 Nwobodo Alexander Kenechukwu1,2†Man Li1,2†Lei An3†Miaomiao Cui1,2†Cailin Wang1,2†Aili Wang4Yulin Chen1,2Saijun Du1,2
Nwobodo Alexander Kenechukwu1,2†Man Li1,2†Lei An3†Miaomiao Cui1,2†Cailin Wang1,2†Aili Wang4Yulin Chen1,2Saijun Du1,2 Chenyao Feng5Sijin Zhong5Yuying Gao5Xueyan Cao5Li Wang5Ezenwali Moses Obinna6Xinyu Mei7*Yuanjian Song1,2*Zongyun Li8*
Chenyao Feng5Sijin Zhong5Yuying Gao5Xueyan Cao5Li Wang5Ezenwali Moses Obinna6Xinyu Mei7*Yuanjian Song1,2*Zongyun Li8* Dashi Qi1,2,4*
Dashi Qi1,2,4*- 1Jiangsu Key Laboratory of Brain Disease Bioinformation, Xuzhou Medical University, Xuzhou, China
- 2Department of Genetics, Research Center for Neurobiology, Xuzhou Medical University, Xuzhou, China
- 3Huaihe Hospital of Henan University, Henan University College of Medicine, Kaifeng, China
- 4Institute of Gastrointestinal Surgery and Translational Medicine, Tongji University School of Medicine, Shanghai, China
- 5Department of Clinical, Xuzhou Medical University, Xuzhou, China
- 6Department of Applied Biochemistry, Enugu State University of Science and Technology, Enugu, Nigeria
- 7Interdisciplinary Research Center on Biology and Chemistry (IRCBC), Chinese Academy of Sciences, Shanghai, China
- 8School of Life Sciences, Jiangsu Normal University, Xuzhou, China
This present research work reports the comparative analysis of the entire nucleotide sequence of mitochondrial genomes of Serranochromis robustus and Buccochromis nototaenia and phylogenetic analyses of their protein-coding genes in order to establish their phylogenetic relationship within Cichlids. The mitochondrial genomes of S. robustus and B. nototaenia are 16,583 and 16,580 base pairs long, respectively, including 13 protein-coding genes (PCGs), 2 ribosomal RNA genes, 22 transfer RNA genes, and one control region (D-loop) which is 888 and 887 base pairs long, respectively, showing the same gene order and identical number of gene or regions with other well-elucidated mitogenomes of Cichlids. However, with exception of cytochrome-c oxidase subunit-1 (COX-1) gene, all the identified PCGs were initiated by ATG-codons. Structurally, 11 tRNA genes in B. nototaenia species and 9 tRNA genes in S. robustus species, folded into typical clover-leaf secondary structure created by the regions of self-complementarity within tRNA. All the 22 tRNA genes in both species lack variable loop. Moreover, 28 genes which include 12-protein-coding genes are encoded on the H-strand and the remaining 9 genes including one protein-coding gene are encoded on the L-strand. Thirteen sequences of concatenated mitochondrial protein-coding genes were aligned using MUSCLE, and the phylogenetic analyses performed using maximum likelihood and Bayesian inference showed that S. robustus and B. nototaenia had a broad phylogenetic relationship. These results may be a useful tool in resolving higher-level relationships in organisms and a useful dataset for studying the evolution of the Cichlidae mitochondrial genome, since Cichlids are well-known model species in the study of evolutionary biology, because of their extreme morphological, biogeographical, parental care behavior for eggs and larvae and phylogenetic diversities.
Introduction
Serranochromis robustus and Buccochromis nototaenia are essential species in commercial fisheries that belong to the family of Cichlidae; both are commonly found in tropical freshwater in estuaries in Lake Malawi, upper Shire River, Luongo River in the Congo, and Zambia. They respond promptly to environmental alterations and are both carnivorous and oviparous maternal mouthbrooder fish. Buccochromis consists of four different subspecies, B. rhoadesii, B. spectalbilis, B. Lepturus, and B. nototaenia, which stay at an average depth of 10 m in offshore sandy beaches. There are two subspecies of serranchromis, S. robustus robustus and S. robustus jallae. The latter is found in Cunene, Okavango, Kafue, upper and the middle Zambezi, Luangwa Luapula-Moero, Lualaba, and kassi-rivers and it has been transferred to Zimbabwe, Limpopo River and Natal, South Africa (Jellum, 1970; Kocher, 2004). The phylogenetic diversity of Cichlid fish, a vital tool in the study of evolutionary biology can be well-apprehended through proper investigation on mitochondrion, an evolutionary endosymbiont derived from bacteria (prokaryotes) (Sagan, 1967). The circular mitochondrial DNA (mtDNA) which can reproduce independent of its cell is an apparent cause of endosymbiosis. This is based on its close similarities with prokaryotes in their circular DNA, 70 ribosomes, 22 transfer RNAs, formyl-methionine initiating amino acid, and their susceptibility to tetracycline. The integral inner membrane proteins are synthesized from circular mtDNA, while proteins of the outer membrane of the mitochondria are translational products of nuclear mRNA. Since mitochondria are evolutionary endosymbionts that were derived from bacteria. The cellular injury will release mitochondria “damage-associated molecular patterns” DAMPs (formyl-peptides and circular mtDNA) into the circulation with functional critical immune consequences, and this is believed to be the link between trauma, inflammation, and systemic inflammatory response syndrome (SIRS) (Zhang et al., 2010). The mitochondrial genome of fish is a circular double-stranded molecule which is about 15 to 19 kb in length (Cui et al., 2017). Mitochondria play a central role in metabolism (Brand, 1997), including oxidative phosphorylation (Smeitink et al., 2001), apoptosis (Kroemer et al., 1998), diseases (Graeber and Muller, 1998), aging (Wei, 1998), and also different other biochemical functions.
