpandauD. F. (2008). UVB-induced senescence in human keratinocytes requires a functional insulin-like growth factor-1 receptor and p53.Mol. Biol. Cell191346–1353. 10.1091/mbc.E07-10-1041
111
LinS.-T.FuY.-H. (2009). miR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination.Dis. Model. Mech.2178–188. 10.1242/dmm.001065
112
LombardD. B.ChuaK. F.MostoslavskyR.FrancoS.GostissaM.AltF. W. (2005). DNA repair, genome stability, and aging.Cell120497–512. 10.1016/j.cell.2005.01.028
113
López-OtínC.BlascoM. A.PartridgeL.SerranoM.KroemerG. (2013). The hallmarks of aging.Cell1531194–1217. 10.1016/j.cell.2013.05.039
114
LukášováE.KovaříkA.BačíkováA.FalkM.KozubekS. (2017). Loss of lamin B receptor is necessary to induce cellular senescence.Biochem. J.474281–300. 10.1042/BCJ20160459
115
MaW.WlaschekM.Tantcheva-PoórI.SchneiderL. A.NaderiL.Razi-WolfZ.et al (2001). Chronological ageing and photoageing of the fibroblasts and the dermal connective tissue.Clin. Exp. Dermatol.26592–599. 10.1046/j.1365-2230.2001.00905.x
116
MaruG. B.GandhiK.RamchandaniA.KumarG. (2014). The role of inflammation in skin cancer.Adv. Exp. Med. Biol.816437–469. 10.1007/978-3-0348-0837-8_17
117
McCartE. A.ThangapazhamR. L.LombardiniE. D.MogS. R.PanganibanR. A. M.DicksonK. M.et al (2017). Accelerated senescence in skin in a murine model of radiation-induced multi-organ injury.J. Radiat. Res.58636–646. 10.1093/jrr/rrx008
118
MendezM. V.StanleyA.ParkH. Y.ShonK.PhillipsT.MenzoianJ. O. (1998). Fibroblasts cultured from venous ulcers display cellular characteristics of senescence.J. Vasc. Surg.28876–883. 10.1016/S0741-5214(98)70064-3
119
MeridethM. A.GordonL. B.ClaussS.SachdevV.SmithA. C. M.PerryM. B.et al (2008). Phenotype and course of hutchinson–gilford progeria syndrome.N. Engl. J. Med.358592–604. 10.1056/NEJMoa0706898
120
MichaloglouC.VredeveldL. C. W.SoengasM. S.DenoyelleC.KuilmanT.van der HorstC. M. A. M.et al (2005). BRAFE600-associated senescence-like cell cycle arrest of human naevi.Nature436720–724. 10.1038/nature03890
121
MimeaultM.BatraS. K. (2010). Recent advances on skin-resident stem/progenitor cell functions in skin regeneration, aging and cancers and novel anti-aging and cancer therapies.J. Cell. Mol. Med.14116–134. 10.1111/j.1582-4934.2009.00885.x
122
MisteliT.ScaffidiP. (2005). Genome instability in progeria: when repair gets old.Nat. Med.11718–719. 10.1038/nm0705-718
123
MitchellJ. R.WoodE.CollinsK. (1999). A telomerase component is defective in the human disease dyskeratosis congenita.Nature402551–555. 10.1038/990141
124
MontagnaW.CarlisleK. (1979). Structural changes in aging human skin.J. Invest. Dermatol.7347–53. 10.1111/1523-1747.ep12532761
125
MoritaA. (2007). Tobacco smoke causes premature skin aging.J. Dermatol. Sci.48169–175. 10.1016/j.jdermsci.2007.06.015
126
MoriwakiS. I.RayS.TaroneR. E.KraemerK. H.GrossmanL. (1996). The effect of donor age on the processing of UV-damaged DNA by cultured human cells: reduced DNA repair capacity and increased DNA mutability.Mutat. Res.364117–123. 10.1016/0921-8777(96)00029-8
127
MunroJ.BarrN. I.IrelandH.MorrisonV.ParkinsonE. K. (2004). Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock.Exp. Cell Res.295525–538. 10.1016/j.yexcr.2004.01.017
128
MuranoS.ThweattR.Shmookler ReisR. J.JonesR. A.MoermanE. J.GoldsteinS. (1991). Diverse gene sequences are overexpressed in werner syndrome fibroblasts undergoing premature replicative senescence.Mol. Cell. Biol.113905–3914. 10.1128/MCB.11.8.3905.Updated
129
