 Christa E. Flück1
Christa E. Flück1 Laura Audí2Mónica Fernández-Cancio2
Laura Audí2Mónica Fernández-Cancio2 Kay-Sara Sauter1
Kay-Sara Sauter1 Idoia Martinez de LaPiscina3Luis Castaño4Isabel Esteva5
Idoia Martinez de LaPiscina3Luis Castaño4Isabel Esteva5 Núria Camats2*
Núria Camats2*- 1Pediatric Endocrinology, Diabetology and Metabolism, Department of Pediatrics and Department of BioMedical Research, Bern University Hospital and University of Bern, Bern, Switzerland
- 2Growth and Development Research Unit, Vall d’Hebron Research Institute (VHIR), Center for Biomedical Research on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Barcelona, Spain
- 3Endocrinology and Diabetes Research Group, BioCruces Bizkaia Health Research Institute, Cruces University Hospital, CIBERDEM, CIBERER, University of the Basque Country (UPV-EHU), Barakaldo, Spain
- 4Pediatric Endocrinology Section, Cruces University Hospital, Endocrinology and Diabetes Research Group, BioCruces Bizkaia Health Research Institute, CIBERDEM, CIBERER, University of the Basque Country (UPV-EHU), Barakaldo, Spain
- 5Endocrinology Section, Gender Identity Unit, Regional University Hospital of Malaga, Málaga, Spain
Disorders/differences of sex development (DSD) are the result of a discordance between chromosomal, gonadal, and genital sex. DSD may be due to mutations in any of the genes involved in sex determination and development in general, as well as gonadal and/or genital development specifically. MAMLD1 is one of the recognized DSD genes. However, its role is controversial as some MAMLD1 variants are present in normal individuals, several MAMLD1 mutations have wild-type activity in functional studies, and the Mamld1-knockout male mouse presents with normal genitalia and reproduction. We previously tested nine MAMLD1 variants detected in nine 46,XY DSD patients with broad phenotypes for their functional activity, but none of the mutants, except truncated L210X, had diminished transcriptional activity on known target promoters CYP17A1 and HES3. In addition, protein expression of MAMLD1 variants was similar to wild-type, except for the truncated L210X. We hypothesized that MAMLD1 variants may not be sufficient to explain the phenotype in 46,XY DSD individuals, and that further genetic studies should be performed to search for additional hits explaining the broad phenotypes. We therefore performed whole exome sequencing (WES) in seven of these 46,XY patients with DSD and in one 46,XX patient with ovarian insufficiency, who all carried MAMLD1 variants. WES data were filtered by an algorithm including disease-tailored lists of MAMLD1-related and DSD-related genes. Fifty-five potentially deleterious variants in 41 genes were identified; 16/55 variants were reported in genes in association with hypospadias, 8/55 with cryptorchidism, 5/55 with micropenis, and 13/55 were described in relation with female sex development. Patients carried 1-16 variants in 1-16 genes together with their MAMLD1 variation. Network analysis of the identified genes revealed that 23 genes presented gene/protein interactions with MAMLD1. Thus, our study shows that the broad phenotypes of individual DSD might involve multiple genetic variations contributing towards the complex network of sexual development.
Introduction
Disorders/differences of sex development (DSD) occur when there is a discordance between chromosomal, gonadal, and genital sex (Ostrer, 2014). DSD may be due to mutations in any of the genes involved in sex determination and development in general, as well as gonadal and/or genital development specifically (Ostrer, 2014).
MAMLD1 (Xq28, OMIM 300120) is one of the recognized DSD-related genes (Fukami et al., 2006; Baxter et al., 2015). Variations in MAMLD1 sequence have been described mainly in 46,XY DSD individuals, mostly associated with hypospadias (Fukami et al., 2006; Kalfa et al., 2008; Chen et al., 2010; Kalfa et al., 2012; Metwalley and Farghaly, 2012; Camats et al., 2015; Igarashi et al., 2015; Eggers et al., 2016), but also with other DSD phenotypes, including micropenis (Chen et al., 2010; Kalfa et al., 2012; Camats et al., 2015; Liu et al., 2017), and/or crypthorchidism (Fukami et al., 2006; Kalfa et al., 2008; Kalfa et al., 2012; Camats et al., 2015), 46,XY with female external genitalia (Fukami et al., 2006; Camats et al., 2015) and 46,XY with complete gonadal dysgenesis (Ruiz-Arana et al., 2015). Furthermore, one homozygous MAMLD1 variant was also reported in a 46,XX patient with gonadal dysgenesis, primary amenorrhea, bilateral streak gonads and clitoromegaly (Brandao et al., 2011).
However, the role of MAMLD1 in sex development is controversial for several reasons: a) some MAMLD1 variants are present in the normal population (Fukami et al., 2006; Chen et al., 2010; Gaspari et al., 2011; Kalfa et al., 2011); b) the same MAMLD1 variant may be present in patients with different phenotypes (Camats et al., 2015); c) MAMLD1 variants are not present in all DSD individuals of the same family (Fukami et al., 2006); d) several MAMLD1 mutations present wild-type activity in functional studies (Camats et al., 2015); and e) the Mamld1-knockout male mouse presents with normal genitalia and reproduction (Miyado et al., 2012; Miyado et al., 2017).
MAMLD1 is expressed in human fetal and adult testis and in human ovaries (Fukami et al., 2006; O’Shaughnessy et al., 2007; Camats et al., 2015), and seems to be involved in sex development in fetal life and in adult reproductive function. Yet its exact role is not clear. It is expressed in mice gonadal cells during start of androgen biosynthesis up to male external genitalia formation and is therefore thought to be involved in the expression of Leydig-cell genes (Miyado et al., 2012), as well as supporting testosterone production in critical periods of male development (Fukami et al., 2008; Nakamura et al., 2011). In contrast, Mamld1-KO mice present normal external genitalia (but small testes and reduced seminiferous tubule size and proliferating germ cells) and reproduce similarly to wild-type mice (Miyado et al., 2012; Miyado et al., 2017). These findings challenge the role of MAMLD1 in sex development.
In a previous study, we tested functional activity of nine MAMLD1 variants detected in nine 46,XY DSD patients with broad phenotypes (Camats et al., 2015). None of the MAMLD1 mutants, except truncated L210X, had diminished transcriptional activity on known target promoters CYP17A1 and HES3. In addition, protein expression of MAMLD1 variants was similar to wild-type, except for the truncated L210X. We therefore hypothesized that MAMLD1 variants may not be sufficient to explain the phenotype in 46,XY DSD carriers, and that further genetic studies should be performed to search for additional hits explaining the broad variability.
In the past decade, High throughput sequencing (HTS) has changed the genetic approach in research and diagnostics. Whole-exome sequencing (WES) has led to the discovery of many new genes and has given insight into complex traits. Oligogenic inheritance is currently discovered for several disorders by HTS. In the field of sex development, digenic inheritance has recently been suggested in a 46,XY DSD patient with gonadal dysgenesis (NR5A1 and MAP3K1 variants) (Mazen et al., 2016); in a family with 46,XY DSD males (NR5A1 variants) and 46,XX POF females (NR5A1 and TBX2) (Werner et al., 2017); as well as in a DSD patient with ambiguous genitalia, micropenis, and inguinal testes (SEMA3A and AKR1C4) (Fan et al., 2017). Similarly, we found oligogenic origin of disease in heterozygous NR5A1 46,XY DSD patients by performing WES (Camats et al., 2018). In addition, in patients with hypospadias, an oligogenic origin was suggested by two other NGS studies (Kon et al., 2015; Eggers et al., 2016).
Therefore, in this study, we performed WES in seven 46,XY patients with DSD (Camats et al., 2015) and one 46,XX patient with ovarian insufficiency, who all carried MAMLD1 variants. WES data were filtered by common tools and a disease-tailored algorithm including MAMLD1-related and DSD-related known and candidate genes. Additional hits in likely disease-causing genes were detected in all eight MAMLD1 carriers. Our results suggest that oligogenic origin of disease may contribute towards the broad phenotypes of human MAMLD1.
Patients and Methods
Patients
The study was approved by the Ethics Committee of Hospital Universitari Vall d'Hebron (Barcelona, Spain) (CEIC: PR(IR)23/2016). Written informed consent was obtained from the patients for the publication of their cases. Eight DSD patients (seven 46,XY and one 46,XX) each carrying one MAMLD1 variant were analyzed using WES. Clinical and genetic characteristics of 46,XY patients were previously reported in detail in Camats et al., (2015 and are summarized in Table 1 together with the 46,XX patient.