However, because of the conserved characteristics of coding content, location on Y-chromosomal DNA, rapid evolution, and low levels of intermolecular genetic recombination, mitogenomes are biomarkers for molecular research in such areas as phylogenetic molecular evolution, population genetics, and evolutionary genomics (Bentzen et al., 1998; Boore, 1999). A typical mitochondrial genome contains typically two ribosomal RNA genes (s-rRNA and l-rRNA), 22 transfer RNA genes (tRNAs), 13 protein-coding genes (PCGs), and two typical non-coding control regions {control region (CR) and origin of the light strand (OL)} with regulatory elements essential for transcription and replication (Miya and Nishida, 1999). In some mitochondrial genomes the genes are located on both strands, whereas in others, all genes are transcribed from one strand (Ojala et al., 1981). Twelve of the 13 protein-coding genes are located on the H-strand, and only ND6 gene is located on the L-strand. The energy values of foods are harnessed in the form of ATP through coupling of energy-releasing activities of electron transport chain, proton pump, and oxidative phosphorylation (Cadenas and Davies, 2000). The electron transport chain has been proven to be “leaky.” The leaky nature of the electron transport chain results in the conversion of molecular oxygen into superoxide anion radical () (www.ncbi.nlm.nih.gov). Dismutation of superoxide produces hydrogen peroxide (H2O2). Hydrogen peroxide may, in turn, be partially reduced to hydroxyl radical (HO−) or entirely reduced to water. These free radicals generated from mitochondria or other sites inside or outside the cell leads to cell (destruction) damage through mtDNA, RNA, and protein modifications and lipid peroxidation (Cline, 2012). Mutated mitochondrial DNA molecules accumulate with age. This age-dependent accumulation of mutated mtDNA (Lagouge and Larsson, 2013), can in principle be explained by two primary mechanisms, replication error, and unrepaired damage. First, it has been suggested that the massive mtDNA replication occurring during embryogenesis will result in replication errors due to the inherent error rate of the mitochondrial DNA polymerase. The mtDNA mutations formed during embryogenesis will be subjected to segregation and clonal expansion in postnatal life. Secondly, the alternative proposal states that damage caused by ROS, may overwhelm the repair machinery and result in accumulation of mutated mtDNA (Larsson, 2010; Lagouge and Larsson, 2013). Over the last 10 years, these properties of mtDNA namely; high mutation rate, fast evolutionary rate, and non-significant genetic recombination have made it universally accepted biomarker for determination of genetic diversity among species (Galtier et al., 2009). Cichlids are universal accepted species model in the study of phylogenetic biology due to their extreme diversified morphological and biogeographic traits, parental care behavior for eggs and larvae (Klett and Meyer, 2002; Kocher, 2004; Salzburger and Meyer, 2004; Seehausen, 2006; Turner, 2007; Kuraku and Meyer, 2008). Fish of this family are popularly remarked with two distinct features; a single opening of the nostrils and an interrupted lateral line (Hartvigsen and Halvorsen, 1994; Khuda-Bukhsh and Chakrabarti, 2000). As economically important fish, some Cichlids are widely used extensively in aquaculture for several reasons. They are good source of “white fish” and fish products, they lack small bones in the muscle, and some species can grow quite large, allowing for the production of value-added products like filets. Most essentially, they depend on the lower food chain (aquatic plants and plankton) reducing their cost of feeding (Pulling, 1991). in this study, we sequenced the complete mitochondrial genome of the two Cichlidae species and investigated the gene content and organization compared with other species. We also reconstructed phylogenetic tree based on PCG sequences for the purpose of analyzing the evolutionary relationships within Cichlids family. These results may provide more insight and a useful dataset for studying the evolution of the Cichlidae mitochondrial genome.
Materials and Methods
Sampling and DNA Extraction
The samples of S. robustus and B. nototaenia, collected from a trawler catch in the Southeast Arm of Lake Malawi (between latitudes 9° and 18°S, and longitudes 32° and 36°E) by the Welcome Trust Sanger Institute (SC) in their private collection, were not endangered or protected species according to the IUCN Red List. The circular mitochondrial genomic DNAs were extracted from dorsal muscle tissue samples using the Animal Tissue Genomic DNA Extraction Kit (SangonBiotech China) according to the manufacturer's instructions. Every protocol were performed in accordance with the international guidelines concerning the care and treatment of experimental animals. A known volume (15 ml) of 95% ethanol were used to preserve all the samples at −80°C until DNA extraction. Polymerase Chain Reaction was used to amplify the complete mitogenomes of the extracted DNA.
Mitochondrial DNA Amplification and Sequencing
Eppendorf Thermal Cycler (Eppendorf, Germany) was used to perform polymerase chain reaction with a fix reaction mixture of 50 μl (microliter) consisting of 2 units of Taq DNA polymerase, 5 μl PCR buffer (Tiangen products, China), 2 μl template DNA (50 ng/ μl), 2 μl dNTP (0.4 mM), 4 μl primers (0.2 μM each), and 35 μl deionized/distilled water. After 3 min the reaction was denatured at 95°C, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 1–5 min. All the PCR products were sequenced using the primer walking method with a 3730XL DNA Analyzer. The obtained sequences had 100% coverage of the PCR products.
Sequence Editing and Analysis
All the reads were mapped to full mitochondrial genome reference sequences of A. geoffreyi (NC_028033) by using SOAPalingner/soap2 (V2.21). Then we assembled the roads which could map to the reference genome by SPAdes3 (V3.1.0) and got the circular mitochondrial genome. Additionally, the location of the 13 PCGs and the two rRNAs for each species were primarily identified through Dual Organellar Genome Annotator (DOGMA) (Wyman et al., 2004). The tRNA-scan-SE1.21 identified most of the transfer RNA (tRNA) genes from the website http://lowelab.ucsc.edu/tRNAscan-SE/, using the default search mode and the “Mito/chloroplast” source (Lowe and Eddy, 1997). To infer the secondary structures of tRNA molecules, we used a widely scientifically accepted comparative approach to correct for unusual pairings with RNA-editing mechanisms that are well-known in fish mitogenomes. The software RNA structure was used in drawing the secondary structure of tRNA genes (Mathews, 2014). The skewness of the nucleotide compositions were measured according to the following formulas: AT skew [(A – T) / (A + T)] and GC skew [(G – C)/ (G + C)] (Perna and Kocher, 1995). The full mitochondrial genomic DNA sequence of the S. robustus and the B. nototaenia were stored in the GenBank database with the following accession numbers, accession KX595333 and KX631426, respectively.
Phylogenetic Analysis
To establish evolutionary relationships between S. robustus and B. nototaenia mitogenomes within the family Cichlidae the complete mitogenome sequence of S. robustus and B. nototaenia and 13 other species available in GenBank were used. Both amino acid and nucleotide sequences for each species of the 13 PCGs were aligned using default settings and concatenated, which were used for phylogenetic analysis through the Maximum Likelihood (ML) and Bayesian inference (BI) methods. Using raxmlGUI v 8.0.26 and MrBayes v 3.2.4, respectively (Ronquist et al., 2012; Silvestro and Michalak, 2012), which allowed different substitution models in individual partitions. Clustal X with default settings were used to align all the genes separately (Thompson et al., 2002). However, GTR + I + G was selected as the appropriate model for the nucleotide sequences by Modeltest 3.7 based on Akaike's information criterion (AIC) (Beier et al., 2004). MtArt + I + G + F were the appropriate model for the amino acid sequence dataset according to ProtTest 3.4 based on AIC (Abascal et al., 2005). The resulting phylogenetic trees were drawn in Molecular Evolutionary Genetics Analysis (MEGA) version 6.0 (Lewis et al., 1994).
Results and Discussion
Mitochondrial Genome Organization and Composition
The structure of the mitochondrial genome of the newly sequenced S. robustus and B. nototaenia are similar to those of other cichlids characterized so far example., F. rostatus, and A. geoffreyi (Qi et al., 2016), they have the same types, number, and genomic features. The sequence data were deposited in GenBank under accession KX595333 and KX631426, respectively. The complete mitochondrial genomes were 16,583 bp long for S. robustus and 16,580 bp long for B. nototaenia, both were closed circular DNAs. The two species contain 13 protein-coding genes (ATP6, ATP8, Cytb, Cox1-3, ND1-6, and ND4L), 22 interspersed transfer RNA (tRNA) genes, 2 ribosomal RNA (rRNA) genes (s-rRNA and l-rRNA), and one control region (CR; also termed displacement loop region or D-Loop) (Table 1). Among these genes, 28 including 12 protein-coding genes are encoded on the H-strand and the remaining 9 genes including one protein-coding gene are encoded on the L-strand (ND6, Gln, Ala, Asn, Cys, Try, Ser, Glu, and Pro). The overall base composition of the mitochondrial genome is highly similar between these two species: A = 4,555 (27.47%), G = 2,599 (15.67%), T = 4,414 (26.62%) in S. robustus and A = 4,555 (27.47%), G = 2,624 (15.83%), T = 4,409 (26.59%) in B. nototaenia, whereas C = 4,993 (30.11%) in both species. However, the C content is relatively lower when compared with P. managuensis (31.0%) (Liu et al., 2016). An illustration of the complete mitochondrial genome of S. robustus and B. nototaenia is shown in Figure 1.
