 Ramón Gómez-Moreno1,2†
Ramón Gómez-Moreno1,2† Rachell Martínez-Ramírez1,2†
Rachell Martínez-Ramírez1,2† Abiel Roche-Lima3
Abiel Roche-Lima3 Kelvin Carrasquillo-Carrión3
Kelvin Carrasquillo-Carrión3 Josué Pérez-Santiago4*
Josué Pérez-Santiago4* Abel Baerga-Ortiz1,2*
Abel Baerga-Ortiz1,2*- 1University of Puerto Rico Medical Sciences Campus, San Juan, Puerto Rico
- 2Molecular Sciences Research Center, San Juan, Puerto Rico
- 3CCRHD–RCMI Program University of Puerto Rico Medical Sciences Campus, San Juan, Puerto Rico
- 4University of Puerto Rico Comprehensive Cancer Center, San Juan, Puerto Rico
The gut microbiota has been implicated in a number of normal and disease biological processes. Recent studies have identified a subset of gut bacterial genes as potentially involved in inflammatory processes. In this work, we explore the sequence variability for some of these bacterial genes using a combination of deep sequencing and oligotyping, a data analysis application that identifies mutational hotspots in short stretches of DNA. The genes for pks island, tcpC and usp, all harbored by certain strains of E. coli and all implicated in inflammation, were amplified by PCR directly from stool samples and subjected to deep amplicon sequencing. For comparison, the same genes were amplified from individual bacterial clones. The amplicons for pks island and tcpC from stool samples showed minimal levels of heterogeneity comparable with the individual clones. The amplicons for usp from stool samples, by contrast, revealed the presence of five distinct oligotypes in two different regions. Of these, the oligotype GT was found to be present in the control uropathogenic clinical isolate and also detected in stool samples from individuals with colorectal cancer (CRC). Mutational hotspots were mapped onto the USP protein, revealing possible substitutions around Leu110, Glu114, and Arg115 in the middle of the pyocin domain (Gln110, Gln114, and Thr115 in most healthy samples), and also Arg218 in the middle of the nuclease domain (His218 in the uropathogenic strain). All of these results suggest that a level of variability within bacterial pro-inflammatory genes could explain differences in bacterial virulence and phenotype.
Introduction
The gut is a rich microbial ecosystem that contains numerous species, many of them implicated in human diseases. Direct relationships have been delineated between the presence of certain microbial species and inflammatory diseases such as Crohn’s disease, ulcerative colitis, and colorectal cancer (CRC) (Kostic et al., 2011; Martinez-Medina and Garcia-Gil, 2014; Brennan and Garrett, 2016). For instance, there are reports of associations between the presence of Fusobacterium nucleatum, Akkermansia muciniphila, enterotoxigenic Bacteroides fragilis, and Streptococcus gallolyticus, and CRC (Sears, 2009; Boleij et al., 2011; Kostic et al., 2011; Weir et al., 2013). More specifically, Escherichia coli has been found in the mucosa of CRC patients in higher numbers than in the mucosa of healthy individuals (Swidsinski et al., 1998; Martin et al., 2004).
It is increasingly clear that the involvement of E. coli in CRC takes place through mechanisms that are encoded by specific sets of genes (Bonnet et al., 2014; Raisch et al., 2014). These genes encoding genotoxic cyclomodulins and other pro-inflammatory molecules have been identified in certain strains of gram-negative bacteria. For instance, the pks island genes, a cluster of genes encoding enzymes for the production of the elusive natural product colibactin, has been found more frequently in mucosa samples from CRC individuals than in healthy donors (Arthur et al., 2012; Gomez-Moreno et al., 2019). Bacteria harboring the pks genes have also been found to induce the formation of tumors in AOX-treated mice (Nougayrede et al., 2006; Arthur et al., 2012). Similarly, the tcpC gene encoding a toll-like receptor antagonist that promotes the formation of kidney abscesses in mice has been found in strains of uropathogenic E. coli (Yadav et al., 2010). Also, the gene for the uropathogenic specific protein (usp) initially identified in E. coli from urinary tract infections encodes a DNAse with genotoxic activity that promotes cell death (Nipic et al., 2013).
Our group had previously established a protocol for the detection of specific pro-inflammatory genes in stool samples and established some preliminary associations with CRC (Gomez-Moreno et al., 2019). In that work, the presence of pks island genes and of tcpC was more common in stool samples from CRC patients and adenoma cases than in healthy controls. Also, the presence of the usp gene (encoding the uropathogenic specific protein) was found to be associated with colorectal neoplasia (Gomez-Moreno et al., 2019). Interestingly, a systematic search through the shotgun metagenomic databases also found the usp gene more frequently in samples or datasets from CRC patients than in the healthy population, suggesting its presence as a possible marker for CRC risk (Roche-Lima and Baerga-Ortiz, unpublished data).
While many of these genes seem to be present predominantly in CRC cases, they are not exclusive to CRC cases. Numerous samples from healthy individuals were found to harbor one or more of these pro-inflammatory genes. One possibility is that there are DNA sequence variants within the population and that some variants are more highly associated with disease or represent bacterial lineages more highly associated with disease.
