 Feng Gao
Feng Gao Jun Tian
Jun Tian- Department of Trauma and Emergency Surgeon, The Second Affiliated Hospital, Harbin Medical University, Harbin, China
Osteoarthritis (OA) is the most common degenerative joint disorder worldwide. To identify more genetic signals, genome-wide association study (GWAS) has been widely used and elucidated some OA susceptibility genes. However, these susceptibility genes could only explain only a small part of heritability of OA. It is suggested that the identification of disease-related pathways may contribute to understand the genomic etiology of OA. Here, we integrated the GWAS into pathway analysis to identify novel OA risk pathways. In this study, we first selected 187 independent genetic variants identified by GWAS (P < 1.00E−05) and found that most of these genetic variants are noncoding mutations. We then conducted an expression quantitative trait loci analysis and found that 165 of these 187 genetic variants could significantly regulate the expression of nearby genes. Third, we identified OA susceptibility genes corresponding to these genetic variants, conducted a pathway analysis, and identified novel OA-related KEGG pathways, GO biological processes, GO molecular functions, and GO cellular components. In KEGG database, transforming growth factor β signaling pathway is the most significant signal (P = 5.98E−05) and is the only pathway after the BH multiple-test adjustment with false discovery rate (FDR) = 0.02. In GO database, we identified 24 statistically significant GO biological processes, one statistically significant GO molecular function, and five statistically significant GO cellular components (FDR < 0.05). These signals are related with chondrocyte differentiation and development, which are all known biological pathways associated with OA. Finally, we conducted an OA case–control gene expression analysis to evaluate the differential expression of these OA risk genes. Using an OA case–control gene expression analysis, we showed that 44 risk genes were suggestively differentially expressed in OA cases compared with controls (P < 0.05). Three genes, WWP2, COG5, and MAPT, were statistically differentially expressed in OA cases compared with controls (P < 0.05/122 = 4.10E−04). Hence, our findings may contribute to understanding the genomic etiology of OA.
Introduction
Osteoarthritis (OA) is the most common degenerative joint disorder worldwide (Martel-Pelletier et al., 2016). Osteoarthritis could cause pain and disability in elderly people (Parmet et al., 2003; Zeggini et al., 2012). It is considered that OA is caused by the combination of risk factors with increasing age and obesity (Parmet et al., 2003; Martel-Pelletier et al., 2016). Osteoarthritis is a complex disease and has a strong genetic component (Zeggini et al., 2012). To identify more genetic signals, genome-wide association study (GWAS) has been widely used and elucidated some OA susceptibility genes (Zeggini et al., 2012; Styrkarsdottir et al., 2014; Styrkarsdottir et al., 2018; Zengini et al., 2018; Tachmazidou et al., 2019). However, these susceptibility genes could explain only a small part of heritability of OA (Zeggini et al., 2012; Styrkarsdottir et al., 2014; Styrkarsdottir et al., 2018; Zengini et al., 2018; Tachmazidou et al., 2019). It is suggested that the identification of disease-related pathways may contribute to understand the genomic etiology of OA (Cibrian Uhalte et al., 2017).
In recent years, kinds of bioinformatics software and databases have been developed, especially for the identification of disease-related pathways such as VEGAS (Versatile Gene-based Association Study) (Liu et al., 2010), INRICH (INterval enRICHment analysis) (Lee et al., 2012a), and GSA-SNP2 (Yoon et al., 2018). Meanwhile, these methods have been widely used in OA-related disease rheumatoid arthritis (RA) (Eleftherohorinou et al., 2009; Eleftherohorinou et al., 2011; Lee et al., 2012b; Song et al., 2013; Zhang et al., 2016), but not in OA. Here, we first selected all significant OA risk variants identified by GWAS and evaluated their distribution in genome coding and noncoding regions. Second, we then conducted an expression quantitative trait loci (eQTLs) analysis to evaluate the effect of these genetic variants on gene expression. Third, we performed a pathway analysis of OA susceptibility genes detected by OA-related genetic pathways. Finally, we conducted an OA case–control gene expression analysis to evaluate the differential expression of these OA risk genes.
Materials and Methods
Selection of OA Risk Variants
We selected the potential OA risk variants identified by GWAS by searching for the NHGRI GWAS catalog database using the keyword “osteoarthritis” (Welter et al., 2014; MacArthur et al., 2017; Buniello et al., 2019). Finally, we selected 187 genetic variants associated with OA (P < 1.00E−05) (Welter et al., 2014; MacArthur et al., 2017; Buniello et al., 2019). These variants are associated with OA, OA of the hip, OA of the knee, or OA of the hand. Here, we provided all related information about these variants in Supplementary Table 1.
Identification of OA Risk Genes
For each of these 187 genetic variants, if this variant is an intronic variant, we select its corresponding gene. If this variant is an intergenic variant, we select its corresponding nearest upstream and downstream genes. The gene set consists of 202 risk genes using all these 187 genetic variants, as provided in Supplementary Table 1.
Function Analysis
We used the HaploReg tool (v4.1) to annotate these 187 genetic variants and evaluate how many genetic variants are coding and noncoding mutations (Ward and Kellis, 2012; Ward and Kellis, 2016). The reference information is from the 1000 Genomes Project (Ward and Kellis, 2012; Ward and Kellis, 2016).
eQTLs Analysis
When noncoding genetic variants are identified, we conducted an eQTLs analysis to evaluate the effect of these genetic variants on gene expression using Phenol Scanner (v2), a database of human genotype–phenotype associations (Staley et al., 2016; Kamat et al., 2019).
