 Julia Dluzewska
Julia Dluzewska Maja Szymanska
Maja Szymanska Piotr A. Ziolkowski
Piotr A. Ziolkowski- Department of Genome Biology, Institute of Molecular Biology and Biotechnology, Adam Mickiewicz University, Poznań, Poland
It is believed that recombination in meiosis serves to reshuffle genetic material from both parents to increase genetic variation in the progeny. At the same time, the number of crossovers is usually kept at a very low level. As a consequence, many organisms need to make the best possible use from the one or two crossovers that occur per chromosome in meiosis. From this perspective, the decision of where to allocate rare crossover events becomes an important issue, especially in self-pollinating plant species, which experience limited variation due to inbreeding. However, the freedom in crossover allocation is significantly limited by other, genetic and non-genetic factors, including chromatin structure. Here we summarize recent progress in our understanding of those processes with a special emphasis on plant genomes. First, we focus on factors which influence the distribution of recombination initiation sites and discuss their effects at both, the single hotspot level and at the chromosome scale. We also briefly explain the aspects of hotspot evolution and their regulation. Next, we analyze how recombination initiation sites translate into the development of crossovers and their location. Moreover, we provide an overview of the sequence polymorphism impact on crossover formation and chromosomal distribution.
Introduction
Crossover recombination lies in the center of sexual reproduction, providing physical connections between homologous chromosomes during meiosis. In most sexually reproducing eukaryotes these connections are required to ensure proper segregation of chromosomes during the first meiotic division (Moore and Orr-Weaver, 1997; Villeneuve and Hillers, 2001). To fulfill this requirement only one crossover per each chromosome pair is needed and many species regulate the crossover number to not exceed this lower functional limit (Mercier et al., 2015; Ritz et al., 2017). In fact, it was estimated that amongst nearly 50 eukaryotes belonging to different kingdoms, 80% of chromosome pairs have fewer than 3 crossovers (Fernandes et al., 2018b). Both indirect and direct data indicate that crossover rate is under selection in both directions (Ritz et al., 2017). The reasons for selection against crossover rate being too low are easy to understand: lack of crossover may lead to chromosome non-disjunction, which can yield in aneuploidy. The causes of constrains against too frequent recombination are less intuitive, as crossovers increase genetic diversity within population by breaking up haplotypes. However, recombination can also break association between beneficial alleles on the same haplotypes, which might lead to reduced progeny fitness (Otto and Lenormand, 2002). In fact, there is still not much empirical evidence that recombination is advantageous for natural population under selection (Otto, 2009). Benefits seem to emerge for finite populations in situations where selection varies over time and/or space (Otto, 2009). As these situations are not permanent, the crossover rate may evolve to be kept at low levels. Direct evidence that crossovers carry more de novo mutations than non-recombinant DNA molecules has been recently reported for human (Arbeithuber et al., 2015). In Arabidopsis, higher mutation rate was reported in regions proximal to crossovers (Yang et al., 2015). This would suggest that crossover repair is more mutagenic than other meiotic repair pathways. Moreover, recent results for Arabidopsis plants, where crossover rate was significantly increased by combining mutations of different anti-crossover factors (Fernandes et al., 2018b) and/or overexpression of pro-crossover factors (Serra et al., 2018b) indicated some fertility defects apparent even in the first generations. Detailed cytological investigation suggested that those defects are likely due to improper repair of a subset of recombination intermediates (Fernandes et al., 2018b). This would potentially result in dangerous accumulation of mutations in subsequent generations. It is possible, however, that the improperly repaired intermediates do not necessarily include or are not limited to crossovers, hence these findings cannot be considered as a prove of crossover genotoxicity.
Whatever are the reasons of restricted crossover numbers, this rises an interesting question: where to put the crossover to get the best from it. Historically, it was believed that crossover distribution is even – actually this assumption stands as a major basis of genetic (recombination) mapping (Sturtevant, 1913). But even in the very first work of Sturtevant (1913) it was suggested that the map distances are not just physical, but are some kind of joint function between length and “strength” over a region of chromosome. With time we have realized that the assumption on random crossover distribution is far from being accurate, though useful for many genetic approaches. We currently know that a large number of different factors is responsible for chromosomal distribution of crossovers. In this review, we discuss different levels of control for crossover chromosomal distribution with a special emphasis on DNA heterozygosity. Mammalian systems are very distinct in this respect, mainly due to the presence of mammalian-specific PRDM9 histone methyltransferase, which is a major determinant of crossover pattern in human and mice (Baudat et al., 2010; Parvanov et al., 2010). Therefore, we will specifically focus on factors determining crossover location in plants and support this view with extensive progress in understanding of the recombination-related processes, which has been achieved in budding yeast.
General Information About Crossover in Plants
The initial step inducing meiotic recombination is the formation of programmed DNA double-strand breaks (DSBs) catalyzed by the conserved topoisomerase-like complex, SPO11/TPOVIBL (Keeney et al., 1997; de Massy, 2013; Robert et al., 2016; Vrielynck et al., 2016). Following formation, DSBs are resected to generate single-stranded DNA (ssDNA), which is bound by the RecA-related recombinases RAD51 and DMC1 (Mercier et al., 2015). As a result, nucleoprotein filaments are created and further invade a sister chromatid or a chromatid located on a homologous chromosome. This results in a displacement loop (D-loop), which could be further processed via second-end capture to form double Holliday junction (dHJ) between the two chromatids (Wang and Copenhaver, 2018). It has been accepted that DMC1, a meiosis-specific homologue of RecA protein, is responsible for interhomolog bias – an increased chance of repair using homolog chromatid (Schwacha and Kleckner, 1997; Kurzbauer et al., 2012). The resolution of duplexes formed between sister chromatid results in DNA molecules, which are undistinguishable from their parental molecules, as they do not differ in DNA sequence. Repair based on non-sister chromatids may proceed via either several various non-crossover or crossover pathways. In contrast to crossovers (COs), where large fragments of DNA are reciprocally exchanged between parental chromosomes, non-crossovers (NCOs) result in gene conversion without affecting the template. The decision, which of DSBs will mature into crossover, and which will be repaired by non-crossovers, is called crossover designation.
In most eukaryotes including plants, two types of crossover pathways exist. The major pathway, named ZMM after the budding yeast genes ZIP1, ZIP2, ZIP3, ZIP4, MSH4, MSH5, and MER3, results in 85–90% of crossovers (called class I crossovers) in Arabidopsis, maize and rice (Higgins et al., 2004; Mercier et al., 2005; Falque et al., 2009; Shen et al., 2012). This pathway is meiosis-specific and depends on synaptonemal complex (SC) formation (Lynn et al., 2007). It is believed that ZMM proteins act to stabilize the interhomolog recombination intermediates to promote resolution by crossover (Lynn et al., 2007). Class I crossovers show interference, i.e., occurrence of a crossover in one location on a chromosome reduces significantly a chance for a second crossover in adjacent region on the same chromosome. Interference is detectable over the scale of megabases in Arabidopsis (Lynn et al., 2007; Mercier et al., 2015). Although interference has a great impact on chromosomal distribution of crossovers, it will not be discussed in this review as there are numerous articles focusing specifically on this phenomenon (Wang et al., 2015; Sun et al., 2017).
The residual, non-ZMM crossovers (class II COs) are interference independent. The best-known player for class II COs is MUS81, an endonuclease which is able to process joint molecules (e.g., D-loops). The null mutation of MUS81 reduces recombination by 10% in wild-type Arabidopsis plants and eliminates ca. 1/3 of the residual COs in zmm mutants (Berchowitz et al., 2007; Higgins et al., 2008a; Macaisne et al., 2011). This suggests that class II crossovers result from several different, partially redundant pathways. Opposite to ZMM pathway, the other pathways usually promote recombination intermediates resolution by non-crossovers.
Impact of Dsb on Crossover Distribution
Distribution of DSBs could be considered as the first level of defining crossover sites. Obviously, crossover can occur only at a DSB site, hence blocking DSBs at one chromosomal location will automatically exclude this region from the pool of potential recombination sites. However, in most organisms including plants, the number of DSBs significantly exceeds the number of crossovers (Mercier et al., 2015). For example, there is about 150–300 DSBs in Arabidopsis thaliana producing only around 10 crossovers (Chelysheva et al., 2010; Kurzbauer et al., 2012; Choi et al., 2013). Similarly in maize, nearly 500 DSBs lead to the formation of about 20 crossovers (Anderson et al., 2003; Pawlowski et al., 2003; Sidhu et al., 2015). As a consequence, a crossover site is selected from a wide range of potential locations. Even though, CO distribution is significantly associated with high levels of DSBs in Arabidopsis, at least at a genome-wide scale (Choi et al., 2018). Similar correlation was not reported for maize, which could be due to a very different, heterochromatin-rich genome architecture of the former species (He et al., 2017).
Recombination Initiation Hotspots in the Context of Chromatin Structure
In many eukaryotes DSBs occur in discrete, non-randomly distributed regions referred to as DSB hotspots. Their distribution at this scale is strictly dependent on chromatin structure, as meiotic chromosomes have specific architecture defined by chromatin loops protruding from proteinaceous axis (Blat et al., 2002; Borde and de Massy, 2013). Hotspots predominantly reside within loop regions, however, the machinery responsible for DSB formation, RMM complex (for budding yeast proteins REC114, MEI4, MER2), is physically located within the axis (Borde and de Massy, 2013). Interestingly, a strong anticorrelation between regions directly interacting with axis and with hotspot is observed, which suggests a repressing activity of some axis components. In addition, DSB hotspot regions frequently overlap with 5′ ends of yeast genes indicating that open chromatin states play a role in hotspot determining within the chromatin loops (Pan et al., 2011).
Trimethylation at histone 3 at lysine 4 (H3K4me3; to a lesser extent also dimethylation H3K4me2) has been found as an important determinant of DSB location in numerous eukaryotes and extensively investigated in budding yeast (Acquaviva et al., 2013; Sommermeyer et al., 2013). This histone mark, which is usually located at 5′-ends of genes, is also associated with high expression levels (Santos-Rosa et al., 2002). H3K4me3 is not considered as instructive for gene expression but rather as its consequence, which may also have a function in epigenetic memory (Howe et al., 2017). In contrary, H3K4 methylation seems to be very important for designation of DSB sites in budding yeast. First, it was observed that in the absence of H3K4me3 the number of meiotic DSB is significantly reduced (Borde et al., 2009). Borde et al. (2009) found also that elevated H3K4me3 levels near DSBs were independent of local transcription levels, replication and, more importantly, of DSB formation. Further work resulted in proposing a model, in which a PHD finger domain protein SPP1 recognizes H3K4me2/3 and tether the corresponding site of chromatin loop to chromosome axis by interaction with MER2 and other factors (Borde et al., 2009; Acquaviva et al., 2013; Sommermeyer et al., 2013). More recent report indicates that SPP1 protein adopts multiple roles in this process including meiosis-specific histone methyltransferase, dedicated for DSB designation (Adam et al., 2018). Whether similar mechanisms exist in plants remains unknown.
In plants, first analyses of DSB sites were performed indirectly by studying CO and NCO pattern within two recombination hotspots in A. thaliana (Drouaud et al., 2013). The authors noticed, however, that NCO tracts were relatively short and, due to limited polymorphisms, a large portion of the NCO events could not be detected. More recently, the DSB distribution was investigated at the genome-wide scale in maize using RAD51-ChIP (He et al., 2017) and in Arabidopsis using SPO11-oligos mapping (Choi et al., 2018). The first method uses antibodies to specifically precipitate DNA fragments associated with RAD51 – a protein that together with DMC1 binds to ssDNA formed at DSB site. SPO11-oligos mapping takes advantage from the fact that SPO11 protein covalently binds a short ssDNA fragment in the process of DSB formation; following SPO11 isolation from meiocytes, the DNA oligonucleotides may be extracted and sequenced to precisely identify DSB sites. Both analyses indicated relatively low correlation between DSBs and H3K4me3 at the chromosome and hotspot level, which is consistent with the data received from budding yeasts and mouse (Lange et al., 2016; Yamada et al., 2017a). However, correlation between COs and H3K4me3 was detected in both species (Choi et al., 2013; Shilo et al., 2015; He et al., 2017), suggesting that this chromatin mark may play similar role for loop tethering to chromosome axis as reported in yeast and mammals (Borde et al., 2009; Acquaviva et al., 2013; Sommermeyer et al., 2013; Adam et al., 2018). On the other hand, H3K4me3 is a universal chromatin mark associated with open chromatin structure, especially with transcription start sites, therefore its location next to CO sites may be purely coincidental.
Plant DSB hotspots have been estimated to be 1.2 kb (maize) and 0.8 kb (Arabidopsis) in size on average, and exhibit the most evident association with open chromatin structure defined as chromosome regions depleted in nucleosomes (He et al., 2017; Choi et al., 2018). This indicates that SPO11 acts in an opportunistic fashion, being able to cut different sequences as long as it has an access to DNA. This resembles the situation in budding yeast (Pan et al., 2011), but is very different from many mammals where DSB hotspots are mainly determined by PRDM9 meiosis-specific histone methyltransferase (Baudat et al., 2010; Lam and Keeney, 2015a). One clear negative correlation was observed between DSB hotspots and DNA methylation, visualized also in met1 background DSB mapping, in which most of CG context DNA methylation is removed (Choi et al., 2018). In this case elevation of DSB levels clearly mirrored decrease in DNA methylation accompanied by reduction of nucleosome occupancy and slight increase in H3K4me3 in pericentromeric regions. At the fine scale, these changes in recombination initiation sites are specifically evident for some transposable element classes (Gypsy LTRs and EnSpm/CACTA), but not all (LINE and SINE), which clearly reflects alteration in CG methylation pattern in those elements (Choi et al., 2018). Furthermore, increase in DSBs was also observed in kyp suvh5 suvh6 triple mutant, in which CHG and CHH methylation as well as H3K9me2 are strongly diminished (Underwood et al., 2018). Here the changes were mainly associated with elevation of DSB levels in centromeric repeats, such as CEN180. On the other hand, DNA methylation is usually associated with heterochromatin and closed chromatin structure (Zhang, 2008). Therefore, it is difficult to conclude whether the inhibition of DSB formation in methylated regions is a direct (e.g., due to physical obstacles during dHJ resolution) or indirect (e.g., secondary effects on chromatin structure) consequence of DNA methylation.
The number of DSBs tends to be modestly diminished in most repetitive sequences such as segmental duplications and repetitive transposons, however a subset of transposons (Stowaway elements in potato, and Helitron, Pogo/Tc1/Mariner DNA transposons in A. thaliana) are enriched in genomic regions spanning crossovers (Marand et al., 2017; Choi et al., 2018). The increased number of SPO11-oligos within Helitron, Pogo/Tc1/Mariner DNA repetitive transposons was observed when adjacent to immunity genes, which may contribute to enhanced favorable recombination within those regions (Choi et al., 2018).
When compared to plants, other eukaryotes have more pronounced DSB hotspots. In mammals this is due to the stringent PRDM9-dependent DSB patterning (Baudat et al., 2010; Parvanov et al., 2010). On the other hand, in budding yeast pronounced DSB peaks may be caused by extremely compact genome: from one side this contributes to significantly lower number of gene-related nucleosome-depleted regions (NDRs), which SPO11 tends to opportunistically bind to, and from the other side this results in a relatively high crossover pressure (de Massy, 2013). As it was mentioned, in plants, SPO11 hotspots are frequently found also in NDRs at the 3′-ends of genes, as well as in introns (Choi et al., 2018). This increases significantly the number of potential SPO11 targets in plant genomes, which may result in the more uniform recombination landscape.
Transcription Factors (TFs)
In most eukaryotes including plants, DSB hotspots do not correlate with transcription in this mean that genes highly expressed in meiosis do not show higher DSB levels (Tischfield and Keeney, 2012; He et al., 2017; Choi et al., 2018; see however Yamada et al., 2017b). In some cases, however, DSB hotspot activity was connected with the binding of sequence-specific transcription factors. For instance, this has been reported at the HIS4 locus in budding yeast and the ade-M26 allele in fission yeast (White et al., 1991; Kon et al., 1997; Steiner et al., 2002). In fission yeast a heterodimeric basic-leucine-zipper transcription factor ATF1-PCR1 was found to recognize hotspot-specific DNA motifs M26 leading to recruitment of DSB-machinery (Kon et al., 1997; Steiner et al., 2002). In the more recent genome-scale analysis of meiotic DSB landscape in Schizosaccharomyces pombe, the positive effect of hotspot activity was detected for less than a quarter of loci containing the motif showing that other factors contribute to the development of the recombination initiation hotspot (Fowler et al., 2014). In many of those loci DSBs seem to be symmetrically arrayed around the TF’s binding sites. Interestingly, about half of the identified M26 hotspots showed increased DSB levels to one side of the motif while the other side exhibited strong transcription. The mechanism by which ATF1-PCR1 affects DSB formation remains elusive (Fowler et al., 2014). In summary, the study showed that binding of ATF1-PCR1 alone is not sufficient to target high levels of DSB formation nearby.
Although, the way how TFs affects recombination is ambiguous (Fowler et al., 2014; Zhu and Keeney, 2015), in some cases (HIS4 and PHO5 in budding yeast) the effect of TF is achieved by changing DNA accessibility in the hotspot region (Wu and Lichten, 1995). Therefore, it has been proposed that transcription factor-induced chromatin modifications underlie activation of recombination breakpoints (Hirota et al., 2008; Wahls and Davidson, 2010). The activating chromatin modifications are likely to belong to different types, however, formation of nucleosome-depleted regions along with histone acetylation and methylation at specific positions are considered as the most universal (Hirota et al., 2008; Getun et al., 2017).
