 Yi-Tian Fu1
Yi-Tian Fu1 Guo-Hua Liu
Guo-Hua Liu- 1College of Veterinary Medicine, Hunan Agricultural University, Changsha, China
- 2Hunan Co-Innovation Center of Animal Production Safety, Changsha, China
Postharmostomum commutatum (Platyhelminthes: Brachylaimoidea), a parasite of the caeca of poultry, has been frequently reported from many countries and regions, including China. However, the molecular epidemiology, population genetics and phylogenetics of this parasite are poorly understood. In the present study, we determined and characterized the complete mitochondrial (mt) genome of P. commutatum, as the first representative from the superfamily Brachylaimoidea. The mt genome of P. commutatum is a circular DNA molecule of 13,799 bp in size and encodes the complete set of 36 genes (12 protein-coding genes, 22 transfer RNA genes, two ribosomal RNA genes) as well as a typical control region. The mt genome of P. commutatum presents a clear bias in nucleotide composition with a negative AT-skew on average (−0.306) and a positive GC-skew on average (0.466). Phylogenetic analyses showed that P. commutatum (superfamily Brachylaimoidea) and other ten members of the order Diplostomida were recovered as sister groups of the order Plagiorchiida, indicating that the order Diplostomida is paraphyletic. This is the first mt genome of any member of the superfamily Brachylaimoidea and should represent a rich source of genetic markers for molecular epidemiological, population genetic and phylogenetic studies of parasitic flukes of socio-economic importance in poultry.
Introduction
Postharmostomum commutatum (= P. gallinum) (Platyhelminthes: Brachylaimoidea) is one of the most common flukes of poultry (Taylor et al., 2016). This parasite inhabits the intestinal caeca of poultry and may be associated with the occurrence of inflammation and hemorrhages in heavily infected animals (Taylor et al., 2016). P. commutatum was mentioned for the first time by Wagener (1852), who found this parasite in the caeca of a young chicken from Italy. To date, this parasite has been frequently reported in Africa, the Americas, Asia and Europe (Valadão et al., 2018).
Metazoan mitochondrial (mt) genome is a biological macromolecule containing 36–37 genes (12–13 protein-coding genes, two ribosomal RNA genes and 22 transfer RNA genes) (Boore, 1999; Hu and Gasser, 2006). This gene content has been shown to vary in cestodes and trematodes and Chromadorea nematodes, which lack atp8 gene (Boore, 1999). Due to its maternal inheritance, high genome copy numbers, fast evolutionary rate, simple genetic structure and lack of recombination, mt genome sequences have been widely used in molecular epidemiological, population genetic and phylogenetic studies at various taxonomic levels of different parasitic worms (Zhang et al., 2017; Wu et al., 2018; Jin et al., 2019).
The digeneans (subclass Digenea) are distributed worldwide and comprise ∼18,000 described species (Kostadinova and Pérez-Del-Olmo, 2019). The phylogeny and classification of digeneans have been substantially revised with analyses of the two nuclear ribosomal RNA genes (Olson et al., 2003; Littlewood et al., 2015; Pérez-Ponce de León and Hernández-Mena, 2019). Recently, mt genomic datasets have also been used to understand the phylogenetic relationships of digeneans (Webster and Littlewood, 2012; Brabec et al., 2015; Briscoe et al., 2016; Chen et al., 2016; Locke et al., 2018). A major difference between the most accepted classification based on nuclear rRNA genes and mt genome phylogenies is the order Diplostomida. All previous phylogenetic analyses based on nuclear rRNA genes have supported the monophyly of this order within the subclass Digenea (Olson et al., 2003; Littlewood et al., 2015; Pérez-Ponce de León and Hernández-Mena, 2019). However, mt genome phylogeny rejected the monophyly of this order because Clinostomum complanatum (Schistosomatoidea), Alaria americana, Hysteromorpha triloba, Tylodelphys immer, Cardiocephaloides medioconiger, Cotylurus marcogliesei, Posthodiplostomum centrarchid, Cyathocotyle prussica (Diplostomoidea) and two Diplostomum species were recovered as sister groups of order Plagiorchiida, not the order Diplostomida (Brabec et al., 2015; Chen et al., 2016; Locke et al., 2018). The order Diplostomida currently consists of three superfamilies (Brachylaimoidea, Diplostomoidea and Schistosomatoidea). The superfamily Brachylaimoidea has potential veterinary importance and complex taxonomic history. Currently, the superfamily Brachylaimoidea contains at least 20 valid species that consist of parasites of mammals and birds (Heneberg et al., 2016). Despite their importance, no mt genome had been sequenced and characterized for any members of the superfamily Brachylaimoidea.
The objectives of the present study were to determine and analyze the complete mt genome of P. commutatum, as the first representative from the superfamily Brachylaimoidea, and to assess the systematic and phylogenetic position of this fluke within the subclass Digenea using concatenated protein sequences derived from all coding genes.
Materials and Methods
Parasites and Total Genomic DNA Isolation
Adult specimens of P. commutatum were collected from a naturally infected chicken in Hunan province of China. Adult worm specimens were washed separately in physiological saline, identified preliminarily to species based on morphological features described previously (Pojmańska, 2002), fixed in 70% (v/v) ethanol and stored at −20°C until further use. DNA extraction was performed from individual flukes using a commercially available kit (Wizard® SV Genomic DNA Purification System, Promega) according to the manufacturer’s instructions. The molecular identity of each specimen was further verified by PCR using an established method and then sequenced (Bowles and McManus, 1994). The mt cox1 sequences of P. commutatum samples showed 99% similarity with that of P. commutatum from Gallus gallus in Brazil (GenBank accession no. MH919409) (Valadão et al., 2018). Our phylogenetic analyses based on mt cox1 sequences of P. commutatum and relatives showed that two P. commutatum isolates grouped together, suggesting that the Postharmostomum isolate from present study represented P. commutatum (Figure S1).
Sequencing and Assembling
High molecular weight genomic DNA was extracted from an adult fluke and agarose-gel electrophoresis (1%) was used to verify DNA integrity. After fragmentation (400–500 bp) of this DNA by shearing using G-tubes (Coavris M220), a paired-end genomic library (about 320 bp inserts) was constructed using TruSeq™ DNA Sample Prep Kit (Illumina). All sequencing was carried out on Illumina Hiseq 4000 platform and data recorded in FASTQ format. The clean reads were obtained from raw reads by removing adaptor sequences, highly redundant sequences, reads that contained more than 10% ambiguous positions (N) and low-quality reads. Clean reads were assembled into contigs with Geneious 11.1.5 (Kearse et al., 2012) based on mt cox1 conserved sequence motifs. The assembly parameters were minimum overlap identity 99.5%, minimum overlap 150 bp and maximum gap size 5 bp. The assembly generated a large contig ending with overlapping fragments. As this structure allowed a single circular organization of the mt genome, we assumed that the complete mt genome had been assembled. The completeness of the mt genome assembly was further verified by long PCR experiment using five pairs of primers (Table S1) which were designed in the conserved regions.
Annotations
The assembled mt genome was annotated with the MITOS webserver (Bernt et al., 2013). The boundaries of protein-coding genes and rRNA genes were determined by alignment with the homologous genes of C. complanatum using the computer program MAFFT 7.122 with the option (L-INS-I) (Katoh and Standley, 2013). Amino acid sequences of 12 protein-coding genes were inferred using MEGA 6.0 (Tamura et al., 2013). Translation start and stop codons were identified based on comparison with those of C. complanatum reported previously (Chen et al., 2016). The identification, boundary delimitation and secondary structure folding of 22 tRNA genes were identified using ARWEN (Laslett and Canbäck, 2008) and the program tRNAscan-SE (Lowe and Chan, 2016) under the default search model, with the “other mitochondrial” sequence source and the “invertebrate mitochondrial” genetic code, and manual adjustment. The Ka/Ks ratio was calculated for nucleotide sequences of all 12 mt protein-coding genes of P. commutatum and other digeneans using DnaSP v5 (Librado and Rozas, 2009).
Phylogenetic Analysis
All mt genome sequences of subclass Digenea (Table 1), along with an outgroup of the subclass Monogenea (Gyrodactylus derjavinoides; GenBank accession number EU293891) (Huyse et al., 2008), were obtained from GenBank and combined for phylogenetic analysis. The deduced amino acid sequences of 12 protein-coding genes were aligned individually using MAFFT 7.122. The well-aligned conserved blocks were identified using Gblocks 0.91b with default parameters using the option for a less stringent selection (Talavera and Castresana, 2007). The individual amino acid or concatenated amino acid alignments and newick trees have been stored in a publicly available data repository (Accession ID: 25084; Study Accession URL: http://purl.org/phylo/treebase/phylows/study/TB2:S25084). Phylogenetic analyses were conducted using Maximum likelihood (ML) and Bayesian inference (BI). ML analysis were computed using PhyML 3.0 (Guindon et al., 2010). For ML analysis, it was partitioned by gene, and bootstrapping frequencies (BS) was performed using the rapid bootstrapping option with 100 iterations, the JTT (genes 1–6; cytb, cox3, nad2, nad4L, nad5 and nad6), LG (genes 7–8; cox2 and nad4), MtArt (genes 9–10; cox1 and nad1) and MtREV (genes 11–12; atp6 and nad3) models were used as selected by ProtTest 2.4 (Abascal et al., 2005) based on the Akaike information criterion (AIC). BI analysis was performed using MrBayes 3.2.6 (Ronquist and Huelsenbeck, 2003), two independent runs with four incrementally heated Metropolis-coupled Markov chains Monte Carlo were run for two million generations, with tree sampling conducted at every 200 generations. The first 25% of the sampled trees were discarded as burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities (Bpp). The potential scale reduction factor approached 1 and the average split frequency of less than 0.01 were used to represent the convergence of the two simultaneous runs. For BI analysis, the dataset was partitioned by gene, and the amino acid model for each gene was estimated from above models with model-given frequencies and gamma distributed rates. PhyloBayes 3.3b (Lartillot and Philippe, 2004) was run using the site-heterogeneous mixture CAT model, and the analysis was stopped when the conditions considered to indicate a good run were reached (maxdiff <0.1 and minimum effective size >300). The phylogenetic trees were visualized using FigTree v.1.42 (http://tree.bio.ed.ac.uk/software/figtree/).
