 Mengli Wang
Mengli Wang Lixia Qin
Lixia Qin Beisha Tang
Beisha Tang- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2Laboratory of Medical Genetics, Central South University, Changsha, China
- 3National Clinical Research Center for Geriatric Disorders, Changsha, China
- 4Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
- 5Parkinson’s Disease Center of Beijing Institute for Brain Disorders, Beijing, China
Alzheimer’s disease (AD) is a progressive and devastating neurodegenerative disorder. It is the leading cause of dementia in the world’s rapidly growing aging population. The characteristics of AD are memory loss and cognitive impairment, meaning patients cannot carry out their daily activities independently. The increase of AD cases poses heavy burdens on families, society and the economy. Despite frequent efforts being made to research the etiology of AD, the causes of AD remain unknown, and no curative treatments are available yet. The pathological hallmarks of AD are amyloid plaques and neurofibrillary tangles in the brain. MicroRNAs are endogenous ∼22 nucleotides non-coding RNAs that could regulate gene expression at a post-transcriptional level by transcript degradation or translation repression. MicroRNAs are involved in many biological processes and diseases, particularly multifactorial diseases, providing an excellent tool with which to research the mechanisms of these diseases. AD is a multifactorial disorder, and accumulating evidence shows that microRNAs play a critical role in the pathogenesis of AD. In this review, we will highlight the effect of microRNAs in different pathological processes throughout AD progression.
Introduction
Alzheimer’s disease (AD) is an irreversible progressive neurodegenerative disorder, and is the main cause of dementia in the global elderly population. In 2010, it was estimated that there were 35.6 million people living with dementia across the globe; these numbers are expected to double every 20 years until 2050 (Prince et al., 2013). The characteristics of AD are cognitive impairment and memory loss, causing most AD patients to lose the ability to perform daily activities independently. The cost of caring for these patients is rising, with increasing numbers of AD incidents around the world, posing heavy burdens for individuals, families and society. Although mounting efforts have been made to research the etiology of AD, the causes remain unknown, and effective treatments are not yet available (Idda et al., 2018). The pathological hallmarks of AD are senile plaques consisting of accumulated β-amyloid peptides (Aβ) and neurofibrillary tangles (NFTs) primarily containing highly phosphorylated Tau (Ballard et al., 2011). The two most commonly accepted hypotheses, the Aβ hypothesis and the Tau hypothesis, are also based on these two pathological characteristics. The amyloid cascade hypothesis (see Figure 1) suggests that the imbalance between the production and clearance of Aβ is the key trigger of a cascade of events that leads to AD (Blennow et al., 2006; Ballard et al., 2011). The Aβs are produced from processing the amyloid precursor protein (Richter et al., 2012) through sequential enzymes digested by β-secretase (beta-site amyloid precursor protein cleaving enzyme1, BACE1) and γ-secretase (consisting of presenilin1 and presenilin2) (Querfurth and LaFerla, 2010). In physiology, Aβ-degrading proteases mediate proteolytic degradation and receptor-modulated endocytosis function to clear the Aβ (Hauser and Ryan, 2013). The proteolytic system includes neprilysin and an insulin-degrading enzyme (Qiu et al., 1998; Iwata et al., 2001; Kanemitsu et al., 2003); the low density lipoprotein receptor family is involved in the receptor system. The most toxic forms of Aβ are soluble oligomers and intermediate amyloids (Walsh and Selkoe, 2007), which can cause synaptic loss, neurotoxicity, neuron apoptosis, inflammation and mitochondrion dysfunction (Walsh et al., 2005; Hauptmann et al., 2006; Klyubin et al., 2008; Reddy and Beal, 2008). Tau are microtubule-associated proteins which stabilize the microtubule and promote vesicle transportation. In neurons, the microtubules are essential for the maintenance of neuronal structure, axonal transportation and neuronal plasticity (Lindwall and Cole, 1984). Highly phosphorylated Tau can lose its stabilization ability and may start to self-form NFTs. Tau are phosphorylated by types of kinases, and the phosphate residues are removed by phosphatases (Iqbal et al., 2005). The imbalance between the hyper-phosphorylated and de-phosphorylated forms of Tau could lead to the formation of NFTs (see Figure 2). Tauopathies are considered to be an indicator of the severity of AD.
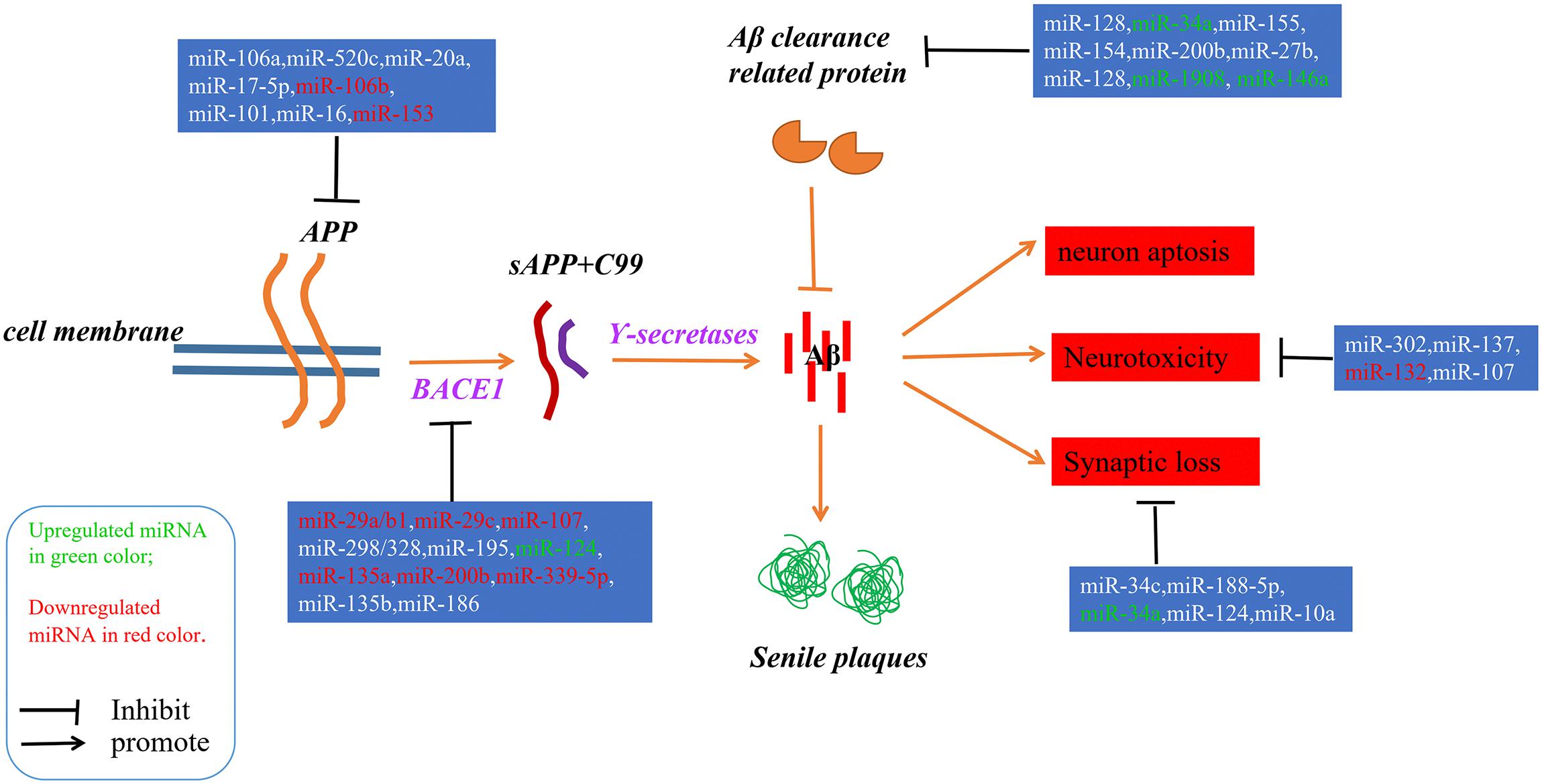
Figure 1. A schematic of the Aβ hypothesis of AD pathogenesis and the microRNAs involved in each step. The Aβs were produced as a result of processing the amyloid precursor protein (Richter et al., 2012) through a sequential enzyme digested by BACE1 and γ-secretase; an imbalance between the production and clearance of Aβ is the key trigger of AD.
