 Ke Han1,2*
Ke Han1,2* Miao Wang3Lei Zhang3Ying Wang1Mian Guo4Ming Zhao1,2Qian Zhao1,2Yu Zhang1,2
Miao Wang3Lei Zhang3Ying Wang1Mian Guo4Ming Zhao1,2Qian Zhao1,2Yu Zhang1,2 Nianyin Zeng5
Nianyin Zeng5 Chunyu Wang6
Chunyu Wang6- 1School of Computer and Information Engineering, Harbin University of Commerce, Harbin, China
- 2Heilongjiang Provincial Key Laboratory of Electronic Commerce and Information Processing, Harbin University of Commerce, Harbin, China
- 3Life Sciences and Environmental Sciences Development Center, Harbin University of Commerce, Harbin, China
- 4Department of Neurosurgery, The Second Affiliated Hospital of Harbin Medical University, Harbin, China
- 5Department of Instrumental and Electrical Engineering, Xiamen University, Xiamen, China
- 6School of Computer Science and Technology, Harbin Institute of Technology, Harbin, China
Motivation: The number of ion channels is increasing rapidly. As many of them are associated with diseases, they are the targets of more than 700 drugs. The discovery of new ion channels is facilitated by computational methods that predict ion channels and their types from protein sequences.
Methods: We used the SVMProt and the k-skip-n-gram methods to extract the feature vectors of ion channels, and obtained 188- and 400-dimensional features, respectively. The 188- and 400-dimensional features were combined to obtain 588-dimensional features. We then employed the maximum-relevance-maximum-distance method to reduce the dimensions of the 588-dimensional features. Finally, the support vector machine and random forest methods were used to build the prediction models to evaluate the classification effect.
Results: Different methods were employed to extract various feature vectors, and after effective dimensionality reduction, different classifiers were used to classify the ion channels. We extracted the ion channel data from the Universal Protein Resource (UniProt, http://www.uniprot.org/) and Ligand-Gated Ion Channel databases (http://www.ebi.ac.uk/compneur-srv/LGICdb/LGICdb.php), and then verified the performance of the classifiers after screening. The findings of this study could inform the research and development of drugs.
Introduction
Ion channels are the pathways for the passive transport of various inorganic ions across a membrane. The structure and function of cellular ion channels are the basis of life-sustaining processes, and their genetic variation, and dysfunction are related to the occurrence and development of many diseases (Gabashvili et al., 2007; Bagal et al., 2013; Cheng et al., 2018a,c). Usually, ion channels are in a closed state. Under particular stimuli, the channel protein conformation changes, and the probability of the ion channels opening increases. Based on their type of gate, ion channels are typically categorized into voltage-gated ion channels and ligand-gated ion channels (Wang et al., 2017a). On the binding of a ligand, a ligand-gated channel undergoes a conformational change that causes opening of the channel gate and ion flux. Voltage-gated ion channels predominantly contain potassium (K+), sodium (Na+), calcium (Ca2+), and anion channels (Shu-An et al., 2011). They are usually surrounded by four transmembrane segments of the same subunit. In these channels, there are some charged groups (potential sensors) that control the gate. When the membrane potential changes, the electric sensors undergo a displacement under the effect of the electric field force, and the gate is opened or closed in response to the change in the membrane potential. Ion channels are expressed in practically all tissues and can cause deafness, renal cysts, cardiac arrhythmias migraines, and epilepsy (Cai et al., 2002a). Therefore, many drugs are found to target ion channels. One example is an antiarrhythmic drug, Lidocaine, which acts as a voltage-gated sodium channel inhibitor (Peters et al., 1993; Tiwari and Srivastava, 2015). The actions of Lidocaine affect the conduction system and muscle cells of the heart, raising its depolarization threshold and making it less likely to initiate or conduct action potentials (Lin et al., 2015). Another example is Ziconotide, which targets calcium channels and is used for pain relief. This compound blocks the calcium influx in the nerve terminals, which results in a reduced release of glutamate and neuropeptides, effectively interrupting the spinal transmission of pain signals (Schmidtko et al., 2010).
Owing to the significance of ion channels in biological processes, researchers have initiated conducting more in-depth research on them to establish the relationships between ion channels and different diseases. Currently, ion channels have become important targets for disease diagnosis and drug development. It is known that many chemicals and genetic disorders can disrupt the normal function of ion channels and have catastrophic consequences for living organisms (Santos et al., 2017). Most animal toxins are used to treat diseases such as chronic pain by modulating ion channels to shut down the nervous system.
In recent years, ion channels have played an increasingly important role in the treatment of diseases and drug research and development. Therefore, several researchers have started to pay attention to the structure and function of ion channels. With the rapid growth of proteomics data, earlier prediction and identification of the type of a particular ion channel has become important. Therefore, researchers have developed various bioinformatics software to predict the identification of ion channels. As researchers are interested in developing drugs that target ion channel and extending ion channel protein annotation, a series of high-throughput computational tools have been developed to predict ion channels and their types directly from protein sequences. In the last decade, many computational methods have been developed based on machine learning algorithms (Yu et al., 2015; Zou et al., 2017a,b; Stephenson et al., 2019), which are used in different fields, such as drug repositioning (Yu et al., 2016, 2017). Increasingly, researchers have applied machine learning algorithms to predict and classify ion channels. Sudipto et al. (2006) used amino acid composition and dipeptide composition as the feature vectors and classified them using a support vector machine (SVM) to predict voltage-gated ion channels and their subtypes. Liu et al. (2010) proposed a voltage-gated potassium channel identification method based on local sequence information. The prediction result of this method was better than that of voltage-gated potassium channel identification based on global sequence information (Lin and Ding, 2011). Zhao et al. (2017) constructed a support vector machine (SVM)-based model to quickly predict ion channels and their types. By considering the residue sequence information and their physicochemical properties, a novel feature-extracted method which combined dipeptide composition with the physicochemical correlation between two residues was employed. Recently, Gao et al. (2016) proposed a model based on a SVM to search for predicted ion channels and their subfamilies using the sequence similarity search feature of the basic local alignment search tool. Although many classifiers have been developed for the identification of ion channels, there are still some unresolved problems. For example, ion channel sequence similarity is very high, which may result in overestimation of the predictive classification performance of the model (Olivier and Du, 2012).
In this study, SVM and random forest classifiers were used to identify ion channels and further classify them. The maximum-relevance-maximum-distance (MRMD) method was introduced for feature selection to improve the prediction accuracy. We followed three steps to predict and classify ion channels. First, a protein sequence was detected to determine if it belonged to an ion channel. If the test results demonstrated that the sequence was an ion channel, then the protein sequence was classified as either a voltage-gated ion channel or ligand-gated ion channel. Finally, if the protein sequence was found to belong to a voltage-gated ion channel, we classified it as a potassium (K+), sodium (Na+), calcium (Ca2+), or anion voltage-gated ion channel.
