Abstract
Cornelia de Lange Syndrome (CdLS) is a well described multiple malformation syndrome caused by alterations in genes encoding subunits or regulators of the cohesin complex. In approximately 70% of CdLS patients, pathogenic NIPBL variants are detected and 15% of them are predicted to affect splicing. Moreover, a large portion of genetic variants in NIPBL was shown to be somatic mosaicism. Here we report two family members with different expression of the CdLS phenotype. In both individuals, a c.869-2A>G (r.869_1495del) substitution was detected, affecting a conserved splice-acceptor site. Deep sequencing revealed the presence of somatic mosaicism in the mother. The substitution was detected in 23% of the sequencing reads using DNA derived from blood samples and 51% in DNA from buccal swabs. The analysis of blood DNA of the son excluded the presence of somatic mosaicism. Correlation of molecular and clinical data revealed that various distribution of genetic alteration in different cell types had an impact on the expression of observed clinical features in both individuals.
Introduction
Cornelia de Lange syndrome (CdLS; OMIM: 1227470, 300590, 610759, 614701, and 300882) is a rare genetic disorder characterized by a highly variable clinical presentation. It is caused by genetic alterations in NIPBL, SMC1A, SMC3, HDAC8, and RAD21 (Krantz et al., 2004; Tonkin et al., 2004; Musio et al., 2006; Deardorff et al., 2007, 2012a,b). To date, genetic alterations in NIPBL have been detected in approximately 70% of CdLS patients (OMIM: 608667) (Nizon et al., 2016). Genetic variants affecting gene splicing account for 15% of NIPBL mutations (Mannini et al., 2013; Teresa-Rodrigo et al., 2016).
Castronovo et al. (2010) reported the first patient with a mosaic alteration in NIPBL. Additional studies have shown that somatic mosaicism is very frequent in CdLS patients (Huisman et al., 2013). More recently, familial mosaic cases have been reported (Parenti et al., 2016).
Here, we report a correlation of clinical and molecular data of two family members with a genetic variant in NIPBL affecting a conserved splice-acceptor site. Both Individuals were previously reported as patients harboring a heterozygous germline mutation (Teresa-Rodrigo et al., 2014). However, additional molecular analysis revealed this variant to be somatic mosaicism in the mother, thus explaining her milder CdLS phenotype in comparison to her son, who is not mosaic for the disease-causing variant.
Background
Case Report
Individual 1 is a 41 years old female admitted to hospital at the age of 36 years. She was the second born child of healthy, unrelated, mid-30 parents with no family history of congenital defects. She was born at 38 weeks of uncomplicated gestation. Her birth weight, birth length, and the head circumference were 2900 g (-0.65 SD), 48 cm (-1.04 SD), and 30 cm (-3.31 SD), respectively. She presented with hirsutism and mild musculoskeletal anomalies were also noted including small hands and feet, together with bilateral clinodactyly of the fifth finger. A mild craniofacial dysmorphism was also observed (Figure 1A and Table 1).
FIGURE 1
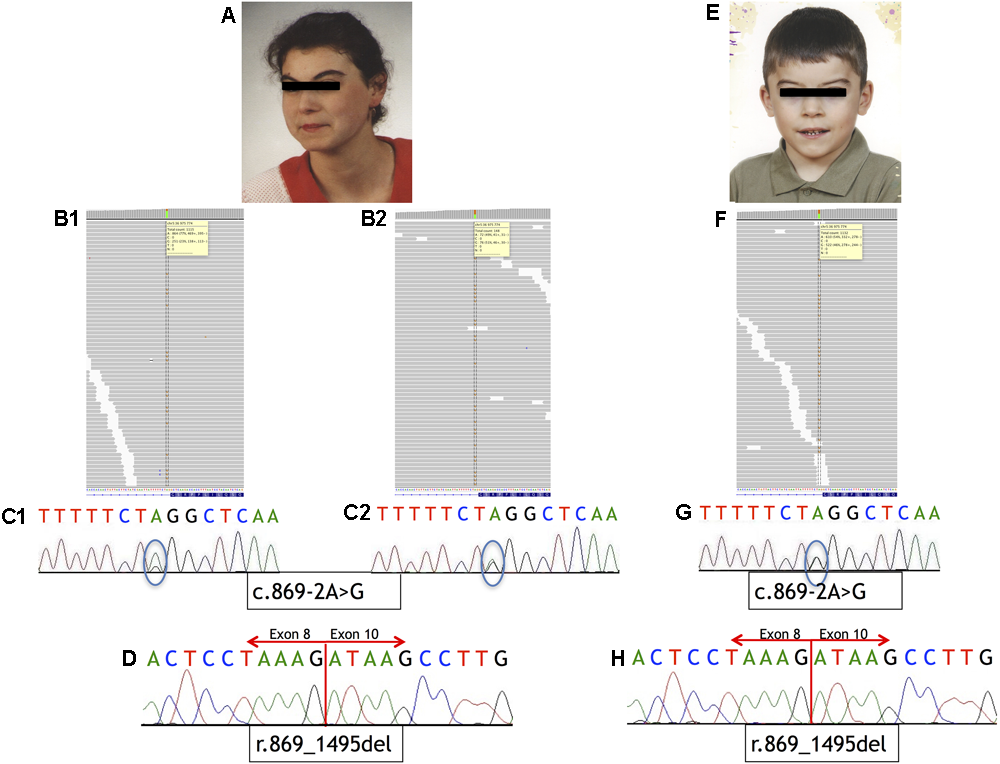
