 José Santiago Ibáñez-Cabellos1,2,3
José Santiago Ibáñez-Cabellos1,2,3 Marta Seco-Cervera1,2,3*
Marta Seco-Cervera1,2,3* Rebeca Osca-Verdegal3
Rebeca Osca-Verdegal3 Federico V. Pallardó1,2,3
Federico V. Pallardó1,2,3 José Luis García-Giménez1,2,3*
José Luis García-Giménez1,2,3*- 1Center for Biomedical Network Research on Rare Diseases (CIBERER), Institute of Health Carlos III, Valencia, Spain
- 2INCLIVA Health Research Institute, Mixed Unit for rare diseases INCLIVA-CIPF, Valencia, Spain
- 3Department of Physiology, Faculty of Medicine and Dentistry, University of Valencia, Valencia, Spain
Autoimmune rheumatic diseases, such as Sjögren syndrome (SS) and rheumatoid arthritis (RA), are characterized by chronic inflammation and autoimmunity, which cause joint tissue damage and destruction by triggering reduced mobility and debilitation in patients with these diseases. Initiation and maintenance of chronic inflammatory stages account for several mechanisms that involve immune cells as key players and the interaction of the immune cells with other tissues. Indeed, the overlapping of certain clinical and serologic manifestations between SS and RA may indicate that numerous immunologic-related mechanisms are involved in the physiopathology of both these diseases. It is widely accepted that epigenetic pathways play an essential role in the development and function of the immune system. Although many published studies have attempted to elucidate the relation between epigenetic modifications (e.g. DNA methylation, histone post-translational modifications, miRNAs) and autoimmune disorders, the contribution of epigenetic regulation to the pathogenesis of SS and RA is at present poorly understood. This review attempts to shed light from a critical point of view on the identification of the most relevant epigenetic mechanisms related to RA and SS by explaining intricate regulatory processes and phenotypic features of both autoimmune diseases. Moreover, we point out some epigenetic markers which can be used to monitor the inflammation status and the dysregulated immunity in SS and RA. Finally, we discuss the inconvenience of using epigenetic data obtained from bulk immune cell populations instead specific immune cell subpopulations.
More than 200 different disorders are considered autoimmune rheumatic diseases, but immune system complexity and the diversity of shared features make diagnosis and prognosis arduous tasks, especially in early follow-up stages (Alarcón et al., 1991). Studies performed using monozygotic twins have revealed an important epigenetic role in the progression of rheumatic diseases in addition to genetic factors (MacGregor et al., 2000;Javierre et al., 2010). Autoimmune rheumatic diseases, such as Sjögren syndrome (SS) and rheumatoid arthritis (RA), are characterized by pain and chronic joint inflammation. In fact, given the overlap of certain clinical features between SS and RA, it is assumed that the genetic contribution to SS and RA may be due to a multigenic contribution, which may affect many immunological-related mechanisms (Table 1). In line with this, Yang et al., suggested that patients with SS should be explored for a second autoimmune associated disease or more, such as RA (Yang et al., 2018), because all autoimmune diseases share a common phenotype and mechanisms. Therefore, genetic and epigenetic factors might influence poly-autoimmunity development (Anaya et al., 2016).

Table 1 Principal and common characteristics of RA and SS.
He et al. described RA as a systemic autoimmune disease that causes extra-articular complications and damage (He et al., 2013a). SS is defined as a chronic autoimmune disease caused by the diminished function of salivary and lacrimal glands due to the lymphocytic infiltration of exocrine glands (Ramos-Casals et al., 2007;He et al., 2013a). SS can be classified as primary or secondary, depending if it coexists with other autoimmune diseases or not, such as RA (Ramos-Casals et al., 2007). Both SS and RA present female predominance and produce chronic inflammation of joints which, in turn, causes pain and impedes normal mobility in affected patients. It is well-known that a person who suffers both pathologies, e.g. secondary SS and RA, has a worse prognosis and faces more comorbidities and increased mortality (He et al., 2013a).