An interesting observation is the formation of ATF-PCR1 heterodimer in response to osmotic stress, which in turn triggers meiosis in fission yeast. In budding yeast activation of HIS4 DSB hotspot requires binding of RAP1, BAS1 and BAS2 transcription factors (White et al., 1993), and a more recent studies showed that a number of other DSB hotspots is dependent on these factors (Mieczkowski et al., 2006; Zhu and Keeney, 2015). Expression of those TFs is strictly linked with cell starvation that also induces meiosis. Thus, the transcription factors involved in preparation for stress response in fission and budding yeasts seem to be reutilized for DSB hotspot generation, resulting in an increased recombination rate. This coincidence could be beneficial from evolutionary point of view. Whether similar mechanisms exist in other organisms remains unknown and there are no clear examples that DSB hotspots might be regulated by TFs in higher eukaryotes. However, existence of similar TF-related modifiers, which would conditionally activate specific recombination hotspots, is an exciting possibility, especially in plants which show gene-located recombination hotspots. For instance, one could easily imagine potential benefits of targeting meiotic recombination nearby genes responsible for resistance to pathogen in biotic stress conditions. Indeed, some R-genes exhibit elevated recombination frequency, which was exemplified both at the historical (based on coalescent analysis) (Choi et al., 2013) and experimental data (Choi et al., 2016; Serra et al., 2018a).
Interestingly, the most important factor that determines recombination hotspot location in many mammals, PRDM9, emerged from a metazoan-specific family of TFs (Vervoort et al., 2016). In this case, however, the PRDM transcription factors are involved in a wide variety of functions during animal development but not in stress response, suggesting an accidental capturing of a new biological function by the TF. PRDM9 possesses an array of zinc-fingers, by which it recognizes specific DNA motifs and incorporates classical H3K4me3 and H4K36me3 marks defining recombination hotpots (Baker et al., 2015). It is currently unknown why modifications by PRDM9 have stronger effect on defining DSB sites than promoter-located H3K4me3 marks, which are formed by other methyltransferase complexes (Brick et al., 2012), however, recent data indicate that concentration of marks may play a role (Diagouraga et al., 2018).
Local Base Composition and DNA Motifs
Local base composition, especially GC-rich regions, was found to be an important factor controlling distribution of recombination initiation hotspots in many organisms, from yeasts to mammals and plants (Blat and Kleckner, 1999; Gerton et al., 2000; Myers et al., 2008; Smeds et al., 2016; He et al., 2017). Several explanations for this phenomenon have been proposed including a higher susceptibility for recombination machinery in GC-enriched regions (Petes, 2001) and biased repair of mismatches in recombining regions toward G/C pairs (Birdsell, 2002). Recent work in budding yeast shows that incorporating GC-rich sequence into URA3 hotspot significantly elevates meiotic and mitotic recombination rate (Kiktev et al., 2018). This would suggest that at least in this case the first hypothesis is correct. This relationship may be a consequence of DSB-formation dependence on chromatin structure, especially nucleosome positioning and specific histone modifications including H3K4me3, which is important for tethering of chromatin loops to chromosome axis.
High GC content has been found also in maize DSB hotspots (He et al., 2017). However, similar relationship was not reported for Arabidopsis (Choi et al., 2018; Underwood et al., 2018). This is surprising in the context of similarities between yeast and plant recombination hotspots, as in both cases nucleosome-depleted regions and open chromatin structures are recalled as the most characteristic features (Choi and Henderson, 2015). However, one should consider significant differences between the structure of Arabidopsis and maize genome: while the first one is very small and compact (0.12 Gb), deprived of transposable elements (TEs) and gene-rich (one gene per 4.5 kb, on average) (The Arabidopsis Genome Initiative, 2000), the other is extremely large (2.3 Gb), TE-rich, with a gene-island organization (single genes separated by very long stretches of non-coding repetitive sequences) (Rodgers-Melnick et al., 2016). As many TE sequences are relatively AT-rich, which is usually considered as a way to escape from silencing via RNA-dependent DNA methylation, from evolutionary perspective it might be beneficial to allocate hotspot in GC-rich regions, which more often belongs to genes. From this point of view, it would be interesting to check the methylation pattern of maize recombination initiation hotspots.
A closer insight into maize recombination landscape indicates that in fact the high GC content of DSB hotspots corresponds to a 20-bp-long sequence motif (He et al., 2017). The motif is present in more than 70% of genic hotspots, however, cannot be detected in repetitive DNA hotspots. In the same work the authors also described a crossover-associated motif, which is similar to DSB motif, however, contains overrepresentation of ‘C’ over ‘G’ (He et al., 2017). Similar, C-rich motifs have been described in Arabidopsis as crossover-associated (Choi et al., 2013; Wijnker et al., 2013; Shilo et al., 2015), although DSB-associated motifs seem to be rather AT-rich (Choi et al., 2018). All those findings suggest that it is not the GC content per se that is responsible for higher DSB formation or crossover formation, but instead the effect is related to specific sequence motifs which are likely recognized by recombination machinery. In addition, as AT-rich regions are known to exclude nucleosomes, it is suggested that those sequences are associated with more open chromatin structure affecting SPO11 accessibility and resulting in elevated DSB levels (Choi et al., 2018).
Effects of Regulatory Circuits on DSB Distribution
Beside chromatin landscape, which can be considered as relatively stable determinant of DSB pattern, a number of regulatory mechanisms actively affect the distribution of DSB hotspots in a cell-to-cell manner. DNA damage-response kinases TEL1ATM and MEC1ATR play a crucial role in this regulation. These proteins are also responsible for DSB interference (Xu and Kleckner, 1995; Lange et al., 2011; Lukaszewicz et al., 2018). Similar to crossover interference, DSB interference reduces the likelihood of DSB formation next to already formed DSB on the chromosome (Garcia et al., 2015). This phenomenon has been examined in details in budding yeast, and it appears to act only at short distances, usually below 100 kb, and is controlled mainly by TEL1ATM (Robine et al., 2007) (Figure 1). It is believed that DSB interference is important to prevent formation of multiple DSBs in the same chromatin loop, which would be hazardous for genome stability (i.e., may cause chromosomal rearrangements). An interesting consequence of DSB interference is hotspot competition. Generation of an artificial DSB hotspot results in reduction of activity of surrounding hotspots, and the opposite can be observed when a hotspot is removed (Wu and Lichten, 1995; Pineda-Krch and Redfield, 2005; Robine et al., 2007; Acquaviva et al., 2013; Cooper et al., 2016).
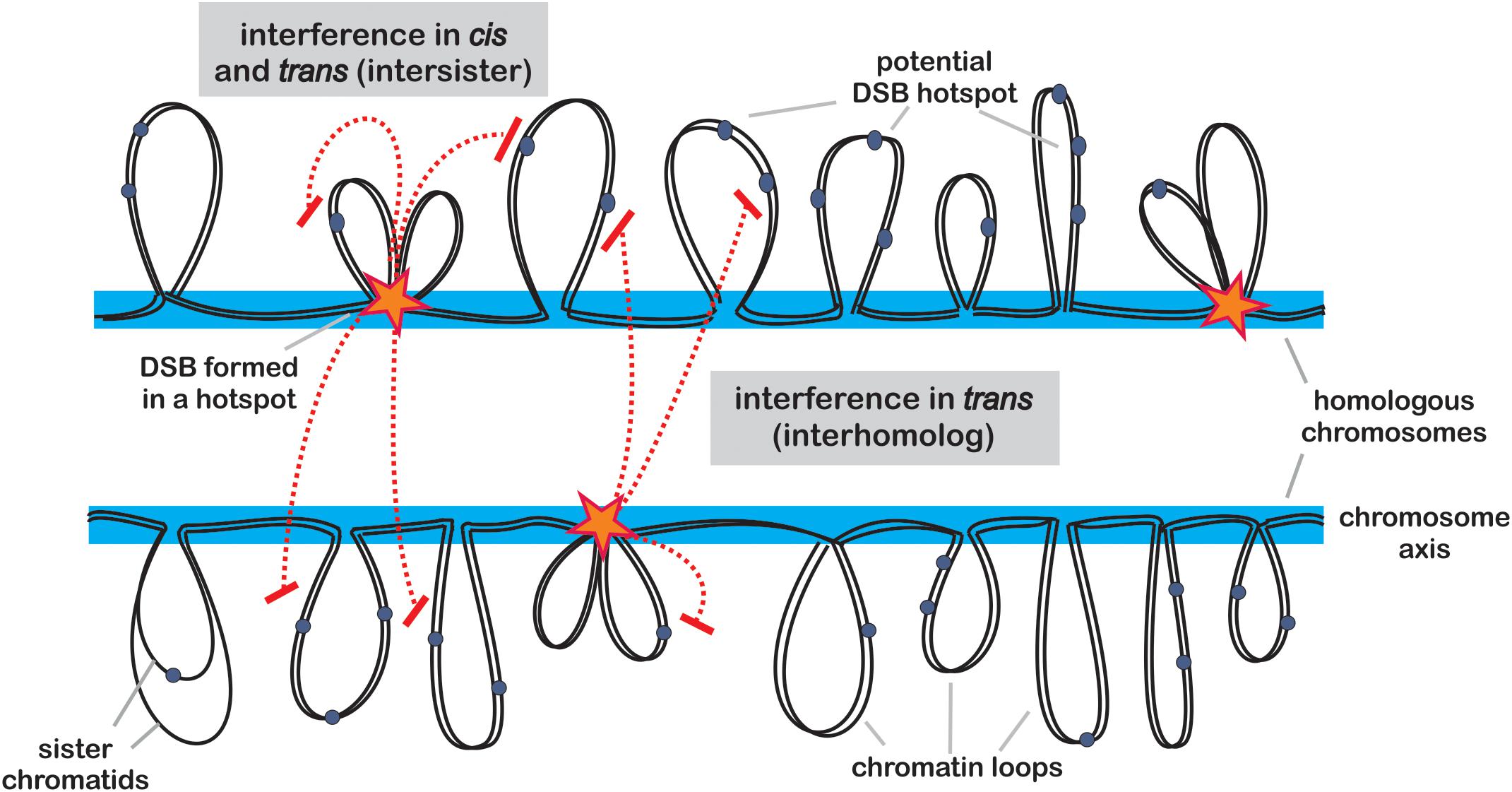
Figure 1. Model of DSB formation and control. Potential hotspot sites are located within chromatin loops (gray) protruding from chromosome axis (blue). Activation of a hotspot (star) requires tethering of the loop to the axis, where SPO11-containing protein complexes are deposited. Once a hotspot is activated, it communicates to other potential hotspots located on the same chromatid (in cis) or on its sister chromatid (in trans) causing their inhibition (red dashed lines). This process of positive DSB interference is dependent on ATM (TEL1) and ATR (MEC1) kinase signaling pathways and acts on distances of 30–100 kb in budding yeast. In addition, another form of interference inhibits formation of DSBs in potential hotspots located in the corresponding regions on homologous chromosome.
In addition, another form of DSB interference has been detected in yeast, acting in trans - between sister chromatid or between homologous chromosomes (Xu and Kleckner, 1995; Rocco and Nicolas, 1996; Zhang et al., 2012) (Figure 1). This mechanism is controlled by TEL1ATM and its partner MEC1ATR, and may act to reduce a chance of two DSBs occurring parallelly in corresponding chromosomal regions on both chromosomes, which would result in difficult to repair, complex double recombination event (Cooper et al., 2016). Recent work shows that MEC1ATR is less sensitive toward DSB numbers than TEL1ATM, therefore it gets activated later during prophase I progression (Joshi et al., 2015). Early DSBs lack homolog bias, however, with progression of meiosis, subsequent MEC1ATR-dependent DSBs are more likely to result in interhomolog COs (Joshi et al., 2015). Currently, it is not clear whether the same mechanisms exist in plants, however, as the DNA damage-response kinases are conserved among all eukaryotes, it seems very probable. In Arabidopsis, null mutants of the TEL1ATM and MEC1ATR homologs, ATM (ataxia telangiectasia mutated) and ATR (ATM and RAD3-related), show significantly decreased fertility, and their double mutant exhibit extensive chromosome fragmentation in meiosis, which results in complete sterility (Garcia et al., 2003; Culligan and Britt, 2008). This indicates that those proteins play an important role in meiotic DSB repair, beside their functions in somatic DSB repair (Garcia et al., 2003; Culligan and Britt, 2008).
Besides ATM/ATR signaling, there are also additional pathways, which are involved in DSB formation. For example, continued DSB formation on unsynapsed chromosomes was observed in male mice with a lowered SPO11 dosage (Kauppi et al., 2013). This indicates that homolog engagement is acting to shut off DSB formation. There are reports suggesting that a similar mechanism can exist also in nematodes and involves meiotic cohesin component REC8 (Hayashi et al., 2007). In the more detailed study on the yeast model, it has been found that the phenomenon is ZMM-dependent with ZIP3 being involved in inhibition of DSBs after homolog engagement (Thacker et al., 2014). Therefore, though at the moment it is not clear whether the same mechanism acts in different species, it is very likely that homolog engagement is an additional level of DSB control conserved in eukaryotes.
Evolution of Recombination Initiation Site Pattern
For a long time, theoretical studies on DSB hotspot evolution enforced a view predicting fast erosion of hotspot sequence leading to its rapid extinction (Boulton et al., 1997; Calabrese, 2007; Coop and Myers, 2007; Latrille et al., 2017). This “hotspot paradox” hypothesis was based on the assumption that a biased gene conversion occurs at a hotspot site, in which the broken chromosome copies DNA sequence from its uncut homolog. In consequence, the recombinationally active allele is replaced with its less active homolog, which results in its overrepresentation in progeny. Even in species where conversion tracts are very short and cannot significantly affect allele frequency (e.g., in Arabidopsis, Drouaud et al., 2013; Wijnker et al., 2013; Li et al., 2015), the active hotspot allele may be rapidly removed from the population simply by accumulation of point mutations. This is because recombination machinery seems to have a mutational effect, at least when crossover recombination is investigated (Lercher and Hurst, 2002; Arbeithuber et al., 2015; Yang et al., 2015). Mutations that reduce or eliminate hotspot activity will be consequently fixed in the population, whereas mutations activating hotspots will be removed.
Meiotic drive from biased conversion was reported in human (Jeffreys and Neumann, 2002), in which SPO11 is targeted to hotspots by PRDM9 protein. PRDM9 targets SPO11 to sites without additional biological functions, which are therefore released from evolutionary constrains other than those, which are recombination-related. Hence, PRDM9 recognition motifs can be subjected to meiotic drive from biased gene conversion leading to their rapid elimination from the population (Baker et al., 2015) (Figure 2A). As PRDM9-recognition motifs constantly disappear from the population, a new version of PRDM9 needs to evolve new combination of zinc-fingers to recognize novel motifs. For this reason PRDM9 belongs to the fastest evolving genes (Ponting, 2011). This phenomenon is observed in many species containing PRDM9-determined recombination initiation hotspots (Myers et al., 2010; Tiemann-Boege et al., 2017).
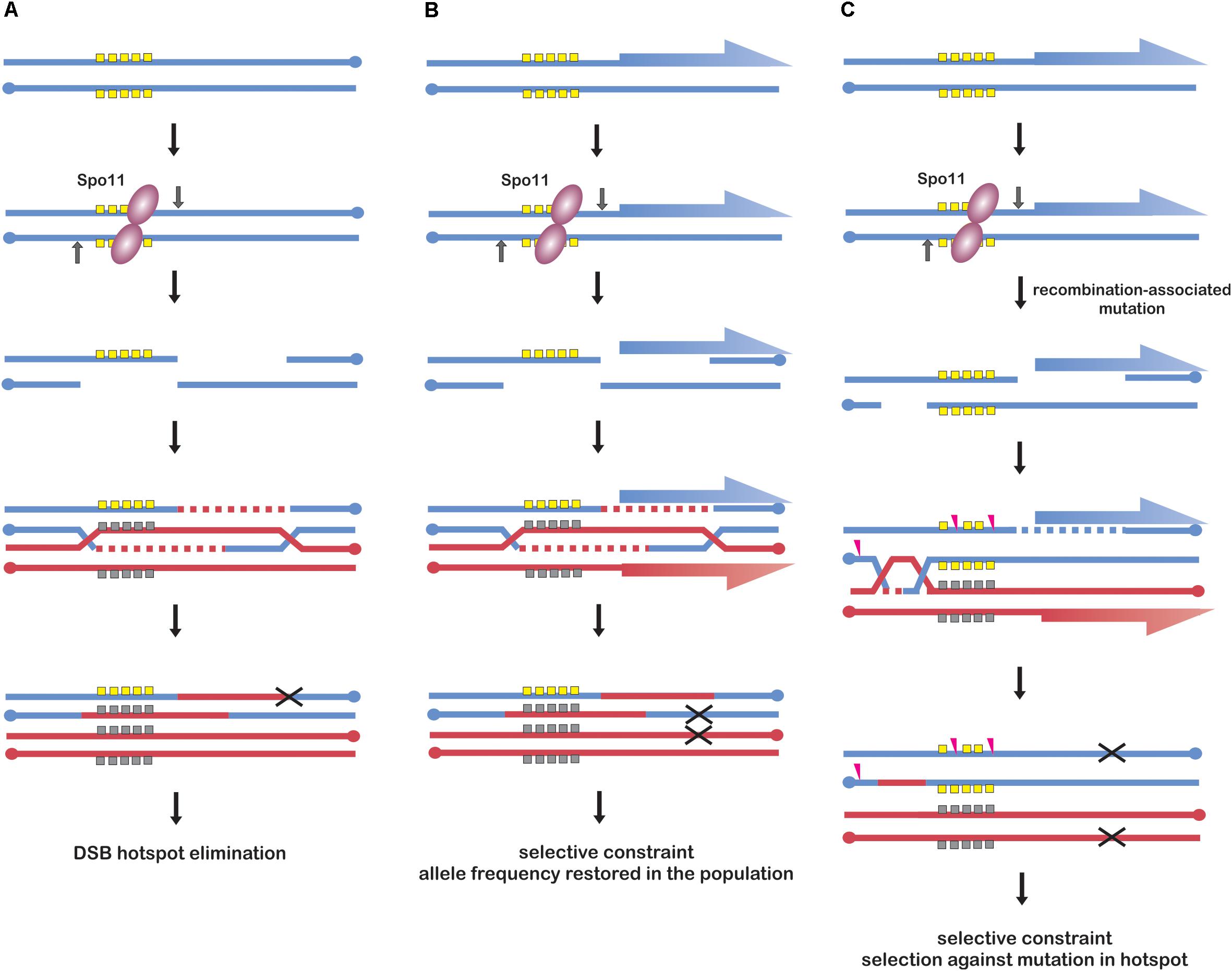
Figure 2. Models of recombination initiation hotspot evolution. (A) Frequent DSB formation in a strong hotspot allele (yellow squares) leads to increased conversion and rapid elimination of the allele from the population (black “X” symbol). This scenario is possible when the hotspot has no additional functions under selection, e.g., in human genome. Models (B,C) illustrate promoter-located hotspots; genes are indicated as blue and red half-arrows. (B) Frequent DSB formation in a hotspot allele (yellow squares) leads to its conversion to another allele (gray squares), however, selective constraint from its effect on promoter function enables its maintenance in the population. The selective constraint causes that particular hotspot alleles maintain comparable DSB activity. This model is likely to occur in yeast. (C) Frequent DSB events in a hotspot allele (yellow) result in an increased mutation rate leading to its erosion (pink arrowheads), however, selective constraint from its effect on regulatory elements enables maintenance of different original alleles in the population. Models (B,C) are not mutually exclusive and are likely to coexist in Arabidopsis.