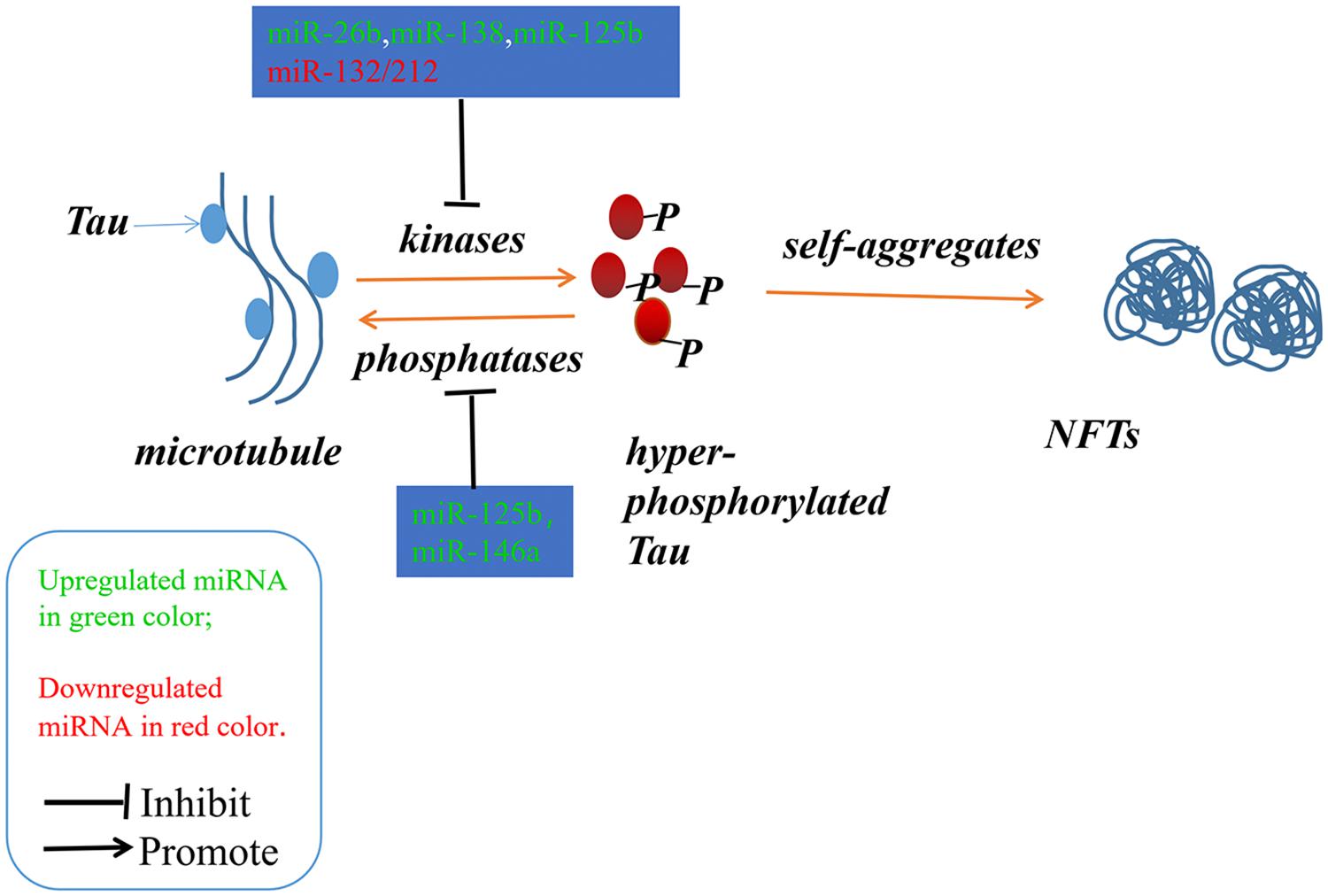
Figure 2. The imbalance between the hyper-phosphorylated and de-phosphorylated processes of Tau could lead to the formation of NFTs. The microRNAs involved in the phosphorylated and de-phosphorylated processes play a role in AD pathogenesis.
MicroRNAs, a class of non-coding RNAs, have been acknowledged as important regulators for post-transcriptional gene expression by either repressing translation or degrading target mRNAs (Maoz et al., 2017). Since their discovery, microRNAs have been identified as the regulators most frequently implicated in many critical biological events, such as development, growth, differentiation and neurodegenerative processes (Huang et al., 2011; Hammond, 2015). One microRNA could target numerous genes and one gene could be regulated by multiple microRNAs, making microRNAs a potential tool to investigate multifactorial diseases, for example AD (Iqbal and Grundke-Iqbal, 2010). The study about examining the 13-brain associated microRNAs abundance in human hippocampus samples from fetal and aged adults, age-matched AD patients suggest that misregulation and alterations of specific microRNAs might make contributions to the process of AD (Lukiw, 2007). Afterward, tremendous researches have demonstrated the alterations of several microRNAs between AD patients and age-matched control, further prove that microRNAs might play an important role in the pathogenesis of AD (Goodall et al., 2013). In this review, we will focus on the microRNAs involved in the two most accepted hypotheses of AD pathogenesis: the Aβ hypothesis and the Tau hypothesis.
MicroRNAs Involved in the Aβ Hypothesis
Since microRNAs have been used to research AD, accumulating microRNAs have been identified as regulators in the process of Aβ production and clearance. The amyloid cascade hypothesis mentions that the imbalance between the production and clearance of Aβ42 could trigger synaptic loss and neurotoxicity. Section “MicroRNAs Involved in the Aβ Hypothesis” describes the microRNAs implicated in the Aβ hypothesis (as shown in Figure 1 and Tables 1, 2).
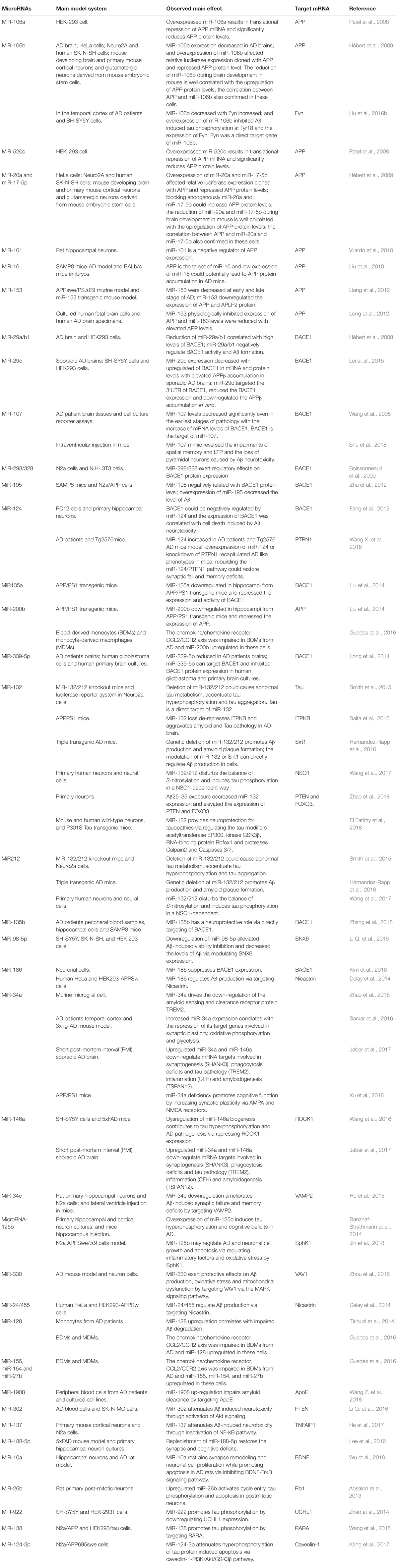
Table 1. A summary of the microRNAs involved in AD.