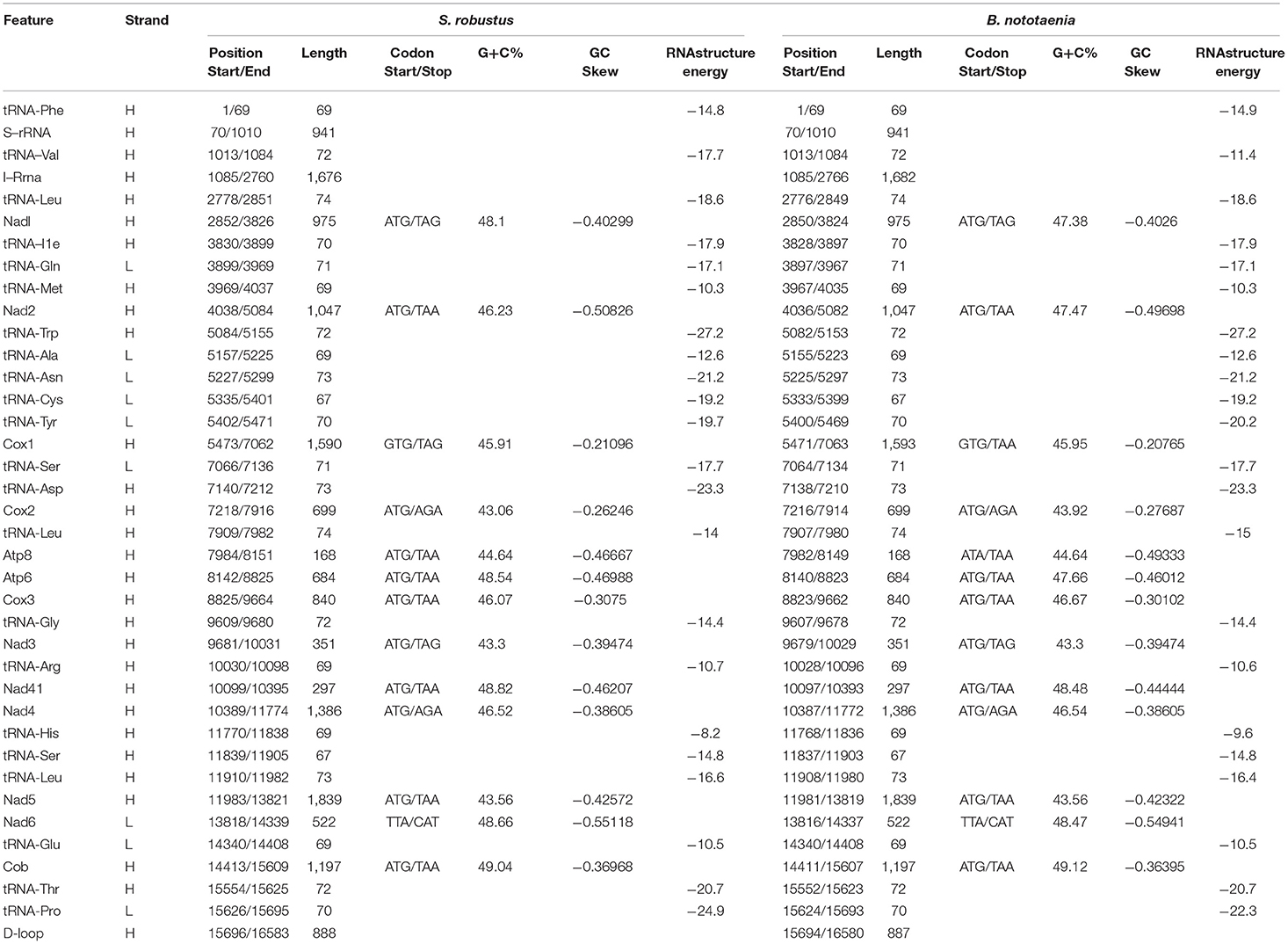
Table 1. Characteristic constituents of the mitochondrial genome organization of S. robustus and in B. nototaenia. H and L refer to the heavy and light strand, respectively.
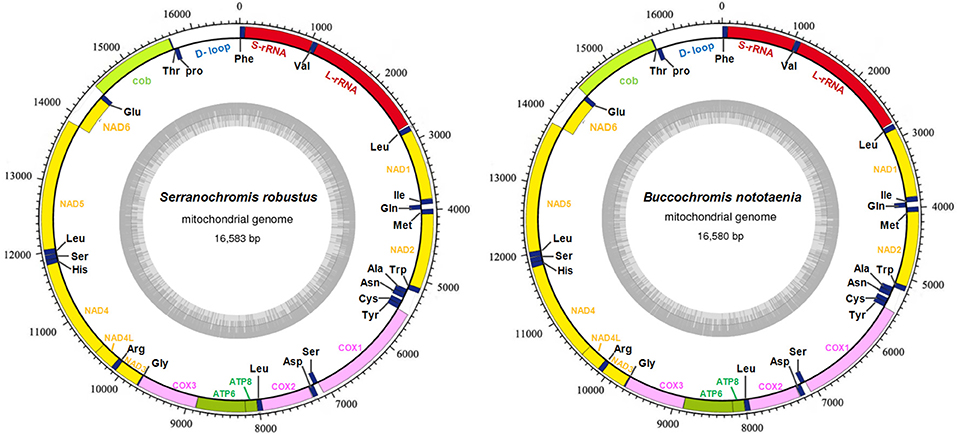
Figure 1. Circular map of the mitogenome of S. robustus and B. nototaenia. Genes encoded on the heavy or light strands are shown outside or inside the circular gene map, respectively, the size of the genome is 16,583 bp for S. robustus and 16,580 bp for B. nototaenia, where both contain the known 13 protein-coding genes of the respiratory chain (ND1, ND2, COX1, COX2, ATP8, ATP6, COX3, ND3, ND4L, ND4, ND5, ND6, and CYTB), 22 interspersed transfer RNA genes, 2 ribosomal RNA genes (12S and 16S rRNA) and one non-coding control region (D-loop region).
