Abstract
Network medicine is an emerging area of research dealing with molecular and genetic interactions, network biomarkers of disease, and therapeutic target discovery. Large-scale biomedical data generation offers a unique opportunity to assess the effect and impact of cellular heterogeneity and environmental perturbations on the observed phenotype. Marrying the two, network medicine with biomedical data provides a framework to build meaningful models and extract impactful results at a network level. In this review, we survey existing network types and biomedical data sources. More importantly, we delve into ways in which the network medicine approach, aided by phenotype-specific biomedical data, can be gainfully applied. We provide three paradigms, mainly dealing with three major biological network archetypes: protein-protein interaction, expression-based, and gene regulatory networks. For each of these paradigms, we discuss a broad overview of philosophies under which various network methods work. We also provide a few examples in each paradigm as a test case of its successful application. Finally, we delineate several opportunities and challenges in the field of network medicine. We hope this review provides a lexicon for researchers from biological sciences and network theory to come on the same page to work on research areas that require interdisciplinary expertise. Taken together, the understanding gained from combining biomedical data with networks can be useful for characterizing disease etiologies and identifying therapeutic targets, which, in turn, will lead to better preventive medicine with translational impact on personalized healthcare.
Introduction
Biological systems are comprised of various molecular entities such as genes, proteins and other biological molecules, as well as interactions between those components. Understanding a given phenotype, the functioning of a cell or tissue, etiology of disease, or cellular organization, requires accurate measurements of the abundance profiles of these molecular entities in the form of biomedical data. Analysis of the biomedical data allows us to explain important features of the interactions leading to a mechanistic understanding of the observed phenotype. The interplay between different components at different levels can be represented in the form of biological networks, for example, protein-protein interactions (PPIs) (Uetz et al., 2000; Cusick et al., 2005) and gene regulatory networks (GRNs) (Davidson, 2006). Different biological networks capture the complex interactions between genes, proteins, RNA molecules, metabolites and genetic variants in the cells of organisms. These networks, also interchangeably known as graphs, are representations in which the complex system components are simplified as nodes that are connected by links (edges) (Vidal et al., 2011). Networks provide a conceptual and intuitive framework to model different components of multiple omics data from the genome, transcriptome, proteome, and metabolome (Figure 1; Liu and Lauffenburger, 2009).
FIGURE 1
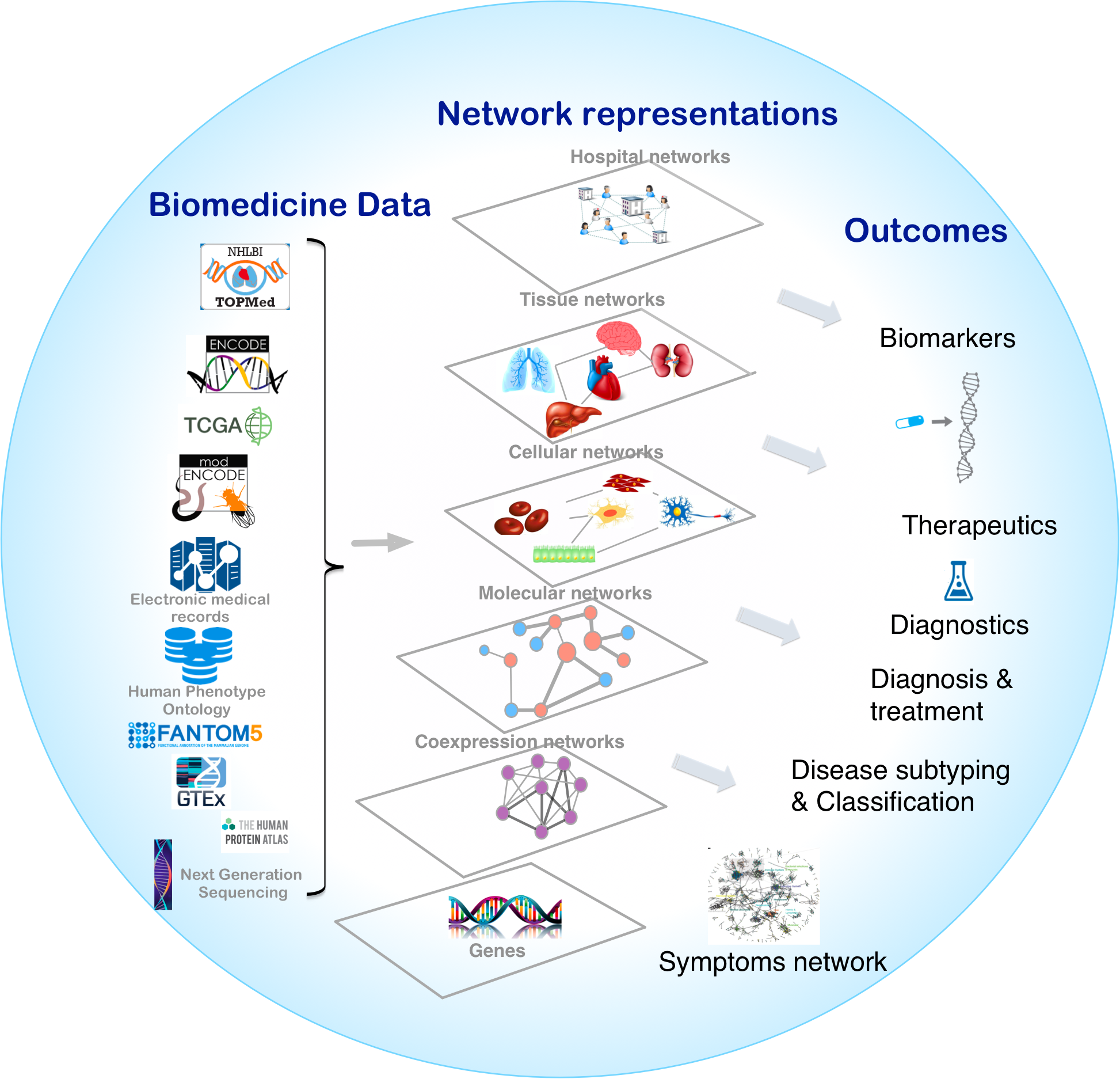
Overview of network medicine approach depicting various biomedical data types discussed at length in the manuscript, along with network representations that simplify different components of multiple omics data from the genome, transcriptome, proteome, and metabolome as nodes that are connected by links (edges). Combining biomedical data with the appropriate network modeling approach allows derivation of disease associated information and outcomes like biomarkers, therapeutics targets, phenotype-specific genes and interactions, and disease subtypes.
The convenient representation of the biological components in graphs led to the field of network biology – a discipline that studies holistic relationships between various biological components by combining graph theory, systems biology, and statistical analyses (Lindfors, 2011; Walhout et al., 2012). Moreover, the quantitative tools of network biology offer the potential to understand cellular organization and capture the impact of perturbations on these complex intracellular networks (Wang et al., 2011). Network Medicine is an extension of network biology with a set of focused goals related to disease biology, including understanding disease etiology, identifying potential biomarkers, and designing therapeutic interventions, including drug targets, dosage, and synergism discovery (Loscalzo et al., 2017). Research in network medicine heavily depends on large datasets for building models, making predictions and assessing their validity. The promise of network medicine research is to develop a more global understanding of how perturbations propagate in the system by identifying the pathways, sub-types of disease states, and key components in the networks that can be targeted in clinical interventions. Moreover, networks are the centerpiece of the “new biology” in the biomedical data revolution and translation to personalized medicine (Schadt and Bjorkegren, 2012).
Advances in high-throughput biotechnologies have led to the generation of massive amounts of biomedical data that provides new research avenues. The rapid decline in costs due to technological advancements such as next generation sequencing (NGS) have provided the necessary impetus to generate multiple large-scale multi-omics biomedical data-sets that characterize various phenotypes. This includes exome and whole genome sequencing, transcriptomics, proteomics, lipidomics, microbiomics, etc. (Schadt and Bjorkegren, 2012). Constructing appropriate network models is a challenging problem that heavily depends on the study design, the phenotype under study, the molecular entities measured, and the type and size of the data. The field of network medicine is largely discovery — rather than hypothesis — driven, uncovering previously unknown relationships and leading to the identification of new biomarkers. The statistical rigor of network predictions comes from the study design and the size of the datasets. Large-scale consortium-based efforts looking at the various aspects of human biology have allowed the application of network-based methods to uncover new insights into the molecular mechanisms of the given phenotype, such as tissue specificity or disease context. In this review, we first examine various large-scale biomedical datasets and types of biological networks as summarized by Figure 1. We then provide three paradigms in which biological networks can be combined with big biomedical data to understand the given phenotype.
Biomedical Data Sources
Recent technological advancements in sequencing technologies, resulting in a reduction in cost per base pair, have heralded an era of massive data generation for different types of molecular profiles across a broad range of phenotypes and diseases. After the completion of the human genome project (Collins et al., 2003), the HapMap project (The International HapMap Consortium, 2003) created an extensive catalog of common human genetic variants, the differences in DNA sequences, based on microarray data. These studies eventually progressed into the “1000 Genomes Project” (The 1000 Genomes Project Consortium, 2015), which leveraged NGS technologies. In cancer research, the cancer genome atlas (TCGA) (Cancer Genome Atlas Research Network, 2008) contains profiles of tumors and matched normal samples from more than 11000 subjects for 33 cancer types. The repertoire of TCGA data includes clinical information (demographic, treatment, and survival information), gene expression profiling, microRNA profiling, copy number variation (CNV) (genomic structural variations) identifications, single nucleotide polymorphism (SNP), DNA methylation (whole genome methylation calls for each CpG site), and exon sequencing (expression signal of particular composite exon of a gene). Together these data have helped in the identification of driver somatic mutations, the molecular basis of cancer progression, and potential therapeutic interventions for cancer subtypes. To understand the role of the epigenetic state in gene regulation and to characterize the functional elements of the transcriptional machinery, the ENCyclopedia Of DNA elements (ENCODE) consortium for humans (ENCODE Project Consortium, 2012), model organism ENCyclopedia Of DNA Elements (modENCODE) for model organisms (Yue et al., 2014), and ROADMAP Epigenomics project (Romanoski et al., 2015) were commissioned to improve the understanding of how epigenomics contributes to disease. The Riken-led Functional ANnoTation Of Mammalian Genome (FANTOM5) (Andersson et al., 2014) project provided cell-type-specific enhancer elements and identified pathobiological regulatory SNPs. To further understand transcriptional patterns in human tissues and their relationship with the genotype, genotype-tissue expression (GTEx) data was generated (GTEx Consortium, 2015; Mele et al., 2015). Trans-omics for precision medicine (TOPMed) (Prokopenko et al., 2018) is another set of multi-omics data on 100k individuals that also includes clinical data and is aimed at understanding the fundamental biological processes that underlie heart, lung, blood, and sleep disorders. The Precision Medicine Initiative or “All of Us” program1 aims to acquire a broad range of data from about 1 million individuals.
Since 2003, the human protein atlas (HPA) (Uhlen et al., 2005; Uhlen et al., 2015), curated by Swedish consortium, has been releasing data on protein expression levels in cells, tissues, and various pathologies, including 17 cancer types. Similarly, the human cell atlas (HCA) (Rozenblatt-Rosen et al., 2017) aims to provide a reference map of single cell omics data in human cells and cell types. The UK-Biobank (Allen et al., 2014; Sudlow et al., 2015) is another commercial resource that has an array of health-related measurements on patients, including biomarkers, images, clinical information, and genetic data. The human microbiome project (HMP) (Turnbaugh et al., 2007) is a categorization of microbiota on different human body sites whose goal is to understand the role of the microbiome and the impact of its dysbiosis on human disease. Apart from these large international databases looking at one or more aspects of health or disease, many other resources from the concerted efforts over decades of data collection are also available. This includes the Nurses’ Health Study (Belanger et al., 1978; Colditz et al., 2016), Health Professionals Follow-up Study (Grobbee et al., 1990), Framingham Heart Study (Dawber et al., 1951; Mahmood et al., 2014), and COPDGene (Pillai et al., 2009). This wealth of biomedical data not only allows for a deeper probing of the underlying biological systems, but also inspires the development of novel methods that can maximize the information that can be extracted from these data. The tools developed within the field of network medicine are highly versatile, enabling their customized application depending on the given biological or disease context.
Collecting large-scale multi-time point data across multiple omics in different disease conditions is expensive and often not feasible, especially for human subjects. However, small-scale longitudinal data for a single omic, such as gene expression, is available in biomedical databases (Jung et al., 2015; Bouquet et al., 2016). High resolution mass spectrometry has also allowed for the collection of longitudinal proteome data, for example to test the effect of drugs (Fournier et al., 2010) or oxidative stress (Vogel et al., 2011) in yeast. A longitudinal multi-omic dataset containing both human transcriptomic and proteomic information has been analyzed to study changes in molecular profiles (Chen et al., 2012). Multi-omic datasets such as this one allows us to probe the relationship between biological molecules based on the central dogma of biology, such as the connection between transcript abundance and protein levels (Marguerat et al., 2012; Liu et al., 2016). Longitudinal data is also amenable to temporal or dynamical network analysis, wherein one can evaluate the statistical dependence of the state of a network on the gene expression patterns from previous time steps (Kim and Kim, 2018; Dondelinger and Mukherjee, 2019). Kim et al. provide a summary of several methods to infer temporal regulatory relationships (Kim et al., 2014).
