 Stephen R. Doyle1*
Stephen R. Doyle1* Geetha Sankaranarayanan1
Geetha Sankaranarayanan1 Fiona Allan2Duncan Berger1Pablo D. Jimenez Castro3,4James Bryant Collins3Thomas Crellen1,5María A. Duque-Correa1Peter Ellis1Tegegn G. Jaleta6Roz Laing7
Fiona Allan2Duncan Berger1Pablo D. Jimenez Castro3,4James Bryant Collins3Thomas Crellen1,5María A. Duque-Correa1Peter Ellis1Tegegn G. Jaleta6Roz Laing7 Kirsty Maitland7Catherine McCarthy1Tchonfienet Moundai8Ben Softley1
Kirsty Maitland7Catherine McCarthy1Tchonfienet Moundai8Ben Softley1 Elizabeth Thiele9Philippe Tchindebet Ouakou8
Elizabeth Thiele9Philippe Tchindebet Ouakou8 John Vianney Tushabe1,10Joanne P. Webster11Adam J. Weiss12
John Vianney Tushabe1,10Joanne P. Webster11Adam J. Weiss12 James Lok6
James Lok6 Eileen Devaney7
Eileen Devaney7 Ray M. Kaplan3James A. Cotton1Matthew Berriman1
Ray M. Kaplan3James A. Cotton1Matthew Berriman1 Nancy Holroyd1*
Nancy Holroyd1*- 1Parasites and Microbes, Wellcome Sanger Institute, Hinxton, United Kingdom
- 2Department of Life Sciences, Natural History Museum, London, United Kingdom
- 3Department of Infectious Diseases, College of Veterinary Medicine, University of Georgia, Athens, GA, United States
- 4Grupo de Parasitologia Veterinaria, Universidad Nacional de Colombia, Bogotá, Colombia
- 5Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
- 6Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 7Institute of Biodiversity Animal Health and Comparative Medicine, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, United Kingdom
- 8Ministry of Public Health, N’Djamena, Chad
- 9Department of Biology, Vassar College, Poughkeepsie, NY, United States
- 10Medical Research Council/Uganda Virus Research Institute and London School of Hygiene & Tropical Medicine Uganda Research Unit, Entebbe, Uganda
- 11Centre for Emerging, Endemic and Exotic Diseases, Department of Pathology and Population Sciences, Royal Veterinary College, University of London, Herts, United Kingdom
- 12Guinea Worm Eradication Program, The Carter Center, Atlanta, GA, United States
Whole-genome sequencing is being rapidly applied to the study of helminth genomes, including de novo genome assembly, population genetics, and diagnostic applications. Although late-stage juvenile and adult parasites typically produce sufficient DNA for molecular analyses, these parasitic stages are almost always inaccessible in the live host; immature life stages found in the environment for which samples can be collected non-invasively offer a potential alternative; however, these samples typically yield very low quantities of DNA, can be environmentally resistant, and are susceptible to contamination, often from bacterial or host DNA. Here, we have tested five low-input DNA extraction protocols together with a low-input sequencing library protocol to assess the feasibility of whole-genome sequencing of individual immature helminth samples. These approaches do not use whole-genome amplification, a common but costly approach to increase the yield of low-input samples. We first tested individual parasites from two species spotted onto FTA cards—egg and L1 stages of Haemonchus contortus and miracidia of Schistosoma mansoni—before further testing on an additional five species—Ancylostoma caninum, Ascaridia dissimilis, Dirofilaria immitis, Strongyloides stercoralis, and Trichuris muris—with an optimal protocol. A sixth species—Dracunculus medinensis—was included for comparison. Whole-genome sequencing followed by analyses to determine the proportion of on- and off-target mapping revealed successful sample preparations for six of the eight species tested with variation both between species and between different life stages from some species described. These results demonstrate the feasibility of whole-genome sequencing of individual parasites, and highlight a new avenue toward generating sensitive, specific, and information-rich data for the diagnosis and surveillance of helminths.
Introduction
Accurate methods for diagnosis and surveillance of helminth infections are of increasing interest in both human and animal health settings. Such approaches are typically proposed to monitor the presence and ultimately decline of populations targeted by large-scale control measures, such as mass drug administration (MDA) for the prevention and/or treatment of human helminth infections, or prophylactic treatment of domesticated animals. An ideal diagnostic will be sensitive to detect the parasite if in fact present, and specific, to identify the targeted parasite species in the presence of non-target material, such as other parasite species or the host. Ideally, samples taken for diagnostic purposes could be used to gather additional information beyond the presence or absence of a specific parasite, so the same material could be used, for example, to predict how well the infection will respond to drug treatment or how the parasite is related to other endemic or imported parasites. As most parasitic stages of helminths of humans and animals are naturally inaccessible in vivo (not accounting for potential availability of some mature stages of helminths following chemo-expulsion, for example, Ascaris lumbricoides and Trichuris trichiura), a diagnostic should also be informative on non-invasive stages of the parasite, such as eggs deposited in feces, or intermediate stages of the parasite’s life cycle that exist in the external environment.
A key challenge of working with environmental stages of helminth parasites is that they are often immature, for example, eggs or early stage larvae, and extremely small (for example, Haemonchus contortus eggs are approximately 75 × 44 μm and Schistosoma mansoni miracidia approximately 140 × 55 μm), limiting the amount of accessible material (e.g., DNA) available to be assayed. They are often environmentally resistant, and the same features that naturally protect the DNA from damage prior to reinfection make it difficult to extract DNA. In many cases, they are isolated from host feces and so are susceptible to bacterial contamination or from host tissues and so become contaminated with host DNA. Furthermore, samples may need to be transported efficiently to a laboratory setting without a significant loss of this already limited material. A number of approaches have been tested to preserve macromolecules from individual parasites for transport and storage, including ethanol, RNAlater, and Whatman® FTA® cards, from which robust PCR and microsatellite data could be profiled (Gower et al., 2007; Webster, 2009; Webster et al., 2012; Xiao et al., 2013; Marek et al., 2014; Boué et al., 2017; Campbell et al., 2017). Although, under ideal conditions, the detection of a single DNA molecule is possible, the limited material available per parasite has, to date, largely restricted assaying to a small number of loci, limiting the amount of information obtained from any individual parasite.
Genomic approaches offer an information-rich technology for diagnostic and surveillance applications. Increasing throughput and decreasing costs of whole-genome sequencing has resulted in the recent and steadily growing application of genomics in helminth parasitology, for example, for diagnostic applications, high-throughput amplicon sequencing for helminth species identification and community composition (Avramenko et al., 2015) and the presence of drug resistance alleles (Avramenko et al., 2019) have been described. Although low DNA concentrations are typically prohibitive for genome-wide approaches on individual parasites, a number of studies have successfully used whole-genome amplification on DNA extracted from single larval stages to perform reduced representation (Shortt et al., 2017) and exome (Le Clec’h et al., 2018; Platt et al., 2019) sequencing on miracidia of Schistosoma spp., and whole-genome sequencing of Haemonchus contortus L3 stage larvae (Doyle et al., 2018) and microfilariae of Wuchereria bancrofti (Small et al., 2018). Whole-genome amplification protocols do, however, add considerable expense per sample and can introduce technical artefacts, such as uneven and/or preferential amplification (potentially of contaminant sequences), chimeric sequences, and allele dropout (Tsai et al., 2014; Sabina and Leamon, 2015), that may lead to a reduction in genetic diversity, and in turn, relevance to the original unamplified material. The field of genomics is, however, rapidly advancing toward very low minimum sample input requirements, and single-cell protocols for DNA and RNA sequencing are now available. Such approaches have begun to be used on parasitic species, such as Plasmodium spp. (Trevino et al., 2017; Ngara et al., 2018; Reid et al., 2018; Howick et al., 2019), but are yet to be adopted by helminth parasitologists. Although these low-input, high-throughput approaches are not designed—and perhaps not currently suitable—for diagnostic applications, the development of molecular biology techniques for low-input sequencing could aid in the use of genomics for helminth applications. Here, we tested a number of low-input DNA extraction approaches for individual helminth samples stored on Whatman® FTA® cards, followed by low-input library preparation without whole-genome amplification and whole-genome sequencing. A total of five DNA extraction approaches were initially tested, after which the most promising approach was applied to multiple life stages from seven helminth species (with an additional species presented for comparison). The results presented here demonstrate the advancement of low-input whole-genome sequencing and may be broadly applicable to helminth and non-helminth species for which low-input whole-genome sequencing is to be performed. Finally, we discuss our results in the context of helminth diagnostics and surveillance.
Methods
Sample Collection
Samples representing accessible, immature life stages of a total of eight helminth species were tested, the collection of which is described below. A number of protocols and substrates or solutions have been described for the safe, effective storage of samples for downstream molecular biology applications; we chose Whatman® FTA® cards as a substrate to store and transport parasite material, as they are a relatively cost-effective method for storing samples without the need for specialized storage and transport conditions, for example, a cold chain from collection to sequencing. Removal of the logistical hurdles and their associated costs is particularly important for collection of specimens from endemic regions, for example, S. mansoni miracidia that were isolated from patients and purified in Africa before transported and stored on FTA cards for down-stream processing the UK as described here.