Identification of OA Risk Pathways
Here, we selected the pathway resources from KEGG and GO databases, as provided in Web-based Gene Set Analysis Toolkit (WebGestalt version 2019) (Wang et al., 2017). To identify a potential OA risk pathway, the hypergeometric test was used to evaluate an overrepresentation of the OA risk genes among all the genes in the selected pathway, as did in previous studies (Bao et al., 2015; Zhao et al., 2015; Jiang et al., 2017; Liu et al., 2017; Wang et al., 2017). For a given pathway, the P value of observing more than K OA risk genes could be calculated by
where N is the total number of genes that are of interest (the reference gene list), S is the number of all OA risk genes (here, n = 202), m is the number of genes in the pathway, and K is the number of OA risk genes in the pathway, as did in previous studies (Liu et al., 2012; Liu et al., 2013; Liu et al., 2014; Quan et al., 2015; Shang et al., 2015; Xiang et al., 2015).
Using WebGestalt version 2019, the minimum number of genes for a category is 5, and the maximum number of genes for a category is 2000. In addition, we used the BH multiple-test adjustment method to adjust the P value of each pathway. A specific pathway with adjusted FDR < 0.05 is considered to be statistically significant. A specific pathway with unadjusted P < 0.05 is considered to be suggestively significant.
OA Case–Control Gene Expression Analysis
We conducted an OA case–control gene expression analysis to evaluate the differential expression of these 202 risk genes, as provided in Supplementary Table 1. The gene expression profile dataset is from peripheral blood mononuclear cells of 106 OA patients and 33 sex and age-matched healthy controls, which is a subset of the Genetics osteoarthritis and Progression study (Ramos et al., 2014). Here, we used the interactive web tool GEO2R to identify genes that are differentially expressed in OA patients and healthy controls. A gene with unadjusted P < 0.05 is considered to be suggestively differentially expressed. In addition, we used the Bonferroni correction method to adjust these P values and define the statistically differential expression, as described in a previous study (Krzywinski and Altman, 2014).
Results
Function Analysis
Function analysis using HaploReg tool (v4.1) showed that eight genetic variants, rs12193102, rs35611929, rs375575359, rs528981060, rs532464664, rs541612392, rs547116051, and rs547181612, were not found in 1000 Genomes Phase 1 data. In the remaining 179 genetic variants, 11 genetic variants are coding mutations, and the other 168 genetic variants are noncoding mutations. More detailed information is provided in Supplementary Table 2.
eQTLs Analysis
Using Phenol Scanner (v2), the eQTLs analysis showed that 165 of these 187 genetic variants could significantly regulate the expression of nearby genes with P < 0.01. We found that the other 22 genetic variants were not eQTLs including rs111427307, rs11335718, rs11409738, rs12028630, rs12193102, rs138063419, rs143083812, rs150198051, rs201708019, rs35087650, rs35912128, rs375575359, rs4867568, rs528981060, rs541612392, rs547116051, rs547181612, rs5834501, rs62262139, rs6557013, rs7510312, and rs9930333. More detailed information is provided in Supplementary Table 3.
Identification of OA-Related KEGG Pathways
Using these 202 OA risk genes, we identified 29 suggestively significant KEGG pathways (unadjusted P < 0.05). Here, we provide the top 20 significant pathways, as provided in Table 1. The transforming growth factor β (TGF-β) signaling pathway is the most significant signal (P = 5.98E−05). Importantly, this is the only pathway after the BH multiple-test adjustment with FDR = 0.02. In addition, we identified other OA risk pathways including inflammatory bowel disease (IBD), human T-cell leukemia virus 1 infection, RA, influenza A, asthma, tuberculosis, prostate cancer, hematopoietic cell lineage, transcriptional misregulation in cancer, allograft rejection, mitogen-activated protein kinase (MAPK) signaling pathway, TH7 cell differentiation, graft-versus-host disease, toxoplasmosis, type 1 diabetes mellitus, cell cycle, intestinal immune network for IgA production, autoimmune thyroid disease, and ubiquitin-mediated proteolysis. More detailed information is described in Table 1.
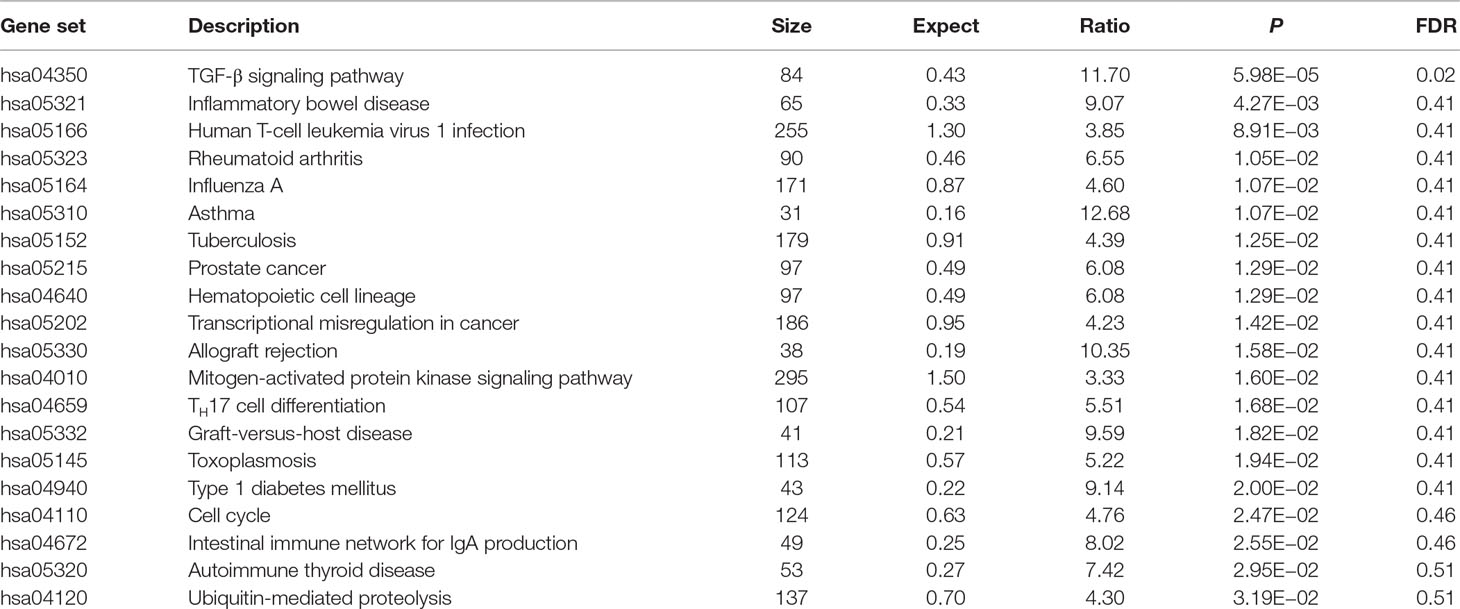
Table 1 Top 20 osteoarthritis risk pathways identified in KEGG database.