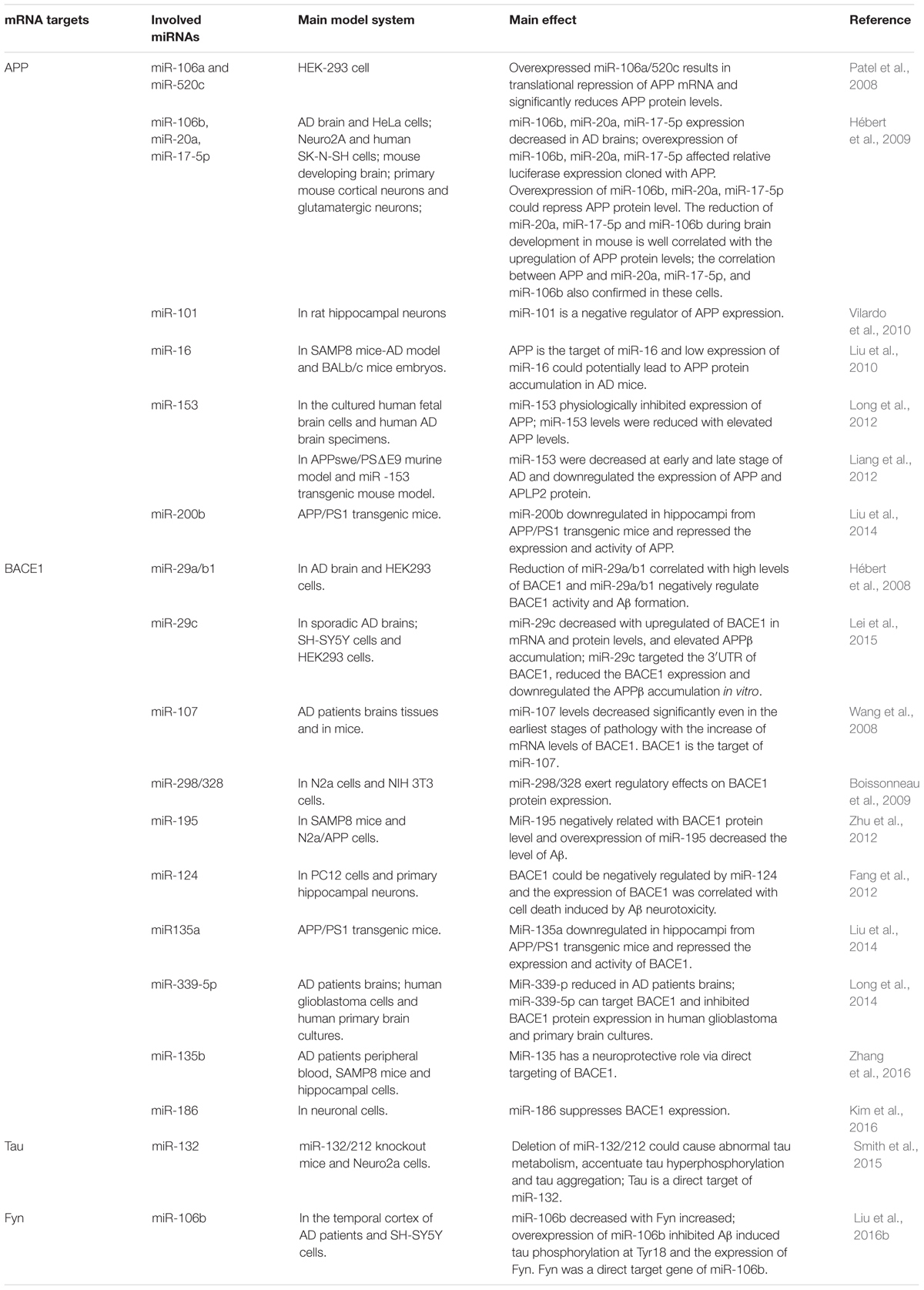
Table 2. microRNAs modulating Aβ plaques formation and tau phosphorylation.
MicroRNAs Involved in Aβ Production
Mounting evidence has shown that specific microRNAs play a key role in regulating the expression of APP and BACE1, which restrict the production of Aβ.
MicroRNAs Regulate APP Expression
Accumulating evidence demonstrates that increased APP expression could promote Aβ production, resulting in neurotoxicity, synaptic failure and, eventually, dementia (Patel et al., 2008). Many microRNAs have participated in the process of regulating APP expression; for example, Patel et al. (2008) found that miR-106a/520c could bind the 3′UTR of APP and repress the expression of APP in human cell lines. Later, the Hebert group found that the miR-20 families (miR-20a, miR-17-5p, and miR-106b) could also regulate APP expression via the binding of APP 3′UTR. In addition, in accordance with miR-20 overexpression in neuron cells, the expression level of APP decreased (Hébert et al., 2009). Similarly, miR-101 (Vilardo et al., 2010), miR-16 (Liu et al., 2010), and miR-153 (Long et al., 2012) have also been identified as negative regulators of APP expression both in vitro and in vivo. Moreover, the low level of miR-153 contributed to the accumulation of Aβ in sporadic AD patients.
MicroRNAs Regulate BACE1 Expression
The division of APP by BACE1 is the first and rate-limit step for Aβ formation, and upregulated BACE1 expression levels and enzymatic activities have been detected in sporadic AD brains (Yang et al., 2003). Several microRNAs regulating BACE1 expression and activity have been found; for example, the miR-29 family implicated in regulating BACE1 expression has been well studied. Hébert et al. (2008) performed the microRNAs expression profiles of sporadic AD patients, and found that the expression levels of the miR29a/b1 cluster decreased significantly with high levels of BACE1 expression in sporadic AD brains. Further analysis showed that miR-29a/b-1 could regulate BACE1 expression in vitro and in vivo. Moreover, it was validated that decreased levels of miR-29a/b-1 could promote the production of Aβ and contribute to the pathogenesis of AD (Hébert et al., 2008). Afterwards, miR-29c, another miR-29 family member, was also found to be downregulated with abnormally high levels of BACE1 in sporadic AD brains; additional validation experiments found that overexpressed miR-29c could induce BACE1 downregulation in human neuroblastoma SH-SY5Y cells via the binding of BACE1 3′UTR (Lei et al., 2015).
Another well studied microRNA is miR-107. The expression of miR-107 decreased significantly with BACE1 increased in AD patients. Then, it was revealed that miR-107 regulated the expression of BACE1 through recognizing and binding the 3′UTR of BACE1 in a cell culture reporter assay (Wang et al., 2008). Afterwards, Nelson and Wang also demonstrated that miR-107 levels negatively correlated with BACE1 mRNA levels, leading to Aβ accumulation (Nelson and Wang, 2010). Moreover, some studies have shown that miR-107 was implicated in preventing the neurotoxicity and blood–brain-barrier dysfunction induced by Aβ (Liu et al., 2016a; Shu et al., 2018), making miR-107 a potential drug target for researchers (Parsi et al., 2015).
Other negative regulators of BACE1 also include miR-298/328 (Boissonneault et al., 2009), microRNA-195 (Zhu et al., 2012), miR-135a (Liu et al., 2014), microRNA-135b (Zhang et al., 2016), miR-339-5p (Long et al., 2014), and miR-186 (Kim et al., 2016), all of which negatively correlated with BACE1 expression and exerted regulatory effects by binding the 3’UTR of BACE1.
