 Mathias Cavaillé1,2*
Mathias Cavaillé1,2* Flora Ponelle-Chachuat1,2
Flora Ponelle-Chachuat1,2 Nancy Uhrhammer1,2
Nancy Uhrhammer1,2 Sandrine Viala1,2Mathilde Gay-Bellile1,2Maud Privat1,2Yannick Bidet1,2
Sandrine Viala1,2Mathilde Gay-Bellile1,2Maud Privat1,2Yannick Bidet1,2 Yves-Jean Bignon1,2*
Yves-Jean Bignon1,2*- 1INSERM, U1240 Imagerie Moléculaire et Stratégies Théranostiques, Université Clermont Auvergne, Clermont-Ferrand, France
- 2Département d’Oncogénétique, Centre Jean Perrin, Clermont-Ferrand, France
A family with an aggregation of rare early onset multiple primary tumors has been managed in our oncogenetics department: the proband developed four early onset carcinomas between ages 31 and 33 years, including acral melanoma, bilateral clear cell renal carcinoma (RC), and follicular variant of papillary thyroid carcinoma. The proband’s parent developed orbital lymphoma and small intestine mucosa-associated lymphoid tissue (MALT) lymphoma between 40 and 50 years old. Whole-exome-sequencing (WES) of the nuclear family (proband, parents, and sibling) identified in the proband a de novo deleterious heterozygous mutation c.1003C > T (p.Arg335∗) in the phosphatase and tensin homolog (PTEN) gene. Furthermore, WES allowed analysis of the nuclear family’s genetic background, and identified deleterious variants in two candidate modifier genes: CEACAM1 and MIB2. CEACAM1, a tumor suppressor gene, presents loss of expression in clear cell RC and is involved in proliferation of B cells. It could explain in part the phenotype of proband’s parent and the occurrence of clear cell RC in the proband. Deleterious mutations in the MIB2 gene are associated with melanoma invasion, and could explain the occurrence of melanoma in the proband. Cowden syndrome is a hereditary autosomal dominant disorder associated with increased risk of muco-cutaneous features, hamartomatous tumors, and cancer. This atypical presentation, including absence of muco-cutaneous lesions, four primary early onset tumors and bilateral clear cell RC, has not been described before. This encourages including the PTEN gene in panel testing in the context of early onset RC, whatever the histological subtype. Further studies are required to determine the implication of CEACAM1 and MIB2 in the severity of Cowden syndrome in our proband and occurrence of early onset MALT lymphoma in a parent.
Background
PTEN hamartoma syndrome (PHTS) is an autosomal dominant disorder characterized by deleterious mutation in the tumor suppressor gene phosphatase and tensin homolog (PTEN) gene (Blumenthal and Dennis, 2008). The clinical presentation is heterogeneous, including Cowden syndrome (CS) (Pilarski, 2009), Bannayan-Riley-Ruvalcaba syndrome (Lachlan et al., 2007),Lhermitte–Duclos disease (Zhou et al., 2003), Segmental outgrowth-lipomatosis-arteriovenous malformation-epidermal nevus (SOLAMEN) syndrome (Caux et al., 2007), and autism-macrocephaly syndrome (Butler et al., 2005).
Cowden syndrome is the most frequent of these entities, with a prevalence of 1 in 250,000 individuals (Nelen et al., 1999), and penetrance of up to 90% in the second decade (Ngeow and Eng, 2015). CS is associated with increased risk of muco-cutaneous features, hamartomatous tumors and cancer, defined by diagnostic and testing criteria (Saslow et al., 2012; Pilarski et al., 2013) (Table 1). Muco-cutaneous lesions including trichilemmoma, acral keratosis and oral papillomatosis, plus macrocephaly are the most frequent features, described in more than 90% of cases after the third decade (Nosé, 2016).

TABLE 1. Testing and clinical diagnosis criteria.
Cowden syndrome also predisposes to cancer, with a high lifetime risk of developing breast or endometrial cancer in women, and thyroid cancer in both sexes. Other cancer risks have recently been identified, including renal carcinoma (RC), gastro-intestinal cancer, and melanoma. RC in CS is predominately of the papillary and chromophobe type, beginning around 40–50 years, while the lifetime risk is unclear. Melanoma is uncommon, with a lifetime risk about 6% (Tan et al., 2012; Bubien et al., 2013). Second cancers are frequent in these patients, but less than 5% develop a third cancer (Ngeow et al., 2014).
Here we described an atypical clinical presentation of CS, which did not meet diagnostic or testing criteria, in a patient who developed four primary early onset carcinomas (one melanoma, two clear cell renal carcinomas, and a follicular variant of papillary thyroid carcinoma), without muco-cutaneous lesions. A heterozygous deleterious mutation in PTEN was identified by whole exome-sequencing (WES).
Case Presentation
The proband presented with a suspect nevus localized on the inner edge of the left foot. Excision revealed acral lentiginous melanoma (Breslow index at 0.41 mm, Clark level II). Extension assessment, which included cerebro-thoraco-abdomino-pelvic computed tomography and inguinal ultrasound, found no secondary lesions, but a small cystic lesion on the upper pole of the left kidney. No complementary treatment was given.
Control tomography 3 months later showed an increase in size of the renal lesion, associated with irregular contours and a heterogeneous aspect, needing expanded nephrectomy. Pathological examination diagnosed clear cell RC, Fuhrman grade II, pT1aNxMx. No complementary treatment was given.
The patient also developed Hashimoto thyroiditis with multinodular goiter of which three nodules measured > 15 mm. Total thyroidectomy revealed follicular variant of papillary carcinoma, pT1NxMx. The patient was treated with Iodine 131.
During surveillance of primary left RC by computed tomography, a cystic lesion of the right kidney with peripheral contrast enhancement was observed, confirmed by magnetic resonance imaging. Gastroendoscopy was performed, removing a single non-inflammatory gastric polyp. No oesophageal glycogenic acanthosis was observed. A partial right nephrectomy was performed. Pathological examination revealed clear cell RC, Furhman grade III, pT1aNxMx. No complementary treatment was given.