In the next section, we will review some of the main types of biological networks constructed using high throughput molecular profiling, literature mining, or manual curation of scientific literature.
Primer on Biological Networks
Each network-based study has to primarily identify two things: what are the critical entities in the system under investigation (nodes), and what is the nature of the interactions between these entities (edges) (de Silva and Stumpf, 2005). This information often comes from multiple different data sources, dealing with the various facets of the biological system. For example, PPIs, also defined as the interactome, is a network of proteins and the physical interactions between them (Cusick et al., 2005). These interactions can be obtained from yeast-2-hybrid assays (Li et al., 2004; Vidal and Fields, 2014), co-immunoprecipitation (Lin and Lai, 2017), literature text-mining (Papanikolaou et al., 2015), 3D structure (Lu et al., 2013), co-expression of genes (Bhardwaj and Lu, 2005), sequence homology (Shen et al., 2007), and other sources. Each of these data sources have both merits and demerits (Cusick et al., 2005). These networks inform us about the overall topological properties of protein interactions as well as the positions of specific proteins within this network. However, extracting phenotype specific (i.e., cell, tissue or disease-specific) information based on the PPI remains an open challenge and requires the development of novel ways of integrating biomedical data with these networks.
Gene co-expression and regulatory networks often make direct use of phenotype-specific gene expression data in the network construction, with additional analysis required to extract meaningful biological information for the underlying phenotype. The availability of transcriptomic data for a wide range of phenotypes presents an opportunity to probe the patterns of molecular co-abundance, albeit with limitations concerning the interpretation of the biology. Gene co-expression networks (GCNs) can be constructed in many ways, including information theoretic, regression-based, and Bayesian approaches (Butte and Kohane, 1999). Several common methods for constructing GCNs include Weighted Gene Co-expression Network Analysis (WGCNA; Langfelder and Horvath, 2008), Context Likelihood of Relatedness (CLR; Faith et al., 2007), Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe; Margolin et al., 2006), Partial Correlation and Information Theory (PCIT; Reverter and Chan, 2008), Gene Network Inference with Ensemble of Trees (GENIE3; Huynh-Thu et al., 2010), Supervised Inference of Regulatory Networks (SIRENE; Mordelet and Vert, 2008), and Gene CO-expression Network method (GeCON; Roy et al., 2014). GRNs are a related type of network that attempts to look beyond the co-abundance of gene expression and instead identify the influencing patterns of transcription factor genes over others in a mechanistic fashion (Marbach et al., 2012). Since transcriptional regulation depends on cis and trans-regulatory elements as well as transcription factor binding, GRNs often incorporate this information during model construction. Many methods with a modified definition of correlations have been proposed to infer GRNs. However, identifying the putative cis-regulatory sequences, such as those found in the promoter regions of genes, that are relevant for a specific biological context is important to enable the understanding of disease, tissue, or cell-specific regulatory perturbations. The location of TF binding to the DNA can be assayed using yeast-1-hybrid (Deplancke et al., 2004), ChIP-Seq (Jaini et al., 2014), or inferred by other means (Mundade et al., 2014). However, the cost and other limitations involved in generating these data in a context-specific manner have meant that incorporating this information when constructing putative regulatory networks remains a challenge.
Other types of biological networks include metabolic networks, which represent a collection of biochemical interactions between metabolites and enzymes (Terzer et al., 2009). Ecological networks, which represent biotic interactions, can also be applied to microbiome data, the collection of microbes’ genes, to construct microbiome networks (Coyte et al., 2015; Layeghifard et al., 2017; Bauer and Thiele, 2018; Rottjers and Faust, 2018). Together, genotype and transcriptomic data can be used to map genetic variants to genes and then summarized in an expression Quantitative Trait Loci (eQTL) network (Platig et al., 2016; Fagny et al., 2017). A network of immune cell communication has been constructed using high-resolution mass spectrometry-based proteomics data and was shown to exhibit social network-like properties. Disease networks, also known as the diseasome, have been proposed; these networks connect diseases and disorders with disease genes based on Online Mendelian Inheritance in Man (OMIM) associations (Boyadjiev and Jabs, 2000; Hamosh et al., 2002; Goh et al., 2007; Wysocki and Ritter, 2011; Zhang et al., 2011). Similarly, networks connecting symptoms with diseases have helped to shed light on the shared genetic associations between diseases (Zhou et al., 2014). Efforts to identify specific disease-causing genes, using genomic intervals obtained from linkage mappings or Genome-Wide Association Studies (GWAS), have been undertaken using hybrid heterogeneous networks. These hybrid networks often include a combination of disease-gene networks, generic or tissue-specific molecular networks such as PPIs or GCNs, and prior knowledge of disease similarities (Navlakha and Kingsford, 2010; Moreau and Tranchevent, 2012; Ni et al., 2016). Various network-based tools have been implemented in the gene prioritization problem (Wu et al., 2008; Li and Patra, 2010; Tian et al., 2017). All these aforementioned types of network biology approaches are particularly useful in understanding complex diseases, which result from multiple genetic factors and environmental influences (Moreau and Tranchevent, 2012).
Analysis of biological networks also necessitates understanding their structural or topological properties. This includes the identification of important modulators, driver nodes, local network structures, and recurrent subgraphs in the network. Local connectivity properties such as degree and other centrality metrics can help to identify key molecular entities that dominate various network neighborhoods, such as hubs, bottlenecks, or core nodes. At the global level, properties like average path length, degree distribution, diameter, clustering coefficients, and controllability (Liu et al., 2011) help with the characterization and comparison of network topologies. Mesoscale measures such subgraphs or network motifs – recurrent patterns connecting a fixed number of nodes (typically 3 or 4) – are considered fundamental components of biological networks (Milo et al., 2002). An extension of network motifs to include more nodes, or graphlets, has been used to analyze the interactome (Przulj et al., 2004; Davis et al., 2015; Malod-Dognin et al., 2017). Identifying the connectivity patterns enriched in a network (i.e., over-represented with respect to a null model) can help to compare, characterize, and discriminate between networks (Shen-Orr et al., 2002; Alon, 2007; Przulj, 2007). These patterns are also commonly associated with control substructures that dominate information flow in the networks, especially in transcriptional regulatory, neuronal, and social networks.
Integrating Biomedical Data With Networks: Challenges and Ways
The ultimate aim of inferring biological networks using biomedical data is to provide lab-testable hypotheses by identifying biomolecular entities that play a crucial role in the observed phenotype (Figure 1). Detecting changes in abundance levels of these biomolecules and their interaction landscape in the context of a tissue, cell, or disease-specific environment requires both relevant data and the application of appropriate network analysis. Each biological network analysis has strengths and limitations based on how it incorporates phenotype specific data, and the research question being addressed (Altaf-Ul-Amin et al., 2014; Kanaya et al., 2014). In some cases, it is possible to identify a baseline network from general physical interactions between proteins, after which disease or phenotype-specific information from specific experiments can be overlaid to generate a more context-specific network.
Protein-protein interaction networks provide a fabric of potential interactions between proteins, but phenotype-specific interactions can only be added as an extra layer from separate biomedical data. The hypothesis behind analyzing such networks, combination of baseline PPI with disease information added as next step, is that the defects or mutations in only a few genes or proteins may propagate to other components in the network, and that this collection of affected genes constitute a critical module in the network (Schadt and Bjorkegren, 2012). Previous work along these lines has shown that these modules are not only structurally related but are also functionally relevant to the observed phenotype. This central tenet of network medicine from the interactome has been successfully tested for many diseases and other phenotypes (Lim et al., 2006; Goh et al., 2007; Taylor et al., 2009; Sharma et al., 2013, 2015, 2018; Menche et al., 2015; Sahni et al., 2015; Huttlin et al., 2017; Huang J.K. et al., 2018; Wang et al., 2018; Willsey et al., 2018) and has also led to novel drug-target discoveries (Yildirim et al., 2007; Guney et al., 2016; Luo et al., 2017) along with novel interactions between genes. Despite recent advances, the PPI is incomplete and inferring disease-specific interactions requires innovative strategies in order to overcome this deficiency.
Gene co-expression networks are by definition context-specific, as they are constructed by calculating correlations in a given gene expression data set. In contrast, GRNs often are built starting from a baseline network composed of all potential interactions between transcription factors and genes. This baseline network can be derived from genetic sequence information and DNA-binding domain sequences within regulatory proteins, such that an interaction is inferred if a given gene’s promoter contains the binding motif of a particular TF. Disease or tissue-specific information then has to be integrated with this baseline prior network to obtain meaningful information about perturbations caused due to the disease.
In this review, we explore the PPI, GCNs, and GRNs, and also provide exemplar methods for each. Based on these three types of networks, we describe three complimentary philosophies and modus-operandi to embed phenotypic specific molecular information from biomedical data into a network framework, as shown in Figure 2. We present these paradigms to demonstrate that applying network phenomenology to big biomedical data requires a nuanced, condition-specific approach. In the following sections, we will focus on each paradigm separately, providing their examples, the questions they intend to answer, and the diagnostics of the outcomes. We mainly focus on reviewing methods to integrate multi-omic data to extract phenotype specific information, specifically disease and tissue specificity in the PPI, GCNs, and GRNs.
FIGURE 2
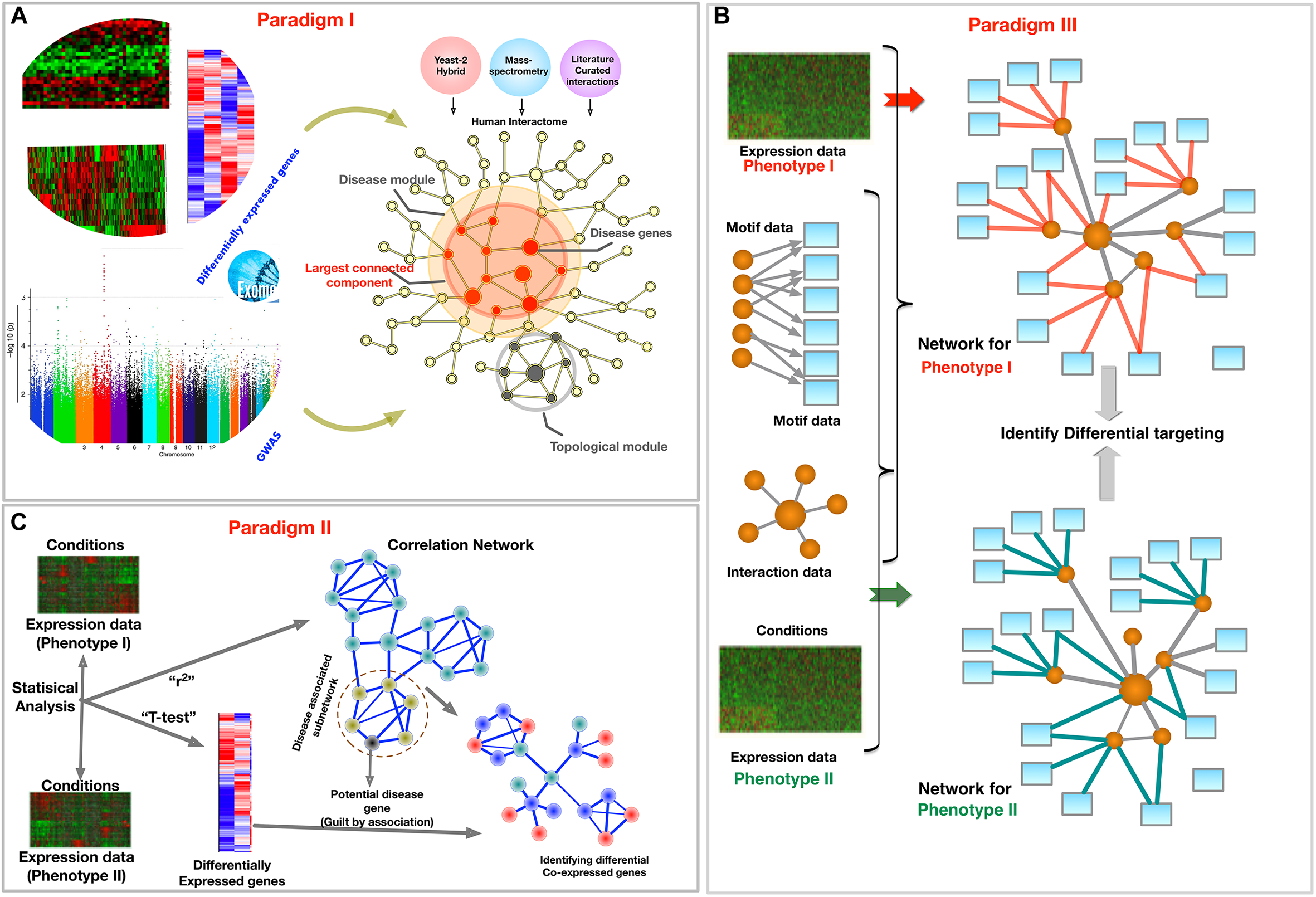
Schematic of three paradigms for combining biological networks with phenotype-specific biomedical data, such as a set of disease genes and transcriptomic profiles for case and control groups. (A) Identification of disease associated network components within the interactome, (B) Co-expression based network modeling to identify disease biomarkers, (C) Constructing phenotype-specific GRNs to identify perturbations and condition-specific regulatory changes.
