 Aarti Rana
Aarti Rana Shweta Thakur
Shweta Thakur Girish Kumar
Girish Kumar Yusuf Akhter
Yusuf Akhter- 1School of Life Sciences, Central University of Himachal Pradesh, Shahpur, India
- 2Department of Biotechnology, Babasaheb Bhimrao Ambedkar University, Lucknow, India
Mycobacterial infections are one of the deadliest infectious diseases still posing a major health burden worldwide. The battle against these pathogens needs to focus on novel approaches and key interventions. In recent times, availability of genome scale data has revolutionized the fields of computational biology and immunoproteomics. Here, we summarize the cutting-edge ‘omics’ technologies and innovative system scale strategies exploited to mine the available data. These may be targeted using high-throughput technologies to expedite the identification of novel antigenic candidates for the rational next generation vaccines and serodiagnostic development against mycobacterial pathogens for which traditional methods have been failing.
Introduction
Despite the massive advancements over the years in the field of effective clinical interventions, a big number of people in the developing countries still suffer from an enormous burden of contagious diseases. Various pathogens such as viruses, bacteria, parasites and fungi are responsible for these widespread infections (Janeway et al., 2001). Over the past decade, among them, mycobacteria are recognized as the most common cause of serious illness and deaths globally (WHO, 2016). The mycobacterial pathogens continually present us with ongoing threats to human and animal health and challenge our endeavors to obstruct and control infectious diseases. Among these, Mycobacterium tuberculosis (Mtb), M. leprae, M. bovis and M. avium subsp. paratuberculosis (MAP) are the four largely known and well established mycobacterial species that can cause a variety of dreadful infectious diseases, such as tuberculosis (TB), leprosy in humans and paratuberculosis in animals (Hoffmann et al., 2018). The overall disease burden posed by these microbes has been constantly on the rise and hence, it is crucial to stop their spread by developing sensitive diagnostic tools for their early detection and design effective vaccines to generate long-term immunoprotection against such infections.
Commonly Available Prophylactic Health Interventions Against Mycobacterial Infections
Foundation of modern medicine has been laid down on valuable anti-infective drugs now in use. However, the rapid evolution of antibiotic resistance has now become a limitingcondition that may impose a considerable economic burden and endanger the efficacy of antibiotics for the control of many infectious diseases (Fair and Tor, 2014). Antibiotic resistance is a disaster which arises due to the excessive exploitation of medications, as well as a lack of new effective vaccines manufactured by the pharmaceutical industry (Ventola, 2015). Therefore, discovering new prophylactic treatments to remedy the infectious diseases has been a major focus of modern medicine. Below, in the next subsections, currently available vaccine candidates and their safety issues have been discussed.
Vaccines
Vaccines were used extensively before the antibiotics became accessible. Vaccination proves to be the most successful available strategy of an integrated prevention/therapeutic toolkit. It has significantly reduced the prevalence of a variety of infectious diseases such as bacterial and viral infections. It has slowed down the rate of development of resistant strains thereby preventing the further spread of several devastating infections globally (Andre et al., 2008; Rana and Akhter, 2016). A vaccine represents a biological formulation which upon administration to a given population can generate life time’s immunity against a particular disease (Mohan et al., 2013). First generation vaccines were developed using attenuated or inactivated strains of microbial pathogens. These have been reported as efficient for inciting both humoral and cellular immune responses (Seder and Hill, 2000). The second generation vaccine is composed of pathogen-derived purified components (devoid of the factors responsible for infection) instead of the whole microbial cells. These have been developed using novel recombinant proteins and DNA molecules (rDNA technology) as well as non-virulent but immunoprotective forms of microbial pathogens. The high-throughput sequencing and availability of complete genomic information have paved the way to a new ‘third generation’ of the vaccines (Seib et al., 2009). On vaccine administration, the vaccinated individual’s immune system encounters antigens expressed by disease-causing foreign pathogen and remembers it in form of immunological memory. This immunological memory, when encounters the real microbe expressing those antigens, there is production and activation of highly specific memory T lymphocytes, B lymphocytes and natural killer cells (Ratajczak et al., 2018). This rapidly generates an effective immune response against the microbial pathogen (Ottenhoff and Kaufmann, 2012). Hence, the most important job of vaccines is to expose the vaccinated individuals with much milder and non-virulent pathogenic antigens to generate immunological memory without actually causing the disease. A brief history of major breakthroughs in vaccine development has been illustrated in Figure 1.
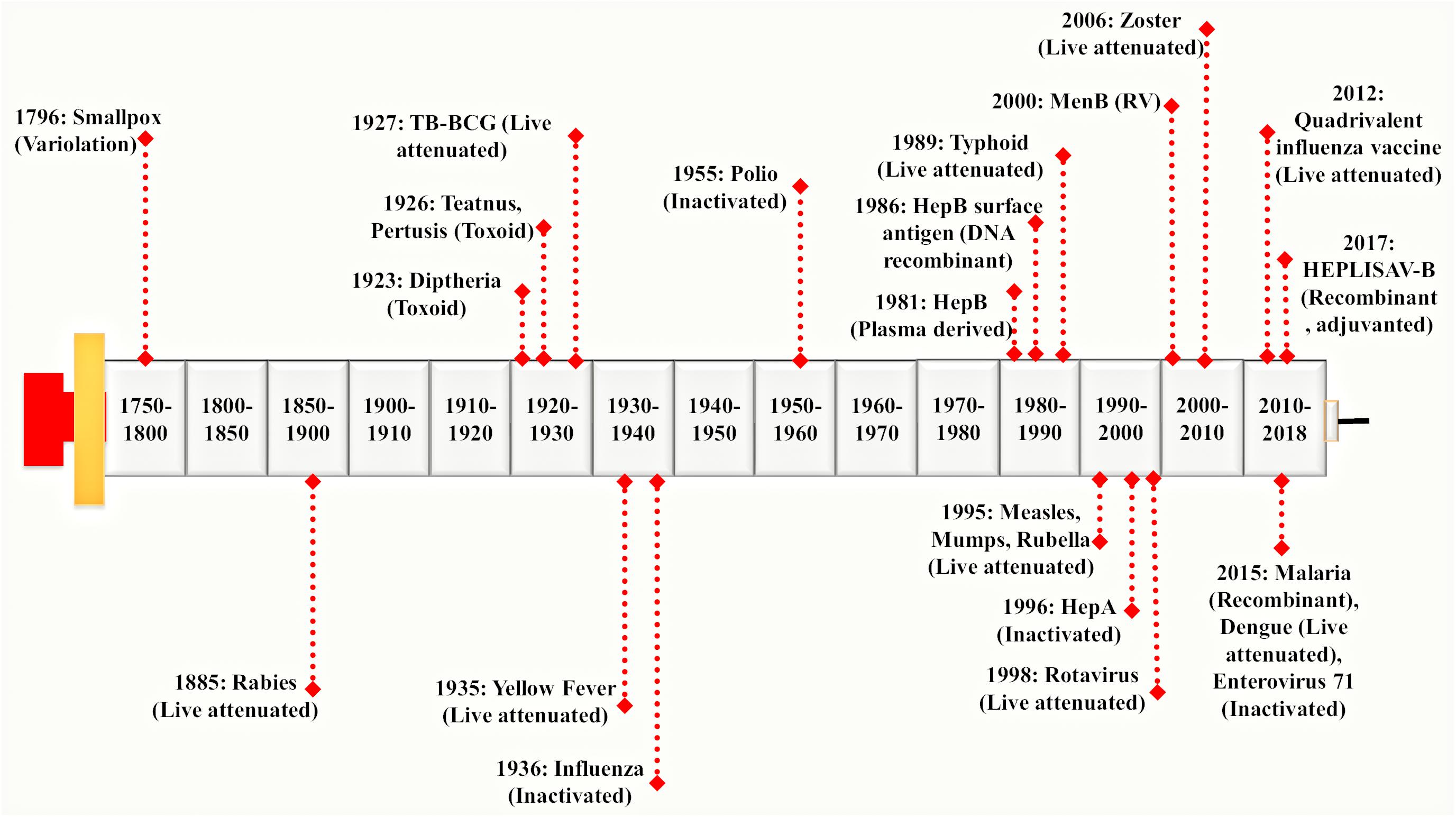
FIGURE 1. Evolution of vaccine development processes: Vaccine development was pioneered by Edward Jenner. He discovered a working vaccine against small pox in 1796 derived by variolation and further work was continued by Louis Pasteur. He has discovered a live attenuated vaccine against Rabies virus in 1885 considered to be one of the 1st generation vaccines. It was followed by a genomic revolution and in the post-genomic era, mankind witnessed the modern sequencing techniques. In early 21st century, Rappuoli introduced Reverse Vaccinology (RV) approach which provided a foundation to the development of 2nd generation vaccines (Rappuoli et al., 2016). Since then, advances in various ‘omics-based’ approaches together with RV led to the development of a much more advanced 3rd generation of vaccines in the present times. Different vaccines derived from variolation, live attenuated, inactivated, toxoid, DNA recombinant have been shown in the timeline.
The most commonly used first generation vaccine against the mycobacterial pathogens is Bacillus Calmette-Guerin (BCG). It is composed of attenuated (non-virulent) strains of M. bovis. In the following subsections, we are summarizing the current use and protection status provided by the BCG vaccine.
The BCG Vaccine
Currently, BCG is the only TB vaccine which is inexpensive, safe and readily available. It is composed of live attenuated strains of M. bovis (Lahey and Von Reyn, 2016). It induces an immune response against the Mycobacterium without actually causing the disease (Trunz et al., 2006). Since it is cheap, it is considered as the most economical way to provide protection to millions of children against TB and leprosy globally (Zwerling et al., 2011). Although the BCG vaccine is one of the oldest extensively used vaccine, it may not be presented as the most successful available strategy. BCG has been reported with incomplete protection against Mtb and M. leprae infection. Over previous decades, different clinical trials and epidemiological studies have been conducted to evaluate the efficacy of BCG in many countries (Trunz et al., 2006). Studies showed that BCG vaccination provides 60–80% protective efficacy to prevent dissemination in children who were otherwise suffering from TB, meningitis, miliary disease and pulmonary TB (Roy et al., 2014). Despite its widespread use, BCG vaccine has been reported to be less effective in TB endemic zones (Brandt et al., 2002).
In the case of leprosy, numerous attempts have been made for the development of a highly specific vaccine against leprosy but still, the efforts have not met with complete success. Currently, the only licensed vaccine administered for protection against M. leprae is the BCG vaccine. This protection has been reported to wane over time as is the case with BCG generated protection against TB infection (Duthie et al., 2011, 2012). Therefore, there is a great need for the discovery of ideal vaccines that may provide better protective efficacy against TB and leprosy. To better understand the mechanistic details about the failures of the BCG vaccine, in the following subsections safety issues, diversity among various BCG strains and their molecular evolution have been discussed.
Safety Issues and Variability in the Efficacy of BCG Vaccine
Bacillus Calmette-Guerin vaccine has been used as a “gold standard” because of its cosmopolitan availability and cost-effectiveness. BCG side-effects are usually very rare and include inflammation at the site of injection among vaccinated individuals (Rowland and McShane, 2011). Another important BCG vaccine safety issue for consideration is its efficacy among the immune-compromised individuals. In the HIV-positive children, an increased risk of diseases was reported which ultimately forced the WHO to put forward the restriction on BCG vaccine administration among HIV-positive children (Brennan and Thole, 2012). The HIV infected immune-compromised individuals administered with BCG vaccine have been observed with an onset of BCG disease because of the primary immunodeficiency. As BCG activates the CD4+ T cells (HIV targeted cells), it may increase the susceptibility of children to HIV infection and accelerate HIV disease progression (Santema et al., 2013). A number of reports have been cited in the literature demonstrating the wide-ranging variability observed in the BCG efficacy. A majority of the reports suggest a nearly 80% BCG efficacy while some of the reports conclude that BCG is completely ineffective (Mangtani et al., 2013). Some studies have reported that BCG administration to children may result in mycobacterial dissemination to various other organs also, which may prove lethal. Moreover, BCG fails to generate complete protection in a patient suffering from adult pulmonary TB (Kernodle, 2010). Some of the major potential reasons responsible for the observed changes in the efficiency of BCG are covered in the following subsections. These include the genetic variability within available versions of attenuated BCG strains and the genetic immuno-polymorphism among the human populations on which the vaccine has been administered. Prior exposure to mycobacterial strains (including environmental mycobacteria) affecting the outcomes of vaccine trials has also been discussed briefly.
BCG strain variation
The M. bovis BCG parent strain was originally developed in 1921 at the Pasteur Institute. The attenuated form of M. bovis was derived through the serial passage of virulent Mycobacterium isolated from a cow suffering from tuberculous mastitis. This attenuated strain was disseminated to several laboratories and developed into different sub-strains possessing different characteristics worldwide (Oettinger et al., 1999). These include evolutionarily early BCG strain: Japan and the evolutionarily late BCG strains: Connaught, Glaxo, Pasteur, Danish and Tice. Some of the commonly used BCG strains for the development of BCG vaccine have been mentioned in Table 1 (Ritz and Curtis, 2009; World Health Organization, Immunization, Vaccines and Biologicals Department, 2012; da Costa et al., 2014).
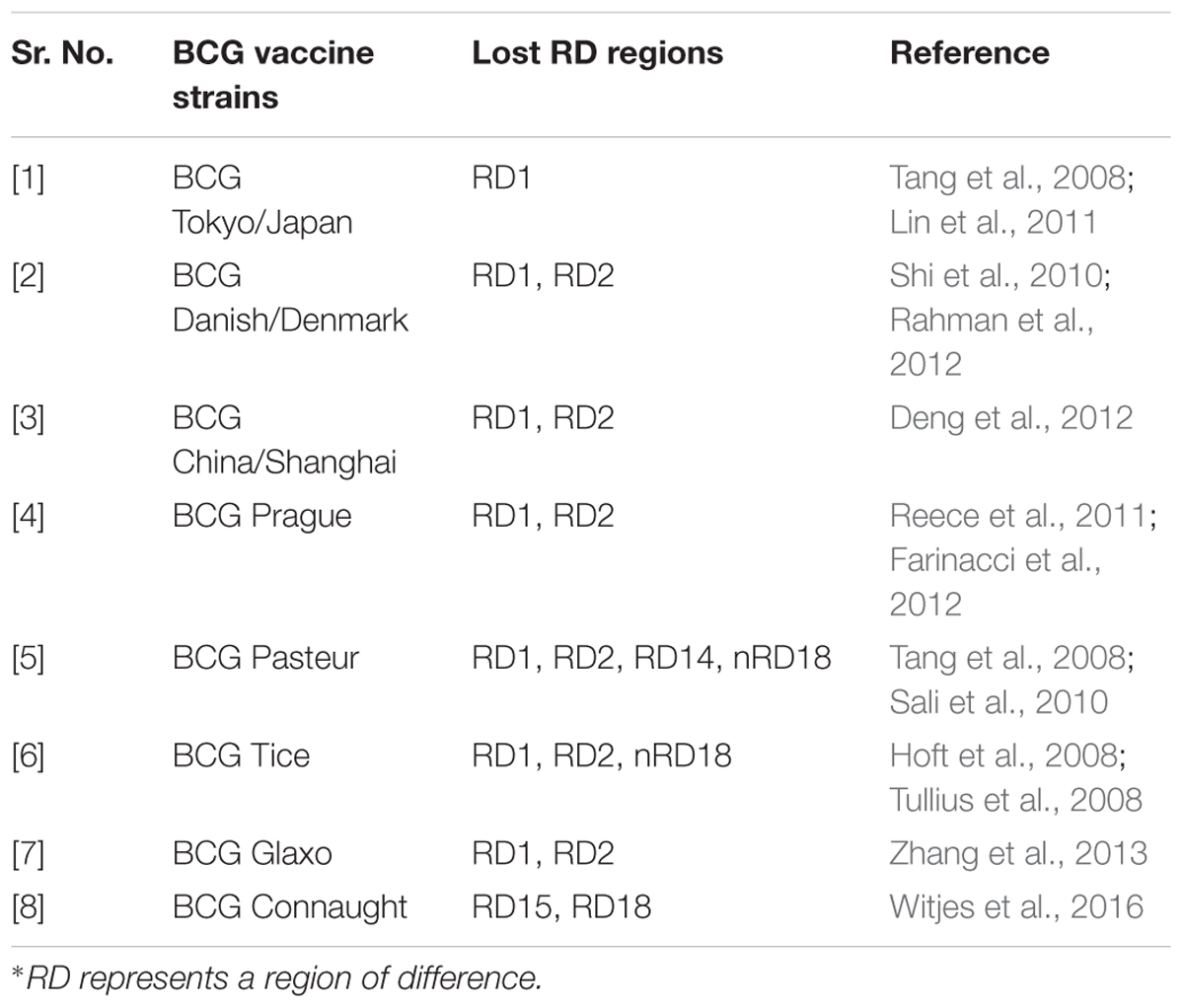
TABLE 1. Different BCG vaccine strains available worldwide.