Examination revealed no other medical antecedents. Identification of risk factors identified moderate obesity [Body Mass Index (BMI) at 33]. The patient had never smoked or been exposed to irradiation. Physical examination by the dermatologist, surgeon and oncogeneticist identified no additional lesions, in particular no cutaneous lesions evocative of CS (trichilemmoma, palmoplantar keratoses, oral papillomatosis, and facial papules). The cranial perimeter was unknown. Brain imaging revealed no Lhermitte–Duclos disease.
Family History
One of the proband’s parents had developed two distinct rare cancers: orbital lymphoma and small intestine mucosa-associated lymphoid tissue (MALT) lymphoma between 40 and 50 years old. No other familial history of cancer or other medical problems were observed (Figure 1).
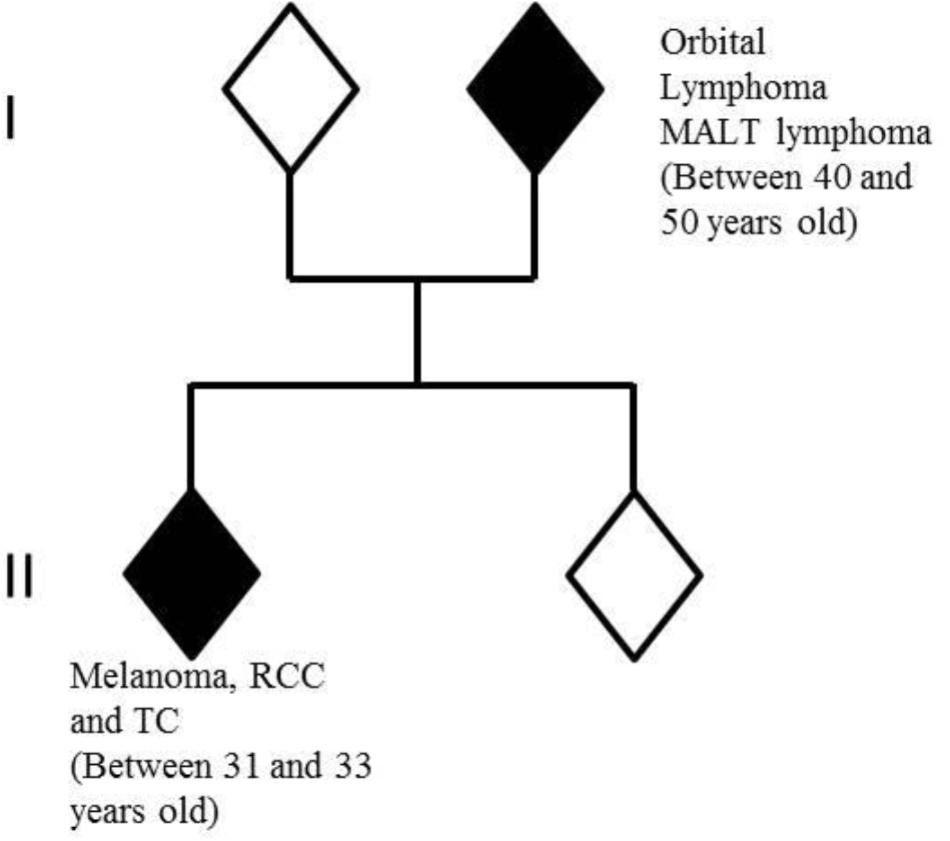
FIGURE 1. Members of nuclear family analyzed by whole-exome-sequencing. In white: healthy individuals; in black: individuals with cancers. RCC, renal cell carcinoma; TC, thyroid carcinoma.
Genetic Exploration
Because of multiple RCs, genetic analysis of VHL was performed in 2009: no qualitative or quantitative mutation was identified. Other genes involved in hereditary RC (MET, FH, FLCN) were not explored because the histological type of RC and examination did not correspond.
Laboratory Investigation
Whole-exome-sequencing of the nuclear family was performed in order to identify constitutional causal mutation(s) for this clinical presentation.
Samples
The nuclear family included the proband, sibling, and parents. Each patient signed informed consent for genetic diagnosis and research for hereditary disease. The local ethics committee at Clermont-Ferrand did not oppose the publication of this case report without patient specific consent. Three samples from the proband’s thyroid tumor were included (cancer cells: 70–100%). Melanoma and renal samples were not available. DNA extraction of tumor samples was performed using Maxwell 16 FFPE Plus LEV kit from formalin-fixed paraffin-embedded blocks.
Whole-Exome-Sequencing
Whole-exome-sequencing was performed on four family members. DNA was extracted from peripheral blood using QIAamp DNA macrokit (Qiagen). Sonic fragmentation was performed on a Bioruptor instrument (Diagencode). Kapa library preparation and SeqCap EZ MedExome kits (Roche) were used for library preparation and capture. Sequencing was performed using NextSeq 500/550 High Output v2 kit (300 cycles) on a NextSeq 500 instrument (Illumina). All steps were performed following providers’ guidelines.
Bioinformatics Analysis
De-multiplexing was performed using bcl2fastq2 Conversion Software (Illumina). Alignment was performed on University of California Santa Cruz human genome reference build 19 using Burrows-Wheeler Aligner. Genome Analysis Toolkit (GATK) and PICARD tools were used for base quality score recalibration (BaseRecalibrator) and realignment (RealignerTargetCreator, IndelRealigner), as recommended by Eurogenetest guidelines (Matthijs et al., 2016). Variant calling was performed using GATK HaplotypeCaller and annotated using EnsemblVariantEffectPredictor. Variants were filtered for quality score ≥ 30, depth ≥ 20x, and present in ≥ 20% of reads.
Biological Filters
Because of familial presentation and to identify variants of interest involved in monogenic hereditary cancer predisposition with high penetrance, biological filters included: minor allele frequency < 1% or unknown as determined by Exome Aggregation Consortium and Exome Sequencing Project, truncating variants (nonsense, frameshift), and splice-site variants with significantly modified Human Splice Finder (HSF) score. Synonymous and missense variants were excluded, except for those with an impact on splice-sites. Analysis of copy number variations was not performed. Genes presenting truncating variants identified by WES in our laboratory in at least 20 persons, including healthy individuals and patients with distinct phenotypes, were excluded.