PARADIGM I: Network-Based Approach to Human Disease Using the Interactome
The high-throughput mapping of the interactome has provided a molecular interaction map of the genes encoding proteins that might drive an underlying pathophenotype (Kamburov et al., 2009; Barabasi et al., 2011; Zhang et al., 2013; Rolland et al., 2014; Hein et al., 2015; Huttlin et al., 2015). Understanding disease associated biomedical data in the context of network principles supports the discovery of more accurate biomarkers, localization of the disease perturbation in the network, personalized networks, better disease sub-type classifications, better targets for drug development, and better drug repurposing. Using this paradigm, one can extract disease-specific signals in a variety of ways. One may consider topological properties of the nodes and assess the functional role of their hubness, i.e., a node property of having a higher number of connections. Alternatively, one can also identify new disease genes in the network by using “guilt-by-association” (Aravind, 2000; Quackenbush, 2003; Stuart et al., 2003; Lage et al., 2007; Sharma et al., 2010; Lee et al., 2011; Sharma et al., 2013; Huang J.K. et al., 2018) — a property ascribed not based on direct evidence but association with other disease genes, albeit with care (Gillis and Pavlidis, 2012). In addition to prioritizing candidate disease genes, molecular interaction networks can assist in identifying the sub-networks that are mechanistically linked to disease phenotypes (Menche et al., 2015; Sharma et al., 2015; Emamjomeh et al., 2017; van Dam et al., 2018). The proteins in these connected subnetworks may have clinical importance by being therapeutic targets and biomarkers (Sharma et al., 2015). Network tools can also provide a framework for disease classification (Halu et al., 2017; Zhou et al., 2018).
Assessing disease genes from other, non-disease genes by their topological properties on the interactome have provided new insight into disease pathobiology. It was found that disease genes tend to have non-hub properties (Goh et al., 2007). Later, it was reported that genes from OMIM and those associated with cancer are more central in a literature-curated interactome (Jonsson and Bates, 2006; Xu and Li, 2006; Ideker and Sharan, 2008). Further, several studies demonstrated that disease genes, in general, mostly have a high-degree and a low clustering coefficient (number of mutual connections with the neighboring nodes) (Feldman et al., 2008; Cai et al., 2010). Moreover, recently it was reported that disease genes have a higher degree, but it was discovered that the cancer-related genes are the primary drivers of this trend (Wachi et al., 2005; Jonsson and Bates, 2006). Genes associated with either Mendelian or complex diseases also have higher degree and lower clustering coefficients compared to non-disease genes (Cai et al., 2010; Pinero et al., 2016). The topological properties of disease-associated genes vary significantly from disease to disease. The factors that influence these discrepancies include the incompleteness of the current interactome, bias toward well-studied genes, and incomplete knowledge about the number genes associated with various diseases (Menche et al., 2015). It is anticipated that the alliance of different technologies like yeast-2-hybrid, affinity purification mass-spectrometry (AP-MS), and cross-linking AP-MS (Schweppe et al., 2018) will provide access to larger data that will be helpful in providing knowledge about the missing interactions. On the disease-gene discovery side, projects like the UK biobank prospective cohort study, which includes in-depth genetic and phenotypic data, will enhance knowledge regarding the missing disease genes (Bycroft et al., 2018).
An important area in which the interactome has helped in understanding complex diseases is the prediction of disease associated genes. The goal is to identify novel genes and proteins, which are involved in the regulation of tissues, or dysregulated in the case of disease, through the association with observed disease candidate genes using the biological hierarchy of molecular interactions. Figure 2A depicts this paradigm where the PPI network serves as map of potential biological interactions between various proteins over which disease associated genes are mapped to uncover relevant biology. The central philosophy in most methods under this paradigm is that the neighbors of the disease associated components or network modules, such as a set of differentially expressed genes (Chuang et al., 2007) or genes with disease-associated SNPs (Oti et al., 2006; Lage et al., 2007; Feldman et al., 2008; Barrenas et al., 2012), could potentially be associated with similar diseases (Goh et al., 2007), and are closer to each other as compared to the other nodes in the network. The definition of this closeness, or vicinity of nodes, just like the definition of modules and clusters, varies with different research strategies. Some methods assume topological closeness in terms of the number of shortest paths connecting given nodes, while others take the similarity of biological function into account. Guilt-by-association methods focus on identifying new disease genes by optimizing based on both the local and global properties of the network and by considering the role of other disease genes and their neighborhood. Network-based strategies to find disease genes and their associated mechanisms can be divided in two types: exploratory and analytic methods (Carter et al., 2013). In exploratory methods one can analyze the biological trends due to perturbations. For example, Chu et al. (2012) expanded on known angiogenesis pathways to construct a PPI network for angiogenesis. In contrast, analytic methods aim to identify specific genes and pathways associated with a disease. For example, Gilman and group developed a method for network-based analysis of genetic associations to identify a biological network of genes affected by rare de novo CNVs in autism (Gilman et al., 2011). Recently, Huang J.K. et al. (2018) systematically evaluated 21 protein-interaction networks for the ability to recover disease genes sets. After correcting for size, they found that the Database for Interacting Proteins (DIP) network (Xenarios et al., 2000) had the highest efficiency in recovering disease genes (Huang J.K. et al., 2018).
In contrast to predicting the disease candidate proteins, finding the associated disease-related network components, or sub-networks, provides a more substantial network space to discover the pathways and mechanisms that influence disease. Goh et al. (2007) proposed a correlation between the location of disease-associated genes and the topology of the molecular interaction network. The tendency of disease-associated genes to interact more often with others compared to random genes in the interactome led to the establishment of the ‘local impact’ hypothesis (Barabasi et al., 2011). According to this hypothesis, molecular entities involved in similar diseases have an increased tendency to interact with each other and to localize in a specific neighborhood of the interactome (Barabasi et al., 2011). The search for these modules involves exploring the structural and topological properties of the PPI network. Community detection algorithms (Spirin and Mirny, 2003), clique percolation (Sun et al., 2011), and genetic algorithms (Liu et al., 2018) have been applied to uncover disease modules using network properties (Vlaic et al., 2018). Module prediction and identifying non-overlapping clusters with the PPI remains challenging since the PPI network has a short diameter, i.e., most nodes are close to all other nodes in terms of network distance. Novel distance metrics and community detection algorithms have been proposed to overcome this problem (Hall-Swan et al., 2018). The recently proposed DIseAse MOdule Detection (DIAMOnD) algorithm (Ghiassian et al., 2015) associates the functional modules of known disease-associated proteins (seed proteins) and identifies the close neighbors of these genes (candidate disease-associated proteins) using topological properties of the interactome. The method suggests that the connectivity significance among the disease-associated proteins is the best predictive quantity to find the disease related components in the interactome. The underlying hypothesis is that close neighbors of known disease proteins may be involved in the disease. The working principle of DIAMOnD is as follows: first, a pool of disease genes encoding proteins is identified for a disease of interest from biological experiments, GWAS, linkage analysis, or other disease associated data sources (Pinero et al., 2017). Next, these disease proteins (seeds) are mapped onto the interactome. Further, neighbor proteins are added iteratively to the set of seed proteins based on the condition that each neighbor added is most significantly connected to the seed proteins. A hypergeometric test assigns a p-value to the proteins that share more connections with seed proteins than expected by chance. Finally, the seed proteins plus the added neighbor proteins are part of network components that represent a disease module, or a subnetwork of proteins in the interactome, the members of which are more functionally and topologically related to each other than to other portions of the network. These subnetworks are designated as disease-specific modules based on the source of initial seed proteins. Disease module identification has also led to endophenotypes, intermediate pathophenotypes, and network modules describing their common and distinctive molecular mediators (Lage et al., 2008; Ghiassian et al., 2016).
As mentioned previously, significant progress has been made in mapping the interactome by high-throughput approaches like yeast-2-hybrid (Rual et al., 2005; Venkatesan et al., 2009; Dreze et al., 2010; Rolland et al., 2014), AP/MS (Hein et al., 2015; Huttlin et al., 2015, 2017) and various literature-curated data sources, such as ConsensusPathDB, STRING, and PCNet, which collate the known and predicted interactions between proteins (Klingstrom and Plewczynski, 2011). Despite these efforts, the current interactome mapping is 80% incomplete (Hart et al., 2006; Venkatesan et al., 2009; Mosca et al., 2013; Menche et al., 2015) and is affected by many experimental and literature biases. Given the incompleteness of the interactome, it is possible that the disease modules are also far from complete. An attempt to overcome this limitation was made using a network-based closeness approach that compares the weighted distance between different disease and seed-gene neighborhoods to random expectation on the network. In the context of Chronic Obstructive Pulmonary Disease (COPD), 140 potential candidate genes (Sharma et al., 2018) were identified. Another shortcoming of disease module detection related to the lack of context-dependence and tissue-specificity within the PPI was studied by Kitsak et al. (2016). They found that the genes expressed in a particular tissue tend to form localized connected subnetworks, which overlap between similar tissues and are situated in the different neighborhoods for pathologically distinct pairs of tissues. The perturbations in tissue-dependent subnetworks may help us understand disease manifestations or pathophenotypes. Integrating multi-omics data, including epigenomics, proteomics, and metabolomics, with PPI analysis remains challenging, but is critical for identifying disease or tissue-specific modules in the interactome.
PARADIGM II: Identifying Important Genes Using Patterns of Co-abundance of Biomolecules
Measuring transcript abundance or gene expression patterns for given phenotypes (case-control) across multiple samples is one of the main research strategies used to probe the system as it is connected to the central dogma of molecular biology. Performing differential gene expression analysis often identifies important genes affected by the disease. However, it does not provide information regarding how these genes are influenced by or influence other genes. It has been observed that genes with similar expression patterns might be part of complexes, influence each other, or be part of the same pathways or mechanisms (Serin et al., 2016). This inspired the construction of GCNs where the patterns of transcript abundance are studied in the context of the disease. The central philosophy of this paradigm is to combine important seed genes with an organic network of co-expression patterns derived from the gene expression data from the same system.
There are many ways to compute co-expression or co-abundance patterns, including using Pearson correlations (Stuart et al., 2003), Spearman rank correlations (Song et al., 2012; Liesecke et al., 2018), mutual information (Butte and Kohane, 1999; Margolin et al., 2006; Meyer et al., 2007), Gaussian graphical models (Toh and Horimoto, 2002), regression-based methods (Yeung et al., 2002; van Someren et al., 2006; Pirgazi and Khanteymoori, 2018), Bayesian approaches (Friedman et al., 2000; Perrin et al., 2003; Li et al., 2007; Xing et al., 2017), random matrix theory (Luo et al., 2007; Jalan et al., 2010; Jalan et al., 2012), and partial correlations (Reverter and Chan, 2008). GCNs identify the functionally coordinated participation of genes in response to an external stimulus or condition. GCNs can be signed or unsigned, weighted or unweighted, and may either be constructed using microarray or RNA-Seq data. Care must be exercised when using thresholding methods to obtain unweighted co-expression networks as these are subjective and can change the network structure and topology (Elo et al., 2007); methods based on the clustering coefficient (Boyadjiev and Jabs, 2000), random matrix theory (Luo et al., 2007), or soft thresholding, which raises the weights by a certain power to penalize weaker edges (Langfelder and Horvath, 2008), have been used to address this limitation. Along with total gene expression levels, isoform abundance and alternative splicing can also be used in constructing GCNs (Saha et al., 2017).
Gene co-expression networks are also used to identify co-expression modules. Clusters, modules, or subgraphs of genes that have similar functions are often highly interconnected in GCNs. These clusters can be identified using network topology-based methods like community detection (Girvan and Newman, 2002), modularity maximization (Newman, 2004), K-means clustering (Stuart et al., 2003), or variants of hierarchical clustering methods (Langfelder and Horvath, 2008; Serin et al., 2016). The genes in the most significant modules are then assessed for their biological importance using functional enrichment methods. The genes in the clusters are also often tested for their enrichment with differentially expressed genes from transcriptomic analysis, as illustrated in Figure 2B. Based on these results, other non-differentially expressed genes in the enriched clusters can be implicated in the disease using ‘guilt-by-association’ approaches. The newly implicated genes may have clinical importance as potential therapeutic targets and biomarkers.
