 Gabrielle Wheway
Gabrielle Wheway Genomics England Research Consortium
Genomics England Research Consortium Hannah M. Mitchison
Hannah M. Mitchison- 1Human Development and Health, Faculty of Medicine, University of Southampton, Southampton General Hospital, Southampton, United Kingdom
- 2Genetics and Genomic Medicine, University College London, UCL Great Ormond Street Institute of Child Health, London, United Kingdom
Cilia are highly specialized cellular organelles that serve multiple functions in human development and health. Their central importance in the body is demonstrated by the occurrence of a diverse range of developmental disorders that arise from defects of cilia structure and function, caused by a range of different inherited mutations found in more than 150 different genes. Genetic analysis has rapidly advanced our understanding of the cell biological basis of ciliopathies over the past two decades, with more recent technological advances in genomics rapidly accelerating this progress. The 100,000 Genomes Project was launched in 2012 in the UK to improve diagnosis and future care for individuals affected by rare diseases like ciliopathies, through whole genome sequencing (WGS). In this review we discuss the potential promise and medical impact of WGS for ciliopathies and report on current progress of the 100,000 Genomes Project, reviewing the medical, technical and ethical challenges and opportunities that new, large scale initiatives such as this can offer.
The 100,000 Genomes Project
The launch of the UK's 100,000 Genomes project was announced in December 2012 as part of the UK's Life Sciences Strategy. This ambitious £300 million national project aimed to sequence 100,000 complete genomes from 70,000 individuals with cancer or rare disease, and their unaffected family members (Turnbull et al., 2018). Unlike other population genomics studies such as those in Iceland, Japan, Finland, Sweden and the Netherlands (An, 2017), the 100,000 Genomes Project is a hybrid clinical/research initiative, with an aim to fully integrate genomic testing for eligible individuals within existing routine healthcare pathways in the UK National Health Service (NHS). Sequence data is linked to longitudinal patient records such as hospital admissions and responses to interventions, providing a rich resource of genomic medical information. In many respects, it is the first scheme of its kind in the world, and one of the largest.
The project built upon the legacy of successful population healthcare genetics studies in the UK, such as the Deciphering Developmental Disorders (DDD) study (Wright et al., 2015) and UK10K (Consortium et al., 2015) which built upon the Avon Longitudinal Study of Parents And Children (ALSPAC) (Boyd et al., 2013) and TwinsUK (Moayyeri et al., 2013). It aims to secure the UK's position as a world-leader in healthcare and genomics.
Recruitment for the 100,000 Genome Project has been coordinated across 13 Genomic Medicine Centers set up across 85 NHS Trusts in England, Northern Ireland and Scotland. Of the estimated 8,000 rare diseases (Boycott et al., 2017), 190 were recruited to the Rare Disease pathway of the 100,000 Genomes Project, from 2015/16 until 31st September 2018. Eligible rare diseases were nominated by clinicians and researchers who felt there was an unmet clinical need, for example, diseases for which there are a sizeable proportion of patients with no known genetic diagnosis. The range of phenotypes and the proportion of patients with different disorders within the rare disease cohort is shown in Figure 1. Where possible, “trios” were recruited, i.e., the affected individual and both parents, with efforts made to recruit both affected and unaffected family members to allow effective variant filtration.
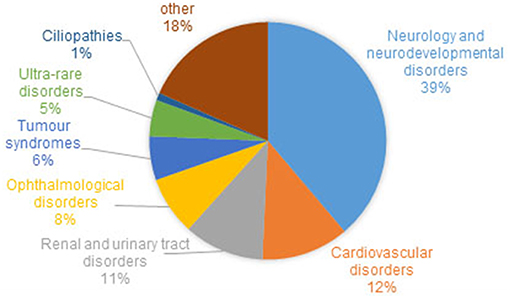
Figure 1. Proportion of families and individuals with different disorders within the rare disease cohort of the 100,000 Genomes Project. Around 1% of recruited families/individuals have a ciliopathy. Cohort summary data courtesy of Genomics England, with permission.
Genetic Basis of Ciliopathies—Diseases Affecting Cilia Structure And Function
Ciliopathies represent one group of rare diseases recruited to the 100,000 Genomes project. The ciliopathies are a diverse grouping but have a common etiology, originating in ciliogenesis (growth and maintenance) and/or structural or functional defects of the cilium (Reiter and Leroux, 2017). Cilia are highly evolved, complex organelles with important roles in motility and signaling (Waters and Beales, 2011; Mitchison and Valente, 2017; Wheway et al., 2018). The extent of the overall medical impact of ciliopathies, which individually most often are rare conditions but collectively affect a large number of individuals, has emerged over the last 20 years as their functions have continued to be revealed through genetic analysis in animal models and human families (Fliegauf et al., 2007; Goetz and Anderson, 2010).
Cilia can broadly be divided into motile and non-motile (primary), each with separate functions, both of which form a unique cellular compartment distinct from the rest of the cell. Primary cilia are more ubiquitous, distributed across most cell types of the body, extending out from the cell surface into their external environment as a single projection with diverse mechanosensory and chemosensory signal transduction roles (Singla and Reiter, 2006). The photoreceptors of the eye have a modified cilium, the outer segment (Wright et al., 2010). In contrast, motile cilia are found only on specialized epithelial surfaces and usually as multiple motile cilia (perhaps 200 per cell). Motile cilia lining notably the respiratory airways, brain ventricles and fallopian tubes, move essential body fluids and gametes, with a structural composition related to that of sperm flagella in male gamete movement (Spassky and Meunier, 2017). Single motile nodal cilia that exist transiently in the embryonic left-right organizer function in laterality determination (Hamada, 2016).
Owing to the central role of the cilium in development and health, ciliopathies often include complex multi-organ developmental phenotypes. The most severe-lethal ciliopathies, Meckel-Gruber syndrome (MKS) (Wheway and Johnson, 2014; Hartill et al., 2017), Joubert syndrome (JBTS) (Parisi and Glass, 1993; Bachmann-Gagescu et al., 2015), orofacial digital syndrome (OFD) (Gurrieri et al., 2007) as well as Bardet-Biedl syndrome (BBS) (Forsythe and Beales, 2013), all include neurodevelopmental features. These are often present alongside other features such as retinal dystrophy, renal dysplasia and skeletal abnormalities (Waters and Beales, 2011). Skeletal ciliopathies range from lethal short-rib thoracic dysplasias to more mild phenotypes such as Jeune syndrome (Huber and Cormier-Daire, 2012; Mitchison and Valente, 2017; Schmidts and Mitchison, 2018). Renal ciliopathies are some of the most common conditions associated with cilia defects. Autosomal polycystic kidney disease (ADPKD) alone affects between 1:2,500 and 1:4,000 individuals in the EU, making it one of the most common genetic diseases in humans, and the most common cause of end–stage renal failure (Willey et al., 2017). Renal malformations can also take the form of nephronophthisis, characterized by chronic tubulointerstitial nephritis, in isolated conditions or part of multiorgan ciliopathies including Senior-Loken syndrome (Wolf and Hildebrandt, 2011). Due to the importance of the highly specialized photoreceptor cilium in the function of the retina, many syndromic ciliopathies include a retinal dystrophy phenotype. Furthermore, around one third of non-syndromic inherited retinal dystrophies, such as retinitiris pigmentosa (RP) and Leber congenital amaurosis (LCA), are associated with a retinal cilium defect (Estrada-Cuzcano et al., 2012). These are termed retinal ciliopathies (Bujakowska et al., 2017).
Rather than being distinct clinical entities, ciliopathies are considered to form a spectrum of disorders, with considerable phenotypic and genotypic overlap between different conditions. In addition to this, there is also extensive phenotypic and genetic heterogeneity amongst ciliopathies (Mitchison and Valente, 2017; Oud et al., 2017). There are currently at least 190 known ciliopathy disease genes causing defects of the primary cilia (Table 1), and mutations in many of the same genes can cause strikingly different phenotypes. A classic example is CEP290, mutations in which can cause perinatal lethal MKS, severe JBTS, or non-syndromic LCA, which only affects the retina (Coppieters et al., 2010; Drivas and Bennett, 2014).

Table 1. Overview of genes mutated in ciliopathies showing the heterogeneity of certain ciliopathies.
Motile ciliopathies are grouped under the name primary ciliary dyskinesia (PCD). A component of PCD manifests with left-right axis abnormalities and this association is also called Kartagener's syndrome, which affects around 50% of PCD patients; additionally there is a more rare disease component arising from defects of ciliogenesis affecting cilia numbers, which is also being called reduced generation of multiple motile cilia (RGMC) (Boon et al., 2014; Lucas et al., 2014; Knowles et al., 2016). Apart from laterality defects that may be linked to cardiac disease (Best et al., 2018), PCD disease features include chronic respiratory infections from earliest life, progressive upper respiratory problems and loss of lung function (bronchiectasis), conductive hearing problems, subfertility, and infrequent hydrocephalus. The same as for the nonmotile ciliopathies, PCD is a genetically and clinically heterogeneous condition, with mutations in around 40 different motile cilia genes currently recognized to cause disease (Table 1). Similarly to the primary ciliopathies, a wider than previously suspected spectrum of motile ciliopathy disease is starting to emerge with greater genetic understanding of these conditions. Gene mutations causing more severe (Davis et al., 2015; Amirav et al., 2016; Irving et al., 2018) and more mild (Knowles et al., 2014; Lucas et al., 2017b; Best et al., 2018; Irving et al., 2018; Shoemark et al., 2018) disease have more recently been recognized, as well as subtypes with features that overlap with the primary ciliopathies such as retinitis pigmentosa and developmental delay (Budny et al., 2006; Moore et al., 2006). These syndromic disease subtypes e.g. Simpson-Golabi-Behmel type 2 syndrome, expand our understanding of the extent of the motile ciliopathy disease spectrum (Mitchison and Shoemark, 2017).
Challenges of Diagnosing Ciliopathies Using Genomics
Despite advances in genetic understanding of these conditions with the advent of next generation sequencing, ciliopathies remain under-diagnosed and poorly recognized due to insensitive and non-specific aspects of available diagnostic tests, compounded by variable disease features. Genetic diagnostic rates of severe primary ciliopathies remain around 62% using targeted gene panel sequencing (Bachmann-Gagescu et al., 2015; Knopp et al., 2015) and 44% using whole exome sequencing (Sawyer et al., 2016). The diagnostic rate of the arguably more uniform motile ciliopathies disease grouping is higher, using well-characterized cohorts, at up to 67% using targeted gene panels (Boaretto et al., 2016; Paff et al., 2018) and 76% using whole exome sequencing with targeted copy number variation (CNV) analysis (Marshall et al., 2015). For both the primary and motile ciliopathies, many genetic causes of these conditions remain unknown. There remain very few, if any, treatment options for the vast majority of these conditions (Lucas et al., 2014, 2017a; Molinari and Sayer, 2017). Thus, there is a pressing clinical need to advance genetic understanding for the purpose of diagnostics, prognostics and development of novel targeted therapies. Clinical genome data from the 100,000 Genomes Project presents exciting opportunities to offer patients genetic diagnoses, gain novel insights into etiology of disease, and uncover new targets for therapies.
Ciliopathy patients and family members account for around 1% of the rare disease cohort recruited to the UK 100,000 Genomes Project (Figure 1). In the majority of cases, patients recruited to the ciliopathy pathway have had the relevant ciliopathy disease genes sequenced and excluded as the cause of their disease. Such testing is provided as a genetic diagnostic service by accredited NHS genomics labs, and involves sequencing the exons and intron/exon boundaries of a panel of currently 123 genes known to be mutated in ciliopathies.
At the time of writing, genomes have been sequenced and analyzed from 274 patients and family members with respiratory ciliopathies (126 PCD patients, 148 non-cystic fibrosis (CF) bronchiectasis patients and family members) and 81 patients and family members with congenitial malformations caused by ciliopathies (45 BBS, 14 JBTS, 22 rare multisystem ciliopathy disorders). These numbers will increase as sequencing and analysis is ongoing. However, it is likely that a much larger number of ciliopathy patients have been recruited to the project within other categories such as “renal and urinary tract disorders,” which includes phenotypic descriptor “cystic kidney disease” which accounts for 1,516 individuals alone. Furthermore, within the “ophthalmological disorders” category there are likely to be many undiagnosed retinal ciliopathy patients. One thousand two hundred and sixty individuals with a diagnosis of rod-cone dystrophy or LCA)/early onset severe retinal dystrophy have been recruited. It can be estimated that around one third of these patients have a retinal dystrophy owing to a retinal photoreceptor cilium defect (Estrada-Cuzcano et al., 2012). There are many patients in the project with other dysmorphic and congenital abnormalities with features overlapping ciliopathy phenotypes, which may be undiagnosed ciliopathies. This includes 19 patients with unexplained monogenic fetal disorders. Similarly, there are more than 6,000 patients (39% of the total rare disease patient cohort, Figure 1) with a general neurology or neurodevelopmental disorder. These include patients with intellectual disability, holoprosencephaly and hereditary ataxia, all of which are ciliopathy phenotypes seen in syndromic ciliopathies. A subset of these patients could also have undiagnosed ciliopathies. Numbers will increase as data from more patients is made available in upcoming data releases. The 100,000 Genomes Project may thus represent a unique opportunity to discover and diagnose orphan ciliopathies in patients whose phenotype does not currently easily fit into existing disease categories.
