 Shalaka Wahane
Shalaka Wahane Dalia Halawani
Dalia Halawani Xiang Zhou1,2
Xiang Zhou1,2 Hongyan Zou
Hongyan Zou- 1Nash Family Department of Neuroscience and Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Department of Orthopedics, Second Affiliated Hospital of Xi’an Jiaotong University, Xi’an, China
- 3Department of Neurosurgery, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Injury to the nervous system triggers a multicellular response in which epigenetic mechanisms play an important role in regulating cell type-specific transcriptional changes. Here, we summarize recent progress in characterizing neuronal intrinsic and extrinsic chromatin reconfigurations and epigenetic changes triggered by axonal injury that shape neuroplasticity and glial functions. We specifically discuss regeneration-associated transcriptional modules comprised of transcription factors and epigenetic regulators that control axon growth competence. We also review epigenetic regulation of neuroinflammation and astroglial responses that impact neural repair. These advances provide a framework for developing epigenetic strategies to maximize adaptive alterations while minimizing maladaptive stress responses in order to enhance axon regeneration and achieve functional recovery after injury.
Introduction
Compared to embryonic state or peripheral nervous system (PNS), adult neurons in mammalian central nervous system (CNS) have limited capacity to regenerate after axonal injury. In contrast, axon regeneration and neural circuit rewiring are more effective in non-mammalian nervous systems, which may reflect an evolutionary price paid in exchange for higher complexity in the mammalian CNS. Mounting evidence supports an important role of epigenetics in neurodevelopment and neurodegeneration (Qureshi and Mehler, 2018), however, our understanding of epigenetic regulation of stress responses and adaptive or maladaptive changes in PNS and CNS injury is still in its nascence (Palmisano and Di Giovanni, 2018).
Since coined by Waddington in the 1940s (Waddington, 1942), epigenetics has evolved in scope to the studies of gene expression changes without altering underlying DNA sequence. Initially referred to as heritable changes of gene expression, epigenetics has since been shown to be highly dynamic, involving writers, readers, and erasers that modify and interpret the epigenetic marks (Kouzarides, 2007). Histone modifications, DNA methylation, and non-coding RNAs are among the best studied, while new modalities continue to emerge. Histones modifications and DNA methylation affect local chromatin structure and protein–DNA interactions, thereby transcriptional output; while non-coding RNAs and modifications of messenger RNA (mRNA) affect RNA metabolism, such as mRNA processing, translation, and decay (Figure 1). Epigenetic mechanisms vastly increase the complexity of gene regulation, allowing cells to fine-tune transcriptional responses to the evolving profiles of intrinsic and extrinsic signals in homeostatic and pathological conditions.
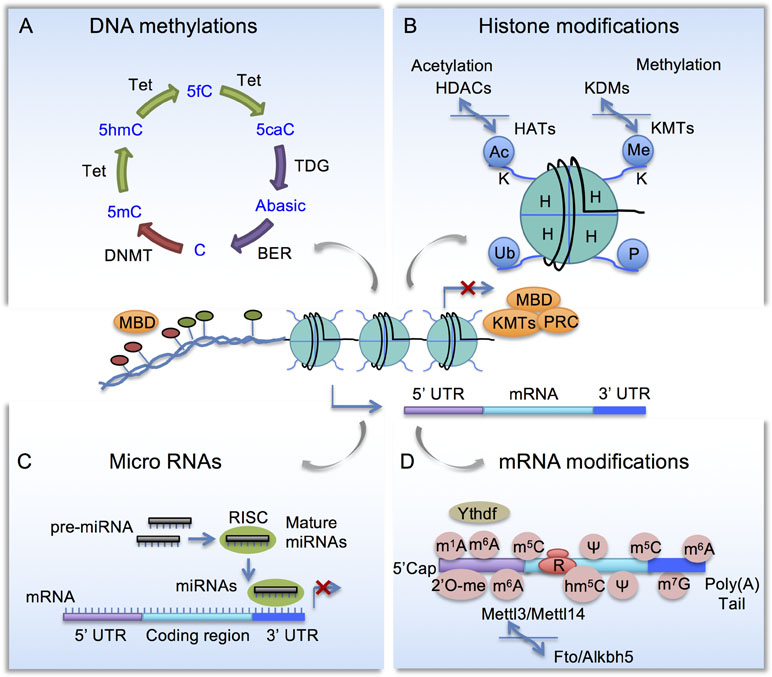
Figure 1 Schematic diagram of epigenetic mechanisms. (A) Cytosine methylation and demethylation process. DNMT, DNA methyltransferase; Tet, Ten-eleven translocation methylcytosine dioxygenase; BER, base excision repair enzymes; MBD, Methyl-CpG-binding domain (MBD) proteins. (B) Histone modifications. HAT, Histone acetyltransferases; HDAC, histone deacetylases; KMT, lysine methyltransferases; KDM, lysine demethylases; PRC, polycomb repressor complex. (C) Micro RNAs are embedded in a multi-protein complex termed RNA-induced silencing complex (RISC) and they repress gene expression by complementary binding to 3’ untranslated region (3’ UTR). (D) mRNA modifications include N6-methyladenosine (m6A), controlled by methyltransferase complex comprised of Mettl3, Mettl14 to install m6A, and demethylases Fto and Alkbh5 to remove m6A. m6A-binding proteins such as YTHDF regulates RNA metabolism.
Here, we summarize recent progress in understanding neural intrinsic and extrinsic epigenetic mechanisms that govern regenerative capacity of neurons and neuroinflammation and glial response after injury. We define specific epigenetic strategies used to enhance axon regeneration and functional recovery after injury. What emerged from these studies is the epigenetic plasticity in shaping both neuroregenerative and neuroinflammatory pathways, and their diverse roles, with some promoting while others hindering axon regeneration depending on the cell types and neuronal tissues that are most affected. What also comes to light is that pro-regenerative transcription factors (TFs) may require interactions with chromatin regulators to maximize the regenerative effect. Finally, epigenetic modifications also influence the inflammatory responses of microglia and astrocytes, and may be employed for neuroprotection and neural repair. Better understanding of the cell type-specific epigenetic plasticity might provide new avenues to harness its vast potential to promote long-lasting functional recovery after injury.
The Conditioning Paradigm of Sensory Neurons in Dorsal Root Ganglion
After neuronal maturation in the mammalian CNS, it is thought that the pro-axon growth gene program is turned off, and it remains inactivated even after axonal injury. Much effort therefore has been devoted to transcriptome profiling across developmental periods, across species, and PNS vs. CNS, in the hope to identify key pro-growth genes that can promote axon regeneration and functional recovery after CNS injury (reviewed in Blackmore, 2012). Sensory neurons in the dorsal root ganglion (DRG) are unique in that they share features with both PNS and CNS neurons. DRG neurons project a unipolar axon with a peripheral branch that innervates peripheral targets and a central branch that projects into the spinal cord (Figure 2). After axotomy, the peripheral, but not the central branch, can initiate robust axon regeneration (McQuarrie and Grafstein, 1973; Richardson and Issa, 1984; Smith and Pate Skene, 1997). Remarkably, peripheral axotomy switches DRG neurons into a growth state that enables not only peripheral but also central branch axon regeneration (Neumann and Woolf, 1999). This so-called conditioning lesion effect is transcription dependent (Smith and Pate Skene, 1997), thus presenting a unique opportunity to study pro-axon growth gene program and the underlying transcriptional mechanisms by comparing gene expression in DRG neurons after peripheral vs. central axotomy.
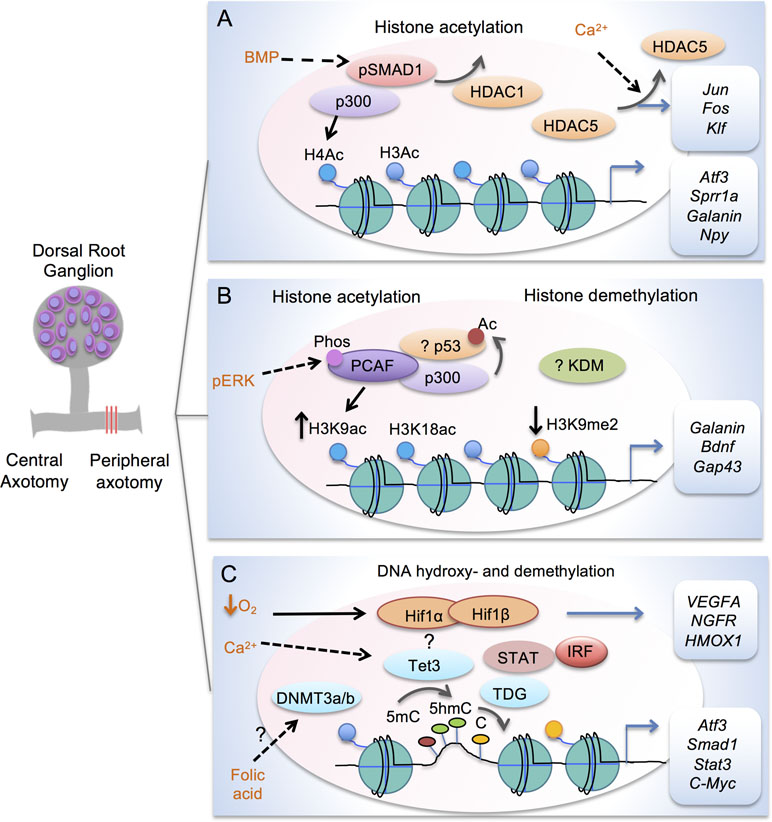
Figure 2 Transcription modules in regulating axon growth potential of DRG neurons after peripheral axotomy. (A) Schematic depiction of transcriptional module comprised of Smad1 and p300 to enhance histone acetylation in conditioned DRG at target loci (Atf3, Sprr1a, Galanin and Npy). Promoter occupancy of Smad1 helps recruitment of p300 and displacement of HDAC1. Peripheral axotomy also triggers nuclear exit of HDAC5 in response to retropropagation of calcium wave, leading to induction of RAGs (Jun, Fos, and Klf). (B) Transcriptional module comprised of p53 and p300/PCAF regulates H3K9ac and H3K18ac at target RAGs (Galanin, Bdnf, Gap43 and Coronin1b) in conditioned DRG. Acetylation of p53 is also increased by HATs in cortical neurons. Retrograde signaling of pERK results in threonine phosphorylation and nuclear localization of PCAF. In conditioned DRG, H3K9me2 is decreased regulated by a yet unknown KDM. (C) Epigenetic factors in mediating DNA hydroxy- and demethylation in conditioned DRG. Peripheral axotomy triggers Tet3 upregulation, which may be dependent on Calcium wave. Tet3 catalyzes conversion of 5mC to 5hmC, while TDG mediates conversion back to C, resulting in RAG induction of Atf3, Smad1, Stat3, and C-Myc. Folate may influence DNA methylation through DNMT3a/3b. Tet3 may partner with HIF, STAT, and IRF for 5hmC reconfigurations. Hypoxia stabilizes HIF complex to induce target genes, e.g. VEGFA, NFGR and HMOX1.
Earlier efforts have been focused on identifying and characterizing differentially regulated genes or regeneration-associated genes (RAGs) in the conditioning lesion paradigm of DRG through transcriptional profiling (Bonilla et al., 2002; Costigan et al., 2002; Michaelevski et al., 2010; Blesch et al., 2012). However, manipulating individual RAGs, such as ATF3 or STAT3, or simultaneously expressing multiple RAGs turned out insufficient to induce a full regenerative program (Seijffers et al., 2007; Bareyre et al., 2011; Fagoe et al., 2015). Clearly, axon regeneration is a complex process involving multiple RAGs that span diverse functional classes, thus understanding the transcriptional mechanisms, in particular epigenetic regulation, may lead to novel strategies to induce an entire class of RAGs. Indeed, several recent studies revealed that peripheral axotomy of DRG neurons elicits unique chromatin alterations with corresponding transcriptional responses (Cho et al., 2013; Finelli et al., 2013b; Puttagunta et al., 2014). In the following sections, we will review progress made in characterizing the multiple modalities of epigenetic mechanisms involved in regulating RAGs expression and regenerative capacity of conditioned DRG neurons, and when applicable, relate and extend the findings to CNS neurons.