Diversified genetic make-up of the test individuals
A vaccine’s efficacy is more or less dependent upon the genetic make-up of the test population. The variation in the form of single nucleotide polymorphisms (SNPs) in the test population genomes can affect their susceptibility to disease and its outcome. It also governs the protective immune response generated by a particular vaccine (MacDonald and Izzo, 2015). The immune response may vary from complete protection to no protection at all. A study demonstrating the genetic variation effect on BCG vaccine efficacy has reported dissemination of disease after BCG administration in patients with mutated IFN-γ receptors (Döffinger et al., 2002). The earlier conducted BCG vaccination clinical trials have shown consistently a suitable immune response against Mtb in UK infants, but on the other hand offered a very little to nil protection among infants of Malawi. This noticeable population variance in the generated immune response against BCG vaccine indicates that it might not be possible to offer equal immunity to the infants from different countries (Lalor et al., 2011). The BCG administered Malawian infants were found to develop T cell immune response with an early cytokine profile that was found to be completely different from that generated among the BCG vaccinated UK infants. This was characterized by the presence of a large population of antigen-specific IFNγ dominated Th1 cells (Lalor et al., 2011). While another study conducted on BCG-vaccinated infants from Indonesia recognized marked induction of not only IFNγ but also of IL-5 and IL-13 in contrast with the findings from the Malawian and UK infants (Djuardi et al., 2010). Hence, the different cytokine bio-signatures generated in the form of immune responses following BCG vaccination in population with differences in their genetic makeup could be attributed as one of the important reasons for observed variability in efficacy of BCG vaccine (Lalor et al., 2009; Dockrell et al., 2012; Kollmann, 2013). Moreover, individuals with observed mutational changes in genes susceptible to a particular disease become highly vulnerable to various other commonly found mycobacterial infections from the environment (Döffinger et al., 2002). Therefore, monitoring of vaccine trials, with appropriate biomarker measurements and genomic diversity of the test individuals must be considered as there is no homogenous population distribution in the world. Therefore, the criteria to carry out a clinical trial for any antimycobacterial vaccine candidate should be laid down carefully.
Pre-exposure to the pathogens and related environmental mycobacteria
Another significant issue of huge importance to be considered while conducting BCG efficacy tests is an individual’s pre-exposure to the pathogen. An individual with a pre-exposure to a particular antigen has a different immune response as compared to someone with no earlier exposure to the antigen. For instance, the children in countries with TB and leprosy-endemic zones have a pre-exposure to Mtb and M. leprae. During various TB and leprosy eradication programs, a huge variability has been observed in the generated immune response on BCG administration among children (Andersen and Woodworth, 2014). Additionally, exposure to the environmental mycobacteria including the non-tuberculous mycobacteria (NTM) found in water and soil generates cross-reactive immune responses which further blocks the BCG activity (Demangel et al., 2005; Halstrom et al., 2015). Hence, a highly efficient and effective vaccine should thus be passed through extremely stringent clinical testing which should consider only those individuals with no pathogen pre-exposure (Mangtani et al., 2013).
Conventional Approaches to Vaccine Development
In 1880, Louis Pasteur when administered Pasteurella septica in chickens, it generated protection against fresh virulent bacterium in the chickens. This demonstrated that the pathogenic bacteria lost disease-causing properties and got completely attenuated (changed into the non-virulent but immunoprotective forms) (Movahedi and Hampson, 2008). Subsequently, a year later, he prepared a vaccine against anthrax using attenuated forms of Bacillus anthracis. His novel approach was further utilized by the scientific community to form the foundation of vaccine discovery. It consists of isolating the pathogen, its attenuation followed by administration of the antigenic pathogen. This approach has allowed the development of vaccines against prevalent diseases in the twentieth century (Serruto and Rappuoli, 2006; Meeusen et al., 2007; Movahedi and Hampson, 2008). The conventionally developed vaccine is based on 2 approaches: attenuating the targeted microbial pathogens in vitro by growing it in growth media several times to obtain a viable non-virulent strain and identifying highly specific potential antigenic components from microbial pathogens (Sette and Rappuoli, 2010). The immunodominant antigenic components of the targeted pathogens are identified by various sera-based methods and molecular genetics based methods. These conventionally available methods are very cumbersome, extremely slow and costly. Moreover, these methods can only be used to identify the highly abundant antigenic components which can be extracted in enough quantities appropriate for vaccine development (Bagnoli et al., 2011). Since the biological methods needed to isolate such components are poor in number, it generally takes decades to identify suitable antigenic molecules for vaccine development. The total number of identified potential immunogens to be used in vaccine development is extremely poor. It is documented that only 25 infections have licensed vaccines (WHO, 2012). These conventional approaches also fail when the microbial pathogens fail to grow in laboratory conditions on available supplemented/not supplemented artificial media (Donati and Rappuoli, 2013).
Current Status of Known Biomarkers for Diagnostic Assays
In order to completely control and eradicate mycobacterial infections globally, accurate diagnosis followed by effective treatment is required. However, there are no gold standard diagnostic tests available against these mycobacterial infections. The available detection tools lack specificity and accuracy. Among the available diagnostic tools for Mtb detection, the tuberculin skin test (TST; standard is the Mantoux test) and interferon (IFN-γ) release assay (IGRA) are widely used. These both are indirect markers for the detection of Mtb infection and measure a cellular immune response to Mtb. Some of the challenges faced by these tools include incompetency to distinguish between active and latent TB, failure to differentiate reinfection from reactivation and poor sensitivity among immunocompromised patients (Pai et al., 2014). In TST, a delayed type 4 hypersensitivity reaction is generated when the purified protein derivative (PPD) obtained from Mtb is injected into the patient. It generally takes 48–72 h for obtaining the final results. This delay may mislay the patient’s compliance and exposure. In addition, the TST as well as some other newly developed serological tests, fail to distinguish between exposure to infectious Mtb and other environmental NTM. Hence, the performance of these diagnostic tools is continuously deteriorating and cannot be relied upon (Doan et al., 2017).
Currently, better serodiagnostic assays with high specificity for pathogenic mycobacterial infections and more sensitive than the available diagnostic tools are needed. One of the newly developed methods for the rapid detection of Mtb includes a nucleic acid amplification assay (NAAA) which targets the insertion sequence (IS) 6110 sequence from Mtb. It combines two PCR techniques: nested polymerase chain reaction (Nested PCR) and real-time polymerase chain reaction (Real-time PCR) in a single tube. The nucleic acid amplification test IS6110 has shown high levels of sensitivity to detect the presence of Mtb. One-tube nested RT-PCR is 100 times more sensitive in comparison to conventional RT-PCR (Choi et al., 2014). In another study, the culture and mpt64RT-PCR demonstrated the same sensitivity (90.3%) in sputum samples. While, mpt64RT-PCR recorded 98.6% specificity in comparison to culture (99.4%) and smear microscopy (99.7%). Hence, this modern day molecular technique NAAA can be utilized in routine laboratories enabling quick and specific TB detection within 5 h (Laux da Costa et al., 2015; Watanabe Pinhata et al., 2015).
In leprosy, the conventional diagnostic tools are usually dependent upon histopathology and bacillary counts of skin smears. Since M. leprae presents tropism for the skin (macrophages) and peripheral nerves (Schwann cells), the slit-skin smear (SSS) still remains the gold standard technique of choice for leprosy diagnosis. Serological tests detecting IgM antibodies against phenolic glycolipid-I (PGL-I; M. leprae cell surface antigen) and IFN-gamma releasing assays (IGRA) detecting IFN-gamma production are also being widely used for diagnosis of M. leprae. These classical methods have been found incompetent to distinguish the active disease from a latent form of M. leprae infection and are inefficient to diagnose the paucibacillary clinical forms of Hansen’s disease. Among the modern-day molecular techniques, especially PCR has emerged as an alternative tool for molecular diagnosis among the hard to diagnose cases of leprosy (neural, paucibacillary and indeterminate leprosy). In fact, the advances in M. leprae structural and functional genomics has allowed the development of highly specific PCR-based gene amplification assays for early rapid M. leprae DNA detection with high sensitivity. PCR has also proved useful in the M. leprae viability determination, identification of routes of transmission and leprosy drug resistance (Geluk et al., 2012; Martinez et al., 2014; Soto and Muñoz, 2015; Maltempe et al., 2016).
In case of Crohn’s disease, the MAP can be detected in infected animal’s milk samples via culture, enzyme-linked immunosorbent assay (ELISA) (Sorge et al., 2011), immunomagnetic separation (IMS) and PCR. For the detection of subclinical MAP infections, various serological methods like agar gel immunodiffusion, complement fixation and ELISA methods have been widely used.
Numerous epidemiological studies are still being carried out to find a reliable molecular method for the rapid and accurate detection of paratuberculosis from clinical samples. These include: real-time PCR, mycobacteria interspersed repetitive units (MIRU) typing, variable number tandem repeat (VNTR) typing, immunomagnetic separation-PCR (IMS-PCR), nested PCR, pulsed-field gel electrophoresis (PFGE), multiplex PCR and IS900 restriction fragment length polymorphism (RFLP) (McKenna et al., 2005; Romano et al., 2005; GÜMÜŞSOY et al., 2015).
In recent times, a number of potential diagnostic biomarkers have also been identified and are under study against mycobacterial infections. The recombinant proteins generated through a combination of secretory proteins from Mtb, Hsp16.3/ESAT6 and Ag85B-Hsp16.3/ESAT6 has been identified as highly potentially antigenic which may be targeted as serodiagnostic biomarkers (Zhang et al., 2015). These may represent the preliminary screening antigens against active TB. Mtb antigens, Rv1681 (Pollock et al., 2013), Rv0444c, Rv3692, and Rv2031c proteins (Zhang et al., 2012) have also been reported with potentials of diagnostic utility and hence these may be exploited as anti-TB biomarkers. The host or pathogen-specific biomarkers in recent times, which remained under investigation for the detection of mycobacterial pathogens, are listed in Table 2.

TABLE 2. Diagnostic biomarkers against mycobacterial pathogens.
An Analytical View of Modern Methodologies That Can Be Used for Efficient Antigen Discovery Against Mycobacterial Pathogens
With the complete sequencing of the human genome, a new era of systems biology known as ‘omics’ technology has emerged. The ‘omics’ technologies represent a holistic view of different molecules that constitute a cell of an organism. They are primarily aimed to explore genes under genomics, protein coding mRNA and non-protein coding RNA under transcriptomics, proteins under proteomics and metabolites under metabolomics in a specific biological sample (Horgan and Kenny, 2011; Tripathi et al., 2017). Currently, prevalent ‘omics’ technologies combined with advanced bioinformatics are constantly putting their efforts to unveil the mechanisms behind molecular pathogenesis of infecting microbes, which may further help us to devise treatment strategies against them. Employing these approaches to vaccine development could actually transform the very expensive purely experimental study of antigen discovery into a cost-effective theoretical and computational one. This scenario will definitely help in enhancing the prospects for novel antigen discovery by selecting the immunodominant epitopes for their use as prime vaccine candidates. Contributions made by various high-throughput technologies are discussed in further subsections.
Genomics
Genomics may be described as a comprehensive analysis of an organism’s complete genome. The genome represents the complete set of DNA/genes (coding and non-coding) present in a cell or organism. There are approximately 3.2 billion bases and an estimated 20000 protein coding genes in humans. Traditionally, genes were analyzed individually but with the advent of microarray technology, genome-wide differential expression studies are made possible in recent years. DNA microarrays measure the subtle differences among DNA sequences (genetic variations) like small-scale insertion/deletions, polymorphic repetitive elements, SNPs and microsatellite variation among different individuals. The most common type of genetic variation is single nucleotide polymorphisms (SNPs). SNP occurs when one nucleotide in the genome is substituted for another and differs between members of the same species (Horgan and Kenny, 2011). This change results in an alternative codon and hence different amino acid which may be of particular interest when associated with complex mycobacterial diseases (Stucki and Gagneux, 2012). Various abnormalities like chromosomal insertions or deletions can be identified with more advanced microarray based comparative genomic hybridization (aCGH). CGH is a popular molecular cytogenetic technique for genome-wide screening of cells for chromosomal copy number variations. It uses two differentially labeled genomic DNAs: test and control sample which are simultaneously cohybridized to metaphase chromosomes. The differentially colored fluorescent signal intensity of the fluorophore labeled test DNA relative to control sample DNA is linearly plotted along the length of each chromosome to provide a cytogenetic representation of copy number variation between the two sources (Kallioniemi et al., 1992). However, CGH shows a very limited resolution of alterations of approximately 5–10 Mb (Kirchhoff et al., 1998; Lichter et al., 2000). To overcome this limitation, a more advanced high-resolution platform is known as array CGH (aCGH) has been developed. Instead of targeting metaphase chromosomes, it utilizes cloned DNA elements (known as probes) arrayed on a slide as the targets for analysis (Lucito et al., 2003). These probes are from different origins and vary in size like oligonucleotides (25–85 base pairs), bacterial artificial chromosomes (BACs; 80,000–200,000 base pairs). The probes used in aCGH are far smaller than the metaphase chromosomes which allows greater mapping resolution in aCGH than the traditional CGH. The mapping resolution depends upon both the probe size and genomic distance between DNA probes (Theisen, 2008).
The human genome project initiated in 1990 annotated the DNA sequence of the complete euchromatic human genome. Since then, the sequencing technologies [Sanger and next-generation sequencing (NGS)] have remained the hottest topic in the field of genomics research (Gasperskaja and Kućinskas, 2017). In the modern DNA sequencing era, with the ongoing technological advancement in the field of genomics, the sequencing technologies are revolutionizing the genome research especially with the high-throughput NGS (HT-NGS). It has a wide range of applications such as: chromatin immunoprecipitation (‘ChIP’) with DNA microarray (‘chip’) also known as ‘ChIP-on-chip’ and ChIp-sequencing (ChIP-seq) (Pareek et al., 2011).
Historically in 1975, the “first generation” DNA sequencing technique, known as ‘Sanger’s method’ or ‘dideoxy chain termination method,’ was developed based on specifically labeled chain terminating dideoxynucleotides (ddNTPs) incorporated by DNA polymerase during in vitro DNA synthesis. The fundamental principle behind this targeted sequencing technique is that the ddNTPs are different from dNTPs at 3′ carbon and fail to make phosphodiester bond with the next nucleotide which terminates the nucleotide chain elongation and hence replication halts. In this way, different bands of varying lengths are generated which are then separated on a polyacrylamide gel. After band separation, a laser reads the gel to detect the fluorescent intensity of each band in the form of colored peaks in a chromatogram. These colored peaks represent the nucleotide in that specific location in the DNA sequence (Russell, 2002).
Although Sanger method has proven useful in performing a thorough analysis of DNA, its use has been limited because of the high cost and size limitation. The Sanger method can only read short pieces of DNA (1000–1200 base pair) and the quality degrades after 700–900 base pairs. More recently, to overcome major stumbling blocks of first generation sequencing, new generations of sequencing techniques have been introduced which include NGS. NGS is capable of sequencing millions of DNA fragments through a massively parallel analysis with much reduced cost producing huge sequencing data. It has proven to be the new game changer for DNA sequencing. Although NGS exploits the principle similar to that of Sanger’s method of sequencing, which relies on the separation of labeled DNA elements by electrophoresis and identification of emitted signals, NGS uses array-based sequencing. It combines Sanger’s techniques (sequencing, separation and detection) for analysis of millions of samples in parallel at reduced cost with high throughput. It involves three steps: library preparation- small fragments of DNA created using random fragmentation (enzymatically or sonification) and ligated with custom linkers, amplification- done by PCR (emulsion PCR or bridge PCR), sequencing- DNA sequenced using “sequencing by synthesis” or “sequencing by ligation” (Zhang et al., 2011; Ari and Arikan, 2016). The ever growing field of sequencing has sparked an enormous range of applications of NGS technology in different research fields such as elucidation of the molecular basis of genetic diseases, infectious diseases and cancer (Del Vecchio et al., 2017).
ChIP assays are the most invaluable methods to identify the protein binding sites on DNA. ChIp-seq couples ChIP assays with NGS to investigate the genome-wide DNA binding sites for physical binding interactions of transcription factors. In ChIP-seq, formaldehyde fixation is used to irreversibly cross-link proteins to their bound DNA. The cross-linked chromatin is sheared with sonication or restriction enzymes to generate small fragments of DNA associated with a particular protein of interest followed by immunoprecipitation with desired antibody-bound magnetic beads. For NGS library preparation, the precipitated genomic DNA is used as input and is sequenced for DNA binding site analysis (Gasperskaja and Kućinskas, 2017). A more recent approach named ‘ChIP-on-chip’ combines ChIP with microarray analysis. In this method, the precipitated DNA fragments are hybridized to a microarray chip for analysis. It generates a global genome-wide chromatin maps depicting genome-wide binding sites of protein which may help to identify the functional elements in the complete genome. While this technique proved to be a revolutionary approach to study large genomic regions, it suffered from certain technical limitations such as high cost and requirement of a large amount of DNA thus extensive amplification leading to biasness and allelic variants hindered by cross-hybridization (Mikkelsen et al., 2007).