However, plants lack PRDM9 protein or analogous system. Plants hotspot arrangement resembles more that of yeast, with recombination initiation hotspots located mostly in nucleosome-depleted regions of promoters (Figure 2). Recent work in budding yeast investigated DSB hotspot location between several different strains and the comparison indicated that yeast hotspots are surprisingly stable (Lam and Keeney, 2015b). Moreover, when hotspot heat was compared, yeast hotspots from different strains appeared to be more similar than human hotspots between men sharing the same PRDM9 alleles, despite much greater sequence diversity (Pratto et al., 2014; Lam and Keeney, 2015b). Similarly, in vertebrate species lacking PRDM9 protein, e.g., birds, crossover hotspots seem to be highly conserved and it is likely that the same is true for recombination initiation hotspots (Singhal et al., 2015). The possible explanation of high hotspot conservancy in yeast could be the selective constraint for additional functions of the underlying sequences, which are usually related to their promoter locations (Nicolas et al., 1989; Pan et al., 2011; Lam and Keeney, 2015b) (Figure 2B). Analogous explanation has been proposed for evolution of avian recombination hotspots (Singhal et al., 2015). Answering the question whether similar, non-paradoxical stability of DSB landscape exists also in plants would require further work, which is now possible with newly developed DSB-mapping approaches in plant systems (He et al., 2017; Choi et al., 2018). However, frequent location of recombination initiation hotspot in gene promoters in plants suggests analogous situations, including also elimination of mutated alleles (Figures 2B,C). In this context, it would be interesting to compare promoter-located hotspots with those located in intergenic regions or transposable elements: according to the proposed scenario, they should evolve much faster/should have shorter life time than promoter-located hotspots.
Recombination Initiation Sites at the Chromosome Scale
Although techniques enabling precise, sequence-based mapping of recombination initiation sites have been developed only recently, identification of those events on the chromosome scale has been possible for a long time based on observation of recombination nodules (RN) (reviewed in Anderson and Stack, 2005). Plant studies of those structures resulted in identification of two types of RN: early (ENs), which are associated with SC from leptotene into pachytene, and late (LNs), observed from pachytene into diplotene. The number of RNs declines in the course of meiosis, therefore ENs are thought to correspond to some stages of DSB repair, while LNs should represent those recombination events that are being repaired via crossover pathways (Zickler and Kleckner, 1998). ENs are randomly distributed along chromosomes at zygotene and do not show interference except at very short distances (in maize ≤ 0.2 μm) (Stack and Anderson, 2002). Nevertheless, ENs tend to be more frequent in distal euchromatic regions of bivalents than in proximal, heterochromatic segments, and the highest concentration of ENs occurs at synaptic forks suggesting their role in homologous synapsis (Stack and Anderson, 2002).
More specific features of recombination initiation sites became apparent with the emergence of high-resolution techniques. At the chromosome scale DSBs exhibit surprisingly uniform pattern of distribution, which has strong negative correlation with nucleosome occupancy in Arabidopsis (Spearman ρ = -0.7 or -0.95 for chromosome arms and pericentromeres, respectively) (Choi et al., 2018). The only regions that show dramatic suppression of recombination initiation are pericentromeres, which also reflects the effect of nucleosome occupancy, as in Arabidopsis heterochromatin is limited almost exclusively to those parts of the genome (The Arabidopsis Genome Initiative, 2000; Simon et al., 2015). All other tested correlations appear to be secondary or derivatives of the nucleosome positioning, especially when compared between chromosome arms and pericentromeres. For example, H3K4me3 was positively correlated with SPO11-oligos in the pericentromeric regions, but negatively in chromosome arms (Spearman ρ = 0.85 or – 0.82, respectively) (Choi et al., 2018). Similar observations were made in maize as assessed by RAD51 ChIP-seq with this difference that DSBs were abundant also in pericentromeric regions (He et al., 2017). Maize genome, however, is characterized by many more TE-rich heterochromatic regions, which show high DNA methylation levels and spread more uniformly along the chromosomes, with the majority of genes (∼85%) positioned within 1 kb of transposons (Schnable et al., 2009; Rodgers-Melnick et al., 2016). Other reasons of differences between Arabidopsis and maize patterns of recombination initiation sites may originate from technical issues applied.
CO Homeostasis
An interesting phenomenon, which links recombination initiation sites (DSBs) with crossover control, is CO homeostasis. This regulatory pathway, which was observed in budding yeast, Caenorhabditis elegans and mice, causes no change in CO numbers even when substantial variation in DSB number is induced (Martini et al., 2006; Rosu et al., 2011; Cole et al., 2012; Yokoo et al., 2012). Recent experiments indicated a limit in CO homeostasis in maize: CO control was robust as long as one crossover per chromosome pair was ensured, but above this threshold the number of COs was linearly correlated to the number of DSBs (Sidhu et al., 2015). In Arabidopsis fas1 mutant, in which a significant increase in DSB number is observed, no change in COs was reported (Varas et al., 2015). However, in hypomorphic A. thaliana spo11 mutants, the reduction in DSB number resulted in proportional, though smaller, reduction in COs (Xue et al., 2018). Those two results are not necessarily mutually excluding, as it is possible that, similarly to maize, in Arabidopsis and other plants CO homeostasis works robustly only in some ranges. Interestingly, a dramatic change in CO distribution was observed in the hypomorphic spo11 mutants, with substantial reduction of recombination in pericentromeres (Xue et al., 2018).
Factors Influencing Spatial Distribution of Crossover
Meiotic DSBs may be repaired by several different mechanisms (see Introduction) and only a minority of them become crossovers (Mercier et al., 2015). The decision, whether the break should be repaired as crossover or non-crossover, is made based on a number of factors, which are largely unknown. Some factors influencing this decision, like modification by stress, have been extensively reviewed in recent works (Modliszewski and Copenhaver, 2017; Morgan et al., 2017) and therefore this topic will not be discussed. However, it should be emphasized that significant differences are observed between recombination initiation site distribution (DSB distribution) and crossover distribution.
In plants, NCO repair leads to minimal exchanges of genetic information between parental genomes, because the conversion tracts are very short and often undetectable (Lu et al., 2012; Sun et al., 2012; Drouaud et al., 2013; Wijnker et al., 2013). Hence, CO remains a major cause of genetic material reshuffling, important for variation in natural plant populations.
Chromosome Level
Similarly to other eukaryotes, the crossover formation in plants is strongly biased toward euchromatic regions, in the contrast to CO inhibition at heterochromatin (Copenhaver et al., 1999; Giraut et al., 2011; Salomé et al., 2011; Mayer et al., 2012; Tomato Genome Consortium, 2012; Yelina et al., 2012; Rodgers-Melnick et al., 2016; He et al., 2017). Crossover suppression in the proximity of centromeres is important for fertility, as recombination events at those sites have been associated with chromosome segregation errors and aneuploidy (Rockmill et al., 2006; Stewart et al., 2008). Plant genomes show strong correlation between gene density and the distribution of genetic crossovers (Erayman et al., 2004; Choi et al., 2013; Wijnker et al., 2013; Rodgers-Melnick et al., 2015). Such fact is especially true for large genome species, like cereals, where crossover events are dramatically skewed toward the distal euchromatic, gene-rich regions of chromosomes. For instance, analyses in barley (Künzel et al., 2000; Mayer et al., 2012; Phillips et al., 2013), maize (Anderson et al., 2003; He et al., 2017) and wheat (Saintenac et al., 2009; Darrier et al., 2017) show elevations in CO number and gene density in subtelomeres and, at the same time, there is a decrease in recombination events and gene number in centromeric and pericentromeric regions (Schnable et al., 2009; Mayer et al., 2012; Tomato Genome Consortium, 2012; Higgins et al., 2014). Cytological studies in barley revealed that observed skewed chiasma distribution reflects polarization in the spatiotemporal initiation of recombination, chromosome pairing, and synapsis. Meiotic progression in distal chromosomal arms occurs in coordination with the chromatin cycles, whilst in interstitial and proximal regions meiotic initiation occurs later, is not coordinated with the cycles, and rarely progresses to form chiasmata. This early meiotic initiation is linked with euchromatic DNA, whilst late replication is observed at heterochromatin, in the centromeric and pericentromeric regions (Higgins et al., 2012).
Association of crossovers with a genic part of the plant genome may be a consequence of a specific, open chromatin structure within promoters and at the 3′-ends of genes, which authorizes the access to DNA. This could be especially important during meiosis, where chromatin is largely condensed and therefore inaccessible for recombination machinery. In concordance with this hypothesis is the intragenic pattern of COs: meta-analyses of both Arabidopsis and maize crossovers showed their underrepresentation within gene bodies and elevation at core promoters and at 3′-UTRs (Wijnker et al., 2013; Rodgers-Melnick et al., 2016). This points out the opportunistic feature of recombination complexes in plants, which is likely a consequence of SPO11 preferences.
Interestingly, the pattern of crossover distribution in some details is different from DSB distribution. For instance, in maize, large DSB numbers are formed in heterochromatic regions, however, they have very low chance for being repaired by crossover (He et al., 2017). This indicates that particular hotpots may significantly differ in CO/NCO ratios. In Arabidopsis, DSBs were not detected within pericentromeric regions, however, their distribution, though correlated to crossovers, shows also some significant differences: DSB levels exhibit more even distribution along the chromosomes than crossovers (Choi et al., 2018). It is unclear what is the reason of this discrepancy, but one factor could be a different genetic material used to achieve both datasets: SPO11-oligos used to map DSBs were obtained from the complemented spo11-1 mutant line in homozygous Col-0 background, while crossover maps were achieved based on recombination events in Col x Ler F1 plants, two different A. thaliana accessions. In consequence, the crossover landscape is shaped by multiple modifiers of recombination, especially the impact of heterozygosity pattern (see below). Investigation of DSBs by SPO11-oligos mapping in the Col x Ler or Ler alone genetic background would provide us a deeper understanding of the relationship between recombination initiation sites and crossover formation. Another explanation could be spatiotemporal characteristics of DSB formation and crossover repair. DSBs are formed within relatively long time during prophase of meiosis I, in parallel to chromosome condensation, however, it is thought that only the late events result in crossover repair (Anderson and Stack, 2005; Joshi et al., 2015). In consequence, the chromatin stage of early and late DSBs may be different, which would result in observed dissimilarities. This hypothesis requires more detailed analysis of chromosome condensation.
Heterochiasmy
Sex differences in recombination, known as heterochiasmy, are a widespread phenomenon described for the first time more than a century ago (Lenormand and Dutheil, 2005). Since that time an extensive set of data from many different species was collected indicating that heterochiasmy is a common feature of eukaryotes, including plants (Lenormand and Dutheil, 2005). Several hypotheses were proposed, however, none of them satisfactorily explain the variation in heterochiasmy in all species (Hedrick, 2007). This may indicate that the causes of heterochiasmy may be different in different taxa.
Heterochiasmy refers to both differences in recombination frequency and its spatial distribution along the chromosomes. We are especially interested in the second aspect of differences in recombination between sexes. In plants, this was extensively studied in Arabidopsis, where dramatic differences in COs, with a very significant, 1.8x higher rates in male compared to female meiosis were observed (Vizir and Korol, 1990). Very similar data were collected by Giraut et al. (2011) who investigated in details chromosomal distribution of crossovers. Although male crossovers are slightly elevated along the whole genome in almost all intervals tested, statistically significant differences were reported only for intervals in subtelomeric regions, and they encompass for the majority of differences in CO rates between sexes (Giraut et al., 2011). Interestingly, the ratio of the male vs. female genetic map length is very similar to the ratio of total SC length between male and female meiosis (Drouaud et al., 2007; Giraut et al., 2011). Similar observations were also made in other species, including mice, Drosophila, human and zebrafish (Kleckner et al., 2003). In C. elegans modification of SC length by a mutation in subunits of condensin results in increased CO rates (Mets and Meyer, 2009). Therefore, it is tempting to speculate that the length of SC determines the crossover number. This hypothesis is supported by recent findings in mice, where a map of recombination initiation sites (based on DMC1 binding) for males and females were achieved (Brick et al., 2018). The authors provided evidence that DSB frequency is not the driver of sex differences in distal crossovers in this species.
Recently, analysis of sex patterns in COs was carried out in maize (Kianian et al., 2018). The authors did not report significant differences in the spatial distribution of COs at the global scale, however, male and female COs differ at the fine scale, in their locations relative to transcription start sites in gene promoters. Differences were also observed with the respect to chromatin marks, including nucleosome occupancy and H3K4me3 (Kianian et al., 2018). This indicates that the sex specific features of crossover distribution could be observed even in species, where the global CO landscape remains the same between male and female meiosis. The mechanisms responsible for those differences are rather complex and currently poorly known. It would be interesting to study how sex differences in COs affect population structure and genome evolution.
DNA Methylation
In plants, DNA methylation occurs in CG, CHG and CHH sequence context (where H = A, T or C) (Law and Jacobsen, 2010). CG methylation is maintained during replication by Methyltransferase1 (Met1) with the help of SWI/SNF chromatin remodeling protein Decreased DNA Methylation1 (DDM1) (Vongs et al., 1993; Saze et al., 2003; Stroud et al., 2012; Zemach et al., 2013). Non-CG methylation is maintained by Chromomethylase2 (CMT2), Chromomethylase3 (CMT3) and Domains Rearranged Mathylase2 (DRM2) (Lindroth et al., 2001; Malagnac et al., 2002; Cao et al., 2003; Du et al., 2012; Stroud et al., 2012). The methylation in non-CG contexts require methylation of histone H3K9 by SET domain methyltransferases (Jackson et al., 2002; Malagnac et al., 2002; Johnson et al., 2007; Du et al., 2014). As the two processes are linked, the non-CG methylation mutants exhibit also reduced H3K9me2.
In Arabidopsis and maize, genome-wide analyses of CO hotspots show low levels of DNA methylation (Choi et al., 2013; Rodgers-Melnick et al., 2015; Kianian et al., 2018). Studies of non-CG methylation mutants, met1 and ddm1, documented that epigenetic crossover remodeling decreases within pericentromeric region and simultaneously increases in gene-rich chromosome arms in Arabidopsis thaliana (Mirouze et al., 2012; Yelina et al., 2012; Melamed-Bessudo and Levy, 2012). This is somehow surprising as significant loss of CG methylation in pericentromeres should result in elevation of recombination in those regions. The mutants did not significantly alter the total number of COs, but rather led to their redistribution along the chromosomes, which suggests the involvement of CO interference (Colomé-Tatché et al., 2012; Melamed-Bessudo and Levy, 2012; Mirouze et al., 2012; Yelina et al., 2012, 2015). In a more recent report Yelina et al. (2015) compared the effect of two crossover pathways in the met1 mutant background and concluded that crossover remodeling is due to the interfering pathway. They proposed that loss of DNA methylation either changes relative timing of DSB formation between arms and pericentromeres, or reduces the chance of crossover designation in the proximity of a centromere. As a consequence of crossover interference, the chromosome arms receive additional COs compared to wild type (Yelina et al., 2015).
Interestingly, very different effect was observed in mutants causing loss of CHG and CHH methylation. Underwood et al. (2018) observed increased CO rate in pericentromeres with simultaneous moderate reduction in chromosome arms. SPO11-oligos mapping revealed a significant increase in DSB levels within centromeres but not adjacent pericentromeric regions in the H3K9me2/non-CG pathway mutant showing that the effect on crossover is not a simple consequence of the DSB level change. Choi et al. (2018) corroborated that a similar increase in DSBs can be observed also in the met1 mutant. Thus, the two types of DNA methylation, CG and non-CG, are able to trigger similar change in DSB pattern, but have almost opposite consequences on CO distribution. The authors proposed that while both CG and non-CG methylation inhibit DSB formation, only non-CG methylation and/or H3K9me2 inhibit crossover (Underwood et al., 2018). In concordance with this hypothesis, euchromatic crossover hotspots in Arabidopsis can be silenced via RNA-directed DNA methylation pathway, which causes both CG and non-CG methylation as well as the increase in H3K9me2 mark (Yelina et al., 2015). It would be interesting to elucidate which of this epigenetic modification is so important for crossover formation.
