 Amanda Brown
Amanda Brown Laura M. Fiori
Laura M. Fiori Gustavo Turecki
Gustavo Turecki- McGill Group for Suicide Studies, Department of Psychiatry, McGill University, Montreal, QC, Canada
Early life adversity (ELA)- including childhood physical, emotional, and sexual abuse, as well as childhood neglect- is an important predictive factor for negative psychopathology, including Major Depressive Disorder (MDD). ELA can epigenetically regulate key emotional and behavioral systems in ways that can stably persist into adulthood and contribute to the development of MDD and other psychopathology. DNA methylation has been one of the most investigated forms of epigenetic regulation in ELA to MDD pathway. From these studies, genes and sites associated with ELA/MDD have been identified and should be further investigated in order to identify potential avenues for intervention.
Introduction
Early life adversity (ELA)- which includes forms of child maltreatment such as physical abuse, sexual abuse, psychological and emotional abuse, and childhood neglect- remains a major public health and welfare issue in high income countries (Krugers et al., 2017). It is defined as any act that is either actively conducted, or neglected to be conducted, by a parent or caregiver that either harms or has potential to harm a child, regardless of intent. These acts include physical abuse such as hitting, punching, beating, strangling, and shaking, sexual abuse such as penetration, sexual contact, and exposure to sexual activity, emotional and psychological abuse such as terrorizing, intimidating, and belittling, and forms of neglect such as failure to provide clothing, food, shelter, or neglect in seeking medical care for a child (Leeb et al., 2008). Overall, the various forms of ELA are estimated to affect approximately 10–15% of children (typically categorized as individuals under the age of 18). Specifically, between 4 and 16% of children experience physical abuse, and approximately 15% of children are subjected to sexual abuse annually (Fergusson et al., 2000; Woodman et al., 2008; Gilbert et al., 2009).
In the United States, approximately 80% of substantiated ELA cases are found to be perpetuated by either one or both parents, with 77% of physical abuse, 26% of sexual abuse, 81% of emotional/psychological abuse, and 87% of neglect cases being perpetrated by a parent (US Department of Health and Human Services, 2008; Jonson-Reid et al., 2012; Nemeroff, 2016). Additionally, 29% of sexual abuse cases are perpetrated by a non-parent relative (Fergusson et al., 2000). It is worth noting that these estimates reflect only officially reported and substantiated cases, and that the actual burden of ELA likely exceeds these estimates.
This substantial phenomenon of ELA is of particular importance as a public health issue considering that repeated exposure to ELA, as well as exposure to multiple types of ELA, has the potential to inflict severe and lasting physical and psychological consequences that represent serious public healthcare and legal costs, as well as increased risk of mortality (Gilbert et al., 2009). ELA has been shown to contribute significantly to the risk of criminal behavior, childhood/adolescent behavioral problems, general physical heath, obesity, promiscuity, prostitution and sex trading, substance use, attempted suicide, and various psychopathologies such as post-traumatic stress disorder (PTSD), and Major Depressive Disorder (MDD), bipolar disorder, and conduct disorder (Thomas et al., 2008; Gilbert et al., 2009; Weder et al., 2014; Nemeroff, 2016; Krugers et al., 2017).
The role of ELA in the development of MDD is of particular interest with regard to public health and welfare, as MDD is the leading cause of global disability, affecting over 300 million people worldwide (World Health Organization [WHO], 2017). It is characterized by persistent depressed mood and loss of interest and pleasure, and may also include symptoms such as weight loss or gain, insomnia or hypersomnia, fatigue, loss of motivation or concentration, recurring thoughts of death, and suicidal ideation (Zajecka et al., 2013).
It is believed that exposure to repeated ELA may increase risk for MDD by cumulating in a hostile and unstable early environment that may trigger adaptive responses in the brain in crucial response systems (such as stress and emotional regulation). There is evidence that these regulatory changes can stably and radically impact aspects of personality development and cognitive functioning in ways that can increase the risk for MDD and associated psychopathologies (Saavedra et al., 2016). The etiology underlying the pathophysiology of MDD is a complex and often varied interplay between genetic, epigenetic, environmental, clinical, and social factors. Research suggests that one of these factors-ELA-can trigger epigenetic alterations (through events such as DNA methylation) in neural systems and genes associated with increased stress response [such as the in the hypothalamic pituitary adrenal axis (HPA)] as a survival mechanism. The persistence of these epigenetic changes often become maladaptive as the early life environment changes, but the altered function of the stress systems does not. While it is not fully understood how these alterations contribute specifically to MDD symptomology, DNA methylation events in these systems in individuals who have experienced ELA have been associated with MDD (Saavedra et al., 2016; Pishva et al., 2017).
It is worth noting that ELA itself is a complex set of phenomena that increases the complexity of MDD pathophysiology, and includes different severities and consequences. There is evidence of a dose-response relationship between ELA and psychopathologies such as MDD, wherein a higher severity of childhood adversity has been linked to increased incidence of psychopathology, higher comorbidity among psychiatric diagnoses, and more severe symptoms within diagnoses (Edwards et al., 2003; Chapman et al., 2004; Heim and Binder, 2012).
With respect to MDD specifically, ELA has been shown to correspond to a more severe course of depression, particularly in terms of increased chronicity, resistance to psychopharmacology and psychotherapy, and greater presentation of atypical symptoms (such as hypersomnia, increased appetite, and increased sensitivity to rejection) (Withers et al., 2013).
There is evidence that certain factors, such as microstructural differences in white matter, as measured by fractional anisotropy, and increased neural fiber connectivity, may be protective against the development of MDD in individuals who experienced ELA. Fractional anisotropy measures the anisotropic movement of water molecules (thought to be important in extrasynaptic communication) associated with fiber bundles (Morgan et al., 2013). One study found that unaffected first degree relatives of individuals with MDD, who also experienced ELA, had significantly increase fractional anisotropy in the right frontal and orbitofrontal lobes, corpus callosum, inferior fronto-occipital fasciculus (IFO), left superior longitudinal fasciculus (SLF) and right fornix (Frodl et al., 2012).
However, resilience is a complex topic in ELA and MDD research, and personal coping mechanisms, behavioral factors, and the presence of positive interpersonal influences concurrent with the ELA were not examined in this study. Epigenetic regulation related to the growth factor brain-derived neurotrophic factor (BDNF) may also play a role in conferring resilience to MDD in these circumstances, and deserves further investigation (Krishnan et al., 2007). Due to all of these intersecting factors, resilience from MDD in the presence of ELA is therefore difficult to study and adequately quantify. Regardless, while certain biological and environmental factors may act as protective factors against MDD development in individuals who have experienced ELA in ways that are not yet fully understood, it remains true that ELA is one of the greatest risk factors for the development of MDD (Saavedra et al., 2016).