Other MicroRNAs Involved in Aβ Production
Recent cases have reported that the miR-132/212 cluster plays an important role in Aβ production; for example, Hernandez-Rapp et al. (2016) found that levels of microRNA-132/212 cluster decreased in AD patients, and the deficiency of microRNA-132/212 could promote Aβ production and senile plaque deposition in AD triple transgenic mice. In another study, Salta et al. (2016) also confirmed that the loss of miR-132 could promote β amyloid accumulation through regulating inositol 1,4,5-trisphosphate 3-kinase B-ITPKB in AD mouse models. Li Q. et al. (2016) found that miR-98-5p could repress the expression of sorting nexin6 (SNX6), which is involved in increasing the levels of Aβ40, Aβ42, BACE1, soluble amyloid precursor protein β (sAPPβ), and membrane-associated APP β-carboxyl terminal fragments in 293T and SK-N-SH cells. Other microRNAs implicated in Aβ production include miR-34a, miR-146 (Jaber et al., 2017), microRNA-125b (Jin et al., 2018), miR-330 (Zhou et al., 2018), miR-24, miR-186, and miR-455 (Delay et al., 2014). All these provide further evidence that specific microRNAs play a critical role in the pathogenesis of AD.
MicroRNAs Involved in Aβ Clearance
Aβ deposits result from an imbalance between the production and clearance of β amyloid peptides. Thus, the dysfunctional clearance of Aβ could also contribute to the accumulation of Aβ peptides. As such, in Section “MicroRNAs Involved in Aβ Clearance” we describe a number of microRNAs involved in Aβ clearance.
The endosomal–lysosomal system degrades accumulated proteins and functions as a protective factor in the central nervous system. Tiribuzi et al. (2014) found that upregulated miR-128 could impair the clearance of Aβ through the targeting of lysosomal system enzymes in monocytes in sporadic AD patients. Aβ degradation improved when miR-128 was inhibited in monocytes from AD patients (Tiribuzi et al., 2014). Another microRNA involved in Aβ clearance was miR-34a which, when upregulated in AD patients, repressed the clearance of Aβ by inhibiting the expression of TREM2 (Zhao et al., 2016). TREM2, a myeloid/microglial cell surface amyloid sensor-receptor implicated in recognizing and digesting Aβ and extracellular amyloidogenic debris, plays a key role in the clearance of over-expressed Aβ (Wang et al., 2015; Song et al., 2016; Ulrich and Holtzman, 2016). The expression of TREM2 also correlated with blood-derived monocytes (BDMs) (Guedes et al., 2016), which have been implicated in the clearance of β amyloid deposits in AD brains (Michaud et al., 2013). Immune-related microRNAs (miR-155,-154,-200b,-27b, and 128) which, when expressed differentially in CCL2/CCR2 (chemokine/chemokine receptor) axis impaired BDMs, reportedly participate in the process of Aβ clearance mediated by BDMs (Guedes et al., 2016). Additionally, several researchers have revealed that differentially expressed apoE isoforms also play a role in Aβ clearance (Deane et al., 2008; Liu et al., 2013). It has been validated that miR-1908 could inhibit ApoE expression in mRNA and protein levels in human macrophage cell line THP-1 and astrocytoma cell line U87. In AD patients, the level of miR-1908 was negatively related to ApoE expression, which suggested that miR-1908 played a crucial role in inhibiting Aβ clearance through repressing ApoE expression (Wang Z. et al., 2018).
MicroRNAs Involved in Aβ Induced Neurotoxicity
Aβ could induce neurotoxicity, and this section describes several microRNAs involved in this process. In 2016, it was discovered that miR-302 could restore Aβ induced neurotoxicity via the PTEN/Akt/Nrf2/HO-1 pathway in AD neurons (Li H. H. et al., 2016). He et al. (2017) found that miR-137 could reduce Aβ induced neurotoxicity by inhibiting the process of NF-κB via repressing TNFAIP1 expression in N2a cells. It has been reported by many groups that miR-132 decreased significantly in AD brains. Zhao et al. (2018) recently discovered that the overexpression of miR-132 in cultured cortical neurons could inhibit the neurotoxicity induced by Aβ via the miR-132/PTEN/AKT/FOXO3a pathway. In the same year, Shu et al. (2018) found that miR-107 could block Aβ induced neurotoxicity in mice; they injected miR-107 mimics, which could intraventricularly generate native miR-107, into mice and found that miR-107 could restore spatial memory impairment and pyramidal neurons loss caused by Aβ. It was reported that MiR-124 could alleviate neurotoxicity via regulating the expression of BACE-1 in AD cellular models (Fang et al., 2012). These microRNAs provide us with a new way to understand the pathogenesis mechanisms of AD.
MicroRNAs Involved in the Aβ Induced Synaptic Failure
Aβ could lead to the synaptic failure and numerous microRNAs involved in this process. It was reported that the level of miR-34c increased significantly in the transgenic mice model, AD patients and Aβ exposed neuron cells. Further investigation demonstrated that inhibiting the expression of miR-34c could rescue the synaptic dysfunction and memory loss induced by Aβ by up-regulating the expression of VAMP2 (Hu et al., 2015). Additionally, Sarkar et al. (2016) identified that miR-34a, another miR-34 family member, could target multiple genes involved in neuronal synaptic deficiency and energy metabolism. Moreover, further research found that the loss of miR-34a could improve cognitive functions through rescuing synaptic dysfunction by repressing the expression of AMPA and NMDA receptors (Xu et al., 2018).
Lee et al. (2016) found that miR-188-5p could restore the synaptic dysfunction and cognitive impairment caused by Aβ in transgenic mice; they also observed that the overexpression of miR-188-5p could alleviate the decrease of dendritic spine density in primary hippocampal neurons exposed to Aβ. Additionally, Wu et al. (2018) found that miR-10a was also a negative regulator in synapse remodeling and cell proliferation as a result of a reduction in BDNF-TrkB signals in AD rats. They confirmed that BDNF was a target of miR-10a using a dual luciferase gene reporter assay (Wu et al., 2018).
Recently, a novel pathway, the miR-124/PTPN1 pathway, has been reported to involve in synaptic transmission deficits. The level of miR-124 increased significantly with the expression of its target, PETN1, decreased dramatically in AD brains. The overexpression of miR-124 or the knockdown of PTPN1 in mice could induce AD-like phenotypes, containing synaptic transmissions and plasticity deficits. Moreover, the inhibition of miR-124 expression or the over-expression of PTPN1 could alleviate the synaptic deficits in AD model mice (Wang X. et al., 2018).
A noticeable thing is that Aβ could induce the synaptic loss, changes in neurotrophin and inflammations, conversely, these deficits could affect the clearance and degradation of Aβ. NF-κB, a pro-inflammatory transcription factor, could be activated by Aβ, and the activation of NF-κB could lead to the upregulated of six inducible miRNAs: miR-7, miR-9, miR-34a, miR-125b, miR-146a, and miR-155 (Zhao et al., 2015). Those NF-κB sensitive upregulated miRNAs in AD brain contributed much to the neuropathological characteristics. For example, upregulated miR-125b may impair the Aβ clearance via repressing the TREM2 expression, induce chronic innate-immune and inflammatory signaling by inhibiting the expression of CFH (complement factor H), repress neurotransmitter package and release, and decrease the neurotrophin within the brain through regulating the expression of SYN-2, 15-LOX, and VDR (synapsin-2, 15-lipoxygenase and vitamin D receptor). Similarly, miR-146a may be responsible for promoting amyloidogenesis through regulating the expression TSPAN12 (tetraspanin12) and/or failing to modulate the NF-κB through regulating IRAK-1 and IRAK-2 (interleukin-1 receptor associated kinase) (Zhao et al., 2015).
MicroRNAs Involved in the Tau Phosphorylated Imbalance
Neurofibrillary tangles were largely composed of hyper-phosphorylated Tau as a result of the imbalance between Tau phosphorylation and de-phosphorylation through regulating the expressions and activities of a number of related kinases and phosphatases (Ballatore et al., 2007). In Section “MicroRNAs Involved in the Tau Phosphorylated Imbalance,” we describe several microRNAs involved in the process of phosphorylation and de-phosphorylation (as shown in Figure 2 and Tables 1, 2).
