 Dóra Tombácz
Dóra Tombácz Zsolt Balázs1
Zsolt Balázs1 Zsolt Csabai
Zsolt Csabai Zsolt Boldogkői
Zsolt Boldogkői- 1Department of Medical Biology, Faculty of Medicine, University of Szeged, Szeged, Hungary
- 2Department of Genetics, School of Medicine, Stanford University, Stanford, CA, United States
Long-read sequencing (LRS) techniques are very recent advancements, but they have already been used for transcriptome research in all of the three subfamilies of herpesviruses. These techniques have multiplied the number of known transcripts in each of the examined viruses. Meanwhile, they have revealed a so far hidden complexity of the herpesvirus transcriptome with the discovery of a large number of novel RNA molecules, including coding and non-coding RNAs, as well as transcript isoforms, and polycistronic RNAs. Additionally, LRS techniques have uncovered an intricate meshwork of transcriptional overlaps between adjacent and distally located genes. Here, we review the contribution of LRS to herpesvirus transcriptomics and present the complexity revealed by this technology, while also discussing the functional significance of this phenomenon.
Introduction
Short-read sequencing (SRS) technologies have revolutionized transcriptome studies because of their high throughput nature, precision, sensitivity, and cost-effectiveness. However, this technology faces some limitations, which include difficulties in the assembly of low-complexity nucleic acid stretches, in the identification of multi-spliced transcripts, in distinguishing between overlapping transcripts, and in the detection of multigenic transcripts (Steijger et al., 2013). Long-read sequencing (LRS) can overcome these problems through its greater efficiency in de novo assembly, in identification of RNA isoforms, including length and splice variants, as well as overlapping and polycistronic transcripts. However, this approach has its own limitations, such as a higher insertion/deletion (indel) error rate, along with lower throughput and higher per base sequencing costs. There are currently two LRS techniques available that are capable of sequencing full-length transcripts, the Single Molecule, Real-time sequencing from Pacific Biosciences (PacBio) and nanopore sequencing from Oxford Nanopore Technologies (ONT). The zero-mode waveguides (ZMW) utilized by PacBio allow for the detection of fluorescent signals emitted during the incorporation of a single labeled nucleotide. The DNA-polymerase, which is fixed to the ZMW, reads the circularized template multiple times. The complete sequence generated from a template is then merged with bioinformatics tools, and as a consequence, the accuracy of the consensus sequence (reads of insert; ROI) is dependent upon the number of passes the polymerase was able to make on the template (Rhoads and Au, 2015). Sequel, the newest platform recently released by PacBio, boasts a much higher throughput than the previous platforms were able to produce (Lin and Liao, 2015). The passive loading of the RSII platform favored reads with lengths of 1–2 kb (Loomis et al., 2013), necessitating size-selection for the extensive characterization of transcriptomes. The Sequel platform has a substantially decreased loading bias compared to its predecessor, and it does not require size-selection (Hon et al., 2017). ONT sequencing is based on measuring the electric current shaped by the nucleotides that occupy the nanopore at a given moment. Nanopore sequencing is capable of sequencing extremely long DNA fragments (Jain et al., 2018) or even native RNA molecules (Garalde et al., 2018). These features allow ONT to cover important niches. Nowadays, ONT sequencing is characterized by higher throughput, but also with a much higher error rate (Weirather et al., 2017). The higher error rate complicates variant calling or the detection of RNA modification events, however, it does not significantly impede the discovery nor the quantification of transcript isoforms. The lower throughput compared to SRS technologies means that LRS is more prone to identify artifacts resulting from template switching or ligation as biological variation. Template switching occurs when the DNA polymerase releases the template strand during synthesis and reinitiates on another template that shares homology with the previous template. Owing to this phenomenon, fusion, and splicing artifacts can be introduced via reverse-transcription (Cocquet et al., 2006) or PCR (Kebschull and Zador, 2015). These should be filtered using bioinformatics tools (Tardaguila et al., 2018). Nevertheless, certain artifacts that contain canonical splice sites might pass through these filters. One of the advantages of direct (d)RNA sequencing (currently available for LRS solely from ONT) is that it is exempt from the artifacts introduced by reverse-transcription and PCR. The ligation of independent sequences during library preparation does not require homologous sequences and (d)RNA library preparation also requires ligases. This complicates the detection of ligation artifacts, which can only be filtered by discarding rare fusion events. Both sequencing platforms excel at the characterization of capped, polyadenylated eukaryotic transcripts for technical reasons. The presence of specific cap and poly(A) sequences facilitate the ascertainment of the integrity of the transcripts, however, theoretically any other specific sequence can be targeted (Yan et al., 2018).
Host contamination is not an important issue because viral-specific transcripts are identified by mapping the sequencing reads to the viral genome. However, the parallel sequencing of host transcripts leads to a decrease in the total output of viral transcripts. In the case of late lytic herpesvirus infections one flow cell on either the MinION or the Sequel platform is sufficient to detect the majority of the expressed viral transcripts, nonetheless increasing the sequencing depth seems to always discover novel isoforms.