Hence, genomic analysis techniques provide an enormous amount of valuable information which may be translated in form of novel biomarkers to expedite antigen discovery. The genomic analysis usually begins with the identification and selection of potential coding regions. Along with this, attribution of functions to the selected novel proteins on the basis of sequence homology followed by a reverse genetic evaluation to characterize the complete repertoire of unannotated hypothetical proteins may be carried out (Geluk et al., 2014). Among the major mycobacterial infections, the complete genome sequence of Mtb H37Rv (Krogh et al., 2001) and CDC-1551 strains (Betts, 2002) and M. bovis AF2122/97 strain (Garnier et al., 2003) have revolutionized a big impact on the pace of anti-mycobacterial drug discovery. The genome sequence of M. leprae strain TN (Singh and Cole, 2011) has also been established. Using various in silico approaches, the whole set of protective antigens can easily be identified from the microbial pathogen’s genome without even cultivating it in the laboratory (Rappuoli, 2000). Hence, genome analysis can circumvent the laborious, costly and time-consuming conventional approaches and may pave the way to a better and faster discovery of antigenic targets against mycobacterial infections.
Transcriptomics
The transcriptome reflects the set of all RNA molecules or transcripts in a cell or organism. Transcriptomics aim to study all species of transcripts including mRNAs, non-coding RNAs and small RNAs produced in a cell of an organism at a specific time (Wang et al., 2009; Kunnath-Velayudhan et al., 2017; Lowe et al., 2017). Transcriptomics analysis has played a central role in unraveling the gene expression during a particular physiological condition and deciphering the intricacies of regulations at the transcriptional level. Expression profiling of transcripts could be targeted to explore the specific genes which show expression or overexpression in host and pathogens simultaneously representing a complete atlas of hot-spots of host-pathogen interactions (Kaiser et al., 2004). Several technologies in the field of transcriptomics have emerged to derive and quantify the RNA content, including hybridization-based and sequence-based approaches. The dominant contemporary techniques like microarrays typically measure the transcripts by hybridization of fluorescently labeled cDNA against a custom-made array of complementary probes or high-density spotted oligonucleotide microarrays. The transcriptional profiling by hybridization-based approaches is labor saving with high throughput and reduced cost. However, these suffer from some limitations such as they can detect only known sequences, high background levels generally lead to cross-hybridization and interfere with detection. Although microarray technology continues to support transcriptomics research, the advent of sequence based approaches have dramatically expanded transcriptomics in the past few years (Wang et al., 2009).
In contrast to classical hybridization techniques, the high-tech sequencing based approaches directly determine the nucleic acid sequence of cDNA. In earlier times, Sanger’s method was used to sequence cDNA or EST libraries (Gerhard et al., 2004), but this method was expensive with relatively low throughput. To overcome this, high throughput tag-based transcriptome profiling methods were developed which included cap analysis gene expression (CAGE), serial analysis gene expression (SAGE), and massively parallel signature sequencing (MPSS). Since, these methods were based on conventional Sanger sequencing technique, these were expensive and failed to map some of the short tags to the reference genome. Additionally, they failed to analyze transcript isoforms which are generally indistinct from each other. These limitations reduced the potential use of conventional sequencing technology as transcriptome profiling method (Wang et al., 2009).
Recently, the newly developed high-throughput DNA sequencing techniques have enabled highly sensitive analysis for mapping, profiling and quantifying RNAs. This rapidly growing transcriptome profiling technique is known as RNA-Seq or whole transcriptome shotgun sequencing (WTSS). RNA-Seq utilizes an NGS platform and is replacing gene expression microarrays at a high rate. For this method, RNA (fractionated or total) is first converted to cDNA molecules with the help of reverse transcriptase followed by PCR amplification. Each molecule is then sequenced using NGS sequencing platform. Following sequencing, a genome-scale transcription map is generated when the output reads are aligned to reference transcripts or reference genome (Wang et al., 2009). RNA-Seq is an effective and excellent approach for transcriptome profiling of host and pathogen simultaneously. Moreover, this technique has also been successfully used to compare HCV- or HIV-infected T-cells to uninfected T-cells in vitro. It has revealed differentially expressed transcripts of the virus and the metabolic effects of viral infection on the target cells (Lefebvre et al., 2011).
Exploiting the above mentioned transcriptomic techniques, a number of studies have been reported describing the identification of various RNA molecules involved in different regulatory networks responsible for the virulence of pathogenic mycobacterial species. RNA-Seq and high-density tiling arrays have deciphered a large repertoire of previously unknown non-coding mycobacterial RNA including novel antisense transcripts, 5’ and 3’ untranslated regions and intergenic small RNAs (sRNAs) (Arnvig and Young, 2012; Michaux et al., 2014).
Non-coding RNA (ncRNA) molecules represent RNA transcripts that are generally not translated into a protein. Although, exceptionally, some ncRNA may contain an ORF and may translate into a polypeptide chain. There are different classes of ncRNA defined on the basis of cellular processes such as ncRNAs involved in mRNA translation (rRNAs and tRNAs), splicing (small nuclear RNAs -snRNAs), modification of rRNAs (small nucleolar RNAs-snoRNAs) and gene expression regulation (microRNAs-miRNAs, piwi-interacting RNAs-piRNAs, long non-coding RNAs-lncRNAs (Arnvig and Young, 2012; Qureshi and Mehler, 2012).
The sRNAs are generally the non-coding small transcripts in the range of 50–250 nucleotides in length. They are involved in gene silencing and post-transcriptional regulation and are generally encoded opposite the ORF (cis-encoded) or between ORF (trans-encoded) (Haning et al., 2014). The first mycobacterial stress regulatory sRNA was identified in 2009. The cDNA libraries of low molecular weight Mtb transcriptomes (exponential and stationary phase) were analyzed to identify 5 trans-encoded and 4 cis-encoded sRNAs in Mtb H37Rv (Arnvig and Young, 2009). Until now, a total of nearly 200 sRNAs have been identified in Mtb (Gerrick et al., 2018). The sRNAs discovered so far have gained significant attention, especially in pathogens as regulators of transcription factors, pathogenic genes, outer membrane adaptation to stress conditions like the variation in environmental pH, temperature and anaerobic stress (Haning et al., 2014; Michaux et al., 2014).
miRNAs are evolutionarily conserved small non-coding RNA molecules of 20–24 nucleotide length. These have been reported to play a regulatory role at the post-transcriptional level by binding to the 3’-UTR of their target mRNAs and inhibiting their translation. In pathogenic mycobacterial species, these miRNAs have been demonstrated to play an important role as immunomodulators by regulating the genes expressed by immune cells of the host and in-turn supporting its growth and survival inside the host. In recent studies, it has been shown that the innate immune response generated against TB is regulated by these miRNAs. Additionally, miRNAs differential expression during TB reflects disease progression and are capable of distinguishing active TB from latent TB (Palazzo and Lee, 2015; Ahluwalia et al., 2017; Sabir et al., 2018).
Hence, the uniquely expressed RNAs identified by high-throughput transcriptomic methods provide new insights into pathogenesis and could be targeted as potential biomarkers or as therapeutic agents against mycobacterial diseases.
Proteomics
Proteome reflects the entire set of expressed proteins in a cell, tissue or organism at any given time (Theodorescu and Mischak, 2007). Proteomics covers a number of different aspects of protein function, including structural proteomics: large-scale analysis of protein structures, expression proteomics: large-scale analysis of protein expression and interaction proteomics: large-scale analysis of protein interactions. The main aim of proteomics is to study and characterize the information flowing within a cell or organism in the form of protein pathways and networks, (Petricoin et al., 2002) in order to understand the functional importance of proteins (Vlahou and Fountoulakis, 2005). Proteomics studies provide a deep understanding of the various virulent factors in different disease causing microorganisms and can aid the discovery of suitable markers as novel therapeutic agents (Fournier and Raoult, 2011).
Conventionally, different chromatographic methods have been used for purification and separation of proteins such as gel filtration/size exclusion chromatography (SEC), ion exchange chromatography (IEC) and affinity chromatography (Jungbauer and Hahn, 2009; Voedisch and Thie, 2010; Hage et al., 2012). To analyze selective proteins, techniques like western blotting and ELISA have been widely used. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), two-dimensional gel electrophoresis (2-DE) and two-dimensional differential gel electrophoresis (2D-DIGE) techniques have also been used to separate complex protein samples (Marouga et al., 2005; Issaq and Veenstra, 2008). An emerging proteomics technique, named as protein microarrays or protein chips provides a versatile platform to analyze proteins on large scale. While mass spectrometry, another analytical technique, is used to analyze complex protein mixtures on the basis of the mass-to-charge ratio of charged particles with high sensitivity (Yates, 2011). Additionally, Edman degradation is used to sequence amino acids in a particular protein (Smith, 2001). To quantify global changes in protein numbers, a number of peptide quantitation techniques have been developed including, metabolic based labeling [stable isotope labeling with amino acids in cell culture (SILAC)] and isotope-coded affinity tag (ICAT) labeling, isobaric mass tagging [isobaric tag for relative and absolute quantitation (iTRAQ)], chemical and enzymatic derivatization [quantitation by isobaric terminal labeling (QIRT)] (Ong and Mann, 2006; Shiio and Aebersold, 2006; Wiese et al., 2007; Kroksveen et al., 2015) etc. The three-dimensional structures of proteins are obtained using two popular experimental high-throughput techniques: nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography (Smyth and Martin, 2000; Aslam et al., 2017).
With the advent of proteomics techniques, their applications have been wide-ranging and expanded in almost every discipline of biological sciences. In silico analysis of the available proteomic data has defined several new ‘omes’ having potential antigenic targets. These include the exportome (Van Ooij et al., 2008), surfome (Sargeant et al., 2006), and interactome (Sanchez et al., 1999). The surfome or surface proteome of several pathogens has been identified using proteolytic shaving (Rodríguez-Ortega et al., 2006) and biotinylation (Cullen et al., 2005). Currently available proteomic techniques exploiting peptide libraries and antibody microarrays have been used to analyze Mtb proteome to identify potential antigen candidates (Kunnath-Velayudhan and Porcelli, 2013). There was a report where workers have annotated most potential subunit vaccine candidates by comparing the mycobacterial proteomes of Mtb and M. bovis BCG. They observed that Rv3407, a DNA vaccine candidate could be used to improve the overall efficacy of the existing BCG vaccine (Mollenkopf et al., 2004). Others have also discovered novel antigenic markers from the identified secreted and transmembrane proteins employing proteomics approach- glutathione S-transferase (GST) fusion protein purification strategy (Zhou et al., 2015). Similarly, Mtb Rv0444c, Rv3692, and Rv2031c have been identified as possible candidate biomarkers from an analysis performed through MALDI-TOF-MS (Zhang et al., 2012). These may be targeted for the development of diagnostic assays against TB in the near future.
Metabolomics
In the present “omics” era, metabolomics is rapidly emerging as a field of science to study the systematic identification, quantification and analysis of cellular metabolites within a given biological system (cell, tissue, organ, biological fluid or organism) at any given time. It is a collection of sophisticated analytical techniques to study the outcome of complex networks of biochemical reactions providing an understanding of the cellular physiology on a global biochemical scale (Mirsaeidi et al., 2015; Nandakumar et al., 2015).
Some of the modern analytical platforms used to study metabolite profiles include proton nuclear magnetic resonance (1H-NMR) spectroscopy, gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectroscopy (LC-MS). These have been used to provide sensitive and reliable detection of metabolites to be exploited in diagnosis and prognosis of several infectious diseases (Weiner et al., 2012; Ghannoum et al., 2013; Mickiewicz et al., 2014). The metabolomics studies of mycobacterial pathogens are still in their nascent period of development. The recent studies about Mtb metabolome have provided unique insights into the biochemical composition, organization, activity and regulation of its physiological network (Nandakumar et al., 2015). The metabolites arising from a mycobacterial pathogen or its host have yielded important information describing undefined metabolism and pathogenic characteristics linked to the pathophysiology of mycobacterial infections (Miyamoto et al., 2016). du Preez and Loots have analyzed the sputum of 34 TB patients with 2D-gas chromatography time-of-flight mass spectrometry (GC-MS) (du Preez and Loots, 2013). They successfully identified 22 metabolites (14 Mtb metabolites and 8 host-related metabolites) as potential biomarkers against TB (du Preez and Loots, 2013). Similarly, in another study, using the same analytical tool, it was reported that 2-acetylamino-2-deoxy-b-D-glucopyranose, a-L-mannopyranose and D-galactose-6-deoxy could be targeted to differentiate TB infected patients from non-infected persons (Cha et al., 2009; O’Sullivan et al., 2012). In a different liquid chromatography-mass spectrometry (LC-MS) based metabolomics study, it was observed that rpoB mutations change the Mtb metabolic profile and it plays an important role in its metabolism. A total of 99 molecular features were found different in the Mtb rifampin-resistant strains (Bisson et al., 2012). In a different study, non-targeted ultrahigh-pressure liquid chromatography time-of-flight mass spectroscopy (UPLC-TOF-MS) was exploited to distinguish a cohort of patients infected with leprosy having bacterial index < 1 from those with a bacterial index > 4 (increased metabolites: polyunsaturated fatty acids, eicosapentaenoic acid and docosahexaenoic acid) (Al-Mubarak et al., 2011).
Compared to the other ‘omics’ technologies, metabolomics has fewer limitations and offers potential advantages in terms of specificity and sensitivity (van Ravenzwaay et al., 2007). As metabolomics captures the snapshot of the metabolic status of the genes providing useful insights about the biochemical networks under study, it allows more complete understanding of cell functions perhaps far more than genomics, transcriptomics or proteomics can (Lindon et al., 2003).
Reverse Vaccinology (RV)
Today, with the advent of genomic technology, the genome-based antigen selection is possible and allows the discovery of antigen and vaccine design. One approach that mines pathogenic bacterial genomes for antigen discovery is known as “Reverse Vaccinology” (RV). RV has emerged as an effective strategy that uses bioinformatics techniques with the aim to identify highly protective and immunogenic peptides encoded by immunologically exposed pathogenicity factors by screening the entire genomes of microbial pathogens (Movahedi and Hampson, 2008; Seib et al., 2012; Donati and Rappuoli, 2013; Figure 2). RV based antigen discovery pipeline involves genome sequence analysis for the identification of antigenic proteins (surface exposed or secreted) expressed by the pathogen, their cloning and expression followed by synthetically producing each protein. The best selected candidates could be tested in the clinical trials for validating their immunogenicity after in vitro immunogenicity examination in cells and animal models. The identified antigens may be targeted for vaccine discovery. To date, RV has been targeted to devise universal and effective vaccines against bacterial pathogens for which the discovery of vaccines was previously impossible. Among these, N. meningitidis serogroup B (MenB) (Pizza et al., 2000), against which there was no effective vaccine, was the first pathogen targeted for the development RV based human vaccine (Delany et al., 2013; Rappuoli et al., 2016).
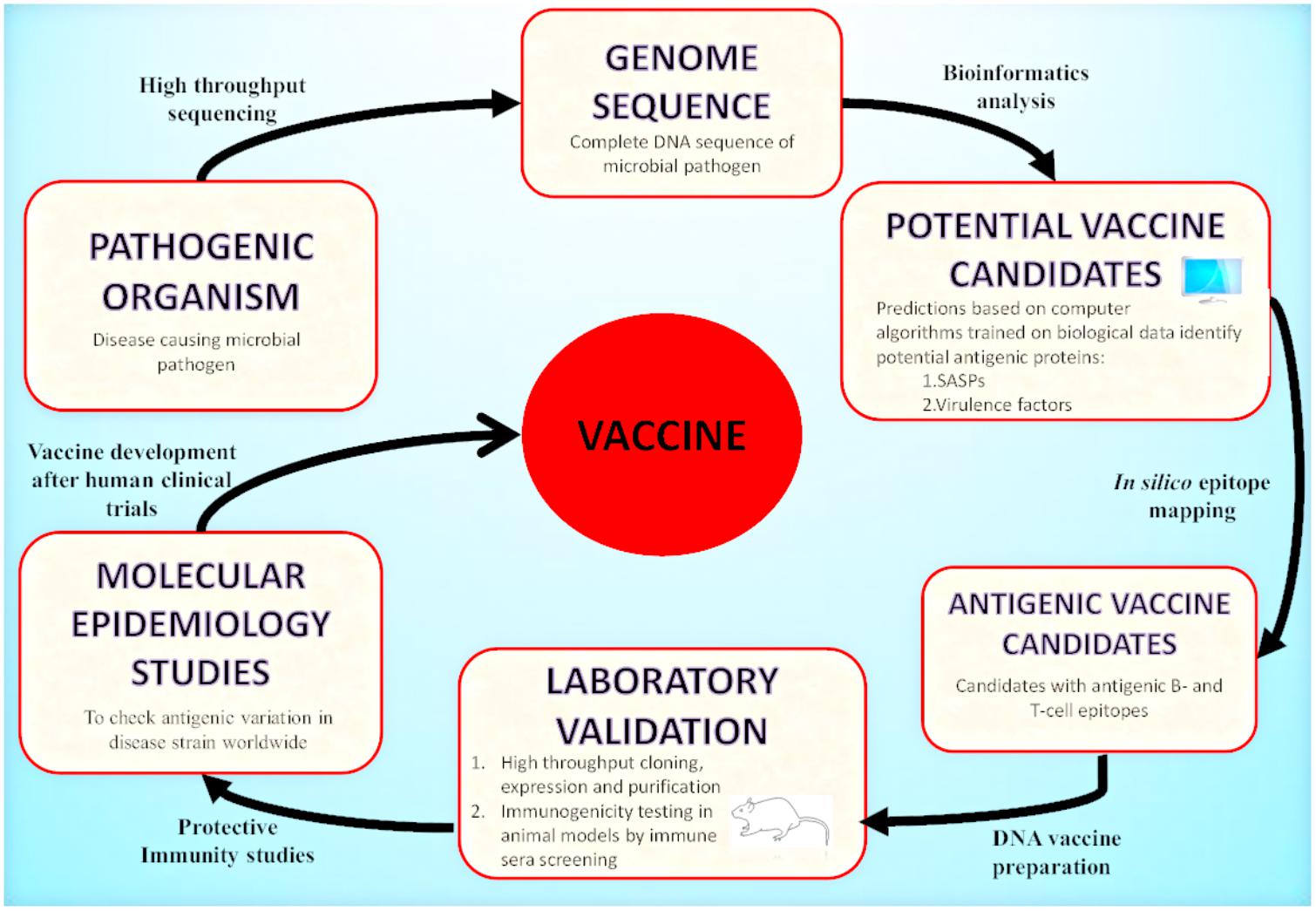
FIGURE 2. Reverse Vaccinology approach: A schematic representation of vaccine development by RV is illustrated in the presented flowchart. RV starts with the computational analysis of the complete genome sequence of the targeted pathogenic organism. Computational predictions are based on algorithms trained on biological data obtained from experimentally carried out studies. The potential vaccine candidates include surface associated and secretory proteins (SASPs) and virulence factors. These are further evaluated to identify protein candidates with antigenic epitopes for B-cells and T-cells. These proteins are then amplified by PCR and expressed in suitable vectors. The recombinant proteins produced are purified and used for immunogenicity testing in animal models (mice). Based on immune sera screening (FACs, Serum Bactericidal Activity), the recombinant proteins capable of inducing sera bactericidal antibodies are selected. The top candidates enter the pre-clinical stage of vaccine development. After the molecular epidemiological studies, the best candidates are used for clinical trials in adults, adolescents and infants and finally they enter the vaccine formulation process.