Histone Modifications in Regulating Axon Growth Gene Program
Histone acetylation is one of the best-studied epigenetic mechanisms that regulate RAGs expression in conditioned DRG (Tables 1 and 2). The basic unit of chromatin is known as nucleosome, with DNA wrapping around an octamer of histones (two copies of H2A, H2B, H3 and H4 each). Post-translational modifications of N-terminal histone tails include acetylation, methylation, phosphorylation, ubiquitination, and deimination, etc., among which the best studied are lysine acetylation and lysine methylation (Huang et al., 2014) (Figure 1B). Histone acetyltransferases (HATs) add an acetyl group to a specific lysine residue of histone tails and histone deacetylases (HDACs) remove it. In mammalian cells, HAT members include three subgroups: the guanine nucleotide-binding protein subunit alpha transducin (GNAT) family, p300/CREB-binding protein (CBP), and the MYST-family, named after founding members MOZ, Ybf2, Sas2, and TIP60 (Lee and Workman, 2007). The HDAC family has 18 members, grouped into four classes based on structural differences and mechanisms of action (Kouzarides, 2007). In general, histone acetylation facilitates transcriptional activity by neutralizing the positive charge of lysine, leading to open chromatin and increased accessibility by transcriptional regulators, while histone deacetylation leads to chromatin compaction and gene silencing (Mellor, 2005). Histone methylation is catalyzed by lysine methyltransferases (KMTs), and demethylation by lysine demethylases (KDMs). Each member of the KMT (33 known members) and KDM families (22 known members) displays specificity for individual lysine residues as well as mono-, di- and/or trimethylation (Black et al., 2012). Histone tails can also be methylated at arginine residues by protein arginine methyl-transferases (PRMTs) and demethylated by Jumonji domain containing lysine demethylases (reviewed in Tewary et al., 2019). In contrast to histone acetylation, histone methylation can result in either transcriptional activation or repression depending on the identity of the targeted histone residues and the type of methylation. Collectively, these histone modifications (“histone codes”) are recognized by different reader proteins that contain specialized domains such as bromodomains and chromodomains (reviewed in Musselman et al., 2012).
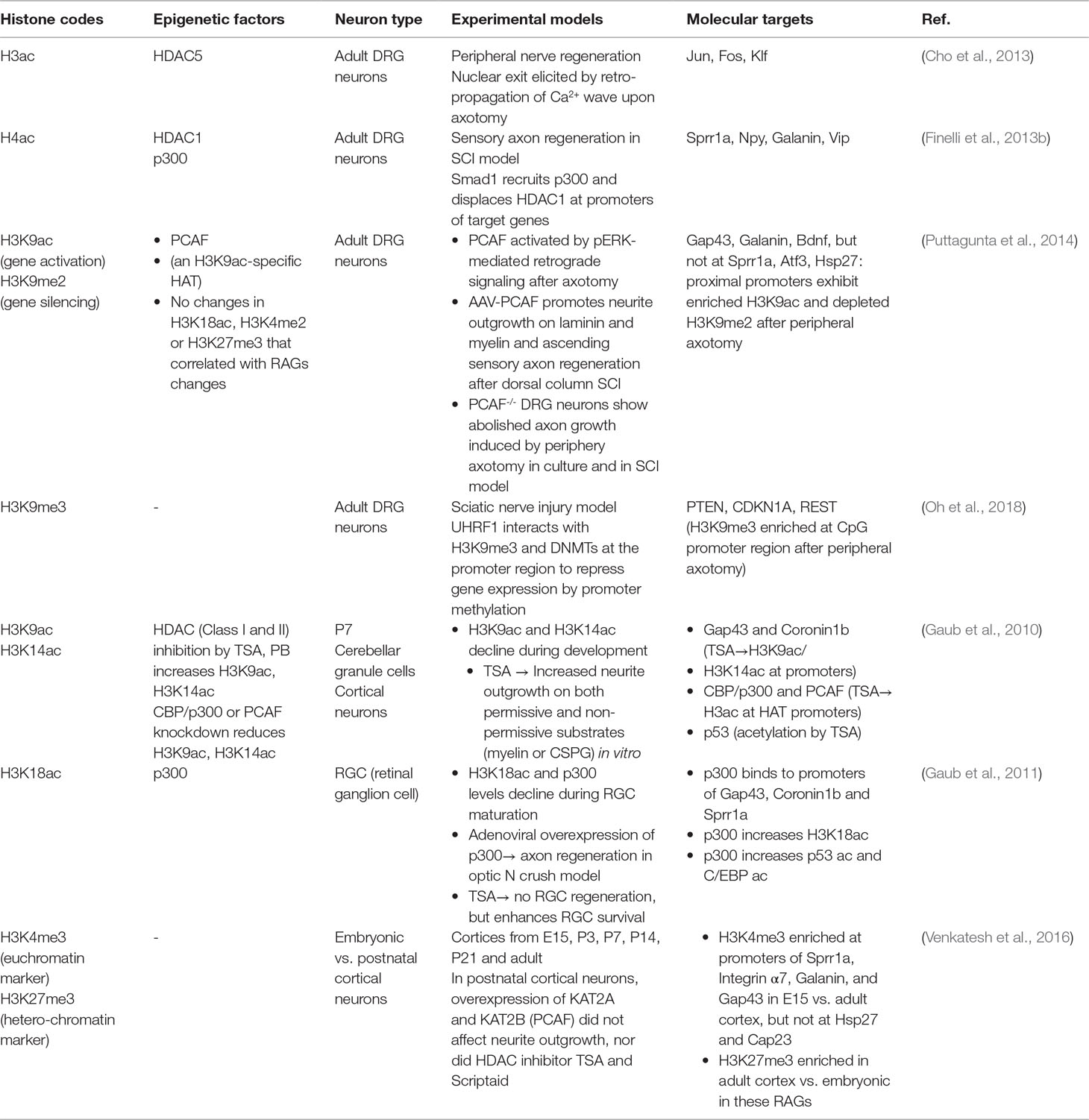
Table 1 Histone modifications associated with axon growth potential.
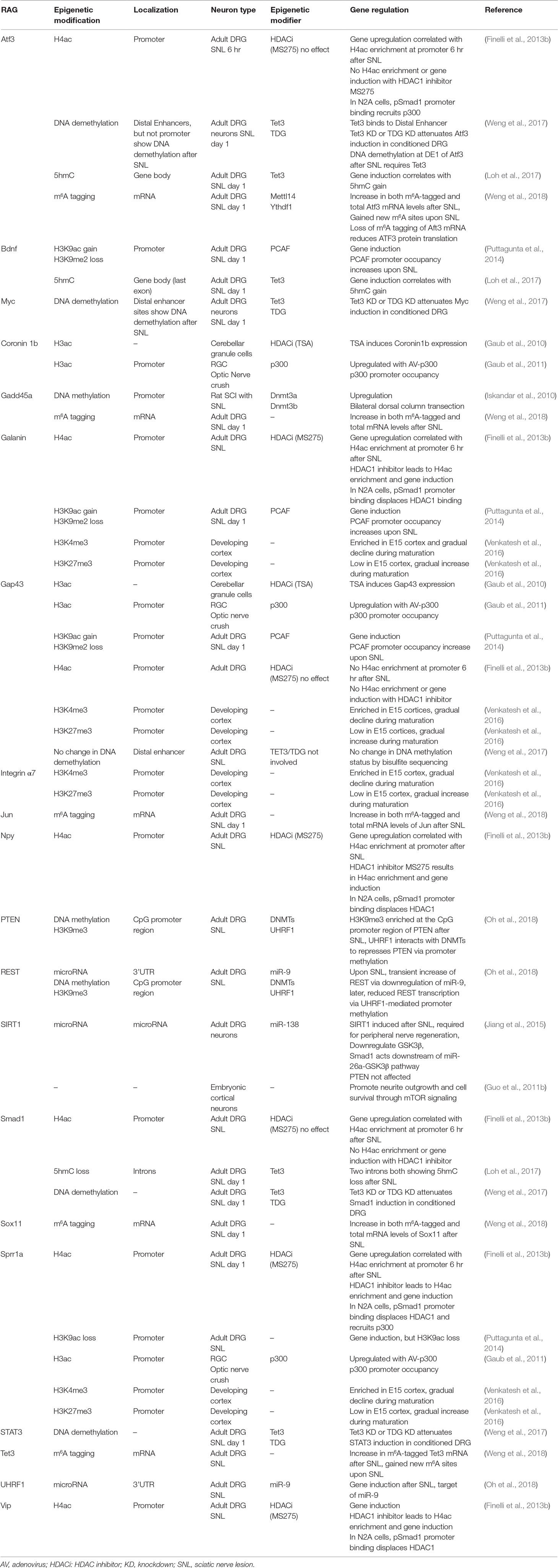
Table 2 Epigenetic regulation of RAG expression.
In the conditional lesion paradigm, it was found that peripheral but not central axotomy results in global enrichment of histone H3 and H4 acetylation (H3ac and H4ac) in DRG neurons (Finelli et al., 2013b; Puttagunta et al., 2014) (Figure 2A and Table 1). Functionally, elevating histone acetylation levels by HDAC inhibitors (HDACi) can induce multiple RAGs in DRG neurons. For instance, administrating MS275, a selective inhibitor of HDAC1, can orchestrate transcriptional change of ∼50% of RAGs examined, and significantly enhance axon growth potential and sensory axon regeneration in a dorsal column spinal cord injury (SCI) model (Finelli et al., 2013b). Moreover, in vitro, axotomy of DRG neurons triggers a retro-propagation of a calcium wave to the soma, which elicits nuclear export of HDAC5, leading to elevated H3 acetylation levels and RAGs induction (Cho et al., 2013).
The next question is which lysine residue modifications and which HAT/HDAC are specifically involved in transcriptional regulation of RAGs. Puttagunta et al. (2014) performed a comprehensive screen by chromatin immunoprecipitation (ChIP) in conditioned DRG to define lysine residue-specific histone modifications at proximal promoters of RAGs, including histone modifications known to correlate with active gene transcription (H3K9ac, H3K18ac, H3K4me2) or with gene silencing (H3K9me2, and H3K27me3). Only H3K9ac and H3K9me2 showed peripheral axotomy-triggered differential changes with consistent transcriptional changes of known RAGs, with H3K9ac enriched and H3K9me2 depleted at the promoter of Galanin, Bdnf and Gap43 (Figure 2B and Table 1). Consistently, ChIP analyses showed that PCAF, also known as K (lysine) acetyltransferase 2B (KAT2B), an H3K9ac-specific acetyltransferase, was enriched at these promoters. Overexpression of PCAF promoted neurite outgrowth from DRG neurons on both laminin and myelin substrates and enhanced ascending sensory axon regeneration in the dorsal column lesion SCI model. Additionally, retrograde transport of pERK after peripheral axotomy was required for PCAF phosphorylation and nuclear localization, thus providing a mechanism of how injury signals from axotomy are linked to nuclear events and chromatin modifications.
A recent study unveiled the involvement of CBP-mediated histone acetylation in the epigenetic reprogramming in proprioceptive DRG neurons triggered by enhanced neuronal activity through environmental enrichment, and this results in increased regenerative potential through RAGs induction (Hutson et al., 2019). Pharmacological activation of CBP/p300 enhances sprouting of both descending motor and ascending sensory axons leading to functional recovery in both mouse and rat SCI models (Hutson et al., 2019).
An important question is whether epigenetic mechanisms that are effective at promoting peripheral axon regeneration of DRG neurons may also work in CNS neurons. In the CNS, efforts to modulate chromatin acetylation status resulted in mixed findings (Table 1). For instance, Trichostatin A (TSA), a broad HDACi, enhanced neurite outgrowth in cultured neonatal cerebellar granule cells (CGNs) (Gaub et al., 2010), but not in retinal ganglion cells (RGCs) (Gaub et al., 2011). Similarly, HDACi alone failed to promote axon growth in postnatal cortical neurons (Venkatesh et al., 2016). These divergent findings indicate that the efficacy of HDACi may depend on the dynamic state of HATs and HDACs in neuronal subtypes. Indeed, DRGs demonstrate basal expression levels of p300 (Finelli et al., 2013b; Usoskin et al., 2015). In RGCs, p300 is developmentally regulated and its expression remained repressed after optic nerve injury (Gaub et al., 2011). Activation of the regeneration program following optic nerve injury requires p300 expression, which increases acetylation of both histone and non-histone target genes (i.e., H3K18, p53, and C/EBP) and RAGs induction (Gap43, Coronin1b, and Sprr1a) (Gaub et al., 2011). Likewise, in cortical neurons and CGNs, TSA can increase the expression of CBP/p300 and PCAF via H3K9ac and H3K14ac enrichment, which in turn mediate acetylation of histones and p53 to induce p53 target genes (Gap43 and Coronin1b) (Gaub et al., 2010). In cortical neurons and facial motoneurons, CBP/p300 works with p53 at promoters of RAGs such as Gap43 (Di Giovanni et al., 2006; Tedeschi et al., 2009). As summarized in Table 2, different RAGs display distinct histone modification patterns and engage different epigenetic modifiers in different neurons. For instance, promoters of Sprr1a and Atf3 showed elevated H4ac but not H3K9ac enrichment in conditioned DRG (Finelli et al., 2013b; Puttagunta et al., 2014).
One caveat of using HDAC inhibitors is that HDACs have non-histone substrates, so their inhibition may affect non-transcriptional pathways that are linked to axon growth. For instance, HDAC5, along with HDAC6 and SIRT2, also have cytoplasmic functions in deacetylating tubulins and microtubules and regulating neurite outgrowth (Cho and Cavalli, 2014). SIRT1 supports neurite outgrowth in the developing cortical neurons, reportedly through inhibiting mTOR signaling (Guo et al., 2011b). In summary, these findings stress the need to fine-tune epigenetic strategies for targeted chromatin remodeling to account for intrinsic cell-type differences in chromatin state and pro-regenerative gene programs.