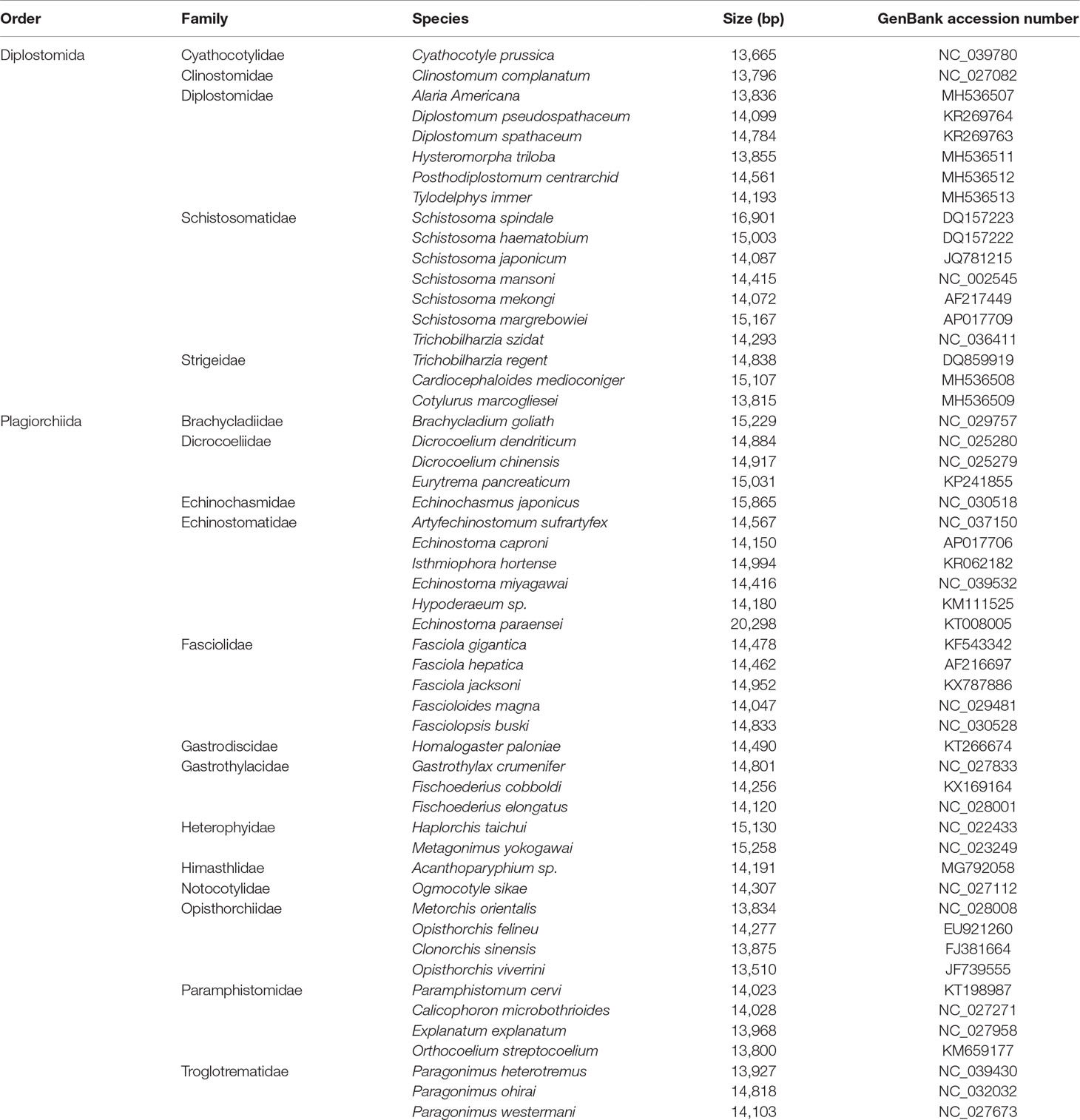
Table 1 Mitochondrial genome sequences of digeneans sequenced completely prior to the present study and used for phylogenetic analysis.
Results and Discussion
Genome Organization and Composition
We sequenced the P. commutatum genome and produced over 3 Gb of Illumina short-read sequence datasets. A total of 14,070,228 × 2 raw reads with the size of 250 bp were generated and 13,411,012 × 2 clean reads were obtained for assembly of the mt genome. The entire mt genome sequence of P. commutatum (GenBank accession no. MN200359) was 13,799 bp in size (Figure 1). We further confirmed the completeness of mt genome assembly using five pairs of primers covering the whole 13,800 bp-long assembled sequence to amplify the entire mt genome of P. commutatum. All five fragments (∼2–4 kb each) were successfully confirmed by long PCR amplification (Figure S2). This complete mt genome was slightly shorter than some other digeneans (such as Echinostoma caproni, Fischoederius elongatus and Schistosoma japonicum) but was slightly longer than some digeneans (such as C. complanatum, C. prussica and Opisthorchis viverrini) (Table 1). This difference is mainly due to the total fraction of non-coding sequences. This circular mt genome contains 12 protein-coding genes (cox1-3, nad1-6, nad4L, atp6 and cytb), 22 tRNA genes, two rRNA genes (rrnL and rrnS) and a non-coding (control or AT-rich) region (Table 2 and Figure 1). The gene orders are the same as those of flukes of the order plagiorchiida, such as O. viverrini, C. complanatum and Fasciola gigantica (Cai et al., 2012; Liu et al., 2014; Chen et al., 2016), but distinct from those of blood flukes, such as S. japonicum (Zhao et al., 2009) and S. turkestanicum (Wang et al., 2011). Additionally, the mt genes of P. commutatum overlap by 47 bp in five locations (1 to 40 bp per location) (Table 2). The mt genome of P. commutatum has 15 intergenic regions, which range from 1 to 24 bp in size. The longest region is between nad4 and tRNA-Gln (Table 2).
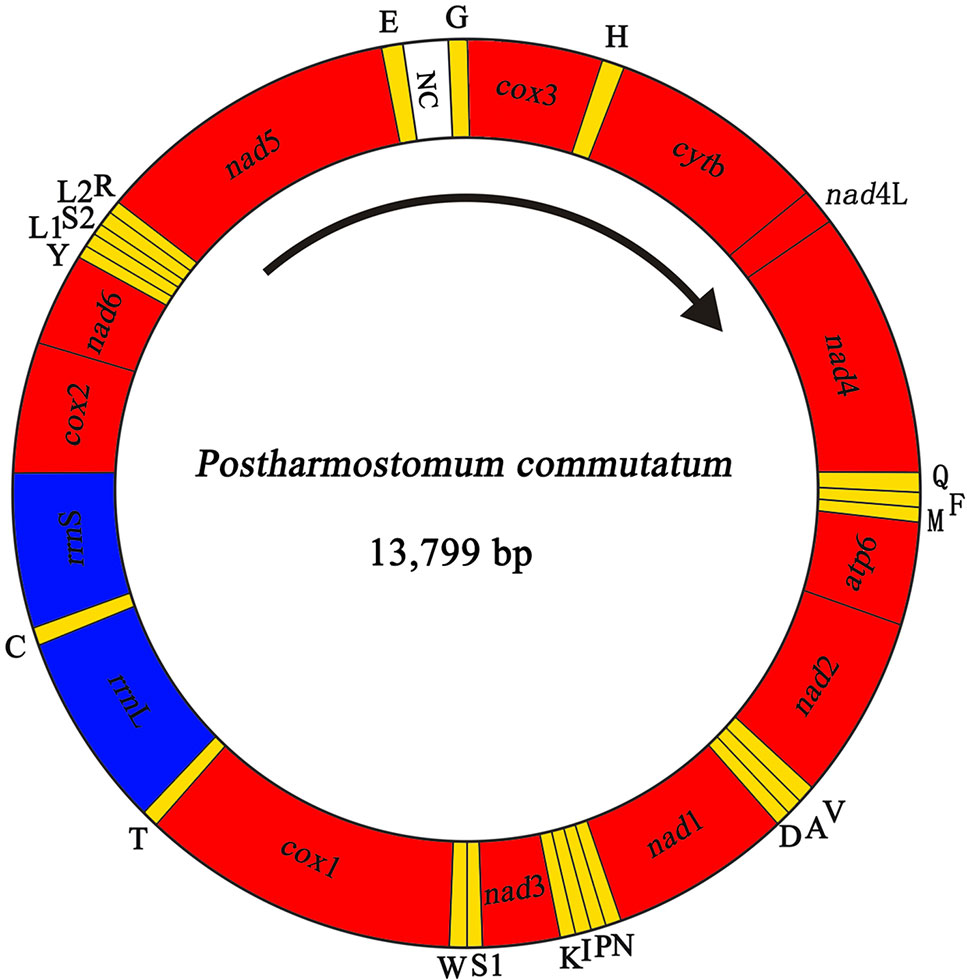
Figure 1 Organization of the mitochondrial genome of Postharmostomum commutatum. Scale is approximate. All genes have standard nomenclature except for the 22 tRNA genes, which are designated by the one-letter code for the corresponding amino acid, with numerals differentiating each of the two leucine- and serine-specifying tRNAs (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families UCN and AGN, respectively). All genes are transcribed in the clockwise direction. ‘NC’ indicates the non-coding region.