All types of variants present in genes known to be involved in hereditary predisposition to cancer or actionable for other hereditary diseases as defined by the American College of Human Genetics (Green et al., 2013) were systematically analyzed.
In silico Analysis
Each variant of interest was annotated and interpreted using ALAMUT (Interactive BioSoftware), which includes splice-site analysis tools (HSF) and protein-function prediction tools (SIFT, Polyphen 2.0). Each variant was confirmed by direct reading of sequence using Integrative Genomics Viewer (IGV, Broad Institute). Gene Expression Profiling Interactive Analysis (GEPIA) was used to identify genes of interest due to their expression profiles in papillary thyroid cancer, RC and/or melanoma.
Sanger Analysis
Constitutional mutations in genes of interest were confirmed in the proband by Sanger Sequencing, using a 3500 × l instrument and BigDye Terminator kit 3.1 (Applied Biosystems). Primers were designed using PrimerBlast to target variants identified by WES. Primers and experimental conditions are available on request.
Loss of heterozygosity (LOH) was studied for these same variants in the proband’s thyroid tumor samples. LOH was defined by ≥80% reduction of an allele, using Seqman software (DNASTAR).
Results
Whole-exome-sequencing presented an average depth of 107 reads (107X). Ninety-four percent of the exome was covered at a depth of ≥20X.
The four persons studied presented an average of 30,794 variants meeting quality criteria, of which an average of 2746 were truncating variants. After further bio-informatics filtering, 42 truncating and splice-site variants present in the proband were selected as potentially disease-causing (cf. Table 2). These included 18 de novo variants of interest, and 24 variants shared with one or the other parent, all of which were heterozygous. The most notable finding was a de novo deleterious mutation in PTEN: c.1003C > T; p.(Arg335∗). Five of the inherited variants in genes associated with cancer or actionable for hereditary diseases have been described in the literature as pathogenic (MUTYH: c.933+3A > C) or of unknown significance (APC, APOB, DSG2, DSP). Unpublished truncating variants in two genes under-expressed in cancer [CEACAM1:c.553_554insAGGC; p.(Leu185Glnfs∗26), and MIB2: c.153C > A; p.(Cys51∗)] were also inherited.
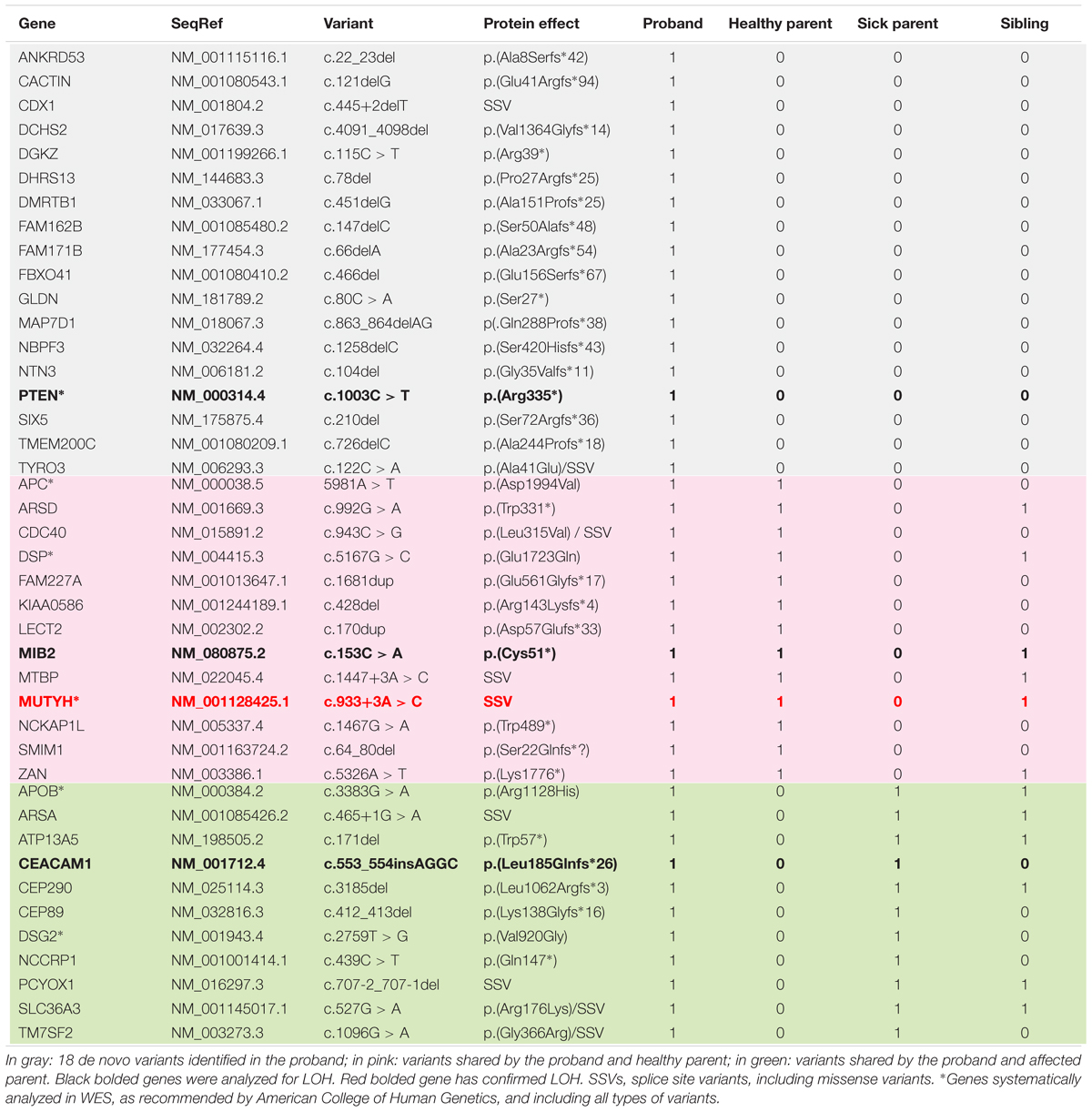
TABLE 2. Variants of interest identified by WES in nuclear family and their segregation.