Considering the prominent role that severe ELA plays in the development of MDD, and the overall prevalence of MDD, these figures indicate that ELA imposes a substantial social, economic, and public health cost upon society. Due to this, research identifying the mechanisms of action in the ELA to MDD pathway could prove to be a great asset in the development of clinical treatments of MDD. One mechanism by which ELA has been shown to influence emotional and stress regulation systems is epigenetic regulation, such as DNA methylation (Cecil et al., 2016). Although other forms of ELA have been examined with respect to the ELA to MDD pathway, DNA methylation remains one of the most predominately studied, and will serve as the focus of this review. Here, we will first explore how DNA methylation has been implicated in ELA and MDD, and then examine how DNA methylation also has the potential to impact clinical research into MDD diagnosis and treatment.
DNA Methylation in ELA/MDD
Epigenetic regulation describes any regulatory event which causes alterations in gene expression without changes to the original DNA sequence. Various forms of epigenetic regulation have been extensively studied as causal factors in the depression endophenotype in individuals who have experienced ELA, including DNA methylation, histone modifications, and microRNA.
DNA methylation is one of the most well-investigated forms of epigenetic regulation and has been studied as a causal factor in the development of MDD in both candidate gene and genome wide studies (Lutz et al., 2015). During DNA methylation, the DNA is covalently modified via addition of a methyl group to a cytosine nucleotide. This phenomenon is particularly common in cytosine-guanine dinucleotides (CpGs) (Jin et al., 2011). The methylation status of the nucleotide is read by methyl CpG-binding domain (MBD) proteins- such as MeCP2 and MBD1-4- which interact with histone deactylases and DNA-methyltransferases that induce chromatin condensation (Chen et al., 2003; Chahrour et al., 2008; Guy et al., 2011). When methylation is found in promoter regions, it can interfere with transcription factor binding and lead to gene silencing. Conversely, site specific dissociation of the MBDs – specifically MeCP2- have been associated with demethylation, and have been implicated to play a major role in maintaining DNA methylation status (Martinowich et al., 2003).
During prenatal development, DNA methylation patterns are crucial to the process of cell differentiation. However, changes in DNA methylation occur beyond this period, as a mechanism that helps the genome adapt to external signals in the environment. This form of DNA methylation allows the genome to adjust its function through diverse -yet stable- changes. This is one possible mechanism by which environmental stressors such as ELA may trigger epigenetic changes that could ultimately contribute to psychopathology such as MDD (Turecki, 2014).
Genome-Wide and Multiple Loci Studies
Human Post Mortem Brain Tissue Studies
In addition to approaches based on the investigation of candidate genes, the role of DNA Methylation in ELA/MDD has also been examined using larger-scale, genome-wide studies. One of the earliest studies examined hippocampal tissue from French Canadian men with a history of severe childhood abuse (who also died by suicide) using meDIP, an antibody targeting methylated cytosines, coupled with an tiled array containing gene promoters. Differential DNA methylation indicated that 248 sites were hypermethylated and 114 were hypomethylated (Labonté et al., 2012). Significant differences involved genes implicated in cellular or neural plasticity, and some of these findings included histone cluster 2 H2ab (HIST2H2AB), nuclear receptor subfamily 1, group D, member 1 (NR1D1); and amyotrophic lateral sclerosis 2 (ALS2). Specifically, in vitro analysis of ALS2 methylation levels indicated functional effects on gene expression- supporting the hypothesis that ELA contributes to cell-type specific reprogramming of the epigenome in ways that may influence emotional and behavioral dysregulation (Lutz et al., 2015).
In another region of the brain that is hypothesized to be implicated in ELA/MDD- the anterior cingulate cortex (ACC) – genome-wide DNA methylation was assessed using reduced representation bisulfite sequencing (RRBS) in human post mortem brain tissue of depressed suicides who experienced ELA (Table 1) (Lutz et al., 2017b). In this study, the most significantly differentially methylated genes were those related to myelin and oligodendrocytes. Specifically, the three most differentially methylated genes were LINGO3 (which codes for LINGO proteins that are implicated in myelination), POU3F1 (a transcription factor that controls myelination), and ITGB1 (an integrin that mediates oligodendrocyte and axonal interactions). These findings suggested that ELA may lead to myelin alterations, a conclusion also supported by both transcriptomic and morphological changes observed in this study using other techniques (Table 1) (Lutz et al., 2017b), and consistent with pre-clinical studies indicating that a critical period in the early social environment regulates myelination that in turn is essential for normal cognitive function (Makinodan et al., 2012).
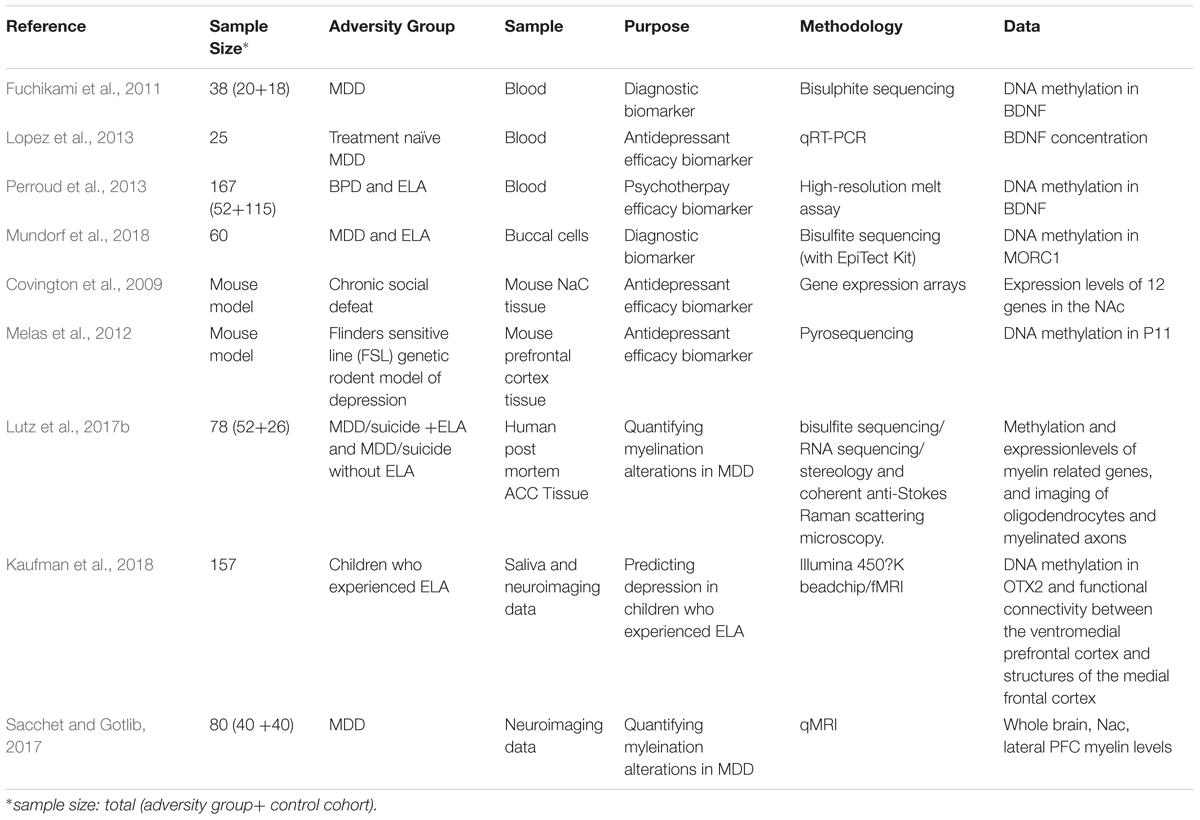
Table 1. Summary of studies investigating clinical applications of ELA and MDD research involving- or related to – DNA methylation in ELA and MDD.