Effects of Heterozygosity on Crossover Distribution
Mismatches between DNA sequences in homologous chromosomes are not likely to be detected at the stage of DSB formation, because this requires strand invasion. However, the heterozygosity has a tremendous impact on crossover distribution by influencing crossover/non-crossover decision. Detection of mismatches during meiotic recombination is possible thanks to mismatch-repair system (MMR) (Manhart and Alani, 2016). In this pathway, heterodimers of MutS homologs (MSH2-MSH3, MSH2-MSH6 or MSH2-MSH7) bind DNA, detect mismatches and recruit heterodimers of MutL homologs (MLH1-MLH3 and MLH1-PMS1) in an ATP-dependent reaction. MLH1-PMS1 complex exhibits strong anti-crossover function, while MLH1-MLH3 complex, in combination with SGS1 and EXO1, is able to resolve double Holliday junctions as crossovers (Börner et al., 2004; Bzymek et al., 2010; Manhart and Alani, 2016). This happens via the major crossover pathway ZMM, which in Arabidopsis involves MSH4, MSH5, MER3, HEI10, ZIP4, SHOC1, PTD) (Copenhaver et al., 2002; Higgins et al., 2004, 2008b; Chen et al., 2005; Macaisne et al., 2008; Chelysheva et al., 2012, 2007). In plants, similarly to many other eukaryotes, strand invasions, which are not resolved by ZMM pathway, can be also repaired by the minor crossover pathway (Berchowitz et al., 2007; Higgins et al., 2008a). This relies on the partially redundant structure-specific nucleases and is not biased toward crossover (De Los Santos et al., 2003; Mercier et al., 2015; Wang and Copenhaver, 2018). The exact mechanism for the ZMM crossover bias is currently not known.
Chromosomal Scale
Early works in bacteria indicated that the recombinant frequencies between mismatched substrates were much lower than those of perfectly matched substrates (Claverys and Lacks, 1986; Shen and Huang, 1986), and that the MMR system establishes a genetic barrier during recombination of diverged sequences (Rayssiguier et al., 1989; Shen and Huang, 1989; Matic et al., 1995). Several studies have demonstrated that also in budding yeast decreased sequence homology between chromosomes significantly reduces meiotic recombination (Nilsson-Tillgren et al., 1981, 1986; Hunter et al., 1996). In the study, where the cross between two Saccharomyces species were investigated, Hunter et al. (1996) found that the resulting interspecific hybrid gave high rate of aneuploidy and low levels of meiotic recombination, but when the same experiment was repeated in the genetic background of MutH and MutL homolog mutants, msh2 and pms1, an increase in recombination and reduction in aneuploidy was observed. Furthermore, in a S. cerevisiae diploid with one copy of chromosome III from Saccharomyces paradoxus, the mismatch repair (MMR)-dependent inhibition of recombination between the homeologous (i.e., heterozygous) chromosomes was also observed, so that in pms1 and msh2 mutants, the recombination was increased between the two chromosomes III leading to reduction in non-disjunction of this chromosome (Chambers et al., 1996). As only one of the 12 yeast chromosomes was homeologous, it is unlikely that the effect observed could be due to any potential trans-acting modifiers. Those studies confirmed that in yeast, similarly to bacteria, heterozygosity suppresses crossover, and that the MMR system acts as a genetic barrier for meiotic recombination between not-perfectly matching chromosomes.
In plants, analysis of the effect of heterozygosity on meiotic recombination in the chromosomal scale is limited due to existence of trans-acting modifiers, which could affect recombination in hybrids. This could lead to results that are difficult to interpret. For instance, in A. thaliana several studies of meiotic recombination in F1 and F2 plants show extensive variation in crossover numbers that does not correlate with sequence differences between parental accessions (Alonso-Blanco et al., 1998; Simon et al., 2008; Salomé et al., 2011; Ziolkowski et al., 2015). Similarly, no such correlation has been reported in maize (Beavis and Grant, 1991; Bauer et al., 2013). This problem could be however partially overcome when chromosome substitution lines would be used for comparison. In chromosome substitution lines a pair of chromosomes in one line or species is replaced by a homeologous pair from another variety/species. Sets of chromosome substitution lines were developed especially for plant crops, though direct comparison of crossover frequencies was not frequent. In tomato, interspecific hybrid between Lycopersicon esculentum and Solanum lycopersicoides shows ca. 27% reduction in meiotic recombination (Chetelat et al., 2000). Interestingly, heterozygous substitution lines containing a single S. lycopersicoides chromosome bred into S. lycopersicum recombine at less than 50% of the rate observed for the same chromosome in the F1 hybrid (Ji and Chetelat, 2003). The fact that most of L. esculentum and S. lycopersicoides chromosomes can be distinguished using genomic in situ hybridization (GISH) suggests that the two genomes have diverged substantially in terms of dispersed repetitive sequences. One of those substitution lines was also analyzed in a background, where MMR system was not fully functional, and this resulted in an increase of crossover frequency (average 17.8% increase when compared to wild type) (Tam et al., 2011). Similar results were obtained in Arabidopsis thaliana, where Col x Ler inter-accession hybrid in msh2 mutant exhibited 1.4-fold increase of CO rate when compared to wild type (Emmanuel et al., 2006). This indicates that observed suppression of recombination is mostly due to heterozygous state in cis.
Suppression of recombination in polymorphic regions is believed to be important for prevention of deleterious ectopic recombination between repetitive sequences in a genome (Modrich and Lahue, 1996; Borts et al., 2000; Evans and Alani, 2000). In general, non-allelic copies of repetitive sequences rapidly accumulate mutations, which help to distinguish them from allelic copies. The conservancy of MMR system and similar effects of its malfunction in different organisms suggest that this effect is universal across eukaryotes.
Local Effects, Hotspot Level
The effect of heterozygosity on meiotic crossover frequency was also analyzed at the recombination-hotspot scale. Borts and Haber (1987) tested the effect of heterozygosity on meiotic recombination products in an artificial MAT-pBR322-URA3-MAT interval. By using yeast strains that contain mismatches within this hotspot (about 0.1% divergence between strains) they showed that the number of crossover events was reduced from 23.4 to 10.1% when compared to fully homozygous strains, and there was a corresponding increase in aberrant events, as detected with the flanking markers. In pms1 mutant recombination was restored, leading the authors to propose that independent repair of these widely spaced mismatches might result in the formation of new double-strand breaks that could in turn stimulate a second round of recombination (Borts et al., 1990). These events were detected because of the presence of flanking repeated MAT sequences and this is a likely reason why they were not detected in other experimental systems (Symington and Petes, 1988; Malone et al., 1994). In mice, highly polymorphic A3 hotspot was repaired mostly via interhomolog NCO pathway and CO refractory zone corresponded to a region containing three indels (Cole et al., 2010). This indicates that also in mammals, polymorphism does not influence DSB formation, however, it affects selection of a repair pathway.
In plants, major work on this subject has been made in maize, where the characterization of strong recombination hotspots a1-sh2 and bronze (Dooner, 1986; Yao et al., 2002), and high genetic diversity between different maize inbred lines (Fu and Dooner, 2002; Lai et al., 2010) provided a perfect experimental system. The 130-kb of the a1-sh2 region exhibits meiotic recombination rate between 0 and 11 cM/Mb, which is significantly more than region average (0.0087 cM/Mb) (Yao et al., 2002). The 1.5-kb long bronze locus has a recombination frequency at least 100 times higher than the average for the maize genome (Dooner, 1986; Dooner and Martínez-Férez, 1997). Both intervals are highly polymorphic between maize haplotypes and reveal both genic and non-genic collinearities (Fu and Dooner, 2002; Yao et al., 2002; Brunner et al., 2005). Even bigger differences have been observed when compared to teosinte haplotypes, which is considered as a wild progenitor of maize (Yao and Schnable, 2005).
In most of the studies the authors concluded that recombination is suppressed by polymorphisms (Dooner and Martínez-Férez, 1997; Yao and Schnable, 2005). Dooner and Martínez-Férez (1997) in an experimental setup, where crosses between maize lines with different number of polymorphisms with a tester line were used, observed a good negative correlation with recombination rate. Similar effects were observed in experiments, where neighboring regions where compared: subintervals that exhibit higher recombination rates per megabase than their juxtaposed subintervals, also exhibit lower levels of polymorphisms (Yao and Schnable, 2005). Less clear relationship was observed for non-adjacent intervals. The general problem with interpretation of those data is that different types of polymorphisms may affect recombination to a different extent, especially SNPs and indels cannot be treated in the same way. In case of experiments with a1-sh2 and bronze hotspots, large transposable elements existing in those lines significantly reduce recombination (Fu et al., 2002; Dooner and He, 2008; He and Dooner, 2009). For instance, a 26 kb retrotransposon cluster located nearby bronze locus suppresses crossover in this hotspot by a factor of two (Dooner and He, 2008), and haplotype structure as defined by the presence of helitrons and retrotransposons in this locus, strongly inhibited occurrence of recombination in heterozygous plants (He and Dooner, 2009).
In A. thaliana, conducting the crossover frequency studies at the hotspot level were not possible for a long time due to the lack of morphological markers similar to those used in maize. However, recent development of pollen typing technique enabled to overcome this limitation (Drouaud and Mézard, 2011; Yelina et al., 2012; Choi et al., 2013; Drouaud et al., 2013). Pollen typing, similarly to sperm typing developed for mammals is based on an allele-specific amplification of a hotspot region from post-meiotic gametes. Subsequent sequencing of the PCR products enables precise determination of recombination breakpoints in regards to polymorphic regions, even though in pollen typing comparison of crossover landscape with completely homozygous line is not possible. Analysis of crossover distribution in relation to SNPs shows that polymorphism suppresses crossover formation at the hotspot scale in a way similar to other eukaryotes (Choi et al., 2013, 2016; Yelina et al., 2015, 2012; Ziolkowski and Henderson, 2017; Lawrence et al., 2018). NCO analysis, as being technically more challenging, was performed only in one study (Drouaud et al., 2013). In other cases, the authors studied only crossover events, therefore it is difficult to conclude on DSB distribution at the hotspots. Fortunately, for direct comparison we can use data from SPO11-oligo sequencing to observe the pattern of DSBs in the crossover hotspots (Choi et al., 2018). Distribution of CO events clearly shows inhibition at polymorphic sites (Choi et al., 2013, 2016). This is especially visible for the highly polymorphic hotspot RAC1-GDSL (Figure 3). As expected, distribution of DSBs is not affected by SNPs and actually the levels of SPO11-oligos are elevated in SNP-rich regions. This suggests that the polymorphism resulted from recombination-associated mutations.
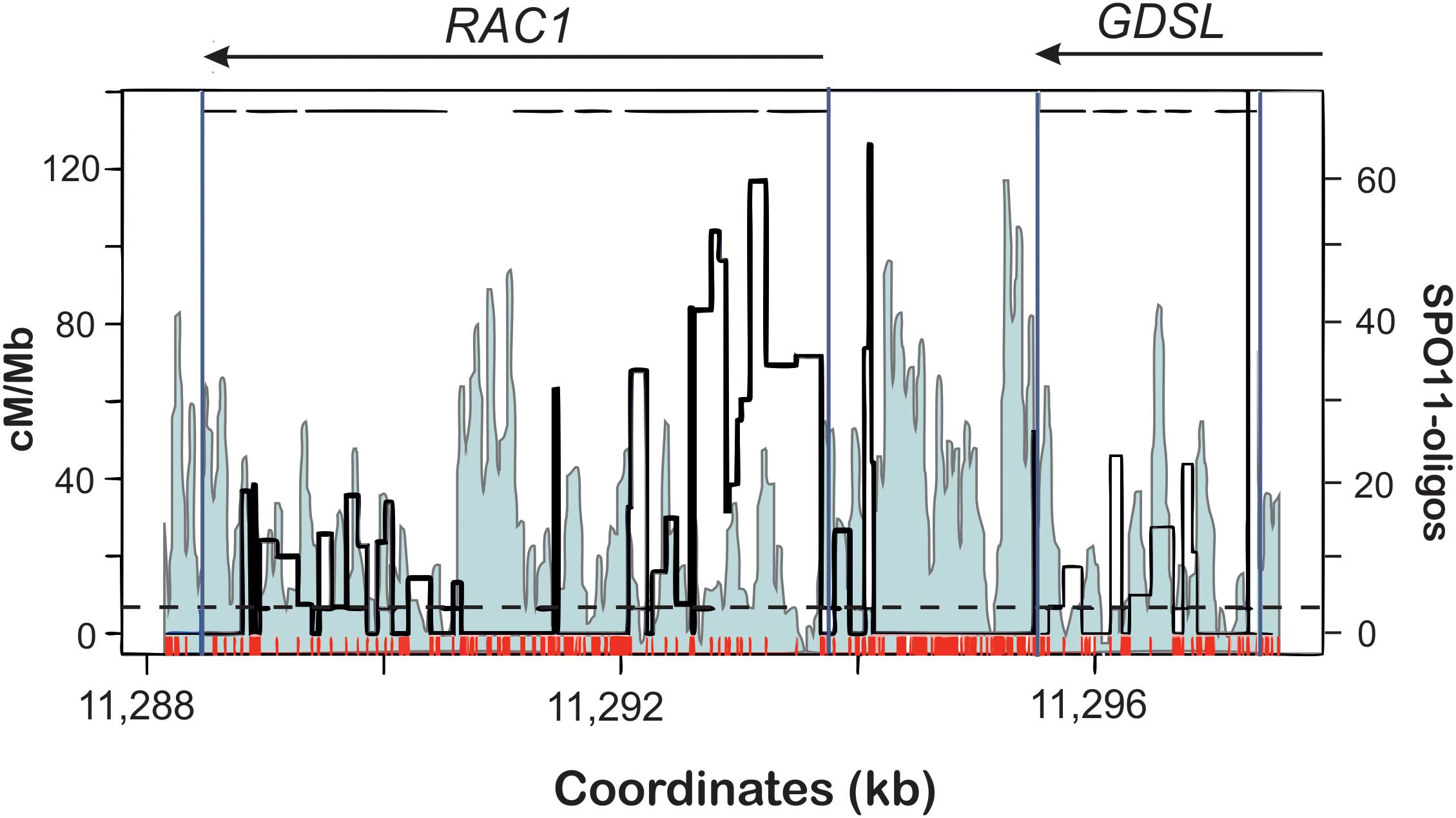
Figure 3. Distributions of DSBs (SPO11-1-oligos; light blue profile) and COs (cM/Mb; thick black line) within the RAC1-GDSL hotspot in A. thaliana. Polymorphism between Col and Ler accessions used for CO mapping is depicted as red ticks. Gene orientation and exon-intron structure is shown at the top of the plot. Note overrepresentation of DSBs at 5′-ends, 3′-ends of the genes, and within some introns. Crossovers appear mostly in SNP-free fragments of the hotspot. Modified from Choi et al. (2018).
Assuming polymorphism-independent distribution of DSB within hotspots, we can conclude that at the kilobase scale polymorphic sites cause inhibition of CO pathways and are repaired mostly by NCOs. If DSB sites compete for CO factors, polymorphism inhibiting CO would lead to recombinational hyperactivity of some hotspots. In other words, polymorphism would act inhibitory at the single hotspot scale, but increase a chance of adjacent polymorphism-free hotspot for entering a crossover repair pathway (Figure 4).