The success of the project may lie in the accuracy and thoroughness of phenotyping information provided i.e., the consistent use of Human Phenotype Ontology (HPO) terms. The HPO system was developed to annotate clinical disease terms and definitions with a standardized phenotypic vocabulary (Robinson et al., 2008; Köhler et al., 2017). A particular difficulty with ciliopathies is that they are extremely heterogeneous conditions and it has been suggested that clinicians should be encouraged to actively involve patients in describing their own phenotype (Gainotti et al., 2018). There is great power in larger collections of well-defined patient cohorts to support better clinical research and diagnostics, with the development of rare disease patient registries being a key component supporting the activities of European Reference Networks (ERNs) on rare diseases (Kodra et al., 2018). The European Organization for Rare Diseases (EURORDIS) has developed recommendations on ethical and responsible international data sharing to help inform a clinical diagnosis (Gainotti et al., 2018).
A wide range of phenotypes of different severities are associated with the ciliopathies, demonstrating their complexity (Lee and Gleeson, 2011; Arts and Knoers, 2013; Mitchison and Valente, 2017). Figure 2 is not exhaustive but shows selected clinical features of the ciliopathies, highlighting those relevant to recruitment criteria for 100,000 Genomes Project. This is a constantly expanding spectrum, with motile and non-motile cilia recently implicated in the etiology of congenital heart disease (You et al., 2015; Best et al., 2018). Situs inversus and associated cardiac malformations found in common between motile and some nonmotile ciliopathies suggest an influence on laterality determination at the embryonic left-right organizer during development from non-motile as well as motile cilia, or else that there are shared motile cilia defects, or a mixture of both. There is growing evidence for respiratory involvement in the non-motile primary ciliopathies, but the molecular basis of these findings remains unclear (Mitchison and Shoemark, 2017).
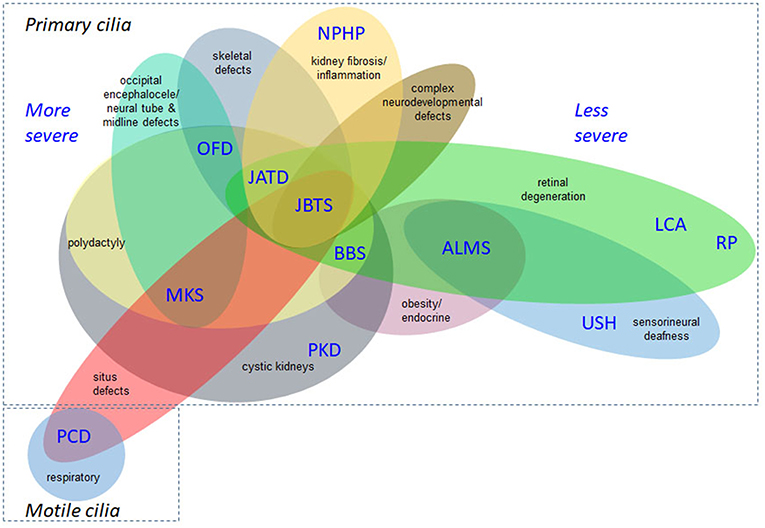
Figure 2. Overlapping disease features of the ciliopathies. This is not an exhaustive list but illustrates the complex phenotypes and overlapping clinical features of ciliopathies. The severe/lethal diseases tend to have more complex combinations of disease features, compared to diseases at the milder end of the clinical spectrum. Situs inversus and associated cardiac malformations are found in common between non-motile and motile ciliopathies and the former can also display respiratory defects. ALMS, Alström syndrome; BBS, Bardet-Biedl syndrome; JATD, Jeune asphyxiating thoracic dysplasia; JBTS, Joubert syndrome; LCA, Leber congenital amaurosis; MKS, Meckel-Gruber syndrome; NPHP, nephronophthisis; OFD, oral-facial-digital syndrome; PCD, primary ciliary dyskinesia; PKD, polycystic kidney disease; RP, retinitis pigmentosa; USH, Usher syndrome.
In terms of genetic heterogeneity, interpreting large volumes of genetic variants from all of the ciliopathy patients in order to identify the truly pathogenic disease-causing variant in each individual represents a major challenge. In addition to primary causal variants, modifier genes and variable mutational load have been proposed to play roles in determining the genetic spectrum for both primary ciliopathies (Katsanis et al., 2001; Davis et al., 2011; Zaki et al., 2011; Lindstrand et al., 2016) and motile ciliopathies (Li et al., 2016). Different mutation types can be expected in different ciliopathies, for example motile cilia disease tends to arise from high impact pathogenicity mutations, most often single base substitutions, small insertions and deletions or larger CNVs, that result in protein frameshifts, premature stop codons, missense changes, or splicing defects giving rise to null alleles effects. In contrast, for the non-motile primary ciliopathies lethality frequently arises as a consequence of such alleles, whilst surviving patients would carry only one or no copies of this type of high impact allele, but instead carry one or two hypomorphic alleles such as milder effect missense changes (Davis et al., 2011; Hildebrandt et al., 2011; Schmidts et al., 2013; Reiter and Leroux, 2017). Some specific mutations may be expected, for example splicing mutations and CNVs are common in retinal degeneration caused by PRPF31 mutations (Buskin et al., 2018). With the superior detection of many mutations through whole genome sequencing, the genetic landscape is expected to greatly expand and may significantly change with implementation of large scale whole genome sequencing (Belkadi et al., 2015). For example the 100,000 Genome Project has already detected a deep intronic mutation in DNAH11 causing PCD which would not have been detected by other current clinical screening methods (Ellingford et al., 2018).
Ciliopathy Genomics Data Analysis in the 100,000 Genomes Project
Genomics England Clinical Interpretation Partnerships (GeCIPs) currently coordinate a crowdsourcing approach to data analysis, building on the success of aggregate consortia such as the Exome Aggregation Consortium (ExAc) (Karczewski et al., 2017) and Genome Aggregation Database (GnomAD) (Lek et al., 2016), and public databases such as ClinVar (Landrum and Kattman, 2018), Human Gene Mutation Database HGMD (Stenson et al., 2017) and the Leiden Open-source Variation Database LOVD (Fokkema et al., 2011). In addition to GeCIPs, formed of individuals from not-for-profit organizations, such as academics and clinicians who must apply for access to anonymized data, private commercial companies also have access to the anonymized data. The project is achieved through public-private partnerships (PPPs) between Genomics England Limited, owned by the UK Government's Department of Health & Social Care, and private companies including Illumina Inc. Illumina is a partner both in sequencing and bioinformatics data analysis (https://www.genomicsengland.co.uk/bioinformatics-partnership-with-illumina/). Other “interpretation partners” include Congenica, ICON, Fabric Genomics and WuxiNextCode (https://www.genomicsengland.co.uk/the-100000-genomes-project/data/current-research/). Further to this, industrial companies have been invited to engage with the project and access data by joining the Genomics Expert Network for Enterprises (GENE) for a fee.
Currently, novel or rare variants identified in rare disease patients in the 100,000 Genomes Project are “tiered” according to predicted pathogenicity, following the Association for Clinical Genetic Science's Best Practice Guidelines for Variant Classification (https://www.acgs.uk.com/quality/best-practice-guidelines/) which builds upon Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer published by the Association for Molecular Pathology (Li et al., 2017). This allows classification of variants into Tier 1, variants with strong clinical significance; Tier 2, variants with potential clinical significance; Tier 3, variants of unknown clinical significance; and Tier 4, variants deemed benign or likely benign. Tiering is achieved using information from PanelApp, an online resource in which clinicians, academic researchers and laboratory scientists pool information about known disease genes, and pathogenic variants within them (https://panelapp.genomicsengland.co.uk/). This crowdsourcing tool enables a “virtual gene panel” approach to the analysis of genomic data; focusing on known or predicted pathogenic genes and variants. Patients' genomes are first analyzed against a panel of genes most closely associated with their disease phenotype (i.e., ciliopathy gene panels), then against other suitable gene panels with features overlapping the phenotype e.g., retinal dystrophy gene panel, neurology panel. Tier 1 variants are protein truncating (frameshift, stop gain, stop loss, splice acceptor variant, or splice donor variant) or de novo (protein truncating, missense, or splice region) variants in at least one transcript of a gene on the diagnostic grade “green” gene list in the virtual gene panel for the disorder in question. Tier 2 variants are protein altering variants, such as missense and splice region variants, in at least one transcript of a gene on the diagnostic grade “green” gene list in the virtual gene panel for the disorder in question. Tier 1 and 2 variants are not commonly found in the general healthy population, the allelic state matches the known mode of inheritance for the gene and disorder, and segregates with disease (where applicable). Protein truncating, de novo or protein altering variants affecting genes not in the virtual gene panel are Tier 3. If a variant does not meet any of these criteria it is untiered1.
To provide some sense of scale of the challenge faced, at the time of going to press, there were 2,605 Tier 3 variants in probands with the phenotype “primary ciliary disorders;” primary ciliary dyskinesia' and “primary ciliary dyskinesia::primary ciliary disorders.” Of these, 174 are frameshift/stop gain/stop loss/splice donor variant/splice acceptor variants i.e., presumed loss-of function, and 1,829 are missense variants, in around 115 genes. Similarly, at the time of going to press, there were 4,518 Tier 3 variants in probands with phenotype “Bardet-Biedl syndrome;” “Joubert syndrome” or “rare multisystem ciliopathy disorders.” Of these, 506 are frameshift/stop gain/stop loss/splice donor variant/splice acceptor variant i.e., presumed loss-of function, and 3,028 are missense variants. Characterizing the effect of these mutations poses a significant challenge in terms of computing power, manpower and bioinformatics expertise.
Molecular MODELING of Ciliopathies
Traditional approaches to modeling non-motile ciliopathies involve 2D ciliated cell line cultures (Table 2) or whole animal studies, typically zebrafish (Marshall and Osborn, 2016; Song et al., 2016), mice (Norris and Grimes, 2012), Xenopus (Walentek and Quigley, 2017; Blum and Ott, 2018), chick (Schock et al., 2016) and C.elegans (Mok and Héon, 2012). Study of motile ciliopathies usually involves studying cells in vivo, or on ex vivo cultures such as mouse tracheal explants or nasal brush samples from patients with PCD, grown at the air-liquid interface (ALI) (Hirst et al., 2010). There are no adherent immortalized cell lines which can grow motile cilia, although a recent paper reported the immortalization of a multiciliated cell line with dyskinetic cilia (Kuek et al., 2018) which is unlikely to be useful as a control for studying motile ciliopathies. Much understanding of motile cilia has been achieved using single celled flagellated organisms such as Chlamydomonas reinhardtii (Harris et al., 2009) and Trypanosoma brucei (Langousis and Hill, 2014) which possess one and two flagella, respectively. More recent models include multiciliates planaria and paramecia (King and Patel-King, 2016; Fassad et al., 2018). These model systems offer numerous advantages, including ease of culture and biochemical purification of motile cilia and ease of genetic manipulation. For a useful review of cilia model organisms, see (Vincensini et al., 2011).