DNA Methylation and Hydroxymethylation in Regulating Axon Regeneration Capacity
DNA methylation status, also known as methylome, refers to cytosine methylation (5mC) that occurs primarily on CpG dinucleotides (Figure 1A). CpG islands (CGI) are genomic areas of high CpG density, typically near gene promoters or enhancers. Generally, CGI are hypomethylated for actively transcribed genes, but heavily methylated for silenced genes. Methyl-CpG-binding domain (MBD) proteins preferentially recognize methylated DNA, serving as readers of the methylome (Klose and Bird, 2006). DNA methyltransferases (DNMTs) use S-adenosyl-L-methionine as methyl group donor to convert C to 5mC, with DNMT1 involved in maintenance of DNA methylation patterns during DNA replication (Smith et al., 1992), and DNMT3a and DNMT3b in de novo DNA methylation (Okano et al., 1999). Cytosine demethylation, on the other hand, occurs through an intermediate base, 5-hydroxymethylcytosine (5hmC), catalyzed by the Ten-eleven translocation (Tet) methylcytosine dioxygenase family (Tet1-3) (Wu and Zhang, 2010). Tet enzymes can iteratively oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxycytosine (5caC), which can then be removed by thymine DNA glycosylase (TDG)-mediated base excision repair pathway through Activation-induced cytidine deaminase (AID) or Apolipoprotein B mRNA editing enzyme catalytic (APOBEC) family proteins (Guo et al., 2011a; Hackett et al., 2013). 5hmC modifications are much more abundant in the CNS than in embryonic stem (ES) cells (Tahiliani et al., 2009; Song et al., 2011), and may have regulatory roles in its own right (Hahn et al., 2013). 5hmC is enriched in gene bodies that are highly expressed, suggesting a potential role for 5hmC in activating and/or maintaining gene expression (Song et al., 2011).
Earlier studies demonstrated that folate can regulate axon regeneration through DNA methylation (Iskandar et al., 2010). In our screen for differentially regulated epigenetic factors in adult DRG after peripheral axotomy, we identified Tet3 as specifically upregulated in conditioned DRG, along with elevated 5hmC levels (Loh et al., 2017). We then conducted a genome-wide survey of 5hmC distribution and dynamics in adult DRG under three different regenerative states: growth state after peripheral axotomy, naïve state with no injury, and a state refractory to axon regeneration after central axotomy (Loh et al., 2017). Genomic analyses revealed that a majority of differentially 5-hydroxymethylated regions (DhMRs) (∼55%) in DRG occur in gene bodies and ∼30% in intergenic regions; moreover, ∼90% of DhMRs were in “open sea” and less than 0.5% on CGIs. The relative importance of DNA hydroxy- or demethylation in CGIs vs. gene bodies in RAGs regulation and axon regeneration of DRG neurons vs. RGCs remains to be elucidated. New tools, such as APOBEC-coupled epigenetic sequencing (ACE-seq), a bisulfite-free method, now allows mapping of 5hmC at single-base resolution with low DNA input and without degrading DNA (Schutsky et al., 2018). Functional annotation of DhMR-associated genes showed that the most divergent functional categories were related to axon growth, neurites outgrowth, and neurite regeneration (Loh et al., 2017), and the top DhMR-associated pathways included phosphatase and tensin homolog (PTEN) and BMP signaling, both important for axon growth and regulation (Liu et al., 2010; Parikh et al., 2011; Finelli et al., 2013a). The epigenomic maps of 5hmC distribution revealed that conditioning lesion resulted in unique acquisition or loss of 5hmC modifications in about half of all RAGs. Unexpectedly, even though central lesion does not upregulate Tet3 expression, it also resulted in widespread 5hmC reconfigurations, but with little overlap with those triggered by peripheral axotomy. This suggests that the central lesion engages distinct 5hmC modifications, which may constitute a roadblock for regeneration. It is worth mentioning that earlier DNA methylation studies have been heavily focused on CGI at gene promoters, which may miss changes occurring elsewhere. Indeed, when DNA methylation microarrays were used to study promoter and CGI DNA methylation in conditioned DRG, only a modest number of genes, and none of the RAGs examined, displayed differential methylation between peripheral vs. central axotomy (Puttagunta et al., 2014).
At the same time, Weng et al. (2017) also discovered induction of Tet3 and elevated 5hmC levels in conditioned adult DRG neurons. Tet3 induction after peripheral axotomy may be dependent on a retrograde Ca2+ wave. Tet3 is required for 5hmC enrichment and induction of RAGs such as Atf3, Smad1, STAT3, and c-Myc, but not Gap43; and it binds to distal enhancer region of Atf3 and c-Myc (Figure 2C and Table 2). TDG knockdown attenuated induction of a similar set of RAGs, suggesting that complete DNA demethylation, not 5hmC increase alone, mediates RAGs induction to unlock axon growth potential of adult DRG neurons. Interestingly, while Tet3 is required for functional peripheral axon regeneration of DRG neurons, Tet1 is required for PTEN-deletion-induced optic nerve regeneration of RGCs (Weng et al., 2017), indicating that RAGs expression may require cell-type specific DNA demethylation gene pathways.
Besides regulating DNA demethylation, Tet3 can also recruit O-linked β-N-acetylglucosamine transferase (OGT) to chromatin (Ito et al., 2014), and OGT catalyzes the addition of N-acetylglucosamine to serine/threonine residues of many proteins, including histones; OGT deletion in DRG neurons results in decreased axonal growth (Su and Schwarz, 2017).
A recent study unveiled that DNA methylation, which generally leads to gene silencing, also contributes to regenerative responses of conditioned DRG neurons (Oh et al., 2018). Treatment with RG108, a direct DNMT inhibitor, reduces peripheral nerve regeneration in a sciatic nerve injury model. Periphery axotomy induces ubiquitin-like containing PHD ring finger 1 (UHRF1) and a transient increase of transcriptional regulator RE1 silencing transcription factor (REST), both downstream targets of miR-9. UHRF1 recognizes methyl groups on histone H3, specifically H3K9me3, and recruits DNMTs to promoter region to silence gene expression of PTEN and restrict REST expression via promoter methylation, leading to enhanced peripheral axon regeneration (Oh et al., 2018). REST upregulation in DRG also contributes to the transition from acute to chronic pain after peripheral nerve injury through repressed expression of Chrm2, a muscarine cholinergic receptor (Zhang et al., 2018a). Together, these studies demonstrate the complex role of DNA (de)methylation pathways in blocking or promoting axon regeneration, depending on target genes. The dynamics of DNA methylation and hydroxymethylation and their roles in CNS axon regeneration are poorly understood. The challenge is to define loci-specific changes in 5C, 5mC and 5hmC patterns and their functions in governing axon regenerative capacity in specific subtypes of CNS neurons. It remains to be seen whether the identification of critical genomic loci may enable the utilization of Cas9-directed DNA methylation editing to enhance axon regeneration in a cell-specific manner (Liu et al., 2016).
microRNAs in Regulating Intrinsic Axon Growth Potential
MicroRNAs (miRNAs) are a class of small (∼22 nucleotides) noncoding RNAs that regulate post-transcriptional gene expression (Bartel, 2004). miRNAs are embedded in a multi-protein complex termed RNA-induced silencing complex (RISC) and bind in a complementary fashion to the 3’ untranslated region (3’ UTR) of mRNAs and suppress protein expression by preventing translation and/or promoting mRNA degradation (Figure 1C). Each individual miRNA has multiple targets, thus capable of concurrently modulating expression of multiple genes. Each cell type in the CNS has a distinct miRNA profile that contributes to the specification and maintenance of distinct neuronal and glial phenotypes (Jovičić et al., 2013).
In animals with deletion of Dicer, which is critical for microRNA processing, sensory neurons show impaired axon regeneration (Wu et al., 2012). Gene profiling revealed differential expression of many microRNAs in adult DRG neurons after peripheral axotomy (Strickland et al., 2011; Zhang et al., 2011; Zhou et al., 2011). For instance, miR-138 controls axon growth of adult DRG neurons through a mutual negative feedback loop with SIRT1, an NAD-dependent HDAC (Liu et al., 2013) (Table 3). In naïve DRG neurons, baseline level of miR-138 is high to inhibit axon growth through suppression of SIRT1 expression. Peripheral axotomy leads to SIRT1 upregulation, which represses miR-138 transcription; this in turn results in sustained elevation of SIRT1 and enhanced axon regeneration in a sciatic nerve injury model (Liu et al., 2013). In another study, miR-26a was found to be required for axon regeneration of adult DRG sensory neurons in vitro and in a sciatic nerve injury model (Jiang et al., 2015) (Table 3). MiR-26a targets specifically glycogen synthase kinase 3β (GSK3β), and Smad1 acts downstream of miR-26a-GSK3β pathway to control sensory axon regeneration.
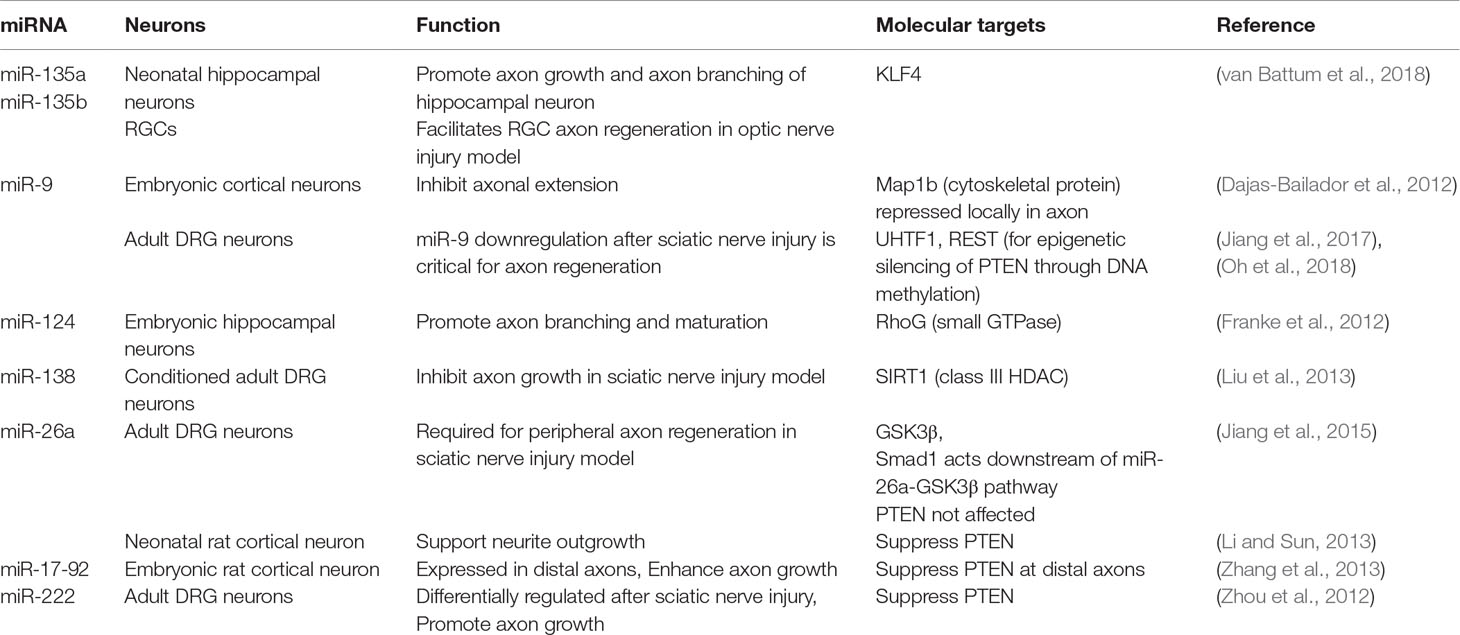
Table 3 miRNA in regulating axon growth and regeneration.
Recent studies reveal a large number of microRNAs with regulatory roles for axonal outgrowth, including miR-9, 17-92, 21, 26a, 30b, 133b, 135a, 135b, 138, 210, 222, and 431 (Table 3, also see review in Weng et al., 2016). For instance, in embryonic cortical neurons, miR-9 inhibits axon extension while augmenting axon branching through translational repression of microtubule-associated protein 1b (Map1b) locally in axon (Dajas-Bailador et al., 2012). In adult DRG neurons, peripheral axotomy results in downregulation of miR-9, which is critical for axon regeneration (Jiang et al., 2017). As mentioned above, REST and UHRF1 are both downstream targets of miR-9, and UHRF1 enhances regenerative capacity of DRG neurons through gene silencing of PTEN and REST via promoter methylation, thus providing an example of interplay between different epigenetic mechanisms (Oh et al., 2018). In embryonic hippocampal neurons, CNS-specific miR-124 promotes axonal branching through suppressing the small GTPase RhoG (Franke et al., 2012). Van Battum et al. recently performed an miRNome-wide functional screen of >1000 miRNAs in a neuronal cell line, SH-SY5Y cells, and identified miR-135a and miR-135b as promoters of axon growth. The promoting effect of these microRNAs were confirmed in primary hippocampal neurons. Intravitreal injection of miR-135s facilitated RGC axon regeneration after optic nerve injury. KLF4, an inhibitor of axon regeneration, was identified as a target of miR-135 (van Battum et al., 2018).