The herpesviruses are a large group of viruses with more than 130 species that infect a wide-range of vertebrate organisms (Carter and Saunders, 2013), and they are responsible for several human and veterinary diseases. The Herpesviridae family is subdivided into three subfamilies: Alphaherpesvirinae [e.g., herpes simplex virus type 1 and 2 (HSV-1 and -2), and pseudorabies virus (PRV)], Betaherpesvirinae [e.g., human cytomegalovirus (HCMV) and human herpesvirus type 6], and Gammaherpesvirinae [e.g., Epstein-Barr virus (EBV), and Kaposi’s sarcoma-associated herpesvirus (KSHV)]. The double-stranded DNA genomes of herpesviruses vary within 125–240 kilobase-pairs (Davison, 2007; Davison and Clements, 2010). The heart of the viral life cycle is the regulation of transcription. The viral genes are classified into three different kinetic groups; immediate-early (IE), early (E), and late (L) genes, which are defined by their peak rates of mRNA synthesis, and how they behave in the presence of protein or DNA synthesis inhibitors. Late genes can be subdivided into leaky late (L1) and true late (L2) groups based on whether they require (L2) the initiation of DNA replication for their expressions or not (L1). IE genes encode regulators of viral transcription; E genes typically specify enzymes needed for the DNA synthesis; while most of the L genes carry information for the structural elements of the virion (Weir, 2001). The herpesvirus genome is organized into polycistronic transcription units, the architecture of which is characterized by varying transcription start sites (TSSs) and shared transcription end sites (TESs).
The annotation of the herpes genomes had earlier been primarily carried out by the detection of open reading frames (ORFs), supplemented with sequencing of cDNAs (McGeoch et al., 1988). Later, next-generation SRS techniques have been applied in some herpesviruses for especially the detection of the TSSs and TESs. The PacBio amplified and non-amplified isoform sequencing (Iso-Seq) and the ONT MinION cDNA and direct dRNA sequencing methods have been applied to investigate the transcriptome of various herpesvirus species, including PRV, EBV, HSV-1 and HCMV (O’Grady et al., 2016; Tombácz et al., 2016, 2017b; Balázs et al., 2017; Moldován et al., 2017). LRS techniques have multiplied the number of previously known herpesvirus transcripts. Besides the precise full-length annotation of the viral transcripts, these studies have identified so far unknown mRNAs, non-coding (nc)RNAs, polycistronic RNAs, and various transcript isoforms including splice as well as TSS and TES variants (Figure 1). LRS has disclosed an immensely greater complexity of herpesvirus transcriptional landscape than had formerly been captured by other techniques.
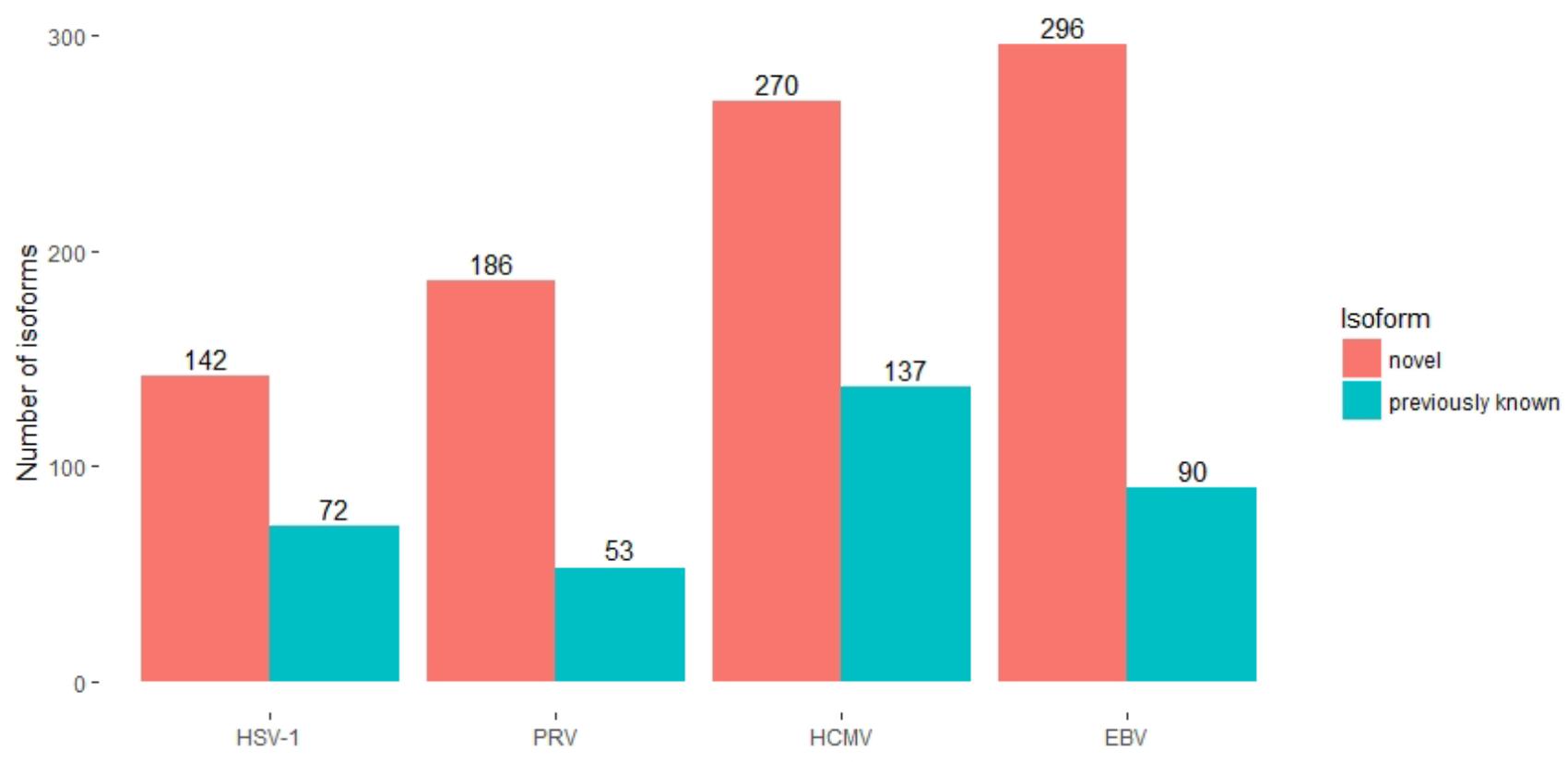
FIGURE 1. Long-read RNA sequencing extended our knowledge of herpesvirus transcriptomes. The numbers of previously known (blue) and novel (red) transcript isoforms, detected by LRS studies are depicted for each examined herpesvirus. The studies examining HSV-1 (Tombácz et al., 2017b), PRV (Tombácz et al., 2016; Moldován et al., 2017) and HCMV (Balázs et al., 2017) considered known isoforms from all strains of the given virus, while the number for EBV is the number of known isoforms in strain Akata (O’Grady et al., 2016). The analyses of the PRV and the EBV transcriptomes combined information from SRS and LRS data.
Putative Coding Transcripts