Oligotyping is a method for the analysis of closely related sequences that is used to identify specific sites of sequence variability in relatively short PCR-amplified fragments. It relies on the parallel sequencing and subsequent comparison of millions of reads per sample to identify nucleotide positions with a phylogenetically relevant signal above the basal variability expected for a large set of information (Eren et al., 2016). The use of this entropy-based decomposition makes it possible to resolve closely related variants that differ by as little as one nucleotide at the amplified region (Eren et al., 2016). Oligotyping has been used to investigate ecological questions in diverse environments including the oral microbiota, gut microbiota, raw sewage, and soil (Mark Welch et al., 2014; Fisher et al., 2015; Turlapati et al., 2015; Vineis et al., 2016).
In an effort to identify sequence variants more associated with disease, we performed oligotyping analysis on DNA fragments amplified from the regions encoding selected pro-inflammatory genes that were found predominantly in CRC cases, but also in healthy controls (Gomez-Moreno et al., 2019).
Methods
Sample selection. Stool samples had been provided by the Early Detection Research Network (EDRN) GLNE Clinical Validation Center to the UPR Stool Sample Repository (IRB Protocol A9560115) for a previous study (Gomez-Moreno et al., 2019). All samples were de-identified and labeled only with a numeric code. The samples chosen for this study were previously found to contain the pks island, tcpC, and the usp genes as reported by our group (Gomez-Moreno et al., 2019). Table 1 summarizes the samples used and links them to the original numeric identifier given by the EDRN repository. The control strain IHE3034 was a generous donation from Dr. Eric Oswald from the University of Toulouse, and the EC640 strain, collected under protocol during a microbiological surveillance protocol, was a generous donation from Dr. Iraida E. Robledo from the University of Puerto Rico Medical Sciences Campus. The stool samples were thawed, and the bacterial DNA was extracted using the QIAgen Stool DNA Kit as described previously (Gómez-Moreno et al., 2014).
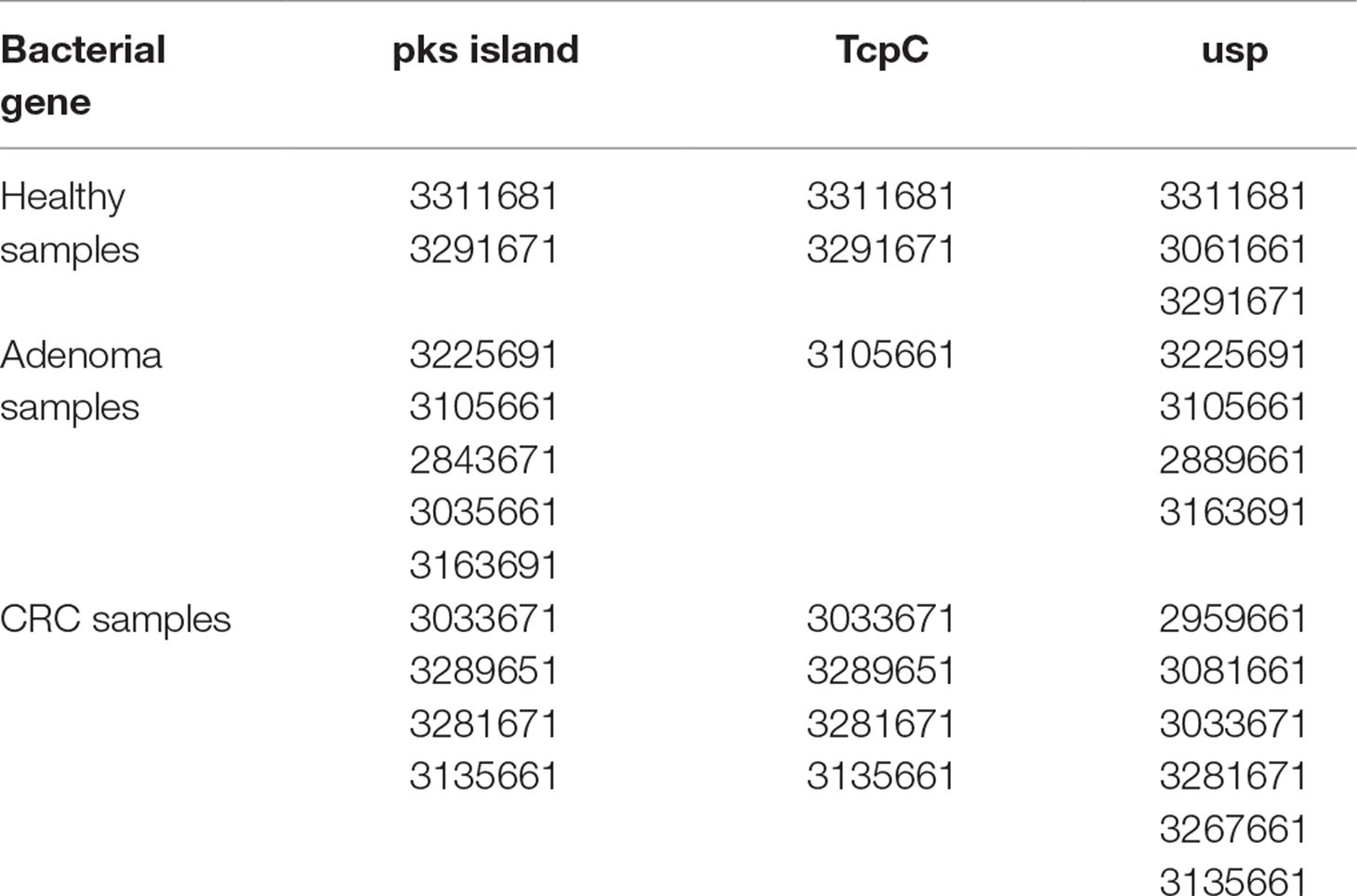
Table 1 Sample selection. Samples that had been previously been found to contain the genes for pks island, tcpC, and usp were selected for oligotyping analysis (Gomez-Moreno et al., 2019). All of the samples generated amplicons that correspond to specific regions of each gene, and those amplicons were subjected to deep sequencing. The number for each sample corresponds to the number from the EDRN (Early Detection Research Network) repository.