Protein Coding Genes
Protein-coding genes (PCGs) of the mitochondrial genome in S. robustus and B. nototaenia include 7 NDH dehydrogenase subunits (NAD1-6, NAD4l), 3 cytochrome c oxidase subunits (Cox1-3), 2 ATPase subunits (ATP6, ATP8), and one cytochrome b gene (Cob), ranging in size from 168 bp (ATPase8) to 1,839 bp (ND5). Frequently methionine (ATG) is the start codon for most PCGs genes, except for cox1, which utilizes GTG; this is an accepted canonical mitochondrial start codon for vertebrate mitogenomes (Yue et al., 2006; Yan et al., 2016; Yang et al., 2016). The termination codons of the 13 PCG can be classified into distinct types, the stop codons (TAA, AGA, CAT, TTA, and TAG) are utilized in these PCGs, seven PCGs (nd2, atp8, atp6, cox3, nad4l, nd5, cob) are terminated with the typical stop codon TAA, while two PCGs (ND1, ND2) has, (COX2, ND4) has AGA stop codon and (ND6) has stop codon in both species and (COX1) has stop codon in S. robustus, TAA in B. nototaenia. The detection of various stop codon types is common among vertebrate mitochondrial genome, and TAA stop codon appears via post-transcription polyadenylation (Ojala et al., 1981). Twelve of the 13 PCGs are encoded on H-stand, whereas only the nd6 gene is encoded on the L-strand (Table 1). Four reading-frame overlaps were observed in the two species. In S. robustus, 10 nucleotides overlap between ATP8 and ATP6, seven nucleotides overlap between ND4L and ND4, four nucleotides between ND5 and ND6 (opposite strands), and one nucleotide overlap between ATP6 and COX3. The same overlaps were also observed in B. nototaenia. The overlapping of protein-coding genes suggests partial sharing of transcripts among neighboring coding regions, which is commonly found in bony fishes (Montoya et al., 1983).
The total nucleotide length of the 13 PCGs is 11,595 bp long for S. robustus, accounts for 69. 92% of the whole lengths, and 11, 598 bp long for B. nototaenia accounting for 69.95% of the whole length. The AT and GC skew values of the PCGs of the two species are shown in Table 2. The A+T base composition is 54.22% for S. robustus and 54.07% for B. nototaenia, respectively. These values are higher than the G+C base compositions (45.78% in S. robustus and 45.93% in B. nototaenia). Additionally, the AT skew (0.0181) for the S. robustus mitogenome and (0.0163) B. nototaenia mitogenome are sliga greater occurrence of As to Ts, and its GC skew (−0.3153) and (−0.3109), respectively are negative, indicating a higher content of Cs than Gs. Among 13 PCGs examined in both species, the length of ND5 gene (1,839 bp) is the longest, whereas the shortest is ATP8 gene (168 bp) (Table 1). Both species contain nucleotide G least frequently in the third codon position. Our result indicates that more Ts and Cs are present in most PCGs, which is consistent with most previous observations (Hwang et al., 2013).
Table 2. List of species used in this study and their corresponding accession numbers included in phylogenetic and comparative analyses.
Transfer RNA (tRNA) and Ribosomal RNA (rRNA)
The secondary structures of 22 tRNA genes (typical clover-leaf secondary structure, including three for Leucine, two for Serine and one for each of the other amino acids) in the two fish mitogenomes are showed in Figures 2 and 3. The H-strand encodes Fourteen tRNAs, and the remaining 8 tRNAs are encoded by the L-strand (Table 1). All tRNAs varied in size from 67 bp (tRNAcys) to 74 bp (tRNAlue) in both species. This tRNA genomic, the molecular structural design is similar in most fish species ever examined such as L. microptera and C. kumu (Cui et al., 2017). Eleven tRNA genes in B. nototaenia species namely; {Ser (7064-7134), Gly (9607-9678), Arg (10028-10096), Phe (1-69), Trp (5082-5153), Gln (3897-3967), Ala (5155-5253), and Tyr (5400-5469)} and nine tRNA genes in S. robustus species namely; {Ser (7066-7136), Gly (9609-9680), Arg (10030-10098), Trp (5084-5155), Tyr (5402-5471), Gln (3899-3969), Ala (5157-5255), Leu (11910-11982), and Val (1013-1084)}, folded into typical clover-leaf secondary structures created by the regions of self-complementarity within tRNA (Figure 2). The data obtained further indicate that all the 22 tRNA genes in both species lack variable loop (Mohanta et al., 2017). The absence of D-arm (D-stem and D-loop) in the secondary structure of these transfer RNA {His (11768-11836), Leu (7907-7980), Asn and (5225-5297)} and [His (11770-11838), and Asn (5227-5299)] in B. nototaenia and S. robustus respectively, invariably altered their recognition potentials (Hardt et al., 1993). The formation of tRNA ribosomal complex by Val (1013-1084) in B. nototaenia and Asn (5227-5299) and Leu (7909-7982) in S. robustus during protein biosynthesis and translation will be greatly hindered because of the absence T-arm (T-stem and T-loop) in their secondary structures, since it serves as specialized recognition site (region) in the ribosome. Organisms with tRNA lacking T-loop exhibit a much lower level of aminoacylation and EF-TU-binding than in organisms which have the native tRNA (Griffith et al., 1999).
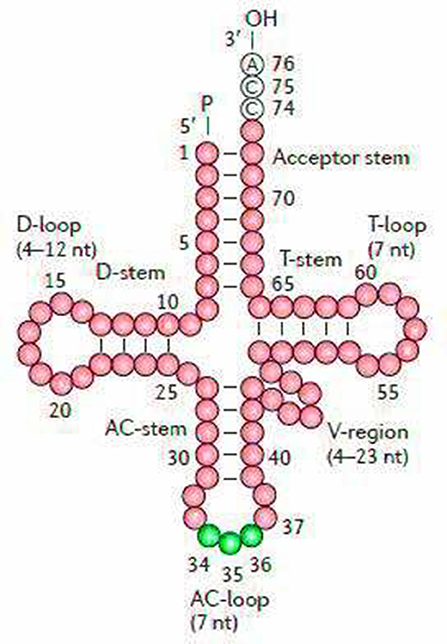
Figure 2. Clover leaf-like structure of tRNA. The tRNA possess the acceptor arm (7 nt), D-arm (3–4 nt), D-loop (4–12 nt), anti-codon arm (5 nt), anti-codon loop (7 nt), variable region (4–23 nt), 9-arm (5 nt), and 9-loop (7 nt). The D-arm, D-loop, and variable region possess variable number of nucleotides whereas the nucleotide number in the acceptor arm, anti-codon arm, anti-codon loop, 9-arm, and 9-loop is always constant.