Despite the aphorism “correlation is not causation”, partial yet informative insights can be gleaned from co-expression networks, such as an underlying regulatory framework mediating the co-expression patterns. New methods based on partial-correlations, Bayesian, and graphical Gaussian models (Werhli et al., 2006) take into account local connectivity when estimating edge strengths and a few methods work by combining prior-knowledge of expression patterns of TFs with co-expression information (Huynh-Thu et al., 2010; Rotival and Petretto, 2014). Gene-gene interaction network methods like ARACNe (Margolin et al., 2006) and CLR (Faith et al., 2007) attempt to better capture these regulatory associations by accounting for connections within a shared neighborhood of genes in order to infer the strength of a link between two genes. Applying these approaches in complex conditions, like a gene being regulated by many regulators, becomes more challenging. Inferring the direct regulatory influence of transcription factors on target genes is central to interpreting the regulatory networks. Concerted efforts to support network-inference, such as the DREAM5 benchmark challenge (Marbach et al., 2012), have summarized different strategies that can be employed to infer regulatory networks. The accuracy of reconstruction approaches is often tested by comparing the predicted networks with high-confidence transcription factor binding data (He and Tan, 2016). However, integrating multi-omic data into these models to understand the pathobiology of disease states is an open challenge. Methods like CellNet (Cahan et al., 2014), an extension of CLR, and MOGRIFY (Rackham et al., 2016) take into account differentially expressed genes within the co-expression network framework in order to predict cellular reprogramming by transcription factors. Thus, co-expression methods have also been used to infer regulatory networks and to delineate the influence of regulatory genes, such as transcription factors, on their targets. However, obtaining condition-specific GRNs requires information regarding transcription factor binding activity in the given context. We will review some of the methods that utilize TF binding information in the next section.
To summarize, inferring disease-specific information from GCN is possible from co-expressed or co-regulated clusters, differentially expressed and co-expressed genes, as well as the topological and functional properties of these. Biomedical big data measuring the transcriptome is highly leveraged by GCNs. For example, human tissue-specific GCNs have been constructed and analyzed (Pierson et al., 2015) using consortium data such as GTEx (Mele et al., 2015). These analyses revealed that genes with tissue-specific function are not hubs but connect to tissue-specific transcription factor hubs. Explorations using relative isoform ratios (RNA transcripts from the same genes with different exons removed) and splicing data revealed distinct co-expression relationships unique to the tissues (Saha et al., 2017). Tissue specificity of GCNs have also been assessed in rats (Xiao et al., 2014), humans (Prieto et al., 2008; Xiao et al., 2014; Kogelman et al., 2016; Ni et al., 2016; Farahbod and Pavlidis, 2018), bats (Rodenas-Cuadrado et al., 2015), and plants (Aravind, 2000). Similarly, TCGA data has been analyzed using WGCNA in order to study the system-level properties of prognostic genes (Yang et al., 2014). Similar to gene co-expression, protein co-abundance networks can also be used to pinpoint influential proteins as potential regulators of the observed phenotype, and have been used to study inflammation (Halu et al., 2018), HCV infections (McDermott et al., 2012), and cancer, including breast cancer (Ryan et al., 2017) and glioblastoma (Kanonidis et al., 2016).
PARADIGM III: Inferring Phenotype Specific Gene Regulatory Networks
In the previous sections, we studied various ways to construct networks and integrate molecular data to extract phenotype-specific biology in the form of gene prioritization, disease modules, or therapeutic targets. Those included immutable PPIs allowing disease-specific information to be embedded onto them and organic ways to model disease-specific information using co-expression networks. Here, separate networks are built for each phenotype which may be case-control, disease-specific, tissue or cell-specific, sex-specific, or for different disease subtypes. The network comparison model stems from the axiom of “differential networking” over “differential expression.” Many examples of differential networking can be found, including the INtegrated DiffErential Expression and Differential network analysis (INDEED) (Zuo et al., 2016) and DICER (Amar et al., 2013) algorithms. In this paradigm, we aim to discuss ways of leveraging phenotype-specific biomedical information to construct condition-specific GRNs. In principle, GCNs can also be phenotype-specific and can be used to infer condition-specific signals, but they lack the underlying set of canonical interactions unlike GRNs which include protein-DNA interaction in the form of TF binding information.
Instead of combining data from cases and controls to obtain key molecular elements, such as differentially expressed genes or genes annotated to GWAS SNPs, in this paradigm the data is used to construct separate networks for each of the conditions. This construction of phenotype specific networks helps to mitigate systematic experimental biases and errors in both conditions (de la Fuente, 2010; Ideker and Krogan, 2012). It allows the comparison of networks to help uncover the specific rewiring of pathways, such as those induced by disease, pharmacological treatment (Bandyopadhyay et al., 2010), or environmental stimuli. GCNs can also be constructed in a phenotype-specific manner, as seen in the previous section. In Figure 2C, we depict an approach where phenotype-specific networks are constructed to uncover differentially targeted interactions. In this section, we focus on transcriptional regulatory networks that depend not only on co-expression, but also on modeling the binding propensities of TFs. These networks may also incorporate other multi-omic data to obtain condition-specific regulatory models.
The primary benefit of comparing phenotype-specific networks, particularly in GRNs, is to better delineate the role of genes in each condition. The “rewiring” of the TFs targeting each of the genes can be tracked and the perturbations leading to these changes can convey information regarding the mechanistic underpinnings of the observed phenotype. An apt extension of “differential networking” to the transcriptional regulatory network framework is “differential targeting,” which captures the highly dynamic nature of gene regulation. Changes in network topology, driven by underlying condition-specific data, can yield valuable insights and help to identify driver nodes and network biomarkers, such as a set of strengthened or weakened interactions between TF and target genes in the context of disease.
We review the Passing Attributes between Networks for Data Assimilation (PANDA) algorithm (Glass et al., 2013) as an exemplary method for constructing condition-specific regulatory networks, allowing for robust differential targeting analysis. PANDA is initiated by constructing a prior regulatory network consisting of potential routes for communication by mapping transcription factor motifs to a reference genome and assigning them to genes if they are in the regulatory region of the genes. PANDA then integrates other sources of information to iteratively optimize the flow of information through the network, modifying the prior to obtain a condition-specific regulatory network. The phenotype-specific regulatory networks are then compared to identify the structures most affected by this “rewiring” and their biological significance. PANDA models the interactions between transcription factors based on the following principles. Firstly, if two transcription factors have a similar targeting profile, i.e., target similar genes or have binding motifs in the promoters of the same genes, they are more likely to physically interact or be members of the same TF complex (Hemberg and Kreiman, 2011; Guo et al., 2016). Cooperative binding of TFs is found to be evolutionarily constrained and conserved (Goke et al., 2011; He et al., 2011), and impacts crucial eukaryotic functions (Hochedlinger and Plath, 2009; Wilson et al., 2010; He et al., 2011; Will and Helms, 2014). Likewise, if two genes are targeted by the same set of TFs, these genes are likely to share similar expression patterns (Yu et al., 2003; Kim et al., 2006; Marco et al., 2009), or be part of the same functional module (Goh et al., 2007; Feldman et al., 2008). For this purpose, PANDA incorporates PPI networks to determine the “responsibility” of TFs co-binding based on shared targets. It also uses GCNs to determine the “availability” of genes to be simultaneously co-regulated, as evidenced by common co-expression. A vital component in PANDA is a “prior” network composed of all potential regulatory routes based on the existence of binding sites for TFs in the regulatory regions of genes. All three ingredients (PPI, GCN, and a network prior) are then assimilated to uncover consistent patterns among these networks using a message-passing framework similar to affinity-propagation (Frey and Dueck, 2007). The outcome is a network elucidating the edges that form self-consistent modules, identifying relevant biological processes.
The phenotype-specific applications of PANDA are broad and include the comparison of disease and control networks in both complex diseases and cancers. For example, PANDA has been used to identify potential drug targets in ovarian cancer subtypes (Glass et al., 2015). Comparing PANDA networks between poor and good responders to asthma therapies identified potential transcriptional mediators of corticosteroid response in asthma (Qiu et al., 2018). The role of serotonin (5HT) dysregulation in mitral valve disease was explored using PANDA to find upregulation in 5HTR2B expression and an increase 5HT receptor signaling (Driesbaugh et al., 2018). The effect of weight-loss on decreased risk of colorectal cancer was evaluated by applying PANDA to gene expression data on rectal mucosa biopsies (Vargas et al., 2016). In cancer research, PANDA network analysis in triple-negative breast cancer (TNBC) identified new core modules of functionally essential TFs and genes in cancer cells (Min et al., 2017). PANDA has also been used to investigate non-epithelial cancers like glioma to identify prognostic biomarkers mainly concerning mesenchymal signatures (Celiku et al., 2017). Sexual dimorphism, where the phenotypes are males and females, is another area where PANDA has been applied extensively, from sex-related targeting differences in COPD (Glass et al., 2014), colorectal cancer (Lopes-Ramos et al., 2018), and understanding crucial sex-related differences in various tissues in the human body (Chen et al., 2016). Differences between cell-lines and their host tissues have also been investigated using PANDA (Lopes-Ramos et al., 2017).
The issue of tissue-specificity can also be addressed by the paradigm of condition-specific networks, where the phenotype is the tissue or cell type. Various methods use gene expression data with regression trees (Huynh-Thu et al., 2010) or consider the context of pathways (Jambusaria et al., 2018). Enhancer and promoter data (Marbach et al., 2016) have been used to construct tissue-specific networks in humans and plants (Huang J. et al., 2018). Using GTEx transcriptome data, PANDA has been used to construct GRNs for 38 distinct human tissues (Sonawane et al., 2017). This analysis assessed the inter-relationship between tissue-specific genes and TFs based on expression data and tissue-specific interactions and the topological positions of functionally important genes in respective tissues. This study also used network centrality measures like betweenness and degree to assess the topological properties of the nodes to identify rewiring around these genes in various tissues. Another significant contribution of this work is the elucidation of the tissue-specific regulatory roles of transcription factors, which were found to be independent of their expression levels. Instead, transcription factors appeared to mediate critical tissue-specific processes through subtle shifts in the GRNs, providing functional redundancy and, as a consequence, phenotypic stability of tissues.
Conclusion and Future Directions
Above we reviewed a limited set of network medicine philosophies that seek to integrate biomedical big data to uncover meaningful biology. Network medicine approaches provide customized and optimized ways to leverage biomedical data. The choice of the appropriate network method is largely dictated by the underlying biological inquiry, hypotheses, study design, and available data. Although this review is not meant to be exhaustive, our intent was to give a essence of how biomedical data requires a nuanced approach when selecting network analyses and provide a resource for both network scientists and biologists to better understand the lexicon of network modeling of biomedical data.
We believe that network medicine approaches will be vital in the future with the increasing emergence of diverse technologies, multi-omic data types, deeper levels of inquiry from tissues to cellular levels, platforms that include large amounts of publicly available biomedical data, and efforts in precision medicine, which aim to find the right drugs for the right patients at the right time. There is a growing realization that genomics is only a part of the story when it comes to cancer and other complex diseases. The field is working to augment genetic information (mutations, deletions, and other somatic genetic alterations) with other omics data, such as epigenomics (methylation, non-coding RNAs, histone modifications, chromatin structures), proteomics (in vitro studies on proteins), and lipidomics (survey of cellular lipids), to name a few. The network medicine framework presents a promising way of thinking about and integrating these heterogeneous data types by elucidating their mutual influences to help explain disease etiologies and cellular functions and providing the basis for personalized therapeutics.
Multi-omics data integration using networks has already started gaining a wide amount of attention in the scientific community (Gligorijevic and Przulj, 2015; Tuncbag et al., 2016; Yugi et al., 2016; Hasin et al., 2017; Huang et al., 2017; Malod-Dognin et al., 2019). Moreover, relatively newer network tools like multiplex networks (Didier et al., 2018), network fusion (Wang et al., 2014), more innovative community detection strategies (Gligorijevic et al., 2016), and higher order structural modularity (Didier et al., 2018), have the potential to be applied to these problems to gain an even deeper and more nuance understanding of biological systems. Multilayer network approaches (De Domenico et al., 2015) for human diseases have unraveled important associations between rare and complex diseases (Halu et al., 2017). Despite several open challenges (Stegle et al., 2015; Ziegenhain et al., 2017), new technologies like single-cell transcriptomics (Hon et al., 2018), have started to be used to construct GRNs (Herbach et al., 2017; Fiers et al., 2018) and cell-specific coactivation networks (Ghazanfar et al., 2016). As the field of network medicine moves forward, one thing that is required more than ever before is the development of methods for systematically validating network predictions. Such validation will provide a greater confidence in network predictions and facilitate their incorporation into translational medicine. We also think active trans-disciplinary collaboration between biologists and scientists from the field of complex networks is required to infuse the field of network medicine with novel algorithms and innovative strategies. The application of network methods to biomedical data presents a great opportunity to test and improve upon the tools originating from the general field of complex networks. We also take this opportunity to thank the many experimental biologists whose operose efforts have led to the generation of the vast amount of invaluable biomedical data, and to the numerous individuals who have donated their data for the sake of science.