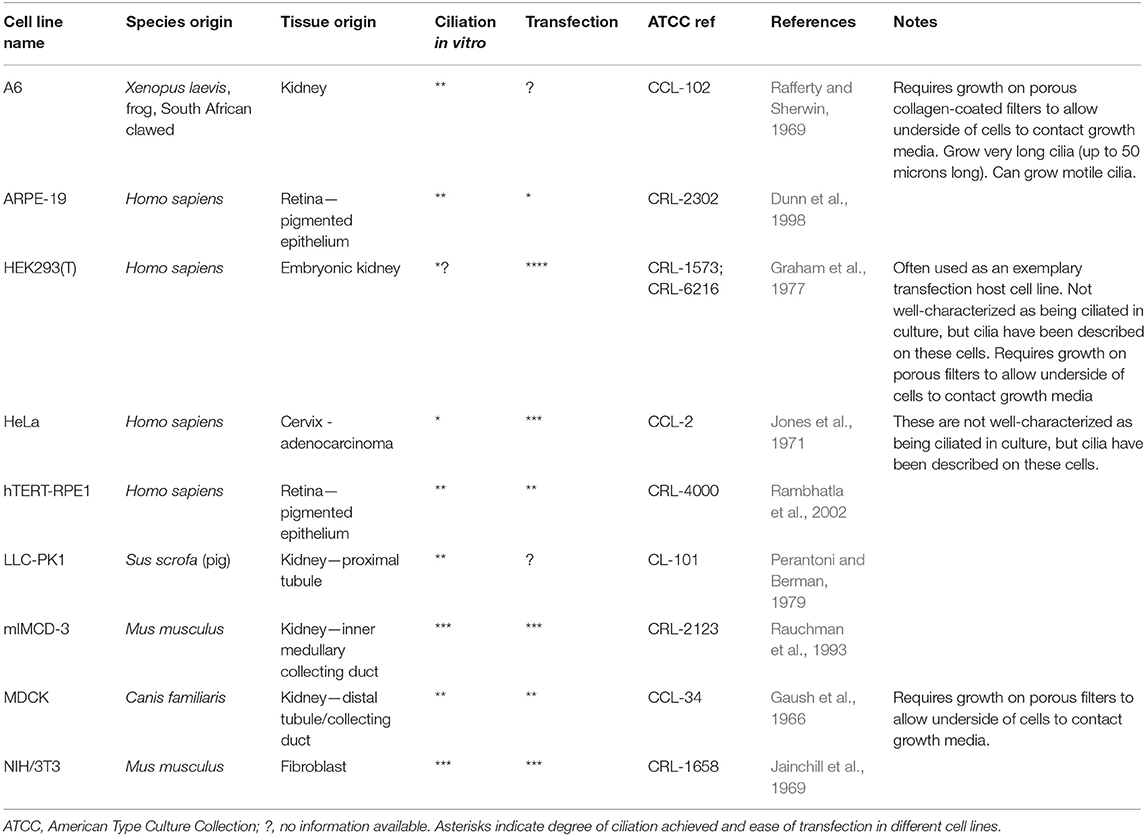
Table 2. Immortalized cell lines used for modeling ciliopathy mutations.
3D organoids derived from human embryonic stem (ES) cells or induced pluripotent stem cell (iPSCs) are increasingly replacing animals in ciliopathy research. These can be derived from patient fibroblasts, or can be genetically engineered to harbor patient mutations, to study effect of mutation and efficacy of possible treatments. In the past decade, techniques have particularly advanced in culture methods for producing in vitro 3D cell culture models to study cilia and ciliopathies. These include urine-derived renal epithelial cells (URECs) (Ajzenberg et al., 2015) and models of mammalian retina for studying retinal ciliopathies. Robust protocols for culture of retinal organoids from human embryonic stem (ES) cells and induced pluripotent stem cells (iPSCs) have been published and refined (Meyer et al., 2011; Kuwahara et al., 2015). These organoids form laminated neural retina with mature photoreceptor cells, which can be selectively isolated using specific cell surface markers (Lakowski et al., 2018). This provides a highly relevant human-derived model for studying retinal development and retinal degeneration. This is particularly useful for studying human retinal dystrophies which are not recapitulated in genetic mouse models, such as knock-in and knock-out mice models of RP associated with pre-mRNA splicing factor 31 mutations, which do show photoreceptor degeneration (Bujakowska et al., 2009). Similarly, culture techniques are advancing toward the ability to grow motile ciliated cells from iPSCs and ES cells, including in 3D spheroids (Firth et al., 2014; Konishi et al., 2016).
Techniques for genetically editing these ciliated cell models are also advancing, where primary patient cells are not suitable or available for research. This field is moving toward a point where investigators can replicate a potentially pathogenic variant of unknown clinical significance in a human ES cell or iPSC, and differentiate these cells toward a cell type relevant to the primary disease tissue to study the effect of mutation and evaluate methods of genetic correction. Since the development of zinc finger nucleases (ZFNs) (Bibikova et al., 2003) and transcription activator-like effector nucleases (TALENs) (Wood et al., 2011) it has been possible to edit the genome with a high degree of accuracy, to introduce DNA double strand breaks at specific genomic locations, which are generally repaired by error-prone non-homologous end joining (NHEJ) introducing small insertions or deletions, in order to create specific genetic knockouts. However, design and production of such zinc finger nucleases was slow and laborious. It is now possible to introduce such specific breaks with one common nuclease; Cas9 nuclease, significantly increasing throughput. Cas9 is targeted to the genome using a specific guide RNA, in a process termed Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 genome editing (Cong et al., 2013). The development of the “dual nickase” approach increases specificity and reduces off-target effects (Ran et al., 2013) but off-target effects remain one of the major challenges of scientists using CRISPR. Whilst knockouts can be generated at high efficiency, relying on NHEJ, the introduction of specific insertions, deletions, transitions or transversions using a specific repair template via homology directed repair (HDR), occurs at significantly lower frequency.
In response to this, investigators have now developed base editing, a modified form of CRISPR/Cas9 genome editing, in which Cas9 nickase (Cas9n) is tethered to a cytidine deaminase enzyme which catalyzes the efficient conversion of C∙G>T∙A changes within a specific window of activity (Komor et al., 2016; Koblan et al., 2018). This refined genetic system can be used to study specific dominant, recessive and compound heterozygous mutations in relevant cells, or animal models. Recently, new adenine base editing tools have been developed, which enable engineering of specific A∙T>G∙C changes in cell lines (Gaudelli et al., 2017; Koblan et al., 2018). Both technologies have been optimized for mutation in mammalian systems (Koblan et al., 2018). C∙G>T∙A and A∙T>G∙C changes can now be efficiently engineered into cells and animals to model dominant disease. However, there is a restricted window of activity 4–8 nucleotides from the first nucleotide of the guide RNA, and guides can only be designed immediately upstream of a relevant protospacer adjacent motif (PAM). Engineering of Cas9 is enabling wider application of this base editing technology by adjusting the PAM sequence recognized by Cas9, so that with the newly engineered Cas9-cytidine deaminase fusions, approximately 2.5x more variants in ClinVar can be targeted using the technology (Kim et al., 2017) (Figure 3). Despite these current limitations, the technology is rapidly advancing and a “double hit” approach provides an efficient new method for modeling compound heterozygous mutations in cell systems. As most ciliopathies are recessive disorders, and compound heterozygosity is common in individuals from non-consanguineous unions, this provides a valuable tool for characterizing variants of unknown significance from the 100,000 Genomes Project.
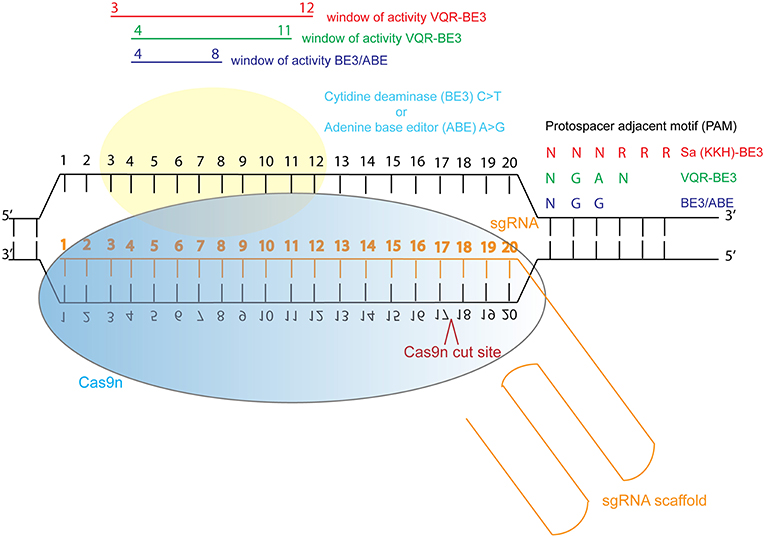
Figure 3. Schematic illustration of cytidine deaminase and adenine base editing of the genome. Abbreviations: sgRNA, single guide RNA; BE3, third generation base editor; ABE, adenine base editor.
Characterizing the genetics of ciliopathies may be a larger challenge than in other rare diseases, due to more complex genetics, for example the putative oligogenic inheritance in BBS (Katsanis et al., 2001) and modifier allele effects in multiple ciliopathies (Khanna et al., 2009; Davis et al., 2011; Cardenas-Rodriguez et al., 2013), but novel genome editing technologies provide solutions to these challenges. In order to process the large volume of variants, high-throughput approaches will need to be employed, for example high-content imaging screens such as previously published screens including a whole genome siRNA knockdown screen in ciliated mouse kidney cell line IMCD3 (Wheway et al., 2015) and a druggable genome siRNA knockdown screen in ciliated human retinal cell line hTERT-RPE1 (Kim et al., 2010).
Health Potential and Future Opportunities of the 100,000 Genomes Project
Despite the challenges, there are undoubtedly enormous opportunities provided by this rich, varied and comprehensively phenotyped dataset. The project represents one of the greatest opportunities for novel disease gene discovery, especially in the case of very rare genes/genetic mutations. The aggregation of many families into the dataset allows multiple families with mutations in the same gene to be identified, leading to disease gene identification. One recent example is PRPS1, a gene in which heterozygous missense mutations were found to be carried by retinal dystrophy patients from 5 families in the 100,000 Genomes Project, providing robust genetic evidence for the cause of this condition. Mutations in PRPS1 are normally found in the more severe Arts syndrome, Charcot-Marie Tooth, and nonsyndromic sensorineural deafness, so if mutations in this gene had only been identified in one family, this may have been disregarded as the pathogenic cause of disease (Fiorentino et al., 2018).
Furthermore, the aggregation of all rare disease patient data into one database may enable diagnosis of novel orphan ciliopathies in a way that was not previously possible. The extensive phenotypic and genotypic heterogeneity of ciliopathies raises the likelihood that there exist individuals or groups of individuals who have a disorder arising from a defect in cilia, but have not had their condition defined as a ciliopathy. The collection of all 60,000 rare disease individuals in the 100,000 Genomes Project may enable identification of novel, previously unrecognized ciliopathies.
In addition to novel disease gene discovery, the project offers many opportunities to gain novel insights into even the most basic gene functions. Around 35% of human genes still have no known function (pantherdb.org) (Figure 4). Functional genetics studies investigating genes and variants of interest from the 100,000 Genomes Project may uncover many novel developmental pathways and gene functionalities.
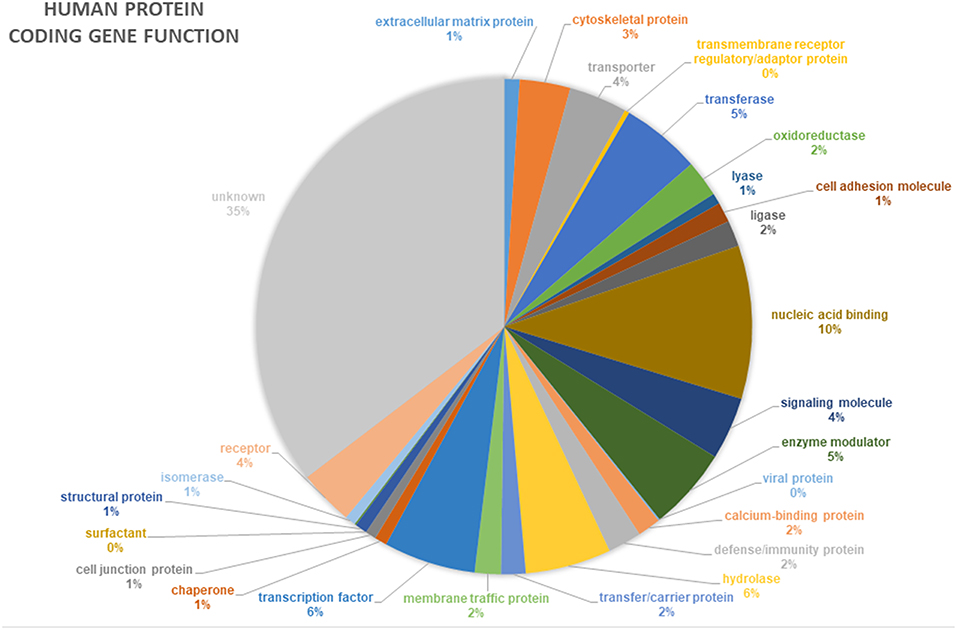
Figure 4. Proportion of annotated human protein coding genes with specific functions. Data from pantherdb.org. Thirty-five percentage of human genes have an unknown function.