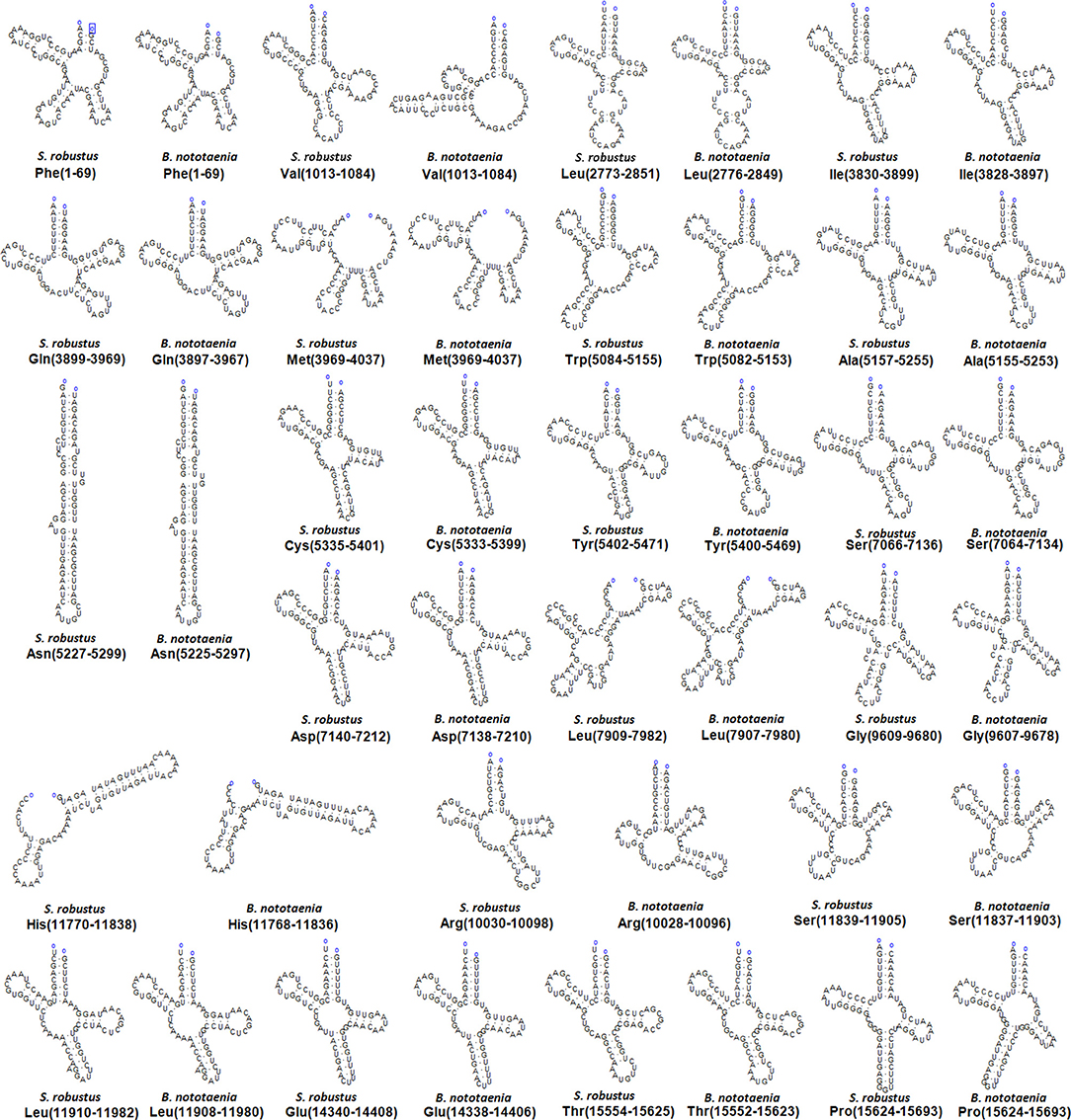
Figure 3. Secondary structures for the tRNA genes in the S. robustus and B. nototania mitogenome.
However, the presence of unpaired 3 bases in the T-loops of {Pro (15624-15693), Cys (5333-5399), and Leu (2776-2849)} and {Cys (5335-5401), and Leu (2778-2851)} in both B. nototaenia and S. robustus, respectively, shows high degree of instability in their T-loops, since the optimal loop length of stability of the T-loop is 7 base pair long (Mohanta et al., 2017). The absence of acceptor's arm in the secondary structures of these transfer RNAs {His (11768-11836) and Met (3967-4035)} and {His (11770-11838), Leu (7909-7982), and Met (3969-4037)} in B. nototeania and S. robustus, respectively and Phe (1-69) with short acceptor arm of 2-base long in S. rubustus may affect their aminoacylation (Schimmel et al., 1993). The presence of unpaired 3-bases in the Anticodon-loops of {Glu (14338-14406), Asp (7138-7210), Pro (15624-15693), Ile (3828-3897), and Cys (5333-5399)} and {Glu(14340-14408), Asp (7140-7212), Pro (15626-15695), Ile (3830-3899), and Cys (5335-5401)} in both B.nototaenia and S. robustus, respectively show the degree of instability in their anticodon-loops, since the optimal loop length for stability of the Anticodon loop is 5 base pair long (Mohanta et al., 2017). The presence of two unusual tRNAs secondary structures, one with 4-loops and another without the T-arm and D loops {Leu (7907-7980), and Asn (5225-5297)} and {Leu (7909-7982), and Asn (5227-5299)} were identified in both B. nototaenia and S. robustus, respectively. The average A+T content of tRNA is 54.07% for B. nototaenia and 54.22% for S. robustus (Figures 4, 5). The mitochondrial genomes of S. robustus and B. nototaenia contain two subunits of ribosomal RNA, a small one (s-rRNA) and a large one (l-rRNA) (Figure 6). The two subunits were encoded in the same H strand, like other typical fishes and are separated by tRNAval. The length of the s-rRNA gene is 941 bp long in the two species; the length of the l-rRNA gene is 1,676 bp long for S. robustus and 1,682 bp long for B. nototaenia.
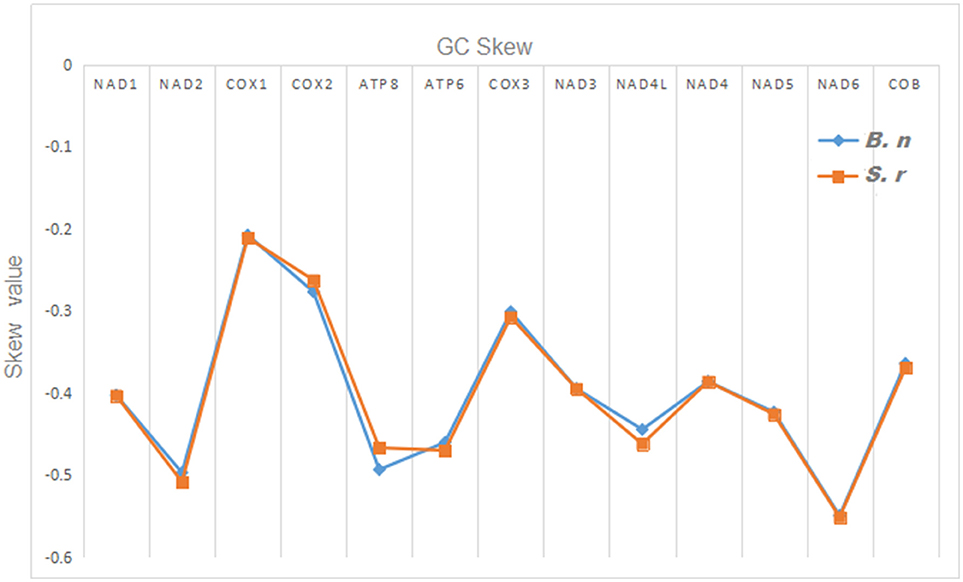
Figure 4. Graphical illustration showing the GC skew in the PCGs of the mitochondrial genome of S. robustus (S.r) and B. nototaenia (B.n).