It is reported that the expression levels of miR-125b are increased significantly in AD patients (Banzhaf-Strathmann et al., 2014; Ma et al., 2017; Jin et al., 2018). In both neuron cells and in mice, over-expression of miR-125b induced Tau hyper-phosphorylation through the targeting of phosphatases DUSP6 and PPP1CA, while inhibition of miR-125b induced a reduction in Tau phosphorylation and kinase expression/activity (Banzhaf-Strathmann et al., 2014). Moreover, Ma et al. (2017) found that overexpression of miR-125b could induce cell apoptosis and Tau hyper-phosphorylation by activating the cyclin-dependent kinase 5 (CDK5) and p35/25 in neuron cells. MiR-125b might be implicated in this process through targeting the forkhead box Q1(FOXQ1) directly (Ma et al., 2017).
Smith et al. (2015) found that the expression of Tau could be directly regulated by miR-132, and the deletion of miR-132/212 in mice could increase the expression, phosphorylation and accumulation of Tau. They also revealed that the delivery of miR-132 mimics into AD mice could reduce the metabolism of Tau (Smith et al., 2015). Wang et al. (2017) also reported that the downregulation of miR-132/212 could promote Tau phosphorylation and break the imbalance between Tau phosphorylation and de-phosphorylation via the NOS1-dependent pathway in primary human neurons and neural cells. miR-132 has been recognized in numerous studies as one of the most significant downregulated microRNAs, and it has been implicated in the process of Tau hyper-phosphorylation through the targeting of EP300, GSK3b, Rbfox1, proteases Calpain2 and caspases 3/7 (El Fatimy et al., 2018).
The expression level of miR-138 is up-regulated in AD patients. Wang et al. (2015) found that miR-138 could directly target the retinoic acid receptor alpha which could repress glycogen synthase kinase-3b (GSK-3β) activity. They further confirmed that miR-138 could promote Tau phosphorylation via the RARA/GSK-3β pathway in N2a/APP and HEK293/tau cells (Wang et al., 2015). The level of miR-26b also increased significantly in the pathological areas of postmortem AD brains. It was confirmed that the elevated levels of miR-26b contributed to the pathology of AD through cell cycle entry, Tau hyper-phosphorylation and apoptosis in post-mitotic neurons (Absalon et al., 2013).
MicroRNA-922 was also reported to contribute to the pathogenesis of AD via regulating the expressions of UCHL1. The levels of UCHL1 were decreased in sporadic AD patients, and the overexpression of UCHL1 rescued the synaptic and cognitive function in AD model mice. And in the AD model mice, the expression levels of miR-922 were upregulated with UCHL1 protein levels decreased. Zhao et al. (2014) found that UCHL1 was one target of miR-922, and the phosphorylation levels of Tau were negatively correlated with the expressions of UCHL1. Thus, they suggested that miR-922 involved in the pathogenesis of AD through regulating the phosphorylation levels of Tau by targeting UCHL1 (Zhao et al., 2014).
The expression levels of microRNA-146a was increased in AD patients’ brains, and the dysregulation of microRNA-146a biogenesis was involved in the tau hyperphosphorylation and AD pathogenesis. In neural cells, it was confirmed that miR-146a targeted the coiled-coil containing protein kinase1 (ROCK1) directly, and the inhibition of ROCK1 could induce abnormal tau phosphorylation, which was related with the low phosphorylation levels of the phosphatase and tensin homolog (PTEN). Additionally, the ROCK1 colocalised with the hyperphosphorylated tau in the early neurofibrillary tangles with decreased ROCK1 protein levels in AD patients. Moreover, in 5xFAD mice, the inhibition of miR-146a through intra-hippocampal delivery induced upregulated ROCK1 protein levels and repressed tau hyperphosphorylation, and restored memory function partly (Wang et al., 2016).
Similarly, the expression level of miR-106b was downregulated with the upregulation of Fyn in the temporal cortex of AD patients. In tau stably expressed SH-SY5Y cells (SH-SY5Y/tau), the overexpression of miR-106b inhibited Aβ42 induced tau phosphorylation at Tyr18 without changes at Ser396/404. It was validated that miR-106b could target Fyn directly and repress the protein levels of Fyn in SH-SY5Y cells. The phosphorylation level of tau at Tyr18 was decreased when Fyn was knockdown, and the inhibitory effects could be rescued when the expression of miR-106b was inhibited. Thus, miR-106b might inhibit Aβ42 induced tau phosphorylation at Tyr18 via regulating the expression of Fyn (Liu et al., 2016b).
The expression levels of miR-124b was decreased in the brain of AD patients and in N2a/APP695swe cells. The overexpression of miR-124-3p could rescue cell apoptosis and attenuate the abnormal hyperphosphorylation of tau with the upregulation of Caveolin-1, phosphoinositide 3-kinase (PI3K), phosphor-Akt (Akt-Ser473)/Akt, phosphor-glycogen synthase kinase-3 beta (GSK-3β-Ser9)/GSK-3β in N2a/APP695swe cells. It was suggested that miR-124-3p could inhibit abnormal hyperphosphorylation of tau through targeting Caveolin-1 and regulating the pathway Caveolin-1-PI3K/Akt/GSK-3β (Kang et al., 2017).
Perspectives
Notably, the pathological functions of multiple miRNAs regulated genes in AD-affected brain are overlapped and highly interactive, and the effects of interactive and overlapped functions network are greater than the individual one. Another noticeable problem is that specific brain enriched miRNAs are relatively labile and short-lived (Sethi and Lukiw, 2009), they may be subject to degradation especially in the highly oxidative environment of an AD brain. Thus, down-regulated microRNAs may be an artifact of the degenerative aspects of the disease. While the up-regulated miRNAs and their down-regulated mRNAs targets are a better bet to study especially in post-mortem tissues (Guo et al., 2010).
In 2018, the National Institute of Aging and Alzheimer’s Association proposed that “Alzheimer’s Disease” was a process of accumulated neuropathological changes, then defined as “AD” by biomarkers in vivo or by postmortem examination, not clinical symptoms (Jack et al., 2018); thus, the search for reliable, effective and timely biomarkers would be of significant value. The most commonly used biomarkers include CSF (cerebrospinal fluid) Aβ42 or Aβ42/Aβ40 ratio, amyloid PET, CSF phosphorylated Tau, CSF total Tau, Tau PET, anatomic MRI and FDG PET, which all focus on Aβ and Tau in bio fluids and neuroimaging techniques (Femminella et al., 2015; Jack et al., 2018). However, none of them could individually diagnose AD, and these methods still arrived too late for early and effective intervention. In recent decades, many studies have shown that microRNAs are implicated in the pathogenesis of AD (described in sections “MicroRNAs Involved in the Aβ Hypothesis” and “MicroRNAs Involved in the Tau Phosphorylated Imbalance” and summarized in Tables 1, 2), and alterations of microRNAs were found in serum, plasma and CSF, meaning that microRNAs are exceptional candidates for AD biomarkers. Most microRNA biomarker lists were produced by conducting microRNA profiles through qPCR (Dong et al., 2015), some of which are even next generation sequences (Guo et al., 2017), and comparing differentially expressed microRNAs between AD patients and control groups. Although mounting microRNAs have been identified as potential biomarkers for AD diagnosis, few have been validated in more than two studies, making their widespread use difficult (Femminella et al., 2015). It was proved that the expression levels of miR-15, miR-181c, miR-125b in AD CSF were up/down-regulated compared with normal controls in more than two studies, and so these specific microRNAs were suggested to be potential biomarkers for AD (Richardson et al., 2008; Danborg et al., 2014; Tan et al., 2014). Despite this, extensive validation and follow-ups in larger cohorts of patients are still necessary.