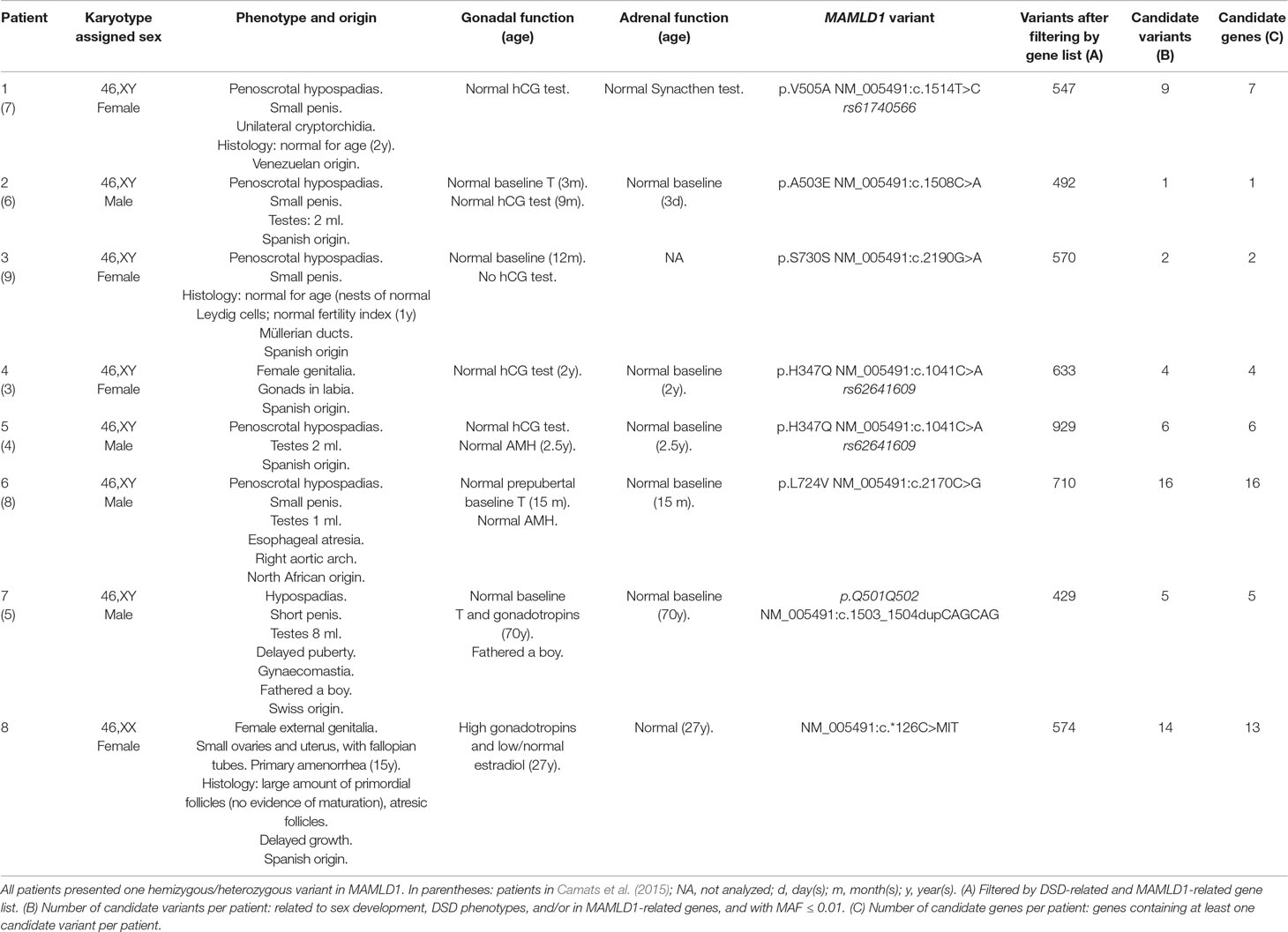
Table 1 Clinical, biochemical and genetic characteristics of the studied MAMLD1 patients.
DNA Extraction, WES and Bioinformatic Analysis
DNA was extracted from blood leukocytes using QiaCube (Qiagen, Hilden, Germany) or manually using a DNA isolation kit (Qiagen). WES was performed by CNAG (Centre Nacional d’Anàlisi Genòmica, Barcelona, Spain). Libraries were prepared with a SureSelect Human All Exon V5 capture kit (Agilent, Santa Clara, CA, USA) and sequenced with a HiSeq™ 2000 sequencing system (v3, 2x100, Illumina, San Diego, CA, USA). Putative candidate variants were confirmed by Sanger sequencing.
The genomic datasets were annotated (alignment with human genome hg19/grch37) and filtered with the functional annotation of genetic variants from HTS data (ANNOVAR; http://annovar.openbioinformatics.org/) (Wang et al., 2010), visualized and explored in Integrative Genomics Viewer (IGV, Broad Institute, Cambridge, MA, USA; https://www.broadinstitute.org/igv/ (Robinson et al., 2011). Frequencies of variants of relevant candidate genes were obtained from the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/about) (Lek et al., 2016) and the Collaborative Spanish Variant Server (CSVS; CIBERER BIER, Valencia, Spain; http://csvs.babelomics.org/; August 2018) (Dopazo et al., 2016). gnomAD includes gene variants from exome and genome sequencing data: 123,136 exomes and 15,496 genomes from unrelated individuals (from population and disease-specific studies). CSVS database includes (among others) exomes from a population of 267 healthy unrelated subjects.
WES data were filtered by a disease-tailored list of MAMLD1-related and DSD-related known and candidate genes (n = 606) similar to the algorithm previously set up for Camats et al., (2018). We generated a project-specific filter for DSD-related and MAMLD1-related genes by searching in published literature and databases. DSD-related genes are part of our DSD-gene database and tools (Camats et al., 2018), which have been currently updated. The DSD-related gene list included genes with reported (potentially) deleterious variants in patients with 46,XY and 46,XX DSD, genes with reported (potentially) disease-causing variants in syndromic patients with involvement of sex development, those “related” to DSD conditions in KO/mutant animal models (mice and rats), and also overexpressed, upregulated or downregulated genes in rodent embryonic gonadal cells (Camats et al., 2018). For the search for functional human partners of MAMLD1 and for possible interactions within interesting genes, the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, http://string-db.org/) (Jensen et al., 2009) and the Biological General Repository for Interaction Datasets (BioGRID, thebiogrid.org) (Stark, 2006) were used.
We used 30 pathogenic predictors to predict possible impact of amino acid substitutions on the structure, function and evolutionary conservation of corresponding human proteins and to predict impact on splicing. These in-silico predictors were accessed through ANNOVAR (Wang et al., 2010) annotation and run through Alamut Visual 2.11 (https://www.interactive-biosoftware.com/es/alamut-visual/). Functional exonic predictors were CADD (Combined Annotation Dependent Depletion of single-nucleotide and insertion/deletion variants, http://cadd.gs.washington.edu/) (Kircher et al., 2014), SIFT (Scale-invariant feature transform; http://sift.jcvi.org/), PolyPhen-2 (Polymorphism Phenotyping v2: HumDiv, HumVar; http://genetics.bwh.harvard.edu/pph2/index.shtml), Provean (http://provean.jcvi.org), MutationAssessor (http://mutationassessor.org/r3/), Mutation Taster (http://www.mutationtaster.org/), LRT, FATHEMM, Fathmm-MKL, PROVEAN, VEST3 (Variant Effect Scoring Tool), MetaSVM, MetaLR, MCAP, DANN and fitCons. Exonic predictors on evolutionary conservation were: GERP++, phyloP (vertebrate and mammalians), phastCons (vertebrate and mammalians) and SiPhy. Splicing predictors were: splicing predictors from dbscSNV ADA and RF and SPIDEX splicing predictor (DPSI), and those splicing predictors from Alamut Visual software were: SSF, MaxEnt, NNSPLICE, GeneSplicer and Ex-Skip.
The following bioinformatics software tools were used for the interpretation and classification of variants: InterVar (http://wintervar.wglab.org/, clinical interpretation of genetic variants by the ACMG/AMP 2015 guideline), VarSome (The Human Genomics Search Engine; https://varsome.com/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and Alamut Visual 2.11 (https://www.interactive-biosoftware.com/es/alamut-visual/). We searched for reported (potentially) disease-causing variants with the Human Gene Mutation Database (HGMD® Professional 2018.2, http://www.biobase-international.com/product/hgmd; Biobase) and dbSNP (http://www.ncbi.nlm.nih.gov/snp/). We used STRING for the search for interactions within genes carriers of interesting variants (DSD-related and/or MAMLD1-related). Data from STRING are extracted from known interactions (curated databases, experimentally determined interactions), predicted interactions (gene neighborhood, gene fusions, gene co-occurrence) and other inferred evidences such as text mining, co-expression and protein homology. We used Pubmed (https://www.ncbi.nlm.nih.gov/pubmed/) and OMIM (https://www.omim.org) to build our DSD gene list and for further data analysis. The datasets generated for this study are publicy available in dbSNP (Sherry, 2001): https://www.ncbi.nlm.nih.gov/projects/SNP/snp_viewBatch.cgi?sbid=1063030.