Such disease gene discoveries and new biological insights may provide opportunities for developing targeted therapies for ciliopathies, which currently have very few, if any, treatment options. A number of studies have investigated the efficacy and safety of gene therapy for treatment of ciliopathies, particularly in retinal ciliopathies. RPGR, mutations in which cause up to 60% of cases of X-linked RP (Vervoort et al., 2000) has been a particular focus for gene therapy development, including tested in large mammalian models, which have shown to be effective in preventing onset of degeneration (Beltran et al., 2012; Deng et al., 2015; Wu et al., 2015) and also successful in preventing progression of established disease (Beltran et al., 2015). More recently, RPGR gene editing approaches have been developed in addition to existing gene augmentation approaches (Deng et al., 2018). However, more than 20 years after the discovery of RPGR, there is still no gene therapy in clinic, although there are three clinical trials currently recruiting, at the time of writing (https://clinicaltrials.gov/ct2/results?cond=&term=rpgr&cntry=&state=&city=&dist=). There are now several more genes being targeted for gene therapy in retinal ciliopathies including CEP290 (mutations in which cause LCA10) (Estrada-Cuzcano et al., 2012; Burnight et al., 2014; Zhang et al., 2018); and LCA5 (Song et al., 2018). Gene therapy is also being investigated as a possible treatment for syndromic ciliopathies. Gene augmentation has been demonstrated to be effective and safe in Bbs4 genetic mouse models of BBS; (McIntyre et al., 2012, 2013; Chamling et al., 2013), but the only clinical phenotype which was rescued was the olfactory sensory defect. It is clear that we need to look beyond gene therapy for treatment options for ciliopathies. The extensive genetic heterogeneity underlying these conditions is a hinderance to development of personalized medicines, but many recurrent and founder effect mutations are also found to underlie the ciliopathies. The future of effective drug development for ciliopathies requires an understanding of the molecular mechanism of disease, which requires genetic and cell biology studies that the 100,000 Genomes Project can accelerate. Much added value will arise from integration of genomics with multi-omics data (proteomics, transcriptomics, metabolomics, epigenomics) and deep phenotyping of ciliopathies, as recently discussed (Kenny et al., 2017).
Re-purposing and re-licensing of FDA-approved drugs could be the most rapid way to bring clinical therapeutics to patients. For example, it has been suggested that re-purposing of cyclin dependent kinase inhibitors could be an appropriate treatment option for a broad range of ciliopathies which have a common basis in replication stress (Slaats et al., 2015). Treatment with CDK inhibitors such as roscovitine has been shown to be effective in rescuing cilia defects in cells derived from patients with Joubert syndrome (Srivastava et al., 2017). Patients with mutations in NEK1, a DNA damage response gene mutated in patients with the ciliopathy short rib polydactyly syndrome (Thiel et al., 2011), could potentially be treated in this manner. NEK1 interacts with other ciliopathy proteins C21orf2 and SPATA7 (Wheway et al., 2015), suggesting that other ciliopathies could likewise be treated with CDK inhibitors. As a center for Hh signaling, cilia defects can also be potentially treated with Hh agonists (Hynes et al., 2014), for example purmorphamine, which has been shown to be effective in rescuing cilia defects in cells derived from patients with Joubert syndrome (Srivastava et al., 2017). Uncovering novel mechanisms of disease through functional characterization of variants in the 100,000 Genomes project ciliopathy patient dataset may well identify common pathways of disease which can be targeted to provide therapies for many individuals. Furthermore, if and when any potential therapies reach clinical trials, the national genomic registry provided by the 100,000 Genomes Project will allow rapid identification of individuals suitable for entering the trial.
Until treatments are developed, perhaps the main advantage of insights provided by the 100,000 Genomes Project is the ability to provide genetic diagnosis, for the purposes of better disease monitoring and management, carrier testing, and family planning. This is a particularly pressing need for rare, poorly recognized and difficult to diagnose conditions such as the ciliopathies. If a genetic diagnosis is made, family members can undergo carrier testing which can also inform marriage planning (Nouri et al., 2017; Komlosi et al., 2018). This is especially useful in communities practicing first cousin marriage (consanguinity) in which ciliopathies are at higher risk. One of the aims of the Human Genome Project in Saudi Arabia, where rates of first cousin marriage are around 40%, is to establish a service whereby every engaged couple can undergo whole genome sequencing to test for pathogenic alleles. Worldwide, couples carrying pathogenic variants can make informed decisions about planning pregnancies, including options for pre-implantation diagnosis (PGD). Genetic diseases require robust genetic information before they will be approved by the Human Fertilization and Embryology Authority (HFEA) for PGD. Many genetic subtypes of ciliopathies are now approved for PGD by HFEA, including BBS1, BBS10 and most genetic subtypes of Joubert syndrome (https://www.hfea.gov.uk/pgd-conditions/).
Future Perspectives for UK Medical Genomics
On 31st September 2018, recruitment to the rare disease pathway of the 100,000 Genomes Project closed. DNA samples from 61,282 rare disease patients and family members have been deposited in the UK Biobank and 87,231 whole genome sequences have been produced, from rare disease and cancer patients.
Data from the project will continue to provide diagnoses and uncover novel biological insights for years to come. But what of the future of genomic testing in the NHS? In place of the project, from 1st October 2018 the new National Genomic Medicine Service was established, with an aim to provide consistent and equitable access to genomic testing services across the NHS. Genetic testing will now be conducted across a Genomic Laboratory Network of seven hubs, with choice of tests available dictated by the National Genomic Test Directory (https://www.england.nhs.uk/publication/national-genomic-test-directories/). Of the 190 rare diseases recruited into the project, neurological, neurodevelopmental conditions and complex congenital malformations, such as ciliopathies, are those most likely to continue to benefit from genome testing. There are now 22 indications for frontline WGS, 12 of which include ciliopathy phenotypes (Table 3). Bardet-Biedl syndrome and respiratory ciliopathies will be initially tested using WES or a large panel test, followed by WGS in year 3 if no results are found (Table 3).
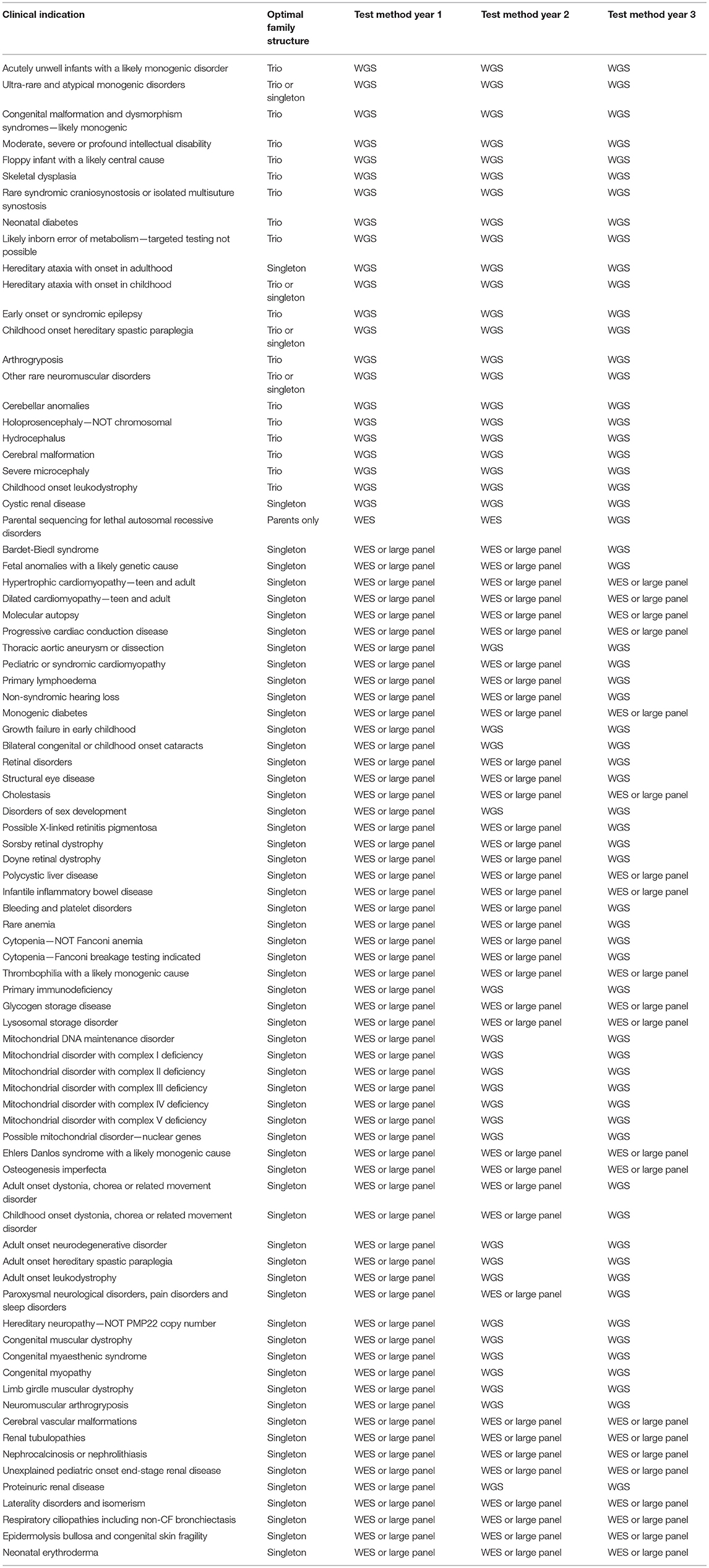
Table 3. Conditions indicated for WGS diagnosis in the UK NHS.
This represents the largest restructuring of genetic testing in the history of the NHS and serves as a model for other countries embarking on similar initiatives. These clinical advances will be of enormous benefit to patients with complex syndromic conditions such as ciliopathies. The aim is that WGS will now be the standard, frontline diagnostic test for many of these families, and hopefully increases in diagnostic rates beyond 60% will be start to be achieved. The medical benefits to families of achieving a rapid diagnosis through WGS for rare diseases like ciliopathies and the costs saved to the NHS can be calculated (Mestek-Boukhibar et al., 2018). Studies in the USA have shown that WGS is by far the most time- and cost-effective way to achieve a diagnosis for complex, heterogeneous disorders such as ciliopathies. In the case of general neurodevelopmental disorders, which are also extensively heterogeneous, WGS can reduce time to diagnosis by 77 months, reduce cost of diagnosis by $11,460, and improve clinical care (Soden et al., 2014). For extremely genetically heterogeneous conditions such as neurodevelopmental disorders and ciliopathies, WGS provides an opportunity to allow families to circumvent the traditional “diagnostic odyssey” (Sawyer et al., 2016). Genomics England now plan to sequence 5 million genomes over next 5 years.
Global Perspective and Future Challenges
There are many other national genome projects around the world, including many with a focus on understanding rare disease, cancer, and genomics for precision medicine. Launched just after the UK's 100,000 Genomes Project, in December 2013, the Saudi Human Genome Program aims to sequence 100,000 individuals, with a specific focus on understanding rare inherited disease, common in Saudi Arabia where first-cousin marriage is common. Two of the largest national genome projects at the time of writing are the USA's $215 million Precision Medicine Initiative announced in January 2015, which aims to sequence 1 million genomes (https://www.nature.com/news/us-precision-medicine-proposal-sparks-questions-1.16774) and China's genomic medicine initiative, launched in 2016, which also aims to sequence 1 million genomes, as part of its 13th five-year plan. Two of the longest running are the Estonia Genome Project (since 2001) and DeCODE project in Iceland, which has been running since 1996 and has sequenced 160,000 individuals, a large proportion of the entire Icelandic population. Others include Genome Russia Project, Proyecto Genoma Navarra (NAGEN) (Spain), Qatar Genome Programme, Belgian Medical Genomics Inititative. More recently, in 2016, France announced its Genomic Medicine Plan 2025 “Médecine Génomique 2025' which involves cooperation with Genomics England, to co-fund analysis of genomes. More local efforts which are large-scale in terms of numbers, but are not nationwide include Geisinger MyCode (Pennsylvania and New Jersey) which has recruited 190,000 patients, and the Utah Genome Project. Transnational efforts include GenomeAsia100K and international efforts include the Personal Genome Project. Transnational and international collaborative efforts may become more commonplace in the future, to increase the power of shared resources.