With the help of RV, whole-genome studies are now being more focused on the development of target specific epitope-based vaccines. An epitope or an antigenic determinant is the specific part of antigen interacting with the immune system (T-cell, B-cell and antibodies). The antigenic epitopes elicit an immune response by interacting with the CD8+ T immune cells and CD4+ T immune cells and may be used in ‘reverse’ to target novel antigens (Sette and Rappuoli, 2010). The immune cells- B and T lymphocyte play a major role in antigen recognition and elicitation of the immune system. The B lymphocytes are the plasma cells that produce antibodies when a foreign antigen triggers immune system and function as humoral immunity component of the adaptive immune system. The epitopes of the antigens are identified by the paratopes of antibody. The T lymphocytes play a central role in cell-mediated immunity. Hence, the prediction of the immunodominant T and B cell epitopes plays an important role in the determination of the peptide-based candidate vaccines (Kanampalliwar et al., 2013).
Based on RV, a number of web-based programs have also been developed to assist the scientific community in identifying potential vaccine candidates against mycobacterial infections. These include MycobacRv, Violin, VaxiJen, and MtbVeb, etc. MycobacRv is an RV based database of potential mycobacterial adhesins vaccine candidates from 23 strains and other species of mycobacteria. It houses detailed epitope information from the predicted adhesins and surface-localized/extracellular proteins which may further facilitate the development of epitope-based mycobacterial vaccines (Chaudhuri et al., 2014). Vaccine Investigation and Online Information Network (VIOLIN) is another web-based database that integrates vaccine literature mining, vaccine data curation and storage. It also provides an analytical platform for potential vaccine target prediction against various infectious agents (He et al., 2014). Likewise, VaxiJen is another useful resource available online for the prediction of protective antigens and subunit vaccines. The predictions are alignment independent and solely based on the physicochemical properties of the target proteins (Doytchinova and Flower, 2007). MtbVeb is a comprehensive database for designing novel vaccines against 59 existing and emerging Mtb strains employing antigen, strain and epitope based approaches (Dhanda et al., 2016). A growing number of studies reporting antigen identification published in the literature have provided valuable insights into RV based vaccine research. Some of them have been discussed in the coming sections of this review paper.
Challenges Faced by Contemporary ‘Omics’ Approaches During Antigen Discovery
The available high-throughput ‘omics’ approaches have made it possible to identify potentially important biomarkers in various microbial pathogens in a much smaller time than the conventional approaches. The wide availability of data generated by these ‘omics’ technologies offer ample opportunities to unravel the disease mechanisms but also present the scientific community with significant challenges to extract the knowledge from such huge data and its application for the welfare of the society.
In genomics, the pathogenic microorganisms with larger genomes, that fails to be cultured in vitro or if there are no animal models available, may not be suitable for antigen discovery utilizing RV because of the huge number of possible targeted proteins with unknown function (Schussek et al., 2014). In the case of transcriptomics, the information generated from deep sequencing studies need in vivo validation and also require validation for multiple isolates of the microbial pathogen (Schussek et al., 2014).
Similarly, although proteomics offers advantages in antigen discovery, it still suffers from certain limitations. While performing proteomics analysis, the organism is allowed to grow in highly favorable conditions (in vitro) and is generally isolated at a specific phase of the cell cycle which certainly does not depict the in vivo environment of that organism (Singh et al., 2015). Furthermore, the proteomics studies may not be suitable enough to identify protein complexes which are resistant to proteases as reported earlier for pili associated proteins, which have been demonstrated as potential vaccine candidates for Staphylococcus aureus and Streptococcus pneumonia (Schussek et al., 2014). Moreover, the proteomics approach gives a limited level of understanding of the protein level events of microorganisms since the mRNA transcription of a gene necessarily does not give an estimation of its translated protein level. The reason could be: the transcribed mRNA might degrade quickly or it might get translated into protein ineffectively or alternative splicing might result in the generation of multiple proteins. Another reason could be the post-translational modifications of proteins which might result in an inactive protein (Kornblihtt et al., 2013). Another major limitation of the proteomics approach is many proteins are involved in complex formation to become completely functional (Srinivas et al., 2002). Additionally, the secondary and tertiary structures of proteins are often difficult to maintain during their analysis. These generally get denatured by the action of enzymes, heat or by external stress. The proteins of low abundance are often found difficult to detect as these cannot be amplified like DNA. Like in plasma, cytokines are present in very low quantity (1–5 pg/mL) and proteomics tools can analyze proteins mostly located at the higher end of the concentration spectrum. Hence, to study these low abundant proteins, the high abundant proteins are removed from plasma. However, this removal is often accompanied by the loss of several potentially important biomarkers resulting from co-removal of antigenically important proteins bound to the high-abundance proteins (Granger et al., 2005; Cho, 2007). For these reasons explained above, very often, the proteomics experiments performed in one laboratory are poorly reproducible in other laboratories.
Nevertheless, the metabolomics key features for several diseases (Monteiro et al., 2013; Aretz and Meierhofer, 2016) have been reported, the potential bottlenecks still exist at various levels of quality biomarker identification. It is hampered by the huge and dynamic variation in the metabolic levels between people, tissues and various time points. The other bio-molecular states like the genome, transcriptome and proteome are comparatively much more stable than the vastly fluctuating metabolites (Aretz and Meierhofer, 2016).
Hence, to fulfill the huge demand for novel robust biomarkers to curb the mycobacterial infections, different ‘omics’ platforms must together be integrated to reveal, assess and track down the novel molecular patterns reflecting the disease-perturbed networks.
Application of Proteome-Scale In Silico Strategies for Discovering Potential Antigens
A number of computational programs exploiting bioinformatics algorithms have been made available for the genome/proteome sequence retrieval, sub-cellular localization of proteins on the basis of the presence of special protein signature sequences (e.g., secretory signal peptide, transmembrane helices, lipoprotein signal peptide, etc.), structural prediction, epitope mapping, virulence prediction and potential vaccine development. Some of the commonly used programs and databases have been summarized in Table 3. By utilizing such tools, numerous in silico studies have reported results deciphering the surface associated and secretory proteins (SASPs) such as OMPs, lipoproteins and secretory proteins. These are the most exposed proteins and may serve as virulence factors for the pathogens (Rana et al., 2014, 2015a,b; Rana and Akhter, 2016). These reports also demonstrate epitope mapping to target the most suitable potential antigens for vaccine development (Figure 3; Fournier and Raoult, 2011; Rana et al., 2014, 2015a,b, 2016; Rana and Akhter, 2016). In the next subsections, we have summarized the utility of the proteome-scale in silico screening strategies based on computational programs (Table 3), to identify the virulence determinants and antigenic targets in microbial pathogens.
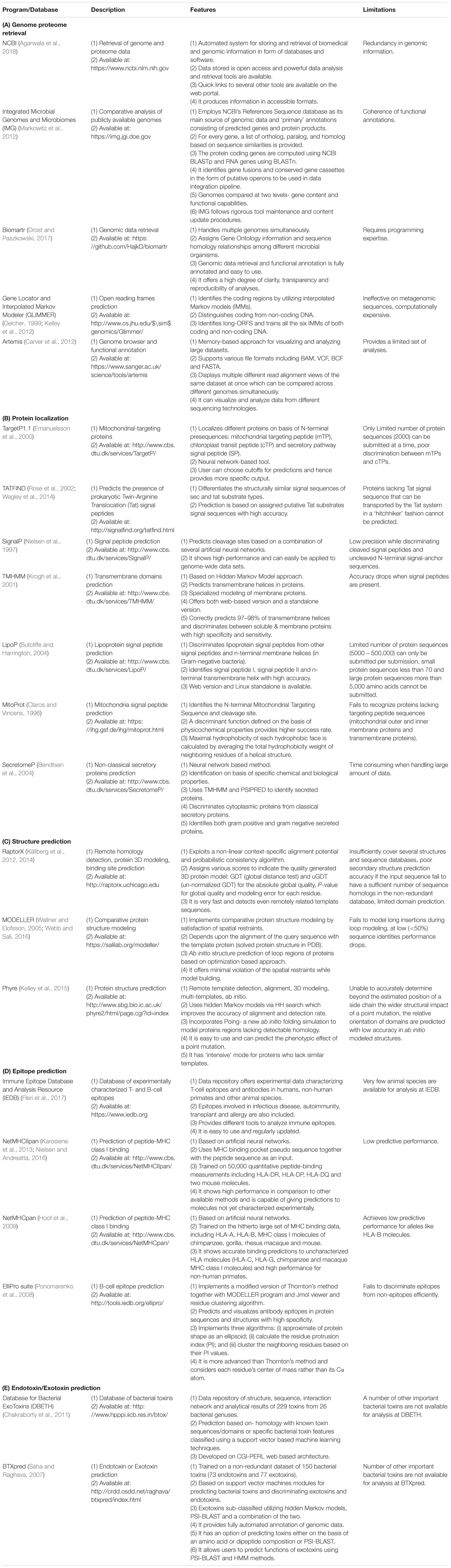
TABLE 3. Commonly used software programs and databases for in silico approaches in antigen discovery.
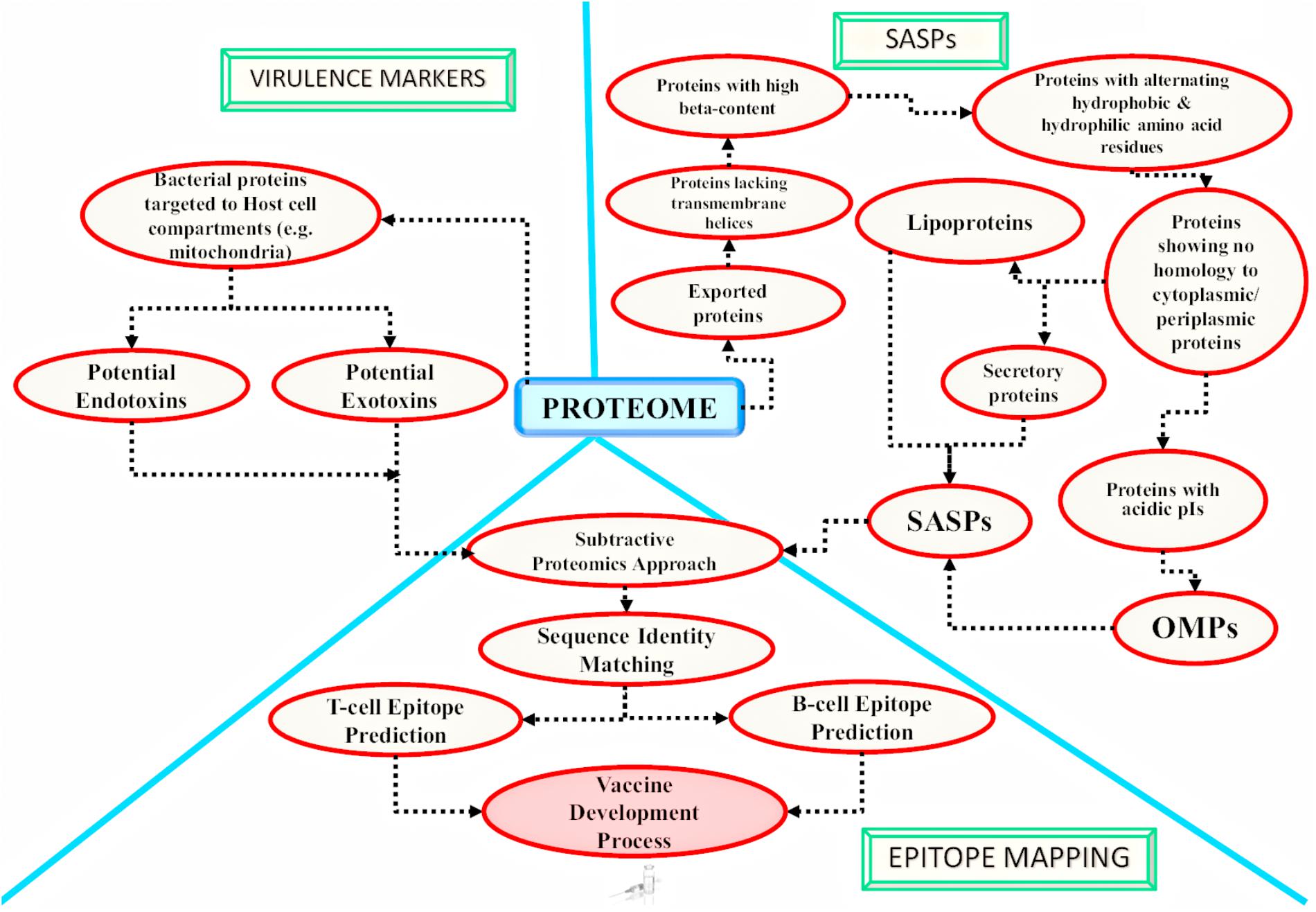
FIGURE 3. Application of in silico approaches for mycobacterial antigen discovery: a schematic overview of the methodologies currently followed using in silico approaches for mycobacterial antigen discovery is shown here. These antigens may be targeted for developing medical interventions against infectious agents. The bacterial factors targeting the host cell compartments are considered as established virulence factors and are reported to be involved in host cell ‘hijacking’ [as reported for mitochondria targeted M. avium subsp. paratuberculosis (MAP) proteins by Rana et al. (2015b)]. There are computational algorithms available which may further identify potential endotoxins and exotoxins from the potential host targeted proteins. The obtained host targeting proteins can further be subjected to epitope mapping analysis. On the other hand, the complete proteome of the pathogen can also be targeted for the identification of potential surface associated and secretory proteins (SASPs), which include lipoproteins, secretory proteins and Outer Membrane Proteins (OMPs). Epitope mapping may be carried out for the identified SASPs (Rana et al., 2015c). The screened epitopes might be utilized for developing next generation vaccines [e.g., chimeric multi-subunit artificial model vaccine as reported by Rana et al. (2016) and novel serodiagnostic markers]. Similar in silico studies may be targeted to identify novel potential antigens against other infectious agents also.