Identification of OA-Related GO Biological Processes
Using these 202 OA risk genes, we identified 24 statistically significant GO biological processes (FDR < 0.05). These biological processes could be mainly divided into two classes including differentiation and development. The differentiation-related biological processes consist of chondrocyte differentiation, regulation of chondrocyte differentiation, positive regulation of chondrocyte differentiation, regulation of epithelial cell proliferation, regulation of cell proliferation, and epithelial cell proliferation. The development-related biological processes consist of regulation of cartilage development, cartilage development, connective tissue development, chondrocyte development, skeletal system development, liver development, hepaticobiliary system development, positive regulation of cartilage development, striated muscle tissue development, and muscle tissue development. Here, we provide the top 20 significant biological processes, as provided in Table 2.
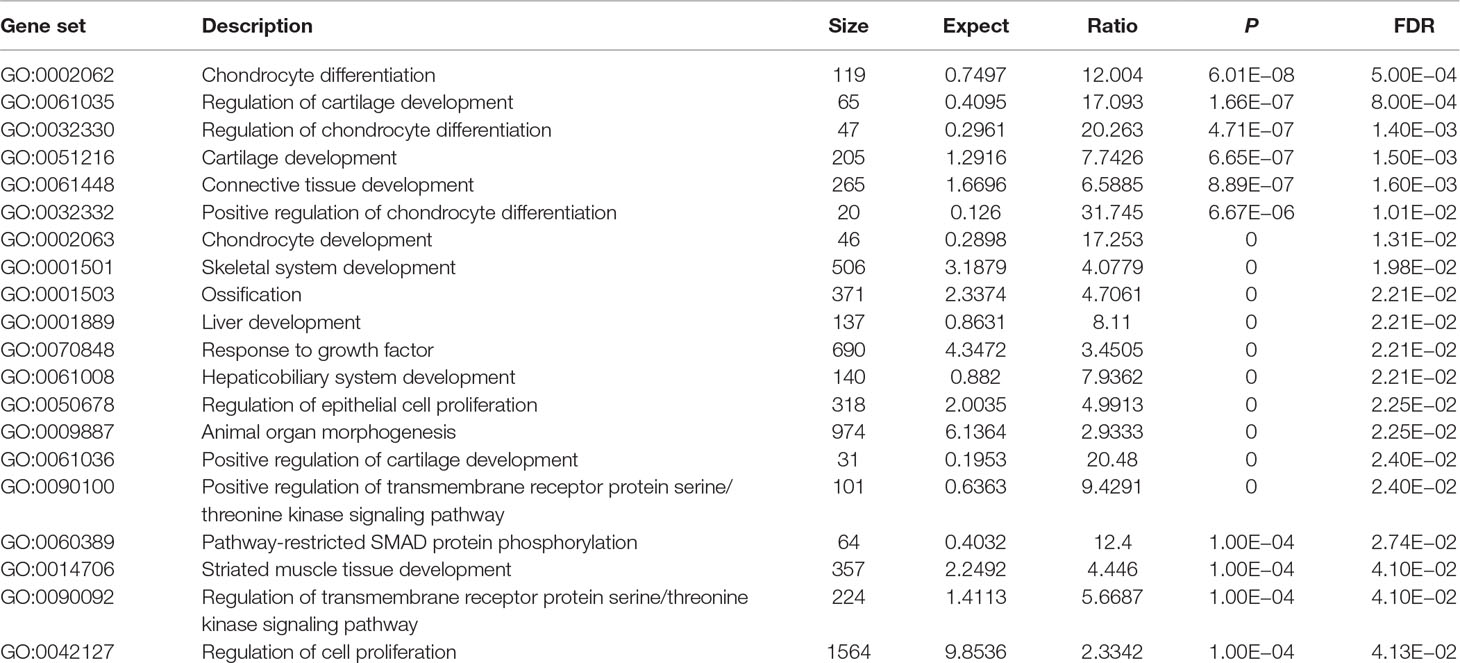
Table 2 Top 20 osteoarthritis risk biological processes identified in GO database.