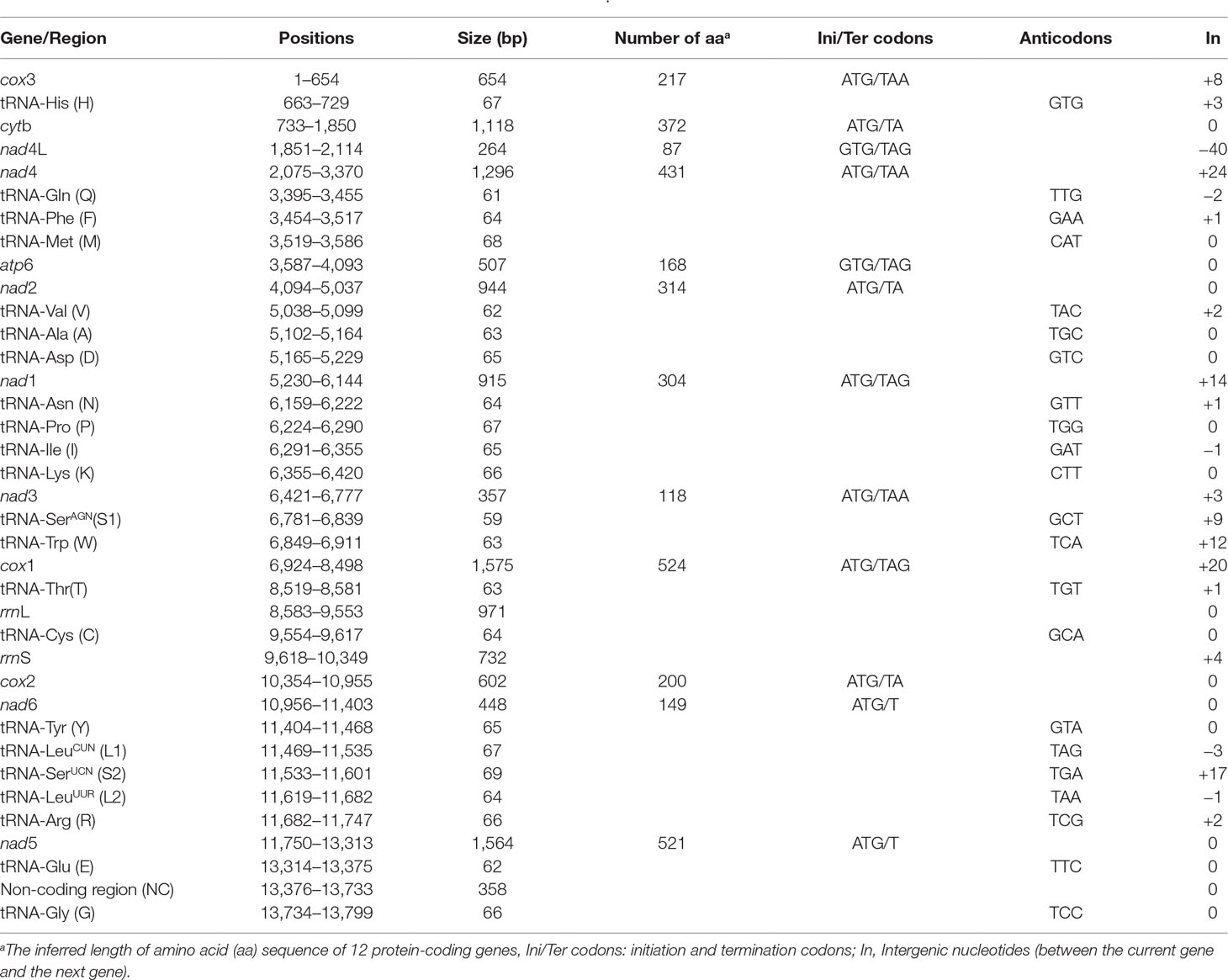
Table 2 The organization of the mt genome of Postharmostomum commutatum.
The nucleotide composition of the complete mt genome of P. commutatum is biased toward A+T (64.8%), in accordance with mt genomes of other digeneans (Zhao et al., 2009; Wang et al., 2011; Suleman et al., 2019b). AT- and GC- skews are a measure of compositional asymmetry. In P. commutatum mt genome, AT- skews values were always negative, while the values of GC- skew were positive (Table 3). The AT- skew value observed is −0.306 on average, ranging from −0.448 (nad6) to −0.140 (rrnS). The average GC- skew value observed is 0.466, ranging from 0.339 (22 tRNA) to 0.737 (nad3) (Table 3). In all mt genome sequences of flatworm (including tapeworms, nematodes and trematodes) reported to date (Liu et al., 2013; Briscoe et al., 2016; Zhao et al., 2016), the GC skew is positive due to the very low C content in mt genomes.
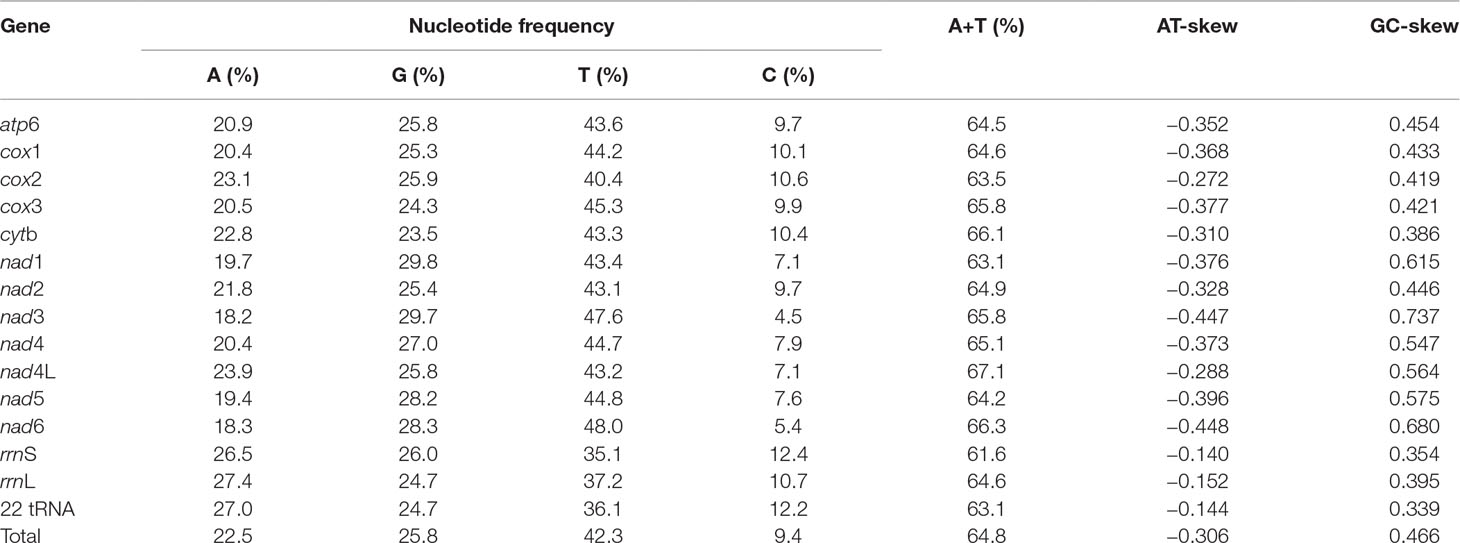
Table 3 Nucleotide composition and skews of Postharmostomum commutatum mitochondrial genome.
Protein-Coding Genes
A total of 3,405 amino acids are encoded by the P. commutatum mt genome. The enriched A+T content was reflected in the codon usage. In P. commutatum mt genome, Leu and Phe are the most frequently encoded amino acids, and Gln is the least frequent (Table 4). Individually, the most frequently used amino acid was TTT (Phe; 8.93%), followed by TTG (Leu; 7.76%), TTA (Leu; 6.91%) and GTT (Val; 5.15%) (Table 4). All of the 12 identified protein-coding genes begin with ATG (cox1, cox2, cox3, cytb, nad1, nad2, nad3, nad4, nad5 and nad6) or GTG (atp6 and nad4L) as their start codons. Seven of the 12 genes appear to use TAA (cox3, nad4 and nad3) or TAG (nad4L, atp6, nad1 and cox1) as the stop codon, while the other genes end with incomplete codon TA (cytb, nad2 and cox2) or T (nad5 and nad6). This is very common in worm mt genomes, such as tapeworms (Nakao et al., 2007), nematodes (Hu et al., 2002; Hu et al., 2003; Kim et al., 2006) and trematodes (Chang et al., 2016). It is hypothesized that the mRNAs ending in T or TA are converted to TAA by post-transcriptional polyadenylation (Ojala et al., 1981).