Earlier studies that were primarily based on ORF analysis revealed that the herpesvirus genomes, depending on the species, contain 70–165 protein-coding genes (Davison, 2007). LRS and ribosome profiling of the herpes transcriptomes have further increased this number with the identification of a number of 5′-truncated ORFs (putative embedded genes), which are located within the ORFs of the larger host genes (Stern-Ginossar et al., 2012; Arias et al., 2014; Moldován et al., 2017; Tombácz et al., 2017b).The tORFs are considered to be separate genes specifying polypeptides with N-terminal deletions compared to the longer protein encoded by the host gene in to which they are embedded. The truncated proteins can have the same or similar function as the full-length proteins, although they might have different localizations (Hagiwara-Komoda et al., 2016; Kuo et al., 2016), or alternatively, they can regulate the function of the host gene (Ménard et al., 2013). LRS cDNA and dRNA sequencing studies have revealed 34 and 20 so far undetected embedded transcripts containing tORFs in HSV-1 (Tombácz et al., 2017b) and in PRV (Moldován et al., 2017), respectively. Ribosome profiling analyses of HCMV and KSHV transcriptome have shown that many tORFs are indeed translated (Stern-Ginossar et al., 2012; Arias et al., 2014). The fORFs are out-of-frame with respect to the host ORFs. These transcripts may be ncRNAs because evolving additional protein-coding information in the same DNA stretch poses an extreme challenge for natural selection, as their sequences are constrained by the overlapping sense sequences. The same problem arises in the antisense (as)ORFs. Indeed, it has been shown that long asORFs at the PRV genome are mere by-products of the selective accumulation of G and C bases at the third codon positions of the viral genes (Boldogköi et al., 1995), and they unlikely specify polypeptides.
Non-Coding Transcripts
Non-coding transcripts are specified by RNA genes that are located within the protein-coding genes or at the intergenic regions. The ncRNAs can be encoded by both the positive and negative DNA strands of protein-coding genes. In this work, we restrict our discussion to the long non-coding (lnc)RNAs (> 200 bp in length), since LRS contributed to their identification, while these techniques are insensitive for shorter sequences, such as micro RNAs, for example.
Antisense lncRNAs
The firstly discovered non-coding herpesvirus RNA was the latency-associated transcript (LAT), which is an antisense (as)RNA overlapping the icp0 gene of HSV-1 and is controlled by its own promoter (LAT promoter) (Zwaagstra et al., 1989). This transcript has also been detected in other alphaherpesviruses (Baxi et al., 1995; Borchers et al., 1999; Inman et al., 2004; Ou et al., 2007). Other examples for the asRNAs include the AZURE transcripts (Tombácz et al., 2016) overlapping the PRV us3 gene, or AST-4 overlapping the HSV-1 ul53 gene transcripts (Tombácz et al., 2016, 2017b). Betaherpesviruses contain several antisense transcripts, including a latency transcript (UL123ast) standing in antisense orientation relative to the IE1 and IE2 genes (Kondo et al., 1996). However, eight other asRNAs have been discovered by LRS in HCMV that are not clustered around the main transactivator genes. These asRNAs contain highly conserved ORFs. The reason for their conservation may simply be the result of negative selection, which had acted to preserve the sequences of their sense partners. Long-read RNA sequencing has shown that the majority of the HCMV asRNAs are represented in multiple isoforms (Balázs et al., 2017).