The potentially disease-causing variants in PTEN, MUTYH, CEACAM1, and MIB2 were confirmed in the proband by Sanger sequencing. Tissue samples from the proband’s thyroid tumor were analyzed in parallel for LOH at these sites. LOH was identified in the MUTYH gene. No LOH was found in the other genes.
Discussion
Whole-exome-sequencing of a patient with early onset multiple primary malignant tumors and his nuclear family identified 42 rare truncating and splice-site variants, including a de novo deleterious heterozygous mutation in PTEN, involved here in atypical presentation of CS.
Clinical Aspects
With only three minor criteria of CS (follicular variant of papillary thyroid cancer, multinodular goiter, and RC), the patient did not meet diagnostic or testing criteria. However, the head circumference and colonic status were unknown.
Macrocephaly is a frequent feature in PTEN mutation carriers, described in 83 to 94% of cases (Mester et al., 2011; Nieuwenhuis et al., 2014). It underlines the importance of cranial perimeter measurement when assessing tumors of the PTEN spectrum. Multiple gastrointestinal intestinal polyps are equally frequent, observed in the colon in up to 93% of cases and the upper gastrointestinal tract in 66–100% of cases, with various histological types (Levi et al., 2011; Stanich et al., 2014). However, macrocephaly and gastrointestinal polyps are not constant, and their absence does not rule out diagnosis of CS.
Our patient had no muco-cutaneous lesions and no evocative upper gastrointestinal lesions. Pathognomonic muco-cutaneous lesions (trichilemmoma, palmoplantar keratosis, oral mucosal papillomatosis, and macular pigmentation of glans penis) are frequent features in CS, with prevalence respectively at 6–38, 82, 85, and 46–53% (Bubien et al., 2013; Pilarski et al., 2013). Lipoma and testicular lipomatosis are described in 30 to 40% (Woodhouse et al., 2005; Gammon et al., 2016). Glycogenic acanthosis and Lhermitte–Duclos disease are present respectively in 48–80 and 2–15% of cases (Levi et al., 2011; Bubien et al., 2013; Pilarski et al., 2013).
Our case was also atypical in regards to the type of cancers diagnosed and age of occurrence. RC presents a lifetime risk of about 1.6% in the general population (Gupta et al., 2017). It is a minor diagnostic criterion of CS with a lifetime maximum risk estimated at 36% (Tan et al., 2012). The prevalence of RC in PTEN mutation carriers is low (1.7 to 4%) in large cohort studies (Pilarski et al., 2011; Mester et al., 2012; Tan et al., 2012; Bubien et al., 2013). It is predominantly papillary (I and II) and chromophobe types, unilateral, and occurs around 40–50 years old. One case of bilateral chromophobe carcinoma has been described, and one case of clear cell carcinoma in a male at 56 years of age (Mester et al., 2012; Shuch et al., 2013).
Here, we describe for the first time a case of bilateral clear cell carcinoma of early onset (between 31 and 33 years of age). These tumors are associated with early diagnosis and good prognosis, which is consistent with the data from the literature (Mester et al., 2012; Shuch et al., 2013). However, the diagnosis was made fortuitously. Current guidelines recommend screening for RC using ultrasound every 1–2 years, according to familial history of RC, starting at age of 40 years of age (Saslow et al., 2012). This screening should be started at age of 30 years of age, as proposed by the French Cowden Disease Network (Bubien et al., 2013), for all patients with constitutional deleterious mutation in the PTEN gene. As RC occurs frequently without familial history of RC, the periodicity of screening for this cancer should not be based on familial history but be proposed annually.
Other genes predispose to RC, including VHL, MET, FH, FLCN, BAP1, and SDHB (Haas and Nathanson, 2014). Thus, PTEN could be included in panel testing of patients with early onset RC, whatever the histological type.
Melanoma presents a lifetime risk of 2.4% in the general population (Apalla et al., 2017). Lentiginous acral melanoma represents 1.5% of melanomas, and is associated with poorer prognosis because of later diagnosis. Most cases appear in the seventh decade on non-sun-exposed sites, which suggested mechanisms of carcinogenesis other than ultraviolet exposure, including genetic factors (Carrera and Puig-Butille, 2018). Lifetime risk is estimated at 6% in PTEN mutation carriers and guidelines do not currently recommend systematic dermatological screening in this population (Saslow et al., 2012). Despite the low risk, annual dermatological screening could be considered for all PTEN mutation carriers. Because pediatric onset has been described, screening could begin upon diagnosis.
Thyroid carcinoma presents a lifetime risk of about 1% in the general population (Nguyen et al., 2015). In PTEN mutation carriers, lifetime risk is estimated at 14 to 38%, with onset in the third decade (Tan et al., 2012; Bubien et al., 2013). Histological sub-types associated with CS include papillary carcinoma (52–60%), follicular carcinoma (14–45%) and follicular variant of papillary carcinoma (4.8–28%) (Laury et al., 2011; Milas et al., 2012). Thyroid carcinoma in our patient is consistent with the data from the literature. He also presented multinodular goiter associated with Hashimoto’s thyroiditis, which is common in case of deleterious mutation in PTEN, about 50% (Pilarski et al., 2013).
The risk of a second malignant neoplasm is higher in PTEN mutation carriers compared to the general population. From a population of 114 patients with deleterious PTEN mutations followed-up during 7 years, a second primary cancer was observed in 40% of cases, with a median interval of 5 years and median age at diagnosis of 50 years. A shorter interval was mostly associated with breast cancer (primary or secondary) (Tan et al., 2012). Of 59 PTEN mutation carriers, 20% had two primary cancers, and 5% had three cancers (Bubien et al., 2013). No patient presenting 4 primary tumors has been described in the literature.
Here, we describe for the first time a patient who developed four primary cancers over a period of 2 years. Obesity, the risk of which is increased by PTEN haploinsufficiency, could have contributed to the development of RC, which increases by 24 to 34% for every 5 kg/m2 rise in BMI (Chow et al., 2010; Pal et al., 2012). However, the change in risk remains low. Another hypothesis is the presence of genetic modifiers. In any case, PTEN mutation carriers should take measures to control BMI and stop smoking.