Statements
Author contributions
ARS wrote the original draft which was reviewed, edited and revised by all the authors. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
KG was supported by the NIH/NHLBI through K25HL133599. We acknowledge the support by National Institutes of Health (NIH) grants R01 HL118455-04-1 and P01 HL13285. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
AS would like to thank John Quackenbush for inspiration of the three paradigms discussed above, along with Trevor R. Leonardo and Rebekka Burkholz for critical reading of the manuscript. The authors thank members of the Quackenbush and Sharma labs for many fruitful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
- CNV
copy number variation
- ENCODE
ENCyclopedia Of DNA elements
- FANTOM5
Functional ANnoTation Of Mammalian Genome
- GCNs
gene co-expression networks
- GRNs
gene regulatory networks
- GTEx
genotype-tissue expression
- HCA
human cell atlas
- HMP
human microbiome project
- HPA
human protein atlas
- modENCODE
model organism ENCyclopedia Of DNA Elements
- NGS
next generation sequencing
- PPIs
protein-protein interactions
- SNP
single nucleotide polymorphism
- TCGA
the cancer genome atlas
- TOPMed
trans-omics for precision medicine
Footnotes
References
1
AllenN. E.SudlowC.PeakmanT.CollinsR.BiobankU. K. (2014). UK biobank data: come and get it.Sci. Transl. Med.6:224ed4. 10.1126/scitranslmed.3008601
2
AlonU. (2007). Network motifs: theory and experimental approaches.Nat. Rev. Genet.8450–461. 10.1038/nrg2102
3
Altaf-Ul-AminM.AfendiF. M.KiboiS. K.KanayaS. (2014). Systems biology in the context of big data and networks.Biomed Res. Int.2014:428570. 10.1155/2014/428570
4
AmarD.SaferH.ShamirR. (2013). Dissection of regulatory networks that are altered in disease via differential co-expression.PLoS Comput. Biol.9:e1002955. 10.1371/journal.pcbi.1002955
5
AnderssonR.GebhardC.Miguel-EscaladaI.HoofI.BornholdtJ.BoydM.et al (2014). An atlas of active enhancers across human cell types and tissues.Nature507455–461. 10.1038/nature12787
6
AravindL. (2000). Guilt by association: contextual information in genome analysis.Genome Res.101074–1077. 10.1101/gr.10.8.1074
7
BandyopadhyayS.MehtaM.KuoD.SungM. K.ChuangR.JaehnigE. J.et al (2010). Rewiring of genetic networks in response to DNA damage.Science3301385–1389. 10.1126/science.1195618
8
BarabasiA. L.GulbahceN.LoscalzoJ. (2011). Network medicine: a network-based approach to human disease.Nat. Rev. Genet.1256–68. 10.1038/nrg2918
9
BarrenasF.ChavaliS.AlvesA. C.CoinL.JarvelinM. R.JornstenR.et al (2012). Highly interconnected genes in disease-specific networks are enriched for disease-associated polymorphisms.Genome Biol.13:R46. 10.1186/gb-2012-13-6-r46
10
BauerE.ThieleI. (2018). From network analysis to functional metabolic modeling of the human gut microbiota.mSystems3:e00209–17. 10.1128/mSystems.00209-17
11
BelangerC. F.HennekensC. H.RosnerB.SpeizerF. E. (1978). The nurses’ health study.Am. J. Nurs.781039–1040. 10.2307/3462013
12
BhardwajN.LuH. (2005). Correlation between gene expression profiles and protein-protein interactions within and across genomes.Bioinformatics212730–2738. 10.1093/bioinformatics/bti398
13
BouquetJ.SoloskiM. J.SweiA.CheadleC.FedermanS.BillaudJ. N.et al (2016). Longitudinal transcriptome analysis reveals a sustained differential gene expression signature in patients treated for acute lyme disease.mBio7:e00100–16. 10.1128/mBio.00100-16
14
BoyadjievS. A.JabsE. W. (2000). Online Mendelian Inheritance in Man (OMIM) as a knowledgebase for human developmental disorders.Clin. Genet.57253–266. 10.1034/j.1399-0004.2000.570403.x
15
ButteA. J.KohaneI. S. (1999). Unsupervised knowledge discovery in medical databases using relevance networks.Proc. AMIA Symp.1999711–715.
16
BycroftC.FreemanC.PetkovaD.BandG.ElliottL. T.SharpK.et al (2018). The UK Biobank resource with deep phenotyping and genomic data.Nature562203–209. 10.1038/s41586-018-0579-z
17
CahanP.LiH.MorrisS. A.Lummertz da RochaE.DaleyG. Q.CollinsJ. J. (2014). CellNet: network biology applied to stem cell engineering.Cell158903–915. 10.1016/j.cell.2014.07.020
18
CaiJ. J.BorensteinE.PetrovD. A. (2010). Broker genes in human disease.Genome Biol. Evol.2815–825. 10.1093/gbe/evq064
19
Cancer Genome Atlas Research Network (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways.Nature4551061–1068. 10.1038/nature07385
20
CarterH.HofreeM.IdekerT. (2013). Genotype to phenotype via network analysis.Curr. Opin. Genet. Dev.23611–621. 10.1016/j.gde.2013.10.003
21
CelikuO.TandleA.ChungJ. Y.HewittS. M.CamphausenK.ShankavaramU. (2017). Computational analysis of the mesenchymal signature landscape in gliomas.BMC Med. Genomics10:13. 10.1186/s12920-017-0252-7
22
ChenC.-Y.KuijjerM. L.PaulsonJ. N.SonawaneA. R.FagnyM.et al (2016). Sexual dimorphism in gene expression and regulatory networks across human tissues.bioRxiv[Preprint]. 10.1101/082289
23
ChenR.MiasG. I.Li-Pook-ThanJ.JiangL.LamH. Y.ChenR.et al (2012). Personal omics profiling reveals dynamic molecular and medical phenotypes.Cell1481293–1307. 10.1016/j.cell.2012.02.009
24
ChuL. H.RiveraC. G.PopelA. S.BaderJ. S. (2012). Constructing the angiome: a global angiogenesis protein interaction network.Physiol. Genomics44915–924. 10.1152/physiolgenomics.00181.2011
25
ChuangH. Y.LeeE.LiuY. T.LeeD.IdekerT. (2007). Network-based classification of breast cancer metastasis.Mol. Syst. Biol.3:140. 10.1038/msb4100180
26
ColditzG. A.PhilpottS. E.HankinsonS. E. (2016). The impact of the nurses’ health study on population health: prevention, translation, and control.Am. J. Public Health1061540–1545. 10.2105/AJPH.2016.303343
27
CollinsF. S.MorganM.PatrinosA. (2003). The Human Genome Project: lessons from large-scale biology.Science300286–290. 10.1126/science.1084564
28
CoyteK. Z.SchluterJ.FosterK. R. (2015). The ecology of the microbiome: networks, competition, and stability.Science350663–666. 10.1126/science.aad2602
29
CusickM. E.KlitgordN.VidalM.HillD. E. (2005). Interactome: gateway into systems biology.Hum. Mol. Genet.14(Suppl. 2), R171–R181. 10.1093/hmg/ddi335
30
DavidsonE. H. (2006). The Regulatory Genome : Gene Regulatory Networks in Development and Evolution.Burlington, MA: Academic Press.
31
DavisD.YaverogluO. N.Malod-DogninN.StojmirovicA.PrzuljN. (2015). Topology-function conservation in protein-protein interaction networks.Bioinformatics311632–1639. 10.1093/bioinformatics/btv026
32
DawberT. R.MeadorsG. F.MooreF. E.Jr. (1951). Epidemiological approaches to heart disease: the Framingham Study.Am. J. Public Health Nations Health41279–281. 10.2105/AJPH.41.3.279
33
De DomenicoM.NicosiaV.ArenasA.LatoraV. (2015). Structural reducibility of multilayer networks.Nat. Commun.6:6864. 10.1038/ncomms7864
34
de la FuenteA. (2010). From ‘differential expression’ to ‘differential networking’ - identification of dysfunctional regulatory networks in diseases.Trends Genet.26326–333. 10.1016/j.tig.2010.05.001
35
de SilvaE.StumpfM. P. (2005). Complex networks and simple models in biology.J. R. Soc. Interface2419–430. 10.1098/rsif.2005.0067
36
DeplanckeB.DupuyD.VidalM.WalhoutA. J. (2004). A gateway-compatible yeast one-hybrid system.Genome Res.142093–2101. 10.1101/gr.2445504
37
DidierG.ValdeolivasA.BaudotA. (2018). Identifying communities from multiplex biological networks by randomized optimization of modularity.F1000Res.7:1042. 10.12688/f1000research.15486.2
38
DondelingerF.MukherjeeS. (2019). Statistical network inference for time-varying molecular data with dynamic bayesian networks.Methods Mol. Biol.188325–48. 10.1007/978-1-4939-8882-2_2
39
DrezeM.MonachelloD.LurinC.CusickM. E.HillD. E.VidalM.et al (2010). High-quality binary interactome mapping.Methods Enzymol.470281–315. 10.1016/S0076-6879(10)70012-4
40
DriesbaughK. H.BranchettiE.GrauJ. B.KeeneyS. J.GlassK.OyamaM. A.et al (2018). Serotonin receptor 2B signaling with interstitial cell activation and leaflet remodeling in degenerative mitral regurgitation.J. Mol. Cell. Cardiol.11594–103. 10.1016/j.yjmcc.2017.12.014
41
EloL. L.JarvenpaaH.OresicM.LahesmaaR.AittokallioT. (2007). Systematic construction of gene coexpression networks with applications to human T helper cell differentiation process.Bioinformatics232096–2103. 10.1093/bioinformatics/btm309
42
EmamjomehA.RobatE. S.ZahiriJ.SoloukiM.KhosraviP. (2017). Gene co-expression network reconstruction: a review on computational methods for inferring functional information from plant-based expression data.Plant Biotechnol. Rep.1171–86. 10.1007/s11816-017-0433-z
43
ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome.Nature48957–74. 10.1038/nature11247
44
FagnyM.PaulsonJ. N.KuijjerM. L.SonawaneA. R.ChenC. Y.Lopes-RamosC. M.et al (2017). Exploring regulation in tissues with eQTL networks.Proc. Natl. Acad. Sci. U.S.A.114E7841–E7850. 10.1073/pnas.1707375114
45
FaithJ. J.HayeteB.ThadenJ. T.MognoI.WierzbowskiJ.CottarelG.et al (2007). Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles.PLoS Biol.5:e8. 10.1371/journal.pbio.0050008
46
FarahbodM.PavlidisP. (2018). Differential coexpression in human tissues and the confounding effect of mean expression levels.Bioinformatics3555–61. 10.1093/bioinformatics/bty538
47
FeldmanI.RzhetskyA.VitkupD. (2008). Network properties of genes harboring inherited disease mutations.Proc. Natl. Acad. Sci. U.S.A.1054323–4328. 10.1073/pnas.0701722105
48
FiersM.MinnoyeL.AibarS.Bravo Gonzalez-BlasC.Kalender AtakZ.AertsS. (2018). Mapping gene regulatory networks from single-cell omics data.Brief. Funct. Genomics17246–254. 10.1093/bfgp/elx046
49
FournierM. L.PaulsonA.PavelkaN.MosleyA. L.GaudenzK.BradfordW. D.et al (2010). Delayed correlation of mRNA and protein expression in rapamycin-treated cells and a role for Ggc1 in cellular sensitivity to rapamycin.Mol. Cell Proteomics9271–284. 10.1074/mcp.M900415-MCP200
50
FreyB. J.DueckD. (2007). Clustering by passing messages between data points.Science315972–976. 10.1126/science.1136800
51
FriedmanN.LinialM.NachmanI.Pe’erD. (2000). Using Bayesian networks to analyze expression data.J. Comput. Biol.7601–620. 10.1089/106652700750050961
52
GhazanfarS.BisogniA. J.OrmerodJ. T.LinD. M.YangJ. Y. (2016). Integrated single cell data analysis reveals cell specific networks and novel coactivation markers.BMC Syst. Biol.10:127. 10.1186/s12918-016-0370-4
53
GhiassianS. D.MencheJ.BarabasiA. L. (2015). A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome.PLoS Comput. Biol.11:e1004120. 10.1371/journal.pcbi.1004120
54
GhiassianS. D.MencheJ.ChasmanD. I.GiulianiniF.WangR.RicchiutoP.et al (2016). Endophenotype network models: common core of complex diseases.Sci. Rep.6:27414. 10.1038/srep27414
55
GillisJ.PavlidisP. (2012). ”Guilt by association” is the exception rather than the rule in gene networks.PLoS Comput. Biol.8:e1002444. 10.1371/journal.pcbi.1002444
56
GilmanS. R.IossifovI.LevyD.RonemusM.WiglerM.VitkupD. (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses.Neuron70898–907. 10.1016/j.neuron.2011.05.021
57
GirvanM.NewmanM. E. (2002). Community structure in social and biological networks.Proc. Natl. Acad. Sci. U.S.A.997821–7826. 10.1073/pnas.122653799
58
GlassK.HuttenhowerC.QuackenbushJ.YuanG. C. (2013). Passing messages between biological networks to refine predicted interactions.PLoS One8:e64832. 10.1371/journal.pone.0064832
59
GlassK.QuackenbushJ.SilvermanE. K.CelliB.RennardS. I.YuanG. C.et al (2014). Sexually-dimorphic targeting of functionally-related genes in COPD.BMC Syst. Biol.8:118. 10.1186/s12918-014-0118-y
60
GlassK.QuackenbushJ.SpentzosD.Haibe-KainsB.YuanG. C. (2015). A network model for angiogenesis in ovarian cancer.BMC Bioinformatics16:115. 10.1186/s12859-015-0551-y
61
GligorijevicV.Malod-DogninN.PrzuljN. (2016). Fuse: multiple network alignment via data fusion.Bioinformatics321195–1203. 10.1093/bioinformatics/btv731
62
GligorijevicV.PrzuljN. (2015). Methods for biological data integration: perspectives and challenges.J. R. Soc. Interface12:20150571. 10.1098/rsif.2015.0571
63
GohK. I.CusickM. E.ValleD.ChildsB.VidalM.BarabasiA. L. (2007). The human disease network.Proc. Natl. Acad. Sci. U.S.A.1048685–8690. 10.1073/pnas.0701361104
64
GokeJ.JungM.BehrensS.ChavezL.O’KeeffeS.TimmermannB.et al (2011). Combinatorial binding in human and mouse embryonic stem cells identifies conserved enhancers active in early embryonic development.PLoS Comput. Biol.7:e1002304. 10.1371/journal.pcbi.1002304
65
GrobbeeD. E.RimmE. B.GiovannucciE.ColditzG.StampferM.WillettW. (1990). Coffee, caffeine, and cardiovascular disease in men.N. Engl. J. Med.3231026–1032. 10.1056/NEJM199010113231504
66
GTEx Consortium (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans.Science348648–660. 10.1126/science.1262110
67
GuneyE.MencheJ.VidalM.BarabasiA. L. (2016). Network-based in silico drug efficacy screening.Nat. Commun.7:10331. 10.1038/ncomms10331
68
GuoY.AlexanderK.ClarkA. G.GrimsonA.YuH. (2016). Integrated network analysis reveals distinct regulatory roles of transcription factors and microRNAs.RNA221663–1672. 10.1261/rna.048025.114
69
Hall-SwanS.CrawfordJ.NewmanR.CowenL. J. (2018). Detangling PPI networks to uncover functionally meaningful clusters.BMC Syst. Biol.12:24. 10.1186/s12918-018-0550-5
70
HaluA.De DomenicoM.ArenasA.SharmaA. (2017). The multiplex network of human diseases.bioRxiv[Preprint]. 10.1101/100370
71
HaluA.WangJ. G.IwataH.MojcherA.AbibA. L.SinghS. A.et al (2018). Context-enriched interactome powered by proteomics helps the identification of novel regulators of macrophage activation.eLife7:e37059. 10.7554/eLife.37059
72
HamoshA.ScottA. F.AmbergerJ.BocchiniC.ValleD.McKusickV. A. (2002). Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders.Nucleic Acids Res.3052–55. 10.1093/nar/30.1.52
73
HartG. T.RamaniA. K.MarcotteE. M. (2006). How complete are current yeast and human protein-interaction networks?Genome Biol.7:120.