Embedded lncRNAs
The embedded lncRNAs can be 3′-truncated forms of mRNAs having no stop codons, such as NCL and NCS transcripts of PRV; or 5′-truncated mRNAs without in-frame ORFs, such as TRL transcripts in PRV (Tombácz et al., 2016, 2017b). The most abundant KSHV lytic transcript, PAN is also a 5′-truncated version of the K7 transcript (Arias et al., 2014).
Intergenic lncRNAs
A number of intergenic lncRNAs, another class of long non-coding transcripts have also been discovered by second (Illumina)-, third (PacBio)- and fourth-generation (ONT) sequencings. For example, the NOIR-2 transcripts of PRV (Tombácz et al., 2016), the LAT 0.7 kb in HSV-1 (Zhu et al., 1999), or RNA2.7, RNA1.2 and RNA4.9 in HCMV (Gatherer et al., 2011; Balázs et al., 2017), BCLT2-4 in EBV (O’Grady et al., 2016). Many intergenic lncRNAs have shorter embedded transcripts, such as the NOIR-1 transcripts of PRV (Tombácz et al., 2016), the AST-2 and LAT 0.7 kb-S of HSV-1 (Tombácz et al., 2017b), as well as the numerous variants of RNA2.7 and RNA4.9 in HCMV (Balázs et al., 2017). Intriguingly, recent ribosome profiling analyses have discovered translated uORFs in various lncRNAs in HCMV (Stern-Ginossar et al., 2012) and in KSHV (Arias et al., 2014), which raises the question of whether lncRNAs are indeed non-coding. Additionally, a novel type of ncRNAs, overlapping the replication origin (Ori) has been discovered in PRV (CTO-S, and CTO-M: (Oláh et al., 2015; Tombácz et al., 2016).
Transcript Isoforms
Splice Isoforms
Splicing enhances the coding potential of the genome by increasing the complexity of the transcriptome and the proteome. Spliced transcripts can contain single or multiple introns. Determination of the splicing patterns of the multiple-intron transcripts is a great challenge by SRS (Figure 2). Most mammalian genes contain multiple introns, whereas splicing is relatively rare in herpesvirus RNAs, and herpesviruses have been shown to produce proteins that retain spliced RNAs and selectively export intronless RNAs from the nucleus (Koffa et al., 2001; Sandri-Goldin, 2004; Boyne et al., 2008; Juillard et al., 2012). However, the expression of spliced and unspliced transcripts during infection is regulated in a complex manner (Sadek and Read, 2016). Several betaherpesvirus (Gatherer et al., 2011) and gammaherpesvirus (O’Grady et al., 2016) mRNAs contain multiple introns, while the large majority of alphaherpesvirus transcripts are intronless (Tombácz et al., 2016, 2017b). LRS has identified numerous novel splice isoforms in herpesviruses.
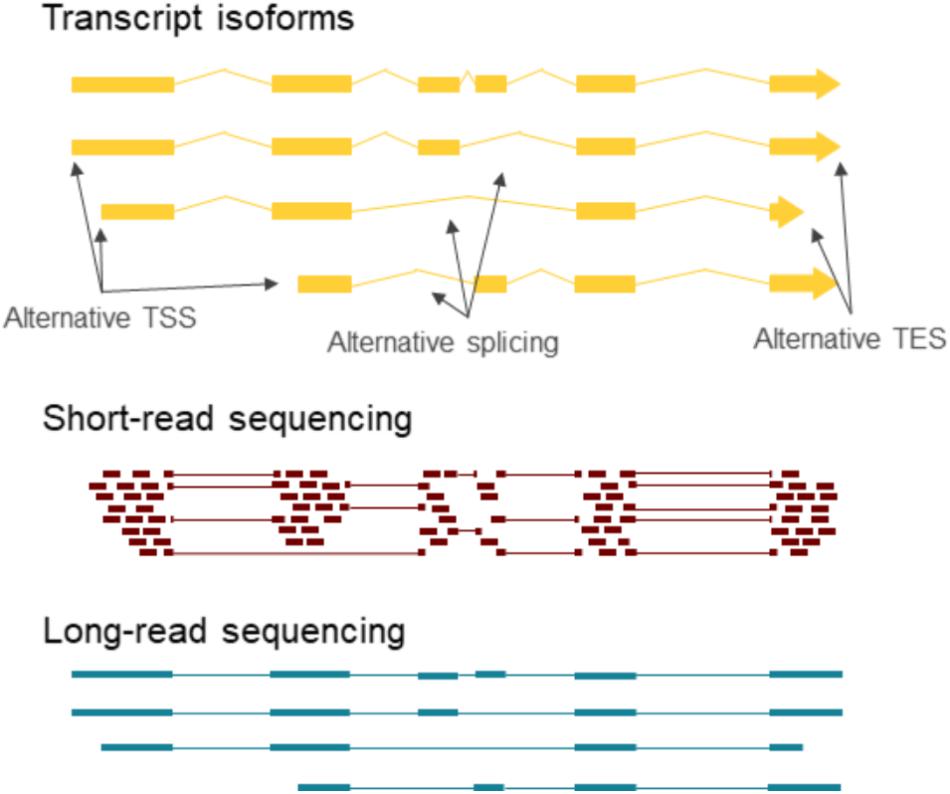
FIGURE 2. Long-read RNA sequencing provides contig information about transcript isoforms. The individual TSSs, TESs and splice junctions can be determined via short-read sequencing, however, the combination of these features is difficult to discern in case of multiple isoforms at the same locus. LRS on the other hand can capture full-length transcripts, which give complete contig information about the exons included in each transcript.