Materials and Methods
Figure 1 shows the basic flow of the processes proposed in this paper. In this section, we introduce in detail the data set, feature extraction method, dimension reduction method, and classifier used in this study.
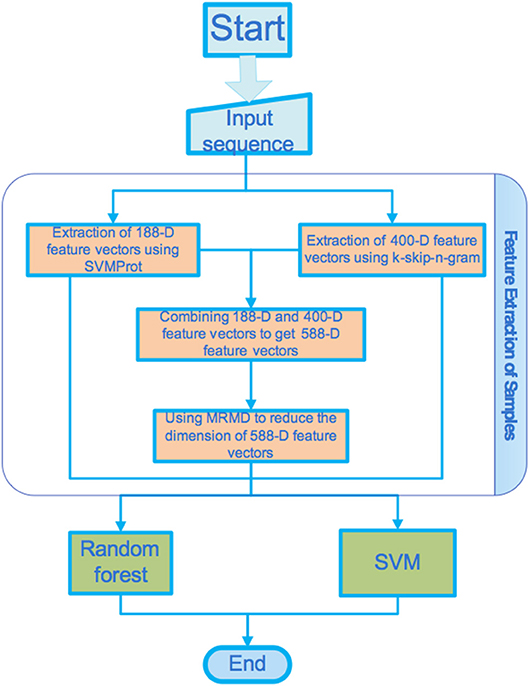
Figure 1. Workflow of the proposed processes.
Benchmark Dataset
The data that we used to establish the prediction model in this study were collected from Lin and Ding (2011). The sequences of ion channels were collected from the Universal Protein Resource (UniProt) and Ligand-Gated Ion channel databases (Marco et al., 2006). The following measures were taken to obtain reliable high-quality datasets. Initially, the protein sequences containing blurred disabilities, such as those with amino acids “X,” “B,” and “Z” were discarded. Then, the sequences of other protein fragments were removed. Proteins that were inferred by homology or prediction were discarded because of their unreliability. Finally, to avoid any homology bias, the CD-HIT (Li and Godzik, 2006) program was used to remove highly homologous sequences, with a 40% sequence identity as the cutoff (Wei et al., 2012; Chen et al., 2016; Zou et al., 2018a).
In strict accordance with the above steps, 148 voltage-gated ion channels, including 81 potassium channels, 29 calcium channels, 12 sodium channels, 26 anion channels, and 150 ligand-gated ion channels were finally extracted. To ensure the reliability and practicability of the ion channel prediction, and classification and maintenance of the balance between the positive and negative data, 300 protein sequences were randomly selected from UniProt as non-ion channels. It was observed that the consistency of these non-ion channel sequences was < 40%.
Feature Extraction of Samples
Section Benchmark dataset mainly discusses the series of preprocessing steps performed for the dataset. The reconstruction provided a reliable database for the study on the positioning method. This section focuses on specific methods of protein subcellular localization based on machine learning.
The first and most important role of a predictor is to extract protein sequences (Liu et al., 2015; Ding et al., 2017a,b; Zou et al., 2018b). We used two feature extraction methods including the SVMProt 188-D feature extraction method, which is based on protein composition and physicochemical properties, and the k-skip-n-gram 400-D feature extraction method.
SVMProt 188-D Feature Extraction
Different types of amino acids possess their own unique physicochemical properties. These characteristics of amino acid sequences can be used to predict types of protein. This method has yielded good predictive results (Cao and Cheng, 2016; Li et al., 2016b). Dubchak et al. (1995) proposed a composition transition distribution model based on the composition, transformation, and distribution of protein sequences, and achieved better results for the prediction of protein folding patterns. The physicochemical properties of protein sequences were fully embodied in this model, where the composition and physical and chemical properties were independent of each other. Cai et al. (2003) extracted 188-dimensional features in combination with amino acid composition and physicochemical characteristics for the characterization of proteins. SVMProt also contains nine physicochemical properties besides amino acid frequencies. The quantities of each of these properties are listed in Table 1 (Zou et al., 2013a,b).
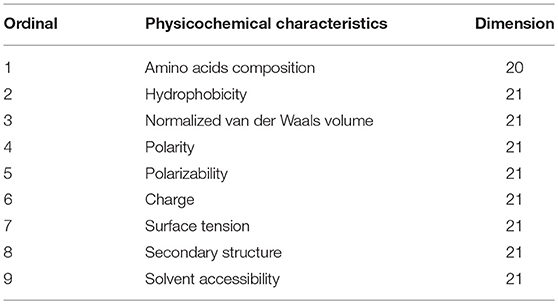
Table 1. Number of features in SVMPRot.
In the model, 20 amino acids in the query protein sequence constitute the first 20-dimensional feature vector. The first 20-dimensional vector is calculated as follows:
where Ai and L denote the number of the amino acids in the sequence and the length of the sequence, respectively, (Zhu et al., 2018b A20). {A1, A2, …, A20} represents the 20 amino acids that form the proteins. According to the physicochemical types, the amino acids can be classified under three categories based on their content (C), distribution (D), and bivalent frequency (F) (Bagal et al., 2013). The features of each of the remaining eight physicochemical properties are obtained using the following formula:
and
where Di (i = c, d, f) and countDidenote the physicochemical properties of the amino acids and number of such properties present in the sequence, respectively. After calculating all the physical and chemical properties, we finally extracted all the 188 (20 + (21 × 8) = 188) feature vectors.
k-skip-n-gram 400-D Feature Extraction
Guthrie et al. (2006) first proposed the k-skip-n-gram model. In protein sequences, the distance between two amino acids Ai and Aj is denoted by DT (Ai, Aj), which is defined as the position interval between two amino acids (Liu et al., 2014). It is calculated as follows:
where i and j are the positions of the amino acids in a sequence.
The k-skip-n-gram model provides the composition of n residues with distances k in a sequence. Its features are calculated as follows:
where N(TSkipGram)and N(am1am2…amn)denote the total number of elements in set TSkipGram and total number of terms am1am2…amn appearing in set TSkipGram, which is formulated as
where
Because only 20 amino acids can form a protein, a sequence has a total of 20n permutations. Therefore, a protein sequence can be transformed into 20n feature vector sets FVSkipGram.