DNA amplification. Polymerase chain reaction (PCR) was performed with sequence specific primers that also contained the overhang adapter sequence for Illumina (Table 2). The first-stage PCR was carried out by heating to 94°C for an initial denaturation step of 1 min, followed by 30 cycles of 94°C for 30 s, 30 s of the corresponding annealing temperature (Table 2), and a 3-min extension at 68°C, and finished with a 10-min extension step at 72°C. The second-stage PCR reaction was performed to add the Illumina index sequences as described in the Illumina protocol (Illumina Corp. Part# 15044223Rev.B)

Table 2 Primers and conditions for the PCR amplification of specific DNA sequences. Each oligonucleotide was synthesized with an additional DNA sequence marked in bold for the purpose of indexing.
Quality assessment. First-stage PCR products were visualized by agarose gel electrophoresis (1%) stained with GelRed™ (Biotium), followed by gel extraction using the QIAquick Gel Extraction kit. The concentration and purity of each amplicon was determined using the nanodrop 2000c (Thermo Scientific). Second-stage PCR quality was tested using the Agilent 2100 Bioanalyzer, and the concentrations were determined using Qubit 2.0 fluorometer (Invitrogen).
Illumina sequencing. DNA sequencing was carried out by paired end sequencing using the MiSeq Sequencing Platform (Illumina, San Diego, CA). All quality controls and pre-processing were performed using the fastx-tool kit (http://hannonlab.cshl.edu/fastx_toolkit/). Reads with a quality score of 30 on at least 80% of the bases were kept for further analysis. For each gene of interest, we combined all samples into a single FASTA file and formatted as required for running the oligotyping pipeline. We added gaps to the shorter reads to match with the longest reads as all reads should have the same length and then performed entropy analysis.
All of the processed sequence data that was used as input for Oligotyping can be found in the NCBI Bioproject database under Accession PRJNA551701.
Oligotyping. We performed oligotyping analysis using the pipeline version 2.1 (available from http://merenlab.org/software/oligotyping/) (Eren et al., 2013). After the initial calculation of Shannon entropy using the analyze-entropy script in the oligotyping pipeline, we ran the method separately for the R1 and R2 reads so as to identify the nucleotide positions with the highest sequence variability. Using the R1 reads generated from the usp amplicon as an example, oligotyping analysis was performed on 5,855,441 reads pooled together using three components automatically selected form the highest entropy values following the initial entropy analysis. To reduce the noise, each oligotype required to have a most abundant unique sequence with a minimum abundance of 10. Oligotypes that did not meet this criterion were removed from the analysis. For the R2 reads, the analysis was performed on 4,412,715 reads as described for R1 reads. The most abundant oligotypes in each sample were quantified and plotted. As a control, all the datasets contained the amplicons generated from a bacterial isolate, which was supposed to show no sequence variation whatsoever.
Searching for usp oligotypes in published metagenomic datasets. We analyzed metagenomic DNA sequence data obtained from the European Nucleotide Archive (https://www.ebi.ac.uk/ena) database (accession no. PRJEB12449). These shotgun gut microbiome datasets were obtained from a case–control study conducted with stool samples collected at two locations: Washington DC and France (Vogtmann et al., 2016). We had already determined that the sequences for pks island, tcpC, and usp were present in these datasets by performing a BLASTN search on the downloaded FASTQ.gz files (Roche-Lima et al., unpublished results). However, the frequency of hits in the Washington DC datasets was very low (in some cases zero). Thus, we decided to carry out the sequence analysis using the datasets from the France population. We used the same BLASTN method to search for the usp-positive reads using the following queries: GAACATGCAGTGGAACGAGCAGACGCAGCG and ACAGGGCGCGGCGTCCCGCTT. These searches were intended as a validation of the sites of sequence variation for this gene locus.
Three-dimensional protein models. A three-dimensional model for USP was built using the I-TASSER (Iterative Threading ASSEmbly Refinement) server and visualized using VMD (Humphrey et al., 1996; Yang et al., 2015). The I-Tasser server selected the crystal structure of the colicin D central domain from Escherichia coli (PDB ID: 5ZNM) as the best template for USP.
Results
For the pks island genes, a fragment of 800 bp from the clbN gene was amplified directly from 11 stool samples that had been found to be positive for this gene (2 healthy, 5 adenomas, and 4 CRC). Similarly for the tcpC gene, a fragment of 283 bp was amplified from seven stool samples (two healthy, one adenoma, and four CRC). Finally, for the usp gene, a fragment of 685 bp was amplified from 13 samples (3 healthy, 4 adenomas, and 6 CRC). As a control, we PCR-amplified the same set of genes using the DNA from single bacterial isolate, IHE3034, which is pks +, and EC640, which is tcpC + and usp +, which was initially obtained from the urine of a 5-month-old patient with a urinary tract infection at the University of Puerto Rico Pediatric Hospital and was found to be positive for all three genes (Wolter et al., 2009).