74
HasinY.SeldinM.LusisA. (2017). Multi-omics approaches to disease.Genome Biol.18:83. 10.1186/s13059-017-1215-1
75
HeB.TanK. (2016). Understanding transcriptional regulatory networks using computational models.Curr. Opin. Genet. Dev.37101–108. 10.1016/j.gde.2016.02.002
76
HeQ.BardetA. F.PattonB.PurvisJ.JohnstonJ.PaulsonA.et al (2011). High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species.Nat. Genet.43414–420. 10.1038/ng.808
77
HeinM. Y.HubnerN. C.PoserI.CoxJ.NagarajN.ToyodaY.et al (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances.Cell163712–723. 10.1016/j.cell.2015.09.053
78
HembergM.KreimanG. (2011). Conservation of transcription factor binding events predicts gene expression across species.Nucleic Acids Res.397092–7102. 10.1093/nar/gkr404
79
HerbachU.BonnaffouxA.EspinasseT.GandrillonO. (2017). Inferring gene regulatory networks from single-cell data: a mechanistic approach.BMC Syst. Biol.11:105. 10.1186/s12918-017-0487-0
80
HochedlingerK.PlathK. (2009). Epigenetic reprogramming and induced pluripotency.Development136509–523. 10.1242/dev.020867
81
HonC. C.ShinJ. W.CarninciP.StubbingtonM. J. T. (2018). The Human Cell Atlas: technical approaches and challenges.Brief. Funct. Genomics17283–294. 10.1093/bfgp/elx029
82
HuangJ.ZhengJ.YuanH.McGinnisK. (2018). Distinct tissue-specific transcriptional regulation revealed by gene regulatory networks in maize.BMC Plant Biol.18:111. 10.1186/s12870-018-1329-y
83
HuangJ. K.CarlinD. E.YuM. K.ZhangW.KreisbergJ. F.TamayoP.et al (2018). Systematic evaluation of molecular networks for discovery of disease genes.Cell Syst.6484–495.e5. 10.1016/j.cels.2018.03.001
84
HuangS.ChaudharyK.GarmireL. X. (2017). More is better: recent progress in multi-omics data integration methods.Front. Genet.8:84. 10.3389/fgene.2017.00084
85
HuttlinE. L.BrucknerR. J.PauloJ. A.CannonJ. R.TingL.BaltierK.et al (2017). Architecture of the human interactome defines protein communities and disease networks.Nature545505–509. 10.1038/nature22366
86
HuttlinE. L.TingL.BrucknerR. J.GebreabF.GygiM. P.SzpytJ.et al (2015). The BioPlex network: a systematic exploration of the human interactome.Cell162425–440. 10.1016/j.cell.2015.06.043
87
Huynh-ThuV. A.IrrthumA.WehenkelL.GeurtsP. (2010). Inferring regulatory networks from expression data using tree-based methods.PLoS One5:e12776. 10.1371/journal.pone.0012776
88
IdekerT.KroganN. J. (2012). Differential network biology.Mol. Syst. Biol.8:565. 10.1038/msb.2011.99
89
IdekerT.SharanR. (2008). Protein networks in disease.Genome Res.18644–652. 10.1101/gr.071852.107
90
JainiS.LyubetskayaA.GomesA.PetersonM.Tae ParkS.RamanS.et al (2014). Transcription factor binding site mapping using ChIP-Seq.Microbiol. Spectr.2:MGM2–MGM0035. 10.1128/microbiolspec.MGM2-0035-2013
91
JalanS.SolymosiN.VattayG.LiB. (2010). Random matrix analysis of localization properties of gene coexpression network.Phys. Rev. E81(4 Pt 2):046118. 10.1103/PhysRevE.81.046118
92
JalanS.UngC. Y.BhojwaniJ.LiB.ZhangL.LanS. H.et al (2012). Spectral analysis of gene co-expression network of Zebrafish.EPL99:48004. 10.1209/0295-5075/99/48004
93
JambusariaA.KlompJ.HongZ.RafiiS.DaiY.MalikA. B.et al (2018). A computational approach to identify cellular heterogeneity and tissue-specific gene regulatory networks.BMC Bioinformatics19:217. 10.1186/s12859-018-2190-6
94
JonssonP. F.BatesP. A. (2006). Global topological features of cancer proteins in the human interactome.Bioinformatics222291–2297. 10.1093/bioinformatics/btl390
95
JungM.JinS. G.ZhangX.XiongW.GogoshinG.RodinA. S.et al (2015). Longitudinal epigenetic and gene expression profiles analyzed by three-component analysis reveal down-regulation of genes involved in protein translation in human aging.Nucleic Acids Res.43:e100. 10.1093/nar/gkv473
96
KamburovA.WierlingC.LehrachH.HerwigR. (2009). ConsensusPathDB–a database for integrating human functional interaction networks.Nucleic Acids Res.37D623–D628. 10.1093/nar/gkn698
97
KanayaS.Altaf-Ul-AminM.KiboiS. K.Mochamad AfendiF. (2014). Big data and network biology.Biomed Res. Int.2014:836708. 10.1155/2014/836708
98
KanonidisE. I.RoyM. M.DeightonR. F.Le BihanT. (2016). Protein co-expression analysis as a strategy to complement a standard quantitative proteomics approach: case of a glioblastoma multiforme study.PLoS One11:e0161828. 10.1371/journal.pone.0161828
99
KimR. S.JiH.WongW. H. (2006). An improved distance measure between the expression profiles linking co-expression and co-regulation in mouse.BMC Bioinformatics7:44. 10.1186/1471-2105-7-44
100
KimY.HanS.ChoiS.HwangD. (2014). Inference of dynamic networks using time-course data.Brief. Bioinform.15212–228. 10.1093/bib/bbt028
101
KimY.KimJ. (2018). Estimation of dynamic systems for gene regulatory networks from dependent time-course data.J. Comput. Biol.25987–996. 10.1089/cmb.2018.0062
102
KitsakM.SharmaA.MencheJ.GuneyE.GhiassianS. D.LoscalzoJ.et al (2016). Tissue specificity of human disease module.Sci. Rep.6:35241. 10.1038/srep35241
103
KlingstromT.PlewczynskiD. (2011). Protein-protein interaction and pathway databases, a graphical review.Brief. Bioinform.12702–713. 10.1093/bib/bbq064
104
KogelmanL. J.FuJ.FrankeL.GreveJ. W.HofkerM.RensenS. S.et al (2016). Inter-tissue gene co-expression networks between metabolically healthy and unhealthy obese individuals.PLoS One11:e0167519. 10.1371/journal.pone.0167519
105
LageK.HansenN. T.KarlbergE. O.EklundA. C.RoqueF. S.DonahoeP. K.et al (2008). A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes.Proc. Natl. Acad. Sci. U.S.A.10520870–20875. 10.1073/pnas.0810772105
106
LageK.KarlbergE. O.StorlingZ. M.OlasonP. I.PedersenA. G.RiginaO.et al (2007). A human phenome-interactome network of protein complexes implicated in genetic disorders.Nat. Biotechnol.25309–316. 10.1038/nbt1295
107
LangfelderP.HorvathS. (2008). WGCNA: an R package for weighted correlation network analysis.BMC Bioinformatics9:559. 10.1186/1471-2105-9-559
108
LayeghifardM.HwangD. M.GuttmanD. S. (2017). Disentangling interactions in the microbiome: a network perspective.Trends Microbiol.25217–228. 10.1016/j.tim.2016.11.008
109
LeeI.BlomU. M.WangP. I.ShimJ. E.MarcotteE. M. (2011). Prioritizing candidate disease genes by network-based boosting of genome-wide association data.Genome Res.211109–1121. 10.1101/gr.118992.110
110
LiP.ZhangC.PerkinsE. J.GongP.DengY. (2007). Comparison of probabilistic Boolean network and dynamic Bayesian network approaches for inferring gene regulatory networks.BMC Bioinformatics8(Suppl. 7):S13. 10.1186/1471-2105-8-S7-S13
111
LiS.ArmstrongC. M.BertinN.GeH.MilsteinS.BoxemM.et al (2004). A map of the interactome network of the metazoan C. elegans.Science303540–543.
112
LiY.PatraJ. C. (2010). Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network.Bioinformatics261219–1224. 10.1093/bioinformatics/btq108
113
LieseckeF.DauduD.Duge de BernonvilleR.BesseauS.ClastreM.CourdavaultV.et al (2018). Ranking genome-wide correlation measurements improves microarray and RNA-seq based global and targeted co-expression networks.Sci. Rep.8:10885. 10.1038/s41598-018-29077-3
114
LimJ.HaoT.ShawC.PatelA. J.SzabóG.RualJ.-F.et al (2006). A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration.Cell125801–814. 10.1016/j.cell.2006.03.032
115
LinJ. S.LaiE. M. (2017). Protein-protein interactions: co-immunoprecipitation.Methods Mol. Biol.1615211–219. 10.1007/978-1-4939-7033-9_17
116
LindforsE. (2011). Network Biology: Applications in Medicine and Biotechnology.Espoo: VTT.
117
LiuE. T.LauffenburgerD. A. (2009). Systems Biomedicine: Concepts and Perspectives ıAmsterdam: Elsevier.
118
LiuW.MaL.JeonB.ChenL.ChenB. (2018). A Network Hierarchy-Based method for functional module detection in protein-protein interaction networks.J. Theor. Biol.45526–38. 10.1016/j.jtbi.2018.06.026
119
LiuY.BeyerA.AebersoldR. (2016). On the dependency of cellular protein levels on mRNA abundance.Cell165535–550. 10.1016/j.cell.2016.03.014
120
LiuY. Y.SlotineJ. J.BarabasiA. L. (2011). Controllability of complex networks.Nature473167–173. 10.1038/nature10011
121
Lopes-RamosC. M.KuijjerM. L.OginoS.FuchsC. S.DeMeoD. L.GlassK.et al (2018). Gene regulatory network analysis identifies sex-linked differences in colon cancer drug metabolism.Cancer Res.785538–5547. 10.1158/0008-5472.CAN-18-0454
122
Lopes-RamosC. M.PaulsonJ. N.ChenC. Y.KuijjerM. L.FagnyM.PlatigJ.et al (2017). Regulatory network changes between cell lines and their tissues of origin.BMC Genomics18:723. 10.1186/s12864-017-4111-x
123
LoscalzoJ.BarabaìsiA. -L. S.SilvermanE. K. (2017). Network Medicine: Complex Systems in Human Disease and Therapeutics.Cambridge, MA: Harvard University Press.