As the number of feature vectors exhibits an exponential distribution, the value of n is quite important. When n = 1, there are only 20 features. If the number of features is quite small, the feature representation of a sequence is negatively affected. In contrast, when the value of n is very high, it affects the calculation efficiency. In this study, the value of n was considered as 2. Finally, we obtained 400 feature vectors.
Feature Selection (MRMD)
Owing to their limitations, the two feature representation methods mentioned above were combined to form a new feature vector containing more than one feature. SVM and random forest classifiers were used to classify the new feature vector set. When multiple feature extraction methods are combined, many dimensions may be generated and the classification result may be affected (Tang et al., 2017; Liu et al., 2018b; Zhu et al., 2018b). Feature selection can alleviate the problem of dimensionality by selecting a subset of features (Zhu et al., 2018c). Therefore, we employed the dimensionality reduction method based on MRMD (http://lab.malab.cn/soft/MRMD/index_en.html) to reduce the dimensionality of the generated feature vectors (Xu et al., 2016; Zou et al., 2016a,b; Zhu et al., 2017, 2018b; Chen et al., 2018; Tang et al., 2018b). MRMD selects the feature with the highest correlation and least redundancy by calculating the maximum relevance and maximum distance. In this study, Pearson's correlation coefficients were used to measure the relevance, and three distance functions were used to calculate the redundancy of the features. As the value of the Pearson correlation coefficient increased, the relationship between the features and target classes became stronger. As the distance between the features increased, the redundancy of the feature vectors decreased. Finally, the sub-features generated after the MRMD dimension reduction were found to possess the characteristics of low redundancy and a strong relationship. This could aid in achieving more accurate classification results.
Classifier Models
Random Forest
A random forest is a classifier that uses multiple trees to train and predict samples; it has been widely used in many bioinformatics tasks (Xu et al., 2013, 2018b; Liu et al., 2018a; Pan et al., 2018; Su et al., 2018; Wei et al., 2018a). It was proposed by Leo Breiman in 2001 and combines the Bagging integrated learning theory with the random subspace method (Verikas et al., 2011). A random forest is an integrated learning model based on a decision tree. It contains multiple decision trees trained by the Bagging integrated learning technology. Samples are input into a random forest for classification. The final classification result is governed by the output of a single decision tree. Since Buntine and Niblett (1992) proposed the random forest algorithm, it has been widely used, owing to its good performance, in many practical fields, such as the classification and regression of gene sequences, action recognition, face recognition, anomaly detection in data mining, and metric learning. In this study, we used a random forest classifier to build a model.
Support Vector Machine
An SVM is a supervised learning model related to learning algorithms and has achieved good performance in several bioinformatics (Momot et al., 2010; Cao et al., 2014; Ding et al., 2016; Li et al., 2016a; Wang et al., 2017b, 2018; Wei et al., 2017a,b, 2018c; Chen and Chuang, 2018; Liu et al., 2018c; Tang et al., 2018a; Shen et al., 2019; Zhu et al., 2019) and biomedicine (Zeng et al., 2018a; Zhang et al., 2018) studies. The dual-classification problem of an SVM can be broadly divided into three cases: linear separable, approximate linear separable, and non-linear separable. The solution for the linear separable problem is an optimal hyperplane that allows two groups of samples to be classified appropriately and to have the largest classification interval. This is shown in Figure 2, where the H plane is the optimal hyperplane. The approximate linear separability problem can be solved by adding a relaxation variable, i, in the optimization function of the linear classification. To solve the non-linear separable problem, we need to select an appropriate kernel function, transform the low-dimensional space into a high-dimensional space, and find the appropriate classification plane in the high-dimensional space so that the two samples can be classified appropriately (Cai et al., 2002b; Yu-Dong et al., 2010; Liu, 2017). Therefore, an SVM can achieve good classification results even when there are few experimental data. In this study, we used LIBSVM 3.23, which was downloaded from https://www.csie.ntu.edu.tw/~cjlin/libsvm/index.html. To obtain the optimal model, we performed a grid search to optimize parameters c and g. Then, the values of c and g were added to the model to obtain the optimal classification result. A combination of different types of features and classifiers can improve the overall performance of the model (Zhu et al., 2016, 2018a).
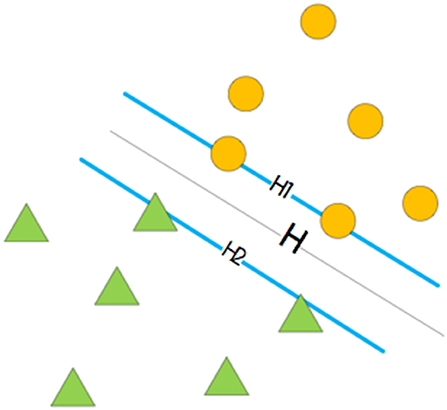
Figure 2. Optimal hyperplane of SVM.
Prediction Assessment
In machine learning, dividing experimental data into training sets is necessary to build a prediction model (Cao et al., 2017; Xu et al., 2017; Cheng et al., 2018b; Hu et al., 2018). Experimental data need to be further divided into test sets so that the final results of the training can be validated. To divide experimental data into training and test sets, a large amount of experimental data is needed. However, in practice, the number of experimental data is often limited. Therefore, researchers often use cross-validation for testing. Three types of cross-validation methods are commonly used in bioinformatics: independent data testing, folding cross-validation, and n-fold cross-validation. Among these, the folding knife test has been widely used in bioinformatics owing to its excellent results. However, this test is time and resource intensive (Lin et al., 2012; Zeng et al., 2016; Lai et al., 2017; Liu et al., 2017b; Manavalan et al., 2018). The n-fold cross-validation is commonly used to test the accuracy of an algorithm. The dataset was divided into 10 parts, nine of which were used as the training data and one as the testing data. After several experiments were conducted using numerous amounts of varied data, the best error estimates were obtained by dividing the dataset into 10 parts. There is sufficient theoretical basis to prove this approach (Chen et al., 2017; Zeng et al., 2018b).
Performance Evaluation
To obtain clearer classification prediction results and estimate the accuracy of the prediction model, we used other evaluation criteria as well (Feng et al., 2013, 2018; Chen et al., 2017; Zhang and Liu, 2017; Dao et al., 2018; Yang et al., 2018). The prediction accuracy was estimated using the sensitivity (Sn), overall accuracy (OA), and average accuracy (AA), which are defined as follows:
and
where TPi and FNi denote the true positives and false positives of the ith class, respectively, (Liu et al., 2017a; Zeng et al., 2017a). N and n are the total number of sequences and number of species, respectively.