Variant Analysis Per Patient
After annotation, variant analysis was performed by the following steps. A) Each patient’s WES data were first filtered by our MAMLD1- and DSD-related known and candidate genes. B) We kept variants with MAF (minor allele frequency) ≤0.015 or not detected in gnomAD, and variants with the following predicted type, consequences and locations: splicing (intronic or exonic), exonic, intergenic, regulatory. C) We confirmed the correct annotation and location of variants by checking their alignment data in IGV (alignment with human genome hg19/grch37) (data not shown). D) We excluded variants that were considered non-relevant for our study: E.g. 1) variants found in more than two patients, 2) variants in repeat regions, 3) variants in genes or gene regions with high variability, 4) variants with low coverage and/or low quality, 5) variants with non-similar allelic depths. E) We revised variants with the annotated pathogenic predictors: functional exonic, evolutionary-conservation and splicing predictors (ANNOVAR and Alamut Visual software), as previously described. F) We run InterVar and VarSome to classify the variants, searched for reported (potentially) human disease-causing variants with the HGMD, and revised evidences of relationship with DSD, sex development and clinical phenotype of each patient with literature and database search. G) We used STRING to find out interactions among genes carriers of interesting variants (DSD-related and/or MAMLD1-related) (Figures 1 and 2). H) We checked MAF in a healthy cohort of Spanish population (CSVS: 267 unrelated healthy controls). I) We rejected variants with MAF ≥ 0.01 (gnomAD, CVSV, August 2018), thus less plausible to be a DSD-causing variant. Importantly, synonymous variants were not rejected because it has been shown that they may affect splicing.
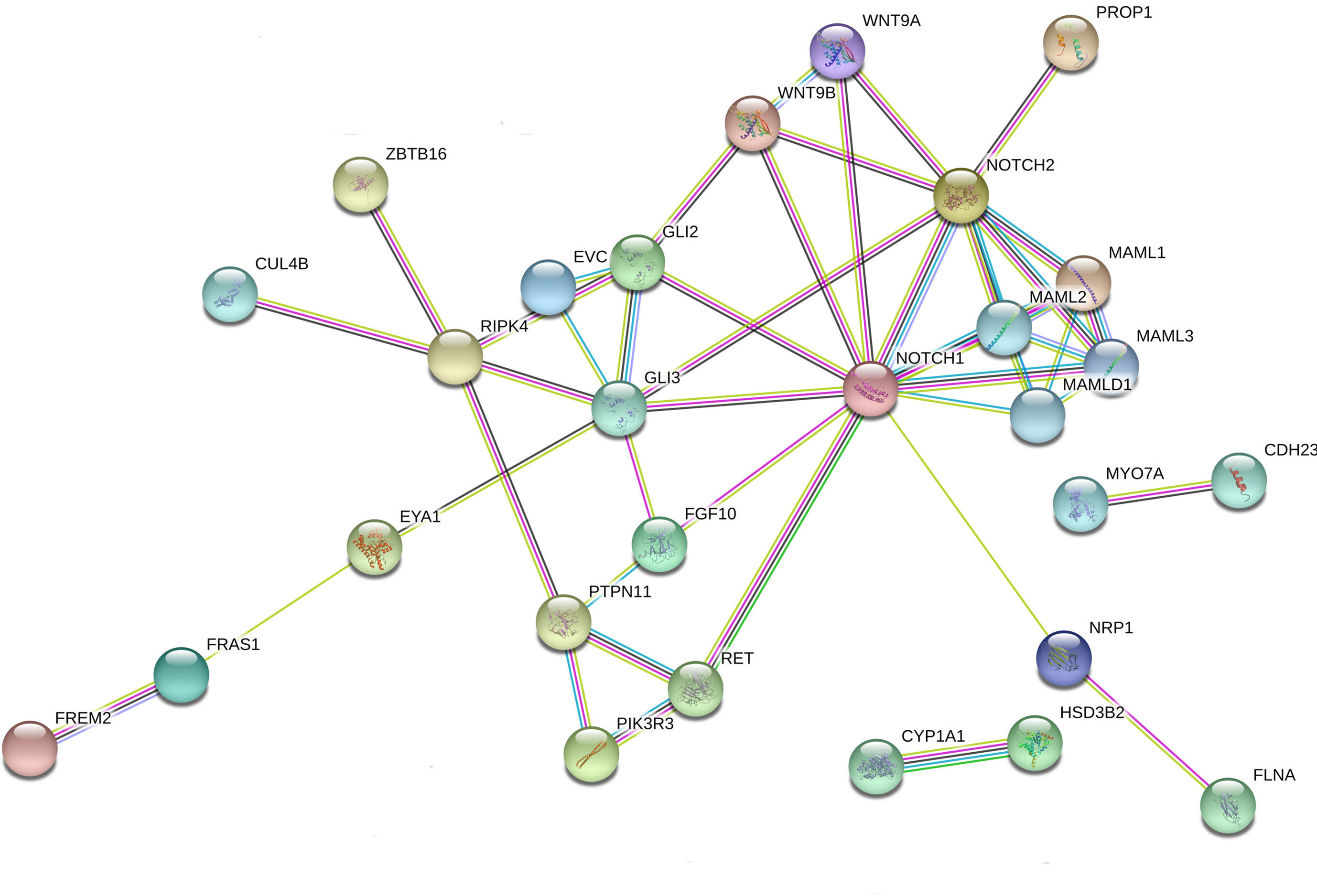
Figure 1 Interaction network of DSD- and MAMLD1-related genes identified in DSD individuals harboring genetic variants in MAMLD1. The scheme depicts an overview of detected genes and their interrelationship. For the search for functional human partners, the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, http://string-db.org/) was used. Nodes represent proteins. Filled nodes show proteins with known or predicted 3D structure. Empty nodes depict proteins with unknown 3D structure. Candidate genes are underlined. Known interactions correspond to curated databases (turquoise lines) and experimentally determined interactions (pink lines). Predicted interactions correspond to gene neighborhood (green lines), gene fusions (red lines) and gene co-occurrence (blue lines). Other interactions correspond to text mining (yellow lines), co-expression (black lines) and protein homology (violet lines).
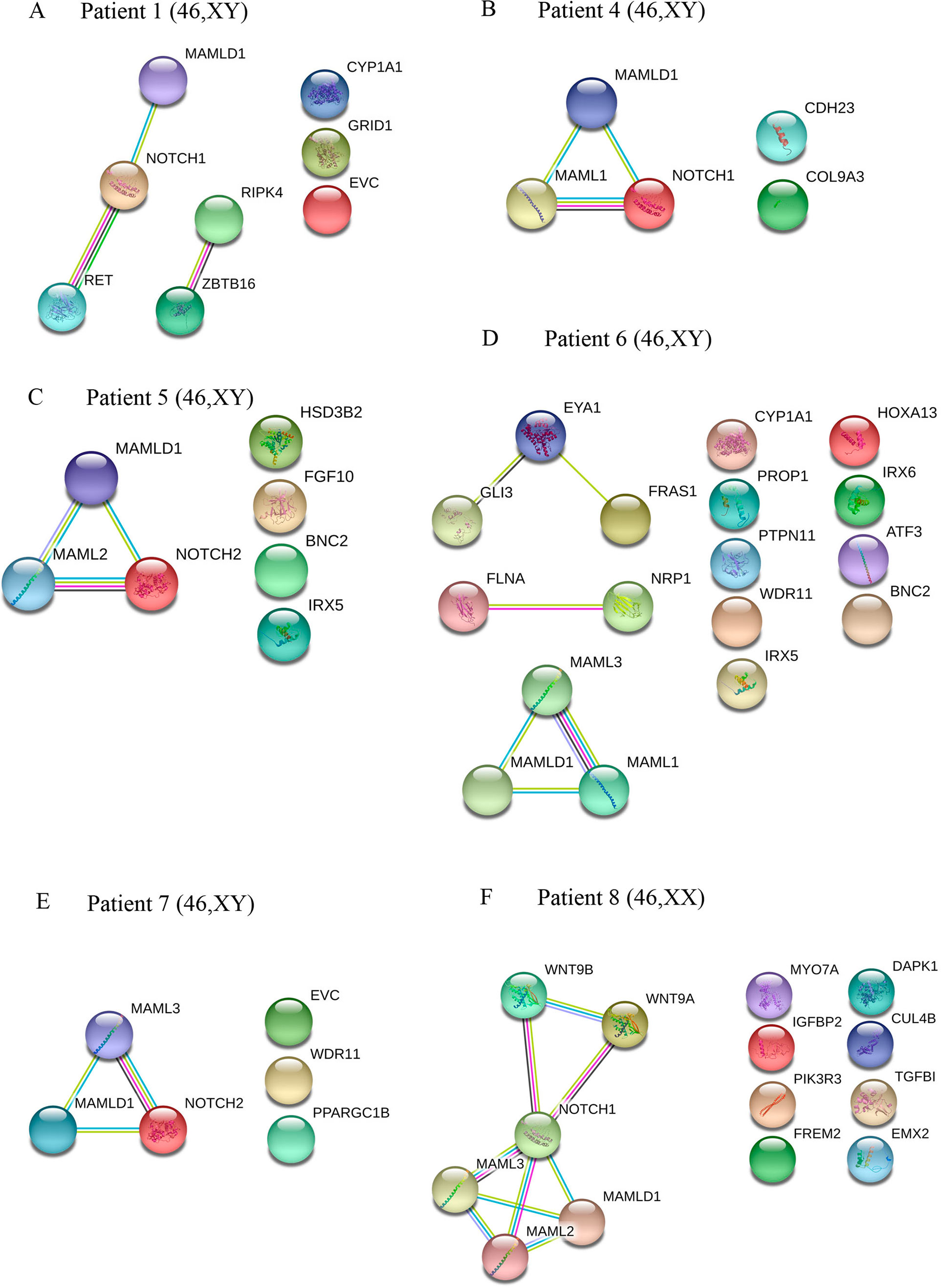
Figure 2 Interaction networks of DSD- and MAMLD1-related genes identified per single MAMLD1 individual. (A) to (F) correspond to the interaction networks per patient. For the search for functional human partner, the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, http://string-db.org/) was used. Nodes represent proteins. Filled nodes show proteins with known or predicted 3D structure. Empty nodes depict proteins with unknown 3D structure. Candidate genes are underlined. Known interactions correspond to curated databases (turquoise lines) and experimentally determined interactions (pink lines). Predicted interactions correspond to gene neighborhood (green lines), gene fusions (red lines) and gene co-occurrence (blue lines). Other interactions correspond to text mining (yellow lines), co-expression (black lines) and protein homology (violet lines). Genes with no interactions are on the right side of each network.