In silico Analysis for the Detection of Virulence Markers
Virulent factors represent the molecules essential for the growth of microbial pathogens which allow them to succeed and establish disease inside the host (Rana et al., 2015b). Earlier, the pathogenicity of bacteria was reported to be linked to toxins (Peterson, 1996) but later, it was considered to originate from the presence of various virulence determinants (Smith, 2003). Thus, it was concluded that targeting these potentially virulent factors would stop the disease establishment and would enable a rapid development of novel vaccines, antibiotics and new screening tests. The three main approaches that have been used for the identification of virulence genes from the complete genome involves: homology search with the experimentally characterized virulent factors (Rana et al., 2015c), identifying genes located in different pathogenic genomic islands (Akhter et al., 2007, 2008, 2012; Che et al., 2014) and the third approach involves identification of the virulence genes by genome comparison of strains having different pathogenicity profiles (virulent versus avirulent strains). Using an in silico approach, a set of 189 putative vaccine candidates have been identified from the complete Mtb genome (3989 gene products) (Zvi et al., 2008). A total of 40 promising therapeutic targets were identified in M. abscessus using novel hierarchical in silico approach and these may be exploited for novel drug discovery (Shanmugham and Pan, 2013). In an another in silico study performed on Mtb, 99 putative lipoproteins, playing important role in virulence, were identified using various bioinformatics utilities like TrEMBL database (Boeckmann et al., 2003), ScanProsite tool (Gattiker et al., 2002), SignalP (Nielsen et al., 1997), and TMHMM program (Krogh et al., 2001; Sutcliffe and Harrington, 2004). Similarly, in 2 different studies performed in silico, a total of 48 lipoproteins in Mtb, 25 lipoproteins in M. leprae, 75 lipoproteins in M. avium, 97 lipoproteins in M. marinum and 61 lipoproteins in M. smegmatis were computationally identified utilizing LipoP (Juncker et al., 2003; Rezwan et al., 2007). The pathogenic proteins targeting the host cell compartments like host mitochondria during infection are also the most commonly targeted virulent factors. A number of in silico proteome-wide studies have reported the potential mitochondria targeting proteins of the microbial pathogens (Moreno-Altamirano et al., 2012; Forrellad et al., 2013; Rana et al., 2015b). Forrellad et al., computationally identified 19 mitochondria targeting proteins from Mtb H37Rv virulent strain by utilizing the MitoProt program (mitochondria targeting proteins prediction) (Claros and Vincens, 1996), PSORT II prediction algorithm (sub-cellular localization) (Horton and Nakai, 1997) and SignalP (signal peptide sequence prediction) (Nielsen et al., 1997; Moreno-Altamirano et al., 2012). In a similar in silico approach, we have reported 46 MAP proteins as potential host mitochondria targeting proteins by employing different bioinformatics algorithms in tandem (Rana et al., 2015b). Firstly, complete MAP proteome was screened to detect the signal peptide sequence utilizing program SignalP and the identified exportome was analyzed for mitochondrial import signal screened through MitoProt II, TargetP and TPpred program (Savojardo et al., 2014). 46 MAP mitochondria targeting proteins were successfully identified. Out of these, 20 MAP proteins were defined as putative endotoxins from DBETH database (Chakraborty et al., 2011) and 14 MAP proteins as exotoxins by BTXpred tool (Saha and Raghava, 2007) which may be acting as potential virulent factors involved in MAP pathogenicity (Rana et al., 2015b).
In silico Analysis for the Detection of Secretory and Surface-Associated Proteins (SASPs)
A ‘secretome’ of an organism represents the total secretory proteins that are being released into the external milieu. This group of proteins is commonly known as excretory/secretory (ES) proteins and is important for the establishment of pathogenic infection within the host (Gomez et al., 2015; Rana et al., 2016). The SASPs include secretory proteins and surface-associated proteins like lipoproteins and OMPs. These SASPs are nowadays considered as promising targets for antigen discovery. These offer ample opportunities for the development of new therapeutic solutions against different clinical infections as the SASPs including ES proteins that are present at the interface of host-pathogen interaction and may also function as immune modulators of the host cells (Zagursky and Russell, 2001). They also help in the pathogen survival inside the host organism and act as virulence factors.
We have earlier reported novel and much advanced in silico approaches (Rana et al., 2014) for the proteome-wide identification of SASPs of MAP, M. leprae and Mtb (Rana et al., 2016). The approach exploits the cardinal sequence and structural features of SASPs from mycobacteria. The exportome of the MAP, M. leprae and Mtb was first identified employing Target P1.1 program followed by transmembrane helix prediction by TMHMM and HMMTOP program. The selected proteins were further analyzed for the presence of α helix and β sheet by utilizing the JPRED3 (Cole et al., 2008) program and amphiphilicity computation using Vogel and Jahnig algorithm (Vogel and Jähnig, 1986). Further, lipoproteins were predicted by PRED-LIPO (Juncker et al., 2003) program and sub-cellular localization of proteins was done using PSORTb followed by identification of non-classical secretory proteins employing SecretomeP program. The performed proteome-wide analysis identified 57 OMPs, 38 lipoproteins, 63 secretory proteins in the MAP; 19 OMPs, 17 lipoproteins, 11 secretory proteins in M. leprae; 36 OMPs, 47 lipoproteins and 49 secretory proteins in Mtb. Similar in silico studies have been conducted on various pathogenic genomes and proteomes to identify the repertoire of SASPs which represented novel candidates as virulence factors. These include: Taenia solium (Gomez et al., 2015), Phytophthora infestans (Raffaele et al., 2010), Yersinia pestis (Yen et al., 2007), Xanthomonas citri (Ferreira et al., 2016), Coxiella burnetii (Ferreira et al., 2016), and enteric pathogens including Shigella spp, E. coli, Vibrio cholerae, Yersinia enterocolitica (Hashmi et al., 2010), Salmonella spp., and Anaplasma marginale (Palmer et al., 2012).
In silico Analysis for Epitope Mapping
Epitope mapping is one of the keystone steps to be considered while designing an effective potent vaccine (Palmer et al., 2012). It has remarkable advantages over the long established conventional methods since it is the most cost effective, highly specific and competent strategy to generate a specific desired long lasting immunity in the host. It also helps to avoid unwanted autoimmune responses. With the advent of diverse bioinformatics tools, epitopes are nowadays can easily be mapped from the whole genomes of microbial pathogens by performing in silico analysis, without immediate reference to the peptide fragments origin. Several immunoinformatics methods have been employed for designing a highly efficient vaccine that must be capable of generating a protective B and T-cell immune response (Davies and Flower, 2007; Rana et al., 2016). Numerous vaccine related studies integrated in silico RV approach to discover putative vaccine candidates against diverse pathogens.
In case of mycobacterial infections, RV studies reported that sxL, PE26, PPE65, PE_PGRS49, PBP1 and Erp were the six proteins identified with antigenic epitopes from Mtb, that could be targeted to design novel and more efficient vaccines against TB (Monterrubio-López, 2015). Eight proteins (MAP2698c, MAP2312c, MAP3651c, MAP2872c, MAP3523c, MAP0187c and the hypothetical proteins MAP3567 and MAP1168c) were also identified with highly immunogenic epitopes in the MAP as potential vaccine candidates for studying antibody and cell-mediated immune responses within infected hosts (Gurung et al., 2012). In our previous work, we have integrated biological knowledge together with bioinformatics tools to design a much more advanced methodology pipeline for epitope mapping of the MAP (Rana et al., 2015b) and M. leprae OMPs (Rana et al., 2016). Moreover, our earlier studies reported 83 potential OMPs from a total of 4356 MAP proteins, out of which 57 MAP proteins were identified as a core set of putative OMPs (Rana et al., 2014). The identified OMPs were first analyzed to identify the host homologous proteins and proteins with significant similarity to closely related Mycobacterium taxa for excluding them to prevent any potential cross-reactivity using BLAST analysis. Further, the non-homologous proteins were subjected to immunoinformatic analyses for the prediction of T-cell (MHC I: artificial neural network approach) (Nielsen et al., 2003; Tenzer et al., 2005); MHC II: consensus approach (Wang et al., 2008) and B-cell epitopes ElliPro suite (Ponomarenko et al., 2008). Similarly, RV has been successfully applied against various other pathogens for identification of suitable antigens for vaccine development such as Dichelobacter nodosus (Myers et al., 2007), Pasteurella multocida (Al-Hasani et al., 2007) and Mtb (Kundu et al., 2016).
Conclusion
In the present post-genomic era, the discovery of novel antigens for vaccines and diagnostics has expedited with the easy accessibility of information about the complete set of different mycobacterial genes and proteins. This offers an enormous amount of knowledge for the development of immunotherapeutics. In particular, the available mycobacterial genomes complemented by state-of-the-art ‘omics’ approaches together with the in silico screening strategies symbolize promising tools to discover potential vaccine candidates and therapeutic targets in diverse pathogenic mycobacterial species. In the modern era, proteomics based approaches are becoming faster and affordable and have shown a significant potential to identify the highly antigenic bacterial SASPs. With the advancement of next-generation sequencing techniques, it is strongly believed that these techniques may shortly be used as standard approaches for the development of medical interventions against mycobacterial pathogens. This will enable the identification of constant and variable genomic regions from thousands of variants, serotypes and isolates recovered from Mycobacterium infected patients. Hence, integrating diverse approaches starting with the various computational studies including comparative genomics within the taxonomic class of the Mycobacterium based on the sequencing data, their epidemiological coverage, functional genomics data and immunoprotective capacities must be utilized to discover excellent mycobacterial antigenic targets. Therefore, presently it is highly important to bridge ‘omics’ fields that are involved in antigen discovery together with system scale in silico methods as a pre-screen and standardization of methods for the flow of information to the in vitro, in vivo and animal model immunoprotection studies of individually selected candidates after utilizing these high-throughput screening methods.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
AR was supported by a research associateship from Indian Council of Medical Research (ICMR). Research in YA lab was supported by extramural research funds from Department of Biotechnology (Govt. of India) and ICMR.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Central University of Himachal Pradesh for providing research infrastructure. We are also thankful to Prof Alfredo Pulvirenti, who has kindly handled this manuscript for the two rounds of editorial reviews, for his insightful suggestions and encouragement.
References
Agarwala, R., Barrett, T., Beck, J., Benson, D. A., Bollin, C., Bolton, E., et al. (2018). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 46, D8–D13. doi: 10.1093/nar/gkx1095
Ahluwalia, P. K., Pandey, R. K., Sehajpal, P. K., and Prajapati, V. K. (2017). Perturbed microRNA expression by Mycobacterium tuberculosis promotes macrophage polarization leading to pro-survival foam cell. Front. Immunol. 8:107. doi: 10.3389/fimmu.2017.00107
Akhter, Y., Ahmed, I., Devi, S. M., and Ahmed, N. (2007). The co-evolved Helicobacter pylori and gastric cancer: trinity of bacterial virulence, host susceptibility and lifestyle. Infect. Agent Cancer 2:2.
Akhter, Y., Ehebauer, M. T., Mukhopadhyay, S., and Hasnain, S. E. (2012). The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie 94, 110–116. doi: 10.1016/j.biochi.2011.09.026
Akhter, Y., Yellaboina, S., Farhana, A., Ranjan, A., Ahmed, N., and Hasnain, S. E. (2008). Genome scale portrait of cAMP-receptor protein (CRP) regulons in mycobacteria points to their role in pathogenesis. Gene 407, 148–158. doi: 10.1016/j.gene.2007.10.017
Al-Hasani, K., Boyce, J., McCarl, V. P., Bottomley, S., Wilkie, I., and Adler, B. (2007). Identification of novel immunogens in Pasteurella multocida. Microb. Cell Fact. 6:3.
Al-Mubarak, R., Vander Heiden, J., Broeckling, C. D., Balagon, M., Brennan, P. J., and Vissa, V. D. (2011). Serum metabolomics reveals higher levels of polyunsaturated fatty acids in lepromatous leprosy: potential markers for susceptibility and pathogenesis. PLoS Negl. Trop. Dis. 5:e1303. doi: 10.1371/journal.pntd.0001303
Andersen, P., and Woodworth, J. S. (2014). Tuberculosis vaccines–rethinking the current paradigm. Trends Immunol. 35, 387–395. doi: 10.1016/j.it.2014.04.006
Andre, F. E., Booy, R., Bock, H. L., Clemens, J., Datta, S. K., John, T. J., et al. (2008). Vaccination greatly reduces disease, disability, death and inequity worldwide. Bull. World Health Organ. 86, 140–146. doi: 10.2471/BLT.07.040089
Aretz, I., and Meierhofer, D. (2016). Advantages and pitfalls of mass spectrometry based metabolome profiling in systems biology. Int. J. Mol. Sci. 17:E632. doi: 10.3390/ijms17050632
Ari,Ş., and Arikan, M. (2016). “Next-generation sequencing: advantages, disadvantages, and future,” in Plant Omics: Trends and Applications, eds K. Hakeem, H. Tombuloðlu, and G. Tombuloðlu (Cham: Springer).
Arnvig, K. B., and Young, D. B. (2009). Identification of small RNAs in Mycobacterium tuberculosis. Mol. Microbiol. 73, 397–408. doi: 10.1111/j.1365-2958.2009.06777.x
Arnvig, K. B., and Young, D. B. (2012). Non-coding RNA and its potential role in Mycobacterium tuberculosis pathogenesis. RNA Biol. 9, 427–436. doi: 10.4161/rna.20105
Aslam, B., Basit, M., Nisar, M. A., Khurshid, M., and Rasool, M. H. (2017). Proteomics: technologies and their applications. J. Chromatogr. Sci. 55, 182–196. doi: 10.1093/chromsci/bmw167
Bagnoli, F., Baudner, B., Mishra, R. P. N., Bartolini, E., Fiaschi, L., Mariotti, P., et al. (2011). Designing the next generation of vaccines for global public health. OMICS 15, 545–566. doi: 10.1089/omi.2010.0127
Banerjee, S., Nandyala, A., Podili, R., Katoch, V. M., Murthy, K. J. R., and Hasnain, S. E. (2004). Mycobacterium tuberculosis (Mtb) isocitrate dehydrogenases show strong B cell response and distinguish vaccinated controls from TB patients. Proc. Natl. Acad. Sci. U.S.A. 101, 12652–12657. doi: 10.1073/pnas.0404347101
Bendtsen, J. D., Jensen, L. J., Blom, N., Von Heijne, G., and Brunak, S. (2004). Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 17, 349–356. doi: 10.1093/protein/gzh037
Betts, J. C. (2002). Transcriptomics and proteomics: tools for the identification of novel drug targets and vaccine candidates for tuberculosis. IUBMB Life 53, 239–242. doi: 10.1080/15216540212651
Bhat, K. H., Ahmed, A., Kumar, S., Sharma, P., and Mukhopadhyay, S. (2012). Role of PPE18 protein in intracellular survival and pathogenicity of Mycobacterium tuberculosis in mice. PLoS One 7:e52601. doi: 10.1371/journal.pone.0052601
Bisson, G. P., Mehaffy, C., Broeckling, C., Prenni, J., Rifat, D., Lun, D. S., et al. (2012). Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, rpoB mutant Mycobacterium tuberculosis. J. Bacteriol. 194, 6441–6452. doi: 10.1128/JB.01013-12
Bodzek, P., Partyka, R., and Damasiewicz-Bodzek, A. (2014). Antibodies against Hsp60 and Hsp65 in the sera of women with ovarian cancer. J. Ovarian Res. 7:30. doi: 10.1186/1757-2215-7-30
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.-C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Brandt, L., Cunha, J. F., Olsen, A. W., Chilima, B., Hirsch, P., Appelberg, R., et al. (2002). Failure of the Mycobacterium bovis BCG vaccine: some species of environmental mycobacteria block multiplication of BCG and induction of protective immunity to tuberculosis. Infect. Immun. 70, 672–678. doi: 10.1128/IAI.70.2.672-678.2002
Brennan, M. J., and Thole, J. (2012). Tuberculosis vaccines: a strategic blueprint for the next decade. Tuberculosis 92, S6–S13. doi: 10.1016/S1472-9792(12)70005-7
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Cha, D., Cheng, D., Liu, M., Zeng, Z., Hu, X., and Guan, W. (2009). Analysis of fatty acids in sputum from patients with pulmonary tuberculosis using gas chromatography-mass spectrometry preceded by solid-phase microextraction and post-derivatization on the fiber. J. Chromatogr. A 1216, 1450–1457. doi: 10.1016/j.chroma.2008.12.039
Chakraborty, A., Ghosh, S., Chowdhary, G., Maulik, U., and Chakrabarti, S. (2011). DBETH: a database of bacterial exotoxins for human. Nucleic Acids Res. 40, D615–D620. doi: 10.1093/nar/gkr942
Chaudhuri, R., Kulshreshtha, D., Raghunandanan, M. V., and Ramachandran, S. (2014). Integrative immunoinformatics for Mycobacterial diseases in R platform. Syst. Synth. Biol. 8, 27–39. doi: 10.1007/s11693-014-9135-9
Che, D., Hasan, M. S., and Chen, B. (2014). Identifying pathogenicity islands in bacterial pathogenomics using computational approaches. Pathogens 3, 36–56. doi: 10.3390/pathogens3010036
Cho, W. C. S. (2007). Proteomics technologies and challenges. Genomics Proteomics Bioinformatics 5, 77–85. doi: 10.1016/S1672-0229(07)60018-7
Choi, Y., Jeon, B. Y., Shim, T. S., Jin, H., Cho, S. N., and Lee, H. (2014). Development of a highly sensitive one-tube nested real-time PCR for detecting Mycobacterium tuberculosis. Diagn. Microbiol. Infect. Dis. 80, 299–303. doi: 10.1016/j.diagmicrobio.2014.08.009
Choudhary, R. K., Mukhopadhyay, S., Chakhaiyar, P., Sharma, N., Murthy, K. J. R., Katoch, V. M., et al. (2003). PPE antigen Rv2430c of Mycobacterium tuberculosis induces a strong B-cell response. Infect. Immun. 71, 6338–6343. doi: 10.1128/IAI.71.11.6338-6343.2003