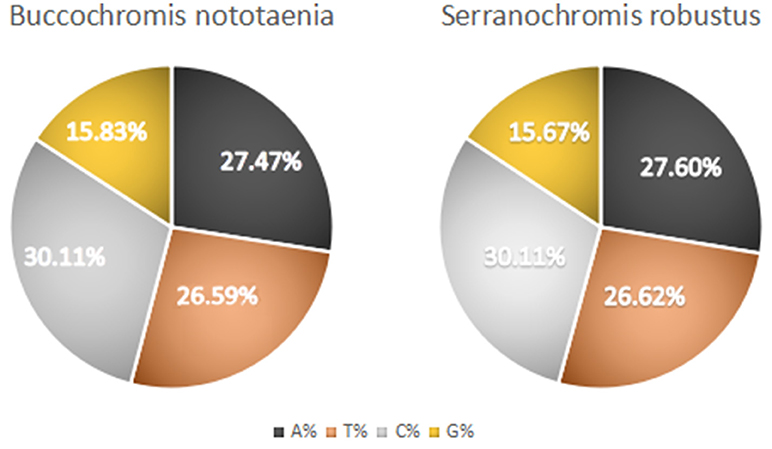
Figure 5. Base composition of the mitochondrial genome.
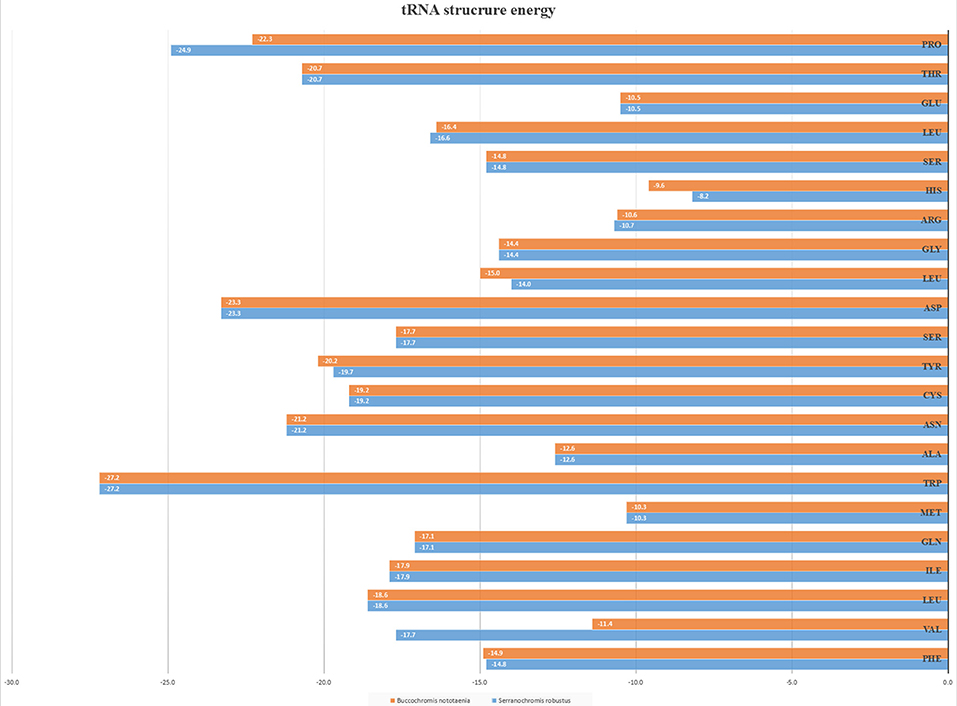
Figure 6. RNA structure energy strand.
Non-coding Regions
The non-coding regions in the mitochondrial genomes of S. robustus and B. nototaenia were flanked by tRNApro and tRNAthr genes (Table 1). This present observation is quite different from most typical mitogenome, in which non-coding (D-loop) region were located between tRNApro and tRNAphe genes (Lee et al., 1995; Yue et al., 2006). In this present study, the D-loop control regions were determined to be 888 and 887 bp in length for S. robustus and B. nototaenia, respectively. The non-coding region of both species is in agreement with observations in other species, with the D-Loop region divided into three primary domains. The first domain is hypervariable and consists of a termination-associated sequence (TAS), the second domain is the central conserved region, and the third domain comprises of three conserved blocks (CSB1, CSB2, and CSB3) (Kartavtsev et al., 2007).
Phylogenetic Analysis
To confirm the evolutionary position of S. robustus and B. nototaenia, we built a phylogenetic tree of 15 species of Cichlidae using published mitogenomes based on the concatenated nucleotide alignment of 13 PCGs via BI and ML methods (Figure 7 and Table 3). The results provide an excellent support for the monophyly of each family. This topology is mainly consistent with previously reported phylogenetic studies (Kartavtsev et al., 2007; Chen et al., 2017). The phylogenetic analysis using 13 concatenated mitochondrial protein-coding genes indicates that S. robustus and B. nototaenia had a broad phylogenetic relationship. However, in comparison with other 15 species of phylum chordate, {(L. lethrinus, H. oxyrhyncha, F. rostratus, B. nototaenia, C. quadrimaculatus, P. longimanus, S. robustus, A. geoffreyi, and C. zilli)} clustered in family Cichlidae, S. robustus and A. geoffreyi formed an independent monophyletic clade; therefore, the relationship between S. robustus and A. geoffreyi calls for further investigation. In further comparison with other established mitogenomes of C. quadrimaculatus and P. longimanus, our findings showed a close relationship between B. nototaenia, C. quadrimaculatus, and P. longimanus and a lineage of S. robustus and A. geoffreyi relatively distinct from B. nototaenia. This result is concordant with the evolutionary relationships inferred based on phylogenetic analysis.
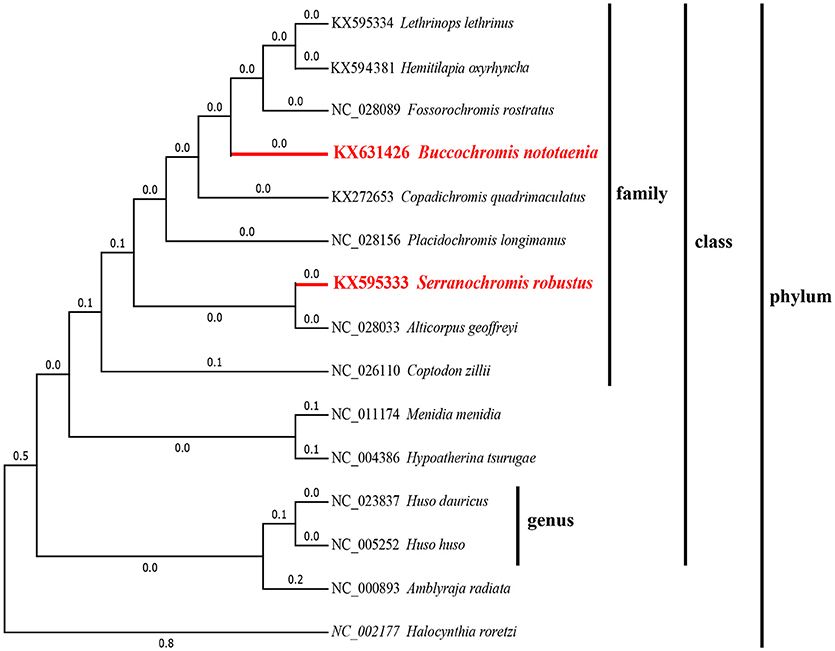
Figure 7. Phylogenetic trees inferred from amino acid and nucleotide sequences of 13 PCGs of the mitogenome. The phylogenetic analyses were conducted with maximum likelihood (ML) and Bayesian inference (BI). The numbers in front of the species are GenBank accession numbers.