Results
Prediction Results of Ion and Non-ion Channels
We compared the predictive effects of the SVM-based and random forest-based methods on both ion and non-ion channels in different dimensions. The results obtained are listed in Table 2. The 10-fold cross-validation results of the 188-dimensional features, 400-dimensional features, and mixed features (188-dimensional features combined with 400-dimensional features) are listed in Table 2. We then applied the MRMD method to reduce the dimensions of the 588-dimensional features to obtain 587-dimensional features. However, the average classification accuracy of the 587-dimensional features was found to be lower than that of the 400-dimensional features. The results also revealed that the SVM classifier was the best method for classifying the 400-dimensional features, with an average overall accuracy (OA) rate of 85.1%. As can be seen in Table 2, 86.6% of the ion channels and 83.7% of the non-ion channels can be appropriately identified using the SVM classifier, with a total accuracy rate of 85.1%. The feature vectors of the 188- and 400- dimensional features yield good prediction results. This result reveals that the SVM can moderately improve the predictive performance of the model. And we also try to use other classifiers to classify ion channels, but the classification effect is obviously worse than that of random forest and SVM classifiers, so we finally choose the two classifiers for comparison.
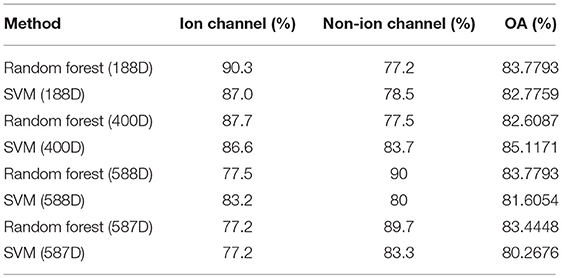
Table 2. Prediction results of ion channels and non-ion channels.
Classification Results of Voltage-Gated and Ligand-Gated Ion Channels
We evaluated the accuracy of the 188-dimensional features, 400-dimensional features, and mixed features (188-dimensional features combined with 400-dimensional features), and the 88-dimensional features obtained after the dimensional reduction using the MRMD method for discriminating between the classification results of voltage-gated and ligand-gated ion channels. The results are tabulated in Table 3. They reveal that the random forest classifier is the best for classifying the 188-dimensional features, with an average overall accuracy rate of 89.9%. As seen in Table 3, 93.9% of the voltage-gated ion channels and 86.0% of the ligand-gated ion channels could be correctly identified using the random forest method. The results reveal that the random forest classifier is better than the SVM classifier in some cases and can improve the prediction performance model.
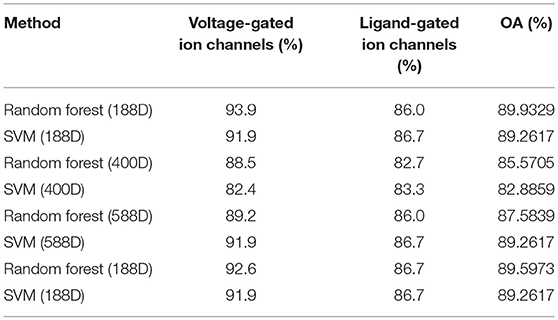
Table 3. Prediction results of voltage-gated and ligand-gated ion channels.
The results listed in Tables 2, 3 reveal that the difference between the voltage-gated and ligand-gated ion channels appears to be more distinct than that between the ion and non-ion channels. This may be due to the obvious differences between voltage-gated ion channels and ligand-gated ion channels with respect to some specific components.
Classification Results of Four Types Voltage-Gated Ion Channels
Finally, we classified the four types of voltage-gated ion channels, i.e., K, Ca, Anion, and Na, using the SVM and random forest methods. The prediction accuracy of the 188-dimension features, 400-dimensional features, 424-dimensional features, and mixed features were calculated individually. The results are listed in Table 4. This table shows that the best classification effect is achieved when the SVM classifier, which had a maximum overall accuracy rate of 72.973%, is used to extract the 188-dimensional features. We applied the MRMD method to reduce the dimensions of the 588-dimensional features to obtain 424-dimensional features. However, the average classification accuracy of the 424-dimensional features was lower than that of the 188-dimensional features. After dimension reduction, the dimension of ion channel feature vectors did not decrease significantly, and the accuracy was even decreasing, which indicates that MRMD was not effective in classifying ion channel feature vectors.
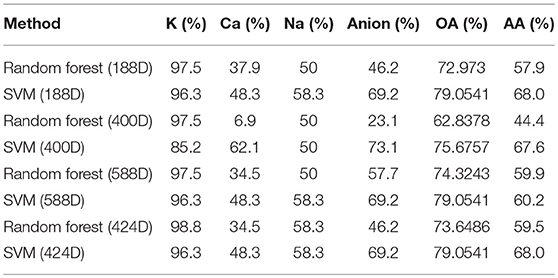
Table 4. Prediction results for four types of voltage-gated ion channels.
In general, the robustness of the results can be improved by using the minimum dimensions of the feature vector data. Therefore, we recommend using 188-dimensional feature vectors to predict the four types of voltage-gated ion channels.
Discussion and Conclusions
In this study, new features were used to extract the features of ion channels, and good prediction results were obtained. To accurately predict and classify ion channels and their types, we constructed SVM-based and random forest-based models that used SVMProt 188- dimensional feature extraction and k-skip-n-gram to extract features. Then, we combined the 188-dimensional features with the 400-dimensional features to obtain 588-dimensional features. To achieve a higher accuracy with fewer features, the MRMD method was used to reduce the dimensions of the 588-dimensional features. Finally, the SVM and random forest models were used to model 188-dimensional features, 400-dimensional features, 588-dimensional features, and the MRMD-reduced features. The experimental results revealed that the features extracted by the SVMProt 188-dimensional feature extraction and k-skip-n-gram methods could effectively predict and classify the ion channels. Such a fast and accurate method can accelerate the prediction of ion channels and promote the discovery of drug targets.
Although this method can guide the study of ion channel discovery, it has some limitations. With the rapid increase in ion channel types and data, more perfect prediction and classification models need to be developed by researchers. We believe that more in-depth research using computational intelligence (Mrozek et al., 2009; Zeng et al., 2014; Cabarle et al., 2017; Xu et al., 2018a) and machine learning (Zeng et al., 2017b; Song et al., 2018; Zhu et al., 2018c) can result in the development of additional feature extraction methods (Wei et al., 2018b) and more accurate prediction classification models (Wang et al., 2016), and contribute to drug research and development.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
KH, MW, LZ, and YW made substantial contributions to the design of the work and drafted and revised the article. MG, MZ, QZ, and YZ focused on the machine learning programs and plotted the figures. NZ and CW mainly made the analysis and interpretation of data for the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the Support Program for Young Academic Key Teachers of Harbin University of Commerce (No. 7XN004).