Results
WES performed in eight unrelated subjects (seven 46,XY and one 46,XX) harboring hemizygous/heterozygous MAMDL1 variants revealed several candidate gene variants that potentially contribute to each patient’s phenotype. A detailed summary of patients’ characteristics and number of variants and genes is shown in Table 1. A list of identified candidate variants and corresponding information from literature is given in Table 2.
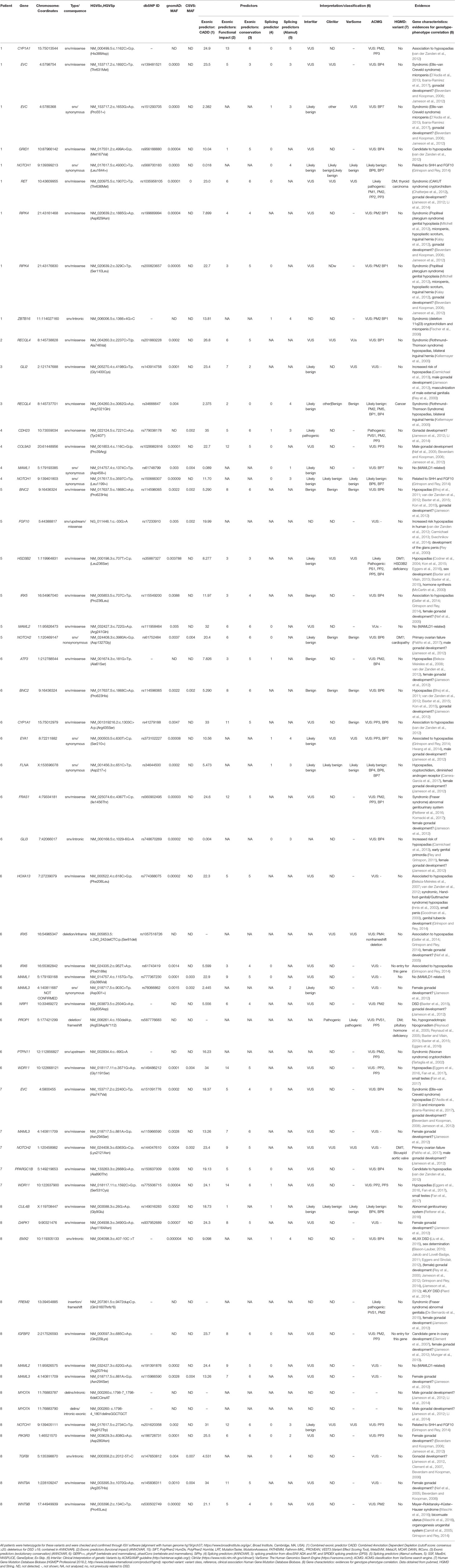
Table 2 Identified genes and variants per patient after specific filtering.
We identified a total of 55 potentially deleterious/candidate heterozygous/hemizygous variants in 41 genes in the eight hemizygous/heterozygous MAMLD1 patients (Tables 1 and 2). In the seven 46,XY patients 1–16 variants were found in a total of 1–16 genes, while the 46,XX MAMLD1 patient revealed 14 additional variants in 13 genes. (Tables 1 and 2).
Patient 1 harbored nine variants in seven genes: CYP1A1, EVC, GRID1, NOTCH1, RET, RIPK4 and ZBTB16, all of them associated to gonadal/genital anomalies (Table 2). Patient 2 carried one variant in RECQL4, associated with syndromic hypospadias (Table 2). Patient 3 presented two variants in two genes: GLI2 (associated to gonadal/genital anomalies) and RECQL4 (associated to syndromic hypospadias) (Table 2). Patient 4 had four variants in four genes: CDH23, COL9A3, MAML1 and NOTCH1; all, except MAML1, have been proposed to be associated with gonadal development (Table 2). In patient 5, six variants in six genes were found: BNC2, FGF10, HSD3B2, IRX5, MAML2 and NOTCH2; all, except MAML2, have also been associated with hypospadias or gonadal development (Table 2). Patient 6 carried 16 variants in 16 genes: ATF3, BNC2, CYP1A1, EYA1, FLNA, FRAS1, GLI3, HOXA13, IRX5, IRX6, MAML1, NRP1, MAML3, PROP1, PTPN11 and WDR11 (Table 2). Thirteen of these genes are associated with risk of hypospadias and/or syndromes that include abnormal gonadal/genital development, whereas MAML1 is unrelated, MAML3 has been proposed to be associated with female gonadal development and PROP1 has only been associated with anterior pituitary insufficiency/hypogonadotropic hypogonadism. In addition, six of these genes have previously been described in patients with aortic diseases and cardiopathies (Table 2). Patient 7 presented five variants in five genes: EVC, MAML3, NOTCH2, PPARGC1B and WDR11; four of them associated with hypospadias or male gonadal development and one, MAML3, with female gonadal development (Table 2). Finally, patient 8 harbored 14 variants in 13 genes: CUL4B, DAPK1, EMX2, FREM2, IGFBP2, MAML2, MAML3, MYO7A, NOTCH1, PIK3R3, TGFBI, WNT9A and WNT9B. Among them, only MAML2 has not been related to gonadal or genitourinary system development (Table 2).
The following genes showed variants in two patients: CYP1A1 in patients 1 and 6; EVC in patients 1 (2 variants) and 7; IRX5 in patients 5 and 6; MAML1 in patients 4 and 6; MAML2 in patients 5 and 8; NOTCH2 in patients 5 and 7; RECQL4 in patients 2 and 3 and WDR11 in patients 6 and 7 (Table 2). In addition, 2 genes presented variants in 3 patients: MAML3 (patients 6, 7 and 8) and NOTCH1 (patients 1, 4 and 8). Furthermore, RIPK4 presented 2 variants in patient 1. Finally, BNC2 variant c.1868C>A:p.(Pro623His) (MAF = 0.002) was detected in 2 patients (patient 1 and 7) and MAML3 variant c.881A>G:p.(Asn294Ser) (MAF = 0.0028) in patients 7 and 8 (Table 2).
We performed interactome analysis for the identified DSD genes using bioinformatic tools for the analysis of possible gene-protein interactions. The network comprising all genes identified is shown in Figure 1. Overall, a connection was found for 27 of the 41 genes. MAMLD1 connects directly to MAML1/2/3. Via NOTCH1/2 8 genes are in connection with MAMLD1, namely WNT9A/9B, GLI2/3, FGF10, RET, PROP1 and NRP1. Some of these genes are also central nodes for further connections; e.g. GLI3 for EVC, FGF10, GLI2, RIPK4 and EYA1; and RET for PIK3R3 with PTPN11, which also is connected with RIPK4. RIPK4 itself is a central node for ZBTB16, CUL4B, GLI3 and PTPN11. NRP1 is connected to FLNA and EYA1 connects with FRAS1 and FREM2. In addition, 2 isolated gene couples have been revealed by our analysis: CYP1A1-HSD3B2 and MYO7A-CDH23. These observations give an idea of the complex interactions among genes related to sex development.
The specific interactome of identified genes in patients 1 and 4 to 8 is shown in Figure 2. In patients 1, 4, 5, 7 and 8, MAMLD1 and MAMLD1-related genes (MAML1, MAML2 or MAML3) are directly related to NOTCH1/2 (Figures 2A–C, E, F). In patient 1, there are 2 networks: ZBTB16-RIPK4 and MAMLD1-NOTCH1-RET (Figure 2A). In patient 6, GLI3, EYA1 and FRAS1 as well as FLNA and NRP1 seem directly related (Figure 2D). In patient 8, NOTCH1 plays a central role connecting to WNT9A, WNT9B and MAMLD1 network (Figure 2F).