There are still no drugs that are both safe and effective for AD use, even though multiple agents have been researched and tested. Studies about microRNAs in AD have provided prominent insights into our understanding of molecular mechanisms, shedding light on potential drugs by targeting specific microRNAs. The regulatory characteristics of microRNAs are sequence-specific and multiple genes regulation, providing an exciting avenue for regulating this complicated disease networks and pathways. Approximately 500 patents were issued and published in United States and European in 2016, suggesting that the therapeutic potentials of microRNAs have attracted a great deal of attention. A number of preclinical studies also have been performed, for example, Parsi et al. (2015) performed a preclinical evaluation and suggested that miR-16 was a good drug candidate for AD. They identified that miR-16 could inhibit the expression of APP and BACE1, repress the production of Aβ and the phosphorylation of Tau in cells. Additionally, the delivery of miR-16 into mice induced a decrease in APP, BACE1, and Tau levels in a region dependent method. Moreover, miR-16 has also been found to associate with oxidative stress and inflammation in AD (Parsi et al., 2015). This successful preclinical study provided support for future microRNA therapy researches. It is important to note that the off target effect of microRNAs should not be ignored at the same time. Although studies about microRNA therapies in AD are still in their infancy, with increasing attention being paid to this field, we believe that researchers will fully understand this at some point, even if there is still a long way to go.
Author Contributions
MW wrote the manuscript. LQ edited it and BT edited the final version of the manuscript.
Funding
This work was supported by the National Key Plan for Scientific Research and Development of China (2016YFC1306000) and the National Natural Science Foundation of China (81430023).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Absalon, S., Kochanek, D. M., Raghavan, V., and Krichevsky, A. M. (2013). MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, Tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 33, 14645–14659. doi: 10.1523/jneurosci.1327-13.2013
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031. doi: 10.1016/s0140-6736(10)61349-9
Ballatore, C., Lee, V. M., and Trojanowski, J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 8, 663–672. doi: 10.1038/nrn2194
Banzhaf-Strathmann, J., Benito, E., May, S., Arzberger, T., Tahirovic, S., Kretzschmar, H., et al. (2014). MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 33, 1667–1680. doi: 10.15252/embj.201387576
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer’s disease. Lancet 368, 387–403. doi: 10.1016/s0140-6736(06)69113-7
Boissonneault, V., Plante, I., Rivest, S., and Provost, P. (2009). MicroRNA-298 and MicroRNA-328 regulate expression of mouse β-Amyloid precursor protein-converting enzyme 1. J. Biol. Chem. 284, 1971–1981. doi: 10.1074/jbc.M807530200
Danborg, P. B., Simonsen, A. H., Waldemar, G., and Heegaard, N. H. (2014). The potential of microRNAs as biofluid markers of neurodegenerative diseases – a systematic review. Biomarkers 19, 259–268. doi: 10.3109/1354750x.2014.904001
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. doi: 10.1172/JCI36663
Delay, C., Dorval, V., Fok, A., Grenier-Boley, B., Lambert, J. C., Hsiung, G. Y., et al. (2014). MicroRNAs targeting Nicastrin regulate Abeta production and are affected by target site polymorphisms. Front. Mol. Neurosci. 7:67. doi: 10.3389/fnmol.2014.00067
Dong, H., Li, J., Huang, L., Chen, X., Li, D., Wang, T., et al. (2015). Serum MicroRNA profiles serve as novel biomarkers for the diagnosis of Alzheimer’s disease. Dis. Markers 2015:625659. doi: 10.1155/2015/625659
El Fatimy, R., Li, S., Chen, Z., Mushannen, T., Gongala, S., Wei, Z., et al. (2018). MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 136, 537–555. doi: 10.1007/s00401-018-1880-5
Fang, M., Wang, J., Zhang, X., Geng, Y., Hu, Z., Rudd, J. A., et al. (2012). The miR-124 regulates the expression of BACE1/β-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 209, 94–105. doi: 10.1016/j.toxlet.2011.11.032
Femminella, G. D., Ferrara, N., and Rengo, G. (2015). The emerging role of microRNAs in Alzheimer’s disease. Front. Physiol. 6:40. doi: 10.3389/fphys.2015.00040
Goodall, E. F., Heath, P. R., Bandmann, O., Kirby, J., and Shaw, P. J. (2013). Neuronal dark matter: the emerging role of microRNAs in neurodegeneration. Front. Cell. Neurosci. 7:178. doi: 10.3389/fncel.2013.00178
Guedes, J. R., Santana, I., Cunha, C., Duro, D., Almeida, M. R., Cardoso, A. M., et al. (2016). MicroRNA deregulation and chemotaxis and phagocytosis impairment in Alzheimer’s disease. Alzheimers Dement. 3, 7–17. doi: 10.1016/j.dadm.2015.11.004
Guo, H., Ingolia, N. T., Weissman, J. S., and Bartel, D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840. doi: 10.1038/nature09267
Guo, R., Fan, G., Zhang, J., Wu, C., Du, Y., Ye, H., et al. (2017). A 9-microRNA signature in serum serves as a noninvasive biomarker in early diagnosis of Alzheimer’s disease. J. Alzheimers Dis. 60, 1365–1377. doi: 10.3233/jad-170343
Hammond, S. W. (2015). An overview of microRNAs. Adv. Drug Deliv. Rev. 87, 3–14. doi: 10.1016/j.addr.2015.05.001
Hauptmann, S., Keil, U., Scherping, I., Bonert, A., Eckert, A., and Müller, W. E. (2006). Mitochon- drial dysfunction in sporadic and genetic Alzheimer’s disease. Exp. Gerontol. 41, 668–673. doi: 10.1016/j.exger.2006.03.012
Hauser, P. S., and Ryan, R. O. (2013). Impact of apolipoprotein E on Alzheimer’s disease. Curr. Alzheimer Res. 10, 809–817. doi: 10.2174/15672050113109990156
He, D., Tan, J., and Zhang, J. (2017). miR-137 attenuates Abeta-induced neurotoxicity through inactivation of NF-kappaB pathway by targeting TNFAIP1 in Neuro2a cells. Biochem. Biophys. Res. Commun. 490, 941–947. doi: 10.1016/j.bbrc.2017.06.144
Hébert, S. S., Horré, K., Nicolaï, L., Bergmans, B., Papadopoulou, A. S., Delacourte, A., et al. (2009). MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 33, 422–428. doi: 10.1016/j.nbd.2008.11.009
Hébert, S. S., Horré, K., Nicolaï, L., Papadopoulou, A. S., Mandemakers, W., Silahtaroglu, A. N., et al. (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105, 6415–6420. doi: 10.1073/pnas.0710263105
Hernandez-Rapp, J., Rainone, S., Goupil, C., Dorval, V., Smith, P. Y., Saint-Pierre, M., et al. (2016). microRNA-132/212 deficiency enhances Abeta production and senile plaque deposition in Alzheimer’s disease triple transgenic mice. Sci. Rep. 6:30953. doi: 10.1038/srep30953
Hu, S., Wang, H., Chen, K., Cheng, P., Gao, S., Liu, J., et al. (2015). MicroRNA-34c downregulation ameliorates amyloid-beta-induced synaptic failure and memory deficits by targeting VAMP2. J. Alzheimers Dis. 48, 673–686. doi: 10.3233/JAD-150432
Huang, Y., Shen, X. J., Zou, Q., Wang, S. P., Tang, S. M., and Zhang, G. Z. (2011). Biological functions of microRNAs: a review. J. Physiol. Biochem. 67, 129–139. doi: 10.1007/s13105-010-0050-6
Idda, M. L., Munk, R., Abdelmohsen, K., and Gorospe, M. (2018). Noncoding RNAs in Alzheimer’s disease. Wiley Interdiscip. Rev. RNA 9:e1463. doi: 10.1002/wrna.1463
Iqbal, K., Alonso Adel, C., Chen, S., Chohan, M. O., El-Akkad, E., Gong, C. X., et al. (2005). Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 1739, 198–210. doi: 10.1016/j.bbadis.2004.09.008
Iqbal, K., and Grundke-Iqbal, I. (2010). Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimers Dement. 6, 420–424. doi: 10.1016/j.jalz.2010.04.006
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., et al. (2001). Metabolic regulation of brain Abeta by neprilysin. Science 292, 1550–1552. doi: 10.1126/science.1059946
Jaber, V., Zhao, Y., and Lukiw, W. J. (2017). Alterations in micro RNA-messenger RNA (miRNA-mRNA) coupled signaling networks in sporadic Alzheimer’s disease (AD) hippocampal CA1. J. Alzheimers Dis. Parkinsonism 7:312. doi: 10.4172/2161-0460.1000312
Jack, C. R. Jr., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., et al. (2018). NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562. doi: 10.1016/j.jalz.2018.02.018
Jin, Y., Tu, Q., and Liu, M. (2018). MicroRNA-125b regulates Alzheimer’s disease through SphK1 regulation. Mol. Med. Rep. 18, 2373–2380. doi: 10.3892/mmr.2018.9156
Kanemitsu, H., Tomiyama, T., and Mori, H. (2003). Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci. Lett. 350, 113–116. doi: 10.1016/S0304-3940(03)00898-X
Kang, Q., Xiang, Y., Li, D., Liang, J., Zhang, X., Zhou, F., et al. (2017). MiR-124-3p attenuates hyperphosphorylation of tau proteininduced apoptosis via caveolin-1-PI3K/Akt/GSK3β pathway in N2a/APP695swe cells. Oncotarget 8, 24314–24326.