To relate the findings in different neurons is challenging as different microRNAs exert different functions and affect different target genes in a cell-type specific manner, with some promoting while others inhibiting axon growth. One example is PTEN: while PTEN reportedly was not affected by miR-26a in adult DRG neurons; but in neonatal rat cortical neurons, miR-26a supported neurite outgrowth by repressing PTEN expression (Li and Sun, 2013) (Table 3). Interestingly, in adult sensory neurons, miR-222 is shown to target PTEN and promotes axon growth (Zhou et al., 2012), while in embryonic rat cortical neurons, the miR-17-92 cluster enhances axon growth through targeting PTEN (Zhang et al., 2013). Clearly, PTEN is regulated by different miRNAs in a cell-type specific manner. Furthermore, miRs are under the regulatory control of long noncoding RNA (LncRNA), which can function to decoy or “sponge” miRs to limit their function in vivo (Ebert and Sharp, 2010). Future characterization of LncRNA-miRNA-mRNA networks in regenerating neurons may provide new avenues to enhance axon growth potential in injured tissue.
Epitranscriptomic Regulation of Axon Regeneration
In analogy to DNA modifications, various covalent modifications are also present on RNA transcripts (Figure 1D). Epitranscriptomic modifications of RNA can influence stability, translation, and non-coding RNA function, thus adding an additional layer of complexity to gene regulation (Zhao et al., 2017). RNA modifications include N6-methyladenosine (m6A), 5-methylcytosine (m5C), N1-methyladenosine (m1A) and pseudouridine (ψ), among others, with m6A being the most abundant in eukaryotic cells, present in over 25% of human mRNAs and being enriched in long exons and near transcription start sites and stop codons (Gilbert et al., 2016; Li et al., 2016). Nearly every gene gives rise to both methylated and unmethylated mRNA (Molinie et al., 2016). In mammals, Mettl3, Mettl14, and other components form a methyltransferase complex that install m6A marks, while demethylases Fto and Alkbh5 remove m6A marks (Zhao et al., 2017). m6A modifications play diverse roles in stem cell function, development, and neuronal physiology by regulating mRNA processing, translation, and decay, through diverse m6A-binding proteins, for example, YT521-B homology domain family (Ythdf) proteins (Zhao et al., 2017).
The role of RNA modifications in CNS injury is largely unknown. A recent study linked m6A methylation to axon regeneration in conditioned DRG neurons (Weng et al., 2018). Peripheral axotomy elevated m6A-tagged mRNA levels in adult DRG neurons, and genome-wide profiles of m6A-tagged mRNA showed that the most enriched gene ontology terms involve translation and metabolism-related processes. A large number of RAGs (129 out of 304 examined) also exhibited significant m6A tagging in conditioned as compared to naïve DRG 24 hr after axotomy, including Atf3, Sox11, Gadd45a, and Tet3 (Weng et al., 2018) (Table 2). Interestingly, single-base mapping of m6A-tagging revealed dynamic changes at m6A-tagged sites, with some transcripts exhibiting a gain or loss of m6A sites across the 5’ UTR, coding regions, and 3’UTR, while other transcripts displaying region-specific changes. The transcripts with newly added m6A site were enriched for genes involved in axonal regulation, while transcripts encoding protein translation machinery components showed elevated m6A-tagged transcript levels, but not new m6A sites. Conditional deletion of Mettl14, a core unit of the mammalian m6A methyltransferase complex, resulted in reduced m6A levels in adult DRGs. Mettl14 and m6A-binding protein YTHDF1 were both required for peripheral axon regeneration in a sciatic nerve injury model (Weng et al., 2018). Finally, in the CNS axon regeneration model of optic nerve injury, PTEN-deletion-induced axon regeneration also required Mettl14 (Weng et al., 2018). Given the high prevalence of RNA modifications, a large amount of work is needed to map out epitranscriptomic changes and define their functional significance in regulating RAGs expression and regenerative capacity in different types of neurons.
Regeneration-Associated Transcriptional Modules for Pro-Axon Growth Gene Programs
In addition to chromatin modifications, full scope activation of the regenerative gene program requires induction of key TFs, which help to recruit chromatin remodelers to specific genomic loci to modify DNA or histones. Indeed, it has been shown that co-expressed genes with coherent functions and similar transcriptional timing and levels may be co-regulated by transcriptional modules consisting of TFs and chromatin remodelers that collaborate with one another (Ram et al., 2011; Ernst et al., 2012). In this regard, TF binding to promoter elements of target genes helps to recruit chromatin modifiers to alter local chromatin structure, which in turn increases TF accessibility. Conversely, chromatin compaction precludes interaction between TFs and target loci. In the context of nerve injury and axon regeneration, specific transcriptional modules may be activated to regulate co-expressed genes for stress response, metabolic adaption, and axon regeneration, etc.
Our laboratory has elucidated one such transcriptional module consisting of HDAC1/p300 and Smad1 that regulates axon growth potential in DRG neurons through histone acetylation (Figure 2A). In embryonic DRG neurons, Smad1-mediated bone morphogenetic protein (BMP) signaling is active and is required for developmental axon growth (Finelli et al., 2013a). Turning down BMP/Smad1 signaling contributes to an age-associated decline of the axon growth potential (Zou et al., 2009; Parikh et al., 2011). Smad1 collaborates with p300 and HDAC1 for RAGs induction: promoter occupancy by Smad1 helps to recruit p300 and displaces HDAC1 at target loci, leading to histone acetylation, and this in turn increases Smad1 promoter binding (Finelli et al., 2013b). Reactivating developmentally regulated transcriptional modules may enable coordinated induction of a large network of pro-growth genes (Weng et al., 2016). Indeed, in the case of HDAC1/Smad1 signaling, both activating BMP/Smad1 signaling in adult DRG neurons by intrathecal delivery of adeno-associated virus 8 (AAV8)-BMP4 and elevating histone acetylation levels by HDAC1 inhibitor MS275 lead to enhanced axon growth potential and sensory axon regeneration in mouse models of SCI (Parikh et al., 2011; Finelli et al., 2013b). It remains to be seen whether combinatorial administration of AAV-BMP4 and MS275 can synergistically promote sensory axon regeneration in SCI models.
How Tet3 is targeted to specific regions of RAGs upon peripheral axotomy of DRG is also unsolved at this point, but may require collaboration with TFs as implicated in the genome-wide 5hmC mapping of conditioned DRG (Loh et al., 2017). Using bioinformatics analyses, we identified enriched TF binding motifs at DhMRs, including HIF (hypoxia-inducible factor), STAT, and IRF families, and these TFs may function as transcriptional coregulators to assist Tet3 for 5hmC modifications (Figure 2C). Notably, in DRG neurons, HIF1α has been shown to regulate multiple RAGs, and its activation is necessary and sufficient to promote peripheral axon regeneration in a sciatic nerve injury model (Cho et al., 2015). In C. elegans, hif-1 mutants show reduced regeneration (Nix et al., 2014). Ongoing work is being conducted to define transcriptional modules composed of Tet3 and TFs such as HIF1α that regulate RAGs through 5mhC modifications. HIF can also form complexes with transcriptional coactivator CBP/p300 (Lisy and Peet, 2008) or chromatin-remodeling complexes (Dekanty et al., 2010). Indeed, HIF1α is required for injury-induced histone H3 acetylation (Cho et al., 2015).
Consistent with the notion of regeneration-associated transcriptional modules, system-level transcriptional network analysis revealed tight co-expression of transcriptional regulators after peripheral axotomy of DRG neurons, which in turn coordinate activation of the regenerative gene program (Chandran et al., 2016). Additionally, transcriptional profiling across early stages of PNS regeneration revealed a clear cascade of target genes activation after axonal injury, starting with a wave of chromatin modification followed by activation of RAGs (Li et al., 2015).
The importance of collaboration between TFs and chromatin remodelers for RAGs induction has been further supported by the finding that epigenetic constraints might actually limit the full efficacy of pro-regenerative TFs (RAG-TFs) even in the setting of overexpression. In a study of ascending sensory axon regeneration after SCI, AAV5 was used to deliver ATF3 alone or in combination with c-JUN, STAT3C (a dominant active mutant form of STAT3) and Smad1-EVE (a constitutively active phospho-mimetic form of Smad1) into lumbar L4 and L5 adult rat DRG (Fagoe et al., 2015). These four RAG-TFs each individually contributes to regenerative axon growth of DRG neurons after peripheral axotomy. It was found that central axon regeneration of DRG neurons was enhanced with ATF3 expression, but no synergistic effect was observed when expressing all the four TFs as compared to ATF3 alone. Thus, overexpressing multiple RAG-TFs was still insufficient for full scope activation of the regeneration program. In another study using dorsal hemisection SCI model, descending corticospinal tract (CST) regeneration was compared with overexpression of SOX11, VP16-KLF7 (addition of a VP16 activation domain), VP16-STAT3, or c-JUN (Venkatesh et al., 2018). While SOX11 and KLF7 stimulated axon regeneration from CST motoneurons, STAT3 and c-JUN did not. It was revealed that the chromatin accessibility of the predicted STAT3 and JUN target genes was lower than that of SOX11 and KLF7 target genes. Thus, to be effective, RAG-TFs either need to have their binding sites sufficiently accessible, or they must be capable of recruiting chromatin regulators to modify chromatin accessibility. Venkatesh et al. (2018) further proposed that a collaboration between pioneer TFs and RAG-TFs might be needed to achieve full efficacy. Pioneer TFs are capable of binding to compacted chromatin and initiate a cascade of molecular events to reshape the chromatin landscape (Zaret and Carroll, 2011; Zaret, 2018). Computational algorithms based on genome-wide chromatin accessibility datasets have recently been developed for chromatin opening index/pioneer index of TFs (Sherwood et al., 2014; Lamparter et al., 2017). Venkatesh et al. went on to use the pipeline to predict pioneer factors that could potentially relieve chromatin constraints for STAT3 or c-JUN target genes. In future studies, it will be exciting to test experimentally whether the rational design of combinatorial activation of TFs and epigenetic factors or pioneering TFs is capable of reversing the seemingly unamendable chromatin constrains of RAGs in adult CNS neurons, leading to synergistic regenerative effect.
Developmental Decline of Axon Growth Potential and Associated Progressive Change of Chromatin Landscape
The regenerative state in adult neurons frequently involves reactivating the developmental program. During neurodevelopment, as embryonic neurons establish axon wiring and synapses mature, a developmental decline in intrinsic axon growth potential occurs (Liu and Snider, 2001; Moore et al., 2009; Parikh et al., 2011; He and Jin, 2016; Tedeschi and Bradke, 2017). During this process, pro-axon growth genes are turned off and growth-inhibitory genes are upregulated. It is now clear that this developmental transition is accompanied by progressive changes of the chromatin landscape to ensure stable gene expression after neuronal maturation (Venkatesh et al., 2018). Unfortunately this restrictive chromatin status becomes an epigenetic barrier for reactivating the pro-growth gene networks, thus finding ways to unlock the epigenetic blockade promises to rejuvenate the growth competence in adult neurons (Wong and Zou, 2014).
Early insights into the epigenetic barriers for regeneration in mature neurons came from studies tracking the changes in histone acetylation levels across developmental stages. In cortical neurons and CGNs, global levels of H3K9ac and H3K14ac decline during development, while in RGCs, H3K18ac level declines as neurons mature (Gaub et al., 2010; Gaub et al., 2011) (Table 1). Consistently, chromatin accessibility at promoter regions of several RAGs displays developmental decline during cortical maturation, as evidenced by depletion of H3K4me3, a marker for euchromatin, and enrichment of H3K27me3, a marker for heterochromatin (Venkatesh et al., 2016) (Table 1). The recent development of the ATAC-seq method (assay for transposase-accessible chromatin using sequencing) allowed direct measurement of chromatin accessibility (Buenrostro et al., 2013), and demonstrated a genome-wide shift in chromatin accessibility in adult mouse cortex as compared to neonatal cortex, with inaccessible regions being highly enriched for gene ontology terms related to axon growth and guidance, cell-cell adhesion and GTPase signaling (Venkatesh et al., 2018). Hence, from a developmental perspective, once axon wiring is completed, chromatin remodeling serves as a safeguard mechanism to restrict accessibility of pro-growth genes. Identifying the underlying epigenetic mechanisms may provide an avenue to promote regeneration through reactivation of developmental programs.
The epigenetic factors that regulate the developmental changes in chromatin landscape are still being investigated. p300 has been shown to be developmentally regulated in RGCs and its expression remains repressed after optic nerve injury (Gaub et al., 2011). By comparison, the developmentally regulated TFs that control intrinsic axon growth capacity comprise an ever expanding list, including ATF3, c-JUN, CREB, Krüppel-like family of TFs (KLFs), Smad1, Sox11, and STAT3 (reviewed in Weng et al., 2016). Many of these TFs also regulate intrinsic growth competence in adult CNS neurons, including Smad1 (Finelli et al., 2013a), c-JUN and KLFs. KLF-based manipulations improved axon regeneration of RGCs after optic nerve injury and CST motor neurons after SCI (Moore et al., 2009). A comprehensive comparison has been conducted across four transcriptomic datasets to identify converging intrinsic axon growth modulators, which identified c-JUN as a common TF to all four datasets, where as ATF3 and members of the KLF and Smad families were found in three datasets and Yy1, Fos, Egr1, Isl2, and members of the STAT, Sox, and Gata families were shared by two datasets (Blackmore, 2012). Whether these developmentally regulated TFs collaborate with epigenetic factors to form pro-growth transcriptional modules remains to be investigated, particularly in the context of CNS neurons.