124
LuH. C.ForniliA.FraternaliF. (2013). Protein-protein interaction networks studies and importance of 3D structure knowledge.Expert Rev. Proteomics10511–520. 10.1586/14789450.2013.856764
125
LuoF.YangY.ZhongJ.GaoH.KhanL.ThompsonD. K.et al (2007). Constructing gene co-expression networks and predicting functions of unknown genes by random matrix theory.BMC Bioinformatics8:299. 10.1186/1471-2105-8-299
126
LuoY.ZhaoX.ZhouJ.YangJ.ZhangY.KuangW.et al (2017). A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information.Nat. Commun.8:573. 10.1038/s41467-017-00680-8
127
MahmoodS. S.LevyD.VasanR. S.WangT. J. (2014). The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective.Lancet383999–1008. 10.1016/S0140-6736(13)61752-3
128
Malod-DogninN.BanK.PrzuljN. (2017). Unified alignment of protein-protein interaction networks.Sci. Rep.7:953. 10.1038/s41598-017-01085-9
129
Malod-DogninN.PetschniggJ.WindelsS. F. L.PovhJ.HemmingwayH.KettelerR.et al (2019). Towards a data–integrated cell.Nat. Commun.10:805. 10.1038/s41467-019-08797-8
130
MarbachD.CostelloJ. C.KuffnerR.VegaN. M.PrillR. J.CamachoD. M.et al (2012). Wisdom of crowds for robust gene network inference.Nat. Methods9796–804. 10.1038/nmeth.2016
131
MarbachD.LamparterD.QuonG.KellisM.KutalikZ.BergmannS. (2016). Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases.Nat. Methods13366–370. 10.1038/nmeth.3799
132
MarcoA.KonikoffC.KarrT. L.KumarS. (2009). Relationship between gene co-expression and sharing of transcription factor binding sites in Drosophila melanogaster.Bioinformatics252473–2477. 10.1093/bioinformatics/btp462
133
MargolinA. A.NemenmanI.BassoK.WigginsC.StolovitzkyG.Dalla FaveraR.et al (2006). ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context.BMC Bioinformatics7(Suppl. 1):S7. 10.1186/1471-2105-7-S1-S7
134
MargueratS.SchmidtA.CodlinS.ChenW.AebersoldR.BahlerJ. (2012). Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells.Cell151671–683. 10.1016/j.cell.2012.09.019
135
McDermottJ. E.DiamondD. L.CorleyC.RasmussenA. L.KatzeM. G.WatersK. M. (2012). Topological analysis of protein co-abundance networks identifies novel host targets important for HCV infection and pathogenesis.BMC Syst. Biol.6:28. 10.1186/1752-0509-6-28
136
MeleM.FerreiraP. G.ReverterF.DeLucaD. S.MonlongJ.SammethM.et al (2015). Human genomics. The human transcriptome across tissues and individuals.Science348660–665. 10.1126/science.aaa0355
137
MencheJ.SharmaA.KitsakM.GhiassianS. D.VidalM.LoscalzoJ.et al (2015). Disease networks. Uncovering disease-disease relationships through the incomplete interactome.Science347:1257601. 10.1126/science.1257601
138
MeyerP. E.KontosK.LafitteF.BontempiG. (2007). Information-theoretic inference of large transcriptional regulatory networks.EURASIP J. Bioinform. Syst. Biol.2007:79879. 10.1155/2007/79879
139
MiloR.Shen-OrrS.ItzkovitzS.KashtanN.ChklovskiiD.AlonU. (2002). Network motifs: simple building blocks of complex networks.Science298824–827. 10.1126/science.298.5594.824
140
MinL.ZhangC.QuL.HuangJ.JiangL.LiuJ.et al (2017). Gene regulatory pattern analysis reveals essential role of core transcriptional factors’ activation in triple-negative breast cancer.Oncotarget821938–21953. 10.18632/oncotarget.15749
141
MordeletF.VertJ. P. (2008). SIRENE: supervised inference of regulatory networks.Bioinformatics24i76–i82. 10.1093/bioinformatics/btn273
142
MoreauY.TrancheventL. C. (2012). Computational tools for prioritizing candidate genes: boosting disease gene discovery.Nat. Rev. Genet.13523–536. 10.1038/nrg3253
143
MoscaR.PonsT.CeolA.ValenciaA.AloyP. (2013). Towards a detailed atlas of protein-protein interactions.Curr. Opin. Struct. Biol.23929–940. 10.1016/j.sbi.2013.07.005
144
MundadeR.OzerH. G.WeiH.PrabhuL.LuT. (2014). Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond.Cell Cycle132847–2852. 10.4161/15384101.2014.949201
145
NavlakhaS.KingsfordC. (2010). The power of protein interaction networks for associating genes with diseases.Bioinformatics261057–1063. 10.1093/bioinformatics/btq076
146
NewmanM. E. (2004). Fast algorithm for detecting community structure in networks.Phys. Rev. E69:066133. 10.1103/PhysRevE.69.066133
147
NiJ.KoyuturkM.TongH.HainesJ.XuR.ZhangX. (2016). Disease gene prioritization by integrating tissue-specific molecular networks using a robust multi-network model.BMC Bioinformatics17:453. 10.1186/s12859-016-1317-x
148
OtiM.SnelB.HuynenM. A.BrunnerH. G. (2006). Predicting disease genes using protein-protein interactions.J. Med. Genet.43691–698. 10.1136/jmg.2006.041376
149
PapanikolaouN.PavlopoulosG. A.TheodosiouT.IliopoulosI. (2015). Protein-protein interaction predictions using text mining methods.Methods7447–53. 10.1016/j.ymeth.2014.10.026
150
PerrinB. E.RalaivolaL.MazurieA.BottaniS.MalletJ.d’Alche-BucF. (2003). Gene networks inference using dynamic Bayesian networks.Bioinformatics19(Suppl. 2), ii138–ii148. 10.1093/bioinformatics/btg1071
151
PiersonE.ConsortiumG. T.KollerD.BattleA.MostafaviS.ArdlieK. G.et al (2015). Sharing and specificity of co-expression networks across 35 human tissues.PLoS Comput. Biol.11:e1004220. 10.1371/journal.pcbi.1004220
152
PillaiS. G.GeD.ZhuG.KongX.ShiannaK. V.NeedA. C.et al (2009). A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci.PLoS Genet.5:e1000421. 10.1371/journal.pgen.1000421
153
PineroJ.BerensteinA.Gonzalez-PerezA.ChernomoretzA.FurlongL. I. (2016). Uncovering disease mechanisms through network biology in the era of Next Generation Sequencing.Sci. Rep.6:24570. 10.1038/srep24570
154
PineroJ.BravoA.Queralt-RosinachN.Gutierrez-SacristanA.Deu-PonsJ.CentenoE.et al (2017). DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants.Nucleic Acids Res.45D833–D839. 10.1093/nar/gkw943
155
PirgaziJ.KhanteymooriA. R. (2018). A robust gene regulatory network inference method base on Kalman filter and linear regression.PLoS One13:e0200094. 10.1371/journal.pone.0200094
156
PlatigJ.CastaldiP. J.DeMeoD.QuackenbushJ. (2016). Bipartite community structure of eQTLs.PLoS Comput. Biol.12:e1005033. 10.1371/journal.pcbi.1005033
157
PrietoC.RisuenoA.FontanilloC.De las RivasJ. (2008). Human gene coexpression landscape: confident network derived from tissue transcriptomic profiles.PLoS One3:e3911. 10.1371/journal.pone.0003911
158
ProkopenkoD.SakornsakolpatP.FierH. L.QiaoD.ParkerM. M.McDonaldM. N.et al (2018). Whole-genome sequencing in severe chronic obstructive pulmonary disease.Am. J. Respir. Cell Mol. Biol.59614–622. 10.1165/rcmb.2018-0088OC
159
PrzuljN. (2007). Biological network comparison using graphlet degree distribution.Bioinformatics23e177–e183. 10.1093/bioinformatics/btl301
160
PrzuljN.CorneilD. G.JurisicaI. (2004). Modeling interactome: scale-free or geometric?Bioinformatics203508–3515.
161
QiuW.GuoF.GlassK.YuanG. C.QuackenbushJ.ZhouX.et al (2018). Differential connectivity of gene regulatory networks distinguishes corticosteroid response in asthma.J. Allergy Clin. Immunol.1411250–1258. 10.1016/j.jaci.2017.05.052
162
QuackenbushJ. (2003). Genomics. Microarrays–guilt by association.Science302240–241. 10.1126/science.1090887
163
RackhamO. J.FirasJ.FangH.OatesM. E.HolmesM. L.KnauppA. S.et al (2016). A predictive computational framework for direct reprogramming between human cell types.Nat. Genet.48331–335. 10.1038/ng.3487
164
ReverterA.ChanE. K. (2008). Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks.Bioinformatics242491–2497. 10.1093/bioinformatics/btn482
165
Rodenas-CuadradoP.ChenX. S.WiegrebeL.FirzlaffU.VernesS. C. (2015). A novel approach identifies the first transcriptome networks in bats: a new genetic model for vocal communication.BMC Genomics16:836. 10.1186/s12864-015-2068-1
166
RollandT.TasanM.CharloteauxB.PevznerS. J.ZhongQ.SahniN.et al (2014). A proteome-scale map of the human interactome network.Cell1591212–1226. 10.1016/j.cell.2014.10.050
167
RomanoskiC. E.GlassC. K.StunnenbergH. G.WilsonL.AlmouzniG. (2015). Epigenomics: roadmap for regulation.Nature518314–316. 10.1038/518314a
168
RotivalM.PetrettoE. (2014). Leveraging gene co-expression networks to pinpoint the regulation of complex traits and disease, with a focus on cardiovascular traits.Brief. Funct. Genomics1366–78. 10.1093/bfgp/elt030
169
RottjersL.FaustK. (2018). From hairballs to hypotheses-biological insights from microbial networks.FEMS Microbiol. Rev.42761–780. 10.1093/femsre/fuy030
170
RoyS.BhattacharyyaD. K.KalitaJ. K. (2014). Reconstruction of gene co-expression network from microarray data using local expression patterns.BMC Bioinformatics15(Suppl. 7):S10. 10.1186/1471-2105-15-S7-S10
171
Rozenblatt-RosenO.StubbingtonM. J. T.RegevA.TeichmannS. A. (2017). The Human Cell Atlas: from vision to reality.Nature550451–453. 10.1038/550451a
172
RualJ. F.VenkatesanK.HaoT.Hirozane-KishikawaT.DricotA.LiN.et al (2005). Towards a proteome-scale map of the human protein-protein interaction network.Nature4371173–1178. 10.1038/nature04209
173
RyanC. J.KennedyS.BajramiI.MatallanasD.LordC. J. (2017). A compendium of co-regulated protein complexes in breast cancer reveals collateral loss events.Cell Syst.5399–409.e5. 10.1016/j.cels.2017.09.011
174
SahaA.KimY.GewirtzA. D. H.JoB.GaoC.McDowellI. C.et al (2017). Co-expression networks reveal the tissue-specific regulation of transcription and splicing.Genome Res.271843–1858. 10.1101/gr.216721.116
175
SahniN.YiS.TaipaleM.Fuxman BassJ. I.Coulombe-HuntingtonJ.YangF.et al (2015). Widespread macromolecular interaction perturbations in human genetic disorders.Cell161647–660. 10.1016/j.cell.2015.04.013
176
SchadtE. E.BjorkegrenJ. L. (2012). NEW: network-enabled wisdom in biology, medicine, and health care.Sci. Transl. Med.4:115rv1. 10.1126/scitranslmed.3002132
177
SchweppeD. K.HuttlinE. L.HarperJ. W.GygiS. P. (2018). BioPlex display: an interactive suite for large-scale AP-MS protein-protein interaction data.J. Proteome Res.17722–726. 10.1021/acs.jproteome.7b00572
178
SerinE. A.NijveenH.HilhorstH. W.LigterinkW. (2016). Learning from Co-expression Networks: possibilities and challenges.Front. Plant Sci.7:444. 10.3389/fpls.2016.00444
179
SharmaA.ChavaliS.TabassumR.TandonN.BharadwajD. (2010). Gene prioritization in Type 2 Diabetes using domain interactions and network analysis.BMC Genomics11:84. 10.1186/1471-2164-11-84
180
SharmaA.GulbahceN.PevznerS. J.MencheJ.LadenvallC.FolkersenL.et al (2013). Network-based analysis of genome wide association data provides novel candidate genes for lipid and lipoprotein traits.Mol. Cell. Proteomics123398–3408. 10.1074/mcp.M112.024851
181
SharmaA.KitsakM.ChoM. H.AmeliA.ZhouX.JiangZ.et al (2018). Integration of molecular interactome and targeted interaction analysis to identify a COPD disease network module.Sci. Rep.8:14439. 10.1038/s41598-018-32173-z
182
SharmaA.MencheJ.HuangC. C.OrtT.ZhouX.KitsakM.et al (2015). A disease module in the interactome explains disease heterogeneity, drug response and captures novel pathways and genes in asthma.Hum. Mol. Genet.243005–3020. 10.1093/hmg/ddv001