TSS Isoforms
The TSS isoforms contain the same ORFs, but differ in the length of their 5′-UTRs and are controlled by distinct promoters. TSS variation represents a common phenomenon in herpesviruses. Alternative promoters can provide differential transcriptional controls for the same gene at distinct stages of infection. For instance, the UL44 gene of the HCMV has three distinct TSSs, two of which are active during the early viral infection and one that is functional after the initiation of viral DNA replication (Isomura et al., 2008).
TES Isoforms
TES variation is less common than the TSS polymorphism in the herpesviruses, for example, in HCMV less than 10% of the genes expressed TES isoforms, while more than half of the genes had different TSS isoforms (Balázs et al., 2017). From a certain point of view, polycistronic transcripts can also be considered as TES isoforms provided that the upstream genes can also be separately transcribed.
Polycistronic and Complex Transcripts
Polycistronic transcription is common in prokaryotic organisms and in certain viruses, but is rare in eukaryotes. In bacteria and bacteriophages the Shine-Dalgarno sequences allow the translation of downstream genes on polycistronic transcripts (Shine and Dalgarno, 1975), while some eukaryotic viruses developed various mechanisms to solve this problem, which includes leaky ribosomal scanning, ribosomal frameshifting, or the use of internal ribosome entry site (IRES) sequences (Firth and Brierley, 2012; Kronstad et al., 2013). Polycistronic RNAs are widespread in herpesviruses, however, there are only few pieces of evidence for the translation of downstream genes. LRS studies have uncovered a large number of polycistronic and complex transcripts, many of them are expressed in low abundance (Tombácz et al., 2016). These works have also revealed that in the majority of polycistronic transcripts of alphaherpesviruses the upstream genes are also transcribed as monocistronic RNA molecules (Tombácz et al., 2016, 2017b; Moldován et al., 2017). Intriguingly, the transactivator genes of α-herpesviruses (e.g., ie180, ep0 and us1 genes of PRV) do not form polycistronic transcripts and are not overlapped by mRNAs encoded by the adjacent genes. Instead, they form overlaps with antisense transcripts (e.g., ie180 and ep0 with LLT, and us1 with PTO-US1 and NCS1 transcripts), which are controlled by their own promoters. Some β-herpesvirus transactivator genes produce monocistronic RNAs (like the RS1 in HCMV or U95 in HHV6-7), while others produce polycistronic transcripts (such as the IE1 and IE2 genes in HCMV and HHV6-7). The EBV transactivator genes are transcribed as a single polycistronic unit, while the KSHV Rta gene is expressed in a bicistronic transcript. Complex transcripts contain gene sequences in opposite polarity of which the sequences standing in antisense orientation are obviously non-coding. Five such transcripts have been described in PRV and 10 in HSV-1 (Tombácz et al., 2016, 2017b; Moldován et al., 2017).
Conclusion
Long-read sequencing has revealed a much greater complexity of the viral transcriptome than it has been known before (Figure 1). It is known that higher order organisms produce multiple transcript isoforms, human genes for example express on average 6.3 isoforms (Encode Project Consortium, 2012). However, until recently, the number of known herpesvirus transcript isoforms was comparable to the number of genes. The complexity of these transcriptomes is even more surprising considering that splicing in herpesviruses is less common than in the host cells. The individual features such as TSSs, TESs, introns and polycistronic transcripts can be investigated by SRS as well; however, the exact transcriptome annotation of high-density genomes such as those of herpesviruses is only feasible by LRS (Figure 2).