Discussion
Sex development is a very complex biological event which requires the concerted collaboration of a large network of genes in a spatial and temporal correct fashion. In the past, much has been learned about human sex development from monogenic DSD, but the broad spectrum of phenotypes in numerous DSD individuals remains a conundrum. Oligogenic disease has been proposed. In fact, multiple genetic hits, which might not be deleterious by themselves, have been found in several individuals with DSD (Kon et al., 2015; Eggers et al., 2016; Mazen et al., 2016; Werner et al., 2017; Camats et al., 2018). In a previous study of 46,XY DSD patients carrying MAMLD1 variants, we showed that none of the variants were functionally pathogenic except for a stop variant (Camats et al., 2015). In the present study, we searched for additional genetic hits in DSD patients harboring MAMLD1 mutations and manifesting with unexplained broad phenotypes. Using HTS and a custom-made algorithm including DSD- and MAMLD1-related genes from literature and databases, we identified potentially deleterious genetic variants in additional genes in all MAMLD1 individuals. Thus, we believe that the broad phenotype of individuals carrying MAMLD1 variants is due to additional genetic hits.
In our study, we identified 55 additional heterozygous/hemizygous variants in 41 genes in seven 46,XY DSD hemizygous and one 46,XX DSD heterozygous MAMLD1 patients. Among the 41 genes, 16 have been previously reported in humans with hypospadias (ATF3, BNC2, CYP1A1, EMX2, EYA1, FLNA, GLI3, GRID1, GLI2,, FGF10, HOXA13, HSD3B2, IRX5, IRX6, PPARGC1B and WDR11 (Table 2); 8 have been related to cryptorchidism (BNC2, FLNA, RET, RECQL4, NRP1, PTPN11, RIPK4 and ZBTB16), and 5 genes have been found in patients with micropenis (BNC2, EVC, FGF10, RIPK4 and ZBTB16). Also, 15 genes have been described in other types of DSD (CUL4B, EMX2, FRAS1, FREM2, HSD3B2, NOTCH2 and NRP1) (Table 2) and/or were reported in different syndromes (CYP1A1, EVC, FRAS1, HOXA13, PTPN11, RECQL4, RET, RIPK4 and ZBTB16) (Table 2). In addition, 27 genes had been previously described in the context of sex or gonadal development (ATF3, BNC2, CDH23, COL9A3, DAPK1, EMX2, EVC, EYA1, FLNA, FRAS1, FREM2, GLI2, GLI3, HOXA13, IGFBP2, IRX5, MAML3, MYO7A, NOTCH1, NOTCH2, NRP1, PIK3R3, RET, RIPK4, TGFBI, WNT9A and WNT9B). Thirteen of these genes have been found involved in female gonadal development and 46,XX DSD (ATF3, DAPK1, EMX2, FLNA, FRAS1, FREM2, GLI3, IGFBP2, IRX5, MAML3, PIK3R3, WNT9A and WNT9B), 8 of which in patient 8 (Table 2).
According to OMIM, almost all of our patients presented at least one variant in a gene with autosomal dominant inheritance (AD) (COL9A3, GLI2, FGF10, FLNA, EYA1, GLI3, HOXA13, NOTCH1, NOTCH2, PTPN11, RET, TGFB and WDR11), while other genes (CDH23, MYO7A and PPARGC1B) may have both AD and autosomal recessive (AR) inheritance. FLNA and CUL4B are X-linked (XLR), while CYP1A1, FREM2, EVC, HSD3B2, IRX5, PROP1, RAS1, RECQL4, RIPK4 and ZBTB16 are known for AR inheritance. No information on inheritance is currently available for the remaining genes including ATF3, BNC2, GRID1, DAPK1, IRX6, IGFBP2, MAML1, MAML2, MAML3, NRP1, PIK3R3, WNT9A and WNT9B.
The seven MAMLD1 patients with 46,XY DSD presented phenotypes from female external genitalia (patient 4) to variable degrees of hypospadias, cryptorchidism and small penis (Table 1). Interestingly, patient 4 with female external genitalia had normal T secretion. Similarly, patient 5 carrying a heterozygous HSD3B2 variant, had normal levels of 17OH-pregnenolone, DHEA and DHEA-S (data not shown). Patient 6, who presented with a right aortic arch, was found to carry variants in five genes (BNC2, FLNA, MAML1, NRP1 and PTPN11) that have been previously described in patients with heart and/or vascular anomalies (Tartaglia et al., 2002; Bhoj et al., 2011; Lee et al., 2014; Shaheen et al., 2015; Preuss et al., 2016; Chen et al., 2018). The 46,XX patient (patient 8), with primary amenorrhea, hypergonadotropic hypogonadism, normal female external genitalia and small uterus harbored gene variants involved in gonadal development and DSD (CUL4B, DAPK1, EMX2, FREM2, IGFBP2, MAML3, MYO7A, NOTCH1, PIK3R3, TGFBI, WNT9A and WNT9B; Table 2). Five of these genes (DAPK1, IGFBP2, MAML3, PIK3R3 and WNT9A) have so far only been related to female gonadal development (Table 2).
Overall, the genes detected in our eight studied patients with MAMLD1 variants have been previously reported in humans with hypospadias, cryptorchidism, micropenis, and other urogenital abnormalities; or they have been found involved in sexual and gonadal development. Also, some of them have been associated with specific syndromes in patients with genitourinary anomalies: CAKUT syndrome, Ellis–van Creveld syndrome, Fraser syndrome 1, Fraser syndrome 2, hand–foot–genital/Guttmacher syndrome, Noonan syndrome, Mayer–Rokitansky–Küster–Hauser syndrome, Popliteal pterygium syndrome and Rothmund–Thomson syndrome (see Table 2). However, none of the present patients presented a complete phenotype for any of these syndromes, maybe because none of the variants completely impairs gene expression and protein function, as inferred by the in silico analyses. Detailed information on these genes from current literature is given in Supplementary Materials (S1).
A search for an underlying network comprising variants in the identified genes related to MAMLD1 revealed a considerable number of genes which showed gene-gene, gene-protein or protein-protein interactions (Figures 1 and 2) suggesting that genetic variations in these genes may affect sex development. In addition, MAML3 was found in a network related to female gonadal development (Jameson et al., 2012). Accordingly, one variant in MAML3 was present in our 46,XX patient. The analysis of gene/protein network interactions per patient gives an idea of the complexity of the interactions among genes related to sex development. The more variants detected in DSD-related genes, the better to build an interaction network searching for clues on genetic relationship(s) for sex development. In our DSD individuals carrying MAMLD1 variants, three genes seemed prominent in the network analysis, NOTCH1, NOTCH2 and GLI3. NOTCH signaling is a highly conserved signaling pathway and comprises 4 transmembrane receptors. It is essential for the regulation of embryonic development of multiple organ systems including gonadal development (Windley and Wilhelm, 2016). NOTCH signaling is implicated in Leydig cell differentiation in an inhibitory regulatory fashion (Windley and Wilhelm, 2016). Autosomal dominant mutations in NOTCH1 cause the Adams–Oliver syndrome (OMIM 616028), while autosomal dominant mutations in NOTCH2 are reported in the Alagille syndrome 2 (OMIM 610205) and in the Hajdu–Cheney syndrome (OMIM 102500). By contrast, GLI3 is a zinc-finger transcription factor belonging to the desert hedgehog (DHH) signal transduction pathway. DHH signaling is essential for driving Leydig cell differentiation (Windley and Wilhelm, 2016). Thus, NOTCH and DHH signaling work together to regulate Leydig cell development (Windley and Wilhelm, 2016). Autosomal dominant mutations in GLI3 are described in the Pallister–Hall syndrome (OMIM 146510) or in the Greig cephalopolysyndactyly syndrome (OMIM 175700).
Taken together, our results expand the landscape of genes possibly involved in DSD by revealing both new and old players. Genetic platforms for DSD diagnostics currently consider about 270 genes that have been identified with monogenetic forms of DSD in (mostly) several independent individuals (Cools et al., 2018). Our eight MAMLD1 individuals share variants in 19 genes comprised in such DSD panels, including ATF3, BNC2, CUL4B, EVC, FLNA, FRAS1, FREM2, GLI3, HOXA13, HSD3B2, IRX5, NOTCH2, PROP1, PTPN11, RECQL4, RET, RIPK4, WDR11 and ZBTB16. By contrast, through our work 22 new genes are now added for considering with differences in sex development: CDH23, COL9A3, CYP1A1, DAPK1, EMX2, EYA1, FGF10, GLI2, GRID1, IGFBP2, IRX6, MAML1, MAML2, MAML3, MYO7A, NOTCH1, NRP1, PIK3R3, PPARGC1B, TGFBI, WNT9A and WNT9B.