Claros, M. G., and Vincens, P. (1996). Computational method to predict mitochondrially imported proteins and their targeting sequences. FEBS J. 241, 779–786. doi: 10.1111/j.1432-1033.1996.00779.x
Cole, C., Barber, J. D., and Barton, G. J. (2008). The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201. doi: 10.1093/nar/gkn238
Cullen, P. A., Xu, X., Matsunaga, J., Sanchez, Y., Ko, A. I., Haake, D. A., et al. (2005). Surfaceome of Leptospira spp. Infect. Immun. 73, 4853–4863. doi: 10.1128/IAI.73.8.4853-4863.2005
da Costa, A. C., Costa-Junior Ade, O., de Oliveira, F. M., Nogueira, S. V., Rosa, J. D., Resende, D. P., et al. (2014). A new recombinant BCG vaccine induces specific Th17 and Th1 effector cells with higher protective efficacy against tuberculosis. PLoS One 9:e112848. doi: 10.1371/journal.pone.0112848
Davies, M. N., and Flower, D. R. (2007). Harnessing bioinformatics to discover new vaccines. Drug Discov. Today 12, 389–395. doi: 10.1016/j.drudis.2007.03.010
Del Vecchio, F., Mastroiaco, V., Di Marco, A., Compagnoni, C., Capece, D., Zazzeroni, F., et al. (2017). Next-generation sequencing: recent applications to the analysis of colorectal cancer. J. Transl. Med. 15:246. doi: 10.1186/s12967-017-1353-y
Delany, I., Rappuoli, R., and Seib, K. L. (2013). Vaccines, reverse vaccinology, and bacterial pathogenesis. Cold Spring Harb. Perspect. Med. 3:a012476. doi: 10.1101/cshperspect.a012476
Delcher, A. (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641. doi: 10.1093/nar/27.23.4636
Demangel, C., Garnier, T., Rosenkrands, I., and Cole, S. T. (2005). Differential effects of prior exposure to environmental mycobacteria on vaccination with Mycobacterium bovis BCG or a recombinant BCG strain expressing RD1 antigens. Infect. Immun. 73, 2190–2196. doi: 10.1128/IAI.73.4.2190-2196.2005
Deng, Y.-H., He, H.-Y., and Zhang, B.-S. (2012). Evaluation of protective efficacy conferred by a recombinant Mycobacterium bovis BCG expressing a fusion protein of Ag85A-ESAT-6. J. Microbiol. Immunol. Infect. 47, 48–56. doi: 10.1016/j.jmii.2012.11.005
Dhanda, S. K., Vir, P., Singla, D., Gupta, S., Kumar, S., and Raghava, G. P. S. (2016). A web-based platform for designing vaccines against existing and emerging strains of Mycobacterium tuberculosis. PLoS One 11:e0153771. doi: 10.1371/journal.pone.0153771
Djuardi, Y., Sartono, E., Wibowo, H., Supali, T., and Yazdanbakhsh, M. (2010). A longitudinal study of BCG vaccination in early childhood: the development of innate and adaptive immune responses. PLoS One 5:e14066. doi: 10.1371/journal.pone.0014066
Doan, T. N., Eisen, D. P., Rose, M. T., Slack, A., Stearnes, G., and McBryde, E. S. (2017). Interferon-gamma release assay for the diagnosis of latent tuberculosis infection: a latent-class analysis. PLoS One 12:e0188631. doi: 10.1371/journal.pone.0188631
Dockrell, H. M., Smith, S. G., and Lalor, M. K. (2012). Variability between countries in cytokine responses to BCG vaccination: what impact might this have on protection? Expert Rev. Vaccines 11, 121–124. doi: 10.1586/erv.11.186
Döffinger, R., Dupuis, S., Picard, C., Fieschi, C., Feinberg, J., Barcenas-Morales, G., et al. (2002). Inherited disorders of IL-12-and IFNγ-mediated immunity: a molecular genetics update. Mol. Immunol. 38, 903–909. doi: 10.1016/S0161-5890(02)00017-2
Donati, C., and Rappuoli, R. (2013). Reverse vaccinology in the 21st century: improvements over the original design. Ann. N. Y. Acad. Sci. 1285, 115–132. doi: 10.1111/nyas.12046
Doytchinova, I. A., and Flower, D. R. (2007). VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:4. doi: 10.1186/1471-2105-8-4
Drost, H. G., and Paszkowski, J. (2017). Biomartr: genomic data retrieval with R. Bioinformatics 33, 1216–1217. doi: 10.1093/bioinformatics/btw821
du Preez, I., and Loots, D. T. (2013). New sputum metabolite markers implicating adaptations of the host to Mycobacterium tuberculosis, and vice versa. Tuberculosis 93, 330–337. doi: 10.1016/j.tube.2013.02.008
Duthie, M. S., Gillis, T. P., and Reed, S. G. (2011). Advances and hurdles on the way toward a leprosy vaccine. Hum. Vaccin. 7, 1172–1183. doi: 10.4161/hv.7.11.16848
Duthie, M. S., Saunderson, P., and Reed, S. G. (2012). The potential for vaccination in leprosy elimination: new tools for targeted interventions. Mem. Inst. Oswaldo Cruz 107, 190–196. doi: 10.1590/S0074-02762012000900027
Emanuelsson, O., Nielsen, H., Brunak, S., and von Heijne, G. (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016. doi: 10.1006/jmbi.2000.3903
Fair, R. J., and Tor, Y. (2014). Antibiotics and bacterial resistance in the 21st century. Perspect. Medicin. Chem. 6, 25–64. doi: 10.4137/PMC.S14459
Farinacci, M., Weber, S., and Kaufmann, S. H. E. (2012). The recombinant tuberculosis vaccine rBCG ΔureC:: hly+ induces apoptotic vesicles for improved priming of CD4+ and CD8+ T cells. Vaccine 30, 7608–7614. doi: 10.1016/j.vaccine.2012.10.031
Ferreira, R. M., Moreira, L. M., Ferro, J. A., Soares, M. R. R., Laia, M. L., Varani, A. M., et al. (2016). Unravelling potential virulence factor candidates in Xanthomonas citri. subsp. citri by secretome analysis. PeerJ 4:e1734. doi: 10.7717/peerj.1734
Fleri, W., Paul, S., Dhanda, S. K., Mahajan, S., Xu, X., Peters, B., et al. (2017). The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 8:278. doi: 10.3389/fimmu.2017.00278
Forrellad, M. A., Klepp, L. I., Gioffré, A., Sabio, Y., Garcia, J., Morbidoni, H. R., et al. (2013). Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4, 3–66. doi: 10.4161/viru.22329
Fournier, P.-E., and Raoult, D. (2011). Prospects for the future using genomics and proteomics in clinical microbiology. Annu. Rev. Microbiol. 65, 169–188. doi: 10.1146/annurev-micro-090110-102922
Garnier, T., Eiglmeier, K., Camus, J.-C., Medina, N., Mansoor, H., Pryor, M., et al. (2003). The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. U.S.A. 100, 7877–7882. doi: 10.1073/pnas.1130426100
Gasperskaja, E., and Kućinskas, V. (2017). The most common technologies and tools for functional genome analysis. Acta Med. Litu. 24, 1–11. doi: 10.6001/actamedica.v24i1.3457
Gattiker, A., Gasteiger, E., and Bairoch, A. M. (2002). ScanProsite: a reference implementation of a PROSITE scanning tool. Appl. Bioinformatics 1, 107–108.
Geluk, A., Bobosha, K., van der Ploeg-van Schip, J. J., Spencer, J. S., Banu, S., Martins, M. V., et al. (2012). New biomarkers with relevance to leprosy diagnosis applicable in areas hyperendemic for leprosy. J. Immunol. 188, 4782–4791. doi: 10.4049/jimmunol.1103452
Geluk, A., van Meijgaarden, K. E., Joosten, S. A., Commandeur, S., and Ottenhoff, T. H. M. (2014). Innovative strategies to identify M. tuberculosis antigens and epitopes using genome-wide analyses. Front. Immunol. 5:256. doi: 10.3389/fimmu.2014.00256
Gerhard, D. S., Wagner, L., Feingold, E. A., Shenmen, C. M., Grouse, L. H., Schuler, G., et al. (2004). The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC). Genome Res. 14, 2121–2127. doi: 10.1101/gr.2596504
Gerrick, E. R., Barbier, T., Chase, M. R., Xu, R., François, J., Lin, V. H., et al. (2018). Small RNA profiling in Mycobacterium tuberculosis identifies MrsI as necessary for an anticipatory iron sparing response. Proc. Natl. Acad. Sci. U.S.A. 115, 6464–6469. doi: 10.1073/pnas.1718003115
Ghannoum, M. A., Mukherjee, P. K., Jurevic, R. J., Retuerto, M., Brown, R. E., Sikaroodi, M., et al. (2013). Metabolomics reveals differential levels of oral metabolites in HIV-infected patients: toward novel diagnostic targets. OMICS 17, 5–15. doi: 10.1089/omi.2011.0035
Goletti, D., Petruccioli, E., Joosten, S. A., and Ottenhoff, T. H. M. (2016). Tuberculosis biomarkers: from diagnosis to protection. Infect. Dis. Rep. 8:6568. doi: 10.4081/idr.2016.6568
Gomez, S., Adalid-Peralta, L., Palafox-Fonseca, H., Cantu-Robles, V. A., Soberon, X., Sciutto, E., et al. (2015). Genome analysis of excretory/secretory proteins in Taenia solium reveals their Abundance of Antigenic Regions (AAR). Sci. Rep. 5:9683. doi: 10.1038/srep09683
Granger, J., Siddiqui, J., Copeland, S., and Remick, D. (2005). Albumin depletion of human plasma also removes low abundance proteins including the cytokines. Proteomics 5, 4713–4718. doi: 10.1002/pmic.200401331
Gurung, R. B., Purdie, A. C., Begg, D. J., and Whittington, R. J. (2012). In silico identification of epitopes in Mycobacterium avium subsp. paratuberculosis proteins that were upregulated under stress conditions. Clin. Vaccine Immunol. 19, 855–864. doi: 10.1128/CVI.00114-12
GÜMÜŞSOY, K. S., İça, T., Abay, S., Aydin, F., and Hizlisoy, H. (2015). Serological and molecular diagnosis of paratuberculosis in dairy cattle. Turk. J. Vet. Anim. Sci. 39, 147–153. doi: 10.3906/vet-1410-96
Hage, D. S., Anguizola, J. A., Bi, C., Li, R., Matsuda, R., Papastavros, E., et al. (2012). Pharmaceutical and biomedical applications of affinity chromatography: recent trends and developments. J. Pharm. Biomed. Anal. 69, 93–105. doi: 10.1016/j.jpba.2012.01.004
Halstrom, S., Price, P., and Thomson, R. (2015). Environmental mycobacteria as a cause of human infection. Int. J. Mycobacteriol. 4, 81–91. doi: 10.1016/j.ijmyco.2015.03.002
Haning, K., Cho, S. H., and Contreras, L. M. (2014). Small RNAs in mycobacteria: an unfolding story. Front. Cell. Infect. Microbiol. 4:96. doi: 10.3389/fcimb.2014.00096
Hashmi, T., Khan, S., Valeed, S. Z., and Bokhari, H. (2010). In silico identification of vaccine candidates against enteric pathogens by a comparative genome sequence approach AsPac. J. Mol. Biol. Biotech. 18, 327–331.
He, Y., Racz, R., Sayers, S., Lin, Y., Todd, T., Hur, J., et al. (2014). Updates on the web-based VIOLIN vaccine database and analysis system. Nucleic Acids Res. 42, D1124–D1132. doi: 10.1093/nar/gkt1133
Hoffmann, E., Machelart, A., Song, O. R., and Brodin, P. (2018). Proteomics of Mycobacterium infection: moving towards a better understanding of pathogen-driven immunomodulation. Front. Immunol. 9:86. doi: 10.3389/fimmu.2018.00086
Hoft, D. F., Blazevic, A., Abate, G., Hanekom, W. A., Kaplan, G., Soler, J. H., et al. (2008). A new recombinant bacille Calmette-Guerin vaccine safely induces significantly enhanced tuberculosis-specific immunity in human volunteers. J. Infect. Dis. 198, 1491–1501. doi: 10.1086/592450
Hoof, I., Peters, B., Sidney, J., Pedersen, L. E., Sette, A., Lund, O., et al. (2009). NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61, 1–13. doi: 10.1007/s00251-008-0341-z
Horgan, R. P., and Kenny, L. C. (2011). ‘Omic’ technologies: genomics, transcriptomics, proteomics and metabolomics. Obstet. Gynaecol. 13, 189–195. doi: 10.1576/toag.13.3.189.27672
Horton, P., and Nakai, K. (1997). Better prediction of protein cellular localization sites with the K nearest neighbors classifier. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5, 147–152.
Hougardy, J.-M., Schepers, K., Place, S., Drowart, A., Lechevin, V., Verscheure, V., et al. (2007). Heparin-binding-hemagglutinin-induced IFN-γ release as a diagnostic tool for latent tuberculosis. PLoS One 2:e926. doi: 10.1371/journal.pone.0000926
Issaq, H., and Veenstra, T. (2008). Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE): advances and perspectives. Biotechniques 44(Suppl. 4), 697–700. doi: 10.2144/000112823
Janeway, C. A. Jr., Travers, P., Walport, M., and Shlomchik, M. J. (2001). “Adaptive immunity to infection,” in Immunobiol: The Immune System in Health and Disease, ed. S. Gibbs (New York, NY: Garland Science), 412–420.
Juncker, A. S., Willenbrock, H., Von Heijne, G., Brunak, S., Nielsen, H., and Krogh, A. (2003). Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12, 1652–1662. doi: 10.1110/ps.0303703
Jungbauer, A., and Hahn, R. (2009). Ion-exchange chromatography. Methods Enzymol. 463, 349–371. doi: 10.1016/S0076-6879(09)63022-6
Kaiser, K., Matuschewski, K., Camargo, N., Ross, J., and Kappe, S. H. I. (2004). Differential transcriptome profiling identifies Plasmodium genes encoding pre-erythrocytic stage-specific proteins. Mol. Microbiol. 51, 1221–1232. doi: 10.1046/j.1365-2958.2003.03909.x
Källberg, M., Margaryan, G., Wang, S., Ma, J., and Xu, J. (2014). RaptorX server: a resource for template-based protein structure modeling. Methods Mol. Biol. 1137, 17–27. doi: 10.1007/978-1-4939-0366-5_2
Källberg, M., Wang, H., Wang, S., Peng, J., Wang, Z., Lu, H., et al. (2012). Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522. doi: 10.1038/nprot.2012.085
Kallioniemi, A., Kallioniemi, O. P., Sudar, D., Rutovitz, D., Gray, J. W., Waldman, F., et al. (1992). Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258, 818–821. doi: 10.1126/science.1359641
Kanampalliwar, A. M., Soni, R., Girdhar, A., and Tiwari, A. (2013). Reverse vaccinology: basics and applications. J. Vaccines Vaccin. 4:194. doi: 10.4172/2157-7560.1000194
Karosiene, E., Rasmussen, M., Blicher, T., Lund, O., Buus, S., and Nielsen, M. (2013). NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65, 711–724. doi: 10.1007/s00251-013-0720-y
Kashyap, R. S., Rajan, A. N., Ramteke, S. S., Agrawal, V. S., Kelkar, S. S., Purohit, H. J., et al. (2007). Diagnosis of tuberculosis in an Indian population by an indirect ELISA protocol based on detection of Antigen 85 complex: a prospective cohort study. BMC Infect. Dis. 7:74. doi: 10.1186/1471-2334-7-74
Kelley, D. R., Liu, B., Delcher, A. L., Pop, M., and Salzberg, S. L. (2012). Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res. 40:e9. doi: 10.1093/nar/gkr1067
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kernodle, D. S. (2010). Decrease in the effectiveness of Bacille Calmette-Guérin vaccine against pulmonary tuberculosis: a consequence of increased immune suppression by microbial antioxidants, not overattenuation. Clin. Infect. Dis. 51, 177–184. doi: 10.1086/653533
Khan, N., Alam, K., Nair, S., Valluri, V. L., Murthy, K. J. R., and Mukhopadhyay, S. (2008). Association of strong immune responses to PPE protein Rv1168c with active tuberculosis. Clin. Vaccine Immunol. 15, 974–980. doi: 10.1128/CVI.00485-07
Kirchhoff, M., Gerdes, T., Rose, H., Maahr, J., Ottesen, A. M., and Lundsteen, C. (1998). Detection of chromosomal gains and losses in comparative genomic hybridization analysis based on standard reference intervals. Cytometry 31, 163–173. doi: 10.1002/(SICI)1097-0320(19980301)31:3<163::AID-CYTO3>3.0.CO;2-M
Kollmann, T. R. (2013). Variation between populations in the innate immune response to vaccine adjuvants. Front. Immunol. 4:81. doi: 10.3389/fimmu.2013.00081
Kornblihtt, A. R., Schor, I. E., Alló, M., Dujardin, G., Petrillo, E., and Muñoz, M. J. (2013). Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 14, 153–165. doi: 10.1038/nrm3525
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kroksveen, A. C., Jaffe, J. D., Aasebo, E., Barsnes, H., Bjorlykke, Y., Franciotta, D., et al. (2015). Quantitative proteomics suggests decrease in the secretogranin-1 cerebrospinal fluid levels during the disease course of multiple sclerosis. Proteomics 15, 3361–3369. doi: 10.1002/pmic.201400142
Kundu, P., Biswas, R., Mukherjee, S., Reinhard, L., Dutta, A., Mueller-Dieckmann, J., et al. (2016). Structure-based epitope mapping of Mycobacterium tuberculosis secretary antigen MTC28. J. Biol. Chem. 291, 13943–13954. doi: 10.1074/jbc.M116.726422
Kunnath-Velayudhan, S., Goldberg, M. F., Saini, N. K., Johndrow, C. T., Ng, T. W., Johnson, A. J., et al. (2017). Transcriptome analysis of mycobacteria-specific CD4+ T cells identified by activation-induced expression of CD154. J. Immunol. 199, 2596–2606. doi: 10.4049/jimmunol.1700654
Kunnath-Velayudhan, S., and Porcelli, S. A. (2013). Recent advances in defining the immunoproteome of Mycobacterium tuberculosis. Front. Immunol. 4:335. doi: 10.3389/fimmu.2013.00335
Lahey, T., and Von Reyn, C. F. (2016). Mycobacterium bovis BCG and new vaccines for the prevention of tuberculosis. Microbiol. Spectr. 4, 187–209. doi: 10.1128/microbiolspec.TNMI7-0003-2016
Lalor, M. K., Ben-Smith, A., Gorak-Stolinska, P., Weir, R. E., Floyd, S., Blitz, R., et al. (2009). Population differences in immune responses to Bacille Calmette-Guerin vaccination in infancy. J. Infect. Dis. 199, 795–800. doi: 10.1086/597069
Lalor, M. K., Floyd, S., Gorak-Stolinska, P., Ben-Smith, A., Weir, R. E., Smith, S. G., et al. (2011). BCG vaccination induces different cytokine profiles following infant BCG vaccination in the UK and Malawi. J. Infect. Dis. 204, 1075–1085. doi: 10.1093/infdis/jir515
Laux da Costa, L., Delcroix, M., Dalla Costa, E. R., Prestes, I. V., Milano, M., Francis, S. S., et al. (2015). A real-time PCR signature to discriminate between tuberculosis and other pulmonary diseases. Tuberculosis 95, 421–425. doi: 10.1016/j.tube.2015.04.008
Lawn, S. D., Dheda, K., Kerkhoff, A. D., Peter, J. G., Dorman, S., Boehme, C. C., et al. (2013). Determine TB-LAM lateral flow urine antigen assay for HIV-associated tuberculosis: recommendations on the design and reporting of clinical studies. BMC Infect. Dis. 13:407. doi: 10.1186/1471-2334-13-407
Lefebvre, G., Desfarges, S., Uyttebroeck, F., Muñoz, M., Beerenwinkel, N., Rougemont, J., et al. (2011). Analysis of HIV-1 expression level and sense of transcription by high-throughput sequencing of the infected cell. J. Virol. 85, 6205–6211. doi: 10.1128/JVI.00252-11
Lichter, P., Joos, S., Bentz, M., and Lampel, S. (2000). Comparative genomic hybridization: uses and limitations. Semin. Hematol. 37, 348–357. doi: 10.1016/S0037-1963(00)90015-5
Lin, C. W., Su, I. J., Chang, J. R., Chen, Y. Y., Lu, J. J., and Douh, Y. (2011). Recombinant BCG coexpressing Ag85B, CFP10, and interleukin-12 induces multifunctional Th1 and memory T cells in mice. APMIS 120, 72–82. doi: 10.1111/j.1600-0463.2011.02815.x
Lindon, J. C., Nicholson, J. K., Holmes, E., Antti, H., Bollard, M. E., Keun, H., et al. (2003). Contemporary issues in toxicology the role of metabonomics in toxicology and its evaluation by the COMET project. Toxicol. Appl. Pharmacol. 187, 137–146. doi: 10.1016/S0041-008X(02)00079-0
Lowe, R., Shirley, N., Bleackley, M., Dolan, S., and Shafee, T. (2017). Transcriptomics technologies. PLoS Comput. Biol. 13:e1005457. doi: 10.1371/journal.pcbi.1005457
Lucito, R., Healy, J., Alexander, J., Reiner, A., Esposito, D., Chi, M., et al. (2003). Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. 13, 2291–2305. doi: 10.1101/gr.1349003
MacDonald, E. M., and Izzo, A. A. (2015). Tuberculosis Vaccine Development—Its History and Future Directions. Tuberculosis Knowledge. London: InTech.