Kim, J., Yoon, H., Chung, D. E., Brown, J. L., Belmonte, K. C., and Kim, J. (2016). miR-186 is decreased in aged brain and suppresses BACE1 expression. J. Neurochem. 137, 436–445. doi: 10.1111/jnc.13507
Klyubin, I., Betts, V., Welzel, A. T., Blennow, K., Zetterberg, H., Wallin, A., et al. (2008). Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immuni- zation. J. Neurosci. 28, 4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008
Lee, K., Kim, H., An, K., Kwon, O. B., Park, S., Cha, J. H., et al. (2016). Replenishment of microRNA-188-5p restores the synaptic and cognitive deficits in 5XFAD mouse model of Alzheimer’s disease. Sci. Rep. 6:34433. doi: 10.1038/srep34433
Lei, X., Lei, L., Zhang, Z., Zhang, Z., and Cheng, Y. (2015). Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 8, 1565–1574.
Li, H. H., Lin, S. L., Huang, C. N., Lu, F. J., Chiu, P. Y., Huang, W. N., et al. (2016). miR-302 attenuates Amyloid-beta-induced neurotoxicity through activation of Akt signaling. J. Alzheimers Dis. 50, 1083–1098. doi: 10.3233/JAD-150741
Li, Q., Li, X., Wang, L., Zhang, Y., and Chen, L. (2016). miR-98-5p acts as a target for Alzheimer’s disease by regulating Abeta production through modulating SNX6 expression. J. Mol. Neurosci. 60, 413–420. doi: 10.1007/s12031-016-0815-7
Liang, C., Zhu, H., Xu, Y., Huang, L., Ma, C., and Deng, W. (2012). MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 1455, 103–113. doi: 10.1016/j.brainres.2011.10.051
Lindwall, G., and Cole, R. D. (1984). Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 259, 5301–5305.
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, C. G., Wang, J. L., Li, L., and Wang, P. C. (2014). MicroRNA-135a and -200b, potential Biomarkers for Alzheimer×s disease, regulate β secretase and amyloid precursor protein. Brain Res. 1583, 55–64. doi: 10.1016/j.brainres.2014.04.026
Liu, W., Cai, H., Lin, M., Zhu, L., Gao, L., Zhong, R., et al. (2016a). MicroRNA-107 prevents amyloid-beta induced blood-brain barrier disruption and endothelial cell dysfunction by targeting Endophilin-1. Exp. Cell Res. 343, 248–257. doi: 10.1016/j.yexcr.2016.03.026
Liu, W., Zhao, J., and Lu, G. (2016b). miR-106b inhibits tau phosphorylation at Tyr18 by targeting Fyn in a model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 478, 852–857. doi: 10.1016/j.bbrc.2016.08.037
Liu, W., Liu, C., Zhu, J., Shu, P., Yin, B., Gong, Y., et al. (2010). MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 33, 522–534. doi: 10.1016/j.neurobiolaging.2010.04.034
Long, J. M., Ray, B., and Lahiri, D. K. (2012). MicroRNA-153 physiologically inhibits expression of Amyloid-β precursor protein in cultured human fetal brain cells and is dysregulated in a Subset of Alzheimer disease patients. J. Biol. Chem. 287, 31298–31310. doi: 10.1074/jbc.M112.366336
Long, J. M., Ray, B., and Lahiri, D. K. (2014). MicroRNA-339-5p down-regulates protein expression of β-Site Amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J. Biol. Chem. 289, 5184–5198. doi: 10.1074/jbc.M113.518241
Lukiw, W. J. (2007). Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 18, 297–300. doi: 10.1097/WNR.0b013e3280148e8b
Ma, X., Liu, L., and Meng, J. (2017). MicroRNA-125b promotes neurons cell apoptosis and Tau phosphorylation in Alzheimer’s disease. Neurosci. Lett. 661, 57–62. doi: 10.1016/j.neulet.2017.09.043
Maoz, R., Garfinkel, B. P., and Soreq, H. (2017). Alzheimer’s disease and ncRNAs. Adv. Exp. Med. Biol. 978, 337–361. doi: 10.1007/978-3-319-53889-1_18
Michaud, J. P., Bellavance, M. A., Préfontaine, P., and Rivest, S. (2013). Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 5, 646–653. doi: 10.1016/j.celrep.2013.10.010
Nelson, P. T., and Wang, W. X. (2010). MiR-107 is reduced in Alzheimer’s disease brain neocortex: validation study. J. Alzheimers Dis. 21, 75–79. doi: 10.3233/JAD-2010-091603
Parsi, S., Smith, P. Y., Goupil, C., Dorval, V., and Hébert, S. S. (2015). Preclinical evaluation of miR-15/107 family members as multifactorial drug targets for Alzheimer’s disease. Mol. Ther. Nucleic Acids 4:e256. doi: 10.1038/mtna.2015.33
Patel, N., Hoang, D., Miller, N., Ansaloni, S., Huang, Q., and Rogers, J. T. (2008). MicroRNAs can regulate human APP levels. Mol. Neurodegener. 3:10. doi: 10.1186/1750-1326-3-10
Prince, M., Bryce, R., Albanese, E., Wimo, A., Ribeiro, W., and Ferri, C. P. (2013). The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 9, 63.e2–75.e2. doi: 10.1016/j.jalz.2012.11.007
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., et al. (1998). Insu- lin-degrading enzyme regulates extracel- lular levels of amyloid beta-protein by deg- radation. J. Biol. Chem. 273, 32730–32738. doi: 10.1074/jbc.273.49.32730
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53. doi: 10.1016/j.molmed.2007.12.002
Richardson, J. C., Cogswell, J. P., Ward, J., Taylor, I. A., Waters, M., Shi, Y., et al. (2008). Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. Alzheimers Dement. 4:T162. doi: 10.1016/j.jalz.2008.05.420
Richter, J., Appenzeller, S., Ammerpohl, O., Deuschl, G., Paschen, S., Brüggemann, N., et al. (2012). No evidence for differential methylation of α-synuclein in leukocyte DNA of Parkinson’s disease patients. Mov. Disord. 27, 590–591. doi: 10.1002/mds.24907
Salta, E., Sierksma, A., Vanden Eynden, E., and De Strooper, B. (2016). miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol. Med. 8, 1005–1018. doi: 10.15252/emmm.201606520
Sarkar, S., Jun, S., Rellick, S., Quintana, D. D., Cavendish, J. Z., and Simpkins, J. W. (2016). Expression of microRNA-34a in Alzheimer’s disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res. 1646, 139–151. doi: 10.1016/j.brainres.2016.05.026
Sethi, P., and Lukiw, W. J. (2009). Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 459, 100–104. doi: 10.1016/j.neulet.2009.04.052
Shu, B., Zhang, X., Du, G., Fu, Q., and Huang, L. (2018). MicroRNA-107 prevents amyloid-beta-induced neurotoxicity and memory impairment in mice. Int. J. Mol. Med. 41, 1665–1672. doi: 10.3892/ijmm.2017.3339
Smith, P. Y., Hernandez-Rapp, J., Jolivette, F., Lecours, C., Bisht, K., Goupil, C., et al. (2015). miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregationin vivo. Hum. Mol. Genet. 24, 6721–6735. doi: 10.1093/hmg/ddv377
Song, F., Qian, Y., Peng, X., Han, G., Wang, J., Bai, Z., et al. (2016). Perturbation of the transcriptome: implications of the innate immune system in Alzheimer’s disease. Curr. Opin. Pharmacol. 26, 47–53. doi: 10.1016/j.coph.2015.09.015
Tan, L., Yu, J. T., Liu, Q. Y., Tan, M. S., Zhang, W., Hu, N., et al. (2014). Circulating miR-125b as a biomarker of Alzheimer’s disease. J. Neurol. Sci. 336, 52–56. doi: 10.1016/j.jns.2013.10.002
Tiribuzi, R., Crispoltoni, L., Porcellati, S., Di Lullo, M., Florenzano, F., Pirro, M., et al. (2014). miR128 up-regulation correlates with impaired amyloid β(1-42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol. Aging 35, 345–356. doi: 10.1016/j.neurobiolaging.2013.08.003
Ulrich, J. D., and Holtzman, D. M. (2016). TREM2 function in Alzheimer’s disease and neurodegeneration. ACS Chem. Neurosci. 7, 420–427. doi: 10.1021/acschemneuro.5b00313
Vilardo, E., Barbato, C., Ciotti, M., Cogoni, C., and Ruberti, F. (2010). MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 285, 18344–18351. doi: 10.1074/jbc.M110.112664
Walsh, D. M., and Selkoe, D. J. (2007). A beta oligomers-a decade of discovery. J. Neuro Chem. 101, 1172–1184.