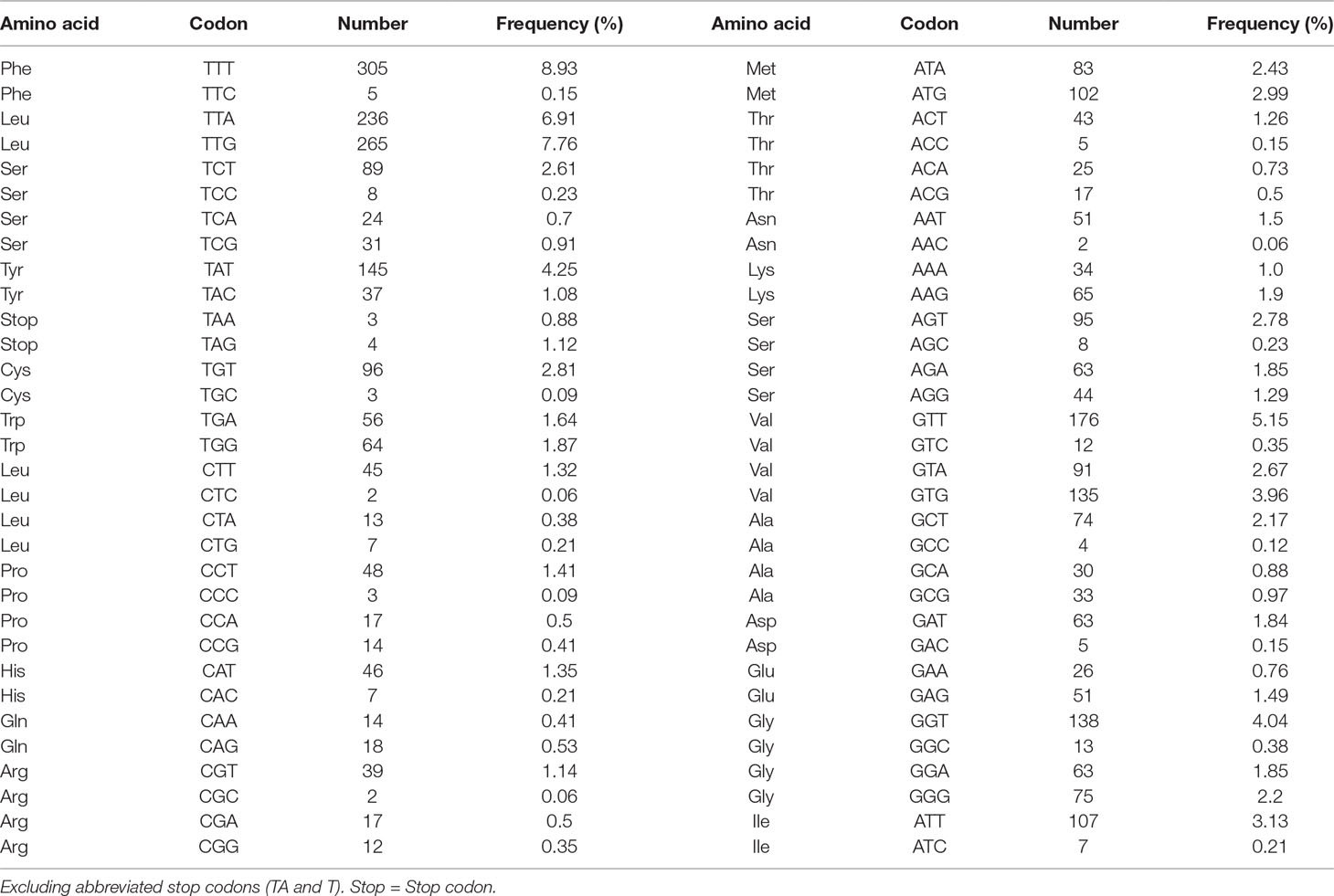
Table 4 Codon usage of Postharmostomum commutatum mitochondrial protein-coding genes.
Transfer RNA Genes and Ribosomal RNA Genes
The sizes of 22 tRNA genes identified in mt genome of P. commutatum, ranged from 59 to 69 bp in length. A standard four-arm cloverleaf structure was inferred for most of the tRNA genes. However, the tRNA-SerAGN (S1) gene shows an unorthodox structure, with the paired dihydrouridine (DHU) arm missing, as usual in all parasitic trematodes (also seen in some cestodes and nematodes) (Nakao et al., 2002; Hu et al., 2003). Structures for tRNA-Cys (C) and tRNA-SerUCN (S2) often vary somewhat among the parasitic trematodes. A paired DHU-arm of these tRNA genes is not seen in Haplorchis taichui, S. mansoni (Blair et al., 1999; Lee et al., 2013), but it is present in P. commutatum. The rrnL gene of P. commutatum is located between tRNA-Thr and tRNA-Cys genes, and rrnS gene is located between tRNA-Cys and cox2genes. The sizes of the rrnL and rrnS genes for P. commutatum were 971 bp and 732 bp, respectively (Table 2). The A+T contents of the rrnL rrnS genes for P. commutatum are 64.6% and 61.6%, respectively. The sizes and A+T contents of the two rRNA genes for P. commutatum are almost similar to those of other digeneans sequenced to date, such as that of Clonorchis sinensis, Paragonimus ohirai and Uvitellina sp. (Cai et al., 2012; Le et al., 2019; Suleman et al., 2019a).
Non-Coding Region
The mt genome sequences of flukes contain usually two non-coding regions (NC) of significant size difference (Yan et al., 2013; Le et al., 2019; Suleman et al., 2019b). However, In P. commutatum mt genome, there is only one NC (Table 2 and Figure 1). The NC is located between the tRNA-Glu and tRNA-Gly, and lacks any tandem repeats. Its size is 358 bp and A+T contents is 76.5%. One NC was also identified in H. taichu and Fasciolopsis buski mt genomes (Lee et al., 2013; Ma et al., 2017). Although the function of non-coding regions is currently unknown, the high A+T content predicts an involvement in the initiation of replication (Keddie et al., 1998).
Non-Synonymous/Synonymous Substitution Ratio of Protein-Coding Genes
The non-synonymous (Ka)/synonymous (Ks) substitutions ratio is particularly useful for characterizing evolutionary relationships between mt protein-coding genes in closely-related species (Fay and Wu, 2003). The Ka/Ks ratio was calculated for nucleotide sequences of all 12 mt protein-coding genes of P. commutatum and other digeneans (Table 1). The Ka/Ks ratio is a measure of selective pressures acting on genes, which indicates either negative (Ka/Ks <1) or positive (Ka/Ks >1) or that positive and negative selection counter-balance each other (Ka/Ks = 1) (Meganathan et al., 2011; Li et al., 2012). In the P. commutatum mt genome, atp6 appeared to have the highest Ka/Ks ratio (2.230), while cox1 is the lowest Ka/Ks ratio (0.154) (Figure 2). Herein, the Ka/Ks ratio of nine protein-coding genes (cox1, cox2, cox3, nad2, nad3, nad4, nad4L, nad6 and cytb) was <1 (range: 0.154 to 0.989), suggesting that these mt protein-coding genes of digeneans are under purifying selection. The Ka/Ks ratio of three protein-coding genes (nad1, nad5 and atp6) was >1 (range: 1.169 to 2.230), indicating that these mt protein-coding genes of digeneans have evolved under positive or Darwinian selection. A similar pattern is also observed for nematode mt genomes (Liu et al., 2013).
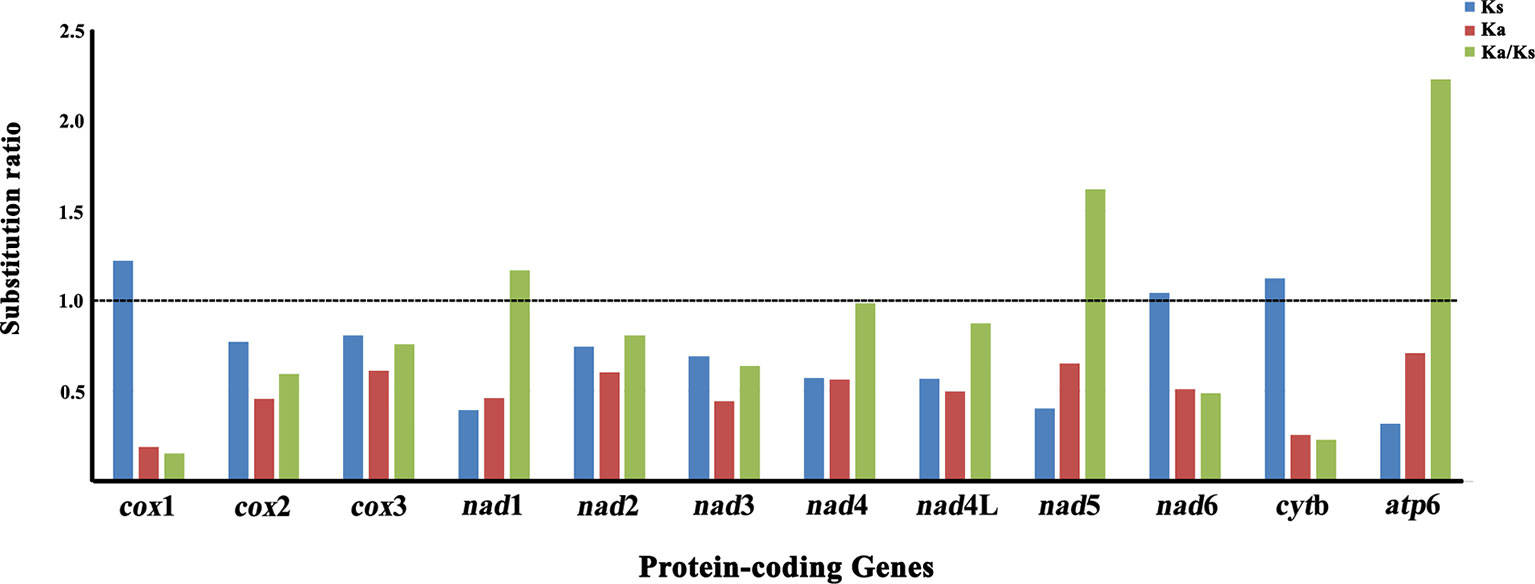
Figure 2 Substitution ratios in the mitochondrial genomes of digeneans. The rate of non-synonymous (Ka), the rate of synonymous (Ks) substitutions, and the respective ratios (Ka/Ks) for individual protein-coding genes are shown.