Non-Hodgkin’s lymphoma (NHL) is a heterogeneous cancer, with a worldwide incidence of 13.2 cases for 100,000 persons (Skrabek et al., 2013). The proband’s parent developed two rare forms in the NHL spectrum: orbital lymphoma and small intestine MALT lymphoma, representing respectively 1 and 1.60% of NHL. Most cases occur from 50 to 60 years old. Risk factors include mainly irradiation and infectious agents (Eckardt et al., 2013; Peng et al., 2015). Age of onset and recurrence of MALT lymphoma in the same patient without any known risk factor could suggest genetic factors.
Molecular Aspects
PTEN is a tumor suppressor gene involved in multiple biological processes of carcinogenesis, including regulation of cell growth, proliferation, angiogenesis, and apoptosis by dysregulation of the PI3K/AKT/mTOR pathway. Currently, therapies targeting this pathway showed benefits in vivo and in vitro. However, efficacies of treatments are temporary and can induce dysmorphologies if administered in early postnatal period (Tachibana et al., 2018).
De novo mutation in PTEN is frequent, ranging from 10.7 to 47.6% (Mester and Eng, 2012). The nonsense mutation c.1003C > T identified here is commonly observed in PTHS. Interestingly, no genotype–phenotype relationship has been found for mutations in PTEN and risk or severity of cancer in CS. Moreover, c.1003C > T can induce the full spectrum of PTHS, even in the same family (Table 3; 12, 14, 26, 40–48). This underlines the importance of genetic background and environmental factors.
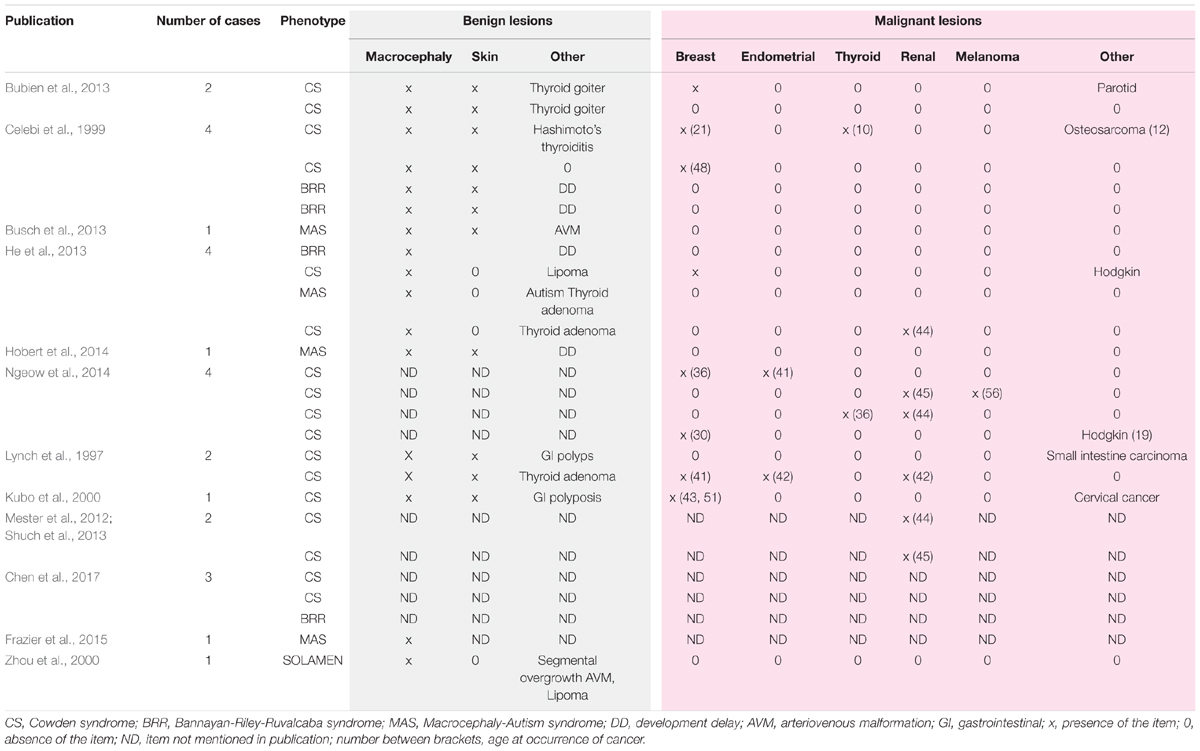
TABLE 3. Phenotypes and cancers associated with mutation c.1003C > T (p.Arg335X) in PTEN gene in literature.
PTEN is known to predispose to thyroid carcinoma. We did not observe LOH in the thyroid tumor samples, although this analysis was limited to the site of the mutation. Promotor methylation or acquired mutation elsewhere in the gene could lead to loss of function of PTEN, frequent in thyroid carcinoma (Alvarez-Nuñez et al., 2006).
Whole-exome-sequencing of the family made it possible to analyze the familial genetic background. Among the variants observed, two occurred in genes of particular interest: CEACAM1 and MIB2.
CEACAM1 encodes an immunoglobulin protein involved in cell–cell adhesion expressed in leukocytes, endothelial and epithelial tissues. CEACAM1 acts as a tumor suppressor gene in the PI3K/AKT pathway in certain epithelial tumors including clear cell RC, while de novo expression is a marker of cancer progression in other tumors including melanoma and thyroid carcinoma. Loss of expression was identified in all clear cell RC samples in a case series study, and in renal adenoma, suggesting early involvement in renal carcinogenesis (Kammerer et al., 2004). Thus, truncating mutations in this gene may contribute to the genesis of clear cell RC and could explain in part the early onset clear cell RC observed in the proband. Interestingly, CEACAM1 is also involved in regulation of lymphocyte B activation and proliferation. CEACAM1-specific monoclonal antibody induces strong B-cell proliferation in mouse, and acts as a negative co-receptor for the B cell receptor, promoting activation-induced cell death in B lymphocytes (Greicius et al., 2003; Lobo et al., 2009). The CEACAM1 mutation in the proband was inherited from a parent affected with MALT lymphoma, and may contribute to their lymphoproliferative B cell disease, and was absent from the two healthy persons in this study.