Peripheral Tissue Studies
Other studies evaluated DNA methylation changes associated with ELA, using peripheral samples. A study investigating methylation in the promoter region of over 20 000 genes, including 489 coding for miRNAs, in the blood DNA of males in the 1958 British Birth Cohort found 997 differentially methylated gene promoters associated with ELA (311 hypermethylated and 686 hypomethylated), which were enriched for genes involved in processes such as transcriptional regulation and development. Additionally, they observed ELA associated methylation in thirty-one miRNA genes, six of which showed hypermethylation consistent with hypomethylation observed in their downstream gene targets (Suderman et al., 2015). Another genome wide study by Weder et al. (2014) used the Illumina 450K BeadChip array to determine potential sites of differential methylation that could predict depression in saliva samples collected from maltreated children compared to non-traumatized. Three genes emerged as predictors of depression in combination with ELA: DNA-Binding Protein Inhibitor ID–3 (ID3); and Tubulin Polymerization Promoting Protein (TPPP) (Weder et al., 2014); and the neurotransmitter gene glutamate receptor, ionotropic N-methyl-D-aspartate 1 (GRIN1) (Weder et al., 2014).
More recently, the Illumina 450K BeadChip array was used by the same group in another study of ELA and MDD that sought to predict differential DNA methylation in maltreated children. In this study (Kaufman et al., 2018), methylation of the Orthodenticle homeobox 2 (OTX2) gene significantly predicted depression in saliva samples of maltreated children compared to controls (Table 1) (Kaufman et al., 2018).
Neuroimaging
Imaging data revealed that methylation of OTX2 is associated with increased functional connectivity between the ventromedial prefrontal cortex and structures of the medial frontal cortex that have been implicated in MDD- such as the paracingulate gyrus, frontal pole, and subcallosal gyrus (Table 1) (Kaufman et al., 2018).
Candidate Genes
HPA Axis
Numerous candidate gene studies have also investigated the relationship between ELA, depressive symptoms, and DNA methylation. The majority of these studies have been performed in peripheral samples such as blood and saliva. However, studies utilizing brain and CNS related tissues and systems have also been widely performed in both animal models and human post mortem tissue studies. One biological system in particular, the hypothalamic-pituitary-adrenal (HPA) axis has been the focus of many of these studies.
The HPA axis is a primary stress response system in mammals that has been extensively studied for its role in various psychopathologies including MDD, bipolar disorder, hypersexual disorder, and behaviors like habitual smoking (Rohleder and Kirschbaum, 2006; Chatzittofis et al., 2016; Murri et al., 2016). The HPA response begins in neurons in the paraventricular nucleus (PVN) of the hypothalamus, where the neurohormones CRF and arginine vasopressin (AVP) are released into blood vessels between the hypothalamus and pituitary gland. The pituitary gland is then stimulated to secrete adrenocorticotropic hormone (ACTH) into the circulation to induce the synthesis and secretion of glucocorticoids like cortisol from the adrenal glands (Pariante and Lightman, 2008; Perroud et al., 2011; Keller et al., 2016).
Studies have demonstrated that ELA can foster HPA axis dysfunction in ways that increase susceptibility to depression. Specifically, this dysfunction may result from disturbances to the negative feedback system of the HPA axis, which is partially controlled by the expression of the glucocorticoid receptor (GR/NR3C1) in the hippocampus, expression of the CRF gene, or changes in FKBP5 expression- a gene responsible for stabilizing the conformation of the GR (Pariante and Lightman, 2008; Keller et al., 2016).
Animal Models
Animal models of maternal care have provided a substantial contribution to the understanding of the relationship between the early environment and functioning of the HPA axis. Rats who receive decreased maternal attention in the form of low licking-grooming (LG) behavior in the first week of life have been found to show increased HPA stress responses and hypothalamic corticotrophin releasing factor (CRF) expression, and decreased GR expression and glucocorticoid feedback sensitivity compared to high LG rats. This is due in large part to differential methylation of the GR/NR3C1 gene which is downregulated in low LG rats, with increased DNA methylation being observed in the promoter region (exon 17) of NR3C1 in the hippocampus, which persists into adulthood (Weaver et al., 2007, 2014).
In addition to NR3C1, hypothalamic corticotrophin releasing factor (CRF), also plays a key role in this feedback system and exerts control over the HPA axis and other stress responses in the brain. Mice subjected to chronic social defeat display decreased DNA methylation in the CRF gene, leading to sustained upregulation of CRF in neurons of the paraventricular nucleus (PVN) in the hypothalamus (Elliott et al., 2010). This ELA model leads to stress-induced phosphorylation of MeCP2, which results in MeCP2 being dissociated from the inhibitory complex of methylated CpG sites in the CRF promotor region, resulting in increased transcription. This mechanism of CRF upregulation is supported by the observation that mice with MeCP2 knockouts in the PVN present with abnormal physiological stress responses (Fyffe et al., 2008). Conversely, a rat model of chronic mild stress actually demonstrated increased DNA methylation of the CRF promoter in the PVN, indicating that sex-specific and/or dose-dependent effects may play a role in altered HPA axis activity (Sterrenburg et al., 2011).
Human Post Mortem Brain Tissue Studies
Human post mortem brain tissue studies have also demonstrated ELA-associated epigenetic changes in the HPA axis, as hippocampal tissue obtained from individuals who died by suicide and experienced ELA (particularly severe childhood abuse) displayed an increase in NR3C1 methylation, and decreased NR3C1 expression (McGowan et al., 2009; Suderman et al., 2012).
Finally, in the neurons of the hypothalamic paraventricular nucleus, increased arginine vasopressin (AVP) release accompanied by HPA axis hypersensitivity has also been linked to hypomethylation in reduced maternal care animals, in a similar mechanism to CRF hypomethylation (Murgatroyd et al., 2009).
Peripheral Tissue Studies
The GR regulating gene, FKBP5, has also been the subject of several studies investigating differential methylation in peripheral tissue. Certain polymorphisms of FKBP5 (rs1360780, rs9296158, rs3800373, and rs9470080) interact with ELA to predict MDD and suicide attempts (Roy et al., 2010; Appel et al., 2011). One study by Klengel et al. (2012) examined FKBP5 methylation levels in the blood of individuals with a history of ELA and found hypomethylation in intron seven. Another study collected saliva samples from children who had experienced ELA, and assessed them for DNA methylation levels at two CpG sites in intron seven of FKBP5 (Tyrka et al., 2015). Once again, hypomethylation was found at both sites in intron seven. It was hypothesized that increased cortisol release following ELA in FKBP5 risk allele carriers would signal differential methylation in FKBP5 and disrupt the feedback loop responsible for FKBP5 and GR activity, leading to stress response dysregulation and increased susceptibility to MDD (Tyrka et al., 2015).