NarayananD. L.SaladiR. N.FoxJ. L. (2010). Ultraviolet radiation and skin cancer.Int. J. Dermatol.49978–986. 10.1111/j.1365-4632.2010.04474.x
130
NaritaM.NũnezS.HeardE.NaritaM.LinA. W.HearnS. A.et al (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence.Cell113703–716. 10.1016/S0092-8674(03)00401-X
131
NavarroC. L.CadiñanosJ.De Sandre-GiovannoliA.BernardR.CourrierS.BoccaccioI.et al (2005). Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors.Hum. Mol. Genet.141503–1513. 10.1093/hmg/ddi159
132
NiedernhoferL. J.BohrV. A.SanderM.KraemerK. H. (2011). Xeroderma pigmentosum and other diseases of human premature aging and DNA repair: molecules to patients.Mech. Ageing Dev.132340–347. 10.1016/j.mad.2011.06.004
133
NovelliG.MuchirA.SangiuoloF.Helbling-LeclercA.D’ApiceM. R.MassartC.et al (2002). Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C.Am. J. Hum. Genet.71426–431. 10.1086/341908
134
NuciforoP. G.LuiseC.CapraM.PelosiG.D’Adda di FagagnaF. (2007). Complex engagement of DNA damage response pathways in human cancer and in lung tumor progression.Carcinogenesis282082–2088. 10.1093/carcin/bgm108
135
OgrodnikM.MiwaS.TchkoniaT.TiniakosD.WilsonC. L.LahatA.et al (2017). Cellular senescence drives age-dependent hepatic steatosis.Nat. Commun.8:15691. 10.1038/ncomms15691
136
OlovnikovA. M. (1973). A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon.J. Theor. Biol.41181–190. 10.1016/0022-5193(73)90198-7
137
OpreskoP. L.ShayJ. W. (2017). Telomere-associated aging disorders.Ageing Res. Rev.3352–66. 10.1016/j.arr.2016.05.009
138
OrtonneJ. P. (1990). Pigmentary changes of the ageing skin.Br. J. Dermatol.122(Suppl.), 21–28. 10.1111/j.1365-2133.1990.tb16121.x
139
OshimaJ.SidorovaJ. M.MonnatR. J. (2017). Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions.Ageing Res. Rev.33105–114. 10.1016/j.arr.2016.03.002
140
PassosJ. F.SaretzkiG.Von ZglinickiT. (2007). DNA damage in telomeres and mitochondria during cellular senescence: is there a connection?Nucleic Acids Res.357505–7513. 10.1093/nar/gkm893
141
PekovicV.HarborthJ.BroersJ. L. V.RamaekersF. C. S.Van EngelenB.LammensM.et al (2007). Nucleoplasmic LAP2α-lamin A complexes are required to maintain a proliferative state in human fibroblasts.J. Cell Biol.176163–172. 10.1083/jcb.200606139
142
PittayapruekP.MeephansanJ.PrapapanO.KomineM.OhtsukiM. (2016). Role of matrix metalloproteinases in photoaging and photocarcinogenesis.Int. J. Mol. Sci.17:E868. 10.3390/ijms17060868
143
PlewigG.KligmanA. M. (1978). Proliferative activity of the sebaceous glands of the aged.J. Invest. Dermatol.70314–317. 10.1111/1523-1747.ep12543478
144
PowellJ. B.DokalI.CarrR.TaibjeeS.CaveB.MossC. (2014). X-linked dyskeratosis congenita presenting in adulthood with photodamaged skin and epiphora.Clin. Exp. Dermatol.39310–314. 10.1111/ced.12272
145
QuanT.FisherG. J. (2015). Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: a mini-review.Gerontology61427–434. 10.1159/000371708
146
QuanT.QinZ.XiaW.ShaoY.VoorheesJ. J.FisherG. J. (2009). Matrix-degrading metalloproteinases in photoaging.J. Investig. Dermatol. Symp. Proc.1420–24. 10.1038/jidsymp.2009.8
147
RaddatzG.HagemannS.AranD.SöhleJ.KulkarniP. P.KaderaliL.et al (2013). Aging is associated with highly defined epigenetic changes in the human epidermis.Epigenetics Chromatin6:36. 10.1186/1756-8935-6-36
148
RavanatJ.-L.DoukiT.CadetJ. (2001). Direct and indirect effects of UV radiation on DNA and its components.J. Photochem. Photobiol. B Biol.6388–102. 10.1016/S1011-1344(01)00206-8
149
RehmanI.QuinnA. G.HealyE.ReesJ. L. (1994). High frequency of loss of heterozygosity in actinic keratoses, a usually benign disease.Lancet344788–789. 10.1016/S0140-6736(94)92343-4