Comparison With Other Selected Digeneans Mt Genomes
The amino acid sequences of P. commutatum were compared with other digeneans (Table 5). In addition, the amino acid sequence similarities between P. commutatum and three species from the order Plagiorchiida ranged from 22.6–72.1% (Table 5). However, the amino acid sequence similarity between P. commutatum and the selected three species from the order Diplostomida ranged from 20.2–71.0% (Table 5). These results show that the superfamily Brachylaimoidea (represented by P. commutatum) was more closely related to the members of order Plagiorchiida than it was to the members of order Diplostomida. Based on identity, COX1 was the most conserved protein, whereas ATP6 was the least conserved (Table 5).
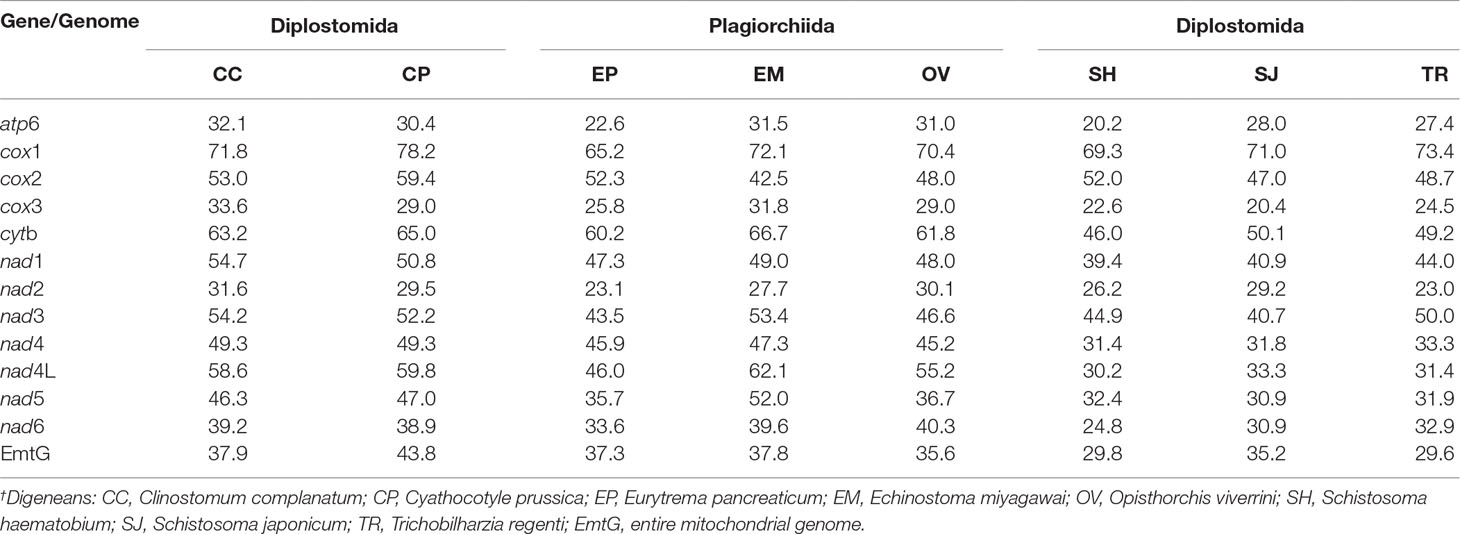
Table 5 Pairwise identities (%) in mitochondrial amino acid sequences between Postharmostomum commutatum and other representative digeneans.
Phylogenetic Analyses
The present study included three superfamilies (Diplostomoidea, Schistosomatoidea and Brachylaimoidea) from the order Diplostomida and phylogenetic analysis showed that the order Diplostomida was paraphyletic with strong support in BI (Bpp= 1.0, Figure 3) and ML (BS = 100, Figure 4), but was weakly supported in PhyloBayes (Bpp = 0.5, Figure 5) analyses. The monophyly of the superfamily Diplostomoidea was weakly supported with BI (Bpp = 0.87, Figure 3), and was strongly supported in ML (BS = 100, Figure 4) and PhyloBayes (Bpp = 1.0, Figure 5) analyses. The superfamily Schistosomatoidea, however, was not monophyletic in all of the three phylogenetic analyses in this study. One species (C. complanatum) from the superfamily Schistosomatoidea was more closely related to C. prussica (Diplostomoidea) (Figure 3) or P. commutatum (Brachylaimoidea) (Figures 4 and 5) than it was to the other 8 species from the superfamily Schistosomatoidea.
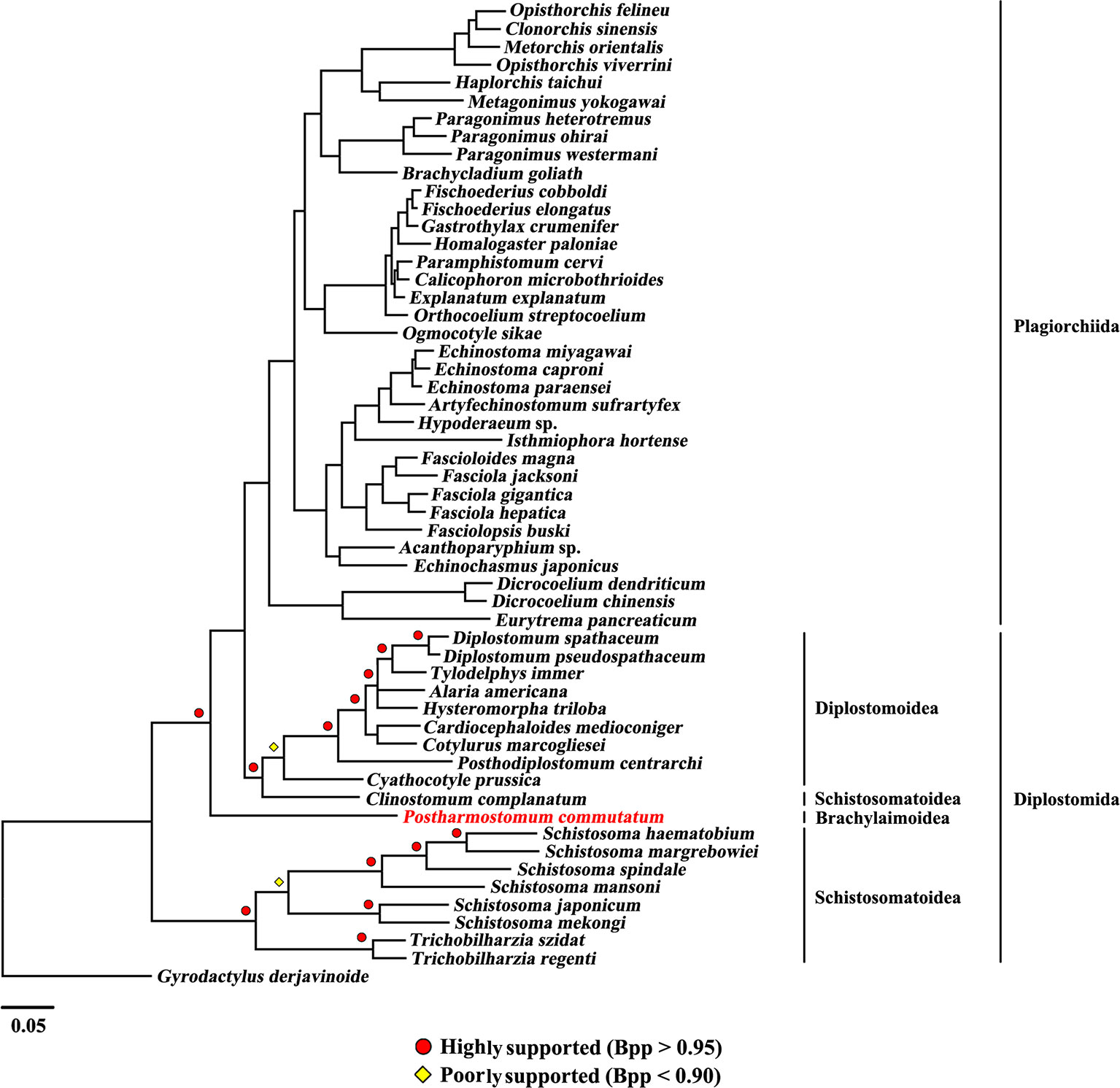
Figure 3 Phylogenetic relationships of Postharmostomum commutatum with other selected digeneans based on mitochondrial sequence data. The concatenated amino acid sequences of 12 protein-coding genes were subjected to analysis by Bayesian inference (BI) using Gyrodactylus derjavinoides as an outgroup. Bayesian posterior probabilities (Bpp) values are indicated.