Ancylostoma caninum: Fresh feces from a research purpose-bred laboratory beagle (University of Georgia, AUP A2017 10-016-Y1-A0) infected with the Barrow isolate (drug-susceptible isolate from Barrow County Georgia, USA) were collected and made into a slurry with water, filtered through 425- and 180-µm sieves, and centrifuged at 2500 rpm for 5 min, after which the supernatant was discarded. Kaolin (Sigma-Aldrich, St. Louis, MO) was then added and resuspended in sodium nitrate (SPG 1.25–1.3) (Feca-Med®; Vedco, Inc. St Joseph, MO, USA). The tube was then centrifuged at 2500 rpm for 5 min, after which the supernatant was passed through a 30-µm sieve and rinsed with distilled water, and reduced to a volume of 10 to 15 mL. The volume was adjusted to one egg per 5 µl using distilled water. The eggs were stored at room temperature for 2 h before placing them onto the Whatman® FTA® cards. Eggs were also placed onto Nematode Growth Medium (NGM) plates (Sulston and Hodgkin, 1988) and incubated at 26°C to obtain the first-stage (L1) larvae. After 48 h, larvae were rinsed off the plate with distilled water and centrifuged at 1000 rpm for 5 min. Larvae were counted, and the concentration adjusted to one larva per 5 µl. The larvae were stored at room temperature for 2 h before placing them onto the Whatman® FTA® cards. To obtain third-stage (L3) larvae, eggs were isolated from fresh feces from a research purpose-bred laboratory beagle (University of Georgia, AUP A2017 10-016-Y1-A0) infected with the Worthy isolate (Worthy 3.1F3Pyr; multiple-drug resistant isolate originally isolated from a greyhound dog, Florida, USA). Eggs were placed onto NGM plates and incubated at 26°C. After 7 days, larvae were rinsed off the plate with distilled water and centrifuged at 1000 rpm for 5 min. Larvae were counted, and the concentration adjusted to one larva per 5 µl. The larvae were stored at room temperature for 2 h before placing them onto the Whatman® FTA® cards.
Ascaridia dissimilis: Eggs of A. dissimilis (Isolate Wi: North Carolina, USA) were isolated from excreta of experimentally infected turkeys. Water was added to the excreta and made into a slurry, which was filtered using a 425-µm and 180-µm sieve to remove large debris. The remaining particulates were placed into 50-mL centrifuge tubes and centrifuged at 433g for 7 min. Supernatant was removed, and the pellet was resuspended in a saturated sucrose solution with a specific gravity of 1.15. The suspension was centrifuged as before, and eggs were isolated from the top layer. Eggs were rinsed over a 20-µm sieve with water to remove residual sucrose and then concentrated to one egg per 5 µl using deionized water. Multiple 5-µl aliquots of the egg solution were dispensed using a micropipette onto the Whatman® FTA® card.
Dirofilaria immitis: Blood was taken from a dog infected with the macrocyclic lactone (ML)-resistant Yazoo strain (Yazoo: originally isolated from a dog in Yazoo City, Mississippi, USA; see Maclean et al. (2017) for complete history). To obtain microfilariae, blood was collected in heparin tubes and centrifuged for 30 min at 2500 rpm, after which the supernatant was discarded. The pellet was suspended in 3.8% sodium citrate (Sigma-Aldrich, St. Louis, MO) and 15% saponin (Sigma-Aldrich) was added in a 1:7 dilution. The tube was then vortexed and centrifuged for 30 min at 2500 rpm, after which the supernatant was discarded and the pellet resuspended in 3.8% sodium citrate to the original blood volume, vortexed, and then centrifuged for 4 min at 2500 rpm. The pellet was then resuspended and mixed in a 1:9 solution of 10𝖷 phosphate-buffered saline (PBS) (Thermo Fisher Scientific, Waltham, MA) and distilled water. The tube was then centrifuged for 4 min at 2500 rpm, and the pellet resuspended in PBS. The microfilariae were then counted and adjusted accordingly to have one microfilaria per 5 µl and stored at room temperature for 2 h before placing them onto the Whatman® FTA® cards.
Dracunculus medinensis: Individual L1 samples were obtained as progeny of an adult female worm manually extracted from an infected dog in Tarangara village, Chad (9.068611 N, 18.708611 E) in 2016. This extraction forms part of the standard containment and treatment procedure for Guinea worm infections, as agreed upon and sanctioned by the World Health Organization and country ministries of health. The adult worm was submerged in ethanol in a microcentrifuge tube for storage; L1 stage progeny that were found settled on the bottom of the tube were collected for analysis.
Haemonchus contortus: Eggs representing the F5 generation of a genetic cross (described in Doyle et al. (2018)) were collected from fresh feces from experimentally infected sheep housed at the Moredun Research Institute, UK. All experimental procedures were examined and approved by the Moredun Research Institute Experiments and Ethics Committee and were conducted under approved UK Home Office licenses (PPL 60/03899) in accordance with the Animals (Scientific Procedures) Act of 1986. Briefly, feces were mixed with tap water and passed through a 210-µm sieve, then centrifuged at 2500 rpm for 5 min in polyallomer tubes. The supernatant was discarded, before adding kaolin to the fecal pellet, vortexing, and resuspending in a saturated salt solution. After centrifugation at 1000 rpm for 10 min, the polyallomer tube was clamped to isolate eggs, which were collected on a 38-µm sieve and rinsed thoroughly with tap water. Eggs were incubated on NGM plates at 20oC for 48 h to hatch to L1 stage larvae. In addition to freshly collected material, eggs collected in the same manner then stored at −20oC, from a previous generation of the cross, were also tested. Eggs and L1 larvae were resuspended in PBS and spotted onto Whatman FTA cards in 3 µl per egg/L1.
Schistosoma mansoni: Three collections of S. mansoni samples were used in this work. The first were field samples collected from humans on Lake Victoria fishing villages in Uganda as part of the LaVIISWA trial (Sanya et al., 2018). Ethical approval for this trail was given by the Uganda Virus Research Institute (reference number GC127), the Uganda National Council for Science and Technology (reference number HS 1183), and the London School of Hygiene & Tropical Medicine (reference number 6187). Parasite eggs were collected from participants’ stool samples using a Pitchford-Visser funnel, washed with mineral water until clean, and transferred into a petri dish with water to be hatched in direct sunlight. After hatching, the miracidia were picked in 2-µl water using a pipette and placed on a Whatman® FTA® card for storage. The second were field samples collected as part of a repeated cross-sectional study of MDA exposure in school children in Uganda. Patient enrolment, including written consent, and sample collection have been described previously (Crellen et al., 2016). Ethical approvals for this study were granted by the Uganda National Council of Science and Technology (MoU sections 1.4, 1.5, 1.6) and the Imperial College Research Ethics Committee (EC NO: 03.36. R&D No: 03/SB/033E). Host stool was sampled 1 to 3 days prior to treatment with praziquantel (40 mg/kg) and albendazole (400 mg). A Pitchford-Visser funnel was used to wash and filter stool to retain parasite eggs. The filtrate was kept overnight in water and hatched the following morning in sunlight. Individual miracidia were isolated with a 20-μL pipette and transferred into petri dishes of nuclease-free water twice before spotting onto Whatman® FTA® cards. The third source of miracidia was derived from the livers of experimentally infected mice kept to maintain the S. mansoni life cycle at Wellcome Sanger Institute. Mouse infection protocols were approved by the Animal Welfare and Ethical Review Body (AWERB) of the Wellcome Sanger Institute, and in accordance with the UK Home Office approved project license P77E8A062. The AWERB is constituted as required by the UK Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012. Balb/C mice (6–8 weeks old) were infected with 250 cercariae, after which livers were collected on day 40 postinfection. Eggs were isolated from the liver tissues using collagenase digestion followed by percoll gradient, and were washed well with sterile PBS, before being hatched in sterile conditioned water. The hatched individual miracidia were spotted onto Whatman® FTA® cards.
Strongyloides stercoralis: The S. stercoralis UPD strain and the isofemale isolate PVO1 were maintained in purpose-bred, prednisone-treated mix breed dogs according to protocol 804883 approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC), USA. IACUC-approved research protocols and all routine husbandry care of the animals were conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, USA. Feces were collected, moisturized, and mixed with equal volume of charcoal and cultured at 22oC in 10-cm plates (Lok, 2007). Post-parasitic stage one and two larvae (L1/L2), free-living male and female adults, and infective third-stage (L3) were isolated by the Baermann technique after 24 and 48 h, and 6 days in these charcoal coprocultures, respectively (Lok, 2007; Jaleta et al., 2017). For this study, free living females (FL), L1 and L3 larvae were collected from the Baermann funnel sediments and washed three times using PBS.