Neurotransmitters
DNA methylation in neurotransmitter genes has also been explored in CNS and peripheral human samples, as well as animal studies, with respect to the ELA to MDD pathway. In addition to the previous discussed Weder et al. study that implicated the neurotransmitter gene glutamate receptor, ionotropic N-methyl-D-aspartate 1 (GRIN1) in ELA/MDD, one gene, 5-HT2A, has been explored in the prefrontal cortex (PFC) for its potential contribution to ELA related MDD and suicide (Du et al., 2000, 2001; De Luca et al., 2007, 2009). One particular 5-HT2A polymorphism, the C allele (C102T), has been associated with depression and suicide, and has been found to be more abundant in the PFC of suicides. Although non-significant hypomethylation differences were found in the C allele in suicides, hypermethylation was reported in suicide ideators, indicating potential differential methylation patterns between these two groups (De Luca et al., 2009).
In the anterior insula, downregulation of the kappa opioid receptor (Kappa) and decreased DNA methylation in the second intron of the Kappa gene has been found in depressed suicides who experienced ELA. This intron serves as a genomic enhancer in GR binding regulated Kappa expression; indicating a mechanism by which endogenous opioids act on stress systems (Lutz et al., 2017a).
Numerous studies have investigated the serotonin transporter gene SLC6A4, which facilitates neurotransmitter reuptake at serotonergic synapses. Hypermethylation has been demonstrated at various CpG sites in ELA cohorts depending on sex, ELA magnitude, and type of ELA experienced (ex. physical versus sexual abuse) (Vijayendran et al., 2012; Alexander et al., 2014; Booij et al., 2015; Frodl et al., 2015). ELA related SLC6A4 promoter DNA methylation status was also found to be significantly associated with more severe pre-treatment presentation of depressive symptoms, elevated stress, and higher family history of psychopathology (Menke et al., 2012).
Growth Factors
Growth factors, such as brain derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor (GDNF), also show differences in expression and DNA methylation related to ELA and MDD. In particular, BDNF has been the subject of significant basic and clinical research with regards to ELA/MDD, as it has been the subject of candidate gene studies, and shows promise as a biomarker and target for epigenome targeted antidepressant therapy.
In the nucleus accumbens (NAc), glial cell-derived neurotrophic factor (GDNF) expression appears to be reduced in chronic early life stressed mice with anxiety- and depressive-like behaviors. This reduction appears to be due to increased DNA methylation related to increased MeCP2 binding (Chahrour et al., 2008; Uchida et al., 2011).
The brain derived neurotrophic factor (BDNF) gene is expressed in the adult PFC, and plays a critical role in neural and behavioral plasticity, and the development of psychiatric disorders such as depression, bipolar disorder, and schizophrenia when coupled with ELA (Kundakovic et al., 2014). Hypomethylation of BDNF has been observed in both human PFC tissue in individuals who have experienced ELA, as well as in the PFC of rats exposed to early life stress (Roth et al., 2009; Roth and Sweatt, 2011).
With a significant amount of research substantiating the biological relevance of genes such as BDNF in the development of ELA related MDD, reliable estimates of methylation at this locus could potentially be used as a diagnostic tool (Table 1) (Fuchikami et al., 2011; Kundakovic et al., 2014).
Taken in context with the established ELA related behavioral vulnerabilities that BDNF methylation alterations represent, blood BDNF expression and methylation levels could potentially be of clinical use as a tool in clinical care of psychopathologies such as MDD, when coupled with the presentation of symptoms and a reported history of ELA (Shonkoff et al., 2009).
One study examined two BDNF CpG islands (I and IV) as potential diagnostic biomarkers of MDD. Examining methylation levels at CpG islands I and IV in human cohorts of MDD patients and healthy controls, it was found that CpG 1, but not IV, levels could accurately discriminate controls from the MDD cohort. While this study utilized a relatively small sample size (20 MDD vs. 18 control individuals), it indicated that DNA methylation levels have the potential to be a valuable resource in clinical diagnosis as a biomarker (Table 1) (Fuchikami et al., 2011).
Clinical research focused on the epigenetic effects of ELA has the potential to not only create new diagnostic procedures, but also to create novel antidepressant therapies that target the epigenome or use the epigenome as a means to evaluate antidepressant efficacy (Qiu et al., 2017). BDNF in particular appears to be influenced by antidepressant treatment (Fuchikami et al., 2016). Notably, in several preclinical animal models, treatment with the histone deacetylase (HDAC) inhibitor sodium butyrate has yielded reduced depressive and manic-like behaviors, as well as reversed decreased expression in BDNF and other neurotrophic factors in chronically stressed animal models (Moretti et al., 2011; Resende et al., 2013). Similarly, animal models treated with the antidepressant imipramine present with reversed behavioral consequences of early life stress in the BDNF promoter region (Tsankova et al., 2006; Hollis et al., 2010, 2011; Covington et al., 2011).
It has been suggested that effective antidepressant treatment should correspond to an increase in peripheral BDNF, and that a sustained lack of increase in BDNF over the first week of treatment likely predicts treatment resistance (Tadić et al., 2011). This finding is based on BDNF exon IV promoter methylation data from an MDD cohort being treated with several antidepressants. This effect was mirrored by findings showing that citalopram treatment in MDD patients increased BDNF expression in treatment responders, and significantly reduced H3K27me3 levels at the BDNF IV promoter (Table 1) (Lopez et al., 2013).
Animal Models of OTX2
In an elegant study, Peña et al. (2017) described that juvenile knockdown of the transcription factor orthodenticle homeobox 2 (Otx2) in the ventral tegmental area (VTA) – a brain reward region- increases stress susceptibility similarly to early life stress. This knockdown of Otx2 was also associated with downregulation of several target genes, many of which play a role in brain development. Animals in this model also exhibited depressive-like behaviors following a second exposure to stress in adulthood. Based on these findings, the researchers proposed that a “two-hit” stress model may be in effect, wherein ELA increases stress susceptibility in the VTA via Otx2 mediation, contributing to a depression-like state and sustained transcriptional alterations in adults following adult social defeat. As the association between ELA/MDD and OTX2 has been also reported in humans (Kaufman et al., 2018), continued investigation into how Otx2 mediates sustained stress susceptibility is warranted.