Identification of OA-Related GO Molecular Functions
Using these 202 OA risk genes, we identified only one statistically significant GO molecular function (FDR < 0.05) TGF-β receptor binding (P = 9.30E−06). Here, we provide the top 20 significant molecular functions, as provided in Table 3. Most of these molecular functions are related with binding including TGF-β receptor binding, phospholipid binding, TGF-β binding, peptide antigen binding, antigen binding, phosphatase binding, type II TGF-β receptor binding, protein phosphatase binding, lipid binding, bridging protein binding, apolipoprotein binding, cytokine receptor binding, and cytokine binding.
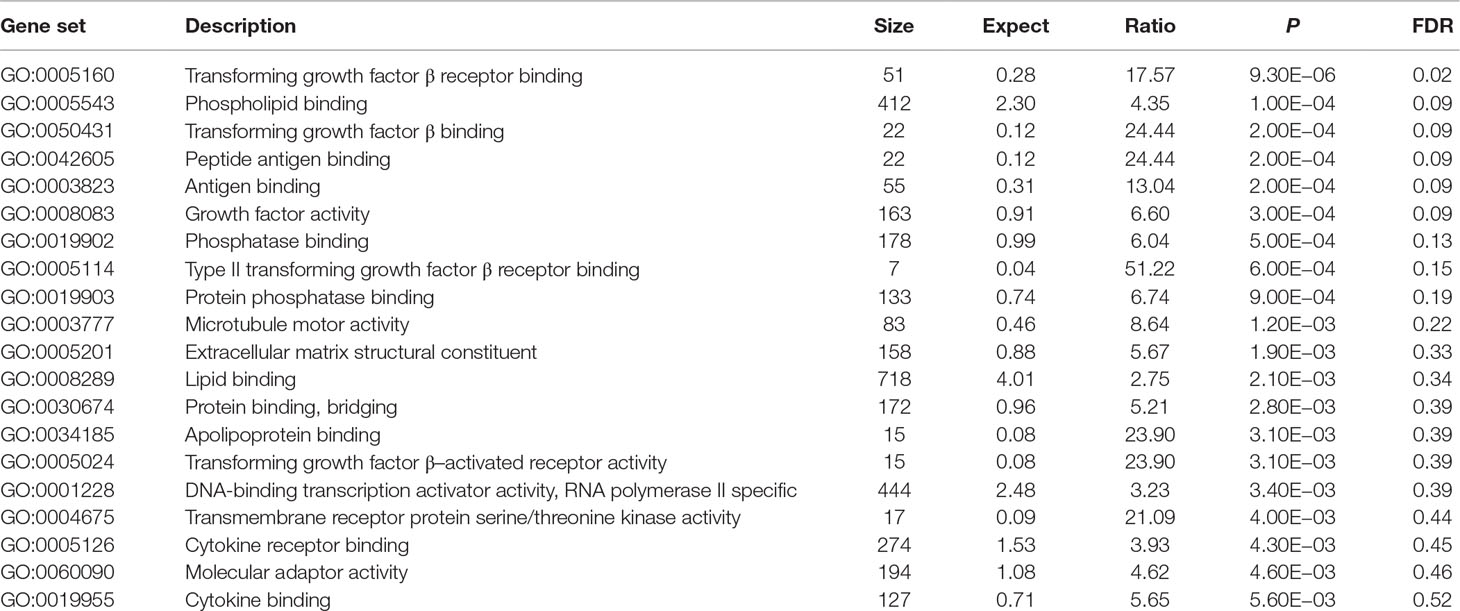
Table 3 Top 20 osteoarthritis risk molecular functions identified in GO database.
Identification of OA-Related GO Cellular Components
Using these 202 OA risk genes, we identified five statistically significant GO cellular components (FDR < 0.05) including extracellular matrix, COPII-coated endoplasmic reticulum (ER) to Golgi transport vesicle, collagen-containing extracellular matrix, extracellular matrix component, and major histocompatibility complex (MHC) protein. In addition, the top 20 significant cellular components also include coated vesicle, nucleoplasm part, endocytic vesicle membrane, fibrillar collagen trimer, banded collagen fibril, Golgi-associated vesicle, MHC class II protein complex, ER to Golgi transport vesicle membrane, cytoplasmic vesicle membrane, ER lumen, vesicle membrane, complex of collagen trimers, ER part, microtubule associated complex, and sarcoplasm. More detailed information is described in Table 4.