Phenotype of the patients and sequencing results. (A–D) Analysis of Individual 1. (A) Phenotype of Individual 1. (B) IGV view of NGS sequence analysis of exon 9 – intron 8 boundaries of the NIPBL gene. (B1) A splice-site variant identified in DNA from blood sample in NIPBL (alternative allele shown in orange) in 23% of the reads (gray bars). (B2) A splice-site variant identified in DNA from buccal swab sample in NIPBL (alternative allele shown in orange) in approximately 51% of the reads (gray bars). (C1) Sequence analysis of DNA from blood sample of NIPBL gene. The mutation is marked with a circle. (C2) Sequence analysis of DNA from buccal swab sample of NIPBL gene. The mutation is marked with a circle. (D) Sequence analysis of cDNA NIPBL gene. Panel exhibit an in-frame deletion of exon 9 of NIPBL gene in Individual 1. (E–H) Analysis of Individual 2. (E) Phenotype of Individual 2. (F) IGV view of NGS sequence analysis of exon 9 – intron 8 boundaries of the NIPBL gene. A splice-site variant identified in DNA from blood sample in NIPBL (alternative allele shown in orange) in 46% of the reads (gray bars). (G) Sequence analysis of DNA NIPBL gene. The mutation is marked with a circle. (H) Sequence analysis of cDNA NIPBL gene. Panel exhibit an in-frame deletion of exon 9 of NIPBL gene in Individual 1.
Table 1
| Individual 1 CdLS02M | Individual 2 CdLS02 | ||
|---|---|---|---|
| Age | 41 years | 8 years | |
| Gender | Female | Male | |
| System | Specific feature | ||
| Facial dysmorphism | Synophrys | + | + |
| Long eyelashes | + | + | |
| Short nose | + | + | |
| Anteverted nostrils | + | + | |
| Long philtrum | + | + | |
| Broad or depressed nasal bridge | + | + | |
| Thin lips with downturned corners | + | + | |
| Low set ears | + | + | |
| Growth | Birth weight (pregnancy week) | 2900 g (38 hbd) | 2870 g (38 hbd) |
| Birth length | 48 cm | 49 cm | |
| Birth head circumference | 30 cm | 32 cm | |
| Postnatal microsomy | + | + | |
| Development | Developmental delays or intellectual disability | + | + |
| Learning disability | + | + | |
| Behavior | Attention deficit disorder | - | + |
| Hyperactivity | - | + | |
| Self-injurious behavior | - | + | |
| Autistic-like features | - | + | |
| Musculoskeletal | Small hands and feet | + | + |
| Fifth finger clinodactyly | + | + | |
| Short first knuckle/proximally placed thumb | + | + | |
| Cardiac Gastrointestinal | Type of malformation | ||
| Poor feeding | + | + | |
| GER | - | + | |
| Constipation | - | + | |
| Skin | Hirsutism | + | + |
| Deafness or hearing loss | + | + (20 dB) | |
| Urogenital | Cryptorchidism | - | + |
Features of two related CdLS patients.
“+,” feature present; “-,” feature not present; GER, gastro-oesophageal reflux; hbd, week of pregnancy.
At infancy she exhibited a slight hypertonia, poor sucking reflex and a poor weight gain. At the age of 2 years she started to suffer from constipation. She was able to sit unsupported around the age of 8 months, walk independently around the age of 13 months. She started to pronounce syllables around the age of 2 years, used simple words around the age of 3 years, but only at the age of 4 years she started to speak with full sentences. She finished regular primary and secondary school with a great help from her parents. She is a very nice person, easily making contact with other people. Currently, with her parents’ help, she is taking care of her son (Individual 2). Her first pregnancy terminated with a miscarriage at the 23rd week of gestation, but prenatal genetic tests were not performed in that case. All clinical features are summarized in Table 1.