Trichuris muris: Infection and maintenance of T. muris was conducted as described (Wakelin, 1967). The care and use of mice were in accordance with the UK Home Office regulations (UK Animals Scientific Procedures Act 1986) under the Project license P77E8A062 and were approved by the institutional AWERB. Female SCID mice (6–10 wk old) were orally infected under anaesthesia with isoflurane with a high dose (n = 400) of embryonated eggs from T. muris E-isolate. Mice were monitored daily for general condition and weight loss. At day 35 postinfection, mice were killed by exsanguination under terminal anesthesia, after which adult worms were harvested from cecums. Adult worms were cultured in RPMI 1640 supplemented with 10% fetal calf serum (v/v), 2 mM L-glutamine, penicillin (100 U/mL), and streptomycin (100 mg/mL; all Invitrogen), for 4 h or overnight, and eggs were collected. The eggs were allowed to embryonate for at least 6 weeks in distilled water, and infectivity was established by worm burden in SCID mice. T. muris eggs were hatched to produce sterile L1 larvae using 32% sodium hypochlorite in sterile water for 2 h at 37°C with 5% CO2. Eggs were washed with RPMI 1640 supplemented with 10% fetal calf serum (v/v), 2 mM l-glutamine, penicillin (100 U/mL), and streptomycin (100 mg/mL; all Invitrogen), and incubated at 37°C with 5% CO2 for 4 to 5 days until they hatched.
For each species, unless otherwise stated, pools of individuals were washed in sterile PBS, before being transferred to a petri dish. Individuals were identified under the microscope, after which 5-µl PBS containing an individual parasite was transferred onto a Whatman® FTA® card and dried for a minimum of 20 min at room temperature prior to storage or shipping to the Wellcome Sanger Institute, UK. The Whatman® FTA® cards with samples spotted were stored in a clean plastic bag in the dark at room temperature prior to analysis.
DNA Extraction
Five DNA extraction methods were tested for their ability to isolate and purify DNA compatible with whole-genome sequencing approaches. The choice of approaches was not systematic or comprehensive, but was available to us at the Wellcome Sanger Institute as low-input DNA extraction approaches that had the potential for high-throughput and/or automatic extraction protocols to generate sequencing libraries suitable for whole-genome sequencing. Despite the arbitrary choice of extraction kits, the comparison of multiple kits with multiple helminth species provides a unique data set that is not easily achieved at this scale outside of a research and development environment, and forms the basis for further comparison with other low-input kits as they become available.
To extract DNA, the sample spots on FTA cards were punched out manually into 96-well plates using either a Harris Punch or autonomously using robotics. The DNA extraction was carried out for each method as described below:
1. Nexttec (NXT): Extraction using the Nexttec 1-step DNA Isolation Kit for Tissues & Cells (cat: 10N.904; Waendel Technology Limited, UK) was performed according to manufacturer’s guidelines. Proteinase lysis buffer (75 µl) was used for digestion.
2. Bloodspot (BSP): The bloodspot extraction was derived from the QIAamp DNA Investigator Kit (cat: 51104/51106; Qiagen), following the “DNA Purification from Dried Blood Spots (QIAamp DNA Mini Kit)” protocol according to manufacturer’s guidelines.
3. CGP (CGP): Extraction using the CGP protocol (Moore et al., 2018) involved adding 30 µl of lysis buffer (1.25 µg/mL of Protease reagent (Qiagen; cat 19155) in Tris HCl pH 8.0, 0.5% Tween 20, 0.5% NP40) to individual Whatman® FTA® punches in a 96-well PCR plate. The samples were incubated at 50°C for an hour followed by protease inactivation by heating the samples to 75°C for 30 min.
4. ForensicGem (FGM): Extraction using the ForensicGEM Universal DNA extraction kit (cat: FUN0100; ZyGem) was performed according to the manufacturer’s guidelines.
5. PicoPure (PIP): Extraction using the ARCTURUS® PicoPure® DNA Extraction Kit (cat: KIT0103; ThermoFisher) was performed according to the manufacturer’s guidelines, using 75 µl of the extraction buffer.
When the extracted DNA from all the above methods was present in a volume greater than 25 µl, samples were cleaned with Agencourt AMPure XP beads (Beckman-Coulter) and eluted in 25-µl nuclease-free H2O. The entire DNA samples were used downstream to make sequencing libraries. A summary of the number of species, life stages, and conditions tested, is presented in Table 1. The differences in the number of samples per species and per assay were largely dependent on the samples available to us, sampled across different times, and sometimes, sampled for different purposes, for example, while most test conditions contained between 6 and 10 replicates, the large number of D. medinensis samples (n = 129 for one condition) were prepared specifically for a different study but we have included it for comparative purposes, whereas the S. mansoni samples (n = 168 for five conditions) were readily available in-house at the Wellcome Sanger Institute, and thus were first used for further validation before samples from other species that required collection from live animal hosts.
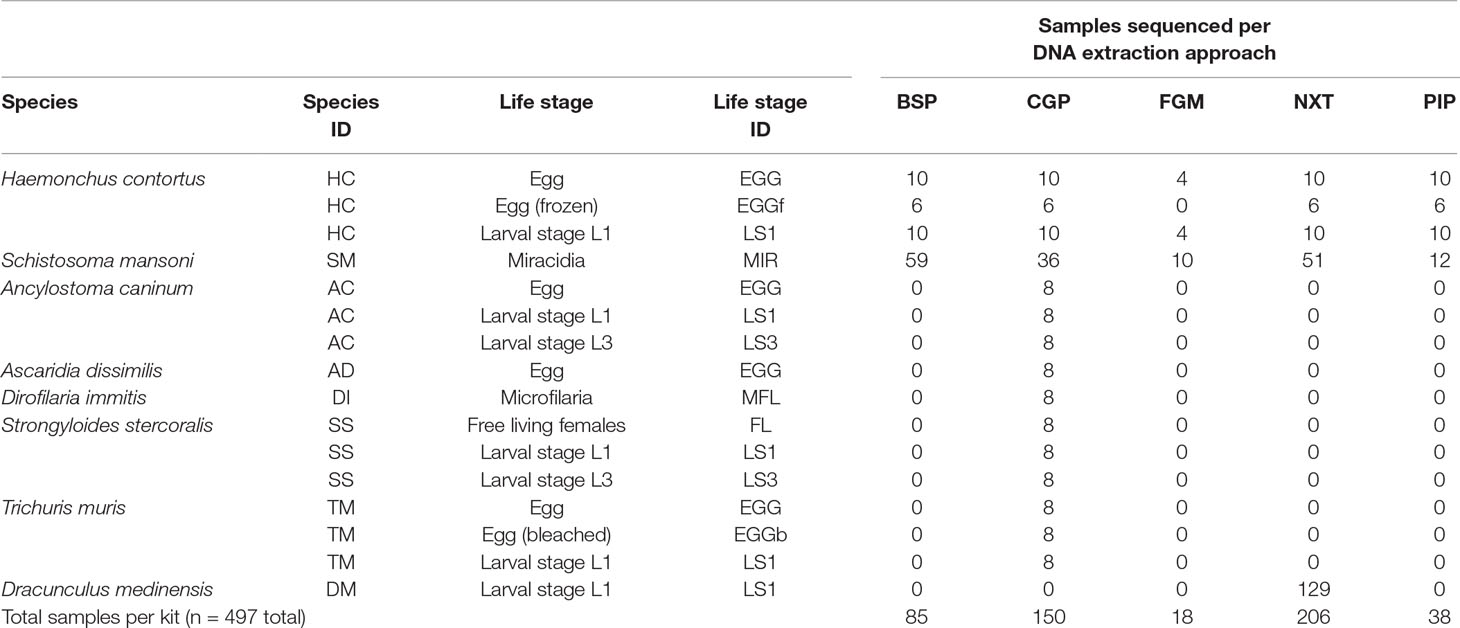
Table 1 Summary of the species, life stages, and conditions tested using one or more of the DNA extraction approaches followed by whole-genome sequencing.
Library Preparation and Sequencing
DNA sequencing libraries for all samples were prepared using a protocol designed for library preparation of laser capture microdissected biopsy (LCMB) samples using the Ultra II FS enzyme (New England Biolabs) for DNA fragmentation as previously described (Lee-Six et al., 2019). A total of 12 cycles of PCR were used (unless otherwise stated in Table S1) to amplify libraries and to add a unique 8-base index sequence for sample multiplexing. Prior to sequencing, library concentration was determined using a fluorometric dsDNA quantification assay (AccuClear® Ultra High Sensitivity dsDNA Quantitation Kit; Biotium) following the manufacturer’s instructions, and measured using a FLUOstar Omega fluorescence plate reader (BMG Labtech). These data were used to normalized the concentration of library DNA for multiplexing.