While LRS has discovered countless novel isoforms and has provided a much more detailed transcriptome annotation of the examined herpesviruses, it has not yet explained the need for such complexity. While certain splice and TSS isoforms increase the coding potential (Balázs et al., 2017), we remain uncertain about the roles of the majority of the novel transcripts. It is possible that some of these transcripts are mere transcriptional noise, however, they could also possess regulatory functions. While certain isoforms, such as those of UL44 of HCMV, have been reported to be differentially expressed (Isomura et al., 2008), an LRS study characterizing the kinetics of multiple PRV isoforms has found that the majority of UTR-isoforms are expressed with similar kinetics and only some cistronic variants showed inverted kinetics (Tombácz et al., 2017a). It is possible though that there are slight differences between the expression patterns of isoforms that would become detectable when observed in higher resolution. Recent studies have uncovered an extensive overlapping pattern of transcriptions in herpesviruses. The function of transcriptional overlaps may be to regulate gene expressions – for example, through giving rise to genome-wide transcriptional interference (Boldogköi, 2012).
Isoform-level time-series studies may clarify the function of the isoforms. The low throughput of LRS platforms limits their quantitative abilities, especially during the early stages of infection when host gene expression exceeds viral transcription. The rapidly increasing throughput of LRS platforms and virus-specific enrichment strategies (Cheng et al., 2017) will facilitate the use of LRS in the quantitative analysis of viral transcriptomes. Precise LRS annotations can also enable isoform-level quantification using SRS data (Trapnell et al., 2012). The exact characterization of the biological importance of each isoform may require molecule modeling or mutational analyses.
Author Contributions
DT and ZBa reviewed the literature. DT, ZBa, ZC, and ZBo wrote the manuscript. MS participated in the coordination of the study. ZBo conceived the project. All authors contributed, read, and approved the manuscript.
Funding
DT was supported by the Bolyai János Scholarship of the Hungarian Academy of Sciences (2015–2018). The study was also supported by the Swiss-Hungarian Cooperation Programme (SH/7/2/8) to ZBo.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arias, C., Weisburd, B., Stern-Ginossar, N., Mercier, A., Madrid, A. S., Bellare, P., et al. (2014). KSHV 2.0: a comprehensive annotation of the kaposi’s sarcoma-associated Herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 10:e1003847. doi: 10.1371/journal.ppat.1003847
Balázs, Z., Tombácz, D., Szûcs, A., Csabai, Z., Megyeri, K., Petrov, A. N., et al. (2017). Long-read sequencing of human cytomegalovirus transcriptome reveals RNA isoforms carrying distinct coding potentials. Sci. Rep. 7:15989. doi: 10.1038/s41598-017-16262-z
Baxi, M. K., Efstathiou, S., Lawrence, G., Whalley, J. M., Slater, J. D., and Field, H. J. (1995). The detection of latency-associated transcripts of equine herpesvirus 1 in ganglionic neurons. J. Gen. Virol. 76(Pt 12), 3113–3118. doi: 10.1099/0022-1317-76-12-3113
Boldogköi, Z. (2012). Transcriptional interference networks coordinate the expression of functionally related genes clustered in the same genomic loci. Front. Genet. 3:122. doi: 10.3389/fgene.2012.00122
Boldogköi, Z., Murvai, J., Fodor, I., Boldogkoi, Z., Murvai, J., and Fodor, I. (1995). G and C accumulation at silent positions of codons produces additional ORFs. Trends Genet. 11, 125–126. doi: 10.1016/S0168-9525(00)89019-8
Borchers, K., Wolfinger, U., and Ludwig, H. (1999). Latency-associated transcripts of equine Herpesvirus type 4 in trigeminal ganglia of naturally infected horses. J. Gen. Virol. 80(Pt 8), 2165–2171. doi: 10.1099/0022-1317-80-8-2165
Boyne, J. R., Colgan, K. J., and Whitehouse, A. (2008). Recruitment of the complete hTREX complex is required for kaposi’s sarcoma–associated Herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog. 4:e1000194. doi: 10.1371/journal.ppat.1000194
Carter, J. B., and Saunders, V. A. (2013). Virology: Principles and Applications. Hoboken, NJ: John Wiley & Sons.
Cheng, S., Caviness, K., Buehler, J., Smithey, M., Nikolich-Žugich, J., and Goodrum, F. (2017). Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc. Natl. Acad. Sci. U.S.A. 114, E10586–E10595. doi: 10.1073/pnas.1710522114
Cocquet, J., Chong, A., Zhang, G., and Veitia, R. A. (2006). Reverse transcriptase template switching and false alternative transcripts. Genomics 88, 127–131. doi: 10.1016/j.ygeno.2005.12.013
Davison, A. J. (2007). “Comparative analysis of the genomes,” in Source Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, eds A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley, et al. (Cambridge: Cambridge University Press).
