 Corina Anastasaki1
Corina Anastasaki1 David H. Gutmann
David H. Gutmann- 1Department of Neurology, Washington University School of Medicine, St. Louis, MO, United States
- 2Department of Surgery, Washington University School of Medicine, St. Louis, MO, United States
A Commentary on:
Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I
by Xu, M., Xiong, H., Han, Y., Li, C., Mai, S., Huang, Z., et al. (2018). Front. Genet. 9:270. doi: 10.3389/fgene.2018.00270
As we begin to consider risk assessment strategies critical to the implementation of precision medicine, it becomes important to identify and evaluate clinically actionable genotype-phenotype associations. Germane to the future management of children with the tumor predisposition syndrome, Neurofibromatosis type 1 (NF1; MIM #162200), the recent report by Xu et al. demonstrates a positive correlation between optic pathway glioma (OPG) and NF1 gene mutation location (Xu et al., 2018). In clinical practice, only 15–20% of these at-risk children with NF1 develop OPGs, low-grade astrocytic tumors of the optic nerve, chiasm, tracts and radiations, which can lead to visual, endocrine, and neurological deficits (Listernick et al., 2007). Currently, the lack of biomarkers predictive of OPG development limits our ability to provide prognostic information to these children and their families.
One such clinically actionable biomarker is the germline NF1 gene mutation. While NF1 is caused by a germline mutation in the NF1 gene, the large number of unique NF1 mutations identified, as well as their lack of spatial clustering, has led to the conclusion that all NF1 mutations are functionally identical. This notion of “mutational equivalency” has been recently challenged by converging data from population-based (Upadhyaya et al., 2007; Pinna et al., 2015; Anastasaki et al., 2017; Kehrer-Sawatzki et al., 2017; Koczkowska et al., 2018a,b; Morris and Gutmann, 2018), human induced pluripotent stem cell (Anastasaki et al., 2015), and genetically engineered mouse studies (Li et al., 2016; Toonen et al., 2016), which have each demonstrated striking differences in the effects of the germline NF1 gene mutation on clinical phenotypes. For instance, large genomic deletions and missense mutations in codons 844-848 are associated with more severe clinical phenotypes in NF1 patients (Kehrer-Sawatzki et al., 2017; Koczkowska et al., 2018b). Conversely, patients with mutations Arg1809Cys and Met992del do not develop dermal and plexiform neurofibromas (Upadhyaya et al., 2007; Pinna et al., 2015).
Adding to these findings, Xu et al. identified two regions of NF1 mutation clustering: (1) the cysteine/serine-rich domain (CSRD, residues 543–909), which was positively associated with OPG, and (2) the HEAT-like repeat region (HLR, residues 1,825–2,428), which negatively correlated with OPG. Additionally, the authors conclude that patients with OPGs were not more likely to harbor mutations within the 5′-tertile of the NF1 gene. Their elegant analysis prompted us to extend our prior analyses using a larger series of previously published and clinically-characterized cohorts of patients with NF1, including their subjects, now with sufficient statistical power to detect potential genotype-phenotype correlations.
To determine whether mutations within specific predicted functional regions were associated with a higher risk of OPG, we employed a combined cohort of 310 NF1 patients who underwent brain magnetic resonance imaging and harbored known NF1 gene (Anastasaki et al., 2017). Using these radiographic criteria, 101 patients harbored an OPG (33%) and 209 did not (67%; no OPG at ≥10 years of age). We then segmented the NF1 gene into nine regions, as described by Xu et al. (CSRD, TBD, GRD, Sec14/PH, HLR, NLS, SBR, CTD and “other” to signify the six regions in between the predicted functional domains), and scored the number of patients with a germline mutation in each region. A separate χ2 test (2 × 2 table) was performed at each of these segments to compare the rates of mutation between patients with and without OPG. As such, we calculated the odds ratios (OR) of presenting with an OPG when the NF1 germline mutation falls within one of the nine described regions, and additionally analyzed the associated sensitivity and specificity for each region. We did not compare each of the regions with the rest of the eight regions. The objective of the manuscript was to confirm associations suggested by previous studies by analyzing the candidate hypotheses, rather than by performing a completely new exploratory analysis. In this regard, we were more concerned about type II errors, especially given the limited sample size caused by the relative rareness of the disease. An adjustment, such as the Bonferroni correction, would reduce type I errors at the expense of increasing type II errors (Rothman, 1990, 2014). Therefore, we did not adjust the resultant p-values for multiple comparisons. Using this approach, we confirmed that patients with OPG in this larger cohort were more likely to harbor mutations within the CSRD domain (OR 2.05; p = 0.028). Conversely, NF1 patients were less likely to develop an OPG if they harbored mutations within the HLR domain (OR = 0.36, p = 0.013) (Table 1A). Importantly, this finding persists (CSRD: OR = 2.21, p = 0.005; HLR: OR = 0.4, p = 0.009) when the analysis was repeated to include the additional 26 OPG (total n = 127) and 45 no-OPG (total n = 254) patients from the Xu study (Table 1B), as well as when the analyses were only performed to include truncating NF1 mutations (data not shown).