The DNA fragments corresponding to the genes pks and tcpC that were amplified from patient-derived stool samples showed a similar oligotype profile as the control isolate strain (Figure 1, the control is the leftmost bar in all panels); a level of sequence heterogeneity consistent with a single population and possibly resulting from method-related phenomena as will be addressed in the discussion section. By contrast, the DNA fragments amplified from stool samples containing usp showed clear hotspots of sequence variation (Figure 2). In the R1 direction, three sites of variability were identified: positions 329, 340, and 344 of the gene (Figure 2A). In the R2 direction, two sites of variation were identified: positions 653 and 662 of the gene (Figure 2B).
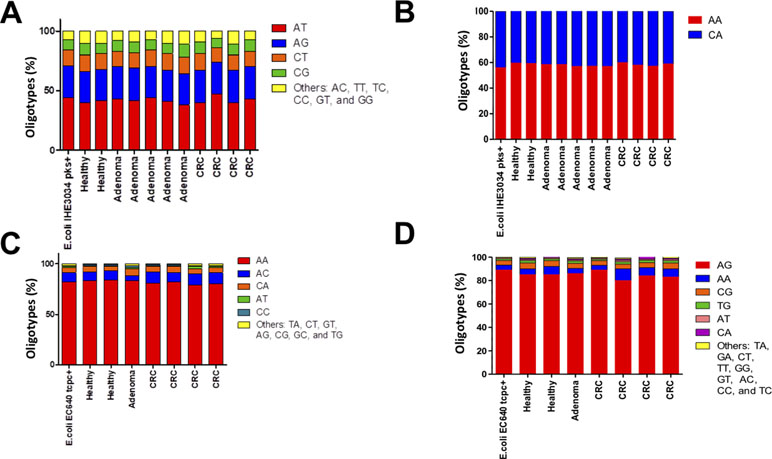
Figure 1 Distribution of oligotypes for the DNA fragments amplified from stool samples. The genes corresponding to (A)pks island R1 (B), pks island R2 (C), tcpC R1, and (D)tcpC R2 were analyzed by oligotyping [Eren et al., 2013]. The distribution of oligotypes amplified from stool samples is similar to the distribution resulting from a single clone for strain IHE3034 (pks+) and strain EC640 (tcpC+), suggesting low sequence variability for these tow genes in the gut microbiome.
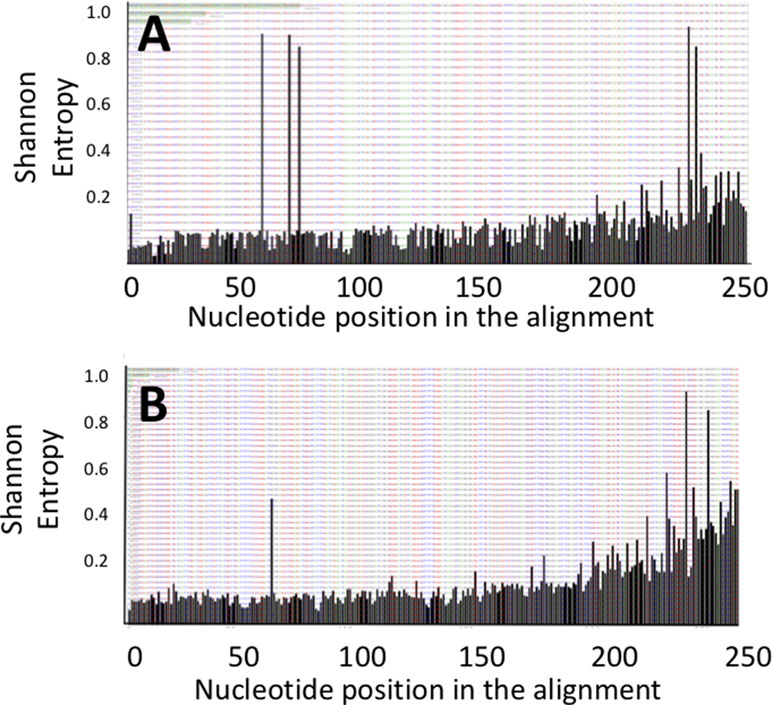
Figure 2 Shannon entropy plots for the usp sequences amplified from stool samples reveal positions of nucleotide variability for the (A) upstream R1 and (B) downstream R2 portions of the usp amplicon.
Sequence analysis revealed two main oligotypes present in different proportions in the R1 (Figure 3A) direction and three main oligotypes in the R2 direction (Figure 3B). The oligotype labeled “TGG” was the most abundant one with a thymine in position 329 and guanosines in positions 340 and 344 of the gene, respectively (Figure 3A). This oligotype was also the most abundant in the EC640 uropathogenic control strain and also present in most of the CRC and adenoma samples (Figure 3A). The other abundant oligotype “ACC” contained adenine in position 329 and cytosines in positions 340 and 344 and was present in most of the healthy samples (Figure 3A).