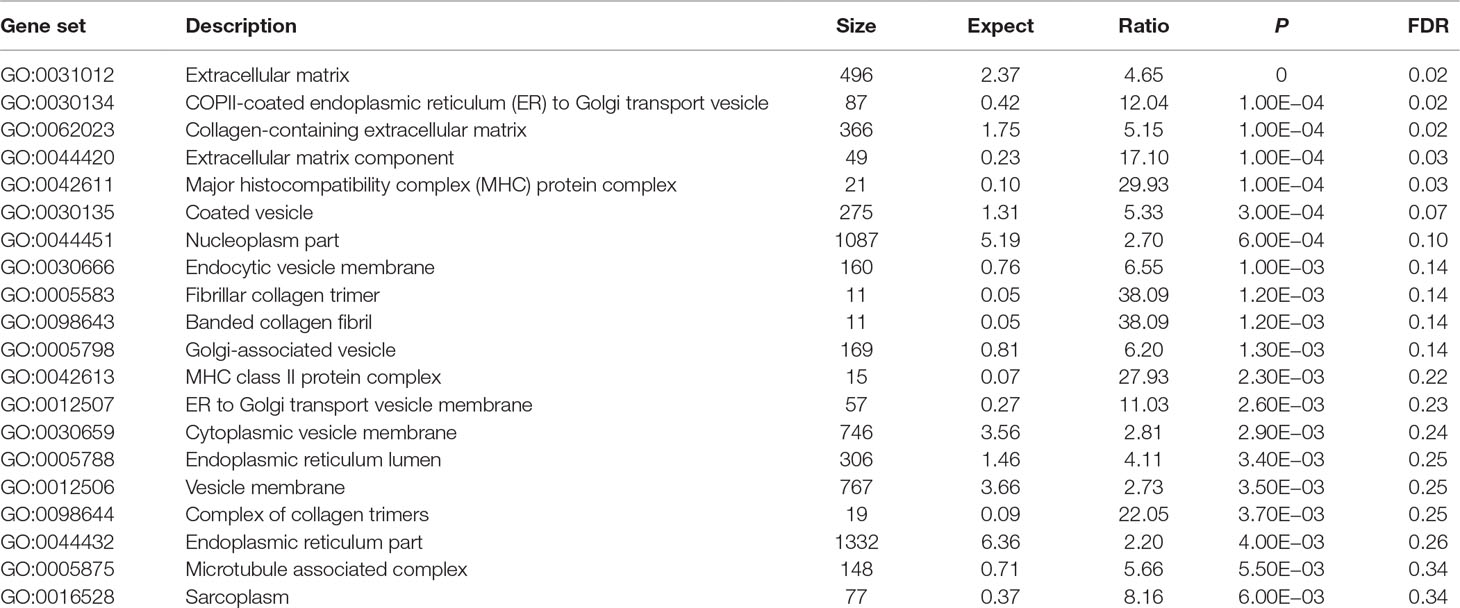
Table 4 Top 20 osteoarthritis risk cellular components identified in GO database.
OA Case–Control Gene Expression Analysis
Of the selected 202 risk genes, 122 risk genes were included in the OA case–control gene expression dataset, as provided in Supplementary Table 4. The results showed that 44 of these 122 risk genes showed suggestively differential expression in OA cases compared with controls (P < 0.05). Importantly, three genes including WWP2, COG5, and MAPT showed statistically different expression in OA cases compared with controls (P < 0.05/122 = 4.10E−04), as described in Table 5.
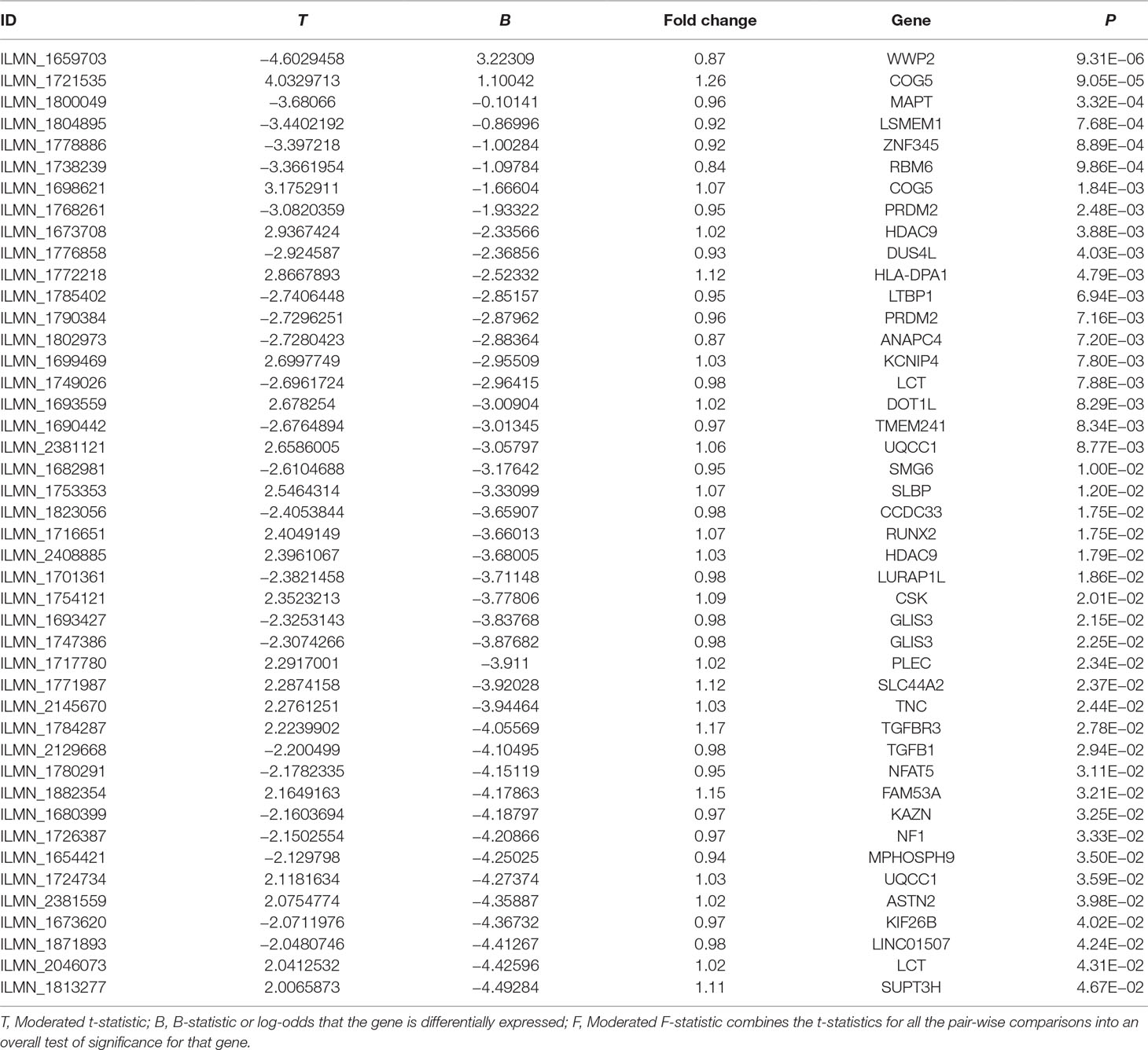
Table 5 Forty-four significantly differentially expressed osteoarthritis risk genes.