Table 3. Base composition of AT and GC skew of the mitochondrial genome.
Conclusion
The mitogenomes sequences of the two species (S. robustus and B. nototaenia) from the family Cichlidae were determined and compared alongside with those of other Chordata Species. Their complete mitogenomes indicate typical circular molecules and had similar genome organization and structure as those found in other Cichlids species. The length of the mitogenome sequences of S. robustus and B. nototaenia were 16,583 and 16,580 bp, respectively. Each mitogenome consists of a typical structure of 13 PCGs, 2 rRNAs, 22 tRNA genes, and one non-coding region. Similar to other vertebrate mitogenomes, most of the PCGs utilized as their initiation codons, except for cox1 which utilizes GTG. Additionally, 11 tRNA genes in B. nototaenia and 9 tRNA genes in S. robustus species, folded correctly into typical clover-leaf secondary structures created by the regions of self-complementarity with the exception of {His (11768-11836) and Met (3967-4035)} and {His (11770-11838), Leu (7909-7982), and Met (3969-4037)} in B. nototeania and S. robustus, respectively that lacked the acceptor arm and Phe (1-69) with short acceptor arm of 2-base long in S. rubustus. The presence of two unusual tRNAs secondary structures, one with 4-loops and another without the T-arm and D-loop {Leu (7907-7980), and Asn (5225-5297)} and {Leu (7909-7982), and Asn (5227-5299)} were identified in both B. nototaenia and S. robustus, respectively. All the 22 tRNA genes in both species lacked variable loop. The phylogenetic analysis using 13 concatenated mitochondrial protein-coding genes indicates that S. robustus and B. nototaenia had wide phylogenetic relationship. Comparison with other established mitogenomes suggests a close relationship between B. nototaenia, C. quadrimaculatus, and P. longimanus and a lineage of S. robustus and A. geoffreyi relatively distinct from B. nototaenia.
Author Contributions
XM, ZL, YS, and DQ conceived and designed the study. MC, AW, LW, NA, CW, ML, MM, and LA conducted the molecular work and data analysis. NA drafted the manuscript. YC, SD, CF, AL, SZ, XC, and YG prepared all figures and tables. ML and LA performed the phylogenetic analyses. EO and ZL analyzed the tRNA and contributed in drafting the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was sponsored by the National Natural Science Foundation of China (NO. 81500977), National Science Foundation of Jiangsu Province (NO. BK20161154) and and the Key Laboratory of Brain Disease Bioinformation of Jiangsu Province (No. JSBL1505). We thank welcome Trust Sanger Institute (SC) for generating the raw sequencing dates in this study.
Abbreviations
PCGs, protein-coding genes; A, adenine; T, thymine; G, guanine; C, cytosine; ML, maximum likelihood; BI, Bayesian inference; BP, Base pair; mtDNA, mitochondrial DNA; Atp6 and Atp8, genes for the ATPase subunits 6 and 8; Cox1-cox3, genes for cytochrome C oxidase subunits I–III; Nad1–nad6 and nad4L, genes for NADH dehydrogenase subunits 1–6 and 4L; rRNA, ribosomal RNA genes subunit; l-rRNA (large), rRNA subunit; s-rRNA, (small); tRNAx, transfer RNA, where X is replaced by three letters amino acid code of the corresponding amino acid.
References
Abascal, F., Zardoya, R., and Posada, D. (2005). ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105. doi: 10.1093/bioinformatics/bti263
Beier, B. A., Nylander, J. A., Chase, M. W., and Thulin, M. (2004). Phylogenetic relationships and biogeography of the desert plant genus Fagonia (Zygophyllaceae), inferred by parsimony and Bayesian model averaging. Mol. Phylogenet. Evol. 33, 91–108. doi: 10.1016/j.ympev.2004.05.010
Bentzen, P., Wright, J. M., Bryden, L. T., Sargent, M., and Zwanenburg, K. C. (1998). Tandem repeat polymorphism and heteroplasmy in the mitochondrial control region of redfishes (Sebastes: Scorpaenidae). J. Hered. 89, 1–7.
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. doi: 10.1016/S0891-5849(00)00317-8
Chen, S., Shi, P., Zhang, D., Lu, Z., and Li, L. (2017). Complete mitochondrial genome of Hepialus xiaojinensis (Lepidoptera: Hepialidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 28, 305–306. doi: 10.3109/19401736.2015.1118089
Cline, S. D. (2012). Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 1819, 979–991. doi: 10.1016/j.bbagrm.2012.06.002
Cui, L., Dong, Y., Liu, F., Gao, X., Zhang, H., Li, L., et al. (2017). The first two complete mitochondrial genomes for the family Triglidae and implications for the higher phylogeny of Scorpaeniformes. Sci. Rep. 7:1553. doi: 10.1038/s41598-017-01654-y
Galtier, N., Nabholz, B., Glemin, S., and Hurst, G. D. (2009). Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol. Ecol. 18, 4541–4550. doi: 10.1111/j.1365-294X.2009.04380.x
Graeber, M. B., and Muller, U. (1998). Recent developments in the molecular genetics of mitochondrial disorders. J. Neurol. Sci. 153, 251–263.
Griffith, J. D., Comeau, L., Rosenfield, S., Stansel, R. M., Bianchi, A., Moss, H., et al. (1999). Mammalian telomeres end in a large duplex loop. Cell 97, 503–514.
Hardt, W. D., Schlegl, J., Erdmann, V. A., and Hartmann, R. K. (1993). Role of the D arm and the anticodon arm in tRNA recognition by eubacterial and eukaryotic RNase P enzymes. Biochemistry 32, 13046–13053.
Hartvigsen, R., and Halvorsen, O. (1994). Spatial patterns in the abundance and distribution of parasites of freshwater fish. Parasitol. Today 10, 28–31.
Hwang, D. S., Byeon, H. K., and Lee, J. S. (2013). Complete mitochondrial genome of the freshwater sculpin Cottus koreanus (Scorpaeniformes, Cottidae). Mitochondrial DNA 24, 490–491. doi: 10.3109/19401736.2013.770500
Jellum, M. D. (1970). Plant introductions of maize as a source of oil with unusual fatty acid composition. J. Agric. Food Chem. 18, 365–370.
Kartavtsev, Y. P., Jung, S. O., Lee, Y. M., Byeon, H. K., and Lee, J. S. (2007). Complete mitochondrial genome of the bullhead torrent catfish, Liobagrus obesus (Siluriformes, Amblycipididae): genome description and phylogenetic considerations inferred from the Cyt b and 16S rRNA genes. Gene 396, 13–27. doi: 10.1016/j.gene.2007.01.027
Khuda-Bukhsh, A. R., and Chakrabarti, C. (2000). Differential C-heterochromatin distribution in two species of freshwater fish, Anabas testudineus (Bloch.) and Puntius sarana (Hamilton.). Indian J. Exp. Biol. 38, 265–268.