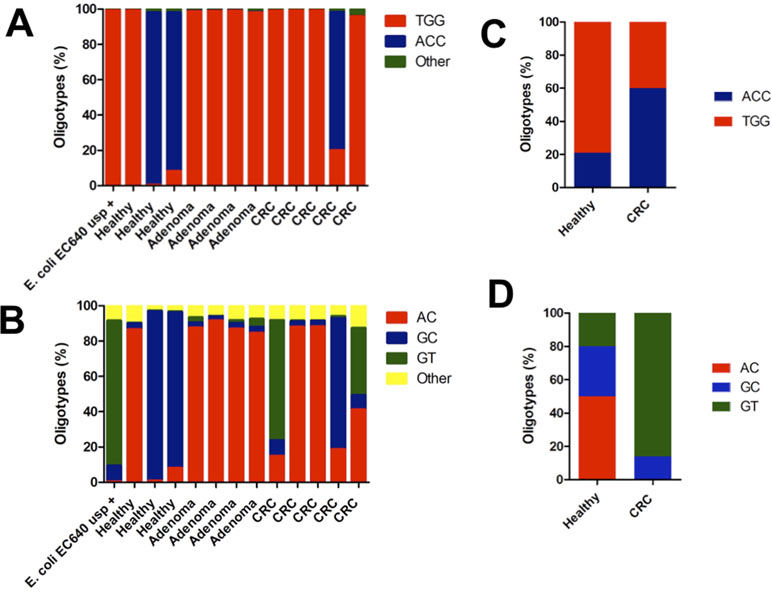
Figure 3 The distribution of oligotypes for the DNA fragments obtained from the amplification of the usp gene reveals patient-specific lineages when sequenced in the (A) forward direction R1 and in (B) reverse direction R2. The control EC640, a uropathogenic strain, is mostly the TGG oligotype in the forward direction and the GT oligotype in the reverse direction. The two patients who harbor this GT oligotype both have CRC, and the GT oligotype is also found more frequently in uropathogenic strains. Oligotyping results compared the data found in the metagenomic datasets corresponding to a CRC case–control study. (C) The same oligotypes, TGG and ACC, were found in the region covered in the R1 reads of the present study. (D) Also, the same oligotypes, AC, GC, and GT, were found in the DNA region covered by the R2 reads in the present study.
Since we performed paired-end sequencing, there was another set of data for the sequence at the opposite end of each of the amplicons. Again, the distribution of oligotypes for pks and tcpC reveals negligible heterogeneity compared to the control (Figure 1C and D). By contrast, the usp sequence reveals three main oligotypes, “AC” being the most abundant one and “GT” being the one associated with the uropathogenic control strain and with two CRC cases (Figure 3B).
In order to validate the presence of these hotspots of sequence variability along the usp gene, we analyzed the sequences from previously published shotgun metagenomic datasets for CRC individuals, compared with age- and gender-matched controls (Vogtmann et al., 2016). To our surprise, we observed in these published datasets the exact same sites of sequence variability that we report for our samples. Despite the fact that the samples were collected at different times and in different continents, they all contain a similar distribution of oligotypes. The sequence datasets from the French population showed no difference in the total number of hits for the usp gene between CRC and healthy controls (16 hits vs 15 hits; data not shown). In terms of oligotype distribution, the TGG oligotype was also found to be the most abundant, with the ACC oligotype marginally associated with disease (Figure 3C). In the other end of the gene, oligotype GT is also most associated with CRC (Figure 3D).
The base pair substitutions in these oligotypes represent a change in amino acid sequences of the resulting USP protein variants, which could result in functional differences. For instance, the abundant TGG oligotype encodes Leu, Glu, and Arg in positions 110, 114, and 115 of the USP protein, whereas the ACC oligotype encodes Gln, Gln, and Thr in those same positions (Figure 4, R1). These amino acids are all located in the N-terminal pyocin-like domain, which is thought to be involved in toxin targeting (Figure 5). At the other end of the protein is the C-terminal DNAase domain, in which the abundant oligotype AC encodes for Arg and Leu in positions 218 and 221, respectively, whereas the CRC to His218 and Pro221 in the uropathogenic oligotype GT (Figure 4, R2). Although these substitutions in the C-terminal domain do not take place near the nuclease active site, they could affect activity through protein structural effects.
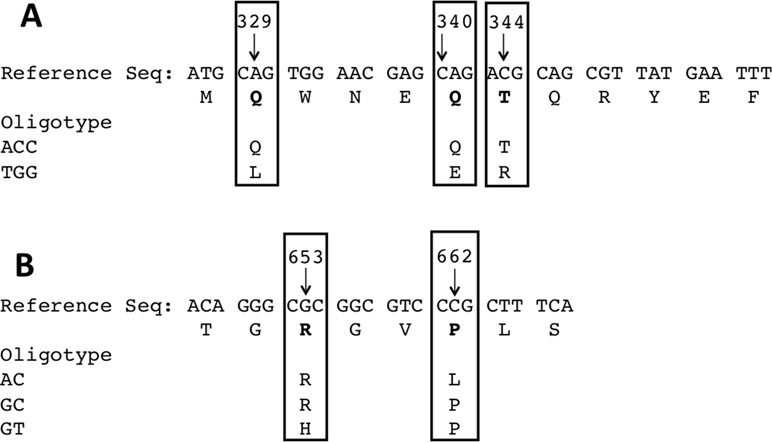
Figure 4 (A) Variability around positions 329, 340, and 344 result in amino acid substitutions along the protein sequence from Leu110/Glu114/Arg115 to Gln110/Gln114/Thr115. (B) Similarly, in the opposite end, the most abundant oligotype encodes Arg218/Leu221, whereas the oligotype most frequently seen in uropathogenic strains encodes His218/Pro221.