150
ResslerS.BartkovaJ.NiedereggerH.BartekJ.Scharffetter-KochanekK.Jansen-DürrP.et al (2006). p16INK4A is a robust in vivo biomarker of cellular aging in human skin.Aging Cell5379–389. 10.1111/j.1474-9726.2006.00231.x
151
RittiéL.FisherG. J. (2015). Natural and sun-induced aging of human skin.Cold Spring Harb. Perspect. Med.5:a015370. 10.1101/cshperspect.a015370
152
RodierF.CoppéJ. P.PatilC. K.HoeijmakersW. A. M.MuñozD. P.RazaS. R.et al (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion.Nat. Cell Biol.11973–979. 10.1038/ncb1909
153
Rothblum-OviattC.WrightJ.Lefton-GreifM. A.McGrath-MorrowS. A.CrawfordT. O.LedermanH. M. (2016). Ataxia telangiectasia: a review.Orphanet J. Rare Dis11:159. 10.1186/s13023-016-0543-7
154
SadaieM.SalamaR.CarrollT.TomimatsuK.ChandraT.YoungA. R.et al (2013). Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence.Genes Dev.271800–1808. 10.1101/gad.217281.113
155
ScaffidiP.MisteliT. (2006). Lamin A-dependent nuclear defects in human aging.Science3121059–1063. 10.1126/science.1127168
156
SchroederP.SchiekeS. M.MoritaA. (2006). “Premature skin aging by infrared radiation, tobacco smoke and ozone,” inSkin Aging, edsGilchrestB. A.KrutmannJ. (Berlin: Springer), 45–53. 10.1007/3-540-32953-6_5
157
SerranoM.LinA. W.McCurrachM. E.BeachD.LoweS. W. (1997). Oncogenic RAS provokes premature cell senescence associated with accumulation of p53 and p16(INK4a).Cell88593–602. 10.1016/S0092-8674(00)81902-9
158
SeverinoJ.AllenR. G.BalinS.BalinA.CristofaloV. J. (2000). Is beta-galactosidase staining a marker of senescence in vitro and in vivo?Exp. Cell Res.257162–171. 10.1006/excr.2000.4875
159
ShahP. P.DonahueG.OtteG. L.CapellB. C.NelsonD. M.CaoK.et al (2013). Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape.Genes Dev.271787–1799. 10.1101/gad.223834.113
160
SharplessN. E.SherrC. J. (2015). Forging a signature of in vivo senescence.Nat. Rev. Cancer15397–408. 10.1038/nrc3960
161
SherrattM. J. (2013). Age-related tissue stiffening: cause and effect.Adv. Wound Care211–17. 10.1089/wound.2011.0328
162
ShimiT.Butin-IsraeliV.AdamS. A.HamanakaR. B.GoldmanA. E.LucasC. A.et al (2011). The role of nuclear lamin B1 in cell proliferation and senescence.Genes Dev.252579–2593. 10.1101/gad.179515.111
163
ShumakerD. K.DechatT.KohlmaierA.AdamS. A.BozovskyM. R.ErdosM. R.et al (2006). Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging.Proc. Natl. Acad. Sci. U.S.A.1038703–8708. 10.1073/pnas.0602569103
164
ShusterS.BlackM. M.McVitieE. (1975). The influence of age and sex on skin thickness, skin collagen and density.Br. J. Dermatol.93639–643. 10.1111/j.1365-2133.1975.tb05113.x
165
SiegelJ. A.KorgavkarK.WeinstockM. A. (2017). Current perspective on actinic keratosis: a review.Br. J. Dermatol.177350–358. 10.1111/bjd.14852
166
SoloveiI.WangA. S.ThanischK.SchmidtC. S.KrebsS.ZwergerM.et al (2013). LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation.Cell152584–598. 10.1016/j.cell.2013.01.009
167
SoufirN.MolèsJ. P.VilmerC.MochC.VerolaO.RivetJ.et al (1999). P16 UV mutations in human skin epithelial tumors.Oncogene185477–5481. 10.1038/sj.onc.1202915
168
StewartC.BurkeB. (1987). Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B.Cell51383–392. 10.1016/0092-8674(87)90634-9
169
SugimotoM.YamashitaR.UedaM. (2006). Telomere length of the skin in association with chronological aging and photoaging.J. Dermatol. Sci.4343–47. 10.1016/j.jdermsci.2006.02.004
170