Signaling Pathways in Organizing Transcriptional Hierarchy of Regenerative Gene Program
Many signaling pathways that regulate axon growth potential have been identified by functional genetic screens, proteomic approaches, and system-level analyses, including PTEN/mTOR, JAK/STAT, and DLK/JNK pathways (Liu et al., 2010; Belin et al., 2015; Chandran et al., 2016) (also see review in Lu et al., 2014). A recent in vivo screen searched for modulators of axon sprouting after SCI by comparing the transcriptomes of intact CST motor neurons that remained quiescent versus those that exhibited injury-induced sprouting, which revealed a set of pro-axon growth pathways including HIPPO signaling, mTOR signaling, and the 3-phosphoinositide degradation pathway (Fink et al., 2017). Signaling pathways might play an active role in organizing transcriptional hierarchy to reestablish axon growth competence (Weng et al., 2016). For instance, ERK-mediated retrograde signaling is required for PCAF-mediated histone acetylation at RAG promoters (Puttagunta et al., 2014) (Figure 2B). One important area of research is to examine whether signaling pathways act in concert with chromatin remodelers and RAG-TFs. A recent review discussed the striking convergence of disparate regeneration-associated biological processes on chromatin accessibility (Danzi et al., 2018). For instance, PTEN deletion promotes axon regeneration, which has been attributed to enhanced mTOR signaling (Park et al., 2008). Interestingly, PTEN can also directly interact with histone H1, and PTEN deletion prevented binding of H1 to DNA and increased H4K16ac, thereby promoting chromatin accessibility (Chen et al., 2014). PTEN also antagonizes insertion of histone variant H3.3, which is associated with transcriptional activity and chromatin accessibility (Benitez et al., 2017). Furthermore, neuronal activity-induced axon growth competence may also involve increased chromatin accessibility, e.g. at AP-1 motif (Su et al., 2017).
It is important to bear in mind that pro-growth epigenetic factors and signaling pathways may not only induce growth-promoting genes, but also suppress growth-restrictive genes. For instance, both PTEN and KLF4 suppress neurite outgrowth in RGCs (Park et al., 2008; Moore et al., 2009). Interestingly, inhibition of S6 kinase I (S6K1), a prominent mTOR target, in fact promotes neurite outgrowth of primary hippocampal neurons, as well as CST regeneration in a dorsal hemisection SCI model in vivo, and this occurs through activation of PI3K/mTOR signaling by way of a negative feedback mechanism (Al-Ali et al., 2017).
Glial Epigenetics in Tissue Repair After CNS Injury
The glial cell compartment, which make up to ∼90% of cells in the CNS, has received little attention in regards to epigenetics in shaping their injury responses. CNS injury triggers a complex, multiphasic glial responses, with both beneficial and detrimental effects (reviewed in Staszewski and Prinz, 2014). Specific patterns of chromatin modifications are initiated in different glial cell types as a result of complex interactions between the type of injury, injury microenvironment, intrinsic factors, and the epigenome unique to each cell types.
Epigenetic Regulation of Neuroinflammation
Inflammatory responses in the CNS are mediated by resident microglia, blood-borne macrophages and many other immune cell types, such as neutrophils, T cells, and B cells, as well as astrocytes (Figure 3). Following injury, resident microglia are activated within minutes, and blood-borne monocytes infiltrate the injury site to differentiate into phagocytic macrophages, which share similar morphology and cell-surface markers, and together they constitute the innate immunity (Beck et al., 2010). In response to injury signals, microglia/macrophages can adopt extensive functional heterogeneity with multitude of roles, including phagocytosis, debris clearing, release of inflammatory mediators, promoting angiogenesis, and supporting neuronal survival (reviewed in Garden, 2013). Each of these tasks engages unique gene networks regulated by distinct epigenetic mechanisms, which may serve as useful therapeutic targets for immunomodulation after CNS injury.
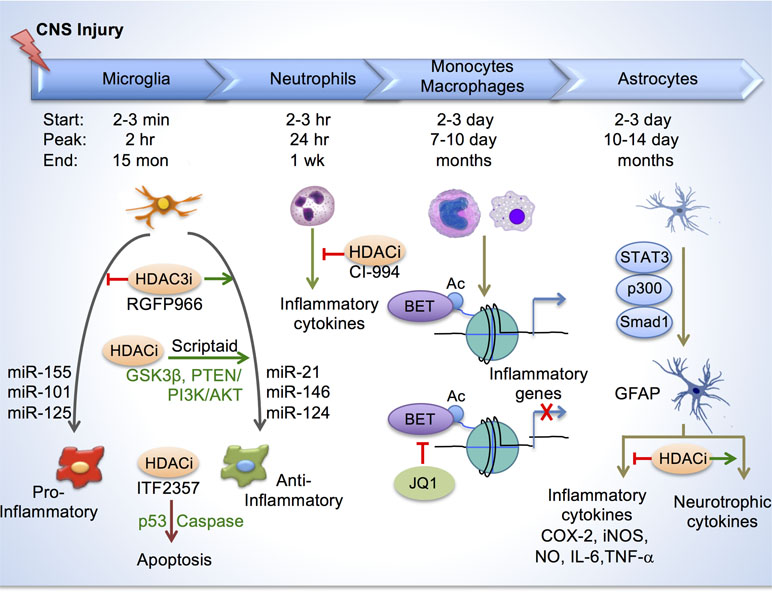
Figure 3 Histone acetylation in regulating glial response after CNS injury. Top, depiction of timelines of activation of different immune cells and astroglia at the injury site after CNS injury. Bottom, in microglia/macrophages, HDAC inhibition reduces inflammation by enhancing anti-inflammatory/pro-repair phenotype and by inducing apoptosis through p53 and caspase. Different micro-RNAs also regulate inflammatory phenotypes of microglia/macrophage. HDAC inhibition by CI-994 suppresses neutrophil accumulation and reduces inflammatory cytokine expression. BETs are epigenetic readers of acetylated histones and promote transcription of inflammatory genes. BET inhibitor JQ1 reduced inflammatory cytokine expression and leukocyte recruitment to the injury site. In astrocytes, transcriptional module consisting of STAT3, p300 and Smad1 induces GFAP expression. HDAC inhibition reduces secretion of inflammatory cytokines, and increases neurotrophic cytokines from reactive astrocytes.
Inflammatory responses compose a spectrum of macrophage activation phenotypes, ranging from a pro-inflammatory, classical activation state (also known as M1 polarization) to an anti-inflammatory, alternative activation state (M2 polarization)(Martinez and Gordon, 2014). M1 polarization is mediated by type 1 T-helper (Th1) cytokines such as interferon-gamma (IFNγ) and Toll-like receptor (TLR) agonists such as lipopolysaccharides (LPS), and is associated with cytotoxic tissue injury. In contrast, M2 polarization is induced by type 2 T-helper (Th2) cytokines such as interleukin-4 (IL-4) and interleukin-13 (IL-13), and is thought to promote tissue repair (Figure 3). While both differentiation states have been reported in CNS injury (Kigerl et al., 2009), the epigenetic mechanisms regulating these activation states and the degree of plasticity between different states are still being investigated.
Histone acetylation plays a critical role in shaping the inflammatory responses of microglia/macrophages. Broad spectrum HDACi have been used to demonstrate the overall impact on improving functional recovery in various CNS injury paradigms, including stroke, experimental autoimmune encephalitis (EAE), SCI, and traumatic brain injury (TBI) (reviewed in Garden, 2013). After CNS injury, H3ac levels decline, thus HDACi-mediated neuroprotection is thought to occur by restoring adequate histone acetylation levels; however, because HDACi affects a variety of cells, and both histone and non-histone targets, including key inflammatory regulators, the underlying mechanisms are likely multifactorial.
In a recent study, we elucidated a role of HDAC3 for epigenetic regulation of inflammatory responses in microglia/macrophages in a contusion SCI model (Kuboyama et al., 2017). We found that HDAC3, but not other class I HDACs (HDAC1, 2, 8), was robustly upregulated in activated microglia/macrophages after SCI. Administration of RGFP966, a small-molecule inhibitor that specifically targets HDAC3, resulted in global suppression of inflammatory cytokines at the injury site, and improved neuroprotection and functional recovery after SCI (Figure 3). Mechanistically, in primary microglia, HDAC3 activity contributes to histone deacetylation and the inflammatory responses to LPS, a classic inflammatory stimulus (Kuboyama et al., 2017). Consistently, in non-CNS injuries, HDAC3 is shown to function as an epigenetic brake in macrophage alternative activation (Mullican et al., 2011). Furthermore, HDAC3 is required for upregulation of an inflammation-associated gene program in macrophages, as macrophages lacking HDAC3 became less responsive to IFNγ stimulation, but hypersensitive to IL-4 stimulation (Chen et al., 2012). Future directions worth pursuing include conditional ablation of HDAC3 in microglia/macrophages in CNS injury models, and identifying specific genomic loci that are under direct regulation of HDAC3.
In TBI, treatment with HDACi Scriptaid also shifted microglia/macrophage towards a protective M2 phenotype and mitigated inflammation through upregulation of GSK3β, which can modulate microglial functions via the PTEN/PI3K/Akt signaling pathway (Wang et al., 2015). In another study, HDACi treatment after TBI attenuated inflammation by promoting apoptosis of activated microglia/macrophages through a mechanism involving p53 upregulation and caspase activation (Shein et al., 2009) (Figure 3).
Recently, another class I HDACi, CI-994, has been shown to promote motor functional recovery following SCI, and this was attributed to a neuroprotective effect since no increased sprouting of CST fibers were observed (Zhang et al., 2018b). In a SCI model, CI-994 treatment resulted in increased H3ac levels in neutrophils and microglia/macrophages at the injury site, and suppressed neutrophil accumulation, reduced inflammatory cytokine expression and decreased neuronal loss as early as three days following injury. Bromodomain and Extra-Terminal Domain-containing proteins (BETs; Brd2, Brd3, Brd4, BrdT) are epigenetic readers that bind acetylated histones and promote transcription of proinflammatory genes. Administration of BET inhibitor JQ1 in SCI model resulted in reduced proinflammatory cytokine expression and leukocyte recruitment to the injury site by three days post injury; however, these changes did not appear to lead to improved functional recovery or smaller lesion size (Rudman et al., 2018).
Other epigenetic modalities are less understood in mediating the innate immune response in CNS injury. In microglia-like BV-2 cells, DNA methylation can influence expression of Alzheimer’s Disease (AD) associated genes by way of hypomethylation of gene promoters (Lin et al., 2009). TBI reportedly induces hypomethylation mainly in ∼10–20% of activated microglia/macrophages in injury area as early as day one (Zhang et al., 2007), but the functional significance is unclear. Micro-RNA expression profiles in microglia are distinct from that of neurons and other glial cells (Jovičić et al., 2013). miRNAs can regulate differentiation and activation of cells of the immune system (Ponomarev et al., 2013). Pro-inflammatory miRNAs included miR-155, miR-101 and miR-125. On the other hand, MiR-21 and miR-146 were also induced in macrophages by the classic proinflammatory stimuli LPS, but they promoted a resolution of inflammation (reviewed in Ponomarev et al., 2013). Brain-specific miR-124 is expressed in homeostatic microglia but absent in peripheral macrophages. miR-124 promotes microglia quiescence; and consistently, it is downregulated in activated microglia in EAE or after treatment with proinflammatory cytokines such as IFNγ and GM-CSF. Treatment of mice with miR-124 inhibits EAE and reduces CNS inflammation (Ponomarev et al., 2011). In the cancer field, advances have been made in understanding epigenetic regulation of T cell dysfunction and therapeutic programmability. For instance, T cells in tumors are dysfunctional due to immune checkpoint blockade. T cells have been shown to differentiate through two distinct chromatin dysfunctional states–a plastic and a fixed state–as a result of heritable, epigenetically imprinted mechanisms (Philip et al., 2017). The functions of these epigenetic mechanisms in influencing immune responses and neural repair in CNS injury awaits future investigation.
Axonal injury also elicits a glial response located near the soma of injured neurons or surround axons. For instance, peripheral but not central axotomy leads to significant macrophage accumulation in the DRG, which increases regenerative capacity of DRG neurons, and this occurs through chemokine CCL2 (Niemi et al., 2013; Niemi et al., 2016).