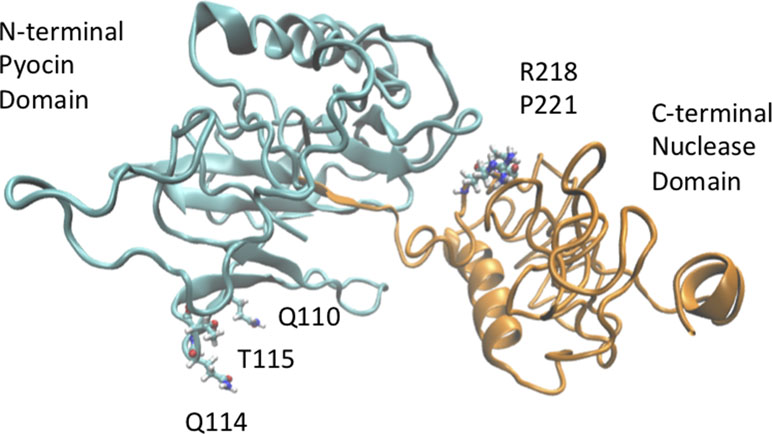
Figure 5 A three-dimensional model of USP was generated using i-TASSER (Eren et al., 2013) and visualized using VMD (Vogtmann et al., 2016) to show the location of possible sites of amino acid variation. Position 110 within the pyocin-like domain of USP (cyan domain) can be occupied either by a Leu or Gln residue. Similarly, positions 218 (Arg/His) and 221 (Leu/Pro) of the nuclease domain (green domain) are also hotspots of sequence variation.
Although the notion that sequence variation can have an effect on USP activity or phenotype remains to be validated, it is clear that there is enough variability in the usp gene to define bacterial lineages that could be further correlated with disease risk or clinical outcome.
Discussion
In previously published work, our group had reported the presence of several bacterial genes known to promote inflammation in stool samples from individuals with CRC (Gomez-Moreno et al., 2019). One of these genes was usp, which encodes a nuclease enzyme typically found in uropathogenic strains of E. coli. In this work, we explore the sequence variability for these genes in human stool and the possible association of specific sequence variants with disease. Interestingly, we found no significant variability in the sequences encoding tcpC or pks island genes, suggesting that at least in those regions that were amplified, only a single sequence or lineage exists within the population tested. By contrast, the usp gene had five main oligotypes, which could, if expressed, give rise to several USP enzyme variants. One of these variants, GT, was present in the clinical isolate that was used as a technical control, which was obtained from a patient with a urinary tract infection. Two samples contained this oligotype predominantly and both were CRC patients. Clearly, the establishment of a correlation between this particular lineage of usp and CRC will require a larger patient sample.
One limitation of the present study is the small number of stool samples that were analyzed, which makes statistical analysis very difficult. Since the original study was a pilot study, it involved a limited number of samples that had been carefully characterized in terms of demographics and diagnosis status (Gomez-Moreno et al., 2019). However, despite the limited number of samples in our study, our results are validated when they are compared with the published sequences in metagenomic (or shotgun) datasets (Vogtmann et al., 2016). The exact same sites of DNA sequence variation were found in the published datasets as were reported in this study. Also, there was a marginal association between the presence of one of the oligotypes, GT, and CRC, as observed in our experiments and confirmed in the published datasets (Figure 3). Our results are also consistent with earlier reports of usp sequences from isolated clinical strains in which they also report a number of sites of DNA sequence variation that coincide with the sites uncovered in this report (Nakano et al., 2001).
Another limitation of this report is that we only measure the sequence variability on a portion of the total sequence space for the genes under study. For instance, the tcpC gene is 924 base pairs long, but we are only amplifying a fragment of 283 base pairs, which was the only PCR product obtained from bacterial DNA purified directly from human stool as a template. We attempted different oligonucleotide combinations to yield longer PCR fragments, but those resulted in mixtures of unrelated DNA sequences that were difficult to deconvolute. Similarly, the usp gene is 1041 bases long, but we only were able to amplify a fragment that was 685 bases long and the coverage of the forward and reverse sequencing was roughly 300 bases on each end. Thus, it is possible that investigating the sequence variability of whole genes will require several amplification reactions to allow for the coverage of the entire gene sequence.
An unexpected finding was the sequence variability of DNA amplified from the individual bacterial clones that we used as controls. We expected these control strains to yield single oligotypes for each of the genes. However, a number of distinct oligotypes were detected for all of the fragments amplified from the control strains (Figures 1 and 3, leftmost bar). This apparently broad distribution of oligotypes in what should be a single clone could be due to a PCR incorporation error or to the formation of sequence chimeras (Haas et al., 2011; Schloss et al., 2011). Also, it cannot be ruled out that the sequence heterogeneity could stem from actual cellular diversification within a single colony, a phenomenon that has been observed as the presence of distinct phenotypes within a bacterial colony (Freeman et al., 1996; Nahku et al., 2011; Saint-Ruf et al., 2014).
Taken together, our results reveal the presence of sequence variants of the usp gene in patient samples, raising the possibility that some variants may have different activity and toxicity profiles.
Data Availability
The data supporting the conclusions of this manuscript is available in the the NCBI Bioproject database under Accession PRJNA551701.