Multiplexed libraries were sequenced using the Illumina MiSeq platform with V2 chemistry 150 bp paired end (PE) reads. The D. medinensis samples were sequenced as part of a different study using the HiSeq 2500 platform with V4 chemistry 125 bp PE reads. Metadata for each sample, including sample IDs, sequencing lane IDs, ENA sample accession numbers, and data generated are described in Table S1. Raw sequence data used in this study is available under the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/en) study ID ERP114942.
Analysis
We performed sufficient low coverage sequencing on each sample to enable us to identify: (i) the proportion of on-target mapped reads, (ii) the proportion of duplicate reads, i.e. library artefacts, and (iii) the proportion of off-target contaminant reads. On-target reads are defined as reads that mapped to the genome of the species from which DNA extraction was performed, and thus, is a proxy for the specificity of the experiment. Similarly off-target reads are defined as sequencing reads that do not map to the genome of the species from which DNA was extracted, but are derived from putative contaminants. The presence and proportion of contaminant-derived sequencing reads in each sequencing library was further analyzed using the kmer-based classification tool, kraken (v0.10.6-a2d113dc8f) (Wood and Salzberg, 2014), which we have used to assign taxonomic labels to raw sequencing data. Kmers from raw reads are compared with a kraken database (custom database built at WSI containing human [GRCh38] and mouse [GRCm38] genomes, all plasmid, bacterial, and viral genomes, as well as all Illumina adapters, from NCBI at the date of generation [v.pi_qc_2015521]); any raw sequencing reads that match kmers present in the database are defined as contaminants, whereas reads that do not match the database are defined as “kraken_unassigned.” As no helminth genomes are present in the kraken database, these reads of interest will be in the kraken_unassigned category, for example, a high proportion of “kraken_unassigned” reads are indicative of low contamination (or at least low known contaminants present in the kraken database).
Reference genomes from each of the test species were obtained from the helminth genome repository WormBase Parasite (Howe et al., 2017) Release 12. Raw sequence data for each species were mapped to their respective reference genome (which included the mitochondrial genome) using bwa (v0.7.17-r1188) (Li, 2013) mem using default parameters (with the inclusion of -Y and -M options to use soft clipping for supplementary alignments, and to mark shorter hits as secondary, respectively), after which duplicate reads were marked using Picard (v2.5.0; https://github.com/broadinstitute/picard). Samtools (v1.3) flagstat and bamtools (v 2.3.0) stats were used to characterise the outcome of the mapping, the results of which were collated using MultiQC v1.3 (Ewels et al., 2016). Data were manipulated and visualized in the R (v3.5.0) environment using the following packages: ggplot2 (https://ggplot2.tidyverse.org/), patchwork (https://github.com/thomasp85/patchwork), and dplyr (https://dplyr.tidyverse.org/).
The relative ratio of the mitochondrial to nuclear genome was calculated using bedtools (v2.17.0) (Quinlan and Hall, 2010) makewindows and samtools (v1.6) bedcov. This was performed for each life stage of each species described in Table 2, whereby average read coverage from the mapped reads (bam files) were determined for the autosomes (characterised as defined chromosomes for S. mansoni and H. contortus, otherwise, the longest 10 scaffolds for the remaining species were used) and mitochondrial genomes. These data were used to compare the theoretical multiplexing that could be performed per species, either for whole-genome sequencing to achieve 30× genome-wide coverage, or alternatively, sufficient sequencing to achieve 100× coverage of the mitochondrial genome (see Table 2 for results). Here, these data were calculated based on Illumina high-throughput sequencing using the NovaSeq 6000 system with S4 2 × 150 bp PE chemistry generating 2.5 terabases (Tb) of data and accounting for 85% mapping rate. However, to aid in the design of experiments for other species and sequencing platforms, the number of samples that can be multiplexed can be determined by:
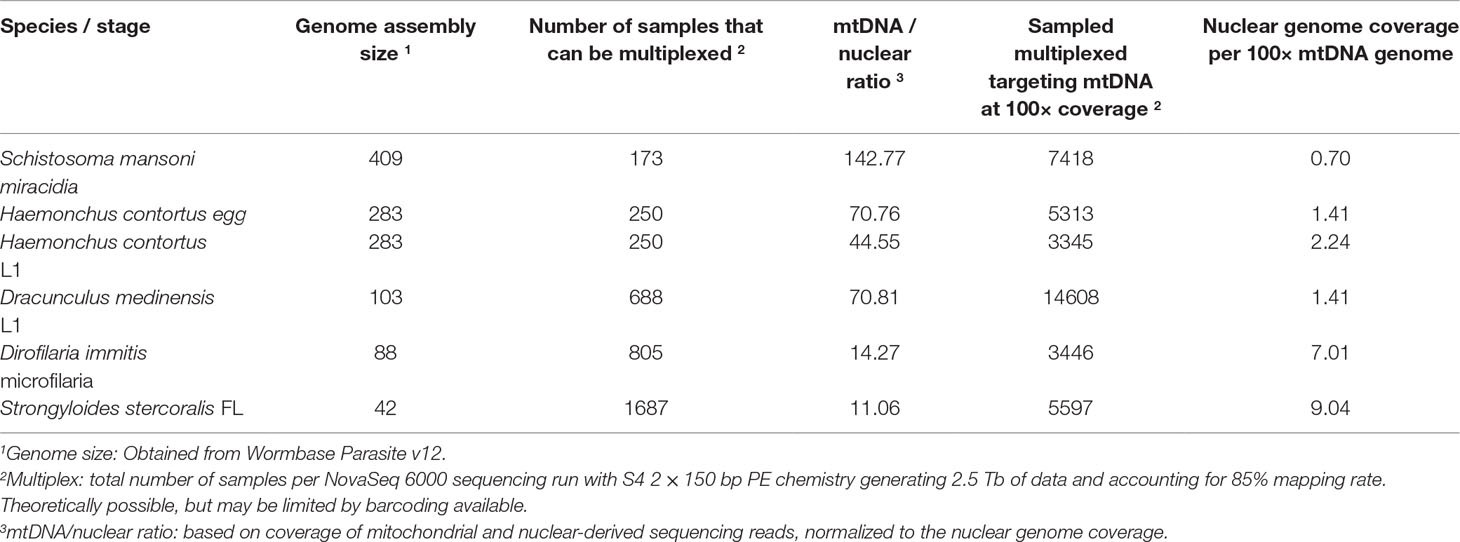
Table 2 Breakdown of sequencing strategies per species based on whole-genome sequencing at 30× coverage and whole-genome sequencing to achieve 100× whole mitochondrial genome coverage.
Where the “genome size” is the estimated number of base pairs in the genome, the “targeted genome coverage” is the intended number of reads covering every base pair of the genome, and the “average mapping efficiency” is the typical proportion of mapped reads from a sequencing library. This latter metric is difficult to predict before performing any sequencing, however, knowledge of this from prior sequencing experiments can improve the actual coverage achieved by applying this scaling factor.
This equation can be extended to low-coverage whole-genome sequencing to target a defined mitochondrial DNA coverage using the following:
where “mtDNA to nuclear ratio” is the number of mitochondrial genomes per nuclear genome, which can be determined by relative reads counts performed here, or by alternative molecular approaches, such as qPCR.
The code to reproduce the analysis and figures for this manuscript is described in https://github.com/stephenrdoyle/helminth_extraction_wgs_test.
Results and Discussion
The aim of this work was to determine the feasibility and efficiency of using a low input DNA extraction and library preparation approach for whole-genome sequencing of individual egg and larval stages of helminth parasites. We have targeted immature life stages that are found in the environment for which it is possible to collect samples non-invasively. We first tested our approaches on the nematode H. contortus and the trematode S. mansoni. Five approaches were tested using single egg (fresh and frozen) and L1 of H. contortus, and miracidia of S. mansoni. Attempts to quantify the raw DNA extractions were inconsistent and largely unsuccessful, due to the extremely low DNA yield per extraction; in the life stages tested, we expect this yield to be in the range of 10 to 100 s of pg of DNA per sample. Quantification of the sequencing libraries of these extractions did however yield sufficient DNA, and revealed significantly more DNA recovered using the CGP and PIP extractions than the other three protocols (Figure 1). Although not a direct measure, sequencing library concentration is likely a sufficient proxy for DNA extraction efficiency, given that the library preparation was standardized across all samples.