A deleterious mutation in MIB2 was transmitted by the proband’s healthy parent. MIB2 encodes an E3 ubiquitin protein ligase that mediates ubiquitination of protein in Notch pathways. LOH or promoter hypermethylation of MIB2 has been associated with melanoma invasion (Takeuchi et al., 2006). A deleterious mutation in this gene could play a role in the development of melanoma in our patient.
The mutation observed in MUTYH is known to be pathogenic and involved in MUTYH-associated polyposis, an autosomal recessive disorder. LOH was identified in the proband’s thyroid tumor sample. However, bi-allelic and mono-allelic carriers of MUTYH mutations are not known to be at risk of melanoma, renal or thyroid carcinoma (Kantor et al., 2017).
Four disease–associated genes in the proband presented VUS: APC, APOB, DSG2, and DSP, respectively associated with familial adenoma polyposis, homozygous familial hypercholesterolemia and hereditary cardiac disease (Awad et al., 2008; Basso et al., 2017; Defesche et al., 2017). The proband did not have signs of those syndromes.
Atypical presentation of CS described here could suggest systematic annual screening of RC and melanoma in PTEN mutation carriers, beginning respectively at 30 years of age and upon diagnosis. Furthermore, PTEN could be included in the panel analysis of precocious RC before 50 years, whatever the histological type, even in the absence of muco-cutaneous signs of CS. Prevention of obesity and smoking should be proposed. Heterogeneous phenotypes associated with mutation c.1003C > T p.(Arg335∗) suggests the presence of modifier genes. WES allowed analysis of genetic background of nuclear family and identified two candidate modifier genes: CEACAM1 and MIB2. Further studies are required to determine the implication of these genes in the severity of CS in the proband and occurrence of early onset MALT lymphoma in one parent.
Author Contributions
MC, YB, and Y-JB designed the study. MC, FP-C, and SV performed the acquisition of data. MC, FP-C, MG-B, MP, NU, YB, and Y-JB performed analysis, interpretation of data, and revised critically the article. MC and NU drafted the article.
Funding
This research has been supported by Centre Jean Perrin, Clermont-Ferrand, France. INSERM, U1240 Imagerie Moléculaire et Stratégies Théranostiques, Université Clermont Auvergne, Clermont-Ferrand, France.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alvarez-Nuñez, F., Bussaglia, E., Mauricio, D., Ybarra, J., Vilar, M., Lerma, E., et al. (2006). PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid 16, 17–23. doi: 10.1089/thy.2006.16.17
Apalla, Z., Lallas, A., Sotiriou, E., Lazaridou, E., and Ioannides, D. (2017). Epidemiological trends in skin cancer. Dermatol. Pract. Concept. 7, 1–6. doi: 10.5826/dpc.0702a01
Awad, M. M., Calkins, H., and Judge, D. P. (2008). Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 5, 258–267. doi: 10.1038/ncpcardio1182
Basso, G., Bianchi, P., Malesci, A., and Laghi, L. (2017). Hereditary or sporadic polyposis syndromes. Best Pract. Res. Clin. Gastroenterol. 31, 409–417. doi: 10.1016/j.bpg.2017.05.011
Blumenthal, G. M., and Dennis, P. A. (2008). PTEN hamartoma tumor syndromes. Eur. J. Hum. Genet. 16, 1289–1300. doi: 10.1038/ejhg.2008.162
Bubien, V., Bonnet, F., Brouste, V., Hoppe, S., Barouk-Simonet, E., David, A., et al. (2013). High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J. Med. Genet. 50, 255–263. doi: 10.1136/jmedgenet-2012-101339
Busch, R. M., Chapin, J. S., Mester, J., Ferguson, L., Haut, J. S., Frazier, T. W., et al. (2013). The cognitive characteristics of pten hamartoma tumor syndromes. Genet. Med. 15, 548–553. doi: 10.1038/gim.2013.1
Butler, M. G., Dasouki, M. J., Zhou, X.-P., Talebizadeh, Z., Brown, M., Takahashi, T. N., et al. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321. doi: 10.1136/jmg.2004.024646
Carrera, C., and Puig-Butille, J. A. (2018). Clinical, epidemiological, and molecular heterogeneity in acral melanoma. J. Investig. Dermatol. 138, 254–255. doi: 10.1016/j.jid.2017.09.027
Caux, F., Plauchu, H., Chibon, F., Faivre, L., Fain, O., Vabres, P., et al. (2007). Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur. J. Hum. Genet. 15, 767–773. doi: 10.1038/sj.ejhg.5201823
Celebi, J. T., Tsou, H., Chen, F. F., Zhang, H., Ping, X. L., Lebwohl, M., et al. (1999). Phenotypic findings of Cowden syndrome and Bannayan-Zonana syndrome in a family associated with a single germline mutation in PTEN. J. Med. Genet. 36, 360–364.