As well as state-funded clinical and research genomics projects, there also exist privately funded genomics projects, including Craig Venter's “Human Longevity,” which aims to sequence 1 million genomes by 2020. AstraZeneca's genomics initiative (in partnership with Human Longevity, Genomics England, Montreal Heart Institute, Sanger Institute and University of Helsinki) uses retrospective patient data from clinical trials over the past 10 years, and will continue to collect data until 2026. In total, up to 2 million genomes will be sequenced, including 500,000 from patients who have taken part in AstraZeneca clinical trials, to significantly enhance drug development for precision medicine approaches.
Each project has different specific aims but they each share many similarities. They all use Illumina short-read sequencing technologies, although it seems likely that in the future at least some will couple these with longer-read sequencing using platforms such as Pacific Biosciences (PacBio) and Oxford Nanopore. Most projects are driven by the same financial initiatives. Since 2008, there has been a global concerted effort to reduce national spending on healthcare, which in developed countries typically accounts for spending equivalent to 10–15% of national GDP (http://www.abpi.org.uk/facts-and-figures/global-pharmaceutical-market/global-health-expenditure-as-a-share-of-gdp/). The Saudi Human Genome Program estimates the total global annual cost of treating rare inherited disease at around US$27 billion (Project Team, 2015). A substantial reduction in children born with genetic disabilities, as a result of improved genetic diagnostics through understanding gained from genome projects, could immediately save over US$270 million. Furthermore, most governments cite the importance of remaining, or becoming, internationally competitive in the nascent field of genome-guided drug development as reasons behind their investment in large-scale genomics projects. The pharmaceutical industry is one of the world's largest industries, worth in excess of $800,000 million annually (http://www.abpi.org.uk/facts-and-figures/global-pharmaceutical-market/top-10-pharmaceutical-markets-by-value-usd/).
In order to achieve the greatest global health benefits from the information in these multiple genomics projects, responsible and secure data sharing is essential. Achieving this whilst maintaining patient confidentiality and data security is a global challenge. The Global Alliance for Genomics and Health (GA4GH) was established in 2013 to develop best-practice guidelines, and develop tools for genome data sharing. Most recently, GA4GH have developed the application programming interface (API) Beacon, which allows organizations to share genomic and health data in a way which preserves patient confidentiality and data anonymity. Through Beacon, individuals who are not registered and approved users of individual genomics datasets, such as a GeCIP member, can still query these datasets to retrieve information about specific alleles. This saves time; there is no need to go through an often lengthy database access protocol and it actually preserves patient confidentiality since no raw genome data is accessed. Beacon is part of the new interoperability standards, contributing to the Framework for Responsible Sharing of Genomic and Health-Related Data developed by GA4GH (https://www.ga4gh.org/genomic-data-toolkit/regulatory-ethics-toolkit/framework-for-responsible-sharing-of-genomic-and-health-related-data/). Congenica and Genomics England are members of the GA4GH and contribute to the Steering Committee of GA4GH, so it seems likely that at some point in the future data will become more widely available for exploitation for the benefit of human health.
Meaningful and effective sharing of data from many genome projects also poses a practical challenge in terms of interoperability of analytical platforms. Whilst all large scale genome projects predominantly use Illumina sequence technologies, each program has bespoke data standards, analytical pipelines, storage and access protocols which are potentially incompatible with each other. There may also be variations in the standards applied to use of phenotypic terms, variant calling and descriptions of pathogenicity of alleles. GA4GH develops tools and defines standards to harmonize data generation, analysis, storage and access. Recently, GA4GH developed a set of standards for variant calling, which allows stratification of performance by variant type and genome context (Krusche et al., 2018). Furthermore, GA4GH develops standards for workflow development, and APIs for packaging of workflows to support their application within multiple different environments. This will allow researchers to run the same analysis workflow on data in multiple clouds and environments in order to standardize data analysis across projects.
Ensuring interoperability of platforms will becoming increasingly important as more genome projects incorporate long-read sequence technologies to complement short-read sequence technologies. This approach allows resolution and alignment of repetitive regions, including regions with many short tandem repeats, or genes with many pseudogenes, phasing of compound heterozygous mutations and provides information about epigenetic marks (Simpson et al., 2017; Pollard et al., 2018). This has clinical relevance in ciliopathies; ADPKD associated with PKD1 mutations is difficult to diagnose genetically due to 6 pseudogenes of PKD1 on chromosome 16 with 97.7% sequence identity (Eisenberger et al., 2015). Similarly, in the X-linked retinal ciliopathy retinitis pigmentosa, diagnostic yields are low due to the fact that most patients have a mutation in the highly repetitive region of RPGR ORF15, which is difficult to amplify by PCR and Sanger sequencing, and difficult to align with Illumina short read sequencing (Vervoort et al., 2000; Branham et al., 2012).
As well as ensuring integration of different sequencing analysis platforms, it is essential to ensure that platforms for sequence storage and analysis integrate properly with electronic clinical data management systems. This is particularly important for reporting and genetic counseling, and clinical management of patients in receipt of a genetic diagnosis (Welch et al., 2018). It is also crucial to the meaningful analysis of genome data, in the context of detailed medical records including family history, phenotype (using standard HPO terms) and treatment history. GA4GH have developed best practice guidelines for using Family Health History patient record systems with genomics data.
Complex challenges remain, including standardizing a definition of what constitutes a pathogenic allele across all genomics projects. The Association for Clinical Genetic Science's Best Practice Guidelines for Variant Classification 2017 (Ellard et al., 2017) forms the basis of the standard for the 100,000 Genomes Project, based upon Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer published by the Association for Molecular Pathology (Li et al., 2017). However, it must be acknowledged that we remain relatively naïve in our understanding of pathogenicity, particularly with regard to variants affecting splicing. It has been calculated that 35-40% of pathogenic variants in non-canonical splice site positions are missing from public databases, suggesting that we currently under-diagnose pathogenic causes of disease associated with splicing (Lord et al., 2018). We do not yet know enough about the functional effect of non-coding variants, subtle missense variants and variants in exonic splice enhancers to be able to report these back to patients without functional validation. This is likely to be the significant bottleneck in exploitation of the expanding wealth of genome and population-genetics data. Most in silico predictors of pathogenicity are notoriously unreliable, and will only improve with machine learning aided by robust in vitro data from functional experiments. This is time-consuming and expensive work. Beyond simple monogenic disorders, we also need frameworks for consistently assigning pathogenicity based on polygenic risk scores, and combined total gene variation scores (Khera et al., 2018; Mossotto et al., 2018). For these reasons, it seems appropriate that this remains an ongoing area of development by GA4GH's Variation Annotation Task Team.
Global Social Challenges
The UK, USA and China all strive to be global leaders in this area, and as a result, have each launched ambitious genome sequencing projects such as the 100,000 Genomes Project, arguably before the necessary skills, understanding or, importantly, public appetite, to manage these projects effectively is in place. Public understanding and trust of genomics remains a global challenge to the success of all genomics projects. Lack of understanding or confidence in genomics in the workforce and the patient population poses a barrier to effective implementation and recruitment to genomics projects. This requires an entire “Workforce transformation” (https://www.genomicseducation.hee.nhs.uk/news/item/357-transforming-healthcare-the-impact-of-genomics-on-the-nhs/). Health Education England, (HEE) is a key delivery partner in the 100,000 Genomes Project and has developed a Genomics Education Programme delivered by higher education providers around the UK as a postgraduate masters (MSc) in Genomic Medicine. Parts of this programme are also possible to study, with a shorter training period, awarded as a PGCert and PGDip, or as standalone modules available as part of continuing professional development (CPD). Courses are designed to fit around work, or to be attended part-time, by staff within the NHS and a One-day Primer In Genomic Medicine is currently offered by University of Southampton (https://www.southampton.ac.uk/medicine/primer-in-genomic-medicine-web-form.page; https://www.genomicseducation.hee.nhs.uk/taught-courses/courses/primer-genomic-medicine/.
Patient education and education of the general public in genomics remains a barrier to greater success and higher recruitment, and impacts on truly “informed consent.” Furthermore, the nature of a consent process in which all individuals must consent to the research element of the project, which includes sharing of information with commercial companies, raises issues around autonomy and consent (Dheensa et al., 2018). The public outreach initiative “Socializing the Genome,” developed by West of England Genomic Medicine Center, reached 19,000 people across the south-west, now launched across Manchester region (https://www.genomicseducation.hee.nhs.uk/news/item/347-how-lego-pro-bots-are-bringing-genomics-to-life-in-the-classroom/). At a time when public understanding, including in the workforce, remains low, there is concern that mainstreaming of genomics services at this point in time may be premature (Ormondroyd et al., 2018). Ethical implications of incidental findings, data security and anonymity, and intellectual property of findings resulting from genome studies also remain unresolved issues across all projects. Many professionals currently believe incidental findings should be reported with caution, with an “approach that is responsive to accumulating evidence.” These issues present a significant challenge (Ormondroyd et al., 2018). The general consensus is that there is not enough evidence to form a robust policy regarding secondary findings yet, so data should be interpreted with caution (Ormondroyd et al., 2018). Similarly, decisions about how often to reanalyze data and the mechanism for reporting data back to patients still requires resolution.
It is of utmost importance for healthcare professionals, researchers, and policymakers to be open and honest about the challenges, in order to avoid “genohype” and setting unrealistic expectations. In the past there has been a tendency to overstate the opportunities and understate the risks associated with genetic testing (Samuel and Farsides, 2017) and this has led to some loss of public confidence in genetics. Time taken to return findings remains a significant issue in genomic testing (Moss and Wernham, 2018). Transparency, clear and ongoing communication between healthcare professionals and patients is essential (Dheensa et al., 2018), communicating the possibility of no certain findings either now, or in the near future. We also need to reconsider our concept of the need for fully informed consent, which may not be possible in genomics. Rather, we must consider the general social contract around healthcare provision in the UK, and commitments to public education in the area of genomics.
Concluding Remarks
The 100,000 Genomes Project provides many opportunities and challenges to improve our understanding of ciliopathies. Perhaps the real power in this dataset will be realized through aggregation of this information with other genome projects worldwide. For this to be a success, data security, regulation and ethics must be at the center of such efforts, to establish and maintain public trust in genome testing. There must be a truly global effort to standardize phenotypic descriptions, data standards, analysis pipelines and mechanisms for data sharing and discovery. Perhaps then, with global cooperation, we can finally increase diagnostic yields, inform better clinical management and translate new understanding into targeted therapies for ciliopathy patients.
Data Availability
All datasets generated for this study are included in the manuscript and/or the supplementary files.
Author Contributions
GERC provided essential data for this publication. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
GW was supported by a Wellcome Trust Seed Award in Science (204378/Z/16/Z) and a University of Southampton Faculty of Medicine Research Management Committee Research Project Award. HM was supported by Great Ormond Street Children's Charity grant Leadership awards (V1299, V2217), the NIHR Biomedical Research Center at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London and the COST Action BEAT-PCD: Better Evidence to Advance Therapeutic options for PCD network (BM1407). This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support.
The Members of the Genomics England Research Consortium
Ambrose J. C.1, Baple E. L.1, Bleda M.1, Boardman-Pretty F.1,2, Boissiere J. M.1, Boustred C. R.1, Caulfield M. J.1,2, Chan G. C.1, Craig C. E. H.1, Daugherty L. C.1, de Burca A.1, Devereau, A.1, Elgar G.1,2, Foulger R. E.1, Fowler T.1, Furió-Tarí P.1, Hackett J. M.1, Halai D.1, Holman J. E.1, Hubbard T. J. P.1, Jackson R.1, Kasperaviciute D.1,2, Kayikci M.1, Lahnstein L.1, Lawson K.1, Leigh S. E. A.1, Leong I. U. S.1, Lopez F. J.1, Maleady-Crowe F.1, Mason J.1, McDonagh E. M.1,2, Moutsianas L.1,2, Mueller M.1,2, Murugaesu N.1, Need A. C.1,2, Odhams C. A.1, Patch C.1,2, Perez-Gil D.1, Polychronopoulos D.1, Pullinger J.1, Rahim T.1, Rendon A.1, Riesgo-Ferreiro P.1, Rogers T.1, Ryten M.1, Savage K.1, Sawant K.1, Scott R. H.1, Siddiq A.1, Sieghart A.1, Smedley D.1,2, Smith K. R.1,2, Sosinsky A.1,2, Spooner W.1, Stevens H. E.1, Stuckey A.1, Sultana R.1, Thomas E. R. A.1,2, Thompson S. R.1, Tregidgo C.1, Tucci A.1,2, Walsh E.1, Watters, S. A.1, Welland M. J.1, Williams E.1, Witkowska K.1,2, Wood S. M.1,2, Zarowiecki M.1. (1) Genomics England, London, UK. (2) William Harvey Research Institute, Queen Mary University of London, London, EC1M 6BQ, UK.