Maltempe, F. G., Baldin, V. P., Lopes, M. A., Siqueira, V. L. D., Scodro, R. B. L., Cardoso, R. F., et al. (2016). Critical analysis: use of polymerase chain reaction to diagnose leprosy. Braz. J. Pharm. Sci. 52, 163–169. doi: 10.1590/S1984-82502016000100018
Mangtani, P., Abubakar, I., Ariti, C., Beynon, R., Pimpin, L., Fine, P. E. M., et al. (2013). Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin. Infect. Dis. 58, 470–480. doi: 10.1093/cid/cit790
Markowitz, V. M., Chen, I. M., Palaniappan, K., Chu, K., Szeto, E., Grechkin, Y., et al. (2012). IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–D122. doi: 10.1093/nar/gkr1044
Marouga, R., David, S., and Hawkins, E. (2005). The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 382, 669–678. doi: 10.1007/s00216-005-3126-3
Martinez, A. N., Talhari, C., Moraes, M. O., and Talhari, S. (2014). PCR-based techniques for leprosy diagnosis: from the laboratory to the clinic. PLoS Negl. Trop. Dis. 8:e2655. doi: 10.1371/journal.pntd.0002655
McKenna, S. L., Keefe, G. P., Barkema, H. W., and Sockett, D. C. (2005). Evaluation of three ELISAs for Mycobacterium avium subsp. paratuberculosis using tissue and fecal culture as comparison standards. Vet. Microbiol. 110, 105–111. doi: 10.1016/j.vetmic.2005.07.010
Meeusen, E. N. T., Walker, J., Peters, A., Pastoret, P.-P., and Jungersen, G. (2007). Current status of veterinary vaccines. Clin. Microbiol. Rev. 20, 489–510. doi: 10.1128/CMR.00005-07
Michaux, C., Verneuil, N., Hartke, A., and Giard, J. C. (2014). Physiological roles of small RNA molecules. Microbiology 160, 1007–1019. doi: 10.1099/mic.0.076208-0
Mickiewicz, B., Duggan, G. E., Winston, B. W., Doig, C., Kubes, P., and Vogel, H. J. (2014). Metabolic profiling of serum samples by 1H nuclear magnetic resonance spectroscopy as a potential diagnostic approach for septic shock. Crit. Care Med. 42, 1140–1149. doi: 10.1097/CCM.0000000000000142
Mihret, A., and Abebe, M. (2013). Cytokines and chemokines as biomarkers of tuberculosis. J. Mycobact. Dis. 3:128.
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. doi: 10.1038/nature06008
Mirsaeidi, M., Banoei, M. M., Winston, B. W., and Schraufnagel, D. E. (2015). Metabolomics: applications and promise in mycobacterial disease. Ann. Am. Thorac. Soc. 12, 1278–1287. doi: 10.1513/AnnalsATS.201505-279PS
Miyamoto, Y., Mukai, T., Matsuoka, M., Kai, M., Maeda, Y., and Makino, M. (2016). Profiling of intracellular metabolites: an approach to understanding the characteristic physiology of Mycobacterium leprae. PLoS Negl. Trop. Dis. 10:e0004881. doi: 10.1371/journal.pntd.0004881
Mohan, T., Verma, P., and Rao, D. N. (2013). Novel adjuvants & delivery vehicles for vaccines development: a road ahead. Indian J. Med. Res. 138, 779–795.
Mollenkopf, H. J., Grode, L., Mattow, J., Stein, M., Mann, P., Knapp, B., et al. (2004). Application of mycobacterial proteomics to vaccine design: improved protection by Mycobacterium bovis BCG prime-Rv3407 DNA boost vaccination against tuberculosis. Infect. Immun. 72, 6471–6479. doi: 10.1128/IAI.72.11.6471-6479.2004
Monteiro, M. S., Carvalho, M., and Bastos, M. L. (2013). Guedes de Pinho P. Metabolomics analysis for biomarker discovery: advances and challenges. Curr. Med. Chem. 20, 257–271. doi: 10.2174/092986713804806621
Monterrubio-López, G. P. (2015). Identification of novel potential vaccine candidates against tuberculosis based on reverse vaccinology. Biomed Res. Int. 2015:483150. doi: 10.1155/2015/483150
Moreno-Altamirano, M. M. B., Paredes-González, I. S., Espitia, C., Santiago-Maldonado, M., Hernández-Pando, R., and Sánchez-García, F. J. (2012). Bioinformatic identification of Mycobacterium tuberculosis proteins likely to target host cell mitochondria: virulence factors? Microb. Inform. Exp. 2:9. doi: 10.1186/2042-5783-2-9
Movahedi, A. R., and Hampson, D. J. (2008). New ways to identify novel bacterial antigens for vaccine development. Vet. Microbiol. 131, 1–13. doi: 10.1016/j.vetmic.2008.02.011
Myers, G. S. A., Parker, D., Al-Hasani, K., Kennan, R. M., Seemann, T., Ren, Q., et al. (2007). Genome sequence and identification of candidate vaccine antigens from the animal pathogen Dichelobacter nodosus. Nat. Biotechnol. 25, 569–575. doi: 10.1038/nbt1302
Nandakumar, M., Prosser, G. A., de Carvalho, L. P., and Rhee, K. (2015). Metabolomics of Mycobacterium tuberculosis. Methods Mol. Biol. 1285, 105–115. doi: 10.1007/978-1-4939-2450-9_6
Nielsen, H., Engelbrecht, J., Brunak, S., and von Heijne, G. (1997). A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8, 581–599. doi: 10.1142/S0129065797000537
Nielsen, M., and Andreatta, M. (2016). NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 8:33. doi: 10.1186/s13073-016-0288-x
Nielsen, M., Lundegaard, C., Worning, P., Lauemøller, S. L., Lamberth, K., Buus, S., et al. (2003). Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017. doi: 10.1110/ps.0239403
Oettinger, T., Jørgensen, M., Ladefoged, A., Hasløv, K., and Andersen, P. (1999). Development of the Mycobacterium bovis BCG vaccine: review of the historical and biochemical evidence for a genealogical tree. Tuber. Lung Dis. 79, 243–250. doi: 10.1054/tuld.1999.0206
Ong, S.-E., and Mann, M. (2006). “Stable isotope labeling by amino acids in cell culture for quantitative proteomics,” in Quantitative Proteomics by Mass Spectrometry, ed. S. Sechi (Totowa, NJ: Humana Press), 37–52. doi: 10.1385/1-59745-255-6:37
O’Sullivan, D. M., Nicoara, S. C., Mutetwa, R., Mungofa, S., Lee, O. Y., Minnikin, D. E., et al. (2012). Detection of Mycobacterium tuberculosis in sputum by gas chromatography-mass spectrometry of methyl mycocerosates released by thermochemolysis. PLoS One 7:e32836. doi: 10.1371/journal.pone.0032836
Ottenhoff, T. H. M., and Kaufmann, S. H. E. (2012). Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog. 8:e1002607. doi: 10.1371/journal.ppat.1002607
Pai, M., Denkinger, C. M., Kik, S. V., Rangaka, M. X., Zwerling, A., Oxlade, O., et al. (2014). Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin. Microbiol. Rev. 27, 3–20. doi: 10.1128/CMR.00034-13
Palazzo, A. F., and Lee, E. S. (2015). Non-coding RNA: what is functional and what is junk? Front. Genet. 6:2. doi: 10.3389/fgene.2015.00002
Palmer, G. H., Brown, W. C., Noh, S. M., and Brayton, K. A. (2012). Genome-wide screening and identification of antigens for rickettsial vaccine development. FEMS Immunol. Med. Microbiol. 64, 115–119. doi: 10.1111/j.1574-695X.2011.00878.x
Pareek, C. S., Smoczynski, R., and Tretyn, A. (2011). Sequencing technologies and genome sequencing. J. Appl. Genet. 52, 413–435. doi: 10.1007/s13353-011-0057-x
Parveen, S., Das, S., Kundra, C. P., and Pereira, B. M. J. (2003). A comprehensive evaluation of the reproductive toxicity of Quassia amara in male rats. Reprod. Toxicol. 17, 45–50. doi: 10.1016/S0890-6238(02)00080-1
Peterson, J. W. (1996). “Bacterial pathogenesis,” in Medical Microbiology, 4th Edn, ed. S. Baron (Galveston, TX: University of Texas Medical Branch at Galveston).
Petricoin, E., Zoon, K., Kohn, E., Barrett, J., and Liotta, L. (2002). Clinical proteomics: translating benchside promise into bedside reality. Nat. Rev. 1, 683–695. doi: 10.1038/nrd891
Pizza, M., Scarlato, V., Masignani, V., Giuliani, M. M., Aricó, B., Comanducci, M., et al. (2000). Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 287, 1816–1820. doi: 10.1126/science.287.5459.1816
Pollock, N. R., Macovei, L., Kanunfre, K., Dhiman, R., Restrepo, B. I., Zarate, I., et al. (2013). Validation of Mycobacterium tuberculosis Rv1681 protein as a diagnostic marker of active pulmonary tuberculosis. J. Clin. Microbiol. 51, 1367–1373. doi: 10.1128/JCM.03192-12
Ponomarenko, J., Bui, H.-H., Li, W., Fusseder, N., Bourne, P. E., Sette, A., et al. (2008). ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 9:514. doi: 10.1186/1471-2105-9-514
Priya, V. H. S., Latha, G. S., Hasnain, S. E., Murthy, K. J. R., and Valluri, V. L. (2010). Enhanced T cell responsiveness to Mycobacterium bovis BCG r32-kDa Ag correlates with successful anti-tuberculosis treatment in humans. Cytokine 52, 190–193. doi: 10.1016/j.cyto.2010.07.001
Qureshi, I. A., and Mehler, M. F. (2012). Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 13, 528–541. doi: 10.1038/nrn3234
Raffaele, S., Win, J., Cano, L. M., and Kamoun, S. (2010). Analyses of genome architecture and gene expression reveal novel candidate virulence factors in the secretome of Phytophthora infestans. BMC Genomics 11:637. doi: 10.1186/1471-2164-11-637
Rahman, S., Magalhaes, I., Rahman, J., Ahmed, R. K., Sizemore, D. R., Scanga, C. A., et al. (2012). Prime-boost vaccination with rBCG/rAd35 enhances CD8+ cytolytic T-cell responses in lesions from Mycobacterium tuberculosis–infected primates. Mol. Med. 18, 647–658. doi: 10.2119/molmed.2011.00222
Rana, A., Ahmed, M., Rub, A., and Akhter, Y. (2015a). A tug-of-war between the host and the pathogen generates strategic hotspots for the development of novel therapeutic interventions against infectious diseases. Virulence 6, 566–580. doi: 10.1080/21505594.2015.1062211
Rana, A., Kumar, D., Rub, A., and Akhter, Y. (2015b). Proteome-scale identification and characterization of mitochondria targeting proteins of Mycobacterium avium subspecies paratuberculosis: potential virulence factors modulating host mitochondrial function. Mitochondrion 23, 42–54. doi: 10.1016/j.mito.2015.05.005
Rana, A., Rub, A., and Akhter, Y. (2015c). Proteome-wide B and T cell epitope repertoires in outer membrane proteins of Mycobacterium avium subsp. paratuberculosis have vaccine and diagnostic relevance: a holistic approach. J. Mol. Recognit. 28, 506–520. doi: 10.1002/jmr.2458
Rana, A., and Akhter, Y. (2016). A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure based approach. Immunobiology 221, 544–557. doi: 10.1016/j.imbio.2015.12.004
Rana, A., Rub, A., and Akhter, Y. (2014). Proteome-scale identification of outer membrane proteins in Mycobacterium avium subspecies paratuberculosis using a structure based combined hierarchical approach. Mol. Biosyst. 10, 2329–2337. doi: 10.1039/c4mb00234b
Rana, A., Thakur, S., Bhardwaj, N., Kumar, D., and Akhter, Y. (2016). Excavating the surface-associated and secretory proteome of Mycobacterium leprae for identifying vaccines and diagnostic markers relevant immunodominant epitopes. Pathog. Dis. 74:ftw110.