Epigenetics of Reactive Astrocytosis for Neural Repair
Astrocytes are also a key glial component that contributes to inflammatory responses (Gao et al., 2013; Goldmann et al., 2016). HDAC activity is increased in astrocytes under inflammatory conditions, and HDAC inhibition reduces glial inflammatory responses in both microglia and astrocytes (Faraco et al., 2009; Suh et al., 2010). During differentiation of neural progenitors into astrocytes, upregulation of GFAP, an astrocyte marker, requires recruitment of transcriptional module comprised of STAT3, p300 and Smad1 to its promoter (Nakashima et al., 1999). During reactive astrocytosis, GFAP is upregulated; and HDAC inhibition reduced GFAP expression in primary human astrocytes (Kanski et al., 2014), but did not reduce astrocyte activation (Xuan et al., 2012). Reactive astrocytes upregulate cyclooxygenase (COX)-2, iNOS, NO, IL-6 and TNF-α, which can be attenuated by HDAC inhibition in different CNS injury scenarios (reviewed in York et al., 2013). Reactive astrocytes also increase production of glycosaminoglycans, such as chondrointin sulfate proteoglygan (CSPG), some of which can act as HAT inhibitors, thereby decreasing acetylation levels in neighboring cells (Buczek-Thomas et al., 2008). In co-cultures, HDAC inhibition increases secretion of neurotrophic cytokines by astrocytes, resulting in neuroprotection of dopaminergic neurons (Chen et al., 2006). Taken together, epigenetic modulation may be a powerful tool to influence astroglial responses, thereby promoting neuroprotection and neuronal repair after CNS injuries (York et al., 2013).
Caveats and Future Directions
One limitation of current approaches is the use of whole neural tissue for epigenomic studies, which does not distinguish cell-type specific epigenetic changes. For instance, whole DRG consists of sensory neurons and a larger population of glial cells, including macrophages and Schwann cells. There are also many different neuronal subtypes in DRG. Indeed, single cell RNA-Seq revealed heterogeneous transcriptional responses of DRG neuronal subtypes after nerve injury (Hu et al., 2016). Another study found that in DRG, DNMT1 is expressed in both glia and neurons, DNMT3a is preferentially expressed in glia, while DNMT3b is preferentially expressed in neurons but absent in glia; and all are upregulated in a model of nerve injury (Pollema-Mays et al., 2014). Hence, different epigenetic factors are involved in injury responses and transcriptional changes in different cell types in neural tissues.
New epigenetic mechanisms continue to come to light, and their roles in CNS injury are poorly understood. For instance, non-canonical histone variants such as H2A.Z and H3.3 can influence chromatin architecture and genomic function (Henikoff and Smith, 2015). Nucleosome repositioning or sliding along DNA as mediated by adenosine triphosphate (ATP)-dependent chromatin-remodeling enzymes can control DNA accessibility by transcriptional machinery (Mueller-Planitz et al., 2013). Higher-order chromatin remodeling, such as DNA looping, chromatin boundary elements, functional nuclear domains also affect chromatin organization (Mercer and Mattick, 2013). Epigenetic mechanisms also regulate mitochondria gene expression (Shock et al., 2011), which may be important for metabolic adaption during axon regeneration and glial activation.
Individual RAGs may employ multiple epigenetic mechanisms for their transcriptional regulation in a cell-type specific manner, and different epigenetic mechanisms can interact with one another (Table 2). For instance, DNA demethylation may coordinate with histone acetylation or other epigenetic mechanism to promote axon regeneration (Trakhtenberg and Goldberg, 2012). CpG methylation can influence histone methylation patterns. For example, binding of MBD proteins to the methylated CpG can recruit histone modifiers to maintain transcriptional repression (Weng et al., 2016). Conversely, H3K4 methyltransferase can be recruited to non-methylated CpG islands to enhance H3K4me3 levels and activate transcription. Epigenetic and epitranscriptomic mechanisms may also interact, e.g. Tet3 and Gadd45a transcripts are both targets of m6A mRNA modifications (Weng et al., 2018).
New methods, such as a CRISPR-dCas9-based system, now allow activation of target genes by locally rewriting gene-specific epigenetic codes, thus circumventing off-target effects or avoiding pharmacological approaches that ubiquitously modify histone codes (Liao et al., 2017; Xie et al., 2018). Combinatorial epigenetic strategies are worth exploring, with the aim to achieve simultaneous enhancement of neuronal intrinsic axon growth potential and modulation of glial inflammatory responses. It is important to first delineate cell type-specific roles of individual member of the epigenetic factors in CNS injury. For instance, HDAC1 and HDAC5 regulate RAG expression and axon growth potential in DRG neurons (Cho et al., 2013; Finelli et al., 2013b), while HDAC3 controls inflammatory gene programs in activated microglia (Kuboyama et al., 2017). It is worth noting that HDAC3 also functions as a key regulator of apoptotic gene silencing in RGCs after optic nerve injury (Pelzel et al., 2010). HDAC3 was shown to translocate to the nuclei of dying RGCs as an early response to axonal injury, and this is associated with widespread H4 deacetylation and transcriptional dysregulation in dying RGCs. Conditional knockout of Hdac3 or blocking HDAC3 activity using RGFP966 in a dose dependent manner prolonged RGC survival through protection against nuclear atrophy and apoptosis (Schmitt et al., 2014; Schmitt et al., 2018).
Conclusion
Here, we summarized recent progress in understanding the roles of chromatin accessibility, microRNAs, and epitranscriptomics in fine-tuning transcriptional changes in neurons and glia in response to axonal injury. These advances provide a framework for defining how adaptive and maladaptive epigenetic changes impact intrinsic axon regeneration competence and glial activation in the context of neural repair. Given the broad scope and complexity of epigenetic mechanisms, more work is needed before epigenetic strategies can be combined with other treatment modalities to promote functional recovery after CNS injury.
Author Contributions
All authors contributed to manuscript preparation. SW, DH, and XZ performed literature search and prepared the diagrams and tables, and HZ, SW, and DH wrote the review.
Funding
This work was supported by grants from the National Institutes of Health (R01/R56 NS073596), Craig H Neilsen Foundation (#476516), and New York State Spinal Cord Injury Research Board (DOH01-C32242-GG, -C33268GG) to HZ, and postdoctoral fellowship grants from New York State Spinal Cord Injury Research Board (DOH01-C32634GG) to SW, and from the Chinese Scholarship Council to XZ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Roland H. Friedel and members of the Zou laboratory for their discussion and editing of the manuscript.
References
Al-Ali, H., Ding, Y., Slepak, T., Wu, W., Sun, Y., Martinez, Y., et al. (2017). The mTOR Substrate S6 Kinase 1 (S6K1) is a negative regulator of axon regeneration and a potential drug target for central nervous system injury. J. Neurosci. 37, 7079–7095. doi: 10.1523/JNEUROSCI.0931-17.2017
Bareyre, F. M., Garzorz, N., Lang, C., Misgeld, T., Buning, H., Kerschensteiner, M. (2011). In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 6282–6287. doi: 10.1073/pnas.1015239108
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Beck, K. D., Nguyen, H. X., Galvan, M. D., Salazar, D. L., Woodruff, T. M., Anderson, A. J. (2010). Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 133, 433–447. doi: 10.1093/brain/awp322
Belin, S., Nawabi, H., Wang, C., Tang, S., Latremoliere, A., Warren, P., et al. (2015). Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron 86, 1000–1014. doi: 10.1016/j.neuron.2015.03.060
Benitez, J. A., Ma, J., D’Antonio, M., Boyer, A., Camargo, M. F., Zanca, C., et al. (2017). PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nat. Commun. 8, 15223. doi: 10.1038/ncomms15223
Black, J. C., Van Rechem, C., Whetstine, J. R. (2012). Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491–507. doi: 10.1016/j.molcel.2012.11.006
Blackmore, M. G. (2012). Molecular control of axon growth: insights from comparative gene profiling and high-throughput screening. Int. Rev. Neurobiol. 105, 39–70. doi: 10.1016/B978-0-12-398309-1.00004-4
Blesch, A., Lu, P., Tsukada, S., Alto, L. T., Roet, K., Coppola, G., et al. (2012). Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to camp-mediated effects. Exp. Neurol. 235, 162–173. doi: 10.1016/j.expneurol.2011.12.037
Bonilla, I. E., Tanabe, K., Strittmatter, S. M. (2002). Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J. Neurosci. 22, 1303–1315. doi: 10.1523/JNEUROSCI.22-04-01303.2002
Buczek-Thomas, J. A., Hsia, E., Rich, C. B., Foster, J. A., Nugent, M. A. (2008). Inhibition of histone acetyltransferase by glycosaminoglycans. J. Cell. Biochem. 105, 108–120. doi: 10.1002/jcb.21803
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. doi: 10.1038/nmeth.2688
Chandran, V., Coppola, G., Nawabi, H., Omura, T., Versano, R., Huebner, E. A., et al. (2016). A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 89, 956–970. doi: 10.1016/j.neuron.2016.01.034
Chen, P. S., Peng, G. S., Li, G., Yang, S., Wu, X., Wang, C. C., et al. (2006). Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol. Psychiatry 11, 1116–1125. doi: 10.1038/sj.mp.4001893
Chen, X., Barozzi, I., Termanini, A., Prosperini, E., Recchiuti, A., Dalli, J., et al. (2012). Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. U.S.A. 109, E2865–E2874. doi: 10.1073/pnas.1121131109
Chen, Z. H., Zhu, M., Yang, J., Liang, H., He, J., He, S., et al. (2014). PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep. 8, 2003–2014. doi: 10.1016/j.celrep.2014.08.008
Cho, Y., Cavalli, V. (2014). HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol. 27C, 118–126. doi: 10.1016/j.conb.2014.03.008
Cho, Y., Shin, J. E., Ewan, E. E., Oh, Y. M., Pita-Thomas, W., Cavalli, V. (2015). Activating injury-responsive genes with hypoxia enhances axon regeneration through neuronal HIF-1alpha. Neuron 88, 720–734. doi: 10.1016/j.neuron.2015.09.050
Cho, Y., Sloutsky, R., Naegle, K. M., Cavalli, V. (2013). Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155, 894–908. doi: 10.1016/j.cell.2013.10.004
Costigan, M., Befort, K., Karchewski, L., Griffin, R. S., D’Urso, D., Allchorne, A., et al. (2002). Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 3, 16. doi: 10.1186/1471-2202-3-16
Dajas-Bailador, F., Bonev, B., Garcez, P., Stanley, P., Guillemot, F., Papalopulu, N. (2012). microRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nat. Neurosci. 15, 697–699. doi: 10.1038/nn.3082
Danzi, M. C., O’Neill, N., Bixby, J. L., Lemmon, V. P. (2018). Can chromatin accessibility be exploited for axon regeneration? Dev. Neurobiol. 78, 991–997. doi: 10.1002/dneu.22598
Dekanty, A., Romero, N. M., Bertolin, A. P., Thomas, M. G., Leishman, C. C., Perez-Perri, J. I., et al. (2010). Drosophila genome-wide RNAi screen identifies multiple regulators of HIF-dependent transcription in hypoxia. PLoS Genet. 6, e1000994. doi: 10.1371/journal.pgen.1000994
Di Giovanni, S., Knights, C. D., Rao, M., Yakovlev, A., Beers, J., Catania, J., et al. (2006). The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 25, 4084–4096. doi: 10.1038/sj.emboj.7601292
Ebert, M. S., Sharp, P. A. (2010). Emerging roles for natural microRNA sponges. Curr. Biol. 20, R858–R861. doi: 10.1016/j.cub.2010.08.052
Ernst, J., Kheradpour, P., Mikkelsen, T. S., Shoresh, N., Ward, L. D., Epstein, C. B., et al. (2012). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49. doi: 10.1038/nature09906