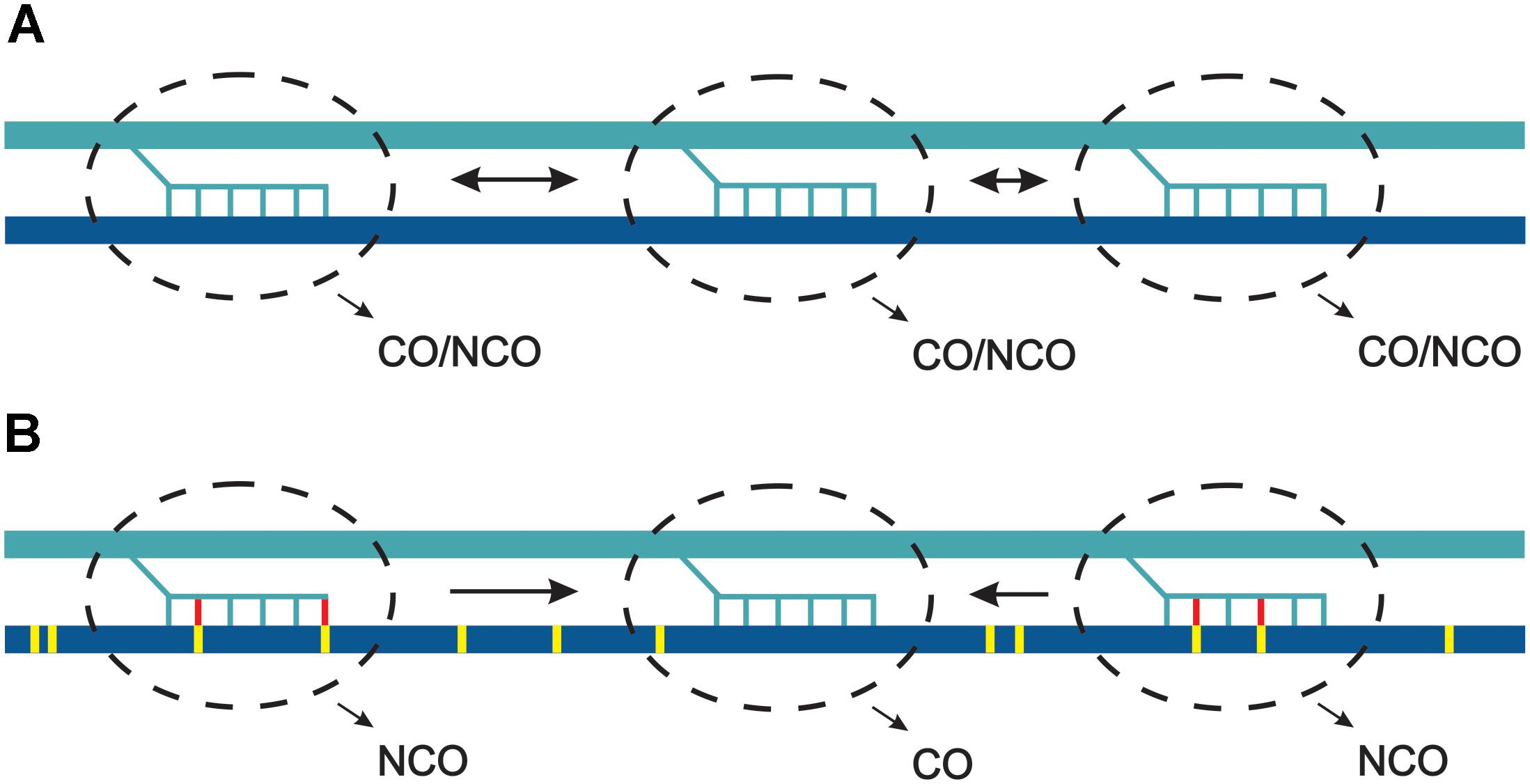
Figure 4. Model of competition between hotspots in response to sequence heterology. (A) Three hotspots (dashed line ovals) become activated and involved in strand invasion into the two homologous chromosomes, which are identical regarding the sequence. Due to scarcity of crossover, they all compete and have similar chances for developing into a crossover. (B) The two homologous chromosomes differ at single base pairs (yellow ticks). During strand invasion mismatches are detected by MMR (yellow-red ticks) and develop into non-crossovers. In consequence, the perfectly matched invasions have higher chance for becoming crossovers. For simplicity, one direction of strand invasion was shown.
Sensitivity of Different Recombination Pathways Toward Heterozygosity
As it was already mentioned, at least two meiotic crossover pathways exist in most eukaryotes including plants (Higgins et al., 2004; Mercier et al., 2005). The major pathway in A. thaliana is responsible for about 85% of crossover events (Chelysheva et al., 2007), is interference-sensitive, and depends on a group of proteins that are collectively called ZMM (Copenhaver et al., 2002; Higgins et al., 2004, 2008b; Chen et al., 2005; Macaisne et al., 2008; Chelysheva et al., 2012, 2007). The remaining crossovers are non-interfering, randomly distributed along the chromosomes, and are dependent on recombinases such as MUS81 that are not meiosis-specific and that have important roles also in somatic cells (Berchowitz et al., 2007; Higgins et al., 2008a). In wild type, the recombination intermediates in non-interfering DSB repair are mostly directed toward NCO pathway by the FANCM helicase (Crismani et al., 2012; Knoll et al., 2012) and the BLM/SGS1 helicase homologs RECQ4A/B (Séguéla-Arnaud et al., 2015, 2017). In addition, strand invasion step is affected by the AAA-ATPase FIGL1, which hinders the interaction with a homologous chromosome (Girard et al., 2015; Fernandes et al., 2018a). Dramatic increases in crossover frequency are observed in mutants of those genes.
The class I and II pathways have been compared with the respect to sensitivity to polymorphism in the chromosomal region scale in Arabidopsis thaliana. Due to lack of proper non-interfering mutants (Mercier et al., 2015) class II behavior can be analyzed only indirectly, e.g., by using fancm mutant, where non-interfering repair is directed toward crossover (Crismani et al., 2012; Knoll et al., 2012). Ziolkowski et al. (2015) reported that in fancm background very little increase is observed in recombination frequency within chromosomal regions in heterozygous state when compared to wild type Arabidopsis. In fancm zip4 double mutant a significant reduction in crossover rate was observed for heterozygous regions, even though the same mutant in homozygous regions shows a dramatic increase. Consistently with this, an increased interference in heterozygous regions is observed in wild type plants (Ziolkowski et al., 2015). Although no direct analysis on how the level of polymorphisms affects the inhibition was carried out, highly polymorphic pericentromeric regions exhibited higher suppressive effect on class II crossover frequency than less polymorphic subtelomeric regions. The authors concluded that both crossover pathways show opposite sensitivity toward heterozygosity, with non-interfering pathways being unable to successfully repair DSBs in such regions, at least in fancm background. Girard et al. (2015) also observed no increase in crossover rate in fancm mutant in hybrids, but a significant increase was observed in fancm figl1 double mutant when compared to either wild type or figl1. FIGL1 is a protein suggested to limit strand invasion step during recombination by regulation of DMC1 and RAD51 proteins (Girard et al., 2015; Fernandes et al., 2018a). Thus, the authors concluded that in the absence of FIGL1 protein the non-interfering FANCM-dependent pathway may successfully repair heterozygous chromosomal regions by crossover. This suggests existence of another unknown mechanism, which impairs the anti-crossover FANCM activity in hybrids (Girard et al., 2015).
In a more recent study Fernandes et al. (2018b) investigated the accumulated effect of A. thaliana mutants in all three anti-recombinational pathways, i.e., recq4, figl1 and fancm, and observed extensive increases in CO rates in inter-accession Col x Ler crosses. However, only marginal increase in CO rate was observed for pericentromeric regions. The authors proposed that this may be due to limited accessibility of pericentromeric chromatin for SPO11, which results in lack of recombination initiation sites (Fernandes et al., 2018b). This explanation seems probable when we consider recent finding of drop in DSBs in Arabidopsis pericentromeres (Choi et al., 2018). Moreover, a strong anticorrelation between recombination and SNP density was reported in recq4 figl1, which was not observed in wild type. This implicates inhibiting effect of polymorphism on crossover rate (Fernandes et al., 2018b). Supporting this observation, significantly lower CO levels where observed in the middle of chromosome 1 right arm, which corresponds to significant elevation of polymorphisms (Ziolkowski et al., 2009; Choi et al., 2016). Therefore, lack of extra COs in pericentromeric regions may be partially due to elevated polymorphisms which seems to discourage CO repair pathway (Fernandes et al., 2018b). To verify this hypothesis an experiment including heterozygosity-homozygosity juxtaposition would be necessary. Further experiments involving the use of proper class II crossover mutants would be required to fully understand the polymorphism-sensitivity of both crossover pathways.
Juxtaposition of Heterozygous and Homozygous Regions Changes the Chromosomal Redistribution of Crossover
The widely documented suppression of crossover frequency at the hotspot level contradicts with the data collected at the chromosomal scale in A. thaliana, when homozygous and heterozygous segments were juxtaposed (Ziolkowski et al., 2015). In such experimental setup a reciprocal crossover increases in heterozygous and decreases in homozygous regions were observed (Figure 5). The total number of crossovers measured by chiasmata counting were not changed, consistent with homeostatic regulation. This phenomenon seems to be independent of chromosomal location as it was shown for two different chromosomes and for both subtelomeric and pericentromeric intervals, and was observed in different A. thaliana crosses (I. R. Henderson, personal communication) Analysis in fancm, zip4 and fancm zip4 mutant background provided strong evidence that the process is interference-dependent.
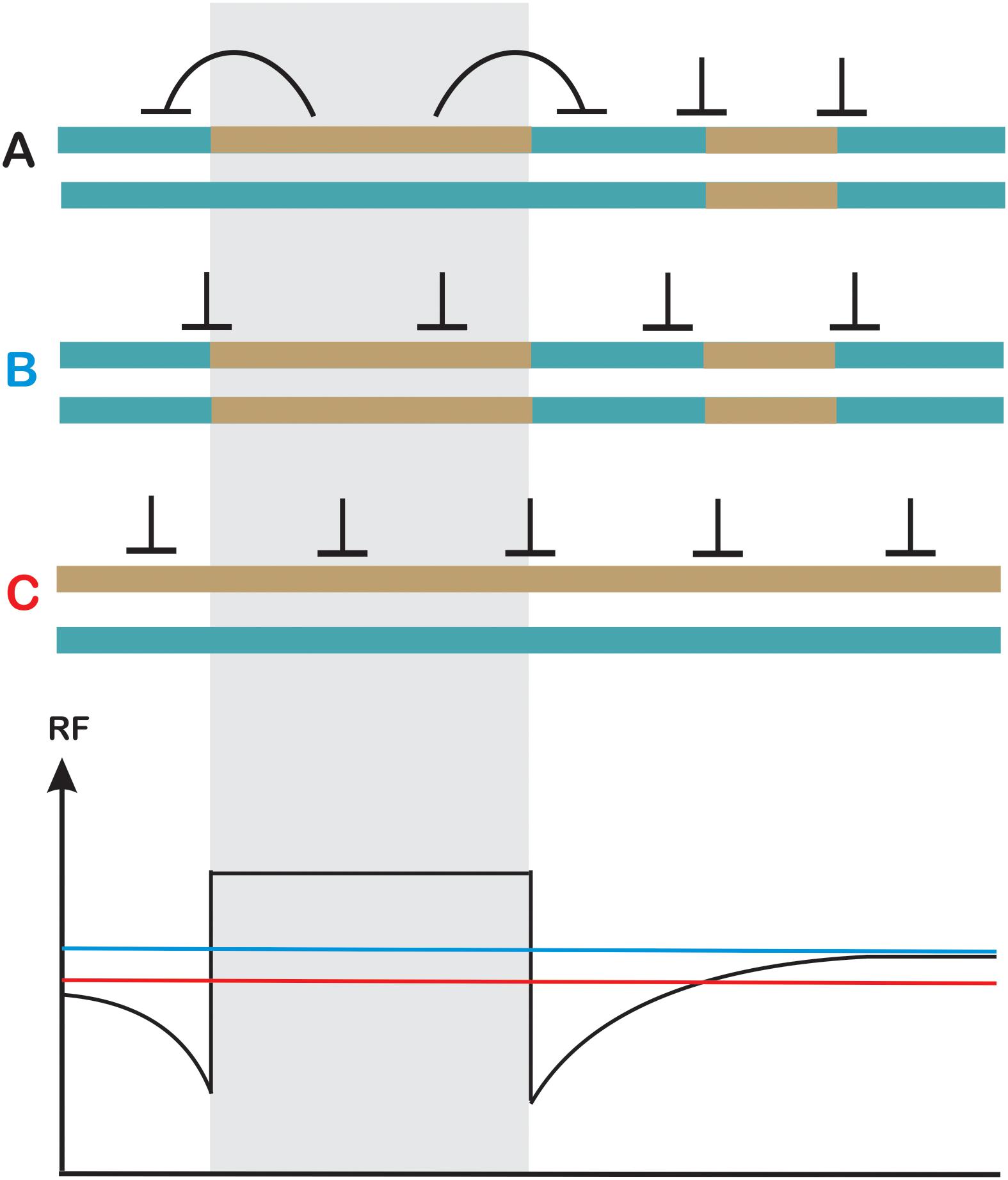
Figure 5. Heterozygosity juxtaposition effect. Two homologous chromosomes in (A–C) differ in the pattern of heterozygosity (turquoise and dark-yellow). (A) Crossover levels get elevated in a heterozygous region at the expense of adjacent homozygous regions on the same chromosome (in cis). (B) Crossovers are evenly spaced in fully homozygous chromosomes (C) Crossovers are evenly spaced in fully heterozygous chromosomes although a reduction in recombination frequency at the chromosome scale is observed. Other effects, which could affect crossover spatial distribution were not shown for simplicity. Recombination levels are schematically shown on the lower panel using the color code for (A–C).
The mechanisms by which juxtaposition effect is executed is not understood, however, it must involve detection of mismatches by MMR proteins, as the effect is dependent mostly on ZMM pathway. It is possible that specific signaling between MMR components and ATM/ATR pathway results in additional DSBs being formed in the region, and this can also include some type of DSB site competition. It is also currently unknown whether this phenomenon is unique to Arabidopsis, or is a general feature of interference-dependent crossover pathway in eukaryotes. Conservation of major components of meiotic DSBs formation and interference-dependent repair pathways suggests that it may exist in other organisms, especially in self-pollinating plant species where situations of adjacent homozygous and heterozygous regions are common. The biological meaning of this process would be to increase the chance to generate novel combinations of genetic material: COs occurring in homozygous regions result in reestablishing parental haplotypes in the next generation, while stimulating recombination in heterozygous segments always result in some new allele assemblies.
Concluding Remarks and Perspectives
Recent discoveries in the field of meiotic recombination significantly changed our understanding of processes responsible for shaping the genome. However, substantial differences have been spotted between mammals and plants. In mammals, PRDM9 histone methyltransferase plays a key role in defining crossover sites, whilst plants the distribution of recombination is dependent on a large number of subtle features, both at the level of genetics and chromatin structure. For instance, it is currently unknown whether H3K4me3 plays a similar function in recombination hotspot tethering to the chromatin loops in plants, as it was shown for budding yeast and animals, as the data are inconsistent. From this perspective further work is needed to define the relationships between particular levels and find rules responsible for priority of some factors over the others.
In comparison with recombination initiation sites, additional regulatory levels of crossover distribution result from CO/NCO decision. Recent developments in plants, especially approaches to asses DSB levels and fine-scale crossover mapping (He et al., 2017; Choi et al., 2018), show that those factors may have also epigenetic origin. The major problem, which researchers meet in their trials to decipher epigenetic factors, lies in the extensive crosstalk between different epigenetic modifications and the fact that they operate on a global scale. Therefore, new targeted approaches will be required to investigate effects of particular alterations locally and at the hotspot scale. Directing particular modifications to specific chromosomal locations, together with targeting recombination events, possibly using CRISPR-dCas9 technology, may provide an attractive strategy for this purpose and should lead to further fascinating discoveries.
Another interesting topic, which requires further investigation, refers to interactions between homologs chromosomes, where local differences in DNA sequence, and probably also local chromatin states, affect the outcomes of strand invasion. This is particularly interesting in self-pollinating plants, which are characterized by a high level of sequence homozygosity. Their rare outcrossing has a result in the existence of heterozygous regions juxtaposed to homozygous ones on the same chromosome, and thereby creates novel chances for genome evolution (Ziolkowski et al., 2015). It would be interesting to investigate the mechanism responsible for these cis effects on crossover stimulation. In this context, questions about potential effects of “epigenetic heterozygosity” and competition between recombination hotspots would be intriguing to answer. Those findings, along with a recent progress in the identification of trans-acting factors responsible for crossover distribution (Ziolkowski et al., 2017; Fernandes et al., 2018b) opens new perspectives for developing novel breeding strategies (Choi, 2017; Lambing and Heckmann, 2018).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The work was supported by EMBO Installation Grant and Polish National Science Centre grants (2016/21/B/NZ2/01757 and 2016/22/E/NZ2/00455) to PAZ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the reviewers for helpful comments and suggestions.
References
Acquaviva, L., Székvölgyi, L., Dichtl, B., Dichtl, B. S., de La Roche Saint André, C., Nicolas, A., et al. (2013). The COMPASS subunit Spp1 links histone methylation to initiation of meiotic recombination. Science 339, 215–218. doi: 10.1126/science.1225739
Adam, C., Guérois, R., Citarella, A., Verardi, L., Adolphe, F., Béneut, C., et al. (2018). The PHD finger protein Spp1 has distinct functions in the Set1 and the meiotic DSB formation complexes. PLoS Genet. 14:e1007223. doi: 10.1371/journal.pgen.1007223
Alonso-Blanco, C., Peeters, A. J., Koornneef, M., Lister, C., Dean, C., van den Bosch, N., et al. (1998). Development of an AFLP based linkage map of Ler, Col and Cvi Arabidopsis thaliana ecotypes and construction of a Ler/Cvi recombinant inbred line population. Plant J. 14, 259–271. doi: 10.1046/j.1365-313X.1998.00115.x
Anderson, L. K., Doyle, G. G., Brigham, B., Carter, J., Hooker, K. D., Lai, A., et al. (2003). High-resolution crossover maps for each bivalent of Zea mays using recombination nodules. Genetics 165, 849–865.