Individual 2, the 8 years old son of Individual 1, was the first-born child of 33-year-old mother and 34-year-old healthy father. The pregnancy was complicated by gestational diabetes, regulated by diet and insulin. He was born at 38 weeks of gestation by cesarean section due to placental insufficiency. His birth weight, birth length, and the head circumference were 2870 g (-1.01 SD), 49 cm (-0.91 SD), and 32 cm (-2.07 SD), respectively. His hands and feet were small but not malformed. Bilateral clinodactyly of the fifth finger was evident. He presented with hirsutism and mild craniofacial dysmorphism (Figure 1E and Table 1).
He demonstrated cryptorchidism and inguinal hernia. In addition, transient hypoglycemia, icterus, and lack of the sucking reflex were observed. During infancy, he had clinical features compatible with gastroesophageal reflux disease (GERD), although it could not be confirmed by pH-metry and endoscopy. The audiometry and auditory brainstem response (ABR) performed at the age of 1 year revealed a mild bilateral sensorineural hearing loss (20 dB). Hand roentgenograms performed in the age of 3 years revealed a delayed bone age.
The patient exhibited motor and language delays. He was able to sit unsupported around the age of 12 months and to walk independently around the age of 18 months. He started to speak syllables around the age of 2 years, simple words around the age of 4 years. At the present time, he still has limited speech abilities. He presents with autistic-like features, some episodes of aggression, and self-injurious behavior. All clinical features are summarized in Table 1.
Materials and Methods
Both blood samples and buccal swabs were obtained after written informed consent from both individuals. Genomic DNA was isolated from peripheral blood lymphocytes and oral mucosa epithelial cells using Genomic DNA from blood kit and Genomic DNA from tissue (Macherey-Nagel) according to the manufacturer’s protocols.
Targeted gene-panel via deep sequencing analysis was performed by using NimbleGen SeqCap EZ HyperCap (Roche Diagnostics) and MiSeq (Illumina). The custom designed panel was spanning 92 kb of the selected genomic sequences including: NIPBL, SMC1A, SMC3, RAD21, HDAC8, STAG1, SGOL1, PDS5A, PTTG1, TAF6, ESCO2, WAPAL, CDCA5, KMT2A, DDX11, ESPL1, PDS5B, PLK1, AURKB, ESCO1, MAU2, ATRX, STAG2, and RECQL4 genes. Probes were designed to enrich exons and 25 bp of flanking introns. The analysis was performed using IGV (Broad Institute) and Alamut (Interactive Biosoftware) softwares. The nomenclature of the alteration was based on NIPBL mRNA sequence NM_133433.3 according to the HGVS recommendations (den Dunnen et al., 2016). Presence of NIPBL alteration was confirmed by independent PCR followed by bidirectional Sanger sequencing.
Total RNA was extracted from blood samples of Individuals 1 and 2 using the Total RNA Mini (A&A Biotechnology). GoScript Reverse Transcriptase (Promega) was applied for cDNA synthesis. For both Individuals, exons 8–10 of NIPBL gene were amplified. PCR products were excised from agarose gel and bidirectionally sequenced. PCR products were sequenced using BigDye Terminator v3.1 Cycle Sequencing Kit and 3100 Series Genetic Analyzer (Thermo Fisher Scientific). Electropherograms were analyzed with Sequencher v.10 DNA Software (Gene Codes).
Results and Discussion
Mutational analysis performed by deep sequencing revealed the presence of a single-nucleotide substitution (c.869-2A>G) in NIPBL in both patients. In Individual 1, this variant was found in 23% of the reads (251/1115) in DNA isolated from blood and in 51% of the reads (76/148) in DNA isolated from buccal swab. From Individual 2, only blood DNA was available for the study, and substitution was detected in 46% of the sequencing reads (522/1132). Additional molecular studies performed on the cDNA of both Individuals confirmed that this variant causes aberrant splicing of NIPBL transcript, as previously reported (Teresa-Rodrigo et al., 2014). Namely, the intronic variant c.869-2A>G resulted in the in-frame deletion of exon 9 [p.Gly290_Lys498del]. Mutational analysis results are shown in Figure 1. Additional molecular analysis could exclude the presence of the NIPBL variant in the father of Individual 2 (data not shown).
Here, we report a pathogenic variant detected within a conserved splice-acceptor of NIPBL in two family members. Although the variant was previously described as heterozygous germline (Teresa-Rodrigo et al., 2014), we could give experimental evidence of somatic mosaicism in the mother (Patient P3B).