Discussion
In this study, we first selected 187 independent genetic variants identified by GWAS (P < 1.00E−05) and found that most of these genetic variants are noncoding mutations. We then conducted an eQTLs analysis and found that 165 of these 187 genetic variants could significantly regulate the expression of nearby genes. Third, we identified OA susceptibility genes corresponding to these genetic variants, conducted a pathway analysis, and identified novel OA-related KEGG pathways, GO biological processes, GO molecular functions, and GO cellular components.
In KEGG database, TGF-β signaling pathway is the most significant signal (P = 5.98E−05) and is the only pathway after the BH multiple-test adjustment with FDR = 0.02. Our finding is consistent with previous findings. Recent findings provide substantial evidence that TGF-β signaling contributes to OA development and progression (Shen et al., 2014; Fang et al., 2016). In addition, we found the association of OA with other human diseases including IBD, RA, asthma, prostate cancer, hematopoietic cell lineage, transcriptional misregulation in cancer, allograft rejection, MAPK signaling pathway, graft-versus-host disease, type 1 diabetes mellitus, and autoimmune thyroid disease. Previous study supported our finding about the association of human T-cell leukemia virus 1 infection pathway with OA. It is reported that human T lymphotropic virus type I retrovirus infection could alter the expression of inflammatory cytokines in primary OA (Yoshihara et al., 2004). Here, we highlighted the association of TH17 cell differentiation pathway with OA. Evidence showed that complement could drive TH17 cell differentiation and trigger autoimmune arthritis (Hashimoto et al., 2010; Li et al., 2017).
In GO database, we identified 24 statistically significant GO biological processes, one statistically significant GO molecular function, and five statistically significant GO cellular components (FDR < 0.05). These signals are related with chondrocyte differentiation and development, which are all known biological pathways associated with OA. Take the chondrocyte differentiation (GO:0002062), for example, previous studies also supported the hypertrophic differentiation of chondrocytes in OA (Dreier, 2010; Goldring, 2012). Some biological pathways are related with TGF-β signaling binding, such as TGF-β receptor binding, TGF-β binding, and type II TGF-β receptor binding. The phospholipid binding (GO:0005543) is the second significant signal among the top 20 OA risk molecular functions identified in GO database. Evidence shows that lipids are important nutrients in chondrocyte metabolism (Villalvilla et al., 2013). The lipid availability is important to keep cartilage status and OA development (Villalvilla et al., 2013).
Using an OA case–control gene expression analysis, we showed that 44 of these 122 risk genes were suggestively differentially expressed in OA cases compared with controls (P < 0.05). Three genes, WWP2, COG5, and MAPT, were statistically differentially expressed in OA cases compared with controls (P < 0.05/122 = 4.10E−04).
In summary, we believe that these findings provide further insight into the underlying genetic mechanisms for these newly identified OA risk genes. Although these are interesting findings, we also realize some limitations to our study. Until now, pathway analyses of RA GWAS datasets have widely been reported, but not OA GWAS datasets (Eleftherohorinou et al., 2009; Eleftherohorinou et al., 2011; Lee et al., 2012b; Song et al., 2013; Zhang et al., 2016). In the future, we will compare our findings using top OA genetic variants with pathway analysis of OA using the whole GWAS datasets and further clarify the differences between top OA genetic variants and the whole OA GWAS datasets. In addition, a gene expression analysis in OA-related tissues is necessary to demonstrate that these pathways are deregulated in OA cases and controls. However, this kind of gene expression datasets in OA-related tissues is not available now. Hence, we will further evaluate our findings using gene expression datasets in OA-related tissues in the future.
Data Availability
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ebi.ac.uk/gwas/downloads/summary-statistics.
Author Contributions
FG and JT conceived and initiated the project. FG analyzed the data. All authors wrote and reviewed the manuscript.