References
Bagal, S. K., Brown, A. D., Cox, P. J., Kiyoyuki, O., Owen, R. M., Pryde, D. C., et al. (2013). Ion channels as therapeutic targets: a drug discovery perspective. J. Med. Chem. 56:593. doi: 10.1021/jm3011433
Buntine, W., and Niblett, T. (1992). A further comparison of splitting rules for decision-tree induction. Mach. Learn. 8, 75–85. doi: 10.1007/BF00994006
Cabarle, F. G. C., Adorna, H. N., Jiang, M., and Zeng, X. (2017). Spiking neural P systems with scheduled synapses. IEEE Trans. Nanobiosci. 16, 792–801. doi: 10.1109/tnb.2017.2762580
Cai, C. Z., Han, L. Y., Ji, Z. L., Chen, X., and Chen, Y. Z. (2003). SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucl. Acids Res. 31, 3692–3697. doi: 10.1093/nar/gkg600
Cai, Y. D., Liu, X. J., Xu, X. B., and Chou, K. C. (2002a). Prediction of protein structural classes by support vector machines. Comput. Chem. 26, 293–296. doi: 10.1016/S0097-8485(01)00113-9
Cai, Y. D., Liu, X. J., Xu, X. B., and Chou, K. C. (2002b). Support vector machines for predicting the specificity of GalNAc-transferase. Peptides 23, 205–208. doi: 10.1016/S0196-9781(01)00597-6
Cao, R. Z., and Cheng, J. L. (2016). Protein single-model quality assessment by feature-based probability density functions. Sci. Rep. 6:23990. doi: 10.1038/srep23990
Cao, R. Z., Freitas, C., Chan, L., Sun, M., Jiang, H. Q., and Chen, Z. X. (2017). ProLanGO: protein function prediction using neural machine translation based on a recurrent neural network. Molecules 22:14. doi: 10.3390/molecules22101732
Cao, R. Z., Wang, Z., Wang, Y. H., and Cheng, J. L. (2014). SMOQ: a tool for predicting the absolute residue-specific quality of a single protein model with support vector machines. BMC Bioinform. 15:120. doi: 10.1186/1471-2105-15-120
Chen, C.-Y. C., and Chuang, T.-J. (2018). Comment on “A comprehensive overview and evaluation of circular RNA detection tools”. PLoS Comput. Biol. 13:e1005420. doi: 10.1371/journal.pcbi.100542
Chen, W., Feng, P., Ding, H., and Lin, H. (2018). Classifying included and excluded exons in exon skipping event using histone modifications. Front. Genet. 9:433. doi: 10.3389/fgene.2018.00433
Chen, W., Feng, P., Tang, H., Ding, H., and Lin, H. (2016). Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics 107, 255-258. doi: 10.1016/j.ygeno.2016.05.003
Chen, W., Yang, H., Feng, P., Ding, H., and Lin, H. (2017). iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 33, 3518–3523. doi: 10.1093/bioinformatics/btx479
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018a). DincRNA: a comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics 34, 1953–1956. doi: 10.1093/bioinformatics/bty002
Cheng, L., Jiang, Y., Ju, H., Sun, J., Peng, J., Zhou, M., et al. (2018b). InfAcrOnt: calculating cross-ontology term similarities using information flow by a random walk. BMC Genomics 19(Suppl. 1):919. doi: 10.1186/s12864-017-4338-6
Cheng, L., Wang, P., Tian, R., Wang, S., Guo, Q., Luo, M., et al. (2018c). LncRNA2Target v2.0: a comprehensive database for target genes of lncRNAs in human and mouse. Nucl. Acids Res. 47, D140–D144. doi: 10.1093/nar/gky1051
Dao, F. Y., Lv, H., Wang, F., Feng, C. Q., Ding, H., Chen, W., et al. (2018). Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics. doi: 10.1093/bioinformatics/bty943
Ding, Y., Tang, J., and Guo, F. (2016). Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinform. 17:398. doi: 10.1186/s12859-016-1253-9
Ding, Y., Tang, J., and Guo, F. (2017a). Identification of drug-target interactions via multiple information integration. Inform. Sci. 418–419, 546–560. doi: 10.1016/j.ins.2017.08.045
Ding, Y., Tang, J., and Guo, F. (2017b). Identification of protein-ligand binding sites by sequence information and ensemble classifier. J. Chem. Inform. Model. 57, 3149–3161. doi: 10.1021/acs.jcim.7b00307
Dubchak, I., Muchnik, I., Holbrook, S. R., and Kim, S. H. (1995). Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. U.S.A. 92, 8700–8704. doi: 10.1073/pnas.92.19.8700
Feng, C. Q., Zhang, Z. Y., Zhu, X. J., Lin, Y., Chen, W., Tang, H., et al. (2018). iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics. doi: 10.1093/bioinformatics/bty827
Feng, P.-M., Chen, W., Lin, H., and Chou, K.-C. (2013). iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 442, 118–125. doi: 10.1016/j.ab.2013.05.024
Gabashvili, I. S., Sokolowski, B. H. A., Morton, C. C., and Giersch, A. B. S. (2007). Ion channel gene expression in the inner ear. J. Assoc. Res. Otolaryngol. 8, 305–328. doi: 10.1007/s10162-007-0082-y
Gao, J., Cui, W., Sheng, Y., Ruan, J., and Kurgan, L. (2016). PSIONplus: accurate sequence-based predictor of ion channels and their types. PLoS ONE 11:e0152964. doi: 10.1371/journal.pone.0152964
Guthrie, D., Allison, B., Liu, W., Guthrie, L., and Wilks, Y. (2006). “A closer look at skip-gram modelling,” in Proceedings of the 5th International Conference on Language Resources and Evaluation (LREC-2006), Vol. 2006 (Magazzini del Cotone), 1222–1225.