Of the common features found in both RA and SS, we underscore chronic inflammation, the interaction of the immune system and other tissues, such as skeletal tissue in RA and exocrine glands in SS, and autoimmunity that produce tissue damage and destruction which, in turn, lead to reduced mobility. Initiation and maintenance of the chronic inflammatory stages accounts for several mechanisms involving immune cells as key players. Pro-inflammatory cytokines, such as interleukin-1 (IL-1), IL-6, IL-8, and tumor necrosis factor α (TNF-α), can be produced by circulating monocytes that differentiate to macrophages or dendritic cells (Arango Duque and Descoteaux, 2014). Monocytes produce reactive oxygen species (ROS) and cyclooxygenase-2 (COX2) as mediators of inflammation (Lu and Wahl, 2005), and attract T- and B-cell chemokines which, in turn, produce pro-inflammatory cytokines. Activated B-cells are able to present autoantigens and generate autoantibodies that preserve inflammation and can, consequently, lead to tissue destruction in these autoimmune rheumatic diseases. In fact, a hallmark of autoimmune rheumatic diseases is the presence of autoantibodies at non organ-specific antigens, specifically antigens occurring in nucleated cells or among circulating plasma proteins (Aggarwal, 2014). Diversity in environmental stimuli requires a quick dynamic response of immune cells. In this fast efficient functional response, epigenetic mechanisms play an essential role in, for example, hematopoietic cell differentiation. Relevant differences in epigenetic regulation between innate and adaptive immune cells have been described. Most are implicated in the hematopoietic process, although other epigenetic alterations are associated with myeloid and lymphoid lineage function (Avgustinova and Benitah, 2016). Inflammation as a transitory physiological process protects from pathogenic invasions, while inflammation malfunction can produce tissue damage and organ dysfunction, and can mediate several pathological processes. Genetic and epigenetic variability in physiological and pathological processes in immune cells is a complex and carefully regulated equilibrium that is difficult to decipher.
This review shows the epigenetic mechanisms related to RA and SS so as to identify the unclear association between SS and RA by elucidating the intricate mechanisms underlying the phenotypic features of both autoimmune diseases.
Epigenetic Regulation of Genetic Programs in Immune Cells
Genome-wide associated studies (GWAS) have identified hundreds of risk loci for autoimmunity (Parkes et al., 2013). Integrated genetic and epigenetic fine mapping may help to identify the causal variants and epigenetic drivers that lead to autoimmune disease-associated loci and, in turn, contribute to explore their role in the control of the immune response (Farh et al., 2015).
Mammals' blood system have highly specialized cell types that derive from hematopoietic stem cells (HSCs). HSCs play the important role of maintaining the homeostasis of specialized cell types' production and self-renewal (Wilson et al., 2008, Wilson et al., 2009). Epigenetic studies have demonstrated that during the differentiation of HSCs, DNA methylation, microARNs (miRNAs), and histone modifications play key roles in expression programs by shifting from pluripotency and proliferation to mature differentiated immune cells (Avgustinova and Benitah, 2016; Mehta and Baltimore, 2016). Some of these mechanisms can fail due to erroneous epigenetic program, which may alter the immune responses in SS and RA. The next part of this review describes some of epigenetic drivers that lead to autoimmunity and altered immune response.
DNA Methylation
DNA methyltransferases and the ten-eleven translocation (TET) families of enzymes regulate DNA methylation and demethylation during hematopoietic differentiation. For this reason, whole genome bisulfite sequencing (WGBS) has been performed to clarify DNA methylation changes in immune cell development (Morales-Nebreda et al., 2019). For example, Cabezas-Wallscheid et al. compared gene expression profiles and differentially methylated regions (DMRs) between HSCs and four multipotent progenitor populations (MPP) in mice. During an early transition of HSC to MPP, they found an association between DMR and inversely correlated gene expression during differentiation. In the most inversely correlated genes, they observed genes related to hematopoietic differentiation and HSCs function, such as Hoxb2, Rorc, and Cd34 (Cabezas-Wallscheid et al., 2014). Differences in the methylation profile have been described as being lineage-specific in innate and adaptive immune systems. HSCs differentiate to multipotent progenitors of lymphoid (CLP) and myeloid (CMP) lineages, which present different DNA methylation patterns. In particular, CLP show increased DNA methylation in a diversity of transcription factors that regulates myeloid differentiation (e.g. Gata2, Tal1, and Lmo2) (Bock et al., 2012).
Both B- and T-cell activation, and proliferation, differentiation, and plasticity, are regulated and mediated by not only epigenetic mechanisms, but also by transcription factors and signaling molecules. The combinational activity of these mechanisms produces a characteristic transcriptome to each subset of differentiated B- and T-cells. The transition from naive to effector B-cells and T-cells exhibits different DNA methylation patterns. In fact, a DNA methylation analysis of B-cells performed in different development stages has shown a hypomethylation tendency after activation, which is reverted in memory B-cells. In addition, DNA methyltransferases (i.e. DNMTs; DNMT1, DNM3A, and DNMT3A) have also shown a shift in their expression levels between naive B-cells and late-stage differentiated B-cells (Lai et al., 2013). Activation of the axis JAK3-STAT6 pathway by IL-4 produces demethylation, which is TET2-dependent in monocytes and necessary for acquiring proper dendritic cell and macrophage identity (Vento-Tormo et al., 2016).