Ideally, genetic variants are tested functionally for proof of their disease-causing effect in model systems. However, when finding multiple variants, which may all contribute only partially, such testing is no longer feasible. Therefore, the likelihood of disease-causing effect of identified variants was assessed in our study by established bioinformatic tools for genetics and by assessing the genotype-phenotype correlation in each patient with current knowledge from literature and databases in the field. In future studies with bigger sample size, next-generation statistical genetic analyses may be employed to identify associations between a group of variants and the complex trait of sex development (Weissenkampen et al., 2019).
In summary, HTS analysis indicates that the broad DSD phenotypes of MAMLD1 patients may be due to additional variants in other DSD-related genes. We found up to 55 additional genetic hits that may contribute to the DSD phenotype making an oligogenic causation plausible. Bioinformatic network analysis can help in interpreting complex genetic data and put identified single candidate genes into a greater perspective to understand their possible role in DSD biology.
Data Availability
The datasets generated for this study are publicly available in dbSNP (Sherry, 2001): https://www.ncbi.nlm.nih.gov/projects/SNP/snp_viewBatch.cgi?sbid=1063030.
Ethics Statement
The study was approved by the Ethics Committee of Hospital Universitari Vall d'Hebron (Barcelona, Spain) (CEIC: PR(IR)23/2016). Written informed consent was obtained from the patients for the publication of their cases.
Author Contributions
CF: Conceptualization, funding acquisition, investigation, methodology, interpretation, project administration, resources, supervision, writing – original draft preparation, writing – review and editing. LA: Interpretation, supervision, resources, visualization, writing – review and editing. MF-C: Investigation, writing – review and editing. K-SS: Validation, interpretation, writing – review and editing. IM: Resources, interpretation, writing – review and editing. LC: Resources, writing – review and editing. IE: Resources, writing – review and editing. NC: Conceptualization, data-curation, formal analysis, investigation, methodology, interpretation, project administration, supervision, visualization, writing – original draft preparation, writing – review and editing.
Funding
This work was supported by grants of the Swiss National Science Foundation (http://www.snf.ch) (320030-146127) to CF, the Instituto de Salud Carlos III (www.isciii.es/; Madrid, Spain) Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER, http://www.ciberer.es/) U-712 to MF-C, the Agency for Management of University and Research Grants (AGAUR; agaur.gencat.cat), Barcelona, Spain (2009SGR31) to LA, and by the Beatriu de Pinós Fellowship 2014 BP-B 00145 (AGAUR, Catalonia, Spain), the Instituto de Salud Carlos III (www.isciii.es/; Madrid, Spain) Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER; http://www.ciberer.es/) U-712 to NC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the patients, families, and their primary physicians for sharing their data for our study. We also thank Ida Paramonov for her help in the bioinformatic analysis. This work was supported by grants of the Swiss National Science Foundation (http://www.snf.ch) (320030-146127) to CF, the Instituto de Salud Carlos III (www.isciii.es/; Madrid, Spain) Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER, http://www.ciberer.es/) U-712 to MF-C, the Agency for Management of University and Research Grants (AGAUR; http://agaur.gencat.cat/en/inici/), Barcelona, Spain (2009SGR31) to LA, and by the Beatriu de Pinós Fellowship 2014 BP-B 00145 (AGAUR, Catalonia, Spain) and the Instituto de Salud Carlos III (www.isciii.es/; Madrid, Spain) Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER; http://www.ciberer.es/) U-712 to NC.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00746/full#supplementary-material
References
Baxter, R. M., Vilain, E. (2013). Translational genetics for diagnosis of human disorders of sex development. Annu. Rev. Genomics Hum. Genet. 14, 371–392. doi: 10.1146/annurev-genom-091212-153417
Baxter, R. M., Arboleda, V. A., Lee, H., Barseghyan, H., Adam, M. P., Fechner, P. Y., et al. (2015). Exome sequencing for the diagnosis of 46,XY disorders of sex development. J. Clin. Endocrinol. Metab. 100, E333–E344. doi: 10.1210/jc.2014-2605
Beleza-Meireles, A., Lundberg, F., Lagerstedt, K., Zhou, X., Omrani, D., Frisen, L., et al. (2007). FGFR2, FGF8, FGF10 and BMP7 as candidate genes for hypospadias. Eur. J. Hum. Genet. 15, 405–410. doi: 10.1038/sj.ejhg.5201777
Beleza-Meireles, A., Töhönen, V., Söderhäll, C., Schwentner, C., Radmayr, C., Kockum, I., et al. (2008). Activating transcription factor 3: a hormone responsive gene in the etiology of hypospadias. Eur. J. Endocrinol. 158 (5), 729–739. doi: 10.1530/EJE-07-0793
Beverdam, A., Koopman, P. (2006). Expression profiling of purified mouse gonadal somatic cells during the critical time window of sex determination reveals novel candidate genes for human sexual dysgenesis syndromes. Hum. Mol. Genet. 15, 417–431. doi: 10.1093/hmg/ddi463
Bhoj, E. J., Ramos, P., Baker, L. A., Cost, N., Nordenskjöld, A., Elder, F. F., et al. (2011). Human balanced translocation and mouse gene inactivation implicate Basonuclin 2 in distal urethral development. Eur. J. Hum. Genet. 19 (5), 540–546. doi: 10.1038/ejhg.2010.245
Biason-Lauber, A. (2010). Control of sex development. Best Pract. Res. Clin. Endocrinol. Metab. 24, 163–186. doi: 10.1016/j.beem.2009.12.002
Brandao, M. P., Costa, E. M., Fukami, M., Gerdulo, M., Pereira, N. P., Domenice, S., et al. (2011). MAMLD1 (mastermind-like domain containing 1) homozygous gain-of-function missense mutation causing 46,XX disorder of sex development in a virilized female. Adv. Exp. Med. Biol. 707, 129–131. doi: 10.1007/978-1-4419-8002-1_28
Camats, N., Fernández-Cancio, M., Audí, L., Mullis, P. E., Moreno, F., González Casado, I., et al. (2015). Human MAMLD1 gene variations seem not sufficient to explain a 46,XY DSD phenotype. PLoS One 10 (11), e0142831. doi: 10.1371/journal.pone.0142831
Camats, N., Fernández-Cancio, M., Audí, L., Schaller, A., Flück, C. E. (2018). Broad phenotypes in heterozygous NR5A1 46,XY patients with a disorder of sex development: an oligogenic origin? Eur. J. Hum. Genet. 26, 749–757. doi: 10.1038/s41431-018-0202-7
Carmichael, S. L., Ma, C., Choudhry, S., Lammer, E. J., Witte, J. S., Shaw, G. M. (2013). Hypospadias and genes related to genital tubercle and early urethral development. J. Urol. 190, 1884–1892. doi: 10.1016/j.juro.2013.05.061
Carrera-García, L., Rivas-Crespo, M. F., Fernández García, M. S. (2017). Androgen receptor dysfunction as a prevalent manifestation in young male carriers of a FLNA gene mutation. Am. J. Med. Genet. Part A. 173 (6), 1710–1713. doi: 10.1002/ajmg.a.38230
Carroll, T. J., Park, J. S., Hayashi, S., Majumdar, A., McMahon, A. P. (2005). Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell. (2), 283–292. doi: 10.1016/j.devcel.2005.05.016
Chatterjee, R., Ramos, E., Hoffman, M., Vanwinkle, J., Martin, D. R., Davis, T. K., et al. (2012). Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Hum. Genet. 131 (11), 1725–1738. doi: 10.1007/s00439-012-1181-3
Chen, M. H., Choudhury, S., Hirata, M., Khalsa, S., Chang, B., Walsh, C. A. (2018). Thoracic aortic aneurysm in patients with loss of function filamin a mutations: clinical characterization, genetics, and recommendations. Am. J. Med. Genet. Part A. 176 (2), 337–350. doi: 10.1002/ajmg.a.38580
Chen, Y., Thai, H. T., Lundin, J., Lagerstedt-Robinson, K., Zhao, S., Markljung, E., et al. (2010). Mutational study of the MAMLD1-gene in hypospadias. Eur. J. Med. Genet. 53, 122–126. doi: 10.1016/j.ejmg.2010.03.005