Davison, A. J., and Clements, J. B. (2010). “Herpesviruses: general properties,” in Topley & Wilson’s Microbiology and Microbial Infections, eds B. W. J. Mahy, V. ter Meulen, S. P. Borriello, P. R. Murray, G. Funke, W. G. Merz, et al. (Chichester: John Wiley & Sons, Ltd.), doi: 10.1002/9780470688618.taw0231
Encode Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Firth, A. E., and Brierley, I. (2012). Non-canonical translation in RNA viruses. J. Gen. Virol. 93, 1385–1409. doi: 10.1099/vir.0.042499-0
Garalde, D. R., Snell, E. A., Jachimowicz, D., Sipos, B., Lloyd, J. H., Bruce, M., et al. (2018). Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206. doi: 10.1038/nmeth.4577
Gatherer, D., Seirafian, S., Cunningham, C., Holton, M., Dargan, D. J., Baluchova, K., et al. (2011). High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. U.S.A. 108, 19755–19760. doi: 10.1073/pnas.1115861108
Hagiwara-Komoda, Y., Choi, S. H., Sato, M., Atsumi, G., Abe, J., Fukuda, J., et al. (2016). Truncated yet functional viral protein produced via RNA polymerase slippage implies underestimated coding capacity of RNA viruses. Sci. Rep. 6:21411. doi: 10.1038/srep21411
Hon, T., Tseng, E., Vedula, A., and Clark, T. A. (2017). Full-Length cDNA Sequencing on the PacBio Sequel Platform. Available at: https://www.pacb.com/wp-content/uploads/Clark-PAG-2017-Full-Length-cDNA-Sequencing-on-the-PacBio-Sequel_Platform.pdf
Inman, M., Zhou, J., Webb, H., and Jones, C. (2004). Identification of a novel bovine Herpesvirus 1 transcript containing a small open reading frame that is expressed in trigeminal ganglia of latently infected cattle. J. Virol. 78, 5438–5447. doi: 10.1128/JVI.78.10.5438-5447.2004
Isomura, H., Stinski, M. F., Kudoh, A., Murata, T., Nakayama, S., Sato, Y., et al. (2008). Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82, 1638–1646. doi: 10.1128/JVI.01917-07
Jain, M., Koren, S., Miga, K. H., Quick, J., Rand, A. C., Sasani, T. A., et al. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345. doi: 10.1038/nbt.4060
Juillard, F., Bazot, Q., Mure, F., Tafforeau, L., Macri, C., Rabourdin-Combe, C., et al. (2012). Epstein–Barr virus protein EB2 stimulates cytoplasmic mRNA accumulation by counteracting the deleterious effects of SRp20 on viral mRNAs. Nucleic Acids Res. 40, 6834–6849. doi: 10.1093/nar/gks319
Kebschull, J. M., and Zador, A. M. (2015). Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res. 43:e143. doi: 10.1093/nar/gkv717
Koffa, M. D., Clements, J. B., Izaurralde, E., Wadd, S., Wilson, S. A., Mattaj, I. W., et al. (2001). Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. EMBO J. 20, 5769–5778. doi: 10.1093/emboj/20.20.5769
Kondo, K., Xu, J., and Mocarski, E. S. (1996). Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. U.S.A. 93, 11137–11142. doi: 10.1073/pnas.93.20.11137
Kronstad, L. M., Brulois, K. F., Jung, J. U., and Glaunsinger, B. A. (2013). Dual short upstream open reading frames control translation of a herpesviral polycistronic mRNA. PLoS Pathog. 9:e1003156. doi: 10.1371/journal.ppat.1003156
Kuo, R.-L., Li, L.-H., Lin, S.-J., Li, Z.-H., Chen, G.-W., Chang, C.-K., et al. (2016). Role of N terminus-truncated NS1 proteins of influenza A virus in inhibiting IRF3 activation. J. Virol. 90, 4696–4705. doi: 10.1128/JVI.02843-15
Lin, H.-H., and Liao, Y.-C. (2015). Evaluation and validation of assembling corrected PacBio long reads for microbial genome completion via hybrid approaches. PLoS One 10:e0144305. doi: 10.1371/journal.pone.0144305
Loomis, E. W., Eid, J. S., Peluso, P., Yin, J., Hickey, L., Rank, D., et al. (2013). Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res. 23, 121–128. doi: 10.1101/gr.141705.112
McGeoch, D. J., Dalrymple, M. A., Davison, A. J., Dolan, A., Frame, M. C., McNab, D., et al. (1988). The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69, 1531–1574. doi: 10.1099/0022-1317-69-7-1531
Ménard, V., Collin, P., Margaillan, G., Guillemette, C., Tephly, T. R., Hum, D. W., et al. (2013). Modulation of the UGT2B7 enzyme activity by C-terminally truncated proteins derived from alternative splicing. Drug Metab. Dispos. 41, 2197–2205. doi: 10.1124/dmd.113.053876
Moldován, N., Tombácz, D., Szûcs, A., Csabai, Z., Snyder, M., and Boldogkõi, Z. (2017). Multi-platform sequencing approach reveals a novel transcriptome profile in pseudorabies virus. Front. Microbiol. 8:2708. doi: 10.3389/fmicb.2017.02708