The single nucleotide substitution (c.869-2A>G) within intron 8 of NIPBL results in the in-frame deletion of exon 9 and is predicted to cause an altered NIPBL protein lacking 209 amino acids. In Individual 1, the variant was detected in 23 and 51% of sequencing reads in DNA derived from blood and buccal swab samples, respectively. In Individual 2, mutational analysis on blood DNA allowed to detect a substitution in 46% of the reads.
Acuna-Hidalgo et al. (2015) proposed two molecular mechanism responsible for induction of somatic mosaicism: the first as inherited from one of the parents with low-level of mosaicism, a second due to post-zygotic change in the embryo.
We were not able to determine underlying mechanism in Individual 1 since molecular analysis was not performed in her parents. However, both parents were healthy individuals with no physical or intellectual symptoms of CdLS. They both have negative family history of congenital defects. Therefore, with high probability we can omit the inherited form of NIPBL alteration.
The more likely mechanism for Individual 1 was a ‘post-zygotic’ mutational event in the embryo. The presence of a genetic alteration in cells from two different germ layers, peripheral blood lymphocytes (mesoderm) and oral mucosa epithelial cells (ectoderm), in mosaic stage indicate that alteration could appear after zygotic stage but before gastrulation process. Because only two tissues were tested, it is difficult to confidently place a time on when and where mosaicism occurred.
In general, various levels of specific alteration in different tissues were previously described as molecular mechanism responsible for phenotypic heterogeneity of CdLS patients (Pozojevic et al., 2017). In Individual 1, it might be possible that low levels of aberrant NIPBL in neural tissue could explain the mild intellectual disability. However, these assumptions are contrast to the embryological data. Indeed, the oral epithelium, heterozygous for the variant, derives from embryonic ectoderm which is also the precursor for the neural tube leading to brain development. Thus, different level of alteration in tissues from the same germ layer cannot be excluded, and such a situation could be observed in Individual 1. However, in particular tissues the presence of a high number of mutant cells might not be tolerated and the number of cells carrying the mutation may be determined by how well mutant cells survived and proliferated during development of that tissue, a phenomenon known as negative selection against the mutation (Baquero-Montoya et al., 2014). In patients, the mosaic NIPBL alteration led to mild intellectual disability in Individual 1, in comparison to the more severely affected Individual 2.
In Individual 2, the inherited substitution is expressed at the same level in all cells resulting in more severe physical and intellectual manifestations than those observed in his mother.
Concluding Remarks
Phenotypic variability among CdLS patients, even in the presence of the same type of pathogenic mutation, can be explained by different levels of expression of a genetic variant in tissues from the same or different germ layers. Based on this study, we conclude that at least in some CdLS patients a high sensitivity molecular techniques should be applied and different tissues should be tested to explain the molecular basis of disease in these individuals.
Statements
Ethics statement
The study was approved by the Ethical Committee of the Medical University of Gdańsk, Poland (NKEBN/395/2014; NKEBN/395-504/2014; and NKEBN/395-288/2014). Written informed consent to participate in the study was obtained from patients’ parents. Written informed consent for publication was obtained from patient’ parents (in the case of Individual 2) and patient (in the case of Individual 1).
Author contributions
JW classified the patients in this study. NK and BW conceived and designed the experiments and analyzed the data. NK and AK performed the experiments. NK, AK, IP, JW, BW, and FK prepared the manuscript. All authors read and approved the final manuscript.
Funding
This study was funded by grant from the National Science Centre of Poland, project no. UMO-2014/15/B/NZ5/03503.