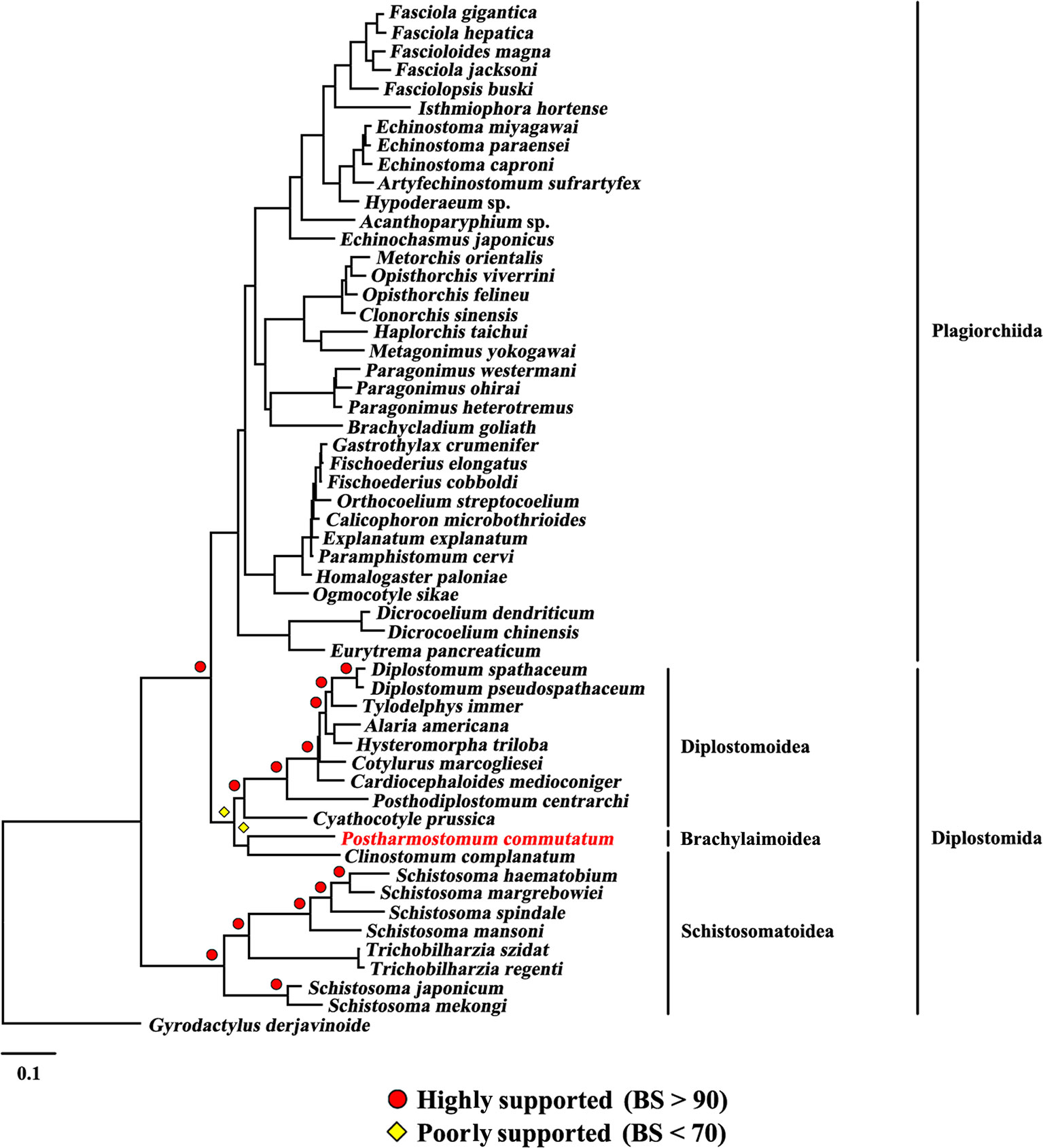
Figure 4 Phylogenetic relationships of Postharmostomum commutatum with other selected digeneans based on mitochondrial sequence data. The concatenated amino acid sequences of 12 protein-coding genes were subjected to analysis by Maximum likelihood (ML) using Gyrodactylus derjavinoides as an outgroup. Bootstrap frequency (BS) values are indicated.
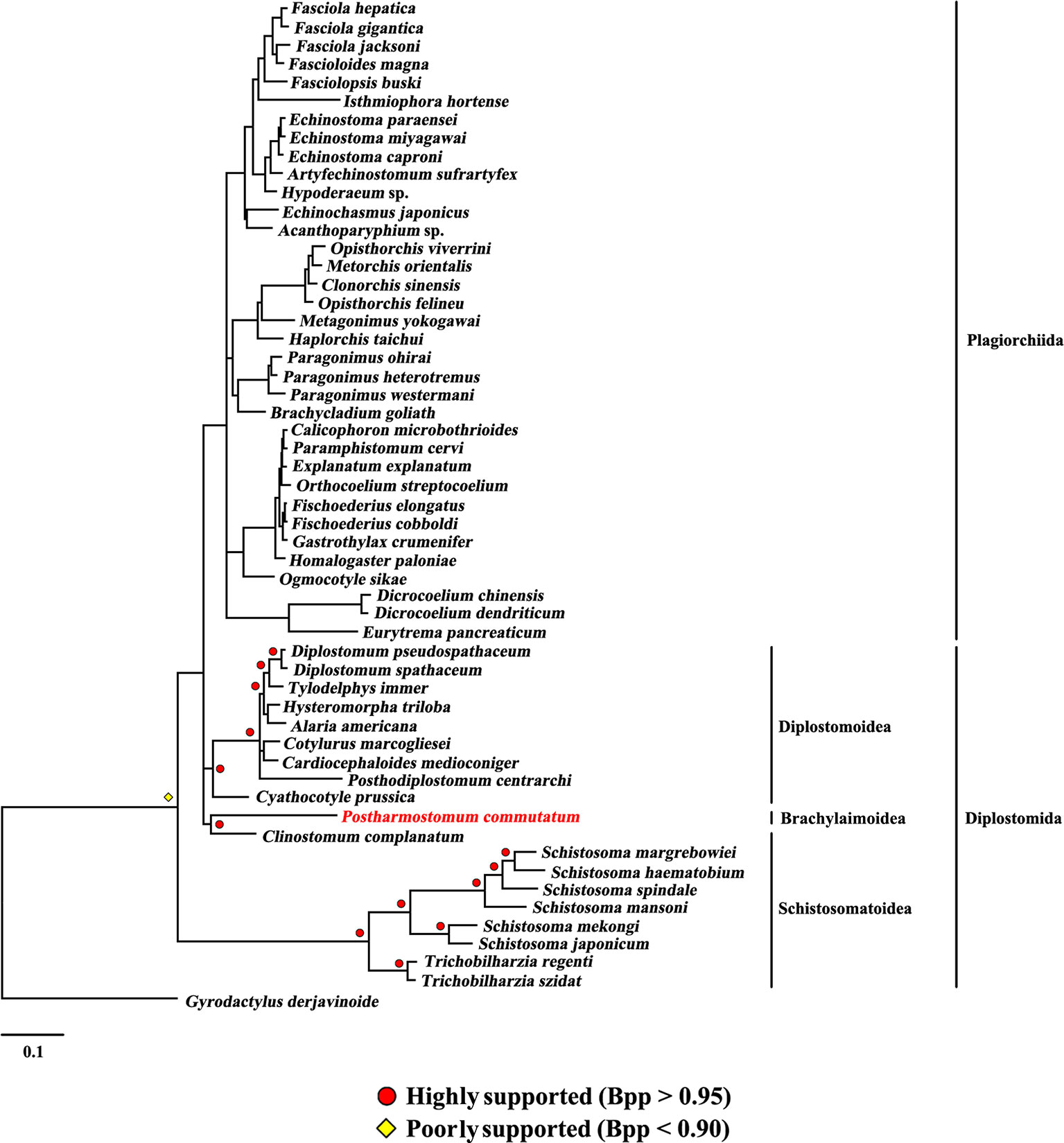
Figure 5 Phylogenetic relationships of Postharmostomum commutatum with other selected digeneans based on mitochondrial sequence data. The concatenated amino acid sequences of 12 protein-coding genes were subjected to analysis by PhyloBayes using Gyrodactylus derjavinoides as an outgroup. Bayesian posterior probabilities (Bpp) values are indicated.
Eleven species representing three superfamilies (Diplostomoidea, Schistosomatoidea and Brachylaimoidea) from the order Diplostomida were more closely related to the members of the order Plagiorchiida than they were to the other eight species from the order Diplostomida. Our results were consistent with those of previous studies from mt genome datasets. For example, Brabec et al. (2015) sequenced the mt genomes of two species of diplostomids, and their phylogenetic analyses recovered the family Diplostomidae as the sister group of the order Plagiorchiida, although those relationships were supported by a low nodal support value. Chen et al. (2016) generated the complete mt genome of C. complanatum and performed a phylogenetic analysis with mt genomes, indicating that C. complanatum is the sister group of the order Plagiorchiida with strong support in ML analyses. Most recently, Locke et al. (2018) determined the complete mt genome of seven diplostomoids representing three families (Diplostomidae, Strigeidae and Cyathocotylidae) and their mt genome phylogenetic tree yielded the order Diplostomida as paraphyletic because strigeids, diplostomids and clinostomids were recovered as sister groups of the order Plagiorchiida, not the order Diplostomida. No species from this superfamily (Brachylaimoidea) of the order Diplostomida were included in previous analyses of mt genome datasets (Brabec et al., 2015; Briscoe et al., 2016; Chen et al., 2016; Locke et al., 2018). In the present study, the determination of the mt genome of P. commutatum allows a reassessment of the phylogenetic relationships of digeneans. Our results confirm and expand on recent analyses showing a paraphyletic pattern of mt genome evolution in the order Diplostomida (Brabec et al., 2015; Briscoe et al., 2016; Chen et al., 2016; Locke et al., 2018).