Hu, Y., Zhao, T., Zhang, N., Zang, T., Zhang, J., and Cheng, L. (2018). Identifying diseases-related metabolites using random walk. BMC Bioinform. 19 (Suppl. 5):116. doi: 10.1186/s12859-018-2098-1
Lai, H. Y., Chen, X. X., Chen, W., Tang, H., and Lin, H. (2017). Sequence-based predictive modeling to identify cancerlectins. Oncotarget 8, 28169–28175. doi: 10.18632/oncotarget.15963
Li, D., Ju, Y., and Zou, Q. (2016a). Protein folds prediction with hierarchical structured SVM. Curr. Proteom. 13, 79–85. doi: 10.2174/157016461302160514000940
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658. doi: 10.1093/bioinformatics/btl158
Li, Y. H., Xu, J. Y., Tao, L., Li, X. F., Li, S., Zeng, X., et al. (2016b). SVM-Prot 2016: A web-server for machine learning prediction of protein functional families from sequence irrespective of similarity. PLoS ONE 11:e0155290. doi: 10.1371/journal.pone.0155290
Lin, H., Ding, C., Song, Q., Yang, P., Ding, H., Deng, K-J., et al. (2012). The prediction of protein structural class using averaged chemical shifts. J. Biomol. Struct. Dynam. 29, 1147–1153. doi: 10.1080/07391102.2011.672628
Lin, H., and Ding, H. (2011). Predicting ion channels and their types by the dipeptide mode of pseudo amino acid composition. J. Theor. Biol. 269, 64–69. doi: 10.1016/j.jtbi.2010.10.019
Lin, H., Liu, W. X., He, J., Liu, X. H., Ding, H., and Chen, W. (2015). Predicting cancerlectins by the optimal g-gap dipeptides. Sci. Rep. 5:16964. doi: 10.1038/srep16964
Liu, B. (2017). BioSeq-Analysis: a platform for DNA, RNA, and protein sequence analysis based on machine learning approaches. Brief. Bioinform. doi: 10.1093/bib/bbx165
Liu, B., Fang, Y., Huang, D.-S., and Chou, K.-C. (2018a). iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformaitcs 34, 33–40. doi: 10.1093/bioinformatics/btx579
Liu, B., Hao, W., Zhang, D., Wang, X., and Chou, K. C. (2017a). Pse-Analysis: a python package for DNA/RNA and protein/peptide sequence analysis based on pseudo components and kernel methods. Oncotarget 8, 13338–13343. doi: 10.18632/oncotarget.14524
Liu, B., Jiang, S., and Zou, Q. (2018b). HITS-PR-HHblits: Protein remote homology detection by combining pagerank and hyperlink-induced topic search. Brief. Bioinform. doi: 10.1093/bib/bby104
Liu, B., Li, K., Huang, D.-S., and Chou, K.-C. (2018c). iEnhancer-EL: Identifying enhancers and their strength with ensemble learning approach. Bioinformatics 34, 3835–3842. doi: 10.1093/bioinformatics/bty458
Liu, B., Liu, F., Wang, X., Chen, J., Fang, L., and Chou, K.-C. (2015). Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucl. Acids Res. 43, W65–W71. doi: 10.1093/nar/gkv458
Liu, B., Xu, J., Zou, Q., Xu, R., Wang, X., and Chen, Q. (2014). Using distances between Top-n-gram and residue pairs for protein remote homology detection. BMC Bioinform. 15:S3. doi: 10.1186/1471-2105-15-S2-S3
Liu, L. X., Meng-Long, L. I., Tan, F. Y., Min-Chun, L. U., and Wang, K. L. (2010). Local sequence information-based support vector machine to classify voltage-gated potassium channels. Acta Biochim. Et Biophys. Sinica 38, 363–371. doi: 10.1111/j.1745-7270.2006.00177.x
Liu, Y., Wang, X., and Liu, B. (2017b). A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief. Bioinform. 20, 1–17. doi: 10.1093/bib/bbx126
Manavalan, B., Subramaniyam, S., Shin, T. H., Kim, M. O., and Lee, G. (2018). Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res. 17:2715–2726. doi: 10.1021/acs.jproteome.8b00148
Marco, D., Marie-Ange, D., and Nicolas, L. N. (2006). LGICdb: a manually curated sequence database after the genomes. Nucl. Acids Res. 34, 267–269. doi: 10.1093/nar/gkj104
Momot, A., Malysiak-Mrozek, B., Kozielski, S., Mrozek, D., Hera, L., Gorczynska-Kosiorz, S., et al. (2010). “Improving performance of protein structure similarity searching by distributing computations in hierarchical multi-agent system,” in Computational Collective Intelligence: Technologies And Applications, eds J. S. Pan, S. M. Chen and N. T. Nguyen (Berlin: Springer-Verlag Berlin), 320. doi: 10.1007/978-3-642-16693-8_34
Mrozek, D., Malysiak-Mrozek, B., Kozielski, S., and Ieee (2009). Alignment of Protein Structure Energy Patterns Represented as Sequences of Fuzzy Numbers. New York, NY: IEEE. doi: 10.1109/NAFIPS.2009.5156391
Olivier, I., and Du, T. L. (2012). A metabolomics approach to characterise and identify various Mycobacterium species. J. Microbiol. Methods 88, 419–426. doi: 10.1016/j.mimet.2012.01.012
Pan, G., Jiang, L., Tang, J., and Guo, F. (2018). A novel computational method for detecting DNA methylation sites with DNA sequence information and physicochemical properties. Int. J. Mol. Sci. 19:511. doi: 10.3390/ijms19020511
Peters, D. J., Spruit, L., Saris, J. J., Ravine, D., Sandkuijl, L. A., Fossdal, R., et al. (1993). Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat. Genet. 5, 359–362. doi: 10.1038/ng1293-359
Santos, R., Ursu, O., Gaulton, A., Bento, A. P., Donadi, R. S., Bologa, C. G., et al. (2017). A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34. doi: 10.1038/nrd.2016.230
Schmidtko, A., Lötsch, J., Freynhagen, R., and Geisslinger, G. (2010) Ziconotide for treatment of severe chronic pain. Lancet 375, 1569–1577. doi: 10.1016/S0140-6736(10)60354-6
Shen, Y., Tang, J., and Guo, F. (2019). Identification of protein subcellular localization via integrating evolutionary and physicochemical information into Chou's general PseAAC. J. Theor. Biol. 462, 230–239. doi: 10.1016/j.jtbi.2018.11.012
Shu-An, C., Yu-Yen, O., Tzong-Yi, L., and, M., and Michael, G. (2011). Prediction of transporter targets using efficient RBF networks with PSSM profiles and biochemical properties. Bioinformatics 27, 2062–2067. doi: 10.1093/bioinformatics/btr340