Acknowledgments
We would like to thank the patients and their family for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Acuna-Hidalgo R. Bo T. Kwint M.P. de Vorst M. Ponelli M. Veltman J.A. et al (2015). Post-zygotic point mutations are an underrecognized source of de novo genomic variantion.Am. J. Med. Genet.9767–74. 10.1016/j.ajhg.2015.05.008
2
Baquero-Montoya C. Gil-Rodríguez M. C. Braunholz D. Teresa-Rodrigo M. E. Obieglo C. Gener B. et al (2014). Somatic mosaicism in Cornelia de Lange syndrome patient with NIPBL mutation identified by different next generation sequencing approaches. Clin. Genet. 86595–597. 10.1111/cge.12333
3
Castronovo P. Delahaye-Duriez A. Gervasini C. Azzollini J. Minier F. Russo S. et al (2010). Somatic mosaicism in Cornelia de Lange syndrome: a further contributor to the wide clinical expressivity?Clin. Genet.78560–564. 10.1111/j.1399-0004.2010.01408.x
4
Deardorff M. A. Bando M. Nakato R. Watrin E. Itoh T. Minamino M. et al (2012a). HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle.Nature489313–317. 10.1038/nature11316
5
Deardorff M. A. Kaur M. Yaeger D. Rampuria A. Korolev S. Pie J. et al (2007). Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de lange syndrome with predominant mental retardation.Am. J. Hum. Genet.80485–494. 10.1086/511888
6
Deardorff M. A. Wilde J. J. Albrecht M. Dickinson E. Tennstedt S. Braunholz D. et al (2012b). RAD21 mutations cause a human cohesinopathy.Am. J. Hum. Genet.901014–1027. 10.1016/j.ajhg.2012.04.019
7
den Dunnen J. T. Dalgleish R. Maglott D. R. Hart R. K. Greenblatt M. S. McGowan-Jordan J. et al (2016). HGVS recommendations for the description of sequence variants: 2016 Update.Hum. Mutat.37564–569. 10.1002/humu.22981
8
Huisman S. Redeker E. Maas S. Mannens M. Hennekam R. (2013). High rate of mosaicism in individuals with Cornelia de Lange syndrome.J. Med. Genet.50339–344. 10.1136/jmedgenet-2012-101477
9
Krantz I. D. McCallum J. DeScipio C. Kaur M. Gillis L. A. Yaeger D. et al (2004). Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B.Nat. Genet.36631–635. 10.1038/ng1364
10
Mannini L. Cucco F. Quarantotti V. Krantz I. D. Musio A. (2013). Mutation spectrum and genotype–phenotype correlation in Cornelia de Lange syndrome.Hum. Mutat.341589–1596. 10.1002/humu.22430
11
Musio A. Selicorni A. Focarelli M. L. Gervasini C. Milani D. Russo S. et al (2006). X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations.Nat. Genet.38528–530. 10.1038/ng1779
12
Nizon M. Henry M. Michot C. Baumann C. Bazin A. Bessières B. et al (2016). A series of 38 novel germline and somatic mutations of NIPBL in Cornelia de Lange syndrome.Clin. Genet.89584–589. 10.1111/cge.12720
13
Parenti I. Gervasini C. Pozojevic J. Wendt K. S. Watrin E. Azzollini J. et al (2016). Expanding the clinical spectrum of the ‘HDAC8-phenotype’ – implications for molecular diagnostics, counseling and risk prediction.Clin. Genet.89564–573. 10.1111/cge.12717
14
Pozojevic J. Parenti I. Graul-Neumann L. Ruiz Gil S. Watrin E. Wendt K.S. et al (2017). Novel mosaic variants in two patients with Cornelia de Lange syndrome.Eur. J. Med. Genet.10.1016/j.ejmg.2017.11.004 [Epub ahead of print].
15
Teresa-Rodrigo M. E. Eckhold J. Puisac B. Dalski A. Gil-Rodríguez M. C. Braunholz D. et al (2014). Functional characterization of NIPBL physiological splice variants and eight splicing mutations in patients with Cornelia de Lange syndrome.Int. J. Mol. Sci.1510350–10364. 10.3390/ijms150610350
16
Teresa-Rodrigo M. E. Eckhold J. Puisac B. Pozojevic J. Parenti I. Baquero-Montoya C. et al (2016). Identification and functional characterization of two intronic NIPBL mutations in two patients with Cornelia de Lange syndrome.Biomed Res. Int.2016:8742939. 10.1155/2016/8742939
17
Tonkin E. T. Wang T. J. Lisqo S. Bamshad M. J. Strachan T. (2004). NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome.Nat. Genet.36636–641. 10.1038/ng1363
Summary
Keywords
Cornelia de Lange syndrome, CdLS, NIPBL gene, mosaic variant, family case, deep sequencing
Citation
Krawczynska N, Kuzniacka A, Wierzba J, Parenti I, Kaiser FJ and Wasag B (2018) Mosaic Intronic NIPBL Variant in a Family With Cornelia de Lange Syndrome. Front. Genet. 9:255. doi: 10.3389/fgene.2018.00255
Received
10 April 2018
Accepted
25 June 2018
Published
13 July 2018
Volume
9 - 2018
Edited by
Ramu Elango, King Abdulaziz University, Saudi Arabia
Reviewed by
Eaaswarkhanth Muthukrishnan, Dasman Diabetes Institute, Kuwait; Julia Horsfield, University of Otago, New Zealand
Updates
Copyright
© 2018 Krawczynska, Kuzniacka, Wierzba, Parenti, Kaiser and Wasag.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bartosz Wasag, bwasag@gumed.edu.pl
This article was submitted to Genetic Disorders, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.