Chen, H. H., Händel, N., Ngeow, J., Muller, J., Hühn, M., Yang, H.-T., et al. (2017). Immune dysregulation in patients with PTEN hamartoma tumor syndrome: analysis of FOXP3 regulatory T cells. J. Allergy Clin. Immunol. 139, 607.e15–620.e15. doi: 10.1016/j.jaci.2016.03.059
Chow, W.-H., Dong, L. M., and Devesa, S. S. (2010). Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 7, 245–257. doi: 10.1038/nrurol.2010.46
Defesche, J. C., Gidding, S. S., Harada-Shiba, M., Hegele, R. A., Santos, R. D., and Wierzbicki, A. S. (2017). Familial hypercholesterolaemia. Nat. Rev. Dis. Primers 3:17093. doi: 10.1038/nrdp.2017.93
Eckardt, A. M., Lemound, J., Rana, M., and Gellrich, N.-C. (2013). Orbital lymphoma: diagnostic approach and treatment outcome. World J. Surg. Oncol. 11, 73. doi: 10.1186/1477-7819-11-73
Frazier, T. W., Embacher, R., Tilot, A. K., Koenig, K., Mester, J., and Eng, C. (2015). Molecular and phenotypic abnormalities in individuals with germline heterozygous pten mutations and autism. Mol. Psychiatry 20, 1132–1138. doi: 10.1038/mp.2014.125
Gammon, A., Jasperson, K., and Champine, M. (2016). Genetic basis of cowden syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 9, 83–92. doi: 10.2147/TACG.S41947
Green, R. C., Berg, J. S., Grody, W. W., Kalia, S. S., Korf, B. R., Martin, C. L., et al. (2013). ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 15, 565–574. doi: 10.1038/gim.2013.73
Greicius, G., Severinson, E., Beauchemin, N., Obrink, B., and Singer, B. B. (2003). CEACAM1 is a potent regulator of B cell receptor complex-induced activation. J. Leukoc. Biol. 74, 126–134. doi: 10.1189/jlb.1202594
Gupta, K., Nizam, A., Millado, K., Liu, J., Guebre-Xabiher, H., and Nguyen, H. (2017). Retrospective review of clear cell and non-clear cell renal carcinomas: characteristics and course in the pre-TKI (tyrosine kinase inhibitor) and post-TKI era. J. Clin. Oncol. 35:e16052.
Haas, N. B., and Nathanson, K. L. (2014). Hereditary kidney cancer syndromes. Adv. Chronic Kidney Dis. 21, 81–90. doi: 10.1053/j.ackd.2013.10.001
He, X., Arrotta, N., Radhakrishnan, D., Wang, Y., Romigh, T., and Eng, C. (2013). Cowden syndrome-related mutations in PTEN associate with enhanced proteasome activity. Cancer Res. 73, 3029–3040. doi: 10.1158/0008-5472.CAN-12-3811
Hobert, J. A., Embacher, R., Mester, J. L., Frazier, T. W., and Eng, C. (2014). Biochemical screening and PTEN mutation analysis in individuals with autism spectrum disorders and macrocephaly. Eur. J. Hum. Genet. 22, 273–276. doi: 10.1038/ejhg.2013.114
Kammerer, R., Riesenberg, R., Weiler, C., Lohrmann, J., Schleypen, J., and Zimmermann, W. (2004). The tumour suppressor gene CEACAM1 is completely but reversibly downregulated in renal cell carcinoma. J. Pathol. 204, 258–267. doi: 10.1002/path.1657
Kantor, M., Sobrado, J., Patel, S., Eiseler, S., and Ochner, C. (2017). Hereditary colorectal tumors: a literature review on mutyh-associated polyposis. Gastroenterol. Res. Pract. 2017, 1–4. doi: 10.1155/2017/8693182
Kubo, Y., Urano, Y., Hida, Y., Ikeuchi, T., Nomoto, M., Kunitomo, K., et al. (2000). A novel PTEN mutation in a Japanese patient with Cowden disease. Br. J. Dermatol. 142, 1100–1105. doi: 10.1046/j.1365-2133.2000.03533.x
Lachlan, K. L., Lucassen, A. M., Bunyan, D., and Temple, I. K. (2007). Cowden syndrome and Bannayan Riley Ruvalcaba syndrome represent one condition with variable expression and age-related penetrance: results of a clinical study of PTEN mutation carriers. J. Med. Genet. 44, 579–585. doi: 10.1136/jmg.2007.049981
Laury, A. R., Bongiovanni, M., Tille, J.-C., Kozakewich, H., and Nosé, V. (2011). Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid 21, 135–144. doi: 10.1089/thy.2010.0226
Levi, Z., Baris, H. N., Kedar, I., Niv, Y., Geller, A., Gal, E., et al. (2011). Upper and lower gastrointestinal findings in pten mutation-positive cowden syndrome patients participating in an active surveillance program. Clin. Transl. Gastroenterol. 2:e5. doi: 10.1038/ctg.2011.4
Lobo, E. O., Zhang, Z., and Shively, J. E. (2009). Pivotal Advance: CEACAM1 is a negative coreceptor for the B cell receptor and promotes CD19-mediated adhesion of B cells in a PI3K-dependent manner. J. Leukoc. Biol. 86, 205–218. doi: 10.1189/jlb.0109037
Lynch, E. D., Ostermeyer, E. A., Lee, M. K., Arena, J. F., Ji, H., Dann, J., et al. (1997). Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am. J. Hum. Genet. 61, 1254–1260. doi: 10.1086/301639
Matthijs, G., Souche, E., Alders, M., Corveleyn, A., Eck, S., Feenstra, I., et al. (2016). Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 24, 2–5. doi: 10.1038/ejhg.2015.226
Mester, J., and Eng, C. (2012). Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet. Med. 14, 819–822. doi: 10.1038/gim.2012.51
Mester, J. L., Tilot, A. K., Rybicki, L. A., Frazier, T. W., and Eng, C. (2011). Analysis of prevalence and degree of macrocephaly in patients with germline PTEN mutations and of brain weight in Pten knock-in murine model. Eur. J. Hum. Genet. 19, 763–768. doi: 10.1038/ejhg.2011.20
Mester, J. L., Zhou, M., Prescott, N., and Eng, C. (2012). Papillary renal cell carcinoma is associated with pten hamartoma tumor syndrome. Urology 79, 1187.e1–1187.e1. doi: 10.1016/j.urology.2011.12.025
Milas, M., Mester, J., Metzger, R., Shin, J., Mitchell, J., Berber, E., et al. (2012). Should patients with Cowden syndrome undergo prophylactic thyroidectomy? Surgery 152, 1201–1210. doi: 10.1016/j.surg.2012.08.055
Nelen, M. R., Kremer, H., Konings, I. B., Schoute, F., van Essen, A. J., Koch, R., et al. (1999). Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur. J. Hum. Genet. 7, 267–273. doi: 10.1038/sj.ejhg.5200289
Ngeow, J., and Eng, C. (2015). PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol. Methods 7, 11–19. doi: 10.1016/j.ymeth.2014.10.011
Ngeow, J., Stanuch, K., Mester, J. L., Barnholtz-Sloan, J. S., and Eng, C. (2014). Second malignant neoplasms in patients with cowden syndrome with underlying germline PTEN mutations. J. Clin. Oncol. 32, 1818–1824. doi: 10.1200/JCO.2013.53.6656
Nguyen, Q. T., Lee, E. J., Huang, M. G., Park, Y. I., Khullar, A., and Plodkowski, R. A. (2015). Diagnosis and treatment of patients with thyroid cancer. Am. Health Drug Benefits 8, 30–40.