183
ShenJ.ZhangJ.LuoX.ZhuW.YuK.ChenK.et al (2007). Predicting protein-protein interactions based only on sequences information.Proc. Natl. Acad. Sci. U.S.A.1044337–4341. 10.1073/pnas.0607879104
184
Shen-OrrS. S.MiloR.ManganS.AlonU. (2002). Network motifs in the transcriptional regulation network of Escherichia coli.Nat. Genet.3164–68. 10.1038/ng881
185
SonawaneA. R.PlatigJ.FagnyM.ChenC. Y.PaulsonJ. N.Lopes-RamosC. M.et al (2017). Understanding tissue-specific gene regulation.Cell Rep.211077–1088. 10.1016/j.celrep.2017.10.001
186
SongL.LangfelderP.HorvathS. (2012). Comparison of co-expression measures: mutual information, correlation, and model based indices.BMC Bioinformatics13:328. 10.1186/1471-2105-13-328
187
SpirinV.MirnyL. A. (2003). Protein complexes and functional modules in molecular networks.Proc. Natl. Acad. Sci. U.S.A.10012123–12128. 10.1073/pnas.2032324100
188
StegleO.TeichmannS. A.MarioniJ. C. (2015). Computational and analytical challenges in single-cell transcriptomics.Nat. Rev. Genet.16133–145. 10.1038/nrg3833
189
StuartJ. M.SegalE.KollerD.KimS. K. (2003). A gene-coexpression network for global discovery of conserved genetic modules.Science302249–255. 10.1126/science.1087447
190
SudlowC.GallacherJ.AllenN.BeralV.BurtonP.DaneshJ.et al (2015). UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age.PLoS Med.12:e1001779. 10.1371/journal.pmed.1001779
191
SunP. G.GaoL.HanS. (2011). Prediction of human disease-related gene clusters by clustering analysis.Int. J. Biol. Sci.761–73. 10.7150/ijbs.7.61
192
TaylorI. W.LindingR.Warde-FarleyD.LiuY.PesquitaC.FariaD.et al (2009). Dynamic modularity in protein interaction networks predicts breast cancer outcome.Nat. Biotechnol.27199–204. 10.1038/nbt.1522
193
TerzerM.MaynardN. D.CovertM. W.StellingJ. (2009). Genome-scale metabolic networks.Wiley Interdiscip. Rev. Syst. Biol. Med.1285–297. 10.1002/wsbm.37
194
The 1000 Genomes Project Consortium (2015). A global reference for human genetic variation.Nature52668–74. 10.1038/nature15393
195
The International HapMap Consortium (2003). The international HapMap project.Nature426789–796. 10.1038/nature02168
196
TianZ.GuoM.WangC.XingL.WangL.ZhangY. (2017). Constructing an integrated gene similarity network for the identification of disease genes.J. Biomed. Semantics8(Suppl. 1):32. 10.1186/s13326-017-0141-1
197
TohH.HorimotoK. (2002). Inference of a genetic network by a combined approach of cluster analysis and graphical Gaussian modeling.Bioinformatics18287–297. 10.1093/bioinformatics/18.2.287
198
TuncbagN.GoslineS. J.KedaigleA.SoltisA. R.GitterA.FraenkelE. (2016). Network-based interpretation of diverse high-throughput datasets through the omics integrator software package.PLoS Comput. Biol.12:e1004879. 10.1371/journal.pcbi.1004879
199
TurnbaughP. J.LeyR. E.HamadyM.Fraser-LiggettC. M.KnightR.GordonJ. I. (2007). The human microbiome project.Nature449804–810. 10.1038/nature06244
200
UetzP.GiotL.CagneyG.MansfieldT. A.JudsonR. S.KnightJ. R.et al (2000). A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae.Nature403623–627. 10.1038/35001009
201
UhlenM.BjorlingE.AgatonC.SzigyartoC. A.AminiB.AndersenE.et al (2005). A human protein atlas for normal and cancer tissues based on antibody proteomics.Mol. Cell. Proteomics41920–1932. 10.1074/mcp.M500279-MCP200
202
UhlenM.FagerbergL.HallstromB. M.LindskogC.OksvoldP.MardinogluA.et al (2015). Proteomics. Tissue-based map of the human proteome.Science347:1260419. 10.1126/science.1260419
203
van DamS.VosaU.van der GraafA.FrankeL.de MagalhaesJ. P. (2018). Gene co-expression analysis for functional classification and gene-disease predictions.Brief. Bioinform.19575–592. 10.1093/bib/bbw139
204
van SomerenE. P.VaesB. L.SteegengaW. T.SijbersA. M.DecheringK. J.ReindersM. J. (2006). Least absolute regression network analysis of the murine osteoblast differentiation network.Bioinformatics22477–484. 10.1093/bioinformatics/bti816
205
VargasA. J.QuackenbushJ.GlassK. (2016). Diet-induced weight loss leads to a switch in gene regulatory network control in the rectal mucosa.Genomics108126–133. 10.1016/j.ygeno.2016.08.001
206
VenkatesanK.RualJ. F.VazquezA.StelzlU.LemmensI.Hirozane-KishikawaT.et al (2009). An empirical framework for binary interactome mapping.Nat. Methods683–90. 10.1038/nmeth.1280
207
VidalM.CusickM. E.BarabasiA. L. (2011). Interactome networks and human disease.Cell144986–998. 10.1016/j.cell.2011.02.016
208
VidalM.FieldsS. (2014). The yeast two-hybrid assay: still finding connections after 25 years.Nat. Methods111203–1206. 10.1038/nmeth.3182
209
VlaicS.ConradT.Tokarski-SchnelleC.GustafssonM.DahmenU.GuthkeR.et al (2018). ModuleDiscoverer: identification of regulatory modules in protein-protein interaction networks.Sci. Rep.8:433. 10.1038/s41598-017-18370-2
210
VogelC.SilvaG. M.MarcotteE. M. (2011). Protein expression regulation under oxidative stress.Mol. Cell. Proteomics10:M111.009217. 10.1074/mcp.M111.009217
211
WachiS.YonedaK.WuR. (2005). Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues.Bioinformatics214205–4208. 10.1093/bioinformatics/bti688
212
WalhoutM.VidalM.DekkerJ. (2012). Handbook of Systems Biology: Concepts and Insights.Oxford: Academic Press.
213
WangB.MezliniA. M.DemirF.FiumeM.TuZ.BrudnoM.et al (2014). Similarity network fusion for aggregating data types on a genomic scale.Nat. Methods11333–337. 10.1038/nmeth.2810
214
WangQ.TanY. X.RenY. B.DongL. W.XieZ. F.TangL.et al (2011). Zinc finger protein ZBTB20 expression is increased in hepatocellular carcinoma and associated with poor prognosis.BMC Cancer11:271. 10.1186/1471-2407-11-271
215
WangS.MaJ.ZhangW.ShenJ. P.HuangJ.PengJ.et al (2018). Typing tumors using pathways selected by somatic evolution.Nat. Commun.9:4159. 10.1038/s41467-018-06464-y
216
WerhliA. V.GrzegorczykM.HusmeierD. (2006). Comparative evaluation of reverse engineering gene regulatory networks with relevance networks, graphical gaussian models and bayesian networks.Bioinformatics222523–2531. 10.1093/bioinformatics/btl391
217
WillT.HelmsV. (2014). Identifying transcription factor complexes and their roles.Bioinformatics30i415–i421. 10.1093/bioinformatics/btu448
218
WillseyA. J.MorrisM. T.WangS.WillseyH. R.SunN.TeerikorpiN.et al (2018). The psychiatric cell map initiative: a convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders.Cell174505–520. 10.1016/j.cell.2018.06.016
219
WilsonN. K.FosterS. D.WangX.KnezevicK.SchutteJ.KaimakisP.et al (2010). Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators.Cell Stem Cell7532–544. 10.1016/j.stem.2010.07.016
220
WuX.JiangR.ZhangM. Q.LiS. (2008). Network-based global inference of human disease genes.Mol. Syst. Biol.4:189. 10.1038/msb.2008.27
221
WysockiK.RitterL. (2011). Diseasome: an approach to understanding gene-disease interactions.Annu. Rev. Nurs. Res.2955–72. 10.1891/0739-6686.29.55
222
XenariosI.RiceD. W.SalwinskiL.BaronM. K.MarcotteE. M.EisenbergD. (2000). DIP: the database of interacting proteins.Nucleic Acids Res.28289–291. 10.1093/nar/28.1.289
223
XiaoX.Moreno-MoralA.RotivalM.BottoloL.PetrettoE. (2014). Multi-tissue analysis of co-expression networks by higher-order generalized singular value decomposition identifies functionally coherent transcriptional modules.PLoS Genet.10:e1004006. 10.1371/journal.pgen.1004006
224
XingL.GuoM.LiuX.WangC.WangL.ZhangY. (2017). An improved Bayesian network method for reconstructing gene regulatory network based on candidate auto selection.BMC Genomics18(Suppl. 9):844. 10.1186/s12864-017-4228-y
225
XuJ.LiY. (2006). Discovering disease-genes by topological features in human protein-protein interaction network.Bioinformatics222800–2805. 10.1093/bioinformatics/btl467
226
YangY.HanL.YuanY.LiJ.HeiN.LiangH. (2014). Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types.Nat. Commun.5:3231. 10.1038/ncomms4231
227
YeungM. K.TegnerJ.CollinsJ. J. (2002). Reverse engineering gene networks using singular value decomposition and robust regression.Proc. Natl. Acad. Sci. U.S.A.996163–6168. 10.1073/pnas.092576199
228
YildirimM. A.GohK. I.CusickM. E.BarabasiA. L.VidalM. (2007). Drug-target network.Nat. Biotechnol.251119–1126. 10.1038/nbt1338
229
YuH.LuscombeN. M.QianJ.GersteinM. (2003). Genomic analysis of gene expression relationships in transcriptional regulatory networks.Trends Genet.19422–427. 10.1016/S0168-9525(03)00175-6
230
YueF.ChengY.BreschiA.VierstraJ.WuW.RybaT.et al (2014). A comparative encyclopedia of DNA elements in the mouse genome.Nature515355–364. 10.1038/nature13992
231
YugiK.KubotaH.HatanoA.KurodaS. (2016). Trans-omics: how to reconstruct biochemical networks across multiple ‘omic’ layers.Trends Biotechnol.34276–290. 10.1016/j.tibtech.2015.12.013
232
ZhangQ. C.PetreyD.GarzonJ. I.DengL.HonigB. (2013). PrePPI: a structure-informed database of protein-protein interactions.Nucleic Acids Res.41D828–D833. 10.1093/nar/gks1231
233
ZhangX.ZhangR.JiangY.SunP.TangG.WangX.et al (2011). The expanded human disease network combining protein-protein interaction information.Eur. J. Hum. Genet.19783–788. 10.1038/ejhg.2011.30
234
ZhouX.LeiL.LiuJ.HaluA.ZhangY.LiB.et al (2018). A systems approach to refine disease taxonomy by integrating phenotypic and molecular networks.EBioMedicine3179–91. 10.1016/j.ebiom.2018.04.002
235
ZhouX.MencheJ.BarabasiA. L.SharmaA. (2014). Human symptoms-disease network.Nat. Commun.5:4212. 10.1038/ncomms5212
236
ZiegenhainC.ViethB.ParekhS.ReiniusB.Guillaumet-AdkinsA.SmetsM.et al (2017). Comparative analysis of single-cell RNA sequencing methods.Mol. Cell65631–643.e4. 10.1016/j.molcel.2017.01.023
237
ZuoY.CuiY.Di PotoC.VargheseR. S.YuG.LiR.et al (2016). INDEED: integrated differential expression and differential network analysis of omic data for biomarker discovery.Methods11112–20. 10.1016/j.ymeth.2016.08.015
Summary
Keywords
network medicine, biological networks, biomedical big data, interactome, co-expression, gene regulations, phenotype-specificity, systems medicine
Citation
Sonawane AR, Weiss ST, Glass K and Sharma A (2019) Network Medicine in the Age of Biomedical Big Data. Front. Genet. 10:294. doi: 10.3389/fgene.2019.00294
Received
26 December 2018
Accepted
19 March 2019
Published
11 April 2019
Volume
10 - 2019
Edited by
Marco Pellegrini, Italian National Research Council (CNR), Italy
Reviewed by
Shailendra Kumar Gupta, University of Rostock, Germany; Adriano Velasque Werhli, Fundação Universidade Federal do Rio Grande, Brazil
Updates
Copyright
© 2019 Sonawane, Weiss, Glass and Sharma.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amitabh Sharma, amitabh.sharma@channing.harvard.edu; amitabhsharmaa@gmail.com
This article was submitted to Bioinformatics and Computational Biology, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.