The work of Olson et al. (2003) created robustness and stability in higher systematics within the subclass Digenea based on nuclear rRNA genes. Monophyly of the order Diplostomida has also been established previously in nuclear rRNA genes (Olson et al., 2003; Littlewood et al., 2015; Locke et al., 2018; Pérez-Ponce de León and Hernández-Mena, 2019). The mt genomic phylogenetic relationships of the order Diplostomida revealed a conflict with the rDNA phylogeny. Locke et al. (2018) discussed the possible causes of the mt genome topology and noted that the discrepancy occurs along short internal branches at the base of longer terminal branches, which could be related to a rapid radiation and incomplete lineage sorting. Along these short internal branches, mt genomes of digeneans may have a lower phylogenetic signal than nuclear genomes, exacerbating effects of incomplete taxon sampling (Hedtke et al., 2006; Philippe et al., 2011). In addition, both mt and nuclear genome data for representatives of the Diplostomida and of the early divergent lineages of the Plagiorchiida are needed to address the relationships of the two major lineages of the Digenea (Locke et al., 2018). Although the number of digenean mt genome sequences is increasing, to date, mt genomes of many lineages of digeneans are underrepresented or not represented. Insufficient taxon sampling for digenean mt genomes might be the cause of the discordance between the mt and nuclear datasets. Therefore, more mt genomes of digenean species representing families that have not yet been sequenced should be included in future analysis to resolve the taxonomic problems of digeneans because mt genome sequences have been shown to resolve deep-level relationships in many metazoan groups (Littlewood, 2008) and the use of mtDNA sequences has been considered promising (Philippe et al., 2011).
Conclusion
The present study determined the complete mt genome sequence of P. commutatum, which shares some similarity with, and interesting differences to, other digeneans. Phylogenetic analyses showed that P. commutatum was recovered as sister group of the order Plagiorchiida, supporting that the order Diplostomida is paraphyletic. The availability of the P. commutatum mt genome should represent a rich source of genetic markers for molecular epidemiological, population genetic and phylogenetic studies of parasitic flukes of socio-economic importance in poultry.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: GenBank accession no. MN200359.
Ethics statement
All procedures involving animals in the present study were approved and this study was approved by the Animal Ethics Committee of Hunan Agricultural University (No. 43321503).
Author Contributions
G-HL and Y-TF conceived and designed the study, and critically revised the manuscript. Y-TF and Y-CJ performed the experiments. Y-TF and Y-CJ analyzed the data. G-HL and Y-TF drafted the manuscript. Y-TF and Y-CJ helped in study design, study implementation, and manuscript preparation. All authors read and approved the final manuscript.
Funding
This study was supported by the Planned Program of Hunan Province Science and Technology Innovation (grant no. 2018RS3085) and the Training Program for Excellent Young Innovators of Changsha (grant No. KQ1802035).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01037/full#supplementary-material
Table S1 | Primers used for assembly validation.
Figure S1 | Inferred phylogenetic relationships among P. commutatum and other relatives based on mitochondrial cox1 sequences utilizing maximum likelihood (ML) using Ornithobilharzia canaliculata as an outgroup.
Figure S2 | PCR amplicons from the mitochondrial genome of Postharmostomum commutatum. Amplicons generated with the P. commutatum primers. M: DL5000 DNA marker, 1: Validation_01, 2: Validation_02, 3: Validation_03, 4: Validation_04, 5: Validation_05, 6: Negative control.
References
Abascal, F., Zardoya, R., Posada, D. (2005). ProtTest: selection of best-fifit models of protein evolution. Bioinformatics 21, 2104–2105. doi: 10.1093/bioinformatics/bti263
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogen. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Blair, D., Le, T. H., Després, L., McManus, D. P. (1999). Mitochondrial genes of Schistosoma mansoni. Parasitology 119, 303–313. doi: 10.1017/S0031182099004709
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic. Acids. Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Bowles, J., McManus, D. P. (1994). Genetic characterization of the Asian Taenia, a newly described taeniid cestode of humans. Am. J. Trop. Med. Hyg. 50, 33–44. doi: 10.4269/ajtmh.1994.50.1.TM0500010033
Brabec, J., Kostadinova, A., Scholz, T. (2015). Complete mitochondrial genomes and nuclear ribosomal RNA operons of two species of Diplostomum (Platyhelminthes: Trematoda): a molecular resource for taxonomy and molecular epidemiology of important fish pathogens. Parasit. Vectors 8, 336. doi: 10.1186/s13071-015-0949-4
Briscoe, A. G., Bray, R. A., Brabec, J., Littlewood, D. T. J. (2016). The mitochondrial genome and ribosomal operon of Brachycladium goliath (Digenea: Brachycladiidae) recovered from a stranded minke whale. Parasitol. Int. 65, 271–275. doi: 10.1016/j.parint.2016.02.004
Cai, X. Q., Liu, G. H., Song, H. Q., Wu, C. Y., Zou, F. C., Yan, H. K., et al. (2012). Sequences and gene organization of the mitochondrial genomes of the liver flukes Opisthorchis viverrini and Clonorchis sinensis (Trematoda). Parasitol. Res. 110, 235–243. doi: 10.1007/s00436-011-2477-2
Chang, Q. C., Liu, G. H., Gao, J. F., Zheng, X., Zhang, Y., Duan, H., et al. (2016). Sequencing and characterization of the complete mitochondrial genome from the pancreatic fluke Eurytrema pancreaticum (Trematoda: Dicrocoeliidae). Gene 576, 160–165. doi: 10.1016/j.gene.2015.09.081
Chen, L., Feng, Y., Chen, H. M., Wang, L. X., Feng, H. L., Yang, X., et al. (2016). Complete mitochondrial genome analysis of Clinostomum complanatum and its comparison with selected digeneans. Parasitol. Res. 115, 3249–3256. doi: 10.1007/s00436-016-5102-6
Fay, J. C., Wu, C. (2003). Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genomics Hum. Genet. 4, 213–235. doi: 10.1146/annurev.genom.4.020303.162528
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hedtke, S. M., Townsend, T. M., Hillis, D. M., Collins, T. (2006). Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Syst. Biol. 55, 522–529. doi: 10.1080/10635150600697358
Heneberg, P., Sitko, J., Bizos, J. (2016). Molecular and comparative morphological analysis of central European parasitic flatworms of the superfamily Brachylaimoidea Allison, 1943 (Trematoda: Plagiorchiida). Parasitology 143, 455–474. doi: 10.1017/S003118201500181X
Hu, M., Chilton, N. B., Gasser, R. B. (2002). The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int. J. Parasitol. 32, 145–158. doi: 10.1016/S0020-7519(01)00316-2
Hu, M., Chilton, N. B., Gasser, R. B. (2003). The mitochondrial genome of Strongyloides stercoralis (Nematoda) idiosyncratic gene order and evolutionary implications. Int. J. Parasitol. 33, 1393–1408. doi: 10.1016/S0020-7519(03)00130-9
Hu, M., Gasser, R. B. (2006). Mitochondrial genomes of parasitic nematodes–progress and perspectives. Trends Parasitol. 22, 78–84. doi: 10.1016/j.pt.2005.12.003
Huyse, T., Buchmann, K., Littlewood, D. T. L. (2008). The mitochondrial genome of Gyrodactylus derjavinoides (Platyhelminthes: Monogenea)–a mitogenomic approach for Gyrodactylus species and strain identification. Gene 417, 27–34. doi: 10.1016/j.gene.2008.03.008
Jin, Y. C., Li, X. Y., Liu, J. H., Zhu, X. Q., Liu, G. H. (2019). Comparative analysis of mitochondrial DNA datasets indicates that Toxascaris leonina represents a species complex. Parasit. Vectors 12, 194. doi: 10.1186/s13071-019-3447-2
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Keddie, E. M., Higazi, T., Unnasch, T. R. (1998). The mitochondrial genome of Onchocerca volvulus: sequence, structure and phylogenetic analysis. Mol. Biochem. Parasitol. 95, 111–127. doi: 10.1016/S0166-6851(98)00102-9
Kim, K. H., Eoma, K. S., Park, J. K. (2006). The complete mitochondrial genome of Anisakis simplex (Ascaridida: Nematoda) and phylogenetic implications. Int. J. Parasitol. 36, 319–328. doi: 10.1016/j.ijpara.2005.10.004