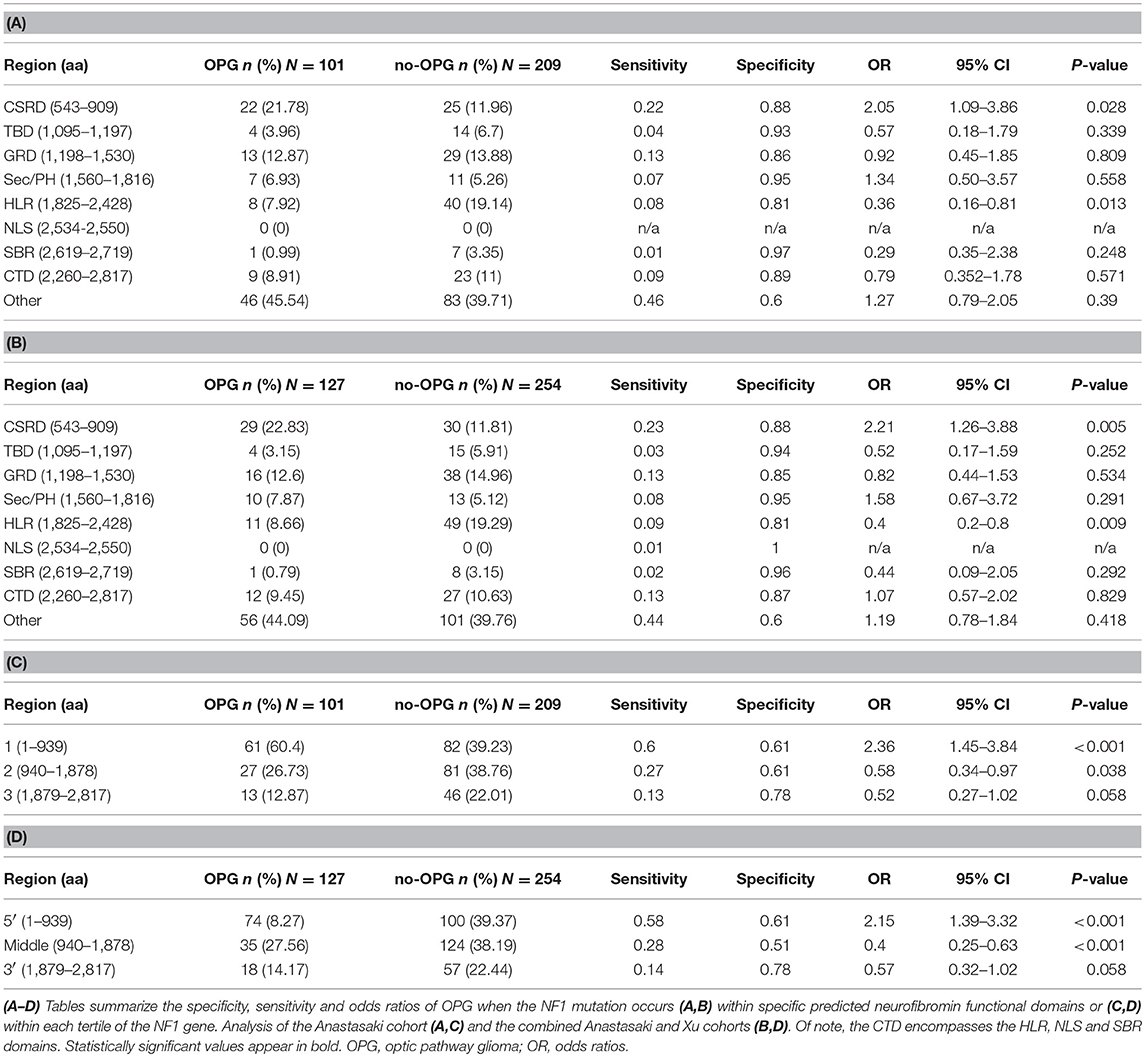
Table 1. Risk of OPG varies with mutations in different regions of the NF1 gene.
We next segmented the NF1 gene into three tertiles (5′-tertile, residues 1–939; middle tertile, residues 940–1,878; 3′-tertile, residues 1,879–2,817), and performed similar χ2 OR analyses to establish whether there was mutational clustering in the 5′-tertile of the NF1 gene in patients with OPG. We have previously reported that cohorts smaller than 307 patients are not sufficiently powered to detect differences in mutational localization across the NF1 locus (Anastasaki et al., 2017). As such, children with OPGs were more likely to harbor mutations in the 5′-tertile in the Anastasaki dataset only (OR = 2.36, p < 0.001, n = 310; Table 1C) or in the combined Anastasaki and Xu dataset (OR = 2.15, p < 0.001, n = 381; Table 1D), but not in the Xu dataset alone (n = 215). Moreover, mutations in the middle tertile of the NF1 gene were associated with a lower risk of OPG both in the Anastasaki dataset (OR = 0.58, p = 0.038) and in the combined dataset (OR = 0.4; p < 0.001), albeit with low sensitivity (0.27 and 0.28, respectively) (Tables 1C,D).