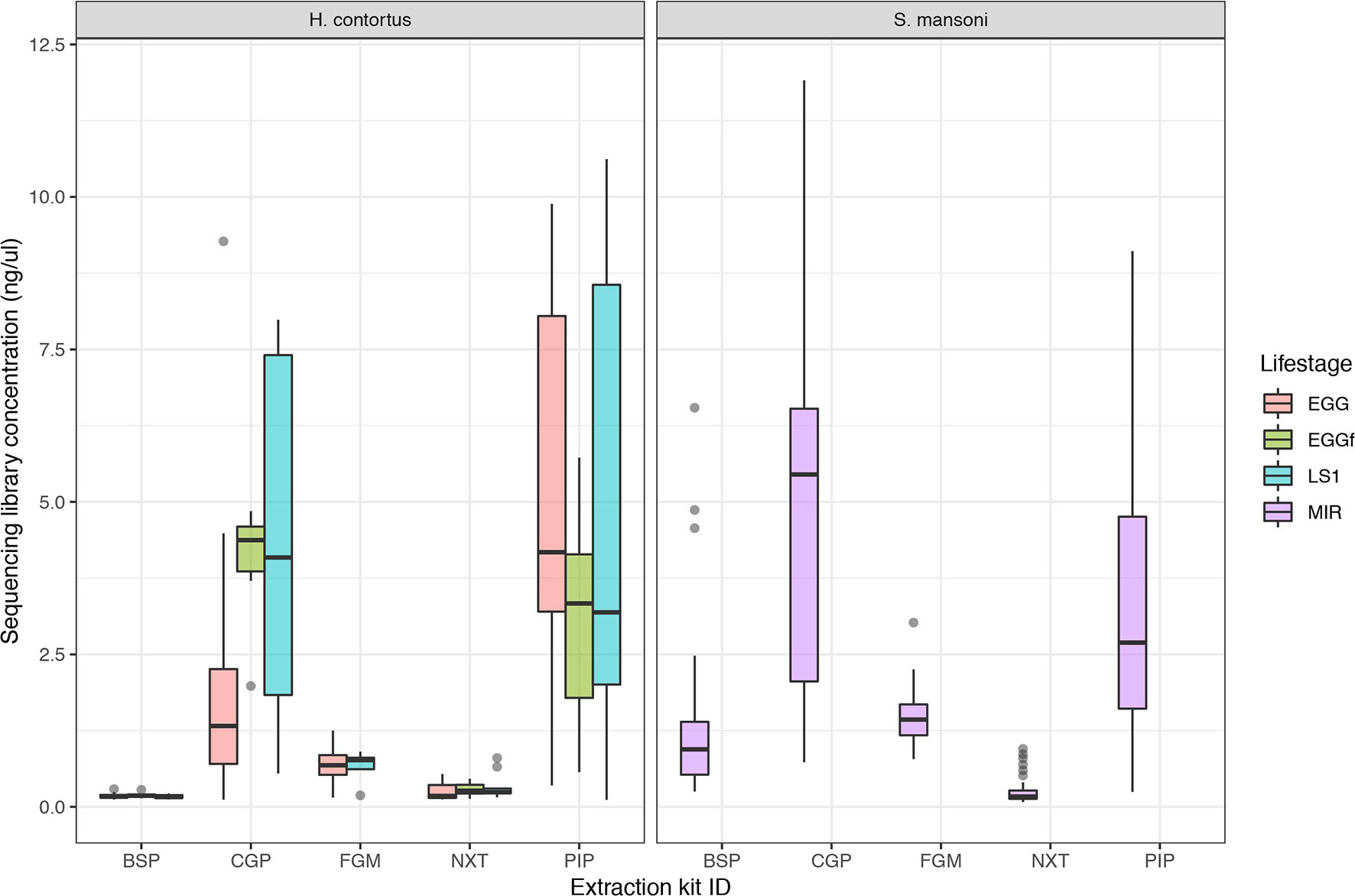
Figure 1 Comparison of sequencing library concentration (ng/µl) as a proxy for relative DNA extraction efficiency for the five DNA extraction protocols tested. DNA from multiple life stages were extracted, including from freshly collected egg (EGG), frozen eggs (EGGf), and L1 stage larvae (LS1) of Haemonchus contortus, and miracidia (MIR) of Schistosoma mansoni. The number of samples in each comparison is presented in Table 1, and the raw data used are described in Table S1.
We determined the success of the library preparation protocols by comparing (i) the proportion of reads mapped to the genome, representing “on-target” mapping as a measure of specificity (Table S1: mapped_reads_percent); (ii) the proportion of reads that matched a “contamination” database, which did not contain helminth genome sequences, and thus represented DNA derived from “off-target” sources such as host or bacterial species commonly present in these samples; those reads that do not match the contamination database are defined as “kraken_unassigned” and are putative helminth-derived reads (Table S1: kraken_unassigned_percent); and (iii) the proportion of duplicate reads, which typically represent library preparation artefacts due to over-amplification of DNA during PCR (Table S1: duplicate_reads_percent).
We examined the impact of library strength on mapping frequency for all DNA extraction protocols tested (Figure 2). For H. contortus, a clear correlation between these two variables were observed, with an inflection point at approximately 0.25 ng/µl, below which the proportion of reads mapped rapidly decreased toward 0 (Figure 2A). Similarly, below this point, the proportion of reads classified as contamination increased in frequency (Figure 2B). There was, however, a distinct difference between H. contortus and S. mansoni in the overall proportion of reads mapped, and the frequency of contaminating reads, with greater variation in both parameters in the S. mansoni samples. While some of this variation may reflect differences in extracting DNA from the two species, the majority of S. mansoni MIR samples were isolated and aliquoted onto FTA cards under less clean conditions in the field and stored for between 2 and 5 years before processing, as compared with the laboratory prepared H. contortus samples that were collected and prepared directly from a current infection (EGG and LS1) or within the previous 6 months (EGGf) of processing. We achieved some on-target mapping for all extraction kits tested (Figure S1A); however, significant variation was observed between approaches. BSP and NXT generally performed poorly, with either low (median = 19.42; median absolute deviation [MAD] = 16.50) or significant variance (median = 66.20; MAD = 26.18) in mapping frequency between samples observed, respectively. PIP performed consistently well with high mapping rates across all stages in H. contortus (median = 90.30; MAD = 3.96); however, it was poor in S. mansoni (median = 11.00; MAD = 12.73). The duplication rate of all conditions were within an acceptable low range (median = 0.46, MAD = 0.4), with only 2% of samples having greater than 5% duplicate reads (Figure S1B). This suggests that the low DNA input did not noticeably impact duplication rates, and therefore, with greater sequencing depth of the same sequencing libraries, we would expect unbiased genome-wide coverage (at least relative to sequencing of libraries derived from higher DNA input). CGP and FGM performed most consistently between stages and species; however, FGM had higher variance and duplication rates relative to CGP across all samples tested. Considering the higher library strength (suggestive of greater DNA recovery efficiency), consistent mapping efficiency, and the cost effective protocol, we chose the CGP extraction to explore further.
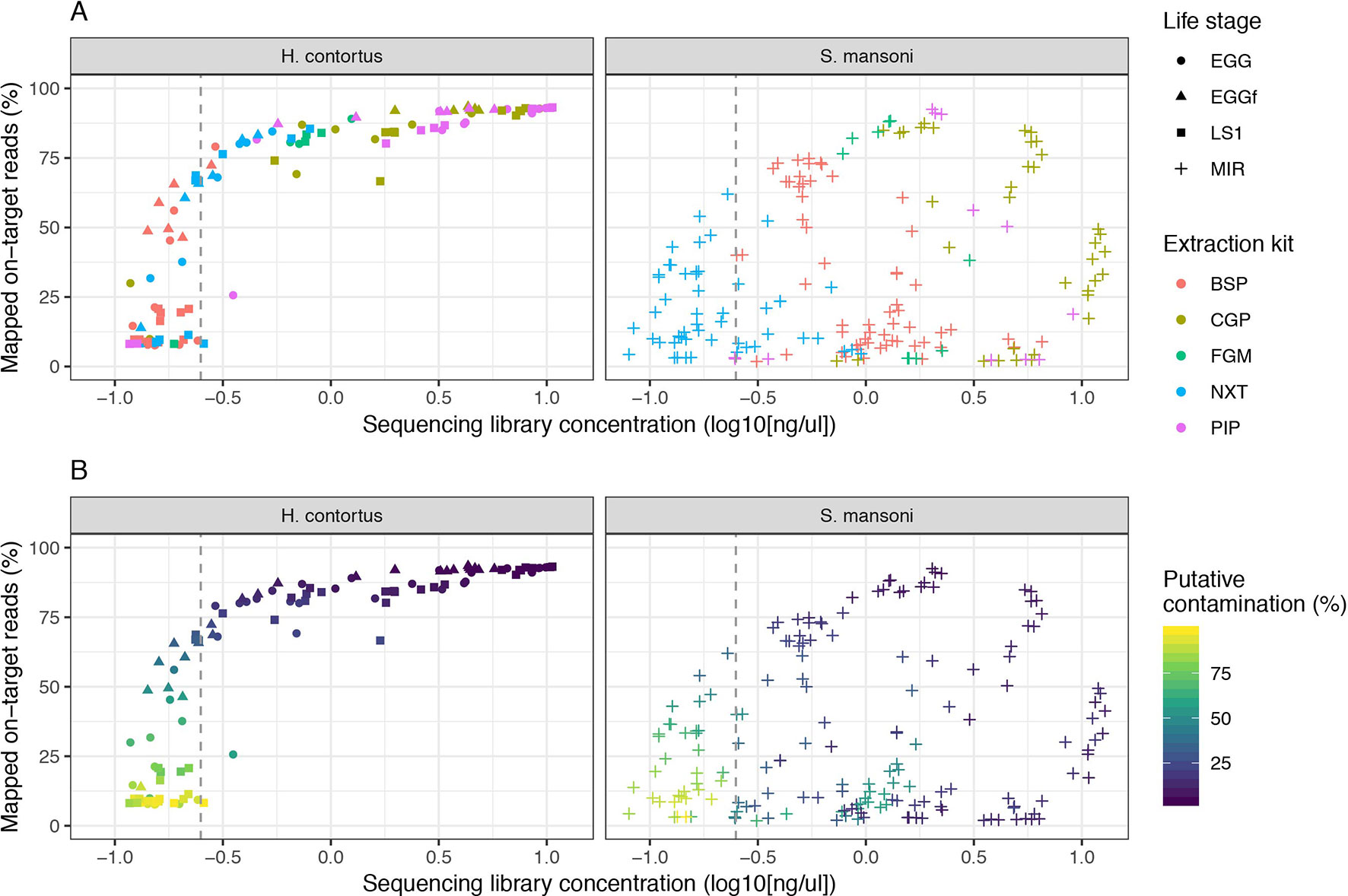
Figure 2 Impact of sequencing library concentration on mapped on-target reads for five low-input DNA extraction protocols. Each point represents an individual sequencing library sample, colored by extraction protocol (A) or proportion of putative contaminating reads (100 - percent_unclassified_reads) (B). The dashed grey vertical line represents a library concentration of 0.25 ng/µl. Extraction protocols were performed on Haemonchus contortus eggs (EGG), frozen eggs (EGGf), and L1 (LS1) stages, and Schistosoma mansoni miracidia (MIR). The number of samples in each comparison is presented in Table 1, and the raw data used are described in Table S1.