Funding
This work was supported by funding from Heilongjiang Natural Science Foundation (grant no. H2016025).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00827/full#supplementary-material
References
Bao, X., Liu, G., Jiang, Y., Jiang, Q., Liao, M., Feng, R., et al. (2015). Cell adhesion molecule pathway genes are regulated by cis-regulatory SNPs and show significantly altered expression in Alzheimer’s disease brains. Neurobiol. Aging 36 (2904), e2901–e2907. doi: 10.1016/j.neurobiolaging.2015.06.006
Buniello, A., MacArthur, J. A. L., Cerezo, M., Harris, L. W., Hayhurst, J., Malangone, C., et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012. doi: 10.1093/nar/gky1120
Cibrian Uhalte, E., Wilkinson, J. M., Southam, L., Zeggini, E. (2017). Pathways to understanding the genomic aetiology of osteoarthritis. Hum. Mol. Genet. 26, R193–R201. doi: 10.1093/hmg/ddx302
Dreier, R. (2010). Hypertrophic differentiation of chondrocytes in osteoarthritis: the developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 12, 216. doi: 10.1186/ar3117
Eleftherohorinou, H., Hoggart, C. J., Wright, V. J., Levin, M., Coin, L. J. (2011). Pathway-driven gene stability selection of two rheumatoid arthritis GWAS identifies and validates new susceptibility genes in receptor mediated signalling pathways. Hum. Mol. Genet. 20, 3494–3506. doi: 10.1093/hmg/ddr248
Eleftherohorinou, H., Wright, V., Hoggart, C., Hartikainen, A. L., Jarvelin, M. R., Balding, D., et al. (2009). Pathway analysis of GWAS provides new insights into genetic susceptibility to 3 inflammatory diseases. PLoS One 4, e8068. doi: 10.1371/journal.pone.0008068
Fang, J., Xu, L., Li, Y., Zhao, Z. (2016). Roles of TGF-beta 1 signaling in the development of osteoarthritis. Histol. Histopathol. 31, 1161–1167. doi: 10.14670/HH-11-779
Goldring, M. B. (2012). Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther. Adv. Musculoskelet. Dis. 4, 269–285. doi: 10.1177/1759720X12448454
Hashimoto, M., Hirota, K., Yoshitomi, H., Maeda, S., Teradaira, S., Akizuki, S., et al. (2010). Complement drives TH17 cell differentiation and triggers autoimmune arthritis. J. Exp. Med. 207, 1135–1143. doi: 10.1084/jem.20092301
Jiang, Q., Jin, S., Jiang, Y., Liao, M., Feng, R., Zhang, L., et al. (2017). Alzheimer’s disease variants with the genome-wide significance are significantly enriched in immune pathways and active in immune cells. Mol. Neurobiol. 54, 594–600. doi: 10.1007/s12035-015-9670-8
Kamat, M. A., Blackshaw, J. A., Young, R., Surendran, P., Burgess, S., Danesh, J., et al. (2019). PhenoScanner V2: an expanded tool for searching human genotype–phenotype associations. Bioinformatics [Epub ahead of print]. doi: 10.1093/bioinformatics/btz469
Krzywinski, M., Altman, N. (2014). Points of significance: comparing samples-part II. Nat. Methods 11, 355–356. doi: 10.1038/nmeth.2900
Lee, P. H., O’dushlaine, C., Thomas, B., Purcell, S. M. (2012a). INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics 28, 1797–1799. doi: 10.1093/bioinformatics/bts191
Lee, Y. H., Bae, S. C., Choi, S. J., Ji, J. D., Song, G. G. (2012b). Genome-wide pathway analysis of genome-wide association studies on systemic lupus erythematosus and rheumatoid arthritis. Mol. Biol. Rep. 39, 10627–10635. doi: 10.1007/s11033-012-1952-x
Li, Y. S., Luo, W., Zhu, S. A., Lei, G. H. (2017). T cells in osteoarthritis: alterations and beyond. Front. Immunol. 8, 356. doi: 10.3389/fimmu.2017.00356
Liu, G., Jiang, Y., Chen, X., Zhang, R., Ma, G., Feng, R., et al. (2013). Measles contributes to rheumatoid arthritis: evidence from pathway and network analyses of genome-wide association studies. PLoS One 8, e75951. doi: 10.1371/journal.pone.0075951
Liu, G., Jiang, Y., Wang, P., Feng, R., Jiang, N., Chen, X., et al. (2012). Cell adhesion molecules contribute to Alzheimer’s disease: multiple pathway analyses of two genome-wide association studies. J. Neurochem. 120, 190–198. doi: 10.1111/j.1471-4159.2011.07547.x
Liu, J. Z., Mcrae, A. F., Nyholt, D. R., Medland, S. E., Wray, N. R., Brown, K. M., et al. (2010). A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet. 87, 139–145. doi: 10.1016/j.ajhg.2010.06.009
Liu, G., Yao, L., Liu, J., Jiang, Y., Ma, G., Chen, Z., et al. (2014). Cardiovascular disease contributes to Alzheimer’s disease: evidence from large-scale genome-wide association studies. Neurobiol. Aging 35, 786–792. doi: 10.1016/j.neurobiolaging.2013.10.084
Liu, G., Zhang, F., Jiang, Y., Hu, Y., Gong, Z., Liu, S., et al. (2017). Integrating genome-wide association studies and gene expression data highlights dysregulated multiple sclerosis risk pathways. Mult. Scler. 23, 205–212. doi: 10.1177/1352458516649038
MacArthur, J., Bowler, E., Cerezo, M., Gil, L., Hall, P., Hastings, E., et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45, D896–D901. doi: 10.1093/nar/gkw1133
Martel-Pelletier, J., Barr, A. J., Cicuttini, F. M., Conaghan, P. G., Cooper, C., Goldring, M. B., et al. (2016). Osteoarthritis. Nat. Rev. Dis. Primers 2, 16072. doi: 10.1038/nrdp.2016.73
Parmet, S., Lynm, C., Glass, R. M. (2003). JAMA patient page. Osteoarthritis of the knee. JAMA 289, 1068. doi: 10.1001/jama.289.8.1068
Quan, B., Qi, X., Yu, Z., Jiang, Y., Liao, M., Wang, G., et al. (2015). Pathway analysis of genome-wide association study and transcriptome data highlights new biological pathways in colorectal cancer. Mol. Genet. Genomics 290, 603–610. doi: 10.1007/s00438-014-0945-y
Ramos, Y. F., Bos, S. D., Lakenberg, N., Bohringer, S., Den Hollander, W. J., Kloppenburg, M., et al. (2014). Genes expressed in blood link osteoarthritis with apoptotic pathways. Ann. Rheum. Dis. 73, 1844–1853. doi: 10.1136/annrheumdis-2013-203405
Shang, H., Liu, G., Jiang, Y., Fu, J., Zhang, B., Song, R., et al. (2015). Pathway analysis of two amyotrophic lateral sclerosis GWAS highlights shared genetic signals with Alzheimer’s disease and Parkinson’s disease. Mol. Neurobiol. 51, 361–369. doi: 10.1007/s12035-014-8673-1
Shen, J., Li, S., Chen, D. (2014). TGF-beta signaling and the development of osteoarthritis. Bone Res. 2014 (2): 14002. doi: 10.1038/boneres.2014.2
Song, G. G., Bae, S. C., Lee, Y. H. (2013). Pathway analysis of genome-wide association studies on rheumatoid arthritis. Clin. Exp. Rheumatol. 31, 566–574.