Author Contributions
RG-M and AB-O designed the study. AB-O obtained the funding necessary for the study. RG-M performed the experiments. RG-M, RM-R, AR-L, KC-C, JP-S, and AB-O performed the data analyses and interpretation of the data. RM-R and AB-O prepared the final figures. All authors contributed in the preparation of tables and figures and in drafting the manuscript. Some of this work was published as part of the doctoral thesis of RG-M.
Funding
This work was supported in part by the National Institutes of Health grant R25GM061838 (NIGMS-RISE Program) to RGM. Oligonucleotides were purchased from an institutional facility supported by NIH Grant G12RR03051 (RCMI Program). Sequencing was carried out at the SGF facility, which is funded through the PR-INBRE program (NIGMS P20 GM103475). The work was also supported by the Center for Collaborative Research in Health Disparities Award Number G12 MD007600 from the National Institute on Minority Health and Health Disparities.
Conflicts of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the support of Dr. Dean Brenner and Dr. Ananda Sen from the University of Michigan and from the Early Detection Research Network (EDRN) and its Great Lakes/New England Clinical Validation Center for providing stool samples. The authors wish to thank Dr. A. Murat Eren at the University of Chicago for providing valuable initial guidance on the use of the Oligotyping software, Dr. Iraida E. Robledo at UPR School of Medicine for providing the control E. coli strain EC640, and Ms. Silvia Planas from the sequencing and genotyping facility for help with the writing of this manuscript. The work was also supported by the Puerto Rico Omics Center at the University of Puerto Rico Comprehensive Cancer Center.
References
Arthur, J. C., Perez-Chanona, E., Mühlbauer, M., Tomkovich, S., Uronis, J. M., Fan, T., et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123. doi: 10.1126/science.1224820
Boleij, A., van Gelder, M. M. H. J., Swinkels, D. W., Tjalsma, H. (2011). Clinical importance of Streptococcus gallolyticus infection among colorectal cancer patients: systematic review and meta-analysis. Clin. Infect. Dis. 53, 870–878. doi: 10.1093/cid/cir609
Bonnet, M., Buc, E., Sauvanet, P., Darcha, C., Dubois, D., Pereira, B., et al. (2014). Colonization of the human gut by E. Clin. Cancer Res. 20, 859–867. doi: 10.1158/1078-0432.CCR-13-1343
Brennan, C. A., Garrett, W. S. (2016). Gut microbiota, inflammation, and colorectal cancer. Annu. Rev. Microbiol. 70, 395–411. doi: 10.1146/annurev-micro-102215-095513
Eren, A. M., Maignien, L., Sul, W. J., Murphy, L. G., Grim, S. L., Morrison, H. G., et al. (2013). Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol. Evol. 4, 1111–1119. doi: 10.1111/2041-210X.12114
Eren, A. M., Sogin, M. L., Maignien, L. (2016). Editorial: New insights into microbial ecology through subtle nucleotide variation. Front. Microbiol. 7, 1318. doi: 10.3389/fmicb.2016.01318
Fisher, J. C., Eren, A. M., Green, H. C., Shanks, O. C., Morrison, H. G., Vineis, J. H., et al. (2015). Comparison of sewage and animal fecal microbiomes by using oligotyping reveals potential human fecal indicators in multiple taxonomic groups. Appl. Environ. Microbiol. 81, 7023–7033. doi: 10.1128/AEM.01524-15
Freeman, R., Sisson, P. R., Burdess, D. (1996). Heterogeneity within apparently pure cultures of Escherichia coli freshly isolated from significant bacteriuria. J. Med. Microbiol. 45, 349–352. doi: 10.1099/00222615-45-5-349
Gómez-Moreno, R., Robledo, I. E., Baerga-Ortiz, A. (2014). Direct detection and quantification of bacterial genes associated with inflammation in DNA isolated from stool. Adv. Microbiol. 4, 1065–1075. doi: 10.4236/aim.2014.415117
Gomez-Moreno, R., Gonzalez-Pons, M., Soto-Salgado, M., Cruz-Correa, M., Baerga-Ortiz, A. (2019). The presence of gut microbial genes encoding bacterial genotoxins or pro-inflammatory factors in stool samples from individuals with colorectal neoplasia. Diseases 7 (1), 16. doi: 10.3390/diseases7010016
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
Humphrey, W., Dalke, A., Schulten, K. (1996). VMD - Visual Molecular Dynamics. J. Molec. Graphics 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Kostic, A. D., Gevers, D., Pedamallu, C. S., Michaud, M., Duke, F., Earl, A. M., et al. (2011). Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298. doi: 10.1101/gr.126573.111
Mark Welch, J. L., Utter, D. R., Rossetti, B. J., Mark Welch, D. B., Eren, A. M., Borisy, G. G. (2014). Dynamics of tongue microbial communities with single-nucleotide resolution using oligotyping. Front. Microbiol. 5, 568. doi: 10.3389/fmicb.2014.00568