SuramA.KaplunovJ.PatelP. L.RuanH.CeruttiA.BoccardiV.et al (2012). Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions.EMBO J.312839–2851. 10.1038/emboj.2012.132
171
TakaiH.SmogorzewskaA.De LangeT. (2003). DNA damage foci at dysfunctional telomeres.Curr. Biol.131549–1556. 10.1016/S0960-9822(03)00542-6
172
TanW. H.BarisH.RobsonC. D.KimonisV. E. (2005). Cockayne syndrome: the developing phenotype.Am. J. Med. Genet. A135214–216. 10.1002/ajmg.a.30731
173
TelgenhoffD.ShrootB. (2005). Cellular senescence mechanisms in chronic wound healing.Cell Death Differ.12695–698. 10.1038/sj.cdd.4401632
174
Tokarsky-AmielR.AzazmehN.HelmanA.SteinY.HassanA.MalyA.et al (2013). Dynamics of senescent cell formation and retention revealed by p14ARFinduction in the epidermis.Cancer Res.732829–2839. 10.1158/0008-5472.CAN-12-3730
175
TsurumiA.LiW. X. (2012). Global heterochromatin loss: a unifying theory of aging?Epigenetics7680–688. 10.4161/epi.20540
176
Vande BergJ. S.RobsonM. C. (2003). Arresting cell cycles and the effect on wound healing.Surg. Clin. North Am.83509–520. 10.1016/S0039-6109(02)00195-0
177
Vande BergJ. S.RoseM. A.Haywood-ReidP. L.RudolphR.PayneW. G.RobsonM. C. (2005). Cultured pressure ulcer fibroblasts show replicative senescence with elevated production of plasmin, plasminogen activator inhibitor-1, and transforming growth factor-β1.Wound Repair Regen.1376–83. 10.1111/j.1067-1927.2005.130110.x
178
VaraniJ.DameM. K.RittieL.FligielS. E. G.KangS.FisherG. J.et al (2006). Decreased collagen production in chronologically aged skin.Am. J. Pathol.1681861–1868. 10.2353/ajpath.2006.051302
179
VaraniJ.SchugerL.DameM. K.LeonardC.FligielS. E. G.KangS.et al (2004). Reduced fibroblast interaction with intact collagen as a mechanism for depressed collagen in synthesis in photodamaged skin.J. Invest. Dermatol.1221471–1479. 10.1111/j.0022-202X.2004.22614.x
180
VidakS.FoisnerR. (2016). Molecular insights into the premature aging disease progeria.Histochem. Cell Biol.145401–417. 10.1007/s00418-016-1411-1
181
VierkötterA.SchikowskiT.RanftU.SugiriD.MatsuiM.KrämerU.et al (2010). Airborne particle exposure and extrinsic skin aging.J. Invest. Dermatol.1302719–2726. 10.1038/jid.2010.204
182
VijgJ.SuhY. (2013). Genome Instability and Aging.Annu. Rev. Physiol.75645–668. 10.1146/annurev-physiol-030212-183715
183
WaaijerM.GunnD. A.HeemstD.Van SlagboomP. E.JohnM.DirksR. W.et al (2018). Do senescence markers correlate in vitro and in situ within individual human donors?Aging10278–289. 10.18632/aging.101389
184
WaaijerM.ParishW. E.StrongitharmB. H.van HeemstD.SlagboomP. E.de CraenA. J. M.et al (2012). The number of p16INK4a positive cells in human skin reflects biological age.Aging Cell11722–725. 10.1111/j.1474-9726.2012.00837.x
185
WajapeyeeN.SerraR. W.ZhuX.MahalingamM.GreenM. R. (2008). Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7.Cell132363–374. 10.1016/j.cell.2007.12.032
186
WangA. S.OngP. F.ChojnowskiA.ClavelC.DreesenO. (2017). Loss of lamin B1 is a biomarker to quantify cellular senescence in photoaged skin.Sci. Rep.7:15678. 10.1038/s41598-017-15901-9
187
WatsonJ. D. (1972). Origin of concatemeric T7DNA.Nat. New Biol.239197–201. 10.1038/newbio239197a0
188
WatsonR. E. B.GibbsN. K.GriffithsC. E. M.SherrattM. J. (2014). Damage to skin extracellular matrix induced by UV exposure.Antioxid. Redox Signal.211063–1077. 10.1089/ars.2013.5653
189
WheatonK.CampuzanoD.MaW.SheinisM.HoB.BrownG. W.et al (2017). Progerin-induced replication stress facilitates premature senescence in hutchinson-gilford progeria syndrome.Mol. Cell. Biol.37e659-16. 10.1128/MCB.00659-16
190