Together with the new data presented herein, the findings reported by Xu et al. have several important implications. First, they underscore the importance of studying a large clinical population in order to generate sufficient statistical power to discover accurate genotype-phenotype associations. Second, they validate the 5′-tertile mutation clustering in NF1 patients with OPG, with mutations preferentially occurring specifically within the CSRD domain. Third, individuals with NF1-OPG are less likely to harbor mutations within the HLR domain, a region associated with increased autism symptomatology (Morris and Gutmann, 2018). Fourth, with domains both upstream and downstream of the only known functional domain (RAS-GAP domain) of the NF1 protein (neurofibromin) exhibiting differential associations with OPG development, it becomes increasingly important to explore additional biological functions for neurofibromin. This is particularly important for a disease in which RAS/RAS effector drugs have been the primary focus of targeted treatment approaches, with variable clinical efficacy. Finally, as these associations lack sufficient sensitivity and the mutation locations are only correlative, future studies should focus on multi-risk factor evaluation relevant to clinical OPG prognostic assessments for children with NF1.
Author Contributions
CA performed the analyses; FG performed secondary statistical analyses; CA and DG wrote the manuscript.
Funding
This work was supported by an R50 Research Specialist Award (1 R50 CA 233164-01 to CA) and a Research Program Award grant from the National Institutes of Health (1-R35-NS07211-01 to DG).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anastasaki, C., Morris, S. M., Gao, F., and Gutmann, D. H. (2017). Children with 5′-end NF1 gene mutations are more likely to have glioma. Neurol. Genet. 3:e192. doi: 10.1212/NXG.0000000000000192
Anastasaki, C., Woo, A. S., Messiaen, L. M., and Gutmann, D. H. (2015). Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 24, 3518–3528. doi: 10.1093/hmg/ddv103
Kehrer-Sawatzki, H., Mautner, V. F., and Cooper, D. N. (2017). Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 136, 349–376. doi: 10.1007/s00439-017-1766-y
Koczkowska, M., Callens, T., Gomes, A., Sharp, A., Chen, Y., Hicks, A. D., et al. (2018a). Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): an update of genotype-phenotype correlation. Genet Med. doi: 10.1038/s41436-018-0269-0 [Epub ahead of print].
Koczkowska, M., Chen, Y., Callens, T., Gomes, A., Sharp, A., Johnson, S., et al. (2018b). Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844-848. Am. J. Hum. Genet. 102, 69–87. doi: 10.1016/j.ajhg.2017.12.001
Li, K., Turner, A. N., Chen, M., Brosius, S. N., Schoeb, T. R., Messiaen, L. M., et al. (2016). Mice with missense and nonsense NF1 mutations display divergent phenotypes compared with human neurofibromatosis type I. Dis. Model. Mech. 9, 759–767. doi: 10.1242/dmm.025783
Listernick, R., Ferner, R. E., Liu, G. T., and Gutmann, D. H. (2007). Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann. Neurol. 61, 189–198. doi: 10.1002/ana.21107
Morris, S. M., and Gutmann, D. H. (2018). A genotype-phenotype correlation for quantitative autistic trait burden in neurofibromatosis 1. Neurology 90, 377–379. doi: 10.1212/WNL.0000000000005000
Pinna, V., Lanari, V., Daniele, P., Consoli, F., Agolini, E., Margiotti, K., et al. (2015). p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 23, 1068–1071. doi: 10.1038/ejhg.2014.243
Rothman, K. J. (1990). No adjustments are needed for multiple comparisons. Epidemiology 1, 43–46. doi: 10.1097/00001648-199001000-00010
Rothman, K. J. (2014). Six persistent research misconceptions. J. Gen. Intern. Med. 29, 1060–1064. doi: 10.1007/s11606-013-2755-z
Toonen, J. A., Anastasaki, C., Smithson, L. J., Gianino, S. M., Li, K., Kesterson, R. A., et al. (2016). NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum. Mol. Genet. 25, 1703–1713. doi: 10.1093/hmg/ddw039
Upadhyaya, M., Huson, S. M., Davies, M., Thomas, N., Chuzhanova, N., Giovannini, S., et al. (2007). An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 80, 140–151. doi: 10.1086/510781
Keywords: neurofibromatosis type 1, genotype-phenotype correlations, optic pathway glioma, mutation location, clinical risk assessment
Citation: Anastasaki C, Gao F and Gutmann DH (2019) Commentary: Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 10:115. doi: 10.3389/fgene.2019.00115
Received: 13 September 2018; Accepted: 31 January 2019;
Published: 01 March 2019.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Mirjana Kocova, Saints Cyril and Methodius University of Skopje, MacedoniaEric Pasmant, Université Paris Descartes, France
Nadia Chuzhanova, Nottingham Trent University, United Kingdom
Copyright © 2019 Anastasaki, Gao and Gutmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David H. Gutmann, Z3V0bWFubmRAd3VzdGwuZWR1 orcid.org/#0000-0002-3127-5045