Martin, H. M., Campbell, B. J., Hart, C. A., Mpofu, C., Nayar, M., Singh, R., et al. (2004). Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer 1. Gastroenterology 127, 80–93. doi: 10.1053/j.gastro.2004.03.054
Martinez-Medina, M., Garcia-Gil, L. (2014). Escherichia coli in chronic inflammatory bowel diseases: an update on adherent invasive Escherichia coli pathogenicity. World J. Gastrointest. Pathophysiol. 5, 213–227. doi: 10.4291/wjgp.v5.i3.213
Nahku, R., Peebo, K., Valgepea, K., Barrick, J. E., Adamberg, K., Vilu, R. (2011). Stock culture heterogeneity rather than new mutational variation complicates short-term cell physiology studies of Escherichia coli K-12 MG1655 in continuous culture. Microbiology 157, 2604–2610. doi: 10.1099/mic.0.050658-0
Nakano, M., Yamamoto, S., Terai, A., Ogawa, O., Makino, S. I., Hayashi, H., et al. (2001). Structural and sequence diversity of the pathogenicity island of uropathogenic Escherichia coli which encodes the USP protein. FEMS Microbiol. Lett. 205, 71–76. doi: 10.1111/j.1574-6968.2001.tb10927.x
Nipic, D., Podlesek, Z., Budic, M., Crnigoj, M., Zgur-Bertok, D. (2013). Escherichia coli uropathogenic-specific protein, Usp, is a bacteriocin-like genotoxin. J. Infect. Dis. 208, 1545–1552. doi: 10.1093/infdis/jit480
Nougayrede, J. P., Homburg, S., Taieb, F., Boury, M., Brzuszkiewicz, E., Gottschalk, G., et al. (2006). Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848–851. doi: 10.1126/science.1127059
Raisch, J., Buc, E., Bonnet, M., Sauvanet, P., Vazeille, E., de Vallée, A., et al. (2014). Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J. Gastroenterol. 20, 6560–6572. doi: 10.3748/wjg.v20.i21.6560
Saint-Ruf, C., Garfa-Traoré, M., Collin, V., Cordier, C., Franceschi, C., Matic, I. (2014). Massive diversification in aging colonies of Escherichia coli. J. Bacteriol. 196, 3059–3073. doi: 10.1128/JB.01421-13
Schloss, P. D., Gevers, D., Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6, e27310. doi: 10.1371/journal.pone.0027310
Sears, C. L. (2009). Enterotoxigenic Bacteroides fragilis: a Rogue among Symbiotes. Clin. Microbiol. Rev. 22, 349–369. doi: 10.1128/CMR.00053-08
Swidsinski, A., Khilkin, M., Kerjaschki, D., Schreiber, S., Ortner, M., Weber, J., et al. (1998). Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology 115, 281–286. doi: 10.1016/S0016-5085(98)70194-5
Turlapati, S. A., Minocha, R., Long, S., Ramsdell, J., Minocha, S. C. (2015). Oligotyping reveals stronger relationship of organic soil bacterial community structure with N-amendments and soil chemistry in comparison to that of mineral soil at Harvard Forest, MA, USA. Front. Microbiol. 6, 49. doi: 10.3389/fmicb.2015.00049
Vineis, J. H., Ringus, D. L., Morrison, H. G., Delmont, T. O., Dalal, S., Raffals, L. H., et al. (2016). Patient-specific bacteroides genome variants in Pouchitis. mBio 7 (6), e01713–16. doi: 10.1128/mBio.01713-16
Vogtmann, E., Hua, X., Zeller, G., Sunagawa, S., Voigt, A. Y., Hercog, R., et al. (2016). Colorectal cancer and the human gut microbiome: reproducibility with whole-genome shotgun sequencing. PLoS ONE 11, e0155362. doi: 10.1371/journal.pone.0155362
Weir, T. L., Manter, D. K., Sheflin, A. M., Barnett, B. A., Heuberger, A. L., Ryan, E. P. (2013). Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE 8, e70803. doi: 10.1371/journal.pone.0070803
Wolter, D. J., Khalaf, N., Robledo, I. E., Vazquez, G. J., Sante, M. I., Aquino, E. E., et al. (2009). Surveillance of carbapenem-resistant Pseudomonas aeruginosa isolates from Puerto Rican Medical Center Hospitals: dissemination of KPC and IMP-18 beta-lactamases. Antimicrob. Agents Chemother. 53, 1660–1664. doi: 10.1128/AAC.01172-08
Yadav, M., Zhang, J., Fischer, H., Huang, W., Lutay, N., Cirl, C., et al. (2010). Inhibition of TIR domain signaling by TcpC: MyD88-dependent and independent effects on Escherichia coli virulence. PLoS Pathog. 6, e1001120. doi: 10.1371/journal.ppat.1001120
Keywords: oligotyping, gut bacteria, colorectal neoplasia, colorectal cancer
Citation: Gómez-Moreno R, Martínez-Ramírez R, Roche-Lima A, Carrasquillo-Carrión K, Pérez-Santiago J and Baerga-Ortiz A (2019) Hotspots of Sequence Variability in Gut Microbial Genes Encoding Pro-Inflammatory Factors Revealed by Oligotyping. Front. Genet. 10:631. doi: 10.3389/fgene.2019.00631
Received: 01 March 2019; Accepted: 17 June 2019;
Published: 09 July 2019.
Edited by:
Jesse Shapiro, Université de Montréal, CanadaReviewed by:
Juan Antonio Ugalde, uBiome, ChileJohn Frederick Pearson, University of Otago, New Zealand
Copyright © 2019 Gómez-Moreno, Martínez-Ramírez, Roche-Lima, Carrasquillo-Carrión, Pérez-Santiago and Baerga-Ortiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abel Baerga-Ortiz, YWJlbC5iYWVyZ2FAdXByLmVkdQ==; Josué Pérez-Santiago, am9wZXJlekBjY2N1cHIub3Jn
†These authors have contributed equally to this work.