The DNA methylation regulation of T-lymphocyte differentiation has been widely described (Kanno et al., 2012; Morales-Nebreda et al., 2019) and is briefly described herein. The development of CD4+ helper T-cells and CD8+ cytotoxic T-cells from CLP is a complex regulated process in which methylation plays an important role. For example, Sellars et al. found that double negative (CD4−CD8−), double positive (CD4+CD8+) timocytes, and mature CD8+ cytotoxic T-cells present hypermethylation in a DMR near the Cd4 gene, while CD4+ helper T-cells showed hypomethylation. These authors also revealed that DNMT1 is responsible for the Cd4 gene silencing in CD8+ cytotoxic T-cells (Sellars et al., 2015). Activated CD4+ helper T-cells are guided to proliferate and differentiate to diverse T-helper cells to perform distinct functions. Many conditional deletion models of different DNMTs have pointed out the importance of methylation in regulating cytokine expression and the stabilization of subset IFN-γ-producing Th1 and IL-4-producing Th1 cell phenotypes (Lee et al., 2001;Gamper et al., 2009). Furthermore, the DNA methylation of many cys-regulatory elements involved in lfng expression in mice becomes demethylated in the IFN-γ-producing subset of Th1 cells, although these regions remain methylated in the subset IL-4-producing Th2 cells (Schoenborn et al., 2007). Another example of the importance of DNA methylation in the regulation of differentiation to various subsets of T-helper cells is regulatory T cells (Treg), which are characterized by Foxp3 expression (Hori et al., 2003). Lal et al. have described demethylated levels of a specific upstream Foxp3 CpG site in the Foxp3 enhancer of naive Treg compared to CD4+ helper T-cells and TGFβ-induced peripheral Treg (Lal et al., 2009). Finally, a recent WGBS study has identified up to 339 DMRs in Treg cells that map to the sites encoding for Ctla4, a gene that transmits an inhibitory signal to T-cells (Delacher et al., 2017).
miRNA Regulation
miRNAs are ≈22 nucleotides noncoding RNAs that post-transcriptionally regulate the gene expression of their mRNA targets by binding to their 3'UTR. In the last decade, increasing knowledge about the role of the miRNAs that control specific transcriptomic programs has demonstrated the relevance of these epigenetic changes during the differentiation, development, and maturation of immune cells (Bissels et al., 2012; Mehta and Baltimore, 2016). For example, the differential expression of miR-155, let-7 and miR-21 may be implicated in altered long-term HSCs proliferation (Gruber et al., 2009). The differential expression of cluster miR-99b-let-7e-miR-125a enhances long-term HSCs self-renewal by conferring protection from apoptosis thanks to BCL-2 homologous antagonist/killer 1 (BAK1) being targeted by miR-125a (Guo et al., 2010). The expression of cluster miR-99a-let-7c-miR-125b promotes HSC differentiation by targeting TGF-ß signaling activators and WNT signaling repressors (Emmrich et al., 2014). In addition, TLR stimulation induces nuclear factor κß (NF-κß) that stimulates inflammamiR miR-146a which, in turn, is a TLR and cytokine signaling negative regulator by targeting TNF receptor-associated factor 6 (TRAF6) and interleukin receptor-associated kinase 1/2 (IRAK1/2) (Taganov et al., 2006). It is noteworthy that the polymorphisms in IRAK1/2 have been considered risk factors for autoimmune diseases, such as RA (Atabaki et al., 2017) and SS. Furthermore, the downregulation of miR-146 brings about the increased secretion of cytokines from hematopoietic progenitors (Zhao et al., 2014), and a polymorphism in the 3'-UTR of IRAK1 in the recognition region of miR-146a, which has been shown to be a factor of susceptibility of RA (Chatzikyriakidou et al., 2010).
The regulation of diverse innate immune cells in several differentiation stages has been reported (Kumar Kingsley and Vishnu Bhat, 2017). Macrophage development and immunity regulation as act as principal players miR-146a and miR-155 (Taganov et al., 2006; O'Connell et al., 2007;Ghani et al., 2011). The development of natural killer cells (NK) to natural killer T-cells (NKT) is controlled by miR-150 (Bezman et al., 2011). Granulocytic differentiation is regulated by the levels of miR-223 whose promoter is, in turn, regulated by either nuclear factor 1 A-type (NFI-A) or CCAAT/enhancer-binding protein-α (C/EBPα) binding (Fazi et al., 2005).