Song, T., Rodríguez-Patón, A., Zheng, P., and Zeng, X. J. (2018). Spiking neural p systems with colored spikes. IEEE Trans. Cogn. Dev. Syst. 10, 1106–1115. doi: 10.1109/TCDS.2017.2785332
Stephenson, N., Shane, E., Chase, J., Rowland, J., Ries, D., Justice, N., et al. (2019). Survey of machine learning techniques in drug discovery. Curr. Drug Metabol. 20, 185–193. doi: 10.2174/1389200219666180820112457
Su, R., Wu, H., Xu, B., Liu, X., and Wei, L. (2018). Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data. IEEE/ACM Trans. Comp. Biol. Bioinform. doi: 10.1109/TCBB.2018.2858756
Sudipto, S., Zack, J., Singh, B., and Raghava, G. P (2006). VGIchan: Prediction and classification of voltage-gated ion channels. Genomics Proteomics Bioinform. 4, 253–258. doi: 10.1016/S1672-0229(07)60006-0
Tang, H., Cao, R. Z., Wang, W., Liu, T. S., Wang, L. M., and He, C. M. (2017). A two-step discriminated method to identify thermophilic proteins. Int. J. Biomathe. 10:1750050. doi: 10.1142/s1793524517500504
Tang, H., Zhao, Y. W., Zou, P., Zhang, C. M., Chen, R., Huang, P., et al. (2018a). HBPred: a tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 14, 957–964. doi: 10.7150/ijbs.24174
Tang, W., Wan, S., Yang, Z., Teschendorff, A. E., and Zou, Q. (2018b). Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 34, 398–406. doi: 10.1093/bioinformatics/btx622
Tiwari, A. K., and Srivastava, R. (2015). An efficient approach for the prediction of ion channels and their subfamilies. Compu. Biol. Chem. 58, 205–221. doi: 10.1016/j.compbiolchem.2015.07.002
Verikas, A., Gelzinis, A., and Bacauskiene, M. (2011). Mining data with random forests: a survey and results of new tests. Patt. Recog. 44, 330–349. doi: 10.1016/j.patcog.2010.08.011
Wang, F., Knutson, K., Alcaino, C., Linden, D. R., Gibbons, S. J., Kashyap, P., et al. (2017a). Mechanosensitive ion channel Piezo2 is important for enterochromaffin cell response to mechanical forces. J. Physiol. 595:79. doi: 10.1113/JP272718
Wang, S. P., Zhang, Q., Lu, J., and Cai, Y. D. (2018). Analysis and prediction of nitrated tyrosine sites with the mrmr method and support vector machine algorithm. Curr. Bioinform. 13, 3–13. doi: 10.2174/1574893611666160608075753
Wang, X., Zeng, X., Ju, Y., Jiang, Y., Zhang, Z., and Chen, W. J. C. B. (2016). A classification method for microarrays based on diversity. Curr. Bioinform. 11, 590–597. doi: 10.2174/1574893609666140820224436
Wang, Y., Ding, Y., Guo, F., Wei, L., and Tang, J. (2017b). Improved detection of DNA-binding proteins via compression technology on PSSM information. PLoS ONE 12:e0185587. doi: 10.1371/journal.pone.0185587
Wei, C., Feng, P., and Hao, L. (2012). Prediction of ketoacyl synthase family using reduced amino acid alphabets. J. Indus. Microbiol. Biotechnol. 39:579. doi: 10.1007/s10295-011-1047-z
Wei, L., Hu, J., Li, F., Song, J., Su, R., and Zou, Q. (2018a). Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief. Bioinform. doi: 10.1093/bib/bby107
Wei, L., Luan, S., Nagai, L. A. E., Su, R., and Zou, Q. J. B. (2018b). Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics. 35, 1326–1333. doi: 10.1093/bioinformatics/bty824
Wei, L., Wan, S., Guo, J., and Wong, K. K. (2017a). A novel hierarchical selective ensemble classifier with bioinformatics application. Artifi. Intell. Med. 83, 82–90. doi: 10.1016/j.artmed.2017.02.005
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017b). Improved prediction of protein-protein interactions using novel negative samples, features, and an ensemble classifier. Artifi. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Wei, L., Zhou, C., Chen, H., Song, J., and Su, R. (2018c). ACPred-FL: a sequence-based predictor based on effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 34, 4007–4016. doi: 10.1093/bioinformatics/bty451
Xu, H., Zeng, W., Zhang, D., and Zeng, X. J. I. T. o.C (2018a). MOEA/HD: A multiobjective evolutionary algorithm based on hierarchical decomposition. IEEE Trans. Cyber. 49, 517–526. doi: 10.1109/TCYB.2017.2779450
Xu, Y., Guo, M., Liu, X., Wang, C., Liu, Y., and Liu, G. (2016). Identify bilayer modules via pseudo-3D clustering: applications to miRNA-gene bilayer networks. Nucl. Acids Res. 44:e152. doi: 10.1093/nar/gkw679
Xu, Y., Guo, M., Shi, W., Liu, X., and Wang, C. (2013). A novel insight into Gene Ontology semantic similarity. Genomics 101, 368–375. doi: 10.1016/j.ygeno.2013.04.010
Xu, Y., Wang, Y., Luo, J., Zhao, W., and Zhou, X. (2017). Deep learning of the splicing (epi)genetic code reveals a novel candidate mechanism linking histone modifications to ESC fate decision. Nucl. Acids Res. 21, 12100–12112. doi: 10.1093/nar/gkx870
Xu, Y., Zhao, W., Olson, S. D., Prabhakara, K. S., and Zhou, X. (2018b). Alternative splicing links histone modifications to stem cell fate decision. Genome Biol. 19:133. doi: 10.1186/s13059-018-1512-3
Yang, H., Lv, H., Ding, H., Chen, W., and Lin, H. (2018). iRNA-2OM: A sequence-based predictor for identifying 2'-O-methylation sites in homo sapiens. J. Comput. Biol. 25, 1266–1277. doi: 10.1089/cmb.2018.0004
Yu, L., Huang, J. B., Ma, Z. X., Zhang, J., Zou, Y. P., and Gao, L. (2015). Inferring drug-disease associations based on known protein complexes. BMC Med. Genomics 8:13. doi: 10.1186/1755-8794-8-s2-s2
Yu, L., Ma, X., Zhang, L., Zhang, J., and Gao, L. (2016). Prediction of new drug indications based on clinical data and network modularity. Sci. Rep. 6:032530. doi: 10.1038/srep32530