Footnotes
1. ^For further information we direct readers to https://panelapp.genomicsengland.co.uk/
References
Ajzenberg, H., Slaats, G. G., Stokman, M. F., Arts, H. H., Logister, I., Kroes, H. Y., et al. (2015). Non-invasive sources of cells with primary cilia from pediatric and adult patients. Cilia 4:8. doi: 10.1186/s13630-015-0017-x
Amirav, I., Wallmeier, J., Loges, N. T., Menchen, T., Pennekamp, P., Mussaffi, H., et al. (2016). Systematic analysis of CCNO variants in a defined population: implications for clinical phenotype and differential diagnosis. Hum. Mutat. 37, 396–405. doi: 10.1002/humu.22957
An, J. Y. (2017). National human genome projects: an update and an agenda. Epidemiol. Health 39:e2017045. doi: 10.4178/epih.e2017045
Arts, H. H., and Knoers, N. V. (2013). Current insights into renal ciliopathies: what can genetics teach us? Pediatr. Nephrol. 28, 863–874. doi: 10.1007/s00467-012-2259-9
Bachmann-Gagescu, R., Dempsey, J. C., Phelps, I. G., O'Roak, B. J., Knutzen, D. M., Rue, T. C., et al. (2015). Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 52, 514–522. doi: 10.1136/jmedgenet-2015-103087
Belkadi, A., Bolze, A., Itan, Y., Cobat, A., Vincent, Q. B., Antipenko, A., et al. (2015). Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. U.S.A. 112, 5473–5478. doi: 10.1073/pnas.1418631112
Beltran, W. A., Cideciyan, A. V., Iwabe, S., Swider, M., Kosyk, M. S., McDaid, K., et al. (2015). Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. Proc. Natl. Acad. Sci. U.S.A. 112, E5844–E5853. doi: 10.1073/pnas.1509914112
Beltran, W. A., Cideciyan, A. V., Lewin, A. S., Iwabe, S., Khanna, H., Sumaroka, A., et al. (2012). Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 109, 2132–2137. doi: 10.1073/pnas.1118847109
Best, S., Shoemark, A., Rubbo, B., Patel, M. P., Fassad, M. R., Dixon, M., et al. (2018). Risk Factors for Situs Defects and congenital heart disease in primary ciliary dyskinesia. Thorax 74, 203–205. doi: 10.1136/thoraxjnl-2018-212104
Bibikova, M., Beumer, K., Trautman, J. K., and Carroll, D. (2003). Enhancing gene targeting with designed zinc finger nucleases. Science 300:764. doi: 10.1126/science.1079512
Blum, M., and Ott, T. (2018). Xenopus: an undervalued model organism to study and model human genetic disease. Cells Tissues Organs. 9, 1–11. doi: 10.1159/000490898
Boaretto, F., Snijders, D., Salvoro, C., Spalletta, A., Mostacciuolo, M. L., Collura, M., et al. (2016). Diagnosis of primary ciliary dyskinesia by a targeted next-generation sequencing panel: molecular and clinical findings in italian patients. J. Mol. Diagn. 18, 912–922. doi: 10.1016/j.jmoldx.2016.07.002
Boon, M., Wallmeier, J., Ma, L., Loges, N. T., Jaspers, M., Olbrich, H., et al. (2014). MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Commun. 5:4418. doi: 10.1038/ncomms5418
Boycott, K. M., Rath, A., Chong, J. X., Hartley, T., Alkuraya, F. S., Baynam, G., et al. (2017). International cooperation to enable the diagnosis of all rare genetic diseases. Am. J. Hum. Genet. 100, 695–705. doi: 10.1016/j.ajhg.2017.04.003
Boyd, A., Golding, J., Macleod, J., Lawlor, D. A., Fraser, A., Henderson, J., et al. (2013). Cohort Profile: the 'children of the 90s'–the index offspring of the avon longitudinal study of parents and children. Int. J. Epidemiol. 42, 111–127. doi: 10.1093/ije/dys064
Branham, K., Othman, M., Brumm, M., Karoukis, A. J., Atmaca-Sonmez, P., Yashar, B. M., et al. (2012). Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Invest. Ophthalmol. Vis. Sci. 53, 8232–8237. doi: 10.1167/iovs.12-11025
Budny, B., Chen, W., Omran, H., Fliegauf, M., Tzschach, A., Wisniewska, M., et al. (2006). A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum. Genet. 120, 171–178. doi: 10.1007/s00439-006-0210-5
Bujakowska, K., Maubaret, C., Chakarova, C. F., Tanimoto, N., Beck, S. C., Fahl, E., et al. (2009). Study of gene-targeted mouse models of splicing factor gene Prpf31 implicated in human autosomal dominant retinitis pigmentosa (RP). Invest. Ophthalmol. Vis. Sci. 50, 5927–5933. doi: 10.1167/iovs.08-3275
Bujakowska, K. M., Liu, Q., and Pierce, E. A. (2017). Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb. Perspect. Biol. 9:a028274. doi: 10.1101/cshperspect.a028274
Burnight, E. R., Wiley, L. A., Drack, A. V., Braun, T. A., Anfinson, K. R., Kaalberg, E. E., et al. (2014). CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 21, 662–672. doi: 10.1038/gt.2014.39
Buskin, A., Zhu, L., Chichagova, V., Basu, B., Mozaffari-Jovin, S., Dolan, D., et al. (2018). Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun. 9:4234. doi: 10.1038/s41467-018-06448-y
Cardenas-Rodriguez, M., Osborn, D. P., Irigoín, F., Graña, M., Romero, H., Beales, P. L., et al. (2013). Characterization of CCDC28B reveals its role in ciliogenesis and provides insight to understand its modifier effect on Bardet-Biedl syndrome. Hum. Genet. 132, 91–105. doi: 10.1007/s00439-012-1228-5
Chamling, X., Seo, S., Bugge, K., Searby, C., Guo, D. F., Drack, A. V., et al. (2013). Ectopic expression of human BBS4 can rescue Bardet-Biedl syndrome phenotypes in Bbs4 null mice. PLoS ONE 8:e59101. doi: 10.1371/journal.pone.0059101
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Consortium, U. K., Walter, K., Min, J. L., Huang, J., Crooks, L., Memari, Y., et al. (2015). The UK10K project identifies rare variants in health and disease. Nature 526, 82–90. doi: 10.1038/nature14962
Coppieters, F., Lefever, S., Leroy, B. P., and De Baere, E. (2010). CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum. Mutat. 31, 1097–1108. doi: 10.1002/humu.21337
Davis, E. E., Zhang, Q., Liu, Q., Diplas, B. H., Davey, L. M., Hartley, J., et al. (2011). TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189–196. doi: 10.1038/ng.756
Davis, S. D., Ferkol, T. W., Rosenfeld, M., Lee, H. S., Dell, S. D., Sagel, S. D., et al. (2015). Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am. J. Respir. Crit. Care Med. 191, 316–324. doi: 10.1164/rccm.201409-1672OC
Deng, W. L., Gao, M. L., Lei, X. L., Lv, J. N., Zhao, H., He, K. W., et al. (2018). Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Rep. 10, 1267–1281. doi: 10.1016/j.stemcr.2018.05.012
Deng, W. T., Dyka, F. M., Dinculescu, A., Li, J., Zhu, P., Chiodo, V. A., et al. (2015). Stability and safety of an AAV vector for treating RPGR-ORF15 X-linked retinitis pigmentosa. Hum. Gene Ther. 26, 593–602. doi: 10.1089/hum.2015.035
Dheensa, S., Samuel, G., Lucassen, A. M., and Farsides, B. (2018). Towards a national genomics medicine service: the challenges facing clinical-research hybrid practices and the case of the 100,000 genomes project. J. Med. Ethics 44, 397–403. doi: 10.1136/medethics-2017-104588
Drivas, T. G. B., and Bennett, J. (2014). “CEP290 and the primary cilium,” in Retinal Degenerative Diseases, eds J. Ash, C. Grimm, J. Hollyfield, R. Anderson, M. LaVail, and C. Bowes Rickman (New York, NY: Springer), 519–525.
Dunn, K. C., Marmorstein, A. D., Bonilha, V. L., Rodriguez-Boulan, E., Giordano, F., and Hjelmeland, L. M. (1998). Use of the ARPE-19 cell line as a model of RPE polarity: basolateral secretion of FGF5. Invest. Ophthalmol. Vis. Sci. 39, 2744–2749.
Eisenberger, T., Decker, C., Hiersche, M., Hamann, R. C., Decker, E., Neuber, S., et al. (2015). An efficient and comprehensive strategy for genetic diagnostics of polycystic kidney disease. PLoS ONE, 10:e0116680. doi: 10.1371/journal.pone.0116680
Ellard, S., Baple, E. L., Owens, M., Eccles, D. M., Abbs, S., Deans, Z. C., et al. (2017). ACGS Best Practice Guidelines for Variant Classification 2017. Association for Clinical Genetic Science. Available online at: http://www.acgs.uk.com/media/1059605/uk_practice_guidelines_for_variant_classification_2017_24_05_17.pdf
Ellingford, J. M., Beaman, G., Webb, K., Callaghan, C., Hirst, R. A., Black, G. C. M., et al. (2018). Whole genomesequencing enables definitive diagnosis of cystic fibrosis and primary ciliary Dyskinesia. bioRxiv[Preprint]:438838. doi: 10.1101/438838
Estrada-Cuzcano, A., Roepman, R., Cremers, F. P., den Hollander, A. I., and Mans, D. A. (2012). Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum. Mol. Genet. 21, R111–R124. doi: 10.1093/hmg/dds298
Fassad, M. R., Shoemark, A., Legendre, M., Hirst, R. A., Koll, F., le Borgne, P., et al. (2018). Mutations in outer dynein arm heavy chain DNAH9 cause motile cilia defects and situs inversus. Am. J. Hum. Genet. 103, 984–994. doi: 10.1016/j.ajhg.2018.10.016
Fiorentino, A., Fujinami, K., Arno, G., Robson, A. G., Pontikos, N., Arasanz Armengol, M., et al. (2018). Missense variants in the X-linked gene PRPS1 cause retinal degeneration in females. Hum. Mutat. 39, 80–91. doi: 10.1002/humu.23349
Firth, A. L., Dargitz, C. T., Qualls, S. J., Menon, T., Wright, R., Singer, O., et al. (2014). Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, E1723–E1730. doi: 10.1073/pnas.1403470111
Fliegauf, M., Benzing, T., and Omran, H. (2007). When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 8, 880–893. doi: 10.1038/nrm2278
Fokkema, I. F., Taschner, P. E., Schaafsma, G. C., Celli, J., Laros, J. F., and den Dunnen, J. T. (2011). LOVD v.2.0: the next generation in gene variant databases. Hum. Mutat. 32, 557–563. doi: 10.1002/humu.21438
Forsythe, E., and Beales, P. L. (2013). Bardet-Biedl syndrome. Eur. J. Hum. Genet. 21, 8–13. doi: 10.1038/ejhg.2012.115
Gainotti, S., Mascalzoni, D., Bros-Facer, V., Petrini, C., Floridia, G., Roos, M., et al. (2018). Meeting patients' right to the correct diagnosis: ongoing international initiatives on undiagnosed rare diseases and ethical and social issues. Int. J. Environ. Res. Public Health 15:E2072. doi: 10.3390/ijerph15102072
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., et al. (2017). Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471. doi: 10.1038/nature24644
Gaush, C. R., Hard, W. L., and Smith, T. F. (1966). Characterization of an established line of canine kidney cells (MDCK). Proc. Soc. Exp. Biol. Med. 122, 931–935. doi: 10.3181/00379727-122-31293
Goetz, S. C., and Anderson, K. V. (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344. doi: 10.1038/nrg2774
Graham, F. L., Smiley, J., Russell, W. C., and Nairn, R. (1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36, 59–74. doi: 10.1099/0022-1317-36-1-59
Gurrieri, F., Franco, B., Toriello, H., and Neri, G. (2007). Oral-facial-digital syndromes: review and diagnostic guidelines. Am. J. Med. Genet. A 143A, 3314–3323. doi: 10.1002/ajmg.a.32032
Hamada, H. (2016). “Roles of motile and immotile cilia in left-right symmetry breaking,” in Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology, eds T. Nakanishi, R. R. Markwald, H. S. Baldwin, B. B. Keller, D. Srivastava, and H. Yamagishi (Tokyo: Springer), 57–65.