Nieuwenhuis, M. H., Kets, C. M., Murphy-Ryan, M., Yntema, H. G., Evans, D. G., Colas, C., et al. (2014). Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam. Cancer 13, 57–63. doi: 10.1007/s10689-013-9674-3
Nosé, V. (2016). Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—pten-hamartoma tumor syndrome/cowden syndrome. Head Neck Pathol. 10, 131–138. doi: 10.1007/s12105-016-0708-7
Pal, A., Barber, T. M., Van de Bunt, M., Rudge, S. A., Zhang, Q., Lachlan, K. L., et al. (2012). PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N. Engl. J. Med. 367, 1002–1011. doi: 10.1056/NEJMoa1113966
Peng, J. C., Zhong, L., and Ran, Z. H. (2015). Primary lymphomas in the gastrointestinal tract. J. Dig. Dis. 16, 169–176. doi: 10.1111/1751-2980.12234
Pilarski, R. (2009). Cowden syndrome: a critical review of the clinical literature. J. Genet. Couns. 18, 13–27. doi: 10.1007/s10897-008-9187-7
Pilarski, R., Burt, R., Kohlman, W., Pho, L., Shannon, K. M., and Swisher, E. (2013). Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 105, 1607–1616. doi: 10.1093/jnci/djt277
Pilarski, R., Stephens, J. A., Noss, R., Fisher, J. L., and Prior, T. W. (2011). Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan–Riley–Ruvalcaba syndrome clinical features. J. Med. Genet. 48, 505–512. doi: 10.1136/jmg.2011.088807
Saslow, D., Solomon, D., Lawson, H. W., Killackey, M., Kulasingam, S. L., Cain, J., et al. (2012). American cancer society, american society for colposcopy and cervical pathology, and american society for clinical pathology screening guidelines for the prevention and early detection of cervical cancer. Am. J. Clin. Pathol. 137, 516–542. doi: 10.1309/AJCPTGD94EVRSJCG
Shuch, B., Ricketts, C. J., Vocke, C. D., Komiya, T., Middelton, L. A., Kauffman, E. C., et al. (2013). Germline PTEN mutation cowden syndrome: an under-appreciated form of hereditary kidney cancer. J. Urol. 190, 1990–1998. doi: 10.1016/j.juro.2013.06.012
Skrabek, P., Turner, D., and Seftel, M. (2013). Epidemiology of non-hodgkin lymphoma. Transfus. Apher. Sci. 49, 133–138. doi: 10.1016/j.transci.2013.07.014
Stanich, P. P., Pilarski, R., Rock, J., Frankel, W. L., El-Dika, S., and Meyer, M. M. (2014). Colonic manifestations of PTEN hamartoma tumor syndrome: case series and systematic review. World J. Gastroenterol. 20, 1833–1838. doi: 10.3748/wjg.v20.i7.1833
Tachibana, N., Touahri, Y., Dixit, R., David, L. A., Adnani, L., Cantrup, R., et al. (2018). Hamartoma-like lesions in the mouse retina: an animal model of Pten hamartoma tumour syndrome. Dis. Model Mech. 11:dmm031005. doi: 10.1242/dmm.031005
Takeuchi, T., Adachi, Y., Sonobe, H., Furihata, M., and Ohtsuki, Y. (2006). A ubiquitin ligase, skeletrophin, is a negative regulator of melanoma invasion. Oncogene 25, 7059–7069. doi: 10.1038/sj.onc.1209688
Tan, M.-H., Mester, J. L., Ngeow, J., Rybicki, L. A., Orloff, M. S., and Eng, C. (2012). Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 18, 400–407. doi: 10.1158/1078-0432.CCR-11-2283
Woodhouse, J. B., Delahunt, B., English, S. F., Fraser, H. H., and Ferguson, M. M. (2005). Testicular lipomatosis in Cowden’s syndrome. Mod. Pathol. 18, 1151–1156. doi: 10.1038/modpathol.3800448
Zhou, X. P., Marsh, D. J., Hampel, H., Mulliken, J. B., Gimm, O., and Eng, C. (2000). Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum. Mol. Genet. 9, 765–768. doi: 10.1093/hmg/9.5.765
Zhou, X.-P., Marsh, D. J., Morrison, C. D., Chaudhury, A. R., Maxwell, M., Reifenberger, G., et al. (2003). Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am. J. Hum. Genet. 73, 1191–1198. doi: 10.1086/379382
Keywords: Cowden syndrome, PTEN, CEACAM1, MIB2, melanoma, renal carcinoma, MALT lymphoma, whole-exome-sequencing
Citation: Cavaillé M, Ponelle-Chachuat F, Uhrhammer N, Viala S, Gay-Bellile M, Privat M, Bidet Y and Bignon Y-J (2018) Early Onset Multiple Primary Tumors in Atypical Presentation of Cowden Syndrome Identified by Whole-Exome-Sequencing. Front. Genet. 9:353. doi: 10.3389/fgene.2018.00353
Received: 05 April 2018; Accepted: 10 August 2018;
Published: 31 August 2018.
Edited by:
Ingrid A. Hedenfalk, Lund University, SwedenReviewed by:
Hernan G. Valdes-Socin, Centre Hospitalier Universitaire de Liège, BelgiumJorge Melendez-Zajgla, Instituto Nacional de Medicina Genómica (INMEGEN), Mexico
Copyright © 2018 Cavaillé, Ponelle-Chachuat, Uhrhammer, Viala, Gay-Bellile, Privat, Bidet and Bignon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathias Cavaillé, bWF0aGlhcy5jYXZhaWxsZUBjbGVybW9udC51bmljYW5jZXIuZnI= Yves-Jean Bignon, eXZlcy1qZWFuLmJpZ25vbkBjbGVybW9udC51bmljYW5jZXIuZnI=