Klett, V., and Meyer, A. (2002). What, if anything, is a Tilapia?-mitochondrial ND2 phylogeny of tilapiines and the evolution of parental care systems in the African cichlid fishes. Mol. Biol. Evol. 19, 865–883. doi: 10.1093/oxfordjournals.molbev.a004144
Kocher, T. D. (2004). Adaptive evolution and explosive speciation: the cichlid fish model. Nat. Rev. Genet. 5, 288–298. doi: 10.1038/nrg1316
Kroemer, G., Dallaporta, B., and Resche-Rigon, M. (1998). The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60, 619–642. doi: 10.1146/annurev.physiol.60.1.619
Kuraku, S., and Meyer, A. (2008). Genomic analysis of cichlid fish 'natural mutants'. Curr. Opin. Genet. Dev. 18, 551–558. doi: 10.1016/j.gde.2008.11.002
Lagouge, M., and Larsson, N. G. (2013). The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 273:529–543. doi: 10.1111/joim.12055
Larsson, N. G. (2010). Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 79, 683–706. doi: 10.1146/annurev-biochem-060408-093701
Lee, W. J., Conroy, J., Howell, W. H., and Kocher, T. D. (1995). Structure and evolution of teleost mitochondrial control regions. J. Mol. Evol. 41, 54–66.
Lewis, P. O., Kumar, S., Tamura, K., Nei, M., and Lewis, P. O. (1994). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Bioinformatics 30, 2725–2729.
Liu, L., He, J., Li, N., Fu, X., Lin, Q., Shi, C., et al. (2016). Complete mitochondrial genome of Parachromis managuensis (Perciformes: Cichlidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 2533–2534. doi: 10.3109/19401736.2015.1038791
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964.
Mathews, D. H. (2014). Using the RNAstructure software package to predict conserved RNA structures. Curr. Protoc. Bioinformatics 46:12 14 11–22. doi: 10.1002/0471250953.bi1204s46
Miya, M., and Nishida, M. (1999). Organization of the mitochondrial genome of a deep-sea fish, Gonostoma gracile (Teleostei: Stomiiformes): first example of transfer RNA gene rearrangements in bony fishes. Mar. Biotechnol. 1, 416–0426.
Mohanta, T. K., Syed, A. S., Ameen, F., and Bae, H. (2017). Novel genomic and evolutionary perspective of cyanobacterial tRNAs. Front. Genet. 8:200. doi: 10.3389/fgene.2017.00200
Montoya, J., Gaines, G. L., and Attardi, G. (1983). The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 34, 151–159.
Ojala, D., Montoya, J., and Attardi, G. (1981). tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474.
Perna, N. T., and Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358.
Pulling, C. A. (1991). The relationship between critical care nurses' knowledge about sleep, and the initiation of sleep promoting nursing interventions. Axone 13, 57–62.
Qi, D. S., Zhang, S. C., Jiang, J., Zhang, L. Q., Fu, Y. Y., Du, Z. M., et al. (2016). The complete mitochondrial genome of the Fossorochromis rostratus. Mitochondrial DNA A DNA Mapp. Seq. Anal. 27, 4284–4285. doi: 10.3109/19401736.2015.1082097
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Salzburger, W., and Meyer, A. (2004). The species flocks of East African cichlid fishes: recent advances in molecular phylogenetics and population genetics. Naturwissenschaften 91, 277–290. doi: 10.1007/s00114-004-0528-6
Schimmel, P., Giege, R., Moras, D., and Yokoyama, S. (1993). An operational RNA code for amino acids and possible relationship to genetic code. Proc. Natl. Acad. Sci. U.S.A. 90, 8763–8768.
Seehausen, O. (2006). African cichlid fish: a model system in adaptive radiation research. Proc. Biol. Sci. 273, 1987–1998. doi: 10.1098/rspb.2006.3539
Silvestro, D., and Michalak, I. (2012). raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12, 1–3. doi: 10.1007/s13127-011-0056-0
Smeitink, J., van den Heuvel, L., and DiMauro, S. (2001). The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2, 342–352. doi: 10.1038/35072063
Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2002). Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics Chapter 2: Unit 2 3. doi: 10.1002/0471250953.bi0203s00
Turner, G. F. (2007). Adaptive radiation of cichlid fish. Curr. Biol. 17, R827–R831. doi: 10.1016/j.cub.2007.07.026
Wei, Y. H. (1998). Oxidative stress and mitochondrial DNA mutations in human aging. Proc. Soc. Exp. Biol. Med. 217, 53–63.
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yan, W., Xiao, X., and Zhang, Y. (2016). Complete genome sequence of the Sporosarcina psychrophila DSM 6497, a psychrophilic Bacillus strain that mediates the calcium carbonate precipitation. J. Biotechnol. 226, 14–15. doi: 10.1016/j.jbiotec.2016.03.036
Yang, X., Wang, L., Feng, H., Qi, M., Zhang, Z., Gao, C., et al. (2016). Characterization of the complete mitochondrial genome sequence of Homalogaster paloniae (Gastrodiscidae, Trematoda) and comparative analyses with selected digeneans. Parasitol. Res. 115, 3941–3949. doi: 10.1007/s00436-016-5160-9
Yue, G. H., Liew, W. C., and Orban, L. (2006). The complete mitochondrial genome of a basal teleost, the Asian arowana (Scleropages formosus, Osteoglossidae). BMC Genomics 7:242. doi: 10.1186/1471-2164-7-242
Keywords: mitochondrial genome, Serranochromis robustus, Buccochromis nototaenia, development application, tRNA
Citation: Alexander Kenechukwu N, Li M, An L, Cui M, Wang C, Wang A, Chen Y, Du S, Feng C, Zhong S, Gao Y, Cao X, Wang L, Obinna EM, Mei X, Song Y, Li Z and Qi D (2019) Comparative Analysis of the Complete Mitochondrial Genomes for Development Application. Front. Genet. 9:651. doi: 10.3389/fgene.2018.00651
Received: 07 April 2018; Accepted: 30 November 2018;
Published: 06 March 2019.
Edited by:
Michael Rossbach, Vela Diagnostics, GermanyReviewed by:
Paola De Sessions, Genome Institute of Singapore, SingaporeShalom Chinedu, Covenant University, Nigeria
Copyright © 2019 Alexander Kenechukwu, Li, An, Cui, Wang, Wang, Chen, Du, Feng, Zhong, Gao, Cao, Wang, Obinna, Mei, Song, Li and Qi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinyu Mei, bXh5MjAxNTEyQDE2My5jb20=
Yuanjian Song, Ymlvc29uZ3l1YW5qaWFuQDEyNi5jb20=
Zongyun Li, em9uZ3l1bmxpQGpzbnUuZWR1LmNu
Dashi Qi, cWlkYXNoaUB4emhtdS5lZHUuY24=
†These authors have contributed equally to this work