O’Grady, T., Wang, X., Höner zu Bentrup, K., Baddoo, M., Concha, M., and Flemington, E. K. (2016). Global transcript structure resolution of high gene density genomes through multi-platform data integration. Nucleic Acids Res. 44:e145. doi: 10.1093/nar/gkw629
Oláh, P., Tombácz, D., Póka, N., Csabai, Z., Prazsák, I., and Boldogkõi, Z. (2015). Characterization of pseudorabies virus transcriptome by Illumina sequencing. BMC Microbiol. 15:130. doi: 10.1186/s12866-015-0470-0
Ou, Y., Davis, K. A., Traina-Dorge, V., and Gray, W. L. (2007). Simian varicella virus expresses a latency-associated transcript that is antisense to open reading frame 61 (ICP0) mRNA in neural ganglia of latently infected monkeys. J. Virol. 81, 8149–8156. doi: 10.1128/JVI.00407-07
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13, 278–289. doi: 10.1016/j.gpb.2015.08.002
Sadek, J., and Read, G. S. (2016). The Splicing history of an mRNA affects its level of translation and sensitivity to cleavage by the virion host shutoff endonuclease during herpes simplex virus infections. J. Virol. 90, 10844–10856. doi: 10.1128/JVI.01302-16
Sandri-Goldin, R. M. (2004). Viral regulation of mRNA export. J. Virol. 78, 4389–4396. doi: 10.1128/JVI.78.9.4389-4396.2004
Shine, J., and Dalgarno, L. (1975). Determinant of cistron specificity in bacterial ribosomes. Nature 254, 34–38. doi: 10.1038/254034a0
Steijger, T., Abril, J. F., Engström, P. G., Kokocinski, F., Hubbard, T. J., Guigó, R., et al. (2013). Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 10, 1177–1184. doi: 10.1038/nmeth.2714
Stern-Ginossar, N., Weisburd, B., Michalski, A., Le, V. T., Hein, M. Y., Huang, S.-X., et al. (2012). Decoding human cytomegalovirus. Science 338, 1088–1093. doi: 10.1126/science.1227919
Tardaguila, M., de la Fuente, L., Marti, C., Pereira, C., Pardo-Palacios, F. J., Del Risco, H., et al. (2018). SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. doi: 10.1101/gr.222976.117 [Epub ahead of print].
Tombácz, D., Balázs, Z., Csabai, Z., Moldován, N., Szûcs, A., Sharon, D., et al. (2017a). Characterization of the dynamic transcriptome of a Herpesvirus with long-read single molecule real-time sequencing. Sci. Rep. 7:43751. doi: 10.1038/srep43751
Tombácz, D., Csabai, Z., Szûcs, A., Balázs, Z., Moldován, N., Sharon, D., et al. (2017b). Long-read isoform sequencing reveals a hidden complexity of the transcriptional landscape of herpes simplex virus type 1. Front. Microbiol. 8:1079. doi: 10.3389/fmicb.2017.01079
Tombácz, D., Csabai, Z., Oláh, P., Balázs, Z., Likó, I., Zsigmond, L., et al. (2016). Full-length isoform sequencing reveals novel transcripts and substantial transcriptional overlaps in a Herpesvirus. PLoS One 11:e0162868. doi: 10.1371/journal.pone.0162868
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Weir, J. P. (2001). Regulation of herpes simplex virus gene expression. Gene 271, 117–130. doi: 10.1016/S0378-1119(01)00512-1
Weirather, J. L., de Cesare, M., Wang, Y., Piazza, P., Sebastiano, V., Wang, X.-J., et al. (2017). Comprehensive comparison of pacific biosciences and Oxford nanopore technologies and their applications to transcriptome analysis. F1000Res. 6:100. doi: 10.12688/f1000research.10571.2
Yan, B., Boitano, M., Clark, T., and Ettwiller, L. (2018). SMRT-Cappable-seq reveals complex operon variants in bacteria. bioRxiv [Preprint]. doi: 10.1101/262964
Zhu, J., Kang, W., Marquart, M. E., Hill, J. M., Zheng, X., Block, T. M., et al. (1999). Identification of a Novel 0.7-kb polyadenylated transcript in the LAT promoter region of HSV-1 that is strain specific and may contribute to virulence. Virology 265, 296–307. doi: 10.1006/viro.1999.0057
Keywords: herpesvirus, transcriptome, long-read sequencing, PacBio sequencing, Oxford Nanopore Technologies, transcript isoforms
Citation: Tombácz D, Balázs Z, Csabai Z, Snyder M and Boldogkői Z (2018) Long-Read Sequencing Revealed an Extensive Transcript Complexity in Herpesviruses. Front. Genet. 9:259. doi: 10.3389/fgene.2018.00259
Received: 23 April 2018; Accepted: 27 June 2018;
Published: 17 July 2018.
Edited by:
Philipp Kapranov, Huaqiao University, ChinaReviewed by:
Richard John Edwards, University of New South Wales, AustraliaOlga Vinnere Pettersson, Science for Life Laboratory (SciLifeLab), Sweden
Weidong Xiao, Temple University, United States
Copyright © 2018 Tombácz, Balázs, Csabai, Snyder and Boldogkői. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zsolt Boldogkői, Ym9sZG9na29pLnpzb2x0QG1lZC51LXN6ZWdlZC5odQ==