Yu, L., Su, R., Wang, B., Zhang, L., Zou, Y., Zhang, J., et al. (2017). Prediction of novel drugs for hepatocellular carcinoma based on multi-source random walk. IEEE-ACM Trans. Comput. Biol. Bioinform. 14, 966–977. doi: 10.1109/TCBB.2016.2550453
Yu-Dong, C., Xiao-Jun, L., Xue-Biao, X., and Kuo-Chen, C. (2010). Support Vector machines for predicting hiv protease cleavage sites in protein. J. Comp. Chem. 23, 267–274. doi: 10.1002/jcc.10017
Zeng, N. Y., Qiu, H., Wang, Z. D., Liu, W. B., Zhang, H., and Li, Y. R. (2018a). A new switching-delayed-PSO-based optimized SVM algorithm for diagnosis of Alzheimer's disease. Neurocomputing 320, 195–202. doi: 10.1016/j.neucom.2018.09.001
Zeng, N. Y., Wang, Z. D., and Zhang, H. (2016). Inferring nonlinear lateral flow immunoassay state-space models via an unscented Kalman filter. Sci. China-Inform. Sci. 59:10. doi: 10.1007/s11432-016-0280-9
Zeng, N. Y., Zhang, H., Li, Y. R., Liang, J. L., and Dobaie, A. M. (2017a). Denoising and deblurring gold immunochromatographic strip images via gradient projection algorithms. Neurocomputing 247, 165–172. doi: 10.1016/j.neucom.2017.03.056
Zeng, N. Y., Zhang, H., Song, B. Y., Liu, W. B., Li, Y. R., and Dobaie, A. M. (2018b). Facial expression recognition via learning deep sparse autoencoders. Neurocomputing 273, 643–649. doi: 10.1016/j.neucom.2017.08.043
Zeng, X., Liao, Y., Liu, Y., and Zou, Q. (2017b). Prediction and validation of disease genes using hetesim scores. IEEE/ACM Trans. Compu. Biol. Bioinform. 14, 687–695. doi: 10.1109/tcbb.2016.2520947
Zeng, X., Pan, L., and Pérez-Jiménez, M. J. (2014). Small universal simple spiking neural P systems with weights. Sci. China Inform. Sci. 57, 1–11. doi: 10.1007/s11432-013-4848-z
Zhang, J., and Liu, B. (2017). PSFM-DBT: Identifying DNA-binding proteins by combing position specific frequency matrix and distance-bigram transformation. Int. J. Mol. Sci. 18:1856. doi: 10.3390/ijms18091856
Zhang, N., Sa, Y., Guo, Y., Lin, W., Wang, P., and Feng, Y. M. (2018). Discriminating ramos and jurkat cells with image textures from diffraction imaging flow cytometry based on a support vector machine. Curr. Bioinform. 13, 50–56. doi: 10.2174/1574893611666160608102537
Zhao, Y. W., Su, Z. D., Yang, W., Lin, H., Chen, W., and Tang, H. (2017). IonchanPred 2.0: a tool to predict ion channels and their types. Int. J. Mol. Sci. 18:1838. doi: 10.3390/ijms18091838
Zhu, P. F., Hu, Q., Hu, Q. H., Zhang, C. Q., and Feng, Z. Z. (2018a). Multi-view label embedding. Patt. Recogn. 84, 126–135. doi: 10.1016/j.patcog.2018.07.009
Zhu, P. F., Hu, Q. H., Han, Y. H., Zhang, C. Q., and Du, Y. (2016). Combining neighborhood separable subspaces for classification via sparsity regularized optimization. Inform. Sci. 370, 270–287. doi: 10.1016/j.ins.2016.08.004
Zhu, P. F., Xu, Q., Hu, Q. H., and Zhang, C. Q. (2018b). Co-regularized unsupervised feature selection. Neurocomputing 275, 2855–2863. doi: 10.1016/j.neucom.2017.11.061
Zhu, P. F., Xu, Q., Hu, Q. H., Zhang, C. Q., and Zhao, H. (2018c). Multi-label feature selection with missing labels. Patt. Recogn.74, 488–502. doi: 10.1016/j.patcog.2017.09.036
Zhu, P. F., Zhu, W. C., Hu, Q. H., Zhang, C. Q., and Zuo, W. M. (2017). Subspace clustering guided unsupervised feature selection. Patt. Recogn. 66, 364–374. doi: 10.1016/j.patcog.2017.01.016
Zhu, X. J., Feng, C. Q., Lai, H. Y., Chen, W., and Lin, H. (2019). Predicting protein structural classes for low-similarity sequences by evaluating different features. Knowledge-Based Syst. 163, 787–793. doi: 10.1016/j.knosys.2018.10.007
Zou, Q., Chen, L., Huang, T., Zhang, Z., and Xu, Y. (2017a). Machine learning and graph analytics in computational biomedicine. Artif Intell Med. 83:1. doi: 10.1016/j.artmed.2017.09.003
Zou, Q., Li, X., Jiang, Y., Zhao, Y., and Wang, G. (2013a). BinMemPredict: a web server and software for predicting membrane protein types. Curr. Proteomics 10, 2–9. doi: 10.2174/1570164611310010002
Zou, Q., Lin, G., Jiang, X., Liu, X., and Zeng, X. (2018a). Sequence clustering in bioinformatics: an empirical study. Brief. Bioinform. doi: 10.1093/bib/bby1090
Zou, Q., Mrozek, D., Ma, Q., and Xu, Y. (2017b). Scalable data mining algorithms in computational biology and biomedicine. Biomed. Res. Int. 2017:5652041. doi: 10.1155/2017/5652041
Zou, Q., Wan, S., Ju, Y., Tang, J., and Zeng, X. (2016a). Pretata: predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 10:114. doi: 10.1186/s12918-016-0353-5
Zou, Q., Wang, Z., Guan, X., Liu, B., Wu, Y., and Lin, Z. (2013b). An approach for identifying cytokines based on a novel ensemble classifier. BioMed. Res. Int. 2013:686090. doi: 10.1155/2013/686090
Zou, Q., Xing, P., Wei, L., and Liu, B. (2018b). Gene2vec: gene subsequence embedding for prediction of mammalian n6-methyladenosine sites from mRNA. RNA 25, 205–218. doi: 10.1261/rna.069112.118
Keywords: ion channel, machine learning, random forest, SVM, feature selection
Citation: Han K, Wang M, Zhang L, Wang Y, Guo M, Zhao M, Zhao Q, Zhang Y, Zeng N and Wang C (2019) Predicting Ion Channels Genes and Their Types With Machine Learning Techniques. Front. Genet. 10:399. doi: 10.3389/fgene.2019.00399
Received: 14 February 2019; Accepted: 12 April 2019;
Published: 03 May 2019.
Edited by:
Dariusz Mrozek, Silesian University of Technology, PolandReviewed by:
Balachandran Manavalan, Ajou University, South KoreaHui Ding, University of Electronic Science and Technology of China, China
Wei Chen, North China University of Science and Technology, China
Copyright © 2019 Han, Wang, Zhang, Wang, Guo, Zhao, Zhao, Zhang, Zeng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Han, dGhydXN0ZXJAMTYzLmNvbQ==