Anderson, L. K., and Stack, S. M. (2005). Recombination nodules in plants. Cytogenet. Genome Res. 109, 198–204. doi: 10.1159/000082400
Arbeithuber, B., Betancourt, A. J., Ebner, T., and Tiemann-Boege, I. (2015). Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc. Natl. Acad. Sci. U.S.A. 112:201416622. doi: 10.1073/pnas.1416622112
Baker, C. L., Kajita, S., Walker, M., Saxl, R. L., Raghupathy, N., Choi, K., et al. (2015). PRDM9 drives evolutionary erosion of hotspots in Mus musculus through haplotype-specific initiation of meiotic recombination. PLoS Genet. 11:e1004916. doi: 10.1371/journal.pgen.1004916
Baudat, F., Buard, J., Grey, C., Fledel-Alon, A., Ober, C., Przeworski, M., et al. (2010). PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840. doi: 10.1126/science.1183439
Bauer, E., Falque, M., Walter, H., Bauland, C., Camisan, C., Campo, L., et al. (2013). Intraspecific variation of recombination rate in maize. Genome Biol. 14:R103. doi: 10.1186/gb-2013-14-9-r103
Beavis, W. D., and Grant, D. (1991). A linkage map based on information from four F2 populations of maize (Zea mays L.). Theor. Appl. Genet. 82, 636–644. doi: 10.1007/BF00226803
Berchowitz, L. E., Francis, K. E., Bey, A. L., and Copenhaver, G. P. (2007). The role of AtMUS81 in interference-insensitive crossovers in A. thaliana. PLoS Genet. 3:e132. doi: 10.1371/journal.pgen.0030132
Birdsell, J. A. (2002). Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol. Biol. Evol. 19, 1181–1197. doi: 10.1093/oxfordjournals.molbev.a004176
Blat, Y., and Kleckner, N. (1999). Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell 98, 249–259. doi: 10.1016/S0092-8674(00)81019-3
Blat, Y., Protacio, R. U., Hunter, N., and Kleckner, N. (2002). Physical and functional interactions among basic chromosome organizational features govern early steps of meiotic chiasma formation. Cell 111, 791–802. doi: 10.1016/S0092-8674(02)01167-4
Borde, V., and de Massy, B. (2013). Programmed induction of DNA double strand breaks during meiosis: setting up communication between DNA and the chromosome structure. Curr. Opin. Genet. Dev. 23, 147–155. doi: 10.1016/j.gde.2012.12.002
Borde, V., Robine, N., Lin, W., Bonfils, S., Géli, V., and Nicolas, A. (2009). Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sites. EMBO J. 28, 99–111. doi: 10.1038/emboj.2008.257
Börner, G. V., Kleckner, N., and Hunter, N. (2004). Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 117, 29–45. doi: 10.1016/S0092-8674(04)00292-2
Borts, R. H., Chambers, S. R., and Abdullah, M. F. (2000). The many faces of mismatch repair in meiosis. Mutat. Res. 451, 129–150. doi: 10.1016/S0027-5107(00)00044-0
Borts, R. H., and Haber, J. E. (1987). Meiotic recombination in yeast: alteration by multiple heterozygosities. Science 237, 1459–1465. doi: 10.1126/science.2820060
Borts, R. H., Leung, W. Y., Kramer, W., Kramer, B., Williamson, M., Fogel, S., et al. (1990). Mismatch repair-induced meiotic recombination requires the pms1 gene product. Genetics 124, 573–584.
Boulton, A., Myers, R. S., and Redfield, R. J. (1997). The hotspot conversion paradox and the evolution of meiotic recombination. Proc. Natl. Acad. Sci. U.S.A. 94, 8058–8063. doi: 10.1073/pnas.94.15.8058
Brick, K., Smagulova, F., Khil, P., Camerini-Otero, R. D., and Petukhova, G. V. (2012). Genetic recombination is directed away from functional genomic elements in mice. Nature 485, 642–645. doi: 10.1038/nature11089
Brick, K., Thibault-Sennett, S., Smagulova, F., Lam, K.-W. G., Pu, Y., Pratto, F., et al. (2018). Extensive sex differences at the initiation of genetic recombination. Nature 561, 338–342. doi: 10.1038/s41586-018-0492-5
Brunner, S., Fengler, K., Morgante, M., Tingey, S., and Rafalski, A. (2005). Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17, 343–360. doi: 10.1105/tpc.104.025627
Bzymek, M., Thayer, N. H., Oh, S. D., Kleckner, N., and Hunter, N. (2010). Double holliday junctions are intermediates of DNA break repair. Nature 464, 937–941. doi: 10.1038/nature08868
Calabrese, P. (2007). A population genetics model with recombination hotspots that are heterogeneous across the population. Proc. Natl. Acad. Sci. U.S.A. 104, 4748–4752. doi: 10.1073/pnas.0610195104
Cao, X., Aufsatz, W., Zilberman, D., Mette, M. F., Huang, M. S., Matzke, M., et al. (2003). Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr. Biol. 13, 2212–2217. doi: 10.1016/j.cub.2003.11.052
Chambers, S. R., Hunter, N., Louis, E. J., and Borts, R. H. (1996). The mismatch repair system reduces meiotic homeologous recombination and stimulates recombination-dependent chromosome loss. Mol. Cell. Biol. 16, 6110–6120. doi: 10.1128/MCB.16.11.6110
Chelysheva, L., Gendrot, G., Vezon, D., Doutriaux, M.-P., Mercier, R., and Grelon, M. (2007). Zip4/Spo22 is required for class I CO formation but not for synapsis completion in Arabidopsis thaliana. PLoS Genet. 3:e83. doi: 10.1371/journal.pgen.0030083
Chelysheva, L., Grandont, L., Vrielynck, N., Le Guin, S., Mercier, R., and Grelon, M. (2010). An easy protocol for studying chromatin and recombination protein dynamics during Arabidopsis thaliana meiosis: immunodetection of cohesins, histones and MLH1. Cytogenet. Genome Res. 129, 143–153. doi: 10.1159/000314096
Chelysheva, L., Vezon, D., Chambon, A., Gendrot, G., Pereira, L., Lemhemdi, A., et al. (2012). The Arabidopsis HEI10 is a new ZMM protein related to Zip3. PLoS Genet. 8:e1002799. doi: 10.1371/journal.pgen.1002799
Chen, C., Zhang, W., Timofejeva, L., Gerardin, Y., and Ma, H. (2005). The Arabidopsis ROCK-N-ROLLERS gene encodes a homolog of the yeast ATP-dependent DNA helicase MER3 and is required for normal meiotic crossover formation. Plant J. 43, 321–334. doi: 10.1111/j.1365-313X.2005.02461.x
Chetelat, R. T., Meglic, V., and Cisneros, P. (2000). A genetic map of tomato based on BC(1) Lycopersicon esculentum x Solanum lycopersicoides reveals overall synteny but suppressed recombination between these homeologous genomes. Genetics 154, 857–867.
Choi, K. (2017). Molecules and cells advances towards controlling meiotic recombination for plant breeding. Mol. Cells 40, 814–822. doi: 10.14348/molcells.2017.0171
Choi, K., and Henderson, I. R. (2015). Meiotic recombination hotspots – a comparative view. Plant J. 83, 52–61. doi: 10.1111/tpj.12870
Choi, K., Reinhard, C., Serra, H., Ziolkowski, P. A., Underwood, C. J., Zhao, X., et al. (2016). Recombination rate heterogeneity within Arabidopsis disease resistance genes. PLoS Genet. 12:e1006179. doi: 10.1371/journal.pgen.1006179
Choi, K., Zhao, X., Kelly, K. A., Venn, O., Higgins, J. D., Yelina, N. E., et al. (2013). Arabidopsis meiotic crossover hot spots overlap with H2A.Z nucleosomes at gene promoters. Nat. Genet. 45, 1327–1336. doi: 10.1038/ng.2766
Choi, K., Zhao, X., Lambing, C., Underwood, C. J., Hardcastle, T. J., Serra, H., et al. (2018). Nucleosomes and DNA methylation shape meiotic DSB frequency in Arabidopsis thaliana transposons and gene regulatory regions. Genome Res. 28, 1–15. doi: 10.1101/gr.225599.117
Claverys, J. P., and Lacks, S. A. (1986). Heteroduplex deoxyribonucleic acid base mismatch repair in bacteria. Microbiol. Rev. 50, 133–165.
Cole, F., Kauppi, L., Lange, J., Roig, I., Wang, R., Keeney, S., et al. (2012). Homeostatic control of recombination is implemented progressively in mouse meiosis. Nat. Cell Biol. 14, 424–430. doi: 10.1038/ncb2451
Cole, F., Keeney, S., and Jasin, M. (2010). Comprehensive, fine-scale dissection of homologous recombination outcomes at a hot spot in mouse meiosis. Mol. Cell 39, 700–710. doi: 10.1016/j.molcel.2010.08.017
Colomé-Tatché, M., Cortijo, S., Wardenaar, R., Morgado, L., Lahouze, B., Sarazin, A., et al. (2012). Features of the Arabidopsis recombination landscape resulting from the combined loss of sequence variation and DNA methylation. Proc. Natl. Acad. Sci. U.S.A. 109, 16240–16245. doi: 10.1073/pnas.1212955109
Coop, G., and Myers, S. R. (2007). Live hot, die young: transmission distortion in recombination hotspots. PLoS Genet. 3:e35. doi: 10.1371/journal.pgen.0030035
Cooper, T. J., Garcia, V., and Neale, M. J. (2016). Meiotic DSB patterning: a multifaceted process. Cell Cycle 15, 13–21. doi: 10.1080/15384101.2015.1093709
Copenhaver, G. P., Housworth, E. A., and Stahl, F. W. (2002). Crossover interference in Arabidopsis. Genetics 160, 1631–1639.
Copenhaver, G. P., Nickel, K., Kuromori, T., Benito, M. I., Kaul, S., Lin, X., et al. (1999). Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286, 2468–2474. doi: 10.1126/science.286.5449.2468
Crismani, W., Girard, C., Froger, N., Pradillo, M., Santos, J. L., Chelysheva, L., et al. (2012). FANCM limits meiotic crossovers. Science 336, 1588–1590. doi: 10.1126/science.1220381
Culligan, K. M., and Britt, A. B. (2008). Both ATM and ATR promote the efficient and accurate processing of programmed meiotic double-strand breaks. Plant J. 55, 629–638. doi: 10.1111/j.1365-313X.2008.03530.x
Darrier, B., Rimbert, H., Balfourier, F., Pingault, L., Josselin, A. A., Servin, B., et al. (2017). High-resolution mapping of crossover events in the hexaploid wheat genome suggests a universal recombination mechanism. Genetics 206, 1373–1388. doi: 10.1534/genetics.116.196014
De Los Santos, T., Hunter, N., Lee, C., Larkin, B., Loidl, J., and Hollingsworth, N. M. (2003). The Mus81/Mms4 endonuclease acts independently of double-holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics 164, 81–94.
de Massy, B. (2013). Initiation of meiotic recombination: how and where? conservation and specificities among eukaryotes. Annu. Rev. Genet. 47, 581–617. doi: 10.1146/annurev-genet-110711-155423
Diagouraga, B., Clément, J. A. J., Duret, L., Kadlec, J., de Massy, B., and Baudat, F. (2018). PRDM9 methyltransferase activity is essential for meiotic DNA double-strand break formation at its binding sites. Mol. Cell 69, 1–13. doi: 10.1016/j.molcel.2018.01.033
Dooner, H. K., and He, L. (2008). Maize genome structure variation: interplay between retrotransposon polymorphisms and genic recombination. Plant Cell 20, 249–258. doi: 10.1105/tpc.107.057596
Dooner, H. K., and Martínez-Férez, I. M. (1997). Recombination occurs uniformly within the bronze gene, a meiotic recombination hotspot in the maize genome. Plant Cell 9, 1633–1646. doi: 10.1105/tpc.9.9.1633
Drouaud, J., Khademian, H., Giraut, L., Zanni, V., Bellalou, S., Henderson, I. R., et al. (2013). Contrasted patterns of crossover and non-crossover at Arabidopsis thaliana meiotic recombination hotspots. PLoS Genet. 9:e1003922. doi: 10.1371/journal.pgen.1003922
Drouaud, J., Mercier, R., Chelysheva, L., Bérard, A., Falque, M., Martin, O., et al. (2007). Sex-specific crossover distributions and variations in interference level along Arabidopsis thaliana chromosome 4. PLoS Genet. 3:e106. doi: 10.1371/journal.pgen.0030106
Drouaud, J., and Mézard, C. (2011). Characterization of meiotic crossovers in pollen from Arabidopsis thaliana. Methods Mol. Biol. 745, 223–249. doi: 10.1007/978-1-61779-129-1_14
Du, J., Hong, X., Bernatavichute, Y. V., Stroud, H., Feng, S., Caro, E., et al. (2012). Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151, 167–180. doi: 10.1016/j.cell.2012.07.034
Du, J., Johnson, L. M., Groth, M., Feng, S., Hale, C. J., Li, S., et al. (2014). Mechanism of DNA methylation-directed histone methylation by KRYPTONITE. Mol. Cell 55, 495–504. doi: 10.1016/j.molcel.2014.06.009
Emmanuel, E., Yehuda, E., Melamed-Bessudo, C., Avivi-Ragolsky, N., and Levy, A. A. (2006). The role of AtMSH2 in homologous recombination in Arabidopsis thaliana. EMBO Rep. 7, 100–105. doi: 10.1038/sj.embor.7400577
Erayman, M., Sandhu, D., Sidhu, D., Dilbirligi, M., Baenziger, P. S., and Gill, K. S. (2004). Demarcating the gene-rich regions of the wheat genome. Nucleic Acids Res. 32, 3546–3565. doi: 10.1093/nar/gkh639
Evans, E., and Alani, E. (2000). Roles for mismatch repair factors in regulating genetic recombination. Mol. Cell. Biol. 20, 7839–7844. doi: 10.1128/MCB.20.21.7839-7844.2000
Falque, M., Anderson, L. K., Stack, S. M., Gauthier, F., and Martin, O. C. (2009). Two types of meiotic crossovers coexist in maize. Plant Cell 21, 3915–3925. doi: 10.1105/tpc.109.071514
Fernandes, J. B., Duhamel, M., Seguéla-Arnaud, M., Froger, N., Girard, C., Choinard, S., et al. (2018a). FIGL1 and its novel partner FLIP form a conserved complex that regulates homologous recombination. PLoS Genet. 14:e1007317. doi: 10.1371/journal.pgen.1007317
Fernandes, J. B., Seguéla-Arnaud, M., Larchevêque, C., Lloyd, A. H., and Mercier, R. (2018b). Unleashing meiotic crossovers in hybrid plants. Proc. Natl. Acad. Sci. U.S.A. 115, 2431–2436. doi: 10.1073/pnas.1713078114
Fowler, K. R., Sasaki, M., Milman, N., Keeney, S., and Smith, G. R. (2014). Evolutionarily diverse determinants of meiotic DNA break and recombination landscapes across the genome. Genome Res. 24, 1650–1664. doi: 10.1101/gr.172122.114
Fu, H., and Dooner, H. K. (2002). Intraspecific violation of genetic colinearity and its implications in maize. Proc. Natl. Acad. Sci. U.S.A. 99, 9573–9578. doi: 10.1073/pnas.132259199
Fu, H., Zheng, Z., and Dooner, H. K. (2002). Recombination rates between adjacent genic and retrotransposon regions in maize vary by 2 orders of magnitude. Proc. Natl. Acad. Sci. U.S.A. 99, 1082–1087. doi: 10.1073/pnas.022635499
Garcia, V., Bruchet, H., Camescasse, D., Granier, F., Bouchez, D., and Tissier, A. (2003). AtATM is essential for meiosis and the somatic response to DNA damage in plants. Plant Cell 15, 119–132. doi: 10.1105/tpc.006577
Garcia, V., Gray, S., Allison, R. M., Cooper, T. J., and Neale, M. J. (2015). Tel1(ATM)-mediated interference suppresses clustered meiotic double-strand-break formation. Nature 520, 114–118. doi: 10.1038/nature13993
Gerton, J. L., DeRisi, J., Shroff, R., Lichten, M., Brown, P. O., and Petes, T. D. (2000). Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 97, 11383–11390. doi: 10.1073/pnas.97.21.11383
Getun, I. V., Wu, Z., Fallahi, M., Ouizem, S., Liu, Q., Li, W., et al. (2017). Functional roles of acetylated histone marks at mouse meiotic recombination hot spots. Mol. Cell. Biol. 37:e00942-15. doi: 10.1128/MCB.00942-15
Girard, C., Chelysheva, L., Choinard, S., Froger, N., Macaisne, N., Lehmemdi, A., et al. (2015). AAA-ATPase FIDGETIN-LIKE 1 and Helicase FANCM antagonize meiotic crossovers by distinct mechanisms. PLoS Genet. 11:e1005369. doi: 10.1371/journal.pgen.1005369
Giraut, L., Falque, M., Drouaud, J., Pereira, L., Martin, O. C., and Mézard, C. (2011). Genome-wide crossover distribution in Arabidopsis thaliana meiosis reveals sex-specific patterns along chromosomes. PLoS Genet. 7:e1002354. doi: 10.1371/journal.pgen.1002354
Hayashi, M., Chin, G. M., and Villeneuve, A. M. (2007). C. elegans germ cells switch between distinct modes of double-strand break repair during meiotic prophase progression. PLoS Genet. 3:e191. doi: 10.1371/journal.pgen.0030191
He, L., and Dooner, H. K. (2009). Haplotype structure strongly affects recombination in a maize genetic interval polymorphic for Helitron and retrotransposon insertions. Proc. Natl. Acad. Sci. U.S.A. 106, 8410–8416. doi: 10.1073/pnas.0902972106
He, Y., Wang, M., Dukowic-Schulze, S., Zhou, A., Tiang, C. L., Shilo, S., et al. (2017). Genomic features shaping the landscape of meiotic double-strand break hotspots in maize. Proc. Natl. Acad. Sci. U.S.A. 114, 12231–12236. doi: 10.1073/pnas.1713225114
Hedrick, P. W. (2007). Sex: differences in mutation, recombination, selection, gene flow, and genetic drift. Evolution 61, 2750–2771. doi: 10.1111/j.1558-5646.2007.00250.x
Higgins, J. D., Armstrong, S. J., Franklin, F. C. H., and Jones, G. H. (2004). The Arabidopsis MutS homolog AtMSH4 functions at an early step in recombination: evidence for two classes of recombination in Arabidopsis. Genes Dev. 18, 2557–2570. doi: 10.1101/gad.317504
Higgins, J. D., Buckling, E. F., Franklin, F. C. H., and Jones, G. H. (2008a). Expression and functional analysis of AtMUS81 in Arabidopsis meiosis reveals a role in the second pathway of crossing-over. Plant J. 54, 152–162. doi: 10.1111/j.1365-313X.2008.03403.x
Higgins, J. D., Vignard, J., Mercier, R., Pugh, A. G., Franklin, F. C. H., and Jones, G. H. (2008b). AtMSH5 partners AtMSH4 in the class I meiotic crossover pathway in Arabidopsis thaliana, but is not required for synapsis. Plant J. 55, 28–39. doi: 10.1111/j.1365-313X.2008.03470.x
Higgins, J. D., Osman, K., Jones, G. H., and Franklin, F. C. H. (2014). Factors underlying restricted crossover localization in barley meiosis. Annu. Rev. Genet. 48, 29–47. doi: 10.1146/annurev-genet-120213-092509
Higgins, J. D., Perry, R. M., Barakate, A., Ramsay, L., Waugh, R., Halpin, C., et al. (2012). Spatiotemporal asymmetry of the meiotic program underlies the predominantly distal distribution of meiotic crossovers in barley. Plant Cell 24, 4096–4109. doi: 10.1105/tpc.112.102483
Hirota, K., Mizuno, K., Shibata, T., and Ohta, K. (2008). Distinct chromatin modulators regulate the formation of accessible and repressive chromatin at the fission yeast recombination hotspot ade6-M26. Mol. Biol. Cell 19, 1162–1173. doi: 10.1091/mbc.E07-04-0377
Howe, F. S., Fischl, H., Murray, S. C., and Mellor, J. (2017). Is H3K4me3 instructive for transcription activation? Bioessays 39, 1–12. doi: 10.1002/bies.201600095
Hunter, N., Chambers, S. R., Louis, E. J., and Borts, R. H. (1996). The mismatch repair system contributes to meiotic sterility in an interspecific yeast hybrid. EMBO J. 15, 1726–1733. doi: 10.1002/j.1460-2075.1996.tb00518.x
Jackson, J. P., Lindroth, A. M., Cao, X., and Jacobsen, S. E. (2002). Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416, 556–560. doi: 10.1038/nature731
Jeffreys, A. J., and Neumann, R. (2002). Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat. Genet. 31, 267–271. doi: 10.1038/ng910
Ji, Y., and Chetelat, R. T. (2003). Homoeologous pairing and recombination in Solanum lycopersicoides monosomic addition and substitution lines of tomato. Theor. Appl. Genet. 106, 979–989. doi: 10.1007/s00122-002-1090-2
Johnson, L. M., Bostick, M., Zhang, X., Kraft, E., Henderson, I., Callis, J., et al. (2007). The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. 17, 379–384. doi: 10.1016/j.cub.2007.01.009
Joshi, N., Brown, M. S., Bishop, D. K., and Börner, G. V. (2015). Gradual implementation of the meiotic recombination program via checkpoint pathways controlled by global DSB levels. Mol. Cell 57, 797–811. doi: 10.1016/j.molcel.2014.12.027
Kauppi, L., Barchi, M., Lange, J., Baudat, F., Jasin, M., and Keeney, S. (2013). Numerical constraints and feedback control of double-strand breaks in mouse meiosis. Genes Dev. 27, 873–886. doi: 10.1101/gad.213652.113
Keeney, S., Giroux, C. N., and Kleckner, N. (1997). Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384. doi: 10.1016/S0092-8674(00)81876-0
Kianian, P. M. A., Wang, M., Simons, K., Ghavami, F., He, Y., and Dukowic-Schulze, S. (2018). High-resolution crossover mapping reveals similarities and differences of male and female recombination in maize. Nat. Commun. 9:2370. doi: 10.1038/s41467-018-04562-5
Kiktev, D. A., Sheng, Z., Lobachev, K. S., and Petes, T. D. (2018). GC content elevates mutation and recombination rates in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 115, E7109–E7118. doi: 10.1073/pnas.1807334115
Kleckner, N., Storlazzi, A., and Zickler, D. (2003). Coordinate variation in meiotic pachytene SC length and total crossover/chiasma frequency under conditions of constant DNA length. Trends Genet. 19, 623–628. doi: 10.1016/j.tig.2003.09.004
Knoll, A., Higgins, J. D., Seeliger, K., Reha, S. J., Dangel, N. J., Bauknecht, M., et al. (2012). The Fanconi anemia ortholog FANCM ensures ordered homologous recombination in both somatic and meiotic cells in Arabidopsis. Plant Cell 24, 1448–1464. doi: 10.1105/tpc.112.096644
Kon, N., Krawchuk, M. D., Warren, B. G., Smith, G. R., and Wahls, W. P. (1997). Transcription factor Mts1/Mts2 (Atf1/Pcr1, Gad7/Pcr1) activates the M26 meiotic recombination hotspot in Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. U.S.A. 94, 13765–13770. doi: 10.1073/pnas.94.25.13765
Künzel, G., Korzun, L., and Meister, A. (2000). Cytologically integrated physical restriction fragment length polymorphism maps for the barley genome based on translocation breakpoints. Genetics 154, 397–412.