WileyC. D.VelardeM. C.LecotP.LiuS.SarnoskiE. A.FreundA.et al (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype.Cell Metab.23303–314. 10.1016/j.cmet.2015.11.011
191
WlaschekM.Tantcheva-PoórI.NaderiL.MaW.SchneiderL. A.Razi-WolfZ.et al (2001). Solar UV irradiation and dermal photoaging.J. Photochem. Photobiol. B Biol.6341–51. 10.1016/S1011-1344(01)00201-9
192
WoodA. M.DanielsenJ. M. R.LucasC. A.RiceE. L.ScalzoD.ShimiT.et al (2015). TRF2 and lamin A/C interact to facilitate the functional organization of chromosome ends.Nat. Commun.5:5467. 10.1038/ncomms6467
193
XuG.SnellmanE.BykovV. J.JansenC. T.HemminkiK. (2000). Effect of age on the formation and repair of UV photoproducts in human skin in situ.Mutat. Res.459195–202. 10.1016/S0921-8777(99)00069-5
194
XuM.PalmerA. K.DingH.WeivodaM. M.PirtskhalavaT.WhiteT. A.et al (2015). Targeting senescent cells enhances adipogenesis and metabolic function in old age.elife4:e12997. 10.7554/eLife.12997
195
YamadaM.UdonoM. U.HoriM.HiroseR.SatoS.MoriT.et al (2006). Aged human skin removes UVB-induced pyrimidine dimers from the epidermis more slowly than younger adult skin in vivo.Arch. Dermatol. Res.297294–302. 10.1007/s00403-005-0618-0
196
YamadaS.MaruyamaI. (2007). HMGB1, a novel inflammatory cytokine.Clin. Chim. Acta37536–42. 10.1016/j.cca.2006.07.019
197
YosefR.PilpelN.Tokarsky-AmielR.BiranA.OvadyaY.CohenS.et al (2016). Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL.Nat. Commun.7:11190. 10.1038/ncomms11190
198
YouY. H.LeeD. H.YoonJ. H.NakajimaS.YasuiA.PfeiferG. P. (2001). Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells.J. Biol. Chem.27644688–44694. 10.1074/jbc.M107696200
199
YuC. E.OshimaJ.FuY. H.WijsmanE. M.HisamaF.AlischR.et al (1996). Positional cloning of the Werner’s syndrome gene.Science72258–262. 10.1126/science.272.5259.258
200
ZhangJ.LianQ.ZhuG.ZhouF.SuiL.TanC.et al (2011). A human iPSC model of hutchinson gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects.Cell Stem Cell831–45. 10.1016/j.stem.2010.12.002
201
ZhangW.LiJ.SuzukiK.QuJ.WangP.ZhouJ.et al (2015). A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging.Science3481160–1163. 10.1126/science.aaa1356
202
ZhuY.TchkoniaT.Fuhrmann-StroissniggH.DaiH. M.LingY. Y.StoutM. B.et al (2016). Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors.Aging Cell15428–435. 10.1111/acel.12445
203
ZindyF.QuelleD. E.RousselM. F.SherrC. J. (1997). Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging.Oncogene15203–211. 10.1038/sj.onc.1201178
204
ZouL. (2007). Single- and double-stranded DNA: building a trigger of ATR-mediated DNA damage response.Genes Dev.21879–885. 10.1101/gad.1550307
205
ZouboulisC. C.BoschnakowA. (2001). Chronological ageing and photoageing of the human sebaceous gland.Clin. Exp. Dermatol.26600–607. 10.1046/j.1365-2230.2001.00894.x
Summary
Keywords
senescence, skin, aging, biomarkers, photoaging, cancer
Citation
Wang AS and Dreesen O (2018) Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 9:247. doi: 10.3389/fgene.2018.00247
Received
26 April 2018
Accepted
22 June 2018
Published
23 August 2018
Volume
9 - 2018
Edited by
Anis Larbi, Singapore Immunology Network (A∗STAR), Singapore
Reviewed by
George A. Garinis, Foundation for Research and Technology – Hellas, Greece; Kaoru Tominaga, Jichi Medical University, Japan
Updates
Copyright
© 2018 Wang and Dreesen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oliver Dreesen, oliver.dreesen@sris.a-star.edu.sg
This article was submitted to Genetics of Aging, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.