Structural Studies of Myelination
The role of myelination in ELA/MDD may also be of potential clinical interest. Myelination is essential in developing and maintaining complex cognitive and behavioral functions, as it increases action potential transmission per unit time, thereby increasing the connectivity and information processing capacity of the human brain (Grydeland et al., 2013). Myelination occurs when oligodendrocytes cells generate myelin, which insulates the neuronal axons and facilitates electrical signal propagation, helping maintain the circuitry of neural networks (such as the axon segments within the cortex), which are crucial to optimizing basic cognitive and behavioral functions (Haroutunian et al., 2014; Long and Corfas, 2014). Due to this, reductions in cortical oligodendrocytes and deficits in myelin gene expression have been associated with cognitive and behavioral dysregulations which include- but are not limited to- schizophrenia, bipolar disorder, and MDD (Uranova et al., 2004). Intracortical myelin abnormalities vary across psychopathologies, with deficits in white matter volume and myelination being more prominent in bipolar disorder and MDD (Mosebach et al., 2013). With respect to MDD specifically, quantitative magnetic resonance imaging (qMRI) analysis has revealed reduced myelin levels in the white matter of brain regions such as the nucleus accumbens (NAcc) and lateral prefrontal cortex (LPFC) (Table 1) (Sacchet and Gotlib, 2017).
Structural alterations in white matter have also been associated with ELA in MRI studies, with more recent MRI strategies being able to more effectively investigate white matter myelin content to the level of bundles with hundreds or thousands of axons (Stikov et al., 2015). Although oligodendrocytes and myelin were long thought to be static, these imaging studies, as well as animal studies, have demonstrated that various forms of learning, as well as social and environmental conditions, correlate with structural changes to white matter. This suggests an important role for myelination in brain plasticity (Long and Corfas, 2014).
In the previously summarized recent study by Lutz et al. (2017b), state of the art microscopy methods were utilized to more precisely measure microstructural changes to white matter myelin that are associated with ELA. They discovered that in post mortem brain tissue from adults who experienced ELA and died by suicide, axonal integrity and myelination of individual fibers was disrupted, particularly in small-diameter axons which may be related to cortico-cortical projections. These structural alterations were found in conjunction with cell type-specific differences in DNA methylation of oligodendrocyte genes such as POU3F1 and LINGO3 in oligodendrocytes (but not neurons), and global decreased expression of a large collection of myelin related genes in the cingulate cortex. These results suggest that ELA may contribute to a neurobiological vulnerability that increases the risk of psychopathology throughout life in affected individuals (Table 1) (Lutz et al., 2017b). These findings may prove of future clinical significance, although further research into the brain region-specific effects of ELA/MDD on myelin is needed before any potential antidepressant therapies or diagnostic procedures related to myelination can be explored further.
Additional DNA Methylation Studies of Clinical Interest
BDNF is not the only gene that has potential as a biomarker for MDD in individuals who have experienced ELA. The MORC family CW-type zinc finger 1 (MORC1) gene, has been found to be hypermethylated in buccal cells of individuals who experienced ELA and scored high on the Beck Depression Inventory. Researchers have suggested that MORC1 could reliably be used as a non-invasive diagnostic biomarker for MDD in individuals who have been affected by ELA (Table 1) (Mundorf et al., 2018).
Other genes implicated in social defeat models of ELA may also serve as promising targets for the future development of novel antidepressant treatments. One mouse model of ELA that presented with altered gene expression in 12 genes in the NAc, including genes coding for actin cytoskeleton reorganization machinery, transcription factors, signaling molecules, and neurotransmitter receptors, that was reversed by treatment with the antidepressant MS-275 (Table 1) (Covington et al., 2009).
In the prefrontal cortex, DNA methylation of the P11 promoter was reduced in MDD patients via administration of escitalopram, increasing P11 and decreasing the presence of DNA methyltransferases (Table 1) (Melas et al., 2012). The epigenetic changes detailed in these studies represent both potential targets for antidepressant therapy, as well as potential biomarkers of drug efficacy.
While there has been an ample amount of basic research on the ELA to MDD pipeline examining regions and genes that are differentially methylated in ELA/MDD, clinical research expanding on these findings has been less bountiful. Further clinical studies that examine expression levels of key genes in peripheral tissue (such as buccal swabs or blood) before, during, and after antidepressant treatment in ELA/MDD subjects could feasibly be undertaken to help evaluate the efficacy of many antidepressant treatments, whether they be pharmacological, psychotherapeutic, or through methods such as brain stimulation- the last two of which (to the authors’ knowledge) – remain to be explored.
Conclusion
The studies described herein have contributed to our understanding of how ELA can epigenetically regulate behavioral and emotional response systems in the brain in ways that contribute to- and increase the vulnerability to- psychopathologies such as MDD. The research indicates that genes that code for the HPA axis, neurotransmitters, growth factors such as BDNF, transcription factors such as OTX2, and myelination/oligodendrocytes may all play a role in this pathway, demonstrating how diverse and complex the epigenetic alterations associated with ELA likely are. Due to this complexity, it is perhaps not surprising that clinical progress-such as the development of biomarkers for ELA/MD and antidepressant therapies that target the epigenome-has thus far been limited and preliminary. This is why further studies will be necessary before the full potential clinical significance of the basic research into the ELA/MDD pathway can be determined.
Author Contributions
AB wrote most of the manuscript, including the entire first draft. LF carried out substantial editing, revisions, and rewrites. GT edited and revised the final draft.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alexander, N., Wankerl, M., Hennig, J., Miller, R., Zänkert, S., Steudte-Schmiedgen, S., et al. (2014). DNA methylation profiles within the serotonin transporter gene moderate the association of 5-HTTLPR and cortisol stress reactivity. Transl. Psychiatry 4:9. doi: 10.1038/tp.2014.88
Appel, K., Schwahn, C., Mahler, J., Schulz, A., Spitzer, C., Fenske, K., et al. (2011). Moderation of adult depression by a polymorphism in the FKBP5 gene and childhood physical abuse in the general population. Neuropsychopharmacology 36, 1982–1991. doi: 10.1038/npp.2011.81
Booij, L., Szyf, M., Carballedo, A., Frey, E., Morris, D., Dymov, S., et al. (2015). DNA methylation of the serotonin transporter gene in peripheral cells and stress-related changes in hippocampal volume: a study in depressed patients and healthy controls. PLoS One 10:3. doi: 10.1371/journal.pone.0119061
Cecil, C. A., Smith, R. G., Walton, E., Mill, J., Mccrory, E. J., and Viding, E. (2016). Epigenetic signatures of childhood abuse and neglect: implications for psychiatric vulnerability. J. Psychiatr. Res. 83, 184–194. doi: 10.1016/j.jpsychires.2016.09.010
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chapman, D. P., Whitfield, C. L., Felitti, V. J., Dube, S. R., Edwards, V. J., and Anda, R. F. (2004). Adverse childhood experiences and the risk of depressive disorders in adulthood. J. Affect. Disord. 82, 217–225. doi: 10.1016/j.jad.2003.12.013
Chatzittofis, A., Arver, S., Öberg, K., Hallberg, J., Nordström, P., and Jokinen, J. (2016). HPA axis dysregulation in men with hypersexual disorder. Psychoneuroendocrinology 63, 247–253. doi: 10.1016/j.psyneuen.2015.10.002
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., et al. (2003). Depression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. doi: 10.1126/science.1086446
Covington, H., Maze, I., LaPlant, Q., Vialou, V., Yoshinori, O., Berton, O., et al. (2009). Antidepressant actions of HDAC inhibitors. J. Neurosci. 29, 11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009
Covington, H. E., Vialou, V. F., Laplant, Q., Ohnishi, Y. N., and Nestler, E. J. (2011). Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci. Lett. 493, 122–126. doi: 10.1016/j.neulet.2011.02.022
De Luca, V. D., Likhodi, O., Kennedy, J. L., and Wong, A. H. (2007). Differential expression and parent-of-origin effect of the5-HT2A receptor gene C102T polymorphism: analysis of suicidality in schizophrenia and bipolar disorder. Am. J. Med. Genet. Part B 144B, 370–374. doi: 10.1002/ajmg.b.30458
De Luca, V. D., Viggiano, E., Dhoot, R., Kennedy, J. L., and Wong, A. H. (2009). Methylation and QTDT analysis of the 5-HT2A receptor 102C allele: analysis of suicidality in major psychosis. J. Psychiatr. Res. 43, 532–537. doi: 10.1016/j.jpsychires.2008.07.007
Du, L., Bakish, D., Lapierre, Y. D., Ravindran, A. V., and Hrdina, P. D. (2000). Association of polymorphism of serotonin 2A receptor gene with suicidal ideation in major depressive disorder. Am. J. Med. Genet. 96, 56–60.