Harris, E. H., Witman, G. B., and Stern, D. (2009). The Chlamydomonas Sourcebook, 2nd Edn, Vol. 1–3. Oxford: Academic Press, Elsevier.
Hartill, V., Szymanska, K., Sharif, S. M., Wheway, G., and Johnson, C. A. (2017). Meckel-gruber syndrome: an update on diagnosis, clinical management, and research advances. Front. Pediatr. 5:244. doi: 10.3389/fped.2017.00244
Hildebrandt, F., Benzing, T., and Katsanis, N. (2011). Ciliopathies. N. Engl. J. Med. 364, 1533–1543. doi: 10.1056/NEJMra1010172
Hirst, R. A., Rutman, A., Williams, G., and O'Callaghan, C. (2010). Ciliated air-liquid cultures as an aid to diagnostic testing of primary ciliary dyskinesia. Chest 138, 1441–1447. doi: 10.1378/chest.10-0175
Huber, C., and Cormier-Daire, V. (2012). Ciliary disorder of the skeleton. Am. J. Med. Genet. C Semin. Med. Genet. 160C, 165–174. doi: 10.1002/ajmg.c.31336
Hynes, A. M., Giles, R. H., Srivastava, S., Eley, L., Whitehead, J., Danilenko, M., et al. (2014). Murine Joubert syndrome reveals Hedgehog signaling defects as a potential therapeutic target for nephronophthisis. Proc. Natl. Acad. Sci. U.S.A. 111, 9893–9898. doi: 10.1073/pnas.1322373111
Irving, S., Dixon, M., Fassad, M. R., Frost, E., Hayward, J., Kilpin, K., et al. (2018). Primary ciliary dyskinesia due to microtubular defects is associated with worse lung clearance index. Lung 196, 231–238. doi: 10.1007/s00408-018-0086-x
Jainchill, J. L., Aaronson, S. A., and Todaro, G. J. (1969). Murine sarcoma and leukemia viruses: assay using clonal lines of contact-inhibited mouse cells. J. Virol. 4, 549–553.
Jones, H. W., McKusick, V. A., Harper, P. S., and Wuu, K. D. (1971). George Otto Gey. (1899-1970). The HeLa cell and a reappraisal of its origin. Obstet Gynecol. 38, 945–949.
Karczewski, K. J., Weisburd, B., Thomas, B., Solomonson, M., Ruderfer, D. M., Kavanagh, D., et al. (2017). The ExAC browser: displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 45, D840–D845. doi: 10.1093/nar/gkw971
Katsanis, N., Ansley, S. J., Badano, J. L., Eichers, E. R., Lewis, R. A., Hoskins, B. E., et al. (2001). Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 293, 2256–2259. doi: 10.1126/science.1063525
Kenny, J., Forsythe, E., Beales, P., and Bacchelli, C. (2017). Toward personalized medicine in Bardet-Biedl syndrome. Per. Med. 14, 447–456. doi: 10.2217/pme-2017-0019
Khanna, H., Davis, E. E., Murga-Zamalloa, C. A., Estrada-Cuzcano, A., Lopez, I., den Hollander, A. I., et al. (2009). A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet. 41, 739–745. doi: 10.1038/ng.366
Khera, A. V., Chaffin, M., Aragam, K. G., Haas, M. E., Roselli, C., Choi, S. H., et al. (2018). Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224. doi: 10.1038/s41588-018-0183-z
Kim, J., Lee, J. E., Heynen-Genel, S., Suyama, E., Ono, K., Lee, K., et al. (2010). Functional genomic screen for modulators of ciliogenesis and cilium length. Nature 464, U1048–U1114. doi: 10.1038/nature08895
Kim, Y. B., Komor, A. C., Levy, J. M., Packer, M. S., Zhao, K. T., and Liu, D. R. (2017). Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376. doi: 10.1038/nbt.3803
King, S. M., and Patel-King, R. S. (2016). Planaria as a model system for the analysis of ciliary assembly and motility. Methods Mol. Biol. 1454, 245–254. doi: 10.1007/978-1-4939-3789-9_16
Knopp, C., Rudnik-Schoneborn, S, Eggermann, T., Bergmann, C., Begemann, M., Schoner, K., et al. (2015). Syndromic ciliopathies: from single gene to multi gene analysis by SNP arrays and next generation sequencing. Mol. Cell. Probes 29, 299–307. doi: 10.1016/j.mcp.2015.05.008
Knowles, M. R., Ostrowski, L. E., Leigh, M. W., Sears, P. R., Davis, S. D., Wolf, W. E., et al. (2014). Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am. J. Respir. Crit. Care Med. 189, 707–717. doi: 10.1164/rccm.201311-2047OC
Knowles, M. R., Zariwala, M., and Leigh, M. (2016). Primary ciliary Dyskinesia. Clin. Chest Med. 37, 449–461. doi: 10.1016/j.ccm.2016.04.008
Koblan, L. W., Doman, J. L., Wilson, C., Levy, J. M., Tay, T., Newby, G. A., et al. (2018). Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 36, 843–846. doi: 10.1038/nbt.4172
Kodra, Y., Weinbach, J., Posada-de-la-Paz, M., Coi, A., Lemonnier, S. L., van Enckevort, D., et al. (2018). Recommendations for improving the quality of rare disease registries. Int. J. Environ. Res. Public Health 15:E1644. doi: 10.3390/ijerph15081644
Köhler, S., Vasilevsky, N. A., Engelstad, M., Foster, E., McMurry, J., Ayme, S., et al. (2017). The human phenotype ontology in 2017. Nucleic Acids Res. 45, D865–D876. doi: 10.1093/nar/gkw1039
Komlosi, K., Diederich, S., Fend-Guella, D. L., Bartsch, O., Winter, J., Zechner, U., and Schweiger, S. (2018). Targeted next-generation sequencing analysis in couples at increased risk for autosomal recessive disorders. Orphanet J. Rare Dis. 13:23. doi: 10.1186/s13023-018-0763-0
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi: 10.1038/nature17946
Konishi, S., Gotoh, S., Tateishi, K., Yamamoto, Y., Korogi, Y., Nagasaki, T., et al. (2016). Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Rep. 6, 18–25. doi: 10.1016/j.stemcr.2015.11.010
Krusche, P., Trigg, L., Boutros, P. C., Mason, C. E., De La Vega, F. M., Moore, B. L., et al. (2018). Best practices for benchmarking germline small variant calls in human genomes. bioRXiv[Preprnt]:270157. doi: 10.1101/270157
Kuek, L. E., Griffin, P., Martinello, P., Graham, A. N., Kalitsis, P., Robinson, P. J., et al. (2018). Identification of an immortalized human airway epithelial cell line with dyskinetic cilia. Am. J. Respir. Cell Mol. Biol. 59, 375–382. doi: 10.1165/rcmb.2017-0188OC
Kuwahara, A., Ozone, C., Nakano, T., Saito, K., Eiraku, M., and Sasai, Y. (2015). Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat. Commun. 6:6286. doi: 10.1038/ncomms7286
Lakowski, J., Welby, E., Budinger, D., Di Marco, F., Di Foggia, V., Bainbridge, J. W. B., et al. (2018). Isolation of human photoreceptor precursors via a cell surface marker panel from stem cell-derived retinal organoids and fetal retinae. Stem Cells 36, 709–722. doi: 10.1002/stem.2775
Landrum, M. J., and Kattman, B. L. (2018). ClinVar at five years: delivering on the promise. Hum. Mutat. 39, 1623–1630. doi: 10.1002/humu.23641
Langousis, G., and Hill, K. L. (2014). Motility and more: the flagellum of Trypanosoma brucei. Nat. Rev. Microbiol. 12, 505–518. doi: 10.1038/nrmicro3274
Lee, J. E., and Gleeson, J. G. (2011). A systems-biology approach to understanding the ciliopathy disorders. Genome Med. 3:59. doi: 10.1186/gm275
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Li, M. M., Datto, M., Duncavage, E. J., Kulkarni, S., Lindeman, N. I., Roy, S., et al. (2017). Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology. American society of clinical oncology, and college of American pathologists. J. Mol. Diagn. 19, 4–23. doi: 10.1016/j.jmoldx.2016.10.002
Li, Y., Yagi, H., Onuoha, E. O., Damerla, R. R., Francis, R., Furutani, Y., et al. (2016). DNAH6 and its interactions with PCD genes in heterotaxy and primary ciliary dyskinesia. PLoS Genet. 12:e1005821. doi: 10.1371/journal.pgen.1005821
Lindstrand, A., Frangakis, S., Carvalho, C. M., Richardson, E. B., McFadden, K. A., Willer, J. R., et al. (2016). Copy-number variation contributes to the mutational load of bardet-biedl syndrome. Am. J. Hum. Genet. 99, 318–336. doi: 10.1016/j.ajhg.2015.04.023
Lord, J., Gallone, G., Short, P. J., McRae, J. F., Ironfield, H., Wynn, E. H., et al. (2018). Pathogenicity and selective constraint on variation near splice sites. Genome Res. 29:159–170. doi: 10.1101/256636
Lucas, J. S., Alanin, M. C., Collins, S., Harris, A., Johansen, H. K., Nielsen, K. G., et al. (2017a). Clinical care of children with primary ciliary dyskinesia. Expert Rev. Respir. Med. 11, 779–790. doi: 10.1080/17476348.2017.1360770
Lucas, J. S., Barbato, A., Collins, S. A., Goutaki, M., Behan, L., Caudri, D., et al. (2017b). European respiratory society guidelines for the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 49:1601090. doi: 10.1183/13993003.01090-2016
Lucas, J. S., Burgess, A., Mitchison, H. M., Moya, E., Williamson, M., Hogg, C., et al. (2014). Diagnosis and management of primary ciliary dyskinesia. Arch. Dis. Child. 99, 850–856. doi: 10.1136/archdischild-2013-304831
Marshall, C. R., Scherer, S. W., Zariwala, M. A., Lau, L., Paton, T. A., Stockley, T., et al. (2015). Whole-exome sequencing and targeted copy number analysis in primary ciliary dyskinesia. G3 (Bethesda). 5, 1775–1781. doi: 10.1534/g3.115.019851
Marshall, R. A., and Osborn, D. P. (2016). Zebrafish: a vertebrate tool for studying basal body biogenesis, structure, and function. Cilia 5:16. doi: 10.1186/s13630-016-0036-2
McIntyre, J. C., Davis, E. E., Joiner, A., Williams, C. L., Tsai, I. C., Jenkins, P. M., et al. (2012). Gene therapy rescues cilia defects and restores olfactory function in a mammalian ciliopathy model. Nat. Med. 18, 1423–1428. doi: 10.1038/nm.2860
McIntyre, J. C., Williams, C. L., and Martens, J. R. (2013). Smelling the roses and seeing the light: gene therapy for ciliopathies. Trends Biotechnol. 31, 355–363. doi: 10.1016/j.tibtech.2013.03.005
Mestek-Boukhibar, L., Clement, E., Jones, W. D., Drury, S., Ocaka, L., Gagunashvili, A., et al. (2018). Rapid Paediatric Sequencing (RaPS): comprehensive real-life workflow for rapid diagnosis of critically ill children. J. Med. Genet. 55, 721–728. doi: 10.1136/jmedgenet-2018-105396
Meyer, J. S., Howden, S. E., Wallace, K. A., Verhoeven, A. D., Wright, L. S., Capowski, E. E., et al. (2011). Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 29, 1206–1218. doi: 10.1002/stem.674
Mitchison, H. M., and Shoemark, A. (2017). Motile cilia defects in diseases other than primary ciliary dyskinesia: The contemporary diagnostic and research role for transmission electron microscopy. Ultrastruct. Pathol. 41, 415–427. doi: 10.1080/01913123.2017.1370050
Mitchison, H. M., and Valente, E. M. (2017). Motile and non-motile cilia in human pathology: from function to phenotypes. J. Pathol. 241, 294–309. doi: 10.1002/path.4843
Moayyeri, A., Hammond, C. J., Valdes, A. M., and Spector, T. D. (2013). Cohort Profile: TwinsUK and healthy ageing twin study. Int. J. Epidemiol. 42, 76–85. doi: 10.1093/ije/dyr207
Mok, C. A., and Héon, E. (2012). Caenorhabditis elegans as a model organism for ciliopathies and related forms of photoreceptor degeneration. Adv. Exp. Med. Biol. 723, 533–538. doi: 10.1007/978-1-4614-0631-0_67
Molinari, E., and Sayer, J. A. (2017). Emerging treatments and personalised medicine for ciliopathies associated with cystic kidney disease. Exp. Opin. Orph. Drugs 5, 785–798. doi: 10.1080/21678707.2017.1372282
Moore, A., Escudier, E., Roger, G., Tamalet, A., Pelosse, B., Marlin, S., et al. (2006). RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J. Med. Genet. 43, 326–333. doi: 10.1136/jmg.2005.034868
Moss, C., and Wernham, A. (2018). The 100 000 Genomes Project: feeding back to patients. BMJ 361:k2441. doi: 10.1136/bmj.k2441
Mossotto, E., Ashton, J. J., Pengelly, R. J., Beattie, R. M., MacArthur, B. D., and Ennis, S. (2018). GenePy - a score for estimating gene pathogenicity in individuals using next-generation sequencing data. bioRXiv[Preprint]:336701. doi: 10.1101/336701
Norris, D. P., and Grimes, D. T. (2012). Mouse models of ciliopathies: the state of the art. Dis. Model. Mech. 5, 299–312. doi: 10.1242/dmm.009340
Nouri, N., Nouri, N., Tirgar, S., Soleimani, E., Yazdani, V., Zahedi, F., et al. (2017). Consanguineous marriages in the genetic counseling centers of Isfahan and the ethical issues of clinical consultations. J. Med. Ethics Hist. Med. 10:12.