Clement, T. M., Anway, M. D., Uzumcu, M., Skinner, M. K. (2007). Regulation of the gonadal transcriptome during sex determination and testis morphogenesis: comparative candidate genes. Reproduction 134, 455–472. doi: 10.1530/REP-06-0341
Codner, E., Okuma, C., Iñiguez, G., Boric, M. A., Avila, A., Johnson, M. C., et al. (2004). Molecular study of the 3β-hydroxysteroid dehydrogenase gene type II in patients with hypospadias. J. Clin. Endocrinol. Metab. 89 (2), 957–964. doi: 10.1210/jc.2002-020873
Cools, M., Nordenström, A., Robeva, R., Hall, J., Westerveld, P., Flück, C., et al. (2018). Caring for individuals with a difference of sex development (DSD): a consensus statement. Nat. Rev. Endocrinol. 14, 415–429. doi: 10.1038/s41574-018-0010-8
D’Asdia, M. C., Torrente, I., Consoli, F., Ferese, R., Magliozzi, M., Bernardini, L., et al. (2013). Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. Eur. J. Med. Genet. 56 (2), 80–87. doi: 10.1016/j.ejmg.2012.11.005
De Bernardo, G., Giordano, M., Di Toro, A., Sordino, D., De Brasi, D. (2015). Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital. J. Pediatr. 41, 86. doi: 10.1186/s13052-015-0195-6
Dopazo, J., Amadoz, A., Bleda, M., Garcia-Alonso, L., Alemán, A., García-García, F., et al. (2016). 267 Spanish exomes reveal population-specific differences in disease-related genetic variation. Mol. Biol. Evol. 33, 1205–1218. doi: 10.1093/molbev/msw005
Eggers, S., Sinclair, A. (2012). Mammalian sex determination-insights from humans and mice. Chromosom. Res. 20 (1), 215–238. doi: 10.1007/s10577-012-9274-3
Eggers, S., Sadedin, S., van den Bergen, J. A., Robevska, G., Ohnesorg, T., Hewitt, J., et al. (2016). Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 17 (1), 243. doi: 10.1186/s13059-016-1105-y
Fan, Y., Zhang, X., Wang, L., Wang, R., Huang, Z., Sun, Y., et al. (2017). Diagnostic application of targeted next-generation sequencing of 80 genes associated with disorders of sexual development. Sci. Rep. 7, 44536. doi: 10.1038/srep44536
Fischer, S., Kohlhase, J., Böhm, D., Schweiger, B., Hoffmann, D., Heitmann, M., et al. (2008). Biallelic loss of function of the promyelocytic leukaemia zinc finger (PLZF) gene causes severe skeletal defects and genital hypoplasia. J. Med. Genet. 45 (11), 731–737. doi: 10.1136/jmg.2008.059451
Fukami, M., Wada, Y., Miyabayashi, K., Nishino, I., Hasegawa, T., Nordenskjold, A., et al. (2006). CXorf6 is a causative gene for hypospadias. Nat. Genet. 38, 1369–1371. doi: 10.1038/ng1900
Fukami, M., Wada, Y., Okada, M., Kato, F., Katsumata, N., Baba, T., et al. (2008). Mastermind-like domain-containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J. Biol. Chem. 283, 5525–5532. doi: 10.1074/jbc.M703289200
Gaspari, L., Paris, F., Philibert, P., Audran, F., Orsini, M., Servant, N., et al. (2011). “Idiopathic” partial androgen insensitivity syndrome in 28 newborn and infant males: impact of prenatal exposure to environmental endocrine disruptor chemicals? Eur. J. Endocrinol. 165, 579–587. doi: 10.1530/EJE-11-0580
Geller, F., Feenstra, B., Carstensen, L., Pers, T. H., Van Rooij, I. A. L. M., Körberg, I. B., et al. (2014). Genome-wide association analyses identify variants in developmental genes associated with hypospadias. Nat. Genet. 46 (9), 957–963. doi: 10.1038/ng.3063
Goodman, F. R., Bacchelli, C., Brady, A. F., Brueton, L. A., Fryns, J.-P., Mortlock, D. P., et al. (2000). Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am. J. Hum. Genet. 67 (1), 197–202. doi: 10.1086/302961
Grinspon, R. P., Rey, R. A. (2014). When hormone defects cannot explain it: malformative disorders of sex development. Birth Defects Res. Part C Embryo Today Rev. 102, 359–373. doi: 10.1002/bdrc.21086
Hwang, D. Y., Dworschak, G. C., Kohl, S., Saisawat, P., Vivante, A., Hilger, A. C., et al. (2014). Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 85 (6), 1429–1433. doi: 10.1038/ki.2013.508
Ibarra-Ramirez, M., Campos-Acevedo, L. D., Lugo-Trampe, J., Martínez-Garza, L. E., Martinez-Glez, V., Valencia-Benitez, M., et al. (2017). Phenotypic Variation in patients with homozygous c.1678G > T mutation in EVC Gene: report of two mexican families with ellis-van creveld syndrome. Am. J. Case Rep. 18, 1325–1329. doi: 10.12659/AJCR.905976
Igarashi, M., Wada, Y., Kojima, Y., Miyado, M., Nakamura, M., Muroya, K., et al. (2015). Novel splice site mutation in MAMLD1 in a patient with hypospadias. Sex Dev. 9 (3), 130–135. doi: 10.1159/000380842
Innis, J. W., Goodman, F. R., Bacchelli, C., Williams, T. M., Mortlock, D. P., Sateesh, P., et al. (2002). A HOXA13 allele with a missense mutation in the homeobox and a dinucleotide deletion in the promoter underlies Guttmacher syndrome. Hum. Mutat. 19 (5), 573–574. doi: 10.1002/humu.9036
Jakob, S., Lovell-Badge, R. (2011). Sex determination and the control of Sox9 expression in mammals. FEBS J. 278, 1002–1009. doi: 10.1111/j.1742-4658.2011.08029.x
Jameson, S. A., Natarajan, A., Cool, J., DeFalco, T., Maatouk, D. M., Mork, L., et al. (2012). Temporal transcriptional profiling of somatic and germ cells reveals biased lineage priming of sexual fate in the fetal mouse gonad. PLoS Genet. 8, e1002575. doi: 10.1371/journal.pgen.1002575
Jensen, L. J., Kuhn, M., Stark, M., Chaffron, S., Creevey, C., Muller, J., et al. (2009). STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416. doi: 10.1093/nar/gkn760
Kalay, E., Sezgin, O., Chellappa, V., Mutlu, M., Morsy, H., Kayserili, H., et al. (2012). Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am. J. Hum. Genet. 90 (1), 76–85 doi: 10.1016/j.ajhg.2011.11.014
Kalfa, N., Cassorla, F., Audran, F., Oulad Abdennabi, I., Philibert, P., Beroud, C., et al. (2011). Polymorphisms of MAMLD1 gene in hypospadias. J. Pediatr. Urol. 7, 585–591. doi: 10.1016/j.jpurol.2011.09.005
Kalfa, N., Fukami, M., Philibert, P., Audran, F., Pienkowski, C., Weill, J., et al. (2012). Screening of MAMLD1 mutations in 70 children with 46,XY DSD: identification and functional analysis of two new mutations. PLoS One 7, e32505. doi: 10.1371/journal.pone.0032505
Kalfa, N., Liu, B., Klein, O., Audran, F., Wang, M. H., Mei, C., et al. (2008). Mutations of CXorf6 are associated with a range of severities of hypospadias. Eur. J. Endocrinol. 159, 453–458. doi: 10.1530/EJE-08-0085
Kellermayer, R., Sütonen, H. A., Hadzsiev, K., Kestilä, M., Kosztolányi, G. (2005). A patient with Rothmund-Thomson syndrome and all features of RAPADILINO. Arch. Dermatol. 141 (5), 617–620. doi: 10.1001/archderm.141.5.617
Kircher, M., Witten, D. M., Jain, P., O’Roak, B. J., Cooper, G. M., Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Kon, M., Suzuki, E., Dung, V. C., Hasegawa, Y., Mitsui, T., Muroya, K., et al. (2015). Molecular basis of non-syndromic hypospadias: systematic mutation screening and genome-wide copy-number analysis of 62 patients. Hum. Reprod. 30, 499–506. doi: 10.1093/humrep/deu364
Kornacki, J., Sowinska-Seidler, A., Socha, M., Ropacka, M., Jamsheer, A. (2017). Prenatal diagnosis of Fraser syndrome using routine ultrasound examination, confirmed by exome sequencing: report of a novel homozygous missense FRAS1 mutation. Congenit. Anom. (Kyoto) 57, 37–38. doi: 10.1111/cga.12177
Lee, H., Deignan, J. L., Dorrani, N., Strom, S. P., Kantarci, S., Quintero-Rivera, F., et al. (2014). Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887. doi: 10.1001/jama.2014.14604
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Li, Y., Zheng, M., Lau, Y. F. (2014). The sex-determining factors SRY and SOX9 regulate similar target genes and promote testis cord formation during testicular differentiation. Cell Rep. 8, 723–733. doi: 10.1016/j.celrep.2014.06.055
Liu, S., Gao, X., Qin, Y., Liu, W., Huang, T., Ma, J., et al. (2015). Nonsense mutation of EMX2 is potential causative for uterus didelphysis: first molecular explanation for isolated incomplete müllerian fusion. Fertil. Steril. 103 (3), 769–774.e2. doi: 10.1016/j.fertnstert.2014.11.030