Du, L., Faludi, G., Palkovits, M., Bakish, D., and Hrdina, P. D. (2001). Serotonergic genes and suicidality. Crisis 22, 54–60. doi: 10.1027//0227-5910.22.2.54
Edwards, V. J., Holden, G. W., Felitti, V. J., and Anda, R. F. (2003). Relationship between multiple forms of childhood maltreatment and adult mental health in community respondents: results from the adverse childhood experiences study. Am. J. Psychiatry 160, 1453–1460. doi: 10.1176/appi.ajp.160.8.1453
Elliott, E., Ezra-Nevo, G., Regev, L., Neufeld-Cohen, A., and Chen, A. (2010). Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat. Neurosci. 13, 1351–1353. doi: 10.1038/nn.2642
Fergusson, D., Horwood, L., and Woodward, L. (2000). The stability of child abuse reports: a longitudinal study of the reporting behaviour of young adults. Psychol Med. 30, 529–544. doi: 10.1017/S0033291799002111
Frodl, T., Carballedo, A., Fagan, A. J., Lisiecka, D., Ferguson, Y., Meaney, J. (2012). Effects of early-life adversity on white matter diffusivity changes in patients at risk for major depression. J. Psychiatry. Neurosci. 37, 37–45. doi: 10.1503/jpn.110028
Frodl, T., Szyf, M., Carballedo, A., Ly, V., Dymov, S., Vaisheva, F., et al. (2015). DNA methylation of the serotonin transporter gene (SLC6A4) is associated with brain function involved in processing emotional stimuli. J. Psychiatry Neurosci. 40, 296–305. doi: 10.1503/jpn.140180
Fuchikami, M., Morinobu, S., Segawa, M., Okamoto, Y., Yamawaki, S., Ozaki, N., et al. (2011). DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS One 6:8. doi: 10.1371/journal.pone.0023881
Fuchikami, M., Yamamoto, S., Morinobu, S., Okada, S., Yamawaki, Y., and Yamawaki, S. (2016). The potential use of histone deacetylase inhibitors in the treatment of depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 64, 320–324. doi: 10.1016/j.pnpbp.2015.03.010
Fyffe, S. L., Neul, J. L., Samaco, R. C., Chao, H., Ben-Shachar, S., Moretti, P., et al. (2008). Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 59, 947–958. doi: 10.1016/j.neuron.2008.07.030
Gilbert, R., Widom, C. S., Browne, K., Fergusson, D., Webb, E., and Janson, S. (2009). Burden and consequences of child maltreatment in high-income countries. Lancet 373, 68–81. doi: 10.1016/S0140-6736(08)61706-7
Grydeland, H., Walhovd, K., Tamnes, C., Westlye, L., and Fjell, A. (2013). Intracortical myelin links with performance variability across the human lifespan: results from T1- and T2-weighted MRI myelin mapping and diffusion tensor imaging. J. Neurosci. 33, 18618–18630. doi: 10.1523/JNEUROSCI.2811-13.2013
Guy, J., Cheval, H., Selfridge, J., and Bird, A. (2011). The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652. doi: 10.1146/annurev-cellbio-092910-154121
Haroutunian, V., Katsel, P., Roussos, P., Davis, K., Altshuler, L., and Bartzokis, G. (2014). Myelination, oligodendrocytes, and serious mental illness. Glia 62, 1856–1877. doi: 10.1002/glia.22716
Heim, C., and Binder, E. B. (2012). Current research trends in early life stress and depression: review of human studies on sensitive periods, gene–environment interactions, and epigenetics. Exp. Neurol. 233, 102–111. doi: 10.1016/j.expneurol.2011.10.032
Hollis, F., Duclot, F., Gunjan, A., and Kabbaj, M. (2011). Individual differences in the effect of social defeat on anhedonia and histone acetylation in the rat hippocampus. Horm. Behav. 59, 331–337. doi: 10.1016/j.yhbeh.2010.09.005
Hollis, F., Wang, H., Dietz, D., Gunjan, A., and Kabbaj, M. (2010). The effects of repeated social defeat on long-term depressive-like behavior and short-term histone modifications in the hippocampus in male Sprague–Dawley rats. Psychopharmacology 211, 69–77. doi: 10.1007/s00213-010-1869-9
Jin, B., Li, Y., and Robertson, K. D. (2011). DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer 2, 607–617. doi: 10.1177/1947601910393957
Jonson-Reid, M., Kohl, P., and Drake, B. (2012). Child and adult outcomes of chronic child maltreatment. Pediatrics 129, 839–845. doi: 10.1542/peds.2011-2529
Kaufman, J., Wymbs, N., Montalvo-Ortiz, J., Orr, C., Albaugh, M., Althoff, R., et al. (2018). Methylation in OTX2 and Related Genes, Maltreatment, and Depression in Children. Available at: https://www.nature.com/articles/s41386-018-0157-y [accessed August 1, 2018].
Keller, J., Gomez, R., Williams, G., Lembke, A., Lazzeroni, L., Murphy, G. M., et al. (2016). HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 22, 527–536. doi: 10.1038/mp.2016.120
Klengel, T., Mehta, D., Anacker, C., Rex-Haffner, M., Pruessner, J. C., Pariante, C. M., et al. (2012). Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions. Nat. Neurosci. 16, 33–41. doi: 10.1038/nn.3275
Krishnan, V., Han, M., Graham, D., Berton, O., Renthal, W., Russo, S., et al. (2007). Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131, 391–404. doi: 10.1016/j.cell.2007.09.018
Krugers, H. J., Arp, J. M., Xiong, H., Kanatsou, S., Lesuis, S. L., Korosi, A., et al. (2017). Early life adversity: lasting consequences for emotional learning. Neurobiol. Stress 6, 14–21. doi: 10.1016/j.ynstr.2016.11.005
Kundakovic, M., Gudsnuk, K., Herbstman, J. B., Tang, D., Perera, F. P., and Champagne, F. A. (2014). DNA methylation of BDNF as a biomarker of early-life adversity. Proc. Natl. Acad. Sci. U.S.A. 112, 6807–6813. doi: 10.1073/pnas.1408355111
Labonté, B., Suderman, M., Maussion, G., Navaro, L., Yerko, V., Mahar, I., et al. (2012). Genome-wide epigenetic regulation by early-life trauma. Arch. Gen. Psychiatry 69:7. doi: 10.1001/archgenpsychiatry.2011.2287
Leeb, R., Paulozzzi, L., Melanson, C., Simon, T., and Arias, I. (2008). Child Maltreatment Surveillance. Uniform Definitions for Public Health and Recommended Data Elements. Atlanta: Centers for Disease Control and Prevention.