Ormondroyd, E., Mackley, M. P., Blair, E., Craft, J., Knight, J. C., Taylor, J. C., et al. (2018). “Not pathogenic until proven otherwise”: perspectives of UK clinical genomics professionals toward secondary findings in context of a Genomic Medicine Multidisciplinary Team and the 100,000 Genomes Project. Genet. Med. 20, 320–328. doi: 10.1038/gim.2017.157
Oud, M. M., Lamers, I. J., and Arts, H. H. (2017). Ciliopathies: genetics in pediatric medicine. J. Pediatr. Genet. 6, 18–29. doi: 10.1055/s-0036-1593841
Paff, T., Kooi, I. E., Moutaouakil, Y., Riesebos, E., Sistermans, E. A., Daniels, H., et al. (2018). Diagnostic yield of a targeted gene panel in primary ciliary dyskinesia patients. Hum. Mutat. 39, 653–665. doi: 10.1002/humu.23403
Parisi, M., and Glass, I. (1993). “Joubert syndrome,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, and A. Amemiya. (Seattle, WA: University of Washington, Seattle), 1–59.
Perantoni, A., and Berman, J. J. (1979). Properties of Wilms' tumor line (TuWi) and pig kidney line (LLC-PK1) typical of normal kidney tubular epithelium. In Vitro 15, 446–454. doi: 10.1007/BF02618414
Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T., and Sandhu, M. S. (2018). Long reads: their purpose and place. Hum. Mol. Genet. 27, R234–R241. doi: 10.1093/hmg/ddy177
Project Team, S. G (2015) The saudi human genome program: an oasis in the desert of Arab medicine is providing clues to genetic disease. IEEE Pulse 6, 22–26 doi: 10.1109/MPUL.2015.2476541.
Rafferty, K. A., and Sherwin, R. W. (1969). The length of secondary chromosomal constrictions in normal individuals and in a nucleolar mutant of Xenopus laevis. Cytogenetics 8, 427–438. doi: 10.1159/000130054
Rambhatla, L., Chiu, C. P., Glickman, R. D., and Rowe-Rendleman, C. (2002). In vitro differentiation capacity of telomerase immortalized human RPE cells. Invest. Ophthalmol. Vis. Sci. 43, 1622–1630.
Ran, F. A., Hsu, P. D., Lin, C. Y., Gootenberg, J. S., Konermann, S., Trevino, A. E., et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. doi: 10.1016/j.cell.2013.08.021
Rauchman, M. I., Nigam, S. K., Delpire, E., and Gullans, S. R. (1993). An osmotically tolerant inner medullary collecting duct cell line from an SV40 transgenic mouse. Am. J. Physiol. 265(3 Pt 2), F416–424. doi: 10.1152/ajprenal.1993.265.3.F416
Reiter, J. F., and Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 18, 533–547. doi: 10.1038/nrm.2017.60
Robinson, P. N., Köhler, S., Bauer, S., Seelow, D., Horn, D., and Mundlos, S. (2008). The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 83, 610–615. doi: 10.1016/j.ajhg.2008.09.017
Samuel, G. N., and Farsides, B. (2017). The UK's 100,000 Genomes Project: manifesting policymakers' expectations. New Genet. Soc. 36, 336–353. doi: 10.1080/14636778.2017.1370671
Sawyer, S. L., Hartley, T., Dyment, D. A., Beaulieu, C. L., Schwartzentruber, J., Smith, A., et al. (2016). Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet. 89, 275–284. doi: 10.1111/cge.12654
Schmidts, M., Arts, H. H., Bongers, E. M., Yap, Z., Oud, M. M., Antony, D., et al. (2013). Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J. Med. Genet. 50, 309–323. doi: 10.1136/jmedgenet-2012-101284
Schmidts, M. M., and Mitchison, H. M. (2018). “Severe skeletal abnormalities caused by defects in retrograde intraflagellar transport dyneins,” in Dyneins: Structure, Biology and Disease, ed S.M. King (Academic Press, Elsevier Inc.), 356–401.
Schock, E. N., Chang, C. F., Youngworth, I. A., Davey, M. G., Delany, M. E., and Brugmann, S. A. (2016). Utilizing the chicken as an animal model for human craniofacial ciliopathies. Dev. Biol. 415, 326–337. doi: 10.1016/j.ydbio.2015.10.024
Shoemark, A., Moya, E., Hirst, R. A., Patel, M. P., Robson, E. A., Hayward, J., et al. (2018). High prevalence of CCDC103 p.His154Pro mutation causing primary ciliary dyskinesia disrupts protein oligomerisation and is associated with normal diagnostic investigations. Thorax 73, 157–166. doi: 10.1136/thoraxjnl-2017-209999
Simpson, J. T., Workman, R. E., Zuzarte, P. C., David, M., Dursi, L. J., and Timp, W. (2017). Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14:407. doi: 10.1038/nmeth.4184
Singla, V., and Reiter, J. F. (2006). The primary cilium as the cell's antenna: signaling at a sensory organelle. Science 313, 629–633. doi: 10.1126/science.1124534
Slaats, G. G., Saldivar, J. C., Bacal, J., Zeman, M. K., Kile, A. C., Hynes, A. M., et al. (2015). DNA replication stress underlies renal phenotypes in CEP290-associated Joubert syndrome. J. Clin. Invest. 125, 3657–3666. doi: 10.1172/JCI80657
Soden, S. E., Saunders, C. J., Willig, L. K., Farrow, E. G., Smith, L. D., Petrikin, J. E., et al. (2014). Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 6:265ra168. doi: 10.1126/scitranslmed.3010076
Song, J. Y., Aravand, P., Nikonov, S., Leo, L., Lyubarsky, A., Bennicelli, J. L., et al. (2018). Amelioration of neurosensory structure and function in animal and cellular models of a congenital blindness. Mol. Ther. 26, 1581–1593. doi: 10.1016/j.ymthe.2018.03.015
Song, Z., Zhang, X., Jia, S., Yelick, P. C., and Zhao, C. (2016). Zebrafish as a model for human ciliopathies. J. Genet. Genomics 43, 107–120. doi: 10.1016/j.jgg.2016.02.001
Spassky, N., and Meunier, A. (2017). The development and functions of multiciliated epithelia. Nat. Rev. Mol. Cell Biol. 18, 423–436. doi: 10.1038/nrm.2017.21
Srivastava, S., Ramsbottom, S. A., Molinari, E., Alkanderi, S., Filby, A., White, K., et al. (2017). A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum. Mol. Genet. 26, 4657–4667. doi: 10.1093/hmg/ddx347
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677. doi: 10.1007/s00439-017-1779-6
Thiel, C., Kessler, K., Giessl, A., Dimmler, A., Shalev, S. A., von der Haar, S., et al. (2011). NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 88, 106–114. doi: 10.1016/j.ajhg.2010.12.004
Turnbull, C., Scott, R. H., Thomas, E., Jones, L., Murugaesu, N., Pretty, F. B., et al. (2018). The 100,000 genomes project: bringing whole genome sequencing to the NHS. BMJ 361:k1687. doi: 10.1136/bmj.k1687
Vervoort, R., Lennon, A., Bird, A. C., Tulloch, B., Axton, R., Miano, M. G., et al. (2000). Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 25, 462–466. doi: 10.1038/78182
Vincensini, L., Blisnick, T., and Bastin, P. (2011). 1001 model organisms to study cilia and flagella. Biol. Cell 103, 109–130. doi: 10.1042/BC20100104
Walentek, P., and Quigley, I. K. (2017). What we can learn from a tadpole about ciliopathies and airway diseases: using systems biology in Xenopus to study cilia and mucociliary epithelia. Genesis 55, 1–2. doi: 10.1002/dvg.23001
Waters, A. M., and Beales, P. L. (2011). Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 26, 1039–1056. doi: 10.1007/s00467-010-1731-7
Welch, B. M., Wiley, K., Pflieger, L., Achiangia, R., Baker, K., Hughes-Halbert, C., et al. (2018). Review and comparison of electronic patient-facing family health history tools. J. Genet. Couns. 27, 381–391. doi: 10.1007/s10897-018-0235-7
Wheway, G., and Johnson, C. A. (2014). “Meckel-Gruber syndrome,” in Ciliopathies, ed T. D. Kenny and P. L. Beales (Oxford: Oxford University Press), 132–149.
Wheway, G., Nazlamova, L., and Hancock, J. T. (2018). Signaling through the primary cilium. Front. Cell Dev. Biol. 6:8. doi: 10.3389/fcell.2018.00008
Wheway, G., Schmidts, M., Mans, D. A., Szymanska, K., Nguyen, T. M., Racher, H., et al. (2015). An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 17, 1074–1087. doi: 10.1038/ncb3201
Willey, C. J., Blais, J. D., Hall, A. K., Krasa, H. B., Makin, A. J., and Czerwiec, F. S. (2017). Prevalence of autosomal dominant polycystic kidney disease in the European Union. Nephrol. Dial. Transpl. 32, 1356–1363. doi: 10.1093/ndt/gfw240
Wolf, M. T., and Hildebrandt, F. (2011). Nephronophthisis. Pediatr. Nephrol. 26, 181–194. doi: 10.1007/s00467-010-1585-z
Wood, A. J., Lo, T. W., Zeitler, B., Pickle, C. S., Ralston, E. J., Lee, A. H., et al. (2011). Targeted genome editing across species using ZFNs and TALENs. Science 333:307. doi: 10.1126/science.1207773
Wright, A. F., Chakarova, C. F., Abd El-Aziz, M. M., and Bhattacharya, S. S. (2010). Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 11, 273–284. doi: 10.1038/nrg2717
Wright, C. F., Fitzgerald, T. W., Jones, W. D., Clayton, S., McRae, J. F., van Kogelenberg, M., et al. (2015). Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 385, 1305–1314. doi: 10.1016/S0140-6736(14)61705-0
Wu, Z., Hiriyanna, S., Qian, H., Mookherjee, S., Campos, M. M., Gao, C., et al. (2015). A long-term efficacy study of gene replacement therapy for RPGR-associated retinal degeneration. Hum. Mol. Genet. 24, 3956–3970. doi: 10.1093/hmg/ddv134
You, L., Klena, N. T., Gabriel, G. C., Liu, X., Kim, A. J., and Lemke, K (2015) Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 521, 520–524. doi: 10.1038/nature14269.
Zaki, M. S., Sattar, S., Massoudi, R. A., and Gleeson, J. G. (2011). Co-occurrence of distinct ciliopathy diseases in single families suggests genetic modifiers. Am. J. Med. Genet. A 155A, 3042–3049. doi: 10.1002/ajmg.a.34173
Keywords: 100,000 Genome Project, ciliopathies, cilia, genomics, genetics
Citation: Wheway G, Genomics England Research Consortium and Mitchison HM (2019) Opportunities and Challenges for Molecular Understanding of Ciliopathies–The 100,000 Genomes Project. Front. Genet. 10:127. doi: 10.3389/fgene.2019.00127
Received: 22 November 2018; Accepted: 05 February 2019;
Published: 11 March 2019.
Edited by:
Carlo Iomini, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
John Andrew Sayer, Newcastle University, United KingdomTheodora Katsila, University of Patras, Greece
Copyright © 2019 Wheway, Genomics England Research Consortium and Mitchison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannah M. Mitchison, aC5taXRjaGlzb25AdWNsLmFjLnVr
†Gabrielle Wheway, orcid.org/0000-0002-0494-0783