Liu, Y., Zhuang, L., Ye, W., Wu, M., Huang, Y. (2017). Association of MAMLD1 single-nucleotide polymorphisms with hypospadias in Chinese Han population. Front. Biosci. 22, 1173–1176. doi: 10.2741/4540
Mazen, I., Abdel-Hamid, M., Mekkawy, M., Bignon-Topalovic, J., Boudjenah, R., El Gammal, M., et al. (2016). Identification of NR5A1 mutations and possible digenic inheritance in 46,XY gonadal dysgenesis. Sex Dev. 10, 147–151. doi: 10.1159/000445983
McCartin, S., Russell, A. J., Fisher, R. A., Wallace, A. M., Arnhold, I. J. P., Mason, J. I., et al. (2000). Phenotypic variability and origins of mutations in the gene encoding 3β-hydroxysteroid dehydrogenase type II. J. Mol. Endocrinol. 24 (1), 75–82. doi: 10.1677/jme.0.0240075
Metwalley, K. A., Farghaly, H. S. (2012). X-linked congenital adrenal hypoplasia associated with hypospadias in an Egyptian baby: a case report. J. Med. Case Rep. 6, 428. doi: 10.1186/1752-1947-6-428
Mitchell, K., O’Sullivan, J., Missero, C., Blair, E., Richardson, R., Anderson, B., et al. (2012). Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am. J. Hum. Genet. 90 (1), 69–75. doi: 10.1016/j.ajhg.2011.11.013
Miyado, M., Nakamura, M., Miyado, K., Morohashi, K., Sano, S., Nagata, E., et al. (2012). Mamld1 deficiency significantly reduces mRNA expression levels of multiple genes expressed in mouse fetal Leydig cells but permits normal genital and reproductive development. Endocrinology 153 (12), 6033–6040. doi: 10.1210/en.2012-1324
Miyado, M., Yoshida, K., Miyado, K., Katsumi, M., Saito, K., Nakamura, S., et al. (2017). Knockout of murine Mamld1 impairs testicular growth and daily sperm production but permits normal postnatal androgen production and fertility. Int. J. Mol. Sci. 18. doi: 10.3390/ijms18061300
Munger, S. C., Natarajan, A., Looger, L. L., Ohler, U., Capel, B. (2013). Fine time course expression analysis identifies cascades of activation and repression and maps a putative regulator of mammalian sex determination. PLoS Genet. 9, e1003630. doi: 10.1371/journal.pgen.1003630
Nakamura, M., Fukami, M., Sugawa, F., Miyado, M., Nonomura, K., Ogata, T. (2011). Mamld1 knockdown reduces testosterone production and Cyp17a1 expression in mouse Leydig tumor cells. PLoS One 6, e19123. doi: 10.1371/journal.pone.0019123
Nef, S., Schaad, O., Stallings, N. R., Cederroth, C. R., Pitetti, J. L., Schaer, G., et al. (2005). Gene expression during sex determination reveals a robust female genetic program at the onset of ovarian development. Dev. Biol. 287, 361–377. doi: 10.1016/j.ydbio.2005.09.008
O’Shaughnessy, P. J., Baker, P. J., Monteiro, A., Cassie, S., Bhattacharya, S., Fowler, P. A. (2007). Developmental changes in human fetal testicular cell numbers and messenger ribonucleic acid levels during the second trimester. J. Clin. Endocrinol. Metab. 92, 4792–4801. doi: 10.1210/jc.2007-1690
Ostrer, H. (2014). Disorders of sex development (DSDs): an update. J. Clin. Endocrinol. Metab. 99, 1503–1509. doi: 10.1210/jc.2013-3690
Patiño, L. C., Beau, I., Carlosama, C., Buitrago, J. C., González, R., Suárez, C. F., et al. (2017). New mutations in non-syndromic primary ovarian insufficiency patients identified via whole-exome sequencing. Hum. Reprod. 32 (7), 1512–1520. doi: 10.1093/humrep/dex089
Piard, J., Mignot, B., Arbez-Gindre, F., Aubert, D., Morel, Y., Roze, V., et al. (2014). Severe sex differentiation disorder in a boy with a 3.8 Mb 10q25.3-q26.12 microdeletion encompassing EMX2. Am. J. Med. Genet. Part A. 164A (10), 2618–2622. doi: 10.1002/ajmg.a.36662
Preuss, C., Capredon, M., Wünnemann, F., Chetaille, P., Prince, A., Godard, B., et al. (2016). Family based whole exome sequencing reveals the multifaceted role of notch signaling in congenital heart disease. PLoS Genet. 12 (10), e1006335. doi: 10.1371/journal.pgen.1006335
Retterer, K., Juusola, J., Cho, M. T., Vitazka, P., Millan, F., Gibellini, F., et al. (2016). Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18 (7), 696–704. doi: 10.1038/gim.2015.148
Rey, R. A., Grinspon, R. P. (2011). Normal male sexual differentiation and aetiology of disorders of sex development. Best Pract. Res. Clin. Endocrinol. Metab. 25, 221–238. doi: 10.1016/j.beem.2010.08.013
Rey, R., Josso, N., Racine, C., (2000). “Sexual Differentiation,” in Endotext. Eds. De Groot, L. J., Chrousos, G., Dungan, K., Feingold, K. R., Grossman, A., Hershman, J. M. [South Dartmouth, (MA)].
Reynaud, R., Barlier, A., Vallette-Kasic, S., Saveanu, A., Guillet, M. P., Simonin, G., et al. (2005). An uncommon phenotype with familial central hypogonadism caused by a novel PROP1 gene mutant truncated in the transactivation domain. J. Clin. Endocrinol. Metab. 90 (8), 4880–4887. doi: 10.1210/jc.2005-0119
Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Ruiz-Arana, I. L., Hubner, A., Cetingdag, C., Krude, H., Gruters, A., Fukami, M., et al. (2015). A novel hemizygous mutation of MAMLD1 in a patient with 46,XY complete gonadal dysgenesis. Sex Dev. 9 (2), 80–85. doi: 10.1159/000371603
Shaheen, R., Al Hashem, A., Alghamdi, M. H., Seidahmad, M. Z., Wakil, S. M., Dagriri, K., et al. (2015). Positional mapping of PRKD1, NRP1 and PRDM1 as novel candidate disease genes in truncus arteriosus. J. Med. Genet. 52 (5), 322–329. doi: 10.1136/jmedgenet-2015-102992
Sherry, S. T. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. doi: 10.1093/nar/29.1.308
Stark, C. (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539. doi: 10.1093/nar/gkj109
Svechnikov, K., Stukenborg, J. B., Savchuck, I., Soder, O. (2014). Similar causes of various reproductive disorders in early life. Asian J. Androl. 16, 50–59. doi: 10.4103/1008-682X.122199
Tartaglia, M., Kalidas, K., Shaw, A., Song, X., Musat, D. L., Van Der Burgt, I., et al. (2002). PTPN11 Mutations in noonan syndrome : molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 70 (6), 1555–1563. doi: 10.1086/340847
van der Zanden, L. F., van Rooij, I. A., Feitz, W. F., Franke, B., Knoers, N. V., Roeleveld, N. (2012). Aetiology of hypospadias: a systematic review of genes and environment. Hum. Reprod. Update 18, 260–283. doi: 10.1093/humupd/dms002
Wang, K., Li, M., Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. 177 (5), 1939–46. doi: 10.1093/nar/gkq603
Wang, Z., Liu, B. C., Lin, G. T., Lin, C. S., Lue, T. F., Willingham, E., et al. (2007). up-regulation of estrogen responsive genes in hypospadias: microarray analysis. J. Urol.177 (5), 1939–1946. doi: 10.1016/j.juro.2007.01.014
Waschk, D. E. J., Tewes, A. C., Römer, T., Hucke, J., Kapczuk, K., Schippert, C., et al. (2016). Mutations in WNT9B are associated with Mayer-Rokitansky-Küster-Hauser syndrome. Clin. Genet. 89 (5), 590–596. doi: 10.1111/cge.12701
Weissenkampen, J. D., Jiang, Y., Eckert, S., Jiang, B., Li, B., Liu, D. J. (2019). Methods for the analysis and interpretation for rare variants associated with complex traits. Curr. Protoc. Hum. Genet. 101 (1), e83. doi: 10.1002/cphg.83
Werner, R., Mönig, I., Lünstedt, R., Wünsch, L., Thorns, C., Reiz, B., et al. (2017). New NR5A1 mutations and phenotypic variations of gonadal dysgenesis. PLoS One 12 (5), e0176720. doi: 10.1371/journal.pone.0176720
Keywords: whole exome sequencing, MAMLD1, disorders/differences of sex development, hypospadias, phenotype variability, oligogenic disorder
Citation: Flück CE, Audí L, Fernández-Cancio M, Sauter K-S, Martinez de LaPiscina I, Castaño L, Esteva I and Camats N (2019) Broad Phenotypes of Disorders/Differences of Sex Development in MAMLD1 Patients Through Oligogenic Disease. Front. Genet. 10:746. doi: 10.3389/fgene.2019.00746
Received: 21 March 2019; Accepted: 16 July 2019;
Published: 29 August 2019.
Edited by:
Jeff Schwartz, Griffith University, AustraliaReviewed by:
Maki Fukami, National Center for Child Health and Development (NCCHD), JapanVincent Russel Harley, Hudson Institute of Medical Research, Australia
Copyright © 2019 Flück, Audí, Fernández-Cancio, Sauter, Martinez de LaPiscina, Castaño, Esteva and Camats. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Núria Camats, bnVyaWEuY2FtYXRzQHZoaXIub3Jn