Kurzbauer, M., Uanschou, C., Chen, D., and Schlögelhofer, P. (2012). The recombinases DMC1 and RAD51 are functionally and spatially separated during meiosis in Arabidopsis. Plant Cell 24, 2058–2070. doi: 10.1105/tpc.112.098459
Lai, J., Li, R., Xu, X., Jin, W., Xu, M., Zhao, H., et al. (2010). Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 42, 1027–1030. doi: 10.1038/ng.684
Lam, I., and Keeney, S. (2015a). Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb. Perspect. Biol. 7:a016634. doi: 10.1101/cshperspect.a016634
Lam, I., and Keeney, S. (2015b). Nonparadoxical evolutionary stability of the recombination initiation landscape in yeast. Science 350, 932–937. doi: 10.1126/science.aad0814
Lambing, C., and Heckmann, S. (2018). Tackling plant meiosis: from model research to crop improvement. Front. Plant Sci. 9:829. doi: 10.3389/fpls.2018.00829
Lange, J., Pan, J., Cole, F., Thelen, M. P., Jasin, M., and Keeney, S. (2011). ATM controls meiotic double-strand-break formation. Nature 479, 237–240. doi: 10.1038/nature10508
Lange, J., Yamada, S., Tischfield, S. E., Pan, J., Kim, S., Zhu, X., et al. (2016). The landscape of mouse meiotic double-strand break formation, processing, and repair. Cell 167, 695–708.e16. doi: 10.1016/j.cell.2016.09.035
Latrille, T., Duret, L., and Lartillot, N. (2017). The red queen model of recombination hot-spot evolution: a theoretical investigation. Philos. Trans. R. Soc. B Biol. Sci. 372:20160463. doi: 10.1098/rstb.2016.0463
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Lawrence, E. J., Griffin, C. H., and Henderson, I. R. (2018). Modification of meiotic recombination by natural variation in plants. J. Exp. Bot. 68, 5471–5483. doi: 10.1093/jxb/erx306
Lenormand, T., and Dutheil, J. (2005). Recombination difference between sexes: a role for haploid selection. PLoS Biol. 3:e63. doi: 10.1371/journal.pbio.0030063
Lercher, M. J., and Hurst, L. D. (2002). Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 18, 337–340. doi: 10.1016/S0168-9525(02)02669-0
Li, X., Li, L., and Yan, J. (2015). Dissecting meiotic recombination based on tetrad analysis by single-microspore sequencing in maize. Nat. Commun. 6:6648. doi: 10.1038/ncomms7648
Lindroth, A. M., Cao, X., Jackson, J. P., and Jacobsen, S. E. (2001). Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292, 2077–2080. doi: 10.1126/science.1059745
Lu, P., Han, X., Qi, J., Yang, J., Wijeratne, A. J., Li, T., et al. (2012). Analysis of Arabidopsis genome-wide variations before and after meiosis and meiotic recombination by resequencing Landsberg erecta and all four products of a single meiosis. Genome Res. 22, 508–518. doi: 10.1101/gr.127522.111
Lukaszewicz, A., Lange, J., Keeney, S., and Jasin, M. (2018). Control of meiotic double-strand-break formation by ATM: local and global views. Cell Cycle 17, 1155–1172. doi: 10.1080/15384101.2018.1464847
Lynn, A., Soucek, R., and Börner, G. V. (2007). ZMM proteins during meiosis: crossover artists at work. Chromosome Res. 15, 591–605. doi: 10.1007/s10577-007-1150-1
Macaisne, N., Novatchkova, M., Peirera, L., Vezon, D., Jolivet, S., Froger, N., et al. (2008). SHOC1, an XPF endonuclease-related protein, is essential for the formation of class I meiotic crossovers. Curr. Biol. 18, 1432–1437. doi: 10.1016/j.cub.2008.08.041
Macaisne, N., Vignard, J., and Mercier, R. (2011). SHOC1 and PTD form an XPF-ERCC1-like complex that is required for formation of class I crossovers. J. Cell Sci. 124, 2687–2691. doi: 10.1242/jcs.088229
Malagnac, F., Bartee, L., and Bender, J. (2002). An Arabidopsis SET domain protein required for maintenance but not establishment of DNA methylation. EMBO J. 21, 6842–6852. doi: 10.1093/emboj/cdf687
Malone, R. E., Kim, S., Bullard, S. A., Lundquist, S., Hutclings-Crow, L., Cramton, S., et al. (1994). Analysis of a recombination hotspot for gene conversion occurring at the HIS2 gene of Saccharomyces cerevisiae. Genetics 137, 5–18.
Manhart, C. M., and Alani, E. (2016). Roles for mismatch repair family proteins in promoting meiotic crossing over. DNA Repair 38, 84–93. doi: 10.1016/j.dnarep.2015.11.024
Marand, A. P., Jansky, S. H., Zhao, H., Leisner, C. P., Zhu, X., Zeng, Z., et al. (2017). Meiotic crossovers are associated with open chromatin and enriched with Stowaway transposons in potato. Genome Biol. 18:203. doi: 10.1186/s13059-017-1326-8
Martini, E., Diaz, R. L., Hunter, N., and Keeney, S. (2006). Crossover homeostasis in yeast meiosis. Cell 126, 285–295. doi: 10.1016/j.cell.2006.05.044
Matic, I., Rayssiguier, C., and Radman, M. (1995). Interspecies gene exchange in bacteria: the role of SOS and mismatch repair systems in evolution of species. Cell 80, 507–515. doi: 10.1016/0092-8674(95)90501-4
Mayer, K. F., Waugh, R., Brown, J. W., Schulman, A., Langridge, P., Platzer, M., et al. (2012). A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716. doi: 10.1038/nature11543
Melamed-Bessudo, C., and Levy, A. A. (2012). Deficiency in DNA methylation increases meiotic crossover rates in euchromatic but not in heterochromatic regions in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 109, E981–E988. doi: 10.1073/pnas.1120742109
Mercier, R., Jolivet, S., Vezon, D., Huppe, E., Chelysheva, L., Giovanni, M., et al. (2005). Two meiotic crossover classes cohabit in Arabidopsis: one is dependent on MER3,whereas the other one is not. Curr. Biol. 15, 692–701. doi: 10.1016/j.cub.2005.02.056
Mercier, R., Mézard, C., Jenczewski, E., Macaisne, N., and Grelon, M. (2015). The molecular biology of meiosis in plants. Annu. Rev. Plant Biol. 66, 297–327. doi: 10.1146/annurev-arplant-050213-035923
Mets, D. G., and Meyer, B. J. (2009). Condensins regulate meiotic DNA break distribution, thus crossover frequency, by controlling chromosome structure. Cell 139, 73–86. doi: 10.1016/j.cell.2009.07.035
Mieczkowski, P. A., Dominska, M., Buck, M. J., Gerton, J. L., Lieb, J. D., and Petes, T. D. (2006). Global analysis of the relationship between the binding of the Bas1p transcription factor and meiosis-specific double-strand DNA breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 26, 1014–1027. doi: 10.1128/MCB.26.3.1014-1027.2006
Mirouze, M., Lieberman-Lazarovich, M., Aversano, R., Bucher, E., Nicolet, J., Reinders, J., et al. (2012). Loss of DNA methylation affects the recombination landscape in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 109, 5880–5885. doi: 10.1073/pnas.1120841109
Modliszewski, J. L., and Copenhaver, G. P. (2017). Meiotic recombination gets stressed out: CO frequency is plastic under pressure. Curr. Opin. Plant Biol. 36, 95–102. doi: 10.1016/j.pbi.2016.11.019
Modrich, P., and Lahue, R. (1996). Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65, 101–133. doi: 10.1146/annurev.bi.65.070196.000533
Moore, D. P., and Orr-Weaver, T. L. (1997). Chromosome segregation during meiosis: building an unambivalent bivalent. Curr. Top. Dev. Biol. 37, 263–299. doi: 10.1016/S0070-2153(08)60177-5
Morgan, C. H., Zhang, H., and Bomblies, K. (2017). Are the effects of elevated temperature on meiotic recombination and thermotolerance linked via the axis and synaptonemal complex? Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 372:20160470. doi: 10.1098/rstb.2016.0470
Myers, S., Bowden, R., Tumian, A., Bontrop, R. E., Freeman, C., MacFie, T. S., et al. (2010). Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327, 876–879. doi: 10.1126/science.1182363
Myers, S., Freeman, C., Auton, A., Donnelly, P., and Mcvean, G. (2008). A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 40, 1124–1129. doi: 10.1038/ng.213
Nicolas, A., Treco, D., Schultes, N. P., and Szostak, J. W. (1989). An initiation site for meiotic gene conversion in the yeast Saccharomyces cerevisiae. Nature 338, 35–39. doi: 10.1038/338035a0
Nilsson-Tillgren, T., Gjermansen, C., Holmberg, S., Litske Petersen, J. G., and Kielland-Brandt, M. C. (1986). Analysis of chromosome V and theILV1 gene from Saccharomyces carlsbergensis. Carlsberg Res. Commun. 51, 309–326. doi: 10.1007/BF02907164
Nilsson-Tillgren, T., Gjermansen, C., Kielland-Brandt, M. C., Petersen, J. G. L., and Holmberg, S. (1981). Genetic differences between Saccharomyces carlsbergensis and S. cerevisiae. Analysis of chromosome III by single chromosome transfer. Carlsberg Res. Commun. 46, 65–76. doi: 10.1007/BF02906199
Otto, S. P., and Lenormand, T. (2002). Resolving the paradox of sex and recombination. Nat. Rev. Genet. 3, 252–261. doi: 10.1038/nrg761
Pan, J., Sasaki, M., Kniewel, R., Murakami, H., Blitzblau, H. G., Tischfield, S. E., et al. (2011). A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144, 719–731. doi: 10.1016/j.cell.2011.02.009
Parvanov, E. D., Petkov, P. M., and Paigen, K. (2010). Prdm9 controls activation of mammalian recombination hotspots. Science 327:835. doi: 10.1126/science.1181495
Pawlowski, W. P., Golubovskaya, I. N., and Cande, W. Z. (2003). Altered nuclear distribution of recombination protein RAD51 in maize mutants suggests the involvement of RAD51 in meiotic homology recognition. Plant Cell 15, 1807–1816. doi: 10.1105/tpc.012898
Petes, T. D. (2001). Meiotic recombination hot spots and cold spots. Nat. Rev. Genet. 2, 360–369. doi: 10.1038/35072078
Phillips, D., Wnetrzak, J., Nibau, C., Barakate, A., Ramsay, L., Wright, F., et al. (2013). Quantitative high resolution mapping of HvMLH3 foci in barley pachytene nuclei reveals a strong distal bias. J. Exp. Bot. 64, 2139–2154. doi: 10.1093/jxb/ert079
Pineda-Krch, M., and Redfield, R. J. (2005). Persistence and loss of meiotic recombination hotspots. Genetics 169, 2319–2333. doi: 10.1534/genetics.104.034363
Ponting, C. P. (2011). What are the genomic drivers of the rapid evolution of PRDM9? Trends Genet. 27, 165–171. doi: 10.1016/j.tig.2011.02.001
Pratto, F., Brick, K., Khil, P., Smagulova, F., Petukhova, G. V., and Camerini-Otero, R. D. (2014). Recombination initiation maps of individual human genomes. Science 346, 826–835. doi: 10.1126/science.1256442
Rayssiguier, C., Thaler, D. S., and Radman, M. (1989). The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342, 396–401. doi: 10.1038/342396a0
Ritz, K. R., Noor, M. A. F., and Singh, N. D. (2017). Variation in recombination rate: adaptive or not? Trends Genet. 33, 364–374. doi: 10.1016/j.tig.2017.03.003
Robert, T., Nore, A., Brun, C., Maffre, C., Crimi, B., Bourbon, H.-M., et al. (2016). The TopoVIB-Like protein family is required for meiotic DNA double-strand break formation. Science 351, 943–949. doi: 10.1126/science.aad5309
Robine, N., Uematsu, N., Amiot, F., Gidrol, X., Barillot, E., Nicolas, A., et al. (2007). Genome-wide redistribution of meiotic double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 1868–1880. doi: 10.1128/MCB.02063-06
Rocco, V., and Nicolas, A. (1996). Sensing of DNA non-homology lowers the initiation of meiotic recombination in yeast. Genes Cells 1, 645–661. doi: 10.1046/j.1365-2443.1996.00256.x
Rockmill, B., Voelkel-Meiman, K., and Roeder, G. S. (2006). Centromere-proximal crossovers are associated with precocious separation of sister chromatids during meiosis in Saccharomyces cerevisiae. Genetics 1754, 1745–1754. doi: 10.1534/genetics.106.058933
Rodgers-Melnick, E., Bradbury, P. J., Elshire, R. J., Glaubitz, J. C., Acharya, C. B., Mitchell, S. E., et al. (2015). Recombination in diverse maize is stable, predictable, and associated with genetic load. Proc. Natl. Acad. Sci. U.S.A. 112, 3823–3828. doi: 10.1073/pnas.1413864112
Rodgers-Melnick, E., Vera, D. L., Bass, H. W., and Buckler, E. S. (2016). Open chromatin reveals the functional maize genome. Proc. Natl. Acad. Sci. U.S.A. 113, E3177–E3184. doi: 10.1073/pnas.1525244113
Rosu, S., Libuda, D. E., and Villeneuve, A. M. (2011). Robust crossover assurance and regulated interhomolog access maintain meiotic crossover number. Science 334, 1286–1289. doi: 10.1126/science.1212424
Saintenac, C., Falque, M., Martin, O. C., Paux, E., Feuillet, C., and Sourdille, P. (2009). Detailed recombination studies along chromosome 3B provide new insights on crossover distribution in wheat (Triticum aestivum L.). Genetics 181, 393–403. doi: 10.1534/genetics.108.097469
Salomé, P. A., Bomblies, K., Fitz, J., Laitinen, R. A. E., Warthmann, N., Yant, L., et al. (2011). The recombination landscape in Arabidopsis thaliana F2 populations. Heredity 108, 1–9. doi: 10.1038/hdy.2011.95
Santos-Rosa, H., Schneider, R., Bannister, A. J., Sherriff, J., Bernstein, B. E., Emre, N. C. T., et al. (2002). Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411. doi: 10.1038/nature01080
Saze, H., Scheid, O. M., and Paszkowski, J. (2003). Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 34, 65–69. doi: 10.1038/ng1138
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115. doi: 10.1126/science.1178534
Schwacha, A., and Kleckner, N. (1997). Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog-only pathway. Cell 90, 1123–1135. doi: 10.1016/S0092-8674(00)80378-5
Séguéla-Arnaud, M., Choinard, S., Larchevêque, C., Girard, C., Froger, N., Crismani, W., et al. (2017). RMI1 and TOP3alpha limit meiotic CO formation through their C-terminal domains. Nucleic Acids Res. 45, 1860–1871. doi: 10.1093/nar/gkw1210
Séguéla-Arnaud, M., Crismani, W., Larchevêque, C., Mazel, J., Froger, N., Choinard, S., et al. (2015). Multiple mechanisms limit meiotic crossovers: TOP3α and two BLM homologs antagonize crossovers in parallel to FANCM. Proc. Natl. Acad. Sci. U.S.A. 112, 4713–4718. doi: 10.1073/pnas.1423107112
Serra, H., Choi, K., Zhao, X., Blackwell, A. R., and Henderson, I. R. (2018a). Interhomolog polymorphism shapes meiotic crossover within RAC1 and RPP13 disease resistance genes. bioRxiv [Preprint]. doi: 10.1101/290478
Serra, H., Lambing, C., Griffin, C. H., Topp, S. D., Nageswaran, D. C., Underwood, C. J., et al. (2018b). Massive crossover elevation via combination of HEI10 and recq4a recq4b during Arabidopsis meiosis. Proc. Natl. Acad. Sci. U.S.A. 115, 2437–2442. doi: 10.1073/pnas.1713071115
Shen, P., and Huang, H. V. (1986). Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics 112, 441–457.