Walsh, D. M., Townsend, M., Podlisny, M. B., Shankar, G. M., Fadeeva, J. V., El Agnaf, O., et al. (2005). Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogen- esis block oligomerization of natural Abeta and thereby rescue long-term poten- tiation. J. Neurosci. 25, 2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005
Wang, G., Huang, Y., Wang, L.-L., Zhang, Y.-F., Xu, J., Zhou, Y., et al. (2016). MicroRNA-146a suppresses ROCK1 allowing hyperphosphorylation of tau in Alzheimer’s disease. Sci. Rep. 6:26697. doi: 10.1038/srep26697
Wang, W. X., Rajeev, B. W., Stromberg, A. J., Ren, N., Tang, G., Huang, Q., et al. (2008). The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 28, 1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008
Wang, X., Liu, D., Huang, H. Z., Wang, Z., Hou, T. Y., Yang, X., et al. (2018). A novel MicroRNA-124/PTPN1 signal pathway mediates synaptic and memory deficits in Alzheimer’s disease. Biol. Psychiatry 83, 395–405. doi: 10.1016/j.biopsych.2017.07.023
Wang, Z., Qin, W., Wei, C. B., Tang, Y., Zhao, L. N., Jin, H. M., et al. (2018). The microRNA-1908 up-regulation in the peripheral blood cells impairs amyloid clearance by targeting ApoE. Int. J. Geriatr. Psychiatry 33, 980–986. doi: 10.1002/gps.4881
Wang, X., Tan, L., Lu, Y., Peng, J., Zhu, Y., Zhang, Y., et al. (2015). MicroRNA-138 promotes tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Lett. 589, 726–729. doi: 10.1016/j.febslet.2015.02.001
Wang, Y., Veremeyko, T., Wong, A. H., El Fatimy, R., Wei, Z., Cai, W., et al. (2017). Downregulation of miR-132/212 impairs S-nitrosylation balance and induces tau phosphorylation in Alzheimer’s disease. Neurobiol. Aging 51, 156–166. doi: 10.1016/j.neurobiolaging.2016.12.015
Wu, B. W., Wu, M. S., and Guo, J. D. (2018). Effects of microRNA-10a on synapse remodeling in hippocampal neurons and neuronal cell proliferation and apoptosis through the BDNF-TrkB signaling pathway in a rat model of Alzheimer’s disease. J. Cell Physiol. 233, 5281–5292. doi: 10.1002/jcp.26328
Xu, Y., Chen, P., Wang, X., Yao, J., and Zhuang, S. (2018). miR-34a deficiency in APP/PS1 mice promotes cognitive function by increasing synaptic plasticity via AMPA and NMDA receptors. Neurosci. Lett. 670, 94–104. doi: 10.1016/j.neulet.2018.01.045
Yang, L. B., Lindholm, K., Yan, R., Citron, M., Xia, W., and Yang, X. L. (2003). Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4. doi: 10.1038/nm0103-3
Zhang, Y., Xing, H., Guo, S., Zheng, Z., Wang, H., and Xu, D. (2016). MicroRNA-135b has a neuroprotective role via targeting of β-site APP-cleaving enzyme 1. Exp. Ther. Med. 12, 809–814. doi: 10.3892/etm.2016.3366
Zhao, Y., Jaber, V., and Lukiw, W. J. (2016). Over-expressed pathogenic miRNAs in Alzheimer’s disease (AD) and Prion disease (PrD) drive deficits in TREM2-mediated Abeta42 peptide clearance. Front. Aging Neurosci. 8:140. doi: 10.3389/fnagi.2016.00140
Zhao, Y., Pogue, A. I., and Lukiw, W. J. (2015). MicroRNA (miRNA) signaling in the human CNS in sporadic Alzheimer’s disease (AD)-novel and unique pathological features. Int. J. Mol. Sci. 16, 30105–30116. doi: 10.3390/ijms161226223
Zhao, Y., Zhao, R., Wu, J., Wang, Q., Pang, K., Shi, Q., et al. (2018). Melatonin protects against Abeta-induced neurotoxicity in primary neurons via miR-132/PTEN/AKT/FOXO3a pathway. Biofactors 44, 609–618. doi: 10.1002/biof.1411
Zhao, Z. B., Wu, L., Xiong, R., Wang, L. L., Zhang, B., Wang, C., et al. (2014). MicroRNA-922 promotes tau phosphorylation by downregulating ubiquitin carboxy-terminal hydrolase L1 (UCHL1) expression in the pathogenesis of Alzheimer’s disease. Neuroscience 275, 232–237. doi: 10.1016/j.neuroscience.2014.06.013
Zhou, Y., Wang, Z. F., Li, W., Hong, H., Chen, J., Tian, Y., et al. (2018). Protective effects of microRNA-330 on amyloid beta-protein production, oxidative stress, and mitochondrial dysfunction in Alzheimer’s disease by targeting VAV1 via the MAPK signaling pathway. J. Cell. Biochem. 119, 5437–5448. doi: 10.1002/jcb.26700
Keywords: Alzheimer’s disease, microRNAs, pathological process, amyloid plaques, neurofibrillary tangles, Aβ, Tau
Citation: Wang M, Qin L and Tang B (2019) MicroRNAs in Alzheimer’s Disease. Front. Genet. 10:153. doi: 10.3389/fgene.2019.00153
Received: 03 November 2018; Accepted: 13 February 2019;
Published: 01 March 2019.
Edited by:
Naoyuki Kataoka, The University of Tokyo, JapanReviewed by:
Walter J. Lukiw, LSU Health Sciences Center New Orleans, United StatesDavide De Pietri Tonelli, Istituto Italiano di Tecnologia, Italy
Copyright © 2019 Wang, Qin and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beisha Tang, YnN0YW5nNzM5OEAxNjMuY29t