Fagoe, N. D., Attwell, C. L., Kouwenhoven, D., Verhaagen, J., Mason, M. R. (2015). Overexpression of ATF3 or the combination of ATF3, c-Jun, STAT3 and Smad1 promotes regeneration of the central axon branch of sensory neurons but without synergistic effects. Hum. Mol. Genet. 24, 6788–6800. doi: 10.1093/hmg/ddv383
Faraco, G., Pittelli, M., Cavone, L., Fossati, S., Porcu, M., Mascagni, P., et al. (2009). Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol. Dis. 36, 269–279. doi: 10.1016/j.nbd.2009.07.019
Finelli, M. J., Murphy, K. J., Chen, L., Zou, H. (2013a). Differential phosphorylation of Smad1 integrates BMP and neurotrophin pathways through Erk/Dusp in axon development. Cell Rep. 3, 1592–1606. doi: 10.1016/j.celrep.2013.04.011
Finelli, M. J., Wong, J. K., Zou, H. (2013b). Epigenetic regulation of sensory axon regeneration after spinal cord injury. J. Neurosci. 33, 19664–19676. doi: 10.1523/JNEUROSCI.0589-13.2013
Fink, K. L., López-Giráldez, F., Kim, I. J., Strittmatter, S. M., Cafferty, W. B. J. (2017). Identification of intrinsic axon growth modulators for intact cns neurons after injury. Cell Rep. 18, 2687–2701. doi: 10.1016/j.celrep.2017.02.058
Franke, K., Otto, W., Johannes, S., Baumgart, J., Nitsch, R., Schumacher, S. (2012). miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling. EMBO J. 31, 2908–2921. doi: 10.1038/emboj.2012.130
Gao, Z., Zhu, Q., Zhang, Y., Zhao, Y., Cai, L., Shields, C. B., et al. (2013). Reciprocal modulation between microglia and astrocyte in reactive gliosis following the CNS injury. Mol. Neurobiol. 48, 690–701. doi: 10.1007/s12035-013-8460-4
Garden, G. A. (2013). Epigenetics and the modulation of neuroinflammation. Neurotherapeutics 10, 782–788. doi: 10.1007/s13311-013-0207-4
Gaub, P., Joshi, Y., Wuttke, A., Naumann, U., Schnichels, S., Heiduschka, P., et al. (2011). The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 134, 2134–2148. doi: 10.1093/brain/awr142
Gaub, P., Tedeschi, A., Puttagunta, R., Nguyen, T., Schmandke, A., Di Giovanni, S. (2010). HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 17, 1392–1408. doi: 10.1038/cdd.2009.216
Gilbert, W. V., Bell, T. A., Schaening, C. (2016). Messenger RNA modifications: Form, distribution, and function. Science 352, 1408–1412. doi: 10.1126/science.aad8711
Goldmann, T., Wieghofer, P., Jordão, M. J., Prutek, F., Hagemeyer, N., Frenzel, K., et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805. doi: 10.1038/ni.3423
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., Song, H. (2011a). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. doi: 10.1016/j.cell.2011.03.022
Guo, W., Qian, L., Zhang, J., Zhang, W., Morrison, A., Hayes, P., et al. (2011b). Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J. Neurosci. Res. 89, 1723–1736. doi: 10.1002/jnr.22725
Hackett, J. A., Sengupta, R., Zylicz, J. J., Murakami, K., Lee, C., Down, T. A., et al. (2013). Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339, 448–452. doi: 10.1126/science.1229277
Hahn, M. A., Qiu, R., Wu, X., Li, A. X., Zhang, H., Wang, J., et al. (2013). Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. 3, 291–300. doi: 10.1016/j.celrep.2013.01.011
He, Z., Jin, Y. (2016). Intrinsic control of axon regeneration. Neuron 90, 437–451. doi: 10.1016/j.neuron.2016.04.022
Henikoff, S., Smith, M. M. (2015). Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 7, a019364. doi: 10.1101/cshperspect.a019364
Hu, G., Huang, K., Hu, Y., Du, G., Xue, Z., Zhu, X., et al. (2016). Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci. Rep. 6, 31851. doi: 10.1038/srep31851
Huang, H., Sabari, B. R., Garcia, B. A., Allis, C. D., Zhao, Y. (2014). SnapShot: histone modifications. Cell 159, 458–458.e451. doi: 10.1016/j.cell.2014.09.037
Hutson, T. H., Kathe, C., Palmisano, I., Bartholdi, K., Hervera, A., De Virgiliis, F., et al. (2019). Cbp-dependent histone acetylation mediates axon regeneration induced by environmental enrichment in rodent spinal cord injury models. Sci. Transl. Med. 11, (487), eaaw2064. doi: 10.1126/scitranslmed.aaw2064
Iskandar, B. J., Rizk, E., Meier, B., Hariharan, N., Bottiglieri, T., Finnell, R. H., et al. (2010). Folate regulation of axonal regeneration in the rodent central nervous system through DNA methylation. J. Clin. Invest. 120, 1603–1616. doi: 10.1172/JCI40000
Ito, R., Katsura, S., Shimada, H., Tsuchiya, H., Hada, M., Okumura, T., et al. (2014). TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes Cells 19, 52–65. doi: 10.1111/gtc.12107
Jiang, J., Hu, Y., Zhang, B., Shi, Y., Zhang, J., Wu, X., et al. (2017). MicroRNA-9 regulates mammalian axon regeneration in peripheral nerve injury. Mol. Pain 13, 1744806917711612. doi: 10.1177/1744806917711612
Jiang, J. J., Liu, C. M., Zhang, B. Y., Wang, X. W., Zhang, M., Saijilafu, et al. (2015). MicroRNA-26a supports mammalian axon regeneration in vivo by suppressing GSK3β expression. Cell Death Dis. 6, e1865. doi: 10.1038/cddis.2015.239
Jovičić, A., Roshan, R., Moisoi, N., Pradervand, S., Moser, R., Pillai, B., et al. (2013). Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J. Neurosci. 33, 5127–5137. doi: 10.1523/JNEUROSCI.0600-12.2013
Kanski, R., Sneeboer, M. A., van Bodegraven, E. J., Sluijs, J. A., Kropff, W., Vermunt, M. W., et al. (2014). Histone acetylation in astrocytes suppresses GFAP and stimulates a reorganization of the intermediate filament network. J. Cell. Sci. 127, 4368–4380. doi: 10.1242/jcs.145912
Kigerl, K. A., Gensel, J. C., Ankeny, D. P., Alexander, J. K., Donnelly, D. J., Popovich, P. G. (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009
Klose, R. J., Bird, A. P. (2006). Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97. doi: 10.1016/j.tibs.2005.12.008
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kuboyama, T., Wahane, S., Huang, Y., Zhou, X., Wong, J. K., Koemeter-Cox, A., et al. (2017). HDAC3 inhibition ameliorates spinal cord injury by immunomodulation. Sci. Rep. 7, 8641. doi: 10.1038/s41598-017-08535-4
Lamparter, D., Marbach, D., Rueedi, R., Bergmann, S., Kutalik, Z. (2017). Genome-wide association between transcription factor expression and chromatin accessibility reveals regulators of chromatin accessibility. PLoS Comput. Biol. 13, e1005311. doi: 10.1371/journal.pcbi.1005311
Lee, K. K., Workman, J. L. (2007). Histone acetyltransferase complexes: one size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 8, 284–295. doi: 10.1038/nrm2145
Li, B., Sun, H. (2013). MiR-26a promotes neurite outgrowth by repressing PTEN expression. Mol. Med. Rep. 8, 676–680. doi: 10.3892/mmr.2013.1534
Li, S., Xue, C., Yuan, Y., Zhang, R., Wang, Y., Wang, Y., et al. (2015). The transcriptional landscape of dorsal root ganglia after sciatic nerve transection. Sci. Rep. 5, 16888. doi: 10.1038/srep16888
Li, X., Xiong, X., Yi, C. (2016). Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods 14, 23–31. doi: 10.1038/nmeth.4110
Liao, H. K., Hatanaka, F., Araoka, T., Reddy, P., Wu, M. Z., Sui, Y., et al. (2017). In Vivo Target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 171, 1495–1507.e1415. doi: 10.1016/j.cell.2017.10.025
Lin, H. C., Hsieh, H. M., Chen, Y. H., Hu, M. L. (2009). S-Adenosylhomocysteine increases beta-amyloid formation in BV-2 microglial cells by increased expressions of beta-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. Neurotoxicology 30, 622–627. doi: 10.1016/j.neuro.2009.03.011
Lisy, K., Peet, D. J. (2008). Turn me on: regulating HIF transcriptional activity. Cell Death Differ. 15, 642–649. doi: 10.1038/sj.cdd.4402315
Liu, C. M., Wang, R. Y., Saijilafu, Jiao, Z. X., Zhang, B. Y., Zhou, F. Q. (2013). MicroRNA-138 and SIRT1 form a mutual negative feedback loop to regulate mammalian axon regeneration. Genes Dev. 27, 1473–1483. doi: 10.1101/gad.209619.112
Liu, K., Lu, Y., Lee, J. K., Samara, R., Willenberg, R., Sears-Kraxberger, I., et al. (2010). PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 13, 1075–1081. doi: 10.1038/nn.2603
Liu, R. Y., Snider, W. D. (2001). Different signaling pathways mediate regenerative versus developmental sensory axon growth. J. Neurosci. 21, RC164. doi: 10.1523/JNEUROSCI.21-17-j0003.2001
Liu, X. S., Wu, H., Ji, X., Stelzer, Y., Wu, X., Czauderna, S., et al. (2016). Editing DNA methylation in the Mammalian genome. Cell 167, 233–247.e217. doi: 10.1016/j.cell.2016.08.056
Loh, Y. E., Koemeter-Cox, A., Finelli, M. J., Shen, L., Friedel, R. H., Zou, H. (2017). Comprehensive mapping of 5-hydroxymethylcytosine epigenetic dynamics in axon regeneration. Epigenetics 12, 77–92. doi: 10.1080/15592294.2016.1264560
Lu, Y., Belin, S., He, Z. (2014). Signaling regulations of neuronal regenerative ability. Curr. Opin. Neurobiol. 27, 135–142. doi: 10.1016/j.conb.2014.03.007
Martinez, F. O., Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13. doi: 10.12703/P6-13
McQuarrie, I. G., Grafstein, B. (1973). Axon outgrowth enhanced by a previous nerve injury. Arch. Neurol. 29, 53–55. doi: 10.1001/archneur.1973.00490250071008
Mellor, J. (2005). The dynamics of chromatin remodeling at promoters. Mol. Cell 19, 147–157. doi: 10.1016/j.molcel.2005.06.023
Mercer, T. R., Mattick, J. S. (2013). Understanding the regulatory and transcriptional complexity of the genome through structure. Genome Res. 23, 1081–1088. doi: 10.1101/gr.156612.113
Michaelevski, I., Segal-Ruder, Y., Rozenbaum, M., Medzihradszky, K. F., Shalem, O., Coppola, G., et al. (2010). Signaling to transcription networks in the neuronal retrograde injury response. Sci. Signal. 3, ra53. doi: 10.1126/scisignal.2000952
Molinie, B., Wang, J., Lim, K. S., Hillebrand, R., Lu, Z. X., Van Wittenberghe, N., et al. (2016). m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat. Methods 13, 692–698. doi: 10.1038/nmeth.3898
Moore, D. L., Blackmore, M. G., Hu, Y., Kaestner, K. H., Bixby, J. L., Lemmon, V. P., et al. (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301. doi: 10.1126/science.1175737
Mueller-Planitz, F., Klinker, H., Becker, P. B. (2013). Nucleosome sliding mechanisms: new twists in a looped history. Nat. Struct. Mol. Biol. 20, 1026–1032. doi: 10.1038/nsmb.2648
Mullican, S. E., Gaddis, C. A., Alenghat, T., Nair, M. G., Giacomin, P. R., Everett, L. J., et al. (2011). Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 25, 2480–2488. doi: 10.1101/gad.175950.111
Musselman, C. A., Lalonde, M. E., Côté, J., Kutateladze, T. G. (2012). Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227. doi: 10.1038/nsmb.2436
Nakashima, K., Yanagisawa, M., Arakawa, H., Kimura, N., Hisatsune, T., Kawabata, M., et al. (1999). Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 284, 479–482. doi: 10.1126/science.284.5413.479
Neumann, S., Woolf, C. J. (1999). Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91. doi: 10.1016/S0896-6273(00)80755-2
Niemi, J. P., Francesco-Lisowitz, A., Cregg, J. M., Howarth, M., Zigmond, R. E. (2016). Overexpression of the monocyte chemokine CCL2 in dorsal root ganglion neurons causes a conditioning-like increase in neurite outgrowth and does so via a STAT3 dependent mechanism. Exp. Neurol. 275 Pt 1, 25–37. doi: 10.1016/j.expneurol.2015.09.018
Niemi, J. P., Francesco-Lisowitz, A., Roldán-Hernández, L., Lindborg, J. A., Mandell, D., Zigmond, R. E. (2013). A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J. Neurosci. 33, 16236–16248. doi: 10.1523/JNEUROSCI.3319-12.2013
Nix, P., Hammarlund, M., Hauth, L., Lachnit, M., Jorgensen, E. M., Bastiani, M. (2014). Axon regeneration genes identified by RNAi screening in C. J. Neurosci. 34, 629–645. doi: 10.1523/JNEUROSCI.3859-13.2014
Oh, Y. M., Mahar, M., Ewan, E. E., Leahy, K. M., Zhao, G., Cavalli, V. (2018). Epigenetic regulator UHRF1 inactivates REST and growth suppressor gene expression via DNA methylation to promote axon regeneration. Proc. Natl. Acad. Sci. U.S.A. 115, E12417–E12426. doi: 10.1073/pnas.1812518115
Okano, M., Bell, D. W., Haber, D. A., Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. doi: 10.1016/S0092-8674(00)81656-6