Long, P., and Corfas, G. (2014). To learn is to myelinate. Science 346, 298–299. doi: 10.1126/science.1261127
Lopez, J. P., Mamdani, F., Labonte, B., Beaulieu, M., Yang, J. P., Berlim, M. T., et al. (2013). Epigenetic regulation of BDNF expression according to antidepressant response. Mol. Psychiatry 18, 398–399. doi: 10.1038/mp.2012.38
Lutz, P., Almeida, D., Fiori, L., and Turecki, G. (2015). Childhood maltreatment and stress-related psychopathology: the epigenetic memory hypothesis. Curr. Pharm. Des. 21, 1413–1417. doi: 10.2174/1381612821666150105124928
Lutz, P., Gross, J. A., Dhir, S. K., Maussion, G., Yang, J., Bramoulle, A., et al. (2017a). Epigenetic regulation of the kappa opioid receptor by child abuse. Biol. Psychiatry 17, 31812–31817. doi: 10.1016/j.biopsych.2017.07.012
Lutz, P., Tanti, A., Gasecka, A., Barnett-Burns, S., Kim, J. J., Zhou, Y., et al. (2017b). Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: convergent epigenetic, transcriptional, and morphological evidence. Am. J. Psychiatry 174, 1185–1194. doi: 10.1176/appi.ajp.2017.16111286
Makinodan, M., Rosen, K., Ito, S., and Corfas, G. (2012). A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 337, 1357–1360. doi: 10.1126/science.1220845
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893. doi: 10.1126/science.1090842
McGowan, P. O., Sasaki, A., Dalessio, A. C., Dymov, S., Labonté, B., Szyf, M., et al. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12, 342–348. doi: 10.1038/nn.2270
Melas, P., Rogdaki, M., Lennartsson, A., Björk, K., Qi, H., Witasp, A., et al. (2012). Antidepressant treatment is associated with epigenetic alterations in the promoter of P11 in a genetic model of depression. Int. J. Neuropsychopharmacol. 15, 669–679. doi: 10.1017/S1461145711000940
Menke, A., Klengel, T., and Binder, E. B. (2012). Epigenetics, depression and antidepressant treatment. Curr. Pharm. Des. 18, 5879–5889. doi: 10.2174/138161212803523590
Moretti, M., Valvassori, S. S., Varela, R. B., Ferreira, C. L., Rochi, N., Benedet, J., et al. (2011). Behavioral and neurochemical effects of sodium butyrate in an animal model of mania. Behav. Pharmacol. 22, 766–772. doi: 10.1097/fbp.0b013e32834d0f1b
Morgan, J., Wu Nordahl, C., and Schumann, C. (2013). “Chapter 3.5 - The amygdala in autism spectrum disorders,” in The Neuroscience of Autism Spectrum Disorders, eds J. D. Buxbaum and P. R. Hof (Cambridge, MA: Academic Press), 297–312. doi: 10.1016/B978-0-12-391924-3.00021-1
Mosebach, J., Keilhoff, G., Gos, T., Schiltz, K., Schoeneck, L., Dobrowolny, H., et al. (2013). Increased nuclear Olig1-expression in the pregenual anterior cingulate white matter of patients with major depression: a regenerative attempt to compensate oligodendrocyte loss? J. Psychiatric Res. 47, 1069–1079. doi: 10.1016/j.jpsychires.2013.03.018
Mundorf, A., Schmitz, J., Güntürkün, O., Freunda, N., and Ocklenburg, S. (2018). Methylation of MORC1: a possible biomarker for depression? J. Psychiatric Res. 103, 208–211. doi: 10.1016/j.jpsychires.2018.05.026
Murgatroyd, C., Patchev, A. V., Wu, Y., Micale, V., Bockmuhl, Y., Fischer, D., et al. (2009). Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 12, 1559–1566. doi: 10.1038/nn.2436
Murri, M., Prestia, D., Mondelli, V., Pariante, C., Patti, S., Olivieri, B., et al. (2016). The HPA axis in bipolar disorder: systematic review and meta-analysis. Psychoneuroendocrinology 63, 327–342. doi: 10.1016/j.psyneuen.2015.10.014
Nemeroff, C. (2016). Paradise lost: the neurobiological and clinical consequences of child abuse and neglect. Neuron 89, 892–909. doi: 10.1016/j.neuron.2016.01.019
Pariante, C. M., and Lightman, S. L. (2008). The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 31, 464–468. doi: 10.1016/j.tins.2008.06.006
Peña, C. J., Kronman, H. G., Walker, D. M., Cates, H. M., Bagot, R. C., Purushothaman, I., et al. (2017). Early life stress confers lifelong stress susceptibility in mice via ventral tegmental area OTX2. Science 356, 1185–1188. doi: 10.1126/science.aan4491
Perroud, N., Paoloni-Giacobino, A., Prada, P., Olié, E., Salzmann, A., Nicastro, R., et al. (2011). Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl. Psychiatry 1, 1–9. doi: 10.1038/tp.2011.60
Perroud, N., Salzmann, A., Prada, P., Nicastro, R., Hoeppli, M., Furrer, S., et al. (2013). Response to psychotherapy in borderline personality disorder and methylation status of the BDNF gene. Transl. Psychiatry 3, 207. doi: 10.1038/tp.2012.140
Pishva, E., Rutten, B., and van den Hove, D. (2017). DNA methylation in major depressive disorder. Neuroepigenom. Aging Dis. 978, 185–196. doi: 10.1007/978-3-319-53889-1_10
Qiu, X., Xiao, X., Li, N., and Li, Y. (2017). Histone deacetylases inhibitors (HDACis) as novel therapeutic application in various clinical diseases. Prog. Neuro Psychopharmacol. Biol. Psychiatry 72, 60–72. doi: 10.1016/j.pnpbp.2016.09.002
Resende, W. R., Valvassori, S. S., Réus, G. Z., Varela, R. B., Arent, C. O., Ribeiro, K. F., et al. (2013). Effects of sodium butyrate in animal models of mania and depression. Behav. Pharmacol. 24, 569–579. doi: 10.1097/FBP.0b013e32836546fc
Rohleder, N., and Kirschbaum, C. (2006). The hypothalamic–pituitary–adrenal (HPA) axis in habitual smokers. Int. J. Psychophysiol. 59, 236–243. doi: 10.1016/j.ijpsycho.2005.10.012
Roth, T. L., Lubin, F. D., Funk, A. J., and Sweatt, J. D. (2009). Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 65, 760–769. doi: 10.1016/j.biopsych.2008.11.028
Roth, T. L., and Sweatt, J. D. (2011). Epigenetic marking of the BDNF gene by early-life adverse experiences. Horm. Behav. 59, 315–320. doi: 10.1016/j.yhbeh.2010.05.005