Rappuoli, R. (2000). Reverse vaccinology. Curr. Opin. Microbiol. 3, 445–450. doi: 10.1016/S1369-5274(00)00119-3
Rappuoli, R., Bottomley, M. J., D’Oro, U., Finco, O., and Gregorio, E. D. (2016). Reverse vaccinology 2.0: human immunology instructs vaccine antigen design. J. Exp. Med. 213, 469–481. doi: 10.1084/jem.20151960
Ratajczak, W., Niedźwiedzka-Rystwej, P., Tokarz-Deptuła, B., and Deptuła, W. (2018). Immunological memory cells. Cent. Eur. J. Immunol. 43, 194–203. doi: 10.5114/ceji.2018.77390
Reece, S. T., Nasser-Eddine, A., Dietrich, J., Stein, M., Zedler, U., Schommer-Leitner, S., et al. (2011). Improved long-term protection against Mycobacterium tuberculosis Beijing/W in mice after intra-dermal inoculation of recombinant BCG expressing latency associated antigens. Vaccine 29, 8740–8744. doi: 10.1016/j.vaccine.2011.07.144
Rezwan, M., Grau, T., Tschumi, A., and Sander, P. (2007). Lipoprotein synthesis in mycobacteria. Microbiology 153, 652–658. doi: 10.1099/mic.0.2006/000216-0
Riaño, F., Arroyo, L., París, S., Rojas, M., Friggen, A. H., van Meijgaarden, K. E., et al. (2012). T cell responses to DosR and Rpf proteins in actively and latently infected individuals from Colombia. Tuberculosis 92, 148–159. doi: 10.1016/j.tube.2011.12.005
Ritz, N., and Curtis, N. (2009). Mapping the global use of different BCG vaccine strains. Tuberculosis 89, 248–251. doi: 10.1016/j.tube.2009.03.002
Rodríguez-Ortega, M. J., Norais, N., Bensi, G., Liberatori, S., Capo, S., Mora, M., et al. (2006). Characterization and identification of vaccine candidate proteins through analysis of the group A Streptococcus surface proteome. Nat. Biotechnol. 24, 191–197. doi: 10.1038/nbt1179
Romano, M. I., Amadio, A., Bigi, F., Klepp, L., Etchechoury, I., Llana, M. N., et al. (2005). Further analysis of VNTR and MIRU in the genome of Mycobacterium avium complex, and application to molecular epidemiology of isolates from South America. Vet. Microbiol. 110, 221–237. doi: 10.1016/j.vetmic.2005.07.009
Rose, R. W., Brüser, T., Kissinger, J. C., and Pohlschröder, M. (2002). Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol. Microbiol. 45, 943–950. doi: 10.1046/j.1365-2958.2002.03090.x
Rowland, R., and McShane, H. (2011). Tuberculosis vaccines in clinical trials. Expert Rev. Vaccines 10, 645–658. doi: 10.1586/erv.11.28
Roy, A., Eisenhut, M., Harris, R. J., Rodrigues, L. C., Sridhar, S., Habermann, S., et al. (2014). Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: systematic review and meta-analysis. BMJ 349:g4643. doi: 10.1136/bmj.g4643
Ruhwald, M., Dominguez, J., Latorre, I., Losi, M., Richeldi, L., Pasticci, M. B., et al. (2011). A multicentre evaluation of the accuracy and performance of IP-10 for the diagnosis of infection with M. tuberculosis. Tuberculosis 91, 260–267. doi: 10.1016/j.tube.2011.01.001
Sabir, N., Hussain, T., Shah, S. Z. A., Peramo, A., Zhao, D., and Zhou, X. (2018). miRNAs in tuberculosis: new avenues for diagnosis and host-directed therapy. Front. Microbiol. 9:602. doi: 10.3389/fmicb.2018.00602
Saha, S., and Raghava, G. P. S. (2007). BTXpred: prediction of bacterial toxins. In Silico Biol. 7, 405–412.
Sali, M., Di Sante, G., Cascioferro, A., Zumbo, A., Nicolò, C., Donà, V., et al. (2010). Surface expression of MPT64 as a fusion with the PE domain of PE_PGRS33 enhances Mycobacterium bovis BCG protective activity against Mycobacterium tuberculosis in mice. Infect. Immun. 78, 5202–5213. doi: 10.1128/IAI.00267-10
Sanchez, C., Lachaize, C., Janody, F., Bellon, B., Röder, L., Euzenat, J., et al. (1999). Grasping at molecular interactions and genetic networks in Drosophila melanogaster using FlyNets, an Internet database. Nucleic Acids Res. 27, 89–94. doi: 10.1093/nar/27.1.89
Santema, W., Rutten, V., Segers, R., Poot, J., Hensen, S., Heesterbeek, H., et al. (2013). Postexposure subunit vaccination against chronic enteric mycobacterial infection in a natural host. Infect. Immun. 81, 1990–1995. doi: 10.1128/IAI.01121-12
Sargeant, T. J., Marti, M., Caler, E., Carlton, J. M., Simpson, K., Speed, T. P., et al. (2006). Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 7:R12. doi: 10.1186/gb-2006-7-2-r12
Savojardo, C., Martelli, P. L., Fariselli, P., and Casadio, R. (2014). TPpred2: improving the prediction of mitochondrial targeting peptide cleavage sites by exploiting sequence motifs. Bioinformatics 30, 2973–2974. doi: 10.1093/bioinformatics/btu411
Schussek, S., Trieu, A., and Doolan, D. L. (2014). Genome-and proteome-wide screening strategies for antigen discovery and immunogen design. Biotechnol. Adv. 32, 403–414. doi: 10.1016/j.biotechadv.2013.12.006
Seder, R. A., and Hill, A. V. S. (2000). Vaccines against intracellular infections requiring cellular immunity. Nature 406, 793–798. doi: 10.1038/35021239
Seib, K. L., Dougan, G., and Rappuoli, R. (2009). The key role of genomics in modern vaccine and drug design for emerging infectious diseases. PLoS Genet. 5:e1000612. doi: 10.1371/journal.pgen.1000612
Seib, K. L., Zhao, X., and Rappuoli, R. (2012). Developing vaccines in the era of genomics: a decade of reverse vaccinology. Clin. Microbiol. Infect. 18, 109–116. doi: 10.1111/j.1469-0691.2012.03939.x
Serruto, D., and Rappuoli, R. (2006). Post-genomic vaccine development. FEBS Lett. 580, 2985–2992. doi: 10.1016/j.febslet.2006.04.084
Sette, A., and Rappuoli, R. (2010). Reverse vaccinology: developing vaccines in the era of genomics. Immunity 33, 530–541. doi: 10.1016/j.immuni.2010.09.017
Shanmugham, B., and Pan, A. (2013). Identification and characterization of potential therapeutic candidates in emerging human pathogen Mycobacterium abscessus: a novel hierarchical in silico approach. PLoS One 8:e59126. doi: 10.1371/journal.pone.0059126
Shi, C., Chen, L., Chen, Z., Zhang, Y., Zhou, Z., Lu, J., et al. (2010). Enhanced protection against tuberculosis by vaccination with recombinant BCG over-expressing HspX protein. Vaccine 28, 5237–5244. doi: 10.1016/j.vaccine.2010.05.063
Shiio, Y., and Aebersold, R. (2006). Quantitative proteome analysis using isotope-coded affinity tags and mass spectrometry. Nat. Protoc. 1, 139–145. doi: 10.1038/nprot.2006.22
Singh, A. K., Pandey, R. K., Siqueira-Neto, J. L., Kwon, Y.-J., Freitas-Junior, L. H., Shaha, C., et al. (2015). Proteomic-based approach to gain insight into reprogramming of THP-1 cells exposed to Leishmania donovani over an early temporal window. Infect. Immun. 83, 1853–1868. doi: 10.1128/IAI.02833-14
Singh, P., and Cole, S. T. (2011). Mycobacterium leprae : genes, pseudogenes and genetic diversity. Future Microbiol. 6, 57–71. doi: 10.2217/fmb.10.153
Smith, I. (2003). Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 16, 463–496. doi: 10.1128/CMR.16.3.463-496.2003
Smith, J. B. (ed.). (2001). “Peptide sequencing by Edman degradation,” in Encyclopedia of Life Sciences, (Hoboken, NJ: Wiley-Blackwell).
Smyth, M. S., and Martin, J. H. (2000). X ray crystallography. Mol. Pathol. 53, 8–14. doi: 10.1136/mp.53.1.8
Sorge, U. S., Lissemore, K., Godkin, A., Hendrick, S., Wells, S., and Kelton, D. (2011). Associations between paratuberculosis milk ELISA result, milk production, and breed in Canadian dairy cows. J. Dairy Sci. 94, 754–761. doi: 10.3168/jds.2010-3404
Soto, A., and Muñoz, P. T. (2015). Leprosy diagnosis: an update on the use of molecular tools Lucrecia. Mol. Biol. 4:139. doi: 10.4172/2168-9547.1000139
Srinivas, P. R., Verma, M., Zhao, Y., and Srivastava, S. (2002). Proteomics for cancer biomarker discovery. Clin. Chem. 48, 1160–1169.
Stucki, D., and Gagneux, S. (2012). Single nucleotide polymorphisms in Mycobacterium tuberculosis and the need for a curated database. Tuberculosis (Edinb.) 93, 30–39. doi: 10.1016/j.tube.2012.11.002
Sutcliffe, I. C., and Harrington, D. J. (2004). Lipoproteins of Mycobacterium tuberculosis: an abundant and functionally diverse class of cell envelope components. FEMS Microbiol. Rev. 28, 645–659. doi: 10.1016/j.femsre.2004.06.002
Tang, C., Yamada, H., Shibata, K., Maeda, N., Yoshida, S., Wajjwalku, W., et al. (2008). Efficacy of recombinant bacille Calmette-Guérin vaccine secreting interleukin-15/antigen 85B fusion protein in providing protection against Mycobacterium tuberculosis. J. Infect. Dis. 197, 1263–1274. doi: 10.1086/586902
Tenzer, S., Peters, B., Bulik, S., Schoor, O., Lemmel, C., Schatz, M. M., et al. (2005). Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. 62, 1025–1037. doi: 10.1007/s00018-005-4528-2
Theodorescu, D., and Mischak, H. (2007). Mass spectrometry based proteomics in urine biomarker discovery. World J. Urol. 25, 435–443. doi: 10.1007/s00345-007-0206-3
Tripathi, R., Chakraborty, P., and Varadwaj, P. K. (2017). Unraveling long non-coding RNAs through analysis of high-throughput RNA-sequencing data. Noncoding RNA Res. 2, 111–118. doi: 10.1016/j.ncrna.2017.06.003
Trunz, B. B., Fine, P. E. M., and Dye, C. (2006). Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet 367, 1173–1180. doi: 10.1016/S0140-6736(06)68507-3
Tullius, M. V., Harth, G., Masleša-Galiæ, S., Dillon, B. J., and Horwitz, M. A. (2008). A replication-limited recombinant Mycobacterium bovis BCG vaccine against tuberculosis designed for human immunodeficiency virus-positive persons is safer and more efficacious than BCG. Infect. Immun. 76, 5200–5214. doi: 10.1128/IAI.00434-08
Tundup, S., Pathak, N., Ramanadham, M., Mukhopadhyay, S., Murthy, K. J. R., Ehtesham, N. Z., et al. (2008). The co-operonic PE25/PPE41 protein complex of Mycobacterium tuberculosis elicits increased humoral and cell mediated immune response. PLoS One 3:e3586. doi: 10.1371/journal.pone.0003586
Van Ooij, C., Tamez, P., Bhattacharjee, S., Hiller, N. L., Harrison, T., Liolios, K., et al. (2008). The malaria secretome: from algorithms to essential function in blood stage infection. PLoS Pathog. 4:e1000084. doi: 10.1371/journal.ppat.1000084
van Ravenzwaay, B., Cunha, G. C., Leibold, E., Looser, R., Mellert, W., Prokoudine, A., et al. (2007). The use of metabolomics for the discovery of new biomarkers of effect. Toxicol. Lett. 172, 21–28. doi: 10.1016/j.toxlet.2007.05.021
Ventola, C. L. (2015). The antibiotic resistance crisis: part 1: causes and threats. P T 40, 277–283.
Vlahou, A., and Fountoulakis, M. (2005). Proteomic approaches in the search for disease biomarkers. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 814, 11–19. doi: 10.1016/j.jchromb.2004.10.024
Voedisch, B., and Thie, H. (2010). “Size exclusion chromatography,” in Antibody Engineering, eds R. Kontermann and S. Dübel (Berlin: Springer),607–612.
Vogel, H., and Jähnig, F. (1986). Models for the structure of outer-membrane proteins of Escherichia coli derived from Raman spectroscopy and prediction methods. J. Mol. Biol. 190, 191–199. doi: 10.1016/0022-2836(86)90292-5
Wagley, S., Hemsley, C., Thomas, R., Moule, M. G., Vanaporn, M., Andreae, C., et al. (2014). The twin arginine translocation system is essential for aerobic growth and full virulence of Burkholderia thailandensis. J. Bacteriol. 196, 407–416. doi: 10.1128/JB.01046-13
Wallner, B., and Elofsson, A. (2005). All are not equal: a benchmark of different homology modeling programs. Protein Sci. 14, 1315–1327. doi: 10.1110/ps.041253405
Wang, P., Sidney, J., Dow, C., Mothe, B., Sette, A., and Peters, B. (2008). A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 4:e1000048. doi: 10.1371/journal.pcbi.1000048
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wassie, L., Demissie, A., Aseffa, A., Abebe, M., Yamuah, L., Tilahun, H., et al. (2008). Ex vivo cytokine mRNA levels correlate with changing clinical status of ethiopian TB patients and their contacts over time. PLoS One 3:e1522. doi: 10.1371/journal.pone.0001522
Watanabe Pinhata, J. M., Cergole-Novella, M. C., Moreira dos Santos Carmo, A., Ruivo Ferro e Silva, R., Ferrazoli, L., Tavares Sacchi, C., et al. (2015). Rapid detection of Mycobacterium tuberculosis complex by real-time PCR in sputum samples and its use in the routine diagnosis in a reference laboratory. J. Med. Microbiol. 64, 1040–1045. doi: 10.1099/jmm.0.000121
Webb, B., and Sali, A. (2016). Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 86, 2.9.1–2.9.37. doi: 10.1002/cpps.20
Weiner, J., Parida, S. K., Maertzdorf, J., Black, G. F., Repsilber, D., Telaar, A., et al. (2012). Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. PLoS One 7:e40221. doi: 10.1371/journal.pone.0040221
WHO (2016). WHO Treatment Guidelines for Drug-Resistant Tuberculosis. Geneva: World Health Organization.
Wiese, S., Reidegeld, K. A., Meyer, H. E., and Warscheid, B. (2007). Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics 7, 340–350. doi: 10.1002/pmic.200600422
Witjes, J. A., Dalbagni, G., Karnes, R. J., Shariat, S., Joniau, S., Palou, J., et al. (2016). The efficacy of BCG TICE and BCG Connaught in a cohort of 2,099 patients with T1G3 non-muscle-invasive bladder cancer. Urol. Oncol. 34, 484.e19–484.e25. doi: 10.1016/j.urolonc.2016.05.033
World Health Organization, Immunization, Vaccines and Biologicals Department (2012). Quality, Safety, and Standards Global Vaccine Safety. Geneva: WHO.
Yates, J. R. III (2011). A century of mass spectrometry: from atoms to proteomes. Nat. Methods 8, 633–637. doi: 10.1038/nmeth.1659
Yen, Y. T., Bhattacharya, M., and Stathopoulos, C. (2007). Genome-wide in silico mapping of the secretome in pathogenic Yersinia pestis KIM. FEMS Microbiol. Lett. 279, 56–63. doi: 10.1111/j.1574-6968.2007.01008.x
Yuan, G., and Xu, G. (2017). Highly unique and stable biomarkers for diagnosis of Mycobacterium tuberculosis pathogens. Biomed. Res. 28, 9633–9637.
Zagursky, R. J., and Russell, D. (2001). Bioinformatics: use in bacterial vaccine discovery. Biotechniques 31, 636–659. doi: 10.2144/01313dd02
Zhang, C., Song, X., Zhao, Y., Zhang, H., Zhao, S., Mao, F., et al. (2015). Mycobacterium tuberculosis secreted proteins as potential biomarkers for the diagnosis of active tuberculosis and latent tuberculosis infection. J. Clin. Lab. Anal. 29, 375–382. doi: 10.1002/jcla.21782
Zhang, J., Chiodini, R., Badr, A., and Zhang, G. (2011). The impact of next-generation sequencing on genomics. J. Genet. Genomics 38, 95–109. doi: 10.1016/j.jgg.2011.02.003
Zhang, L., Wang, Q., Wang, W., Liu, Y., Wang, J., Yue, J., et al. (2012). Identification of putative biomarkers for the serodiagnosis of drug-resistant Mycobacterium tuberculosis. Proteome Sci. 10:12. doi: 10.1186/1477-5956-10-12
Zhang, W., Zhang, Y., Zheng, H., Pan, Y., Liu, H., Du, P., et al. (2013). Genome sequencing and analysis of BCG vaccine strains. PLoS One 8:e71243. doi: 10.1371/journal.pone.0071243
Zhou, F., Xu, X., Wu, S., Cui, X., Fan, L., and Pan, W. (2015). Protein array identification of protein markers for serodiagnosis of Mycobacterium tuberculosis infection. Sci. Rep. 5:15349. doi: 10.1038/srep15349
Zvi, A., Ariel, N., Fulkerson, J., Sadoff, J. C., and Shafferman, A. (2008). Whole genome identification of Mycobacterium tuberculosis vaccine candidates by comprehensive data mining and bioinformatic analyses. BMC Med. Genomics 1:18. doi: 10.1186/1755-8794-1-18
Keywords: Mycobacterium, vaccine, diagnostic markers, reverse vaccinology, antigen discovery
Citation: Rana A, Thakur S, Kumar G and Akhter Y (2018) Recent Trends in System-Scale Integrative Approaches for Discovering Protective Antigens Against Mycobacterial Pathogens. Front. Genet. 9:572. doi: 10.3389/fgene.2018.00572
Received: 26 September 2018; Accepted: 06 November 2018;
Published: 27 November 2018.
Edited by:
Alfredo Pulvirenti, Università degli Studi di Catania, ItalyReviewed by:
Marco Ragusa, Università degli Studi di Catania, ItalySandeep Kumar Dhanda, La Jolla Institute for Allergy and Immunology (LJI), United States
Copyright © 2018 Rana, Thakur, Kumar and Akhter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yusuf Akhter, eXVzdWZAZGFhZC1hbHVtbmkuZGU=; eXVzdWYuYWtodGVyQGdtYWlsLmNvbQ==