Palmisano, I., Di Giovanni, S. (2018). Advances and limitations of current epigenetic studies investigating mammalian axonal regeneration. Neurotherapeutics 15, 529–540. doi: 10.1007/s13311-018-0636-1
Parikh, P., Hao, Y., Hosseinkhani, M., Patil, S. B., Huntley, G. W., Tessier-Lavigne, M., et al. (2011). Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc. Natl. Acad. Sci. 108, E99–E107. doi: 10.1073/pnas.1100426108
Park, K. K., Liu, K., Hu, Y., Smith, P. D., Wang, C., Cai, B., et al. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966. doi: 10.1126/science.1161566
Pelzel, H. R., Schlamp, C. L., Nickells, R. W. (2010). Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 11, 62. doi: 10.1186/1471-2202-11-62
Philip, M., Fairchild, L., Sun, L., Horste, E. L., Camara, S., Shakiba, M., et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. doi: 10.1038/nature22367
Pollema-Mays, S. L., Centeno, M. V., Apkarian, A. V., Martina, M. (2014). Expression of DNA methyltransferases in adult dorsal root ganglia is cell-type specific and up regulated in a rodent model of neuropathic pain. Front. Cell. Neurosci. 8, 217. doi: 10.3389/fncel.2014.00217
Ponomarev, E. D., Veremeyko, T., Barteneva, N., Krichevsky, A. M., Weiner, H. L. (2011). MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nat. Med. 17, 64–70. doi: 10.1038/nm.2266
Ponomarev, E. D., Veremeyko, T., Weiner, H. L. (2013). MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 61, 91–103. doi: 10.1002/glia.22363
Puttagunta, R., Tedeschi, A., Soria, M. G., Hervera, A., Lindner, R., Rathore, K. I., et al. (2014). PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat. Commun. 5, 3527. doi: 10.1038/ncomms4527
Qureshi, I. A., Mehler, M. F. (2018). Epigenetic mechanisms underlying nervous system diseases. Handb. Clin. Neurol. 147, 43–58. doi: 10.1016/B978-0-444-63233-3.00005-1
Ram, O., Goren, A., Amit, I., Shoresh, N., Yosef, N., Ernst, J., et al. (2011). Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147, 1628–1639. doi: 10.1016/j.cell.2011.09.057
Richardson, P. M., Issa, V. M. (1984). Peripheral injury enhances central regeneration of primary sensory neurones. Nature 309, 791–793. doi: 10.1038/309791a0
Rudman, M. D., Choi, J. S., Lee, H. E., Tan, S. K., Ayad, N. G., Lee, J. K. (2018). Bromodomain and extraterminal domain-containing protein inhibition attenuates acute inflammation after spinal cord injury. Exp. Neurol. 309, 181–192. doi: 10.1016/j.expneurol.2018.08.005
Schmitt, H. M., Pelzel, H. R., Schlamp, C. L., Nickells, R. W. (2014). Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol. Neurodegener. 9, 39. doi: 10.1186/1750-1326-9-39
Schmitt, H. M., Schlamp, C. L., Nickells, R. W. (2018). Targeting HDAC3 activity with rgfp966 protects against retinal ganglion cell nuclear atrophy and apoptosis after optic nerve injury. J. Ocul. Pharmacol. Ther. 34, 260–273. doi: 10.1089/jop.2017.0059
Schutsky, E. K., DeNizio, J. E., Hu, P., Liu, M. Y., Nabel, C. S., Fabyanic, E. B., et al. (2018). Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat. Biotechnol. 36, 1083–1090. doi: 10.1038/nbt.4204
Seijffers, R., Mills, C. D., Woolf, C. J. (2007). ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 27, 7911–7920. doi: 10.1523/JNEUROSCI.5313-06.2007
Shein, N. A., Grigoriadis, N., Alexandrovich, A. G., Simeonidou, C., Lourbopoulos, A., Polyzoidou, E., et al. (2009). Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 23, 4266–4275. doi: 10.1096/fj.09-134700
Sherwood, R. I., Hashimoto, T., O’Donnell, C. W., Lewis, S., Barkal, A. A., van Hoff, J. P., et al. (2014). Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 32, 171–178. doi: 10.1038/nbt.2798
Shock, L. S., Thakkar, P. V., Peterson, E. J., Moran, R. G., Taylor, S. M. (2011). DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, 3630–3635. doi: 10.1073/pnas.1012311108
Smith, D. S., Pate Skene, J. H. (1997). A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 17, 646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997
Smith, S. S., Kaplan, B. E., Sowers, L. C., Newman, E. M. (1992). Mechanism of human methyl-directed DNA methyltransferase and the fidelity of cytosine methylation. Proc. Natl. Acad. Sci. U.S.A. 89, 4744–4748. doi: 10.1073/pnas.89.10.4744
Song, C. X., Szulwach, K. E., Fu, Y., Dai, Q., Yi, C., Li, X., et al. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72. doi: 10.1038/nbt.1732
Staszewski, O., Prinz, M. (2014). Glial epigenetics in neuroinflammation and neurodegeneration. Cell Tissue Res. 356, 609–616. doi: 10.1007/s00441-014-1815-y
Strickland, I. T., Richards, L., Holmes, F. E., Wynick, D., Uney, J. B., Wong, L. F. (2011). Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS One 6, e23423. doi: 10.1371/journal.pone.0023423
Su, C., Schwarz, T. L. (2017). -GlcNAc transferase is essential for sensory neuron survival and maintenance. J. Neurosci. 37, 2125–2136. doi: 10.1523/JNEUROSCI.3384-16.2017
Su, Y., Shin, J., Zhong, C., Wang, S., Roychowdhury, P., Lim, J., et al. (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat. Neurosci. 20, 476–483. doi: 10.1038/nn.4494
Suh, H. S., Choi, S., Khattar, P., Choi, N., Lee, S. C. (2010). Histone deacetylase inhibitors suppress the expression of inflammatory and innate immune response genes in human microglia and astrocytes. J. Neuroimmune Pharmacol. 5, 521–532. doi: 10.1007/s11481-010-9192-0
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Tedeschi, A., Bradke, F. (2017). Spatial and temporal arrangement of neuronal intrinsic and extrinsic mechanisms controlling axon regeneration. Curr. Opin. Neurobiol. 42, 118–127. doi: 10.1016/j.conb.2016.12.005
Tedeschi, A., Nguyen, T., Puttagunta, R., Gaub, P., Di Giovanni, S. (2009). A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 16, 543–554. doi: 10.1038/cdd.2008.175
Tewary, S. K., Zheng, Y. G., Ho, M. C. (2019). Protein arginine methyltransferases: insights into the enzyme structure and mechanism at the atomic level. Cell. Mol. Life Sci. 1–16. doi: 10.1007/s00018-019-03145-x
Trakhtenberg, E. F., Goldberg, J. L. (2012). Epigenetic regulation of axon and dendrite growth. Front. Mol. Neurosci. 5, 24. doi: 10.3389/fnmol.2012.00024
Usoskin, D., Furlan, A., Islam, S., Abdo, H., Lönnerberg, P., Lou, D., et al. (2015). Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153. doi: 10.1038/nn.3881
van Battum, E. Y., Verhagen, M. G., Vangoor, V. R., Fujita, Y., Derijck, A. A. H. A., O’Duibhir, E., et al. (2018). An image-based miRNA screen identifies miRNA-135s as regulators of CNS axon growth and regeneration by targeting Krüppel-like factor 4. J. Neurosci. 38, 613–630. doi: 10.1523/JNEUROSCI.0662-17.2017
Venkatesh, I., Mehra, V., Wang, Z., Califf, B., Blackmore, M. G. (2018). Developmental chromatin restriction of pro-growth gene networks acts as an epigenetic barrier to axon regeneration in cortical neurons. Dev. Neurobiol. 78 (10), 960977. doi: 10.1101/259408
Venkatesh, I., Simpson, M. T., Coley, D. M., Blackmore, M. G. (2016). Epigenetic profiling reveals a developmental decrease in promoter accessibility during cortical maturation. Neuroepigenetics 8, 19–26. doi: 10.1016/j.nepig.2016.10.002
Wang, G., Shi, Y., Jiang, X., Leak, R. K., Hu, X., Wu, Y., et al. (2015). HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc. Natl. Acad. Sci. U.S.A. 112, 2853–2858. doi: 10.1073/pnas.1501441112
Weng, Y. L., An, R., Cassin, J., Joseph, J., Mi, R., Wang, C., et al. (2017). An intrinsic epigenetic barrier for functional axon regeneration. Neuron 94, 337–346.e336. doi: 10.1016/j.neuron.2017.03.034
Weng, Y. L., Joseph, J., An, R., Song, H., Ming, G. L. (2016). Epigenetic regulation of axonal regenerative capacity. Epigenomics 8, 1429–1442. doi: 10.2217/epi-2016-0058
Weng, Y. L., Wang, X., An, R., Cassin, J., Vissers, C., Liu, Y., et al. (2018). Epitranscriptomic m. Neuron 97, 313–325.e316. doi: 10.1016/j.neuron.2017.12.036
Wong, J. K., Zou, H. (2014). Reshaping the chromatin landscape after spinal cord injury. Front. Biol. (Beijing) 9, 356–366. doi: 10.1007/s11515-014-1329-8
Wu, D., Raafat, A., Pak, E., Clemens, S., Murashov, A. K. (2012). Dicer-microRNA pathway is critical for peripheral nerve regeneration and functional recovery in vivo and regenerative axonogenesis in vitro. Exp. Neurol. 233, 555–565. doi: 10.1016/j.expneurol.2011.11.041
Wu, S. C., Zhang, Y. (2010). Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 11, 607–620. doi: 10.1038/nrm2950
Xie, N., Zhou, Y., Sun, Q., Tang, B. (2018). Novel Epigenetic techniques provided by the CRISPR/Cas9 system. Stem Cells Int. 2018, 7834175. doi: 10.1155/2018/7834175
Xuan, A., Long, D., Li, J., Ji, W., Hong, L., Zhang, M., et al. (2012). Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 90, 463–468. doi: 10.1016/j.lfs.2012.01.001
York, E. M., Petit, A., Roskams, A. J. (2013). Epigenetics of neural repair following spinal cord injury. Neurotherapeutics 10, 757–770. doi: 10.1007/s13311-013-0228-z
Zaret, K. S. (2018). Pioneering the chromatin landscape. Nat. Genet. 50, 167–169. doi: 10.1038/s41588-017-0038-z
Zaret, K. S., Carroll, J. S. (2011). Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 25, 2227–2241. doi: 10.1101/gad.176826.111
Zhang, H. Y., Zheng, S. J., Zhao, J. H., Zhao, W., Zheng, L. F., Zhao, D., et al. (2011). MicroRNAs 144, 145, and 214 are down-regulated in primary neurons responding to sciatic nerve transection. Brain Res. 1383, 62–70. doi: 10.1016/j.brainres.2011.01.067
Zhang, J., Chen, S. R., Chen, H., Pan, H. L. (2018a). RE1-silencing transcription factor controls the acute-to-chronic neuropathic pain transition and. J. Biol. Chem. 293, 19078–19091. doi: 10.1074/jbc.RA118.005846
Zhang, S., Fujita, Y., Matsuzaki, R., Yamashita, T. (2018b). Class I histone deacetylase (HDAC) inhibitor CI-994 promotes functional recovery following spinal cord injury. Cell Death Dis. 9, 460. doi: 10.1038/s41419-018-0543-8
Zhang, Y., Ueno, Y., Liu, X. S., Buller, B., Wang, X., Chopp, M., et al. (2013). The MicroRNA-17-92 cluster enhances axonal outgrowth in embryonic cortical neurons. J. Neurosci. 33, 6885–6894. doi: 10.1523/JNEUROSCI.5180-12.2013
Zhang, Z. Y., Zhang, Z., Fauser, U., Schluesener, H. J. (2007). Global hypomethylation defines a sub-population of reactive microglia/macrophages in experimental traumatic brain injury. Neurosci. Lett. 429, 1–6. doi: 10.1016/j.neulet.2007.09.061
Zhao, B. S., Roundtree, I. A., He, C. (2017). Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42. doi: 10.1038/nrm.2016.132
Zhou, S., Shen, D., Wang, Y., Gong, L., Tang, X., Yu, B., et al. (2012). microRNA-222 targeting PTEN promotes neurite outgrowth from adult dorsal root ganglion neurons following sciatic nerve transection. PLoS One 7, e44768. doi: 10.1371/journal.pone.0044768
Zhou, S., Yu, B., Qian, T., Yao, D., Wang, Y., Ding, F., et al. (2011). Early changes of microRNAs expression in the dorsal root ganglia following rat sciatic nerve transection. Neurosci. Lett. 494, 89–93. doi: 10.1016/j.neulet.2011.02.064
Keywords: neuroepigenetics, axon regeneration, epigenetic regulation, chromatin accessibility, neuroinflammation, spinal cord injury, CNS injury, neural repair
Citation: Wahane S, Halawani D, Zhou X and Zou H (2019) Epigenetic Regulation Of Axon Regeneration and Glial Activation in Injury Responses. Front. Genet. 10:640. doi: 10.3389/fgene.2019.00640
Received: 30 November 2018; Accepted: 18 June 2019;
Published: 09 July 2019.
Edited by:
Trygve Tollefsbol, University of Alabama at Birmingham, United StatesReviewed by:
Valeria Cavalli, Washington University School of Medicine in St. Louis, St. Louis, United StatesDietmar Spengler, Max Planck Institute of Psychiatry,Germany
Copyright © 2019 Wahane, Halawani, Zhou and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyan Zou, aG9uZ3lhbi56b3VAbXNzbS5lZHU=
†ORCID: Hongyan Zou, orcid.org/0000-0003-4216-5607; Shalaka Wahane, orcid.org/0000-0002-3795-6407; Dalia Halawani, orcid.org/0000-0003-4738-0656
‡These authors have contributed equally to this work.