Roy, A., Gorodetsky, E., Yuan, Q., Goldman, D., and Enoch, M. (2010). Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology 35, 1674–1683. doi: 10.1038/npp.2009.236
Saavedra, K., Molina-Márquez, A., Saavedra, N., Zambrano, T., and Salazar, L. (2016). Epigenetic modifications of major depressive disorder. Int. J. Mol. Sci. 17:1279. doi: 10.3390/ijms17081279
Sacchet, M., and Gotlib, H. (2017). Myelination of the brain in major depressive disorder: an in vivo quantitative magnetic resonance imaging study. Sci. Rep. 7:1. doi: 10.1038/s41598-017-02062-y
Shonkoff, J. P., Boyce, W. T., and Mcewen, B. S. (2009). Neuroscience, molecular biology, and the childhood roots of health disparities. JAMA 301:2252. doi: 10.1001/jama.2009.754
Sterrenburg, L., Gaszner, B., Boerrigter, J., Santbergen, L., Bramini, M., Elliott, E., et al. (2011). Chronic stress induces sex-specific alterations in methylation and expression of corticotropin-releasing factor gene in the rat. PLoS One 6:11. doi: 10.1371/journal.pone.0028128
Stikov, N., Campbell, J., Stroh, T., Lavelée, M., Frey, S., Novek, J., et al. (2015). In vivo histology of the myelin g-ratio with magnetic resonance imaging. NeuroImage 118, 397–405. doi: 10.1016/j.neuroimage.2015.05.023
Suderman, M., Borghol, N., Pappas, J., Pereira, S., Pembrey, M., Hertzman, C., et al. (2015). Childhood abuse is associated with methylation of multiple loci in adult DNA. Soc. Burden Child Abuse 7, 247–274. doi: 10.1201/b18768-19
Suderman, M., Mcgowan, P. O., Sasaki, A., Huang, T. C., Hallett, M. T., Meaney, M. J., et al. (2012). Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl. Acad. Sci. U.S.A. 109, 17266–17272. doi: 10.1073/pnas.1121260109
Tadić, A., Wagner, S., Schlicht, K. F., Peetz, D., Borysenko, L., Dreimüller, N., et al. (2011). The early non-increase of serum BDNF predicts failure of antidepressant treatment in patients with major depression: a pilot study. Prog. Neuro Psychopharmacol. Biol. Psychiatry 35, 415–420. doi: 10.1016/j.pnpbp.2010.08.011
Thomas, C., Hyponnen, E., and Power, C. (2008). Obesity and type 2 diabetes risk in mid-adult life: the role of childhood adversity. Pediatrics 121, e1240–e1249. doi: 10.1542/peds.2007-2403
Tsankova, N. M., Berton, O., Renthal, W., Kumar, A., Neve, R. L., and Nestler, E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9, 519–525. doi: 10.1038/nn1659
Turecki, G. (2014). The molecular bases of the suicidal brain. Nat. Rev. Neurosci. 15, 802–816. doi: 10.1038/nrn3839
Tyrka, A. R., Ridout, K. K., Parade, S. H., Paquette, A., Marsit, C. J., and Seifer, R. (2015). Childhood maltreatment and methylation of FK506 binding protein 5 gene (FKBP5). Dev. Psychopathol. 27, 1637–1645. doi: 10.1017/S0954579415000991
Uchida, S., Hara, K., Kobayashi, A., Otsuki, K., Yamagata, H., Hobara, T., et al. (2011). Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69, 359–372. doi: 10.1016/j.neuron.2010.12.023
Uranova, N., Vostrikov, V., Orlovskaya, D., and Rachmanova, V. (2004). Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the stanley neuropathology consortium. Schizophr. Res. 67, 269–275. doi: 10.1016/S0920-9964(03)00181-6
US Department of Health, and Human Services (2008). Administration on Children youth, and Families. Child Maltreatment 2006. Washington, DC: US Government Printing Office.
Vijayendran, M., Cutrona, C., Beach, S. R., Brody, G. H., Russell, D., and Philibert, R. A. (2012). The relationship of the serotonin transporter (SLC6A4) extra long variant to gene expression in an African American sample. Am. J. Med. Genet. Part B 159B, 611–612. doi: 10.1002/ajmg.b.32054
Weaver, I. C., D’Alessio, A. C., Brown, S. E., Hellstrom, I. C., Dymov, S., and Sharma, S. (2007). The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J. Neurosci. 27, 1756–1768. doi: 10.1523/JNEUROSCI.4164-06.2007
Weaver, I. C., Hellstrom, I. C., Brown, S. E., Andrews, S. D., Dymov, S., Diorio, J., et al. (2014). The methylated-DNA binding protein MBD2 enhances NGFI-A (egr-1)-mediated transcriptional activation of the glucocorticoid receptor. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130513. doi: 10.1098/rstb.2013.0513
Weder, N., Zhang, H., Jensen, K., Yang, B. Z., Simen, A., Jackowski, A., et al. (2014). Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J. Am. Acad. Child Adolesc. Psychiatry 53:4. doi: 10.1016/j.jaac.2013.12.025
Withers, A. C., Tarasoff, J. M., and Stewart, J. W. (2013). Is depression with atypical features associated with trauma history? J. Clin. Psychiatry. 74, 500–506. doi: 10.4088/JCP.12m07870
Woodman, J., Pitt, M., Wentz, R., Taylor, B., Hodes, D., and Gilbert, R. (2008). Performance of screening tests for child physical abuse in accident and emergency departments. Health Technol. Assess. 12:118. doi: 10.3310/hta12330
World Health Organization [WHO] (2017). Depression and Other Common Mental: Disorders Global Health Estimates. Geneva: World Health Organization.
Keywords: epigenetics, major depressive disorder, early life adversity, DNA methylation, biomarkers, antidepressant therapies
Citation: Brown A, Fiori LM and Turecki G (2019) Bridging Basic and Clinical Research in Early Life Adversity, DNA Methylation, and Major Depressive Disorder. Front. Genet. 10:229. doi: 10.3389/fgene.2019.00229
Received: 17 August 2018; Accepted: 01 March 2019;
Published: 22 March 2019.
Edited by:
Tanja Maria Michel, University of Southern Denmark, DenmarkReviewed by:
Manish Kumar Jha, University of Texas Southwestern Medical Center, United StatesGopalkumar Rakesh, Duke University, United States
Copyright © 2019 Brown, Fiori and Turecki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gustavo Turecki, Z3VzdGF2by50dXJlY2tpQG1jZ2lsbC5jYQ==