Shen, P., and Huang, H. V. (1989). Effect of base mismatches on recombination via the RecBCD pathway. Mol. Genet. Genomics 218, 359–360. doi: 10.1007/BF00331291
Shen, Y., Tang, D., Wang, K., Wang, M., Huang, J., Luo, W., et al. (2012). ZIP4 in homologous chromosome synapsis and crossover formation in rice meiosis. J. Cell Sci. 125, 2581–2591. doi: 10.1242/jcs.090993
Shilo, S., Melamed-Bessudo, C., Dorone, Y., Barkai, N., and Levy, A. A. (2015). DNA crossover motifs associated with epigenetic modifications delineate open chromatin regions in Arabidopsis. Plant Cell 27, 2427–2436. doi: 10.1105/tpc.15.00391
Sidhu, G. K., Fang, C., Olson, M. A., Falque, M., Martin, O. C., and Pawlowski, W. P. (2015). Recombination patterns in maize reveal limits to crossover homeostasis. Proc. Natl. Acad. Sci. U.S.A. 112, 15982–15987. doi: 10.1073/pnas.1514265112
Simon, L., Voisin, M., Tatout, C., and Probst, A. V. (2015). Structure and function of centromeric and pericentromeric heterochromatin in Arabidopsis thaliana. Front. Plant Sci. 6:1049. doi: 10.3389/fpls.2015.01049
Simon, M., Loudet, O., Durand, S., Bérard, A., Brunel, D., Sennesal, F.-X., et al. (2008). Quantitative trait loci mapping in five new large recombinant inbred line populations of Arabidopsis thaliana genotyped with consensus single-nucleotide polymorphism markers. Genetics 178, 2253–2264. doi: 10.1534/genetics.107.083899
Singhal, S., Leffler, E. M., Sannareddy, K., Turner, I., Venn, O., Hooper, D. M., et al. (2015). Stable recombination hotspots in birds. Science 350, 928–932. doi: 10.1126/science.aad0843
Smeds, L., Mugal, C. F., Qvarnström, A., and Ellegren, H. (2016). High-resolution mapping of crossover and non-crossover recombination events by whole-genome re-sequencing of an avian pedigree. PLoS Genet. 12:e1006044. doi: 10.1371/journal.pgen.1006044
Sommermeyer, V., Benéut, C., Chaplais, E., Serrentino, M. E., and Borde, V. (2013). Spp1, a member of the Set1 complex, promotes meiotic DSB formation in promoters by tethering histone H3K4 methylation sites to chromosome axes. Mol. Cell 49, 43–54. doi: 10.1016/j.molcel.2012.11.008
Stack, S. M., and Anderson, L. K. (2002). Crossing over as assessed by late recombination nodules is related to the pattern of synapsis and the distribution of early recombination nodules in maize. Chromosome Res. 10, 329–345. doi: 10.1023/A:1016575925934
Steiner, W. W., Schreckhise, R. W., and Smith, G. R. (2002). Meiotic DNA breaks at the S. pombe recombination hot spot M26. Mol. Cell 9, 847–855. doi: 10.1016/S1097-2765(02)00489-6
Stewart, M. N., Dawson, D. S., and Darion, J. (2008). Changing partners: moving from non- homologous to homologous centromere pairing in meiosis. Trends Genet. 24, 564–573. doi: 10.1016/j.tig.2008.08.006
Stroud, H., Greenberg, M. V. C., Feng, S., Bernatavichute, Y. V., and Jacobsen, S. E. (2012). Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364. doi: 10.1016/j.cell.2012.10.054
Sturtevant, A. H. (1913). The linear arrangement of six sex-linked factors in Drosophila, as shown by their mode of association. J. Exp. Zool. 14, 43–59. doi: 10.1002/jez.1400140104
Sun, L., Wang, J., Sang, M., Jiang, L., Zhao, B., Cheng, T., et al. (2017). Landscaping crossover interference across a genome. Trends Plant Sci. 22, 894–907. doi: 10.1016/j.tplants.2017.06.008
Sun, Y., Ambrose, J. H., Haughey, B. S., Webster, T. D., Pierrie, S. N., Muńoz, D. F., et al. (2012). Deep genome-wide measurement of meiotic gene conversion using tetrad analysis in Arabidopsis thaliana. PLoS Genet. 8:e1002968. doi: 10.1371/journal.pgen.1002968
Symington, L. S., and Petes, T. D. (1988). Expansions and contractions of the genetic map relative to the physical map of yeast chromosome III. Mol. Cell. Biol. 8, 595–604. doi: 10.1128/MCB.8.2.595
Tam, S. M., Hays, J. B., and Chetelat, R. T. (2011). Effects of suppressing the DNA mismatch repair system on homeologous recombination in tomato. Theor. Appl. Genet. 123, 1445–1458. doi: 10.1007/s00122-011-1679-4
Thacker, D., Mohibullah, N., Zhu, X., and Keeney, S. (2014). Homologue engagement controls meiotic DNA break number and distribution. Nature 510, 241–246. doi: 10.1038/nature13120
The Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
Tiemann-Boege, I., Schwarz, T., Striedner, Y., Heissl, A., and Tiemann-boege, I. (2017). The consequences of sequence erosion in the evolution of recombination hotspots. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 372:20160462. doi: 10.1098/rstb.2016.0462
Tischfield, S. E., and Keeney, S. (2012). Scale matters. The spatial correlation of yeast meiotic DNA breaks with histone H3 trimethylation is driven largely by independent colocalization at promoters. Cell Cycle 11, 1496–1503. doi: 10.4161/cc.19733
Tomato Genome Consortium (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi: 10.1038/nature11119
Underwood, C. J., Choi, K., Lambing, C., Zhao, X., Serra, H., Borges, F., et al. (2018). Epigenetic activation of meiotic recombination near Arabidopsis thaliana centromeres via loss of H3K9me2 and non-CG DNA methylation. Genome Res. 28, 519–531. doi: 10.1101/gr.227116.117
Varas, J., Sánchez-Morán, E., Copenhaver, G. P., Santos, J. L., and Pradillo, M. (2015). Analysis of the relationships between DNA double-strand breaks, synaptonemal complex and crossovers using the Atfas1-4 mutant. PLoS Genet. 11:e1005301. doi: 10.1371/journal.pgen.1005301
Vervoort, M., Meulemeester, D., Béhague, J., and Kerner, P. (2016). Evolution of Prdm genes in animals: insights from comparative genomics. Mol. Biol. Evol. 33, 679–696. doi: 10.1093/molbev/msv260
Vizir, I. Y., and Korol, A. B. (1990). Sex difference in recombination frequency in Arabidopsis. Heredity 65, 379–383. doi: 10.1038/hdy.1990.107
Vongs, A., Kakutani, T., Martienssen, R. A., and Richards, E. J. (1993). Arabidopsis thaliana DNA methylation mutants. Science 260, 1926–1928. doi: 10.1126/science.8316832
Vrielynck, N., Chambon, A., Vezon, D., Pereira, L., Chelysheva, L., De Muyt, A., et al. (2016). A DNA topoisomerase VI-like complex initiates meiotic recombination. Science 351, 939–943. doi: 10.1126/science.aad5196
Wahls, W. P., and Davidson, M. K. (2010). Discrete DNA sites regulate global distribution of meiotic recombination. Trends Genet. 26, 202–208. doi: 10.1016/j.tig.2010.02.003
Wang, S., Zickler, D., Kleckner, N., and Zhang, L. (2015). Meiotic crossover patterns: obligatory crossover, interference and homeostasis in a single process. Cell Cycle 14, 305–314. doi: 10.4161/15384101.2014.991185
Wang, Y., and Copenhaver, G. P. (2018). Meiotic recombination: mixing it up in plants. Annu. Rev. Plant Biol. 29, 577–609. doi: 10.1146/annurev-arplant-042817-040431
White, M. A., Dominska, M., and Petes, T. D. (1993). Transcription factors are required for the meiotic recombination hotspot at the HIS4 locus in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 90, 6621–6625. doi: 10.1073/pnas.90.14.6621
White, M. A., Wierdl, M., Detloff, P., and Petes, T. D. (1991). DNA-binding protein RAP1 stimulates meiotic recombination at the HIS4 locus in yeast. Proc. Natl. Acad. Sci. U.S.A. 88, 9755–9759. doi: 10.1073/pnas.88.21.9755
Wijnker, E., Velikkakam James, G., Ding, J., Becker, F., Klasen, J. R., Rawat, V., et al. (2013). The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana. eLife 2:e01426. doi: 10.7554/eLife.01426
Wu, T. C., and Lichten, M. (1995). Factors that affect the location and frequency of meiosis-induced double- strand breaks in Saccharomyces cerevisiae. Genetics 140, 55–66.
Xu, L., and Kleckner, N. (1995). Sequence non-specific double-strand breaks and interhomolog interactions prior to double-strand break formation at a meiotic recombination hot spot in yeast. EMBO J. 14, 5115–5128. doi: 10.1002/j.1460-2075.1995.tb00194.x
Xue, M., Wang, J., Jiang, L., Wang, M., Wolfe, S., Pawlowski, W. P., et al. (2018). The number of meiotic double-strand breaks influences crossover distribution in Arabidopsis. Plant Cell 30, 2628–2638. doi: 10.1105/tpc.18.00531
Yamada, S., Kim, S., Tischfield, S. E., Jasin, M., Lange, J., and Keeney, S. (2017a). Genomic and chromatin features shaping meiotic double-strand break formation and repair in mice. Cell Cycle 16, 1870–1884. doi: 10.1080/15384101.2017.1361065
Yamada, S., Okamura, M., Oda, A., Murakami, H., Ohta, K., and Yamada, T. (2017b). Correlation of meiotic DSB formation and transcription initiation around fission yeast recombination hotspots. Genetics 206, 801–809. doi: 10.1534/genetics.116.197954
Yang, S., Wang, L., Huang, J., Zhang, X., Yuan, Y., Chen, J.-Q., et al. (2015). Parent–progeny sequencing indicates higher mutation rates in heterozygotes. Nature 523, 463–467. doi: 10.1038/nature14649
Yao, H., and Schnable, P. S. (2005). Cis-effects on meiotic recombination across distinct a1-sh2 intervals in a common Zea genetic background. Genetics 170, 1929–1944. doi: 10.1534/genetics.104.034454
Yao, H., Zhou, Q., Li, J., Smith, H., Yandeau, M., Nikolau, B. J., et al. (2002). Molecular characterization of meiotic recombination across the 140-kb multigenic a1-sh2 interval of maize. Proc. Natl. Acad. Sci. U.S.A. 99, 6157–6162. doi: 10.1073/pnas.082562199
Yelina, N. E., Choi, K., Chelysheva, L., Macaulay, M., de Snoo, B., Wijnker, E., et al. (2012). Epigenetic remodeling of meiotic crossover frequency in Arabidopsis thaliana DNA methyltransferase mutants. PLoS Genet. 8:e1002844. doi: 10.1371/journal.pgen.1002844
Yelina, N. E., Lambing, C., Hardcastle, T. J., Zhao, X., Santos, B., and Henderson, I. R. (2015). DNA methylation epigenetically silences crossover hot spots and controls chromosomal domains of meiotic recombination in Arabidopsis. Genes Dev. 29, 2183–2202. doi: 10.1101/gad.270876.115
Yokoo, R., Zawadzki, K. A., Nabeshima, K., Drake, M., Arur, S., and Villeneuve, A. M. (2012). COSA-1 reveals robust homeostasis and separable licensing and reinforcement steps governing meiotic crossovers. Cell 149, 75–87. doi: 10.1016/j.cell.2012.01.052
Zemach, A., Kim, M. Y., Hsieh, P.-H., Coleman-Derr, D., Eshed-Williams, L., Thao, K., et al. (2013). The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153, 193–205. doi: 10.1016/j.cell.2013.02.033
Zhang, L., Kim, K. P., Kleckner, N. E., and Storlazzi, A. (2012). Meiotic double-strand breaks occur once per pair of (sister) chromatids and, via Mec1/ATR and Tel1/ATM, once per quartet of chromatids. Proc. Natl. Acad. Sci. U.S.A. 109, 1353–1353. doi: 10.1073/pnas.1117937108
Zhang, X. (2008). The epigenetic landscape of plants. Science 320, 489–492. doi: 10.1126/science.1153996
Zhu, X., and Keeney, S. (2015). High-resolution global analysis of the influences of Bas1 and Ino4 transcription factors on meiotic DNA break distributions in Saccharomyces cerevisiae. Genetics 201, 525–542. doi: 10.1534/genetics.115.178293
Zickler, D., and Kleckner, N. (1998). The leptotene-zygotene transition of meiosis. Annu. Rev. Genet. 32, 619–697. doi: 10.1146/annurev.genet.32.1.619
Ziolkowski, P. A., Berchowitz, L. E., Lambing, C., Yelina, N. E., Zhao, X., Kelly, K. A., et al. (2015). Juxtaposition of heterozygous and homozygous regions causes reciprocal crossover remodelling via interference during Arabidopsis meiosis. eLife 4:e03708. doi: 10.7554/eLife.03708
Ziolkowski, P. A., and Henderson, I. R. (2017). Interconnections between meiotic recombination and sequence polymorphism in plant genomes. New Phytol. 213, 1022–1029. doi: 10.1111/nph.14265
Ziolkowski, P. A., Koczyk, G., Galganski, L., and Sadowski, J. (2009). Genome sequence comparison of Col and Ler lines reveals the dynamic nature of Arabidopsis chromosomes. Nucleic Acids Res. 37, 3189–3201. doi: 10.1093/nar/gkp183
Keywords: meiotic crossover, recombination hot spot, double-strand break (DSB), heterozygosity, polymorphism (genetic), DNA methylation, plants, chromatin
Citation: Dluzewska J, Szymanska M and Ziolkowski PA (2018) Where to Cross Over? Defining Crossover Sites in Plants. Front. Genet. 9:609. doi: 10.3389/fgene.2018.00609
Received: 31 July 2018; Accepted: 19 November 2018;
Published: 12 December 2018.
Edited by:
Carina Farah Mugal, Uppsala University, SwedenReviewed by:
Takeshi Kawakami, Uppsala University, SwedenMateusz Zelkowski, Cornell University, United States
Raphael Mercier, Institut National de la Recherche Agronomique (INRA), France
Copyright © 2018 Dluzewska, Szymanska and Ziolkowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Piotr A. Ziolkowski, cHppb0BhbXUuZWR1LnBs
†These authors have contributed equally to this work