Staley, J. R., Blackshaw, J., Kamat, M. A., Ellis, S., Surendran, P., Sun, B. B., et al. (2016). PhenoScanner: a database of human genotype–phenotype associations. Bioinformatics 32, 3207–3209. doi: 10.1093/bioinformatics/btw373
Styrkarsdottir, U., Lund, S. H., Thorleifsson, G., Zink, F., Stefansson, O. A., Sigurdsson, J. K., et al. (2018). Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat. Genet. 50, 1681–1687. doi: 10.1038/s41588-018-0247-0
Styrkarsdottir, U., Thorleifsson, G., Helgadottir, H. T., Bomer, N., Metrustry, S., Bierma-Zeinstra, S., et al. (2014). Severe osteoarthritis of the hand associates with common variants within the ALDH1A2 gene and with rare variants at 1p31. Nat. Genet. 46, 498–502. doi: 10.1038/ng.2957
Tachmazidou, I., Hatzikotoulas, K., Southam, L., Esparza-Gordillo, J., Haberland, V., Zheng, J., et al. (2019). Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet. 51, 230–236. doi: 10.1038/s41588-018-0327-1
Villalvilla, A., Gomez, R., Largo, R., Herrero-Beaumont, G. (2013). Lipid transport and metabolism in healthy and osteoarthritic cartilage. Int. J. Mol. Sci. 14, 20793–20808. doi: 10.3390/ijms141020793
Ward, L. D., Kellis, M. (2012). HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934. doi: 10.1093/nar/gkr917
Ward, L. D., Kellis, M. (2016). HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881. doi: 10.1093/nar/gkv1340
Wang, J., Vasaikar, S., Shi, Z., Greer, M., Zhang, B. (2017). WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45, W130–W137. doi: 10.1093/nar/gkx356
Welter, D., MacArthur, J., Morales, J., Burdett, T., Hall, P., Junkins, H., et al. (2014). The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006. doi: 10.1093/nar/gkt1229
Xiang, Z., Xu, M., Liao, M., Jiang, Y., Jiang, Q., Feng, R., et al. (2015). Integrating genome-wide association study and brain expression data highlights cell adhesion molecules and purine metabolism in Alzheimer’s disease. Mol. Neurobiol. 52, 514–521. doi: 10.1007/s12035-014-8884-5
Yoon, S., Nguyen, H. C. T., Yoo, Y. J., Kim, J., Baik, B., Kim, S., et al. (2018). Efficient pathway enrichment and network analysis of GWAS summary data using GSA-SNP2. Nucleic Acids Res. 46, e60. doi: 10.1093/nar/gky175
Yoshihara, Y., Tsukazaki, T., Osaki, M., Nakashima, M., Hasui, K., Shindo, H. (2004). Altered expression of inflammatory cytokines in primary osteoarthritis by human T lymphotropic virus type I retrovirus infection: a cross-sectional study. Arthritis Res. Ther. 6, R347–R354. doi: 10.1186/ar1193
Zeggini, E., Panoutsopoulou, K., Southam, L., Rayner, N. W., Day-Williams, A. G., Lopes, M. C., et al. (2012). Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet 380, 815–823. doi: 10.1016/S0140-6736(12)60681-3
Zengini, E., Hatzikotoulas, K., Tachmazidou, I., Steinberg, J., Hartwig, F. P., Southam, L., et al. (2018). Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat. Genet. 50, 549–558. doi: 10.1038/s41588-018-0079-y
Zhang, M., Mu, H., Lv, H., Duan, L., Shang, Z., Li, J., et al. (2016). Integrative analysis of genome-wide association studies and gene expression analysis identifies pathways associated with rheumatoid arthritis. Oncotarget 7, 8580–8589. doi: 10.18632/oncotarget.7390
Keywords: osteoarthritis, genome-wide association study, pathway analysis, expession quantitative trait loci analysis, gene expression analysis
Citation: Gao F, Yao Y, Zhang Y and Tian J (2019) Integrating Genome-Wide Association Studies With Pathway Analysis and Gene Expression Analysis Highlights Novel Osteoarthritis Risk Pathways and Genes. Front. Genet. 10:827. doi: 10.3389/fgene.2019.00827
Received: 28 May 2019; Accepted: 12 August 2019;
Published: 13 September 2019.
Edited by:
Guiyou Liu, Tianjin Institute of Industrial Biotechnology (CAS), ChinaReviewed by:
Shuilin Jin, Harbin Institute of Technology, ChinaYing Hong Li, Chongqing University of Posts and Telecommunications, China
Copyright © 2019 Gao, Yao, Zhang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Tian, dGlhbmp1bjE5NjEwNTE1QDE2My5jb20=