Kostadinova, A., Pérez-Del-Olmo, A. (2019). The Systematics of the Trematoda. Adv. Exp. Med. Biol. 1154, 21–42. doi: 10.1007/978-3-030-18616-6_2
Lartillot, N., Philippe, H. (2004). A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21, 1095–1109. doi: 10.1093/bioinformatics/btm573
Laslett, D., Canbäck, B. (2008). ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175. doi: 10.1093/bioinformatics/btm573
Le, T. H., Nguyen, K. T., Nguyen, N. T. B., Doan, H. T. T., Agatsuma, T., Blair, D. (2019). The complete mitochondrial genome of Paragonimus ohirai (Paragonimidae: Trematoda: Platyhelminthes) and its comparison with P. westermani congeners and other trematodes. PeerJ 7, e7031. doi: 10.7717/peerj.7031
Lee, D., Choe, S., Park, H., Jeon, H. K., Chai, J. Y., Sohn, W. M., et al. (2013). Complete mitochondrial genome of Haplorchis taichui and comparative analysis with other trematodes. Korean J. Parasitol. 51, 719–726. doi: 10.3347/kjp.2013.51.6.719
Li, H., Liu, H., Shi, A., Stys, P., Zhou, X. G., Cai, W. (2012). The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS One 7, e29419. doi: 10.1371/journal.pone.0029419
Librado, P., Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Littlewood, D. T. J. (2008). Platyhelminth systematics and the emergence of new characters. Parasite 15, 333–341. doi: 10.1051/parasite/2008153333
Littlewood, D. T. J., Bray, R. A., Waeschenbach, A. (2015). Parasite Diversity and Diversification: Phylogenetic patterns of diversity in the cestodes and trematodes. London: Cambridge University Press, 304–319. doi: 10.1017/CBO9781139794749
Liu, G. H., Gasser, R. B., Young, N. D., Song, H. Q., Ai, L., Zhu, X. Q. (2014). Complete mitochondrial genomes of the ‘intermediate form’ of Fasciola and Fasciola gigantica, and their comparison with F. hepatica.Parasit. Vectors 7, 150. doi: 10.1186/1756-3305-7-150
Liu, G. H., Gasser, R. B., Otranto, D., Xu, M. J., Shen, J. L., Mohandas, N., et al. (2013). Mitochondrial genome of the eyeworm, Thelazia callipaeda (Nematoda: Spirurida), as the first representative from the family Thelaziidae. PLoS Negl. Trop. Dis. 7, e2029. doi: 10.1371/journal.pntd.0002029
Locke, S. A., Van Dam, A., Caffara, M., Pinto, H. A., López-Hernández, D., Blanar, C. (2018). A. Validity of the Diplostomoidea and Diplostomida (Digenea, Platyhelminthes) upheld in phylogenomic analysis. Int. J. Parasitol. 48, 1043–1059. doi: 10.1016/j.ijpara.2018.07.001
Lowe, T. M., Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Ma, J., Sun, M. M., He, J. J., Liu, G. H., Ai, L., Chen, M. X., et al. (2017). Fasciolopsis buski (Digenea: Fasciolidae) from China and India may represent distinct taxa based on mitochondrial and nuclear ribosomal DNA sequences. Parasit. Vectors 10, 101. doi: 10.1186/s13071-017-2039-2
Meganathan, P. R., Dubey, B., Batzer, M. A., Ray, D. A., Haque, I. (2011). Complete mitochondrial genome sequences of three Crocodylus species and their comparison within the order Crocodylia. Gene 478, 35–41. doi: 10.1016/j.gene.2011.01.012
Nakao, M., Abmed, D., Yamasaki, H., Ito, A. (2007). Mitochondrial genomes of the human broad tapeworms Diphyllobothrium latum and Diphyllobothrium nihonkaiense (Cestoda: Diphyllobothriidae). Parasitol. Res. 101, 233–236. doi: 10.1007/s00436-006-0433-3
Nakao, M., Yokoyama, N., Sako, Y., Fukunaga, M., Ito, A. (2002). The complete mitochondrial DNA sequence of the cestode Echinococcus multilocularis (Cyclophyllidea: Taeniidae). Mitochondrion 1, 497–509. doi: 10.1016/S1567-7249(02)00040-5
Ojala, D., Crews, S., Montoya, J., Gelfand, R., Attardi, G. (1981). A small polyadenylated RNA (7S RNA), containing a putative ribosome attachment site, maps near the origin of human mitochondrial DNA replication. J. Mol. Biol. 150, 303–314. doi: 10.1016/0022-2836(81)90454-X
Olson, P. D., Cribb, T. H., Tkach, V. V., Bray, R. A., Littlewood, D. T. J. (2003). Phylogeny and classification of the Digenea (Platyhelminthes: Trematoda). Int. J. Parasitol. 33, 733–755. doi: 10.1016/S0020-7519(03)00049-3
Pérez-Ponce de León, G., Hernández-Mena, D. I. (2019). Testing the higher-level phylogenetic classification of Digenea (Platyhelminthes, Trematoda) based on nuclear rDNA sequences before entering the age of the ‘next-generation’ Tree of Life. J. Helminthol. 93, 260–276. doi: 10.1017/S0022149X19000191
Philippe, H., Brinkmann, H., Lavrov, D. V., Littlewood, D. T. J., Manuel, M., Wörheide, G., et al. (2011). Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 9, e1000602. doi: 10.1371/journal.pbio.1000602
Pojmańska, T. (2002). “Family Brachylaimidae Joyeux & Foley 1930,” in Keys to the Trematoda. Eds. Gibson, D. I., Jones, A., Bray, R. A. (London: CABI), 37–43. doi: 10.1079/9780851995472.0037
Ronquist, F., Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Suleman, Khan, M. S., Heneberg, P., Zhou, C. Y., Muhammad, N., Zhu, X. Q., et al. (2019a). Characterization of the complete mitochondrial genome of Uvitellina sp., representative of the family Cyclocoelidae and Phylogenetic implications. Parasitol. Res. 118, 2203–2211. doi: 10.1007/s00436-019-06358-y
Suleman, Ma, J., Khan, M. S., Tkach, V. V., Muhammad, N., Zhang, D., et al. (2019b). Characterization of the complete mitochondrial genome of Plagiorchis maculosus (Digenea, Plagiorchiidae), Representative of a taxonomically complex digenean family. Parasitol. Int. 71, 99–105. doi: 10.1016/j.parint.2019.04.001
Talavera, G., Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Taylor, M. A., Coop, R. L., Wall, R. L. (2016). Veterinary Parasitology (Taylor/Veterinary) || Laboratory Diagnosis of Parasitism. Hoboken, Wiley Blackwell, 259–312. doi: 10.1002/9781119073680.ch4
Valadão, M. C., Silva, B. C. M., López-Hernández, D., Araújo, J. V., Locke, S. A., Pinto, H. A. (2018). A molecular phylogenetic study of the caecal fluke of poultry, Postharmostomum commutatum (= P. gallinum) (Trematoda: Brachylaimidae). Parasitol. Res. 117, 3927–3934. doi: 10.1007/s00436-018-6102-5
Wang, Y., Wang, C. R., Zhao, G. H., Gao, J. F., Li, M. W., Zhu, X. Q. (2011). The complete mitochondrial genome of Orientobilharzia turkestanicum supports its affinity with African Schistosoma spp. Infect. Genet. Evol. 11, 1964–1970. doi: 10.1016/j.meegid.2011.08.030
Webster, B. L., Littlewood, D. T. J. (2012). Mitochondrial gene order change in Schistosoma (Platyhelminthes: Digenea: Schistosomatidae). Int. J. Parasitol. 42, 313–321. doi: 10.1016/j.ijpara.2012.02.001
Wu, Y., Li, L., Zhu, G., Li, W., Zhang, N., Li, S., et al. (2018). Mitochondrial genome data confirm that yaks can serve as the intermediate host of Echinococcus canadensis (G10) on the Tibetan Plateau. Parasit. Vectors 11, 166. doi: 10.1186/s13071-018-2684-0
Yan, H. B., Wang, X. Y., Lou, Z. Z., Li, L., Blair, D., Yin, H., et al. (2013). The mitochondrial genome of Paramphistomum cervi (Digenea), the first representative for the family Paramphistomidae. PLoS One 8, e71300. doi: 10.1371/journal.pone.0071300
Zhang, D., Zou, H., Wu, S. G., Li, M., Jakovlić, I., Zhang, J., et al. (2017). Sequencing of the complete mitochondrial genome of a fish-parasitic flatworm Paratetraonchoides inermis (Platyhelminthes: Monogenea): tRNA gene arrangement reshuffling and implications for phylogeny. Parasit. Vectors 10, 462. doi: 10.1186/s13071-017-2404-1
Zhao, G. H., Mo, X. H., Zou, F. C., Li, J., Weng, Y. B., Lin, R. Q., et al. (2009). Genetic variability among Schistosoma japonicum isolates from different endemic regions in China revealed by sequences of three mitochondrial DNA genes. Vet. Parasitol. 162, 67–74. doi: 10.1016/j.vetpar.2009.02.022
Keywords: Postharmostomum commutatum, mitochondrial genome, mitochondrial DNA, phylogenetic analyses, paraphyly
Citation: Fu Y-T, Jin Y-C and Liu G-H (2019) The Complete Mitochondrial Genome of the Caecal Fluke of Poultry, Postharmostomum commutatum, as the First Representative from the Superfamily Brachylaimoidea. Front. Genet. 10:1037. doi: 10.3389/fgene.2019.01037
Received: 16 July 2019; Accepted: 27 September 2019;
Published: 25 October 2019.
Edited by:
Denis Baurain, University of Liège, BelgiumReviewed by:
Francisco J. Ruiz-Ruano, Uppsala University, SwedenCheng Sun, Chinese Academy of Agricultural Sciences, China
Copyright © 2019 Fu, Jin and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guo-Hua Liu, bGl1Z3VvaHVhNTIwMjAwOEAxNjMuY29t