We expanded our analysis of the CGP protocol to a total of seven helminth species for which samples were available, including a total of five distinct life stages (Figure 3). High variability in mapping was observed between species, with 50% or greater mapping frequency achieved in at least one life stage of five of the seven species tested (Figure 3A, Figure S2A). Clear differences were observed between multiple life stages tested within a species, likely reflecting differences in extraction efficiency per life stage, for example: for A. canium, reads from eggs (median = 54.98) mapped much more effectively than L1 (median = 3.61) or L3 stages (median = 7.57), and in S. stercoralis, full-length females (median = 67.84) and L1 (median = 47.19) performed better than L3 (median = 9.01). The proportion of contaminating reads tended to increase as library concentration decreased for all species (Figure 3B); however, some samples for which mapping was poor but higher libraries concentrations were obtained were highly enriched for contaminants, for example, A. canium L1 larvae and T. muris eggs. Interestingly, T. muris L1 larvae (median = 41.85) and bleached eggs (median = 51.35) performed much better than untreated eggs (median = 2.72); bleaching is experimentally used for promoting hatching of T. muris, by dissolving the egg shell layers and in turn improving access to DNA within. However, bleached eggs were embryonated and developmentally more advanced than untreated unembryonated eggs, and therefore would have more DNA available for library preparation. Similar to untreated T. muris eggs, A. dissimilis eggs performed poorly (median = 0.16); for both species, few if any nematode sequences were recovered and the majority of sequencing reads were contaminating bacterial-derived contaminants, perhaps indicative of the challenge of accessing material with the environmentally resistant egg. Analysis of A. dissimilis was further limited by the lack of a reference genome for this species; we mapped against the available genome of Ascaris lumbricoides, the nearest species for which a reference was available, and therefore at best, would have expected suboptimal mapping due to sequence divergence. The duplication rates remain low for all species and conditions tested (Figure S2B), consistent with our initial observation that library input did not significantly influence duplication rates (Figure S1B).
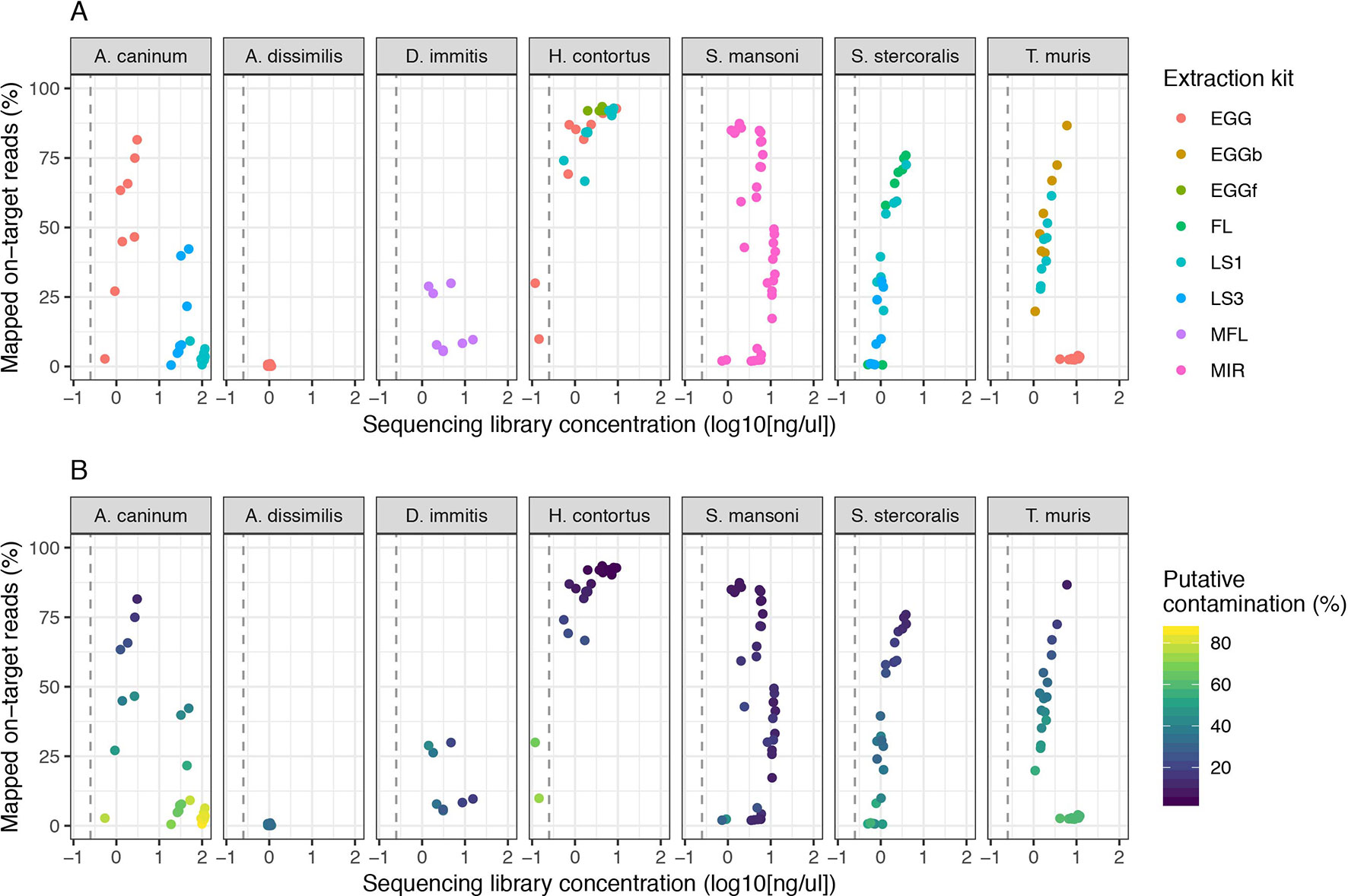
Figure 3 Impact of sequencing library concentration on mapped on-target reads from multiple life stages of seven helminth species. Each point represents an individual sequencing library samples, colored by life stage (A) or proportion of putative contaminating reads (100 - percent_unclassified_reads) (B). DNA was extracted from all samples using the CGP protocol. Consistent with Figure 2, the dashed grey vertical line represents 0.25 ng/µl. Egg, untreated egg; EGGb, bleached egg; EGGf, frozen egg; FL, full-length female; LS1, larval stage L1; LS3, larval stage L3; MFL, microfilaria; MIR, miracidia. The number of samples in each comparison is presented in Table 1.
We also generated data from microfilaria of D. medinensis; these samples were extracted using NXT rather than CGP as part of a separate study; however, we have included these for comparison (Figure S3). High mapping rates were observed for almost all libraries generated; however, the duplication rates per library were also high (median = 36.90); this was unexpected, given NXT did not produce high duplication rates under the initial conditions tested with H. contortus or S. mansoni, nor were there excessive PCR cycles used in this instance. Duplication rates were not correlated with library concentration.
In summary, we present successful DNA extraction followed by whole-genome sequencing of individual egg and larval stages from six of eight parasites species examined. These results significantly extend the possibility of genomic analyses for life stages for which, at best, were limited to low-resolution, low-throughput PCR based assays without the addition of whole-genome amplification. Whatman® FTA® cards provide a convenient substrate for sample collection and storage, and do not limit the application of direct DNA extraction and whole-genome sequencing of parasite samples, even for field samples as demonstrated for S. mansoni miracidia that were collected and processed in Uganda before they were transported to the UK. Further optimization is required to improve the DNA recovery from eggs, for example, from A. dissimilis and T. muris, to provide greater applicability of our approaches to species that generate particularly environmentally resistant stages, such as the soil transmitted helminths. The application of whole-genome sequencing to diagnose and monitor helminth infections at scale is largely limited by the costs of library preparation and sequencing, and therefore, will be restricted to niche applications of the technology. However, the use of low-coverage whole-genome sequencing with the specific aim to target the mitochondrial genome, which is present at a higher copy number than the nuclear genome (Table 2), may be a cost-effective alternative and potentially provides greater diagnostic information than low-throughput PCR-based diagnostics. Continued development of genomic technologies and the associated reduction in sequencing and library preparation costs will make screening large samples by genome sequencing more routine as in viral (Dudas et al., 2017) and bacterial (Domman et al., 2018) population studies. In doing so, the ability to derive high resolution data may provide insight into, for example, (i) within host reproductive dynamics, including within host population size, differential fecundity (Hildebrandt et al., 2014), and reproductive traits such as polyandry (Doyle et al., 2018), (ii) defining parasite transmission zones and rates of transmission between zones to prioritize treatment foci (Crawford et al., 2019; this special edition, in review), (iii) defining effective population sizes of parasite populations (Sallé et al., 2019), and using this to estimate the impact of control strategies over time, and/or (iv) discrimination between reintroduction and recrudescence of parasites in regions where parasite control has been successful (Koala et al., 2019). The use of genomics to provide information-rich data will be increasingly important for diagnostic and surveillance purposes broadly (Cotton et al., 2018), and will be particularly informative as efforts to control human infective helminths using MDA move from control to elimination.
Data Availability
The datasets generated for this study can be found in ENA, ERP114942.
Ethics Statement
Given a number of host/parasite systems are described, we have provided information about the individual ethical approvals obtained associated with each species in the “Sample Collection” subheading of the Methods section.
Author Contributions
Conceptualization: SRD, JAC, NH; Methodology: SRD, GS, NH; Software: SRD; Formal analysis: SRD; Investigation: SRD, GS; Resources: FA, TJ, JL, JBC, PC, JW, TC, ET, MD-C, PE, RL, KM, CM, TM, BS, PO, JT, AW, ED, RK, DB, MB; Data curation: SRD, NH; Writing – original draft preparation: SRD; Writing – review and editing: All authors; Visualization: SRD; Supervision: SRD, JAC, NH; Project administration: NH.
Funding
Work performed at the Wellcome Sanger Institute is supported by the Wellcome Trust (grant 206194) and by the Biotechnology and Biological Sciences Research Council (BB/M003949/1). We thank Alison Elliot for access to S. mansoni samples collected from Uganda, the collection of which was supported by the Wellcome Trust (grant 095778/Z/11/Z). S. mansoni samples were also obtained from the Schistosomiasis Collection at the Natural History Museum (NHM) [SCAN], which is funded with support from the Wellcome Trust (grant no. 104958/Z/14/Z).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Carter Center for supporting molecular work on D. medinensis, and the Guinea Worm Eradication Program for making samples available. We also thank Ian Still from DNA Pipelines at Wellcome Sanger Institute for his expertise in protocol development.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00826/full#supplementary-material
Table S1 | Complete metadata per sample (excel workbook)
Figure S1 | Summary boxplots of mapped on target reads (A) and duplicate reads (B) for all extraction kits tested. The number of samples tested per kit, per life stage is shown at the top of (A).
Figure S2 | Summary boxplots for mapped on target reads (A) and duplicate reads (B) for all species tested using the CGP protocol. The number of samples tested per species, per life stage is shown at the top of (A).
Figure S3 | Analysis of D. medinensis using the NXT protocol. (A) Mapped “on-target” reads. (B) Duplicate reads. (C) Comparison of the effect of sequencing library concentration on mapping efficiency, colored by the proportion of duplicate reads. (D) Comparison of the effect of sequencing library concentration on mapping efficiency, colored by the proportion of putative contaminant reads identified using Kraken (100 - percent_unclassified_reads).
References
Avramenko, R. W., Redman, E. M., Lewis, R., Yazwinski, T. A., Wasmuth, J. D., Gilleard, J. S. (2015). Exploring the gastrointestinal ‘nemabiome’: deep amplicon sequencing to quantify the species composition of parasitic nematode communities. PLoS One 10(12), e0143559. doi: 10.1371/journal.pone.0143559
Avramenko, R. W., Redman, E. M., Melville, L., Bartley, Y., Wit, J., Queiroz, C., et al. (2019). Deep amplicon sequencing as a powerful new tool to screen for sequence polymorphisms associated with anthelmintic resistance in parasitic nematode populations. Int. J. Parasitol. 49 (1), 13–26. doi: 10.1016/j.ijpara.2018.10.005
Boué, F., El Berbri, I., Hormaz, V., Boucher, J.-M., El Mamy, A. B., Traore, A., et al. (2017). Use of FTA® Card methodology for sampling and molecular characterization of Echinococcus granulosus sensu lato in Africa. Exp. Parasitol 173, 29–33. doi: 10.1016/j.exppara.2016.12.016
Campbell, S. J., Stothard, J. R., O’Halloran, F., Sankey, D., Durant, T., Ombede, D. E., et al. (2017). Urogenital Schistosomiasis and soil-transmitted helminthiasis (STH) in Cameroon: an epidemiological update at Barombi Mbo and Barombi Kotto Crater Lakes assessing prospects for intensified control interventions. Infect. Dis. Poverty 6 (1), 49. doi: 10.1186/s40249-017-0264-8
Cotton, J. A., Berriman, M., Dalén, L., Barnes, I. (2018). Eradication genomics—lessons for parasite control. Science 361 (6398), 130–131. doi: 10.1126/science.aar6609
Crawford, K. E., Hedtke, S. M., Doyle, S. R., Kuesel, A. C., Armoo, S., Osei-Atweneboana, M., et al. (2019). Utility of the Onchocerca volvulus mitochondrial genome for delineation of parasite transmission zones. bioRxiv 732446. doi: 10.1101/732446
Crellen, T., Walker, M., Lamberton, P. H. L., Kabatereine, N. B., Tukahebwa, E. M., Cotton, J. A., et al. (2016). Reduced efficacy of praziquantel against Schistosoma mansoni is associated with multiple rounds of mass drug administration. Clin. Infect. Dis. 63 (9), 1151–1159. doi: 10.1093/cid/ciw506
Domman, D., Chowdhury, F., Khan, A. I., Dorman, M. J., Mutreja, A., Uddin, M. I., et al. (2018). Defining endemic cholera at three levels of spatiotemporal resolution within Bangladesh. Nat. Genet. 50 (7), 951–955. doi: 10.1038/s41588-018-0150-8
Doyle, S. R., Laing, R., Bartley, D. J., Britton, C., Chaudhry, U., Gilleard, J. S., et al. (2018). A genome resequencing-based genetic map reveals the recombination landscape of an outbred parasitic nematode in the presence of polyploidy and polyandry. Genome Biol. Evol. 10 (2), 396–409. doi: 10.1093/gbe/evx269
Dudas, G., Carvalho, L. M., Bedford, T., Tatem, A. J., Baele, G., Faria, N. R., et al. (2017). Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 544 (7650), 309–315. doi: 10.1038/nature22040
Ewels, P., Magnusson, M., Lundin, S., Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32 (19), 3047–3048. doi: 10.1093/bioinformatics/btw354
Gower, C. M., Shrivastava, J., Lamberton, P. H. L., Rollinson, D., Webster, B. L., Emery, A., et al. (2007). Development and application of an ethically and epidemiologically advantageous assay for the multi-locus microsatellite analysis of Schistosoma mansoni. Parasitology 134 (Pt 4), 523–536. doi: 10.1017/S0031182006001685
Hildebrandt, J. C., Eisenbarth, A., Renz, A., Streit, A. (2014). Reproductive biology of Onchocerca ochengi, a nodule forming filarial nematode in Zebu cattle. Vet. Parasitol. 205 (1-2), 318–329. doi: 10.1016/j.vetpar.2014.06.006
Howe, K. L., Bolt, B. J., Shafie, M., Kersey, P., Berriman, M. (2017). WormBase ParaSite—a comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 215, 2–10. doi: 10.1016/j.molbiopara.2016.11.005
Howick, V. M., Russell, A., Andrews, T., Heaton, H., Reid, A. J., Natarajan, K. N., et al. (2019). The Malaria Cell Atlas: a comprehensive reference of single parasite transcriptomes across the complete plasmodium life cycle. Science 365 (6455), eaaw2619. doi: 10.1126/science.aaw2619
Jaleta, T. G., Zhou, S., Bemm, F. M., Schär, F., Khieu, V., Muth, S., et al. (2017). Different but overlapping populations of Strongyloides stercoralis in dogs and humans-dogs as a possible source for zoonotic strongyloidiasis. PLoS Negl. Trop. Dis. 11 (8), e0005752. doi: 10.1371/journal.pntd.0005752
Koala, L., Nikièma, A. S., Paré, A. B., Drabo, F., Toé, L. D., Belem, A. M. G., et al. (2019). Entomological assessment of the transmission following recrudescence of onchocerciasis in the Comoé Valley, Burkina Faso. Parasit. Vectors 12 (1), 34. doi: 10.1186/s13071-019-3290-5
Le Clec’h, W., Chevalier, F. D., Dew-White, M., Allan, F., Webster, B. L., Gouvras, A. N., et al. (2018). Whole genome amplification and exome sequencing of archived Schistosome miracidia. Parasitology 145 (13), 1739–1747. doi: 10.1017/S0031182018000811
Lee-Six, H., Ellis, P., Osborne, R. J., Sanders, M. A., Moore, L., Georgakopoulos, N., et al. (2019). The landscape of somatic mutation in normal colorectal epithelial cells. bioRxiv 416800. doi: 10.1101/416800
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN].
Lok, J. B. (2007). “Strongyloides stercoralis: a model for translational research on parasitic nematode biology.” The Online Review of C. Elegans Biology. (WormBook) 1–18. doi: 10.1895/wormbook.1.134.1
Maclean, M. J., Savadelis, M. D., Coates, R., Dzimianski, M. T., Jones, C., Benbow, C., et al. (2017). Does evaluation of in vitro microfilarial motility reflect the resistance status of dirofilaria immitis isolates to macrocyclic lactones? Parasit. Vectors 10 (Suppl 2), 480. doi: 10.1186/s13071-017-2436-6
Marek, M., Zouhar, M., Douda, O., Maňasová, M., Ryšánek, P. (2014). Exploitation of FTA cartridges for the sampling, long-term storage, and DNA-based analyses of plant-parasitic nematodes. Phytopathology 104 (3), 306–312. doi: 10.1094/PHYTO-03-13-0067-R
Moore, L., Leongamornlert, D., Coorens, T. H. H., Sanders, M. A., Ellis, P., Dawson, K., et al. (2018). The mutational landscape of normal human endometrial epithelium. bioRxiv 505685. doi: 10.1101/505685
Ngara, M., Palmkvist, M., Sagasser, S., Hjelmqvist, D., Björklund, Å K., Wahlgren, M., et al. (2018). Exploring parasite heterogeneity using single-cell RNA-Seq reveals a gene signature among sexual stage Plasmodium falciparum parasites. Exp. Cell Res. 371 (1), 130–138. doi: 10.1016/j.yexcr.2018.08.003
Platt, R. N., Dew-White, M., Le Clec’h, W., Chevalier, F. D., Allan, F., Emery, A. M., et al. (2019). Ancient hybridization and adaptive introgression of an invadolysin gene in schistosome parasites. Mol. Biol. Evol. 2019. msz154. doi: 10.1093/molbev/msz154
Quinlan, A. R., Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 (6), 841–842. doi: 10.1093/bioinformatics/btq033
Reid, A. J., Talman, A. M., Bennett, H. M., Gomes, A. R., Sanders, M. J., Illingworth, C. J. R., et al. (2018). Single-Cell RNA-Seq Reveals Hidden Transcriptional Variation in Malaria Parasites. eLife. 7, e33105. doi: 10.7554/eLife.33105
Sabina, J., Leamon, J. H. (2015). Bias in whole genome amplification: causes and considerations. Methods Mol. Biol. 1347, 15–41. doi: 10.1007/978-1-4939-2990-0_2
Sallé, G., Doyle, S. R., Cortet, J., Cabaret, J., Berriman, M., Holroyd, N., et al. (2019). The global diversity of the major parasitic nematode Haemonchus contortus is shaped by human intervention and climate. bioRxiv 450692. doi: 10.1101/450692
Sanya, R. E., Nkurunungi, G., Spaans, R. H., Nampijja, M., O’Hara, G., Kizindo, R., et al. (2018). The impact of intensive versus standard anthelminthic treatment on allergy-related outcomes, helminth infection intensity and helminth-related morbidity in lake victoria fishing communities, Uganda: results from the LaVIISWA Cluster Randomised Trial. Clin. Infect. Dis. 68 (10), 1665–1674. doi: 10.1093/cid/ciy761
Shortt, J. A., Card, D. C., Schield, D. R., Liu, Y., Zhong, B., Castoe, T. A., et al. (2017). Whole genome amplification and reduced-representation genome sequencing of Schistosoma japonicum miracidia. PLoS Negl. Trop. Dis. 11 (1), e0005292. doi: 10.1371/journal.pntd.0005292
Small, S. T., Labbé, F., Coulibaly, Y. I., Nutman, T. B., King, C. L., Serre, D., et al. (2018). Human migration and the spread of the nematode parasite Wuchereria bancrofti. Mol. Biol. Evol. doi: 10.1093/molbev/msz116
Sulston, J., Hodgkin, J. (1988). “Methods,” in The Nematode Caenorhabditis Elegans. Ed. Wood, W. B. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
Trevino, S. G., Nkhoma, S. C., Nair, S., Daniel, B. J., Moncada, K., Khoswe, S., et al. (2017). High-resolution single-cell sequencing of malaria parasites. Genome Biol. Evol. 9 (12), 3373–3383. doi: 10.1093/gbe/evx256
Tsai, I. J., Hunt, M., Holroyd, N., Huckvale, T., Berriman, M., Kikuchi, T. (2014). Summarizing specific profiles in illumina sequencing from whole-genome amplified DNA. DNA Res. 21 (3), 243–254. doi: 10.1093/dnares/dst054
Wakelin, D. (1967). Acquired immunity to Trichuris muris in the albino laboratory mouse. Parasitology 57 (3), 515–524. doi: 10.1017/S0031182000072395
Webster, B. L. (2009). Isolation and preservation of schistosome eggs and larvae in RNAlater® facilitates genetic profiling of individuals. Parasit. Vectors 2 (1), 50. doi: 10.1186/1756-3305-2-50
Webster, B. L., Emery, A. M., Webster, J. P., Gouvras, A., Garba, A., Diaw, O., et al. (2012). Genetic diversity within Schistosoma haematobium: DNA barcoding reveals two distinct groups. PLoS Negl. Trop. Dis. 6 (10), e1882. doi: 10.1371/journal.pntd.0001882
Wood, D. E., Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15 (3), R46. doi: 10.1186/gb-2014-15-3-r46
Keywords: helminths, genomics, whole-genome sequencing, DNA extraction, low input, diagnostics
Citation: Doyle SR., Sankaranarayanan G, Allan F, Berger D, Jimenez Castro PD., Collins JB, Crellen T, Duque-Correa MA., Ellis P, Jaleta TG., Laing R, Maitland K, McCarthy C, Moundai T, Softley B, Thiele E, Ouakou PT, Tushabe JV, Webster JP., Weiss AJ., Lok J, Devaney E, Kaplan RM., Cotton JA., Berriman M and Holroyd N (2019) Evaluation of DNA Extraction Methods on Individual Helminth Egg and Larval Stages for Whole-Genome Sequencing. Front. Genet. 10:826. doi: 10.3389/fgene.2019.00826
Received: 26 April 2019; Accepted: 12 August 2019;
Published: 20 September 2019.
Edited by:
Jose F. Tort, University of the Republic, UruguayReviewed by:
Masoud Zamani Esteki, Maastricht University Medical Centre, NetherlandsNeil David Young, The University of Melbourne, Australia
Guilherme Corrêa De Oliveira, Vale Technological Institute (ITV), Brazil
Copyright © 2019 Doyle, Sankaranarayanan, Allan, Berger, Jimenez Castro, Collins, Crellen, Duque-Correa, Ellis, Jaleta, Laing, Maitland, McCarthy, Moundai, Softley, Thiele, Ouakou, Tushabe, Webster, Weiss, Lok, Devaney, Kaplan, Cotton, Berriman and Holroyd. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen R. Doyle, c3RlcGhlbi5kb3lsZUBzYW5nZXIuYWMudWs=; Nancy Holroyd, bmVoQHNhbmdlci5hYy51aw==