Lots of reviews have described the important role of miRNAs in adaptive immunity (Dooley et al., 2013; Marques et al., 2015; Mehta and Baltimore, 2016; Zhang et al., 2018). miRNAs play an essential role in B-cell development and function, and one example of such is miR-181a-5p (Georgantas et al., 2007; Jensen et al., 2013). Other examples include "inflamma-miRs" miR-155, miR-146a, and miR-150, which are key regulators of the B-cell function. The germinal center response involves miR-155 as an essential player for B-cell maturation and proliferation (Vigorito et al., 2013) and miR-146 deletion produces autoantibodies from B-cells (Boldin et al., 2011). Low expression miR-150 levels in pro-B cells inhibits the progression of pre-B cells differentiation in bone marrow, probably by the repression of its target gene, the MYB transcription factor (Xiao et al., 2007).
As in B-cell development and function, diverse and cell‐specific expression patterns of miRNAs have been described in T-cell subsets (Monticelli et al., 2005; Wu et al., 2007; Sethi et al., 2013). Some relevant miRNAs found in lymphoid cell lines are miR-150, miR-155, miR-146a, and cluster miR-17-92. miR-150 controls early T-cell development inhibition via the downregulation of Notch (Ghisi et al., 2011). miR-155 inhibits Th2 cell proliferation by targeting c-Maf, which is a transactivator of IL-4 (Rodriguez et al., 2007). At the same time, miR-155 is able to lead the differentiation of the Th1 response (Banerjee et al., 2010) and plays a special role regulating the differentiation of Treg and T-helper 17 cells by targeting SOCS1 and controlling JAK/STAT signaling (Yao et al., 2012). Guerau-de-Arellano et al. have described how miR-128 and miR-340 are both implicated in the inhibition of Th2 cell lineage by directing differentiation to Th1 cells (Guerau-de-Arellano et al., 2011). miR-126 has been correlated with the promotion of Th2 cells (Mattes et al., 2009) and there are reports that miR-17-92 cluster deficiency reduces T-bet and IFN-γ expression, which are characteristic of Th1 lineage, and promotes Treg cell differentiation (Jiang et al., 2011). Here it is stressed that T-bet plays key role in autoimmunity development. Finally, another important miRNA in different subsets of T-cells is miR-146a, whose increased expression has been observed in effector Th1 (Monticelli et al., 2005) and Treg (Cobb et al., 2006), but not in Th2 cells and naive CD4+ T cells (Monticelli et al., 2005). Furthermore, as in myeloid cells, miR-146a regulates NF-kß signaling (Taganov et al., 2006) and targets STAT1 to thus control the Treg function (Lu et al., 2010).
Histone Modification
The conformational chromatin changes produced by histone post-translational modifications (PTMs) (e.g., acetylation, methylation, phosphorylation, etc.) could result in easy or blocked DNA accessibility. These reversible modifications have different consequences depending on the affected histone protein or specific residue, which is known as the histone code. Generally speaking, acetylation of histone lysine residues (H3K9, H3K14, H4K5, and H4K16) and methylation (H2BK5, H3K4, H3K36, and H3K79); phosphorylation of histone H3 threonine 3 (H3T3) and serine (H3S10 and H3S28), as well as of histone 4 serine 1 (H4S1) and H2BK120 ubiquitinylation, are marks of open chromatin that increase DNA accessibility to transcription factors and produce increased gene expression. On the contrary, other marks, like the methylation (specifically di-methylation and tri-methylation) of H3K9, H3K27, and H4K20, the ubiquitination of H2AK119, and the sumoylation of H2AK126, H2BK6, and H2BK7, are associated with compacted chromatin and gene repression. In contrast to DNA methylation and miRNA regulation, fewer studies have been conducted into histone modifications in immune cells because of their relative complexity and the difficulty to transfer them to clinical practice. However, alterations of different histone PTMs, in polycomb repressive complexes, and also in histone modifying enzymes, have been described in immune system development and immune disorders (Loizou et al., 2009; Wilting et al., 2010;He et al., 2013b; Vidal and Starowicz, 2017; Di Carlo et al., 2018). Chen and coworkers described how the histone H3 lysine 79 di/trimethylated (H3K79me2/3) modifications by the disruptor of telomeric silencing-1-like (Dot1l) methyltransferase at the Il6 and ifnb1 promoters are abundant in active macrophages (Chen et al., 2018). Kdm6a demethylase promotes Il6 and ifnb1 overexpression in primary macrophages by lowering the H3K27me2/3 levels at their promoters (Li et al., 2017b). B-cell activation increases the levels of H3K4me3, H3K9ac, and H3K14ac in the promoter region of activation-induced cytidine deaminase enzyme (AID) by inducing the DNA cleavage of the regions in the immunoglobulin heavy chain (IgH) locus during class-switch recombination (CSR) (Chowdhury et al., 2008). H3K4me3, H3K9ac, H3K14ac, and H4K8ac are apparently essential for somatic hypermutation in AID target loci (Begum et al., 2012). Increased marks of H3K4me3 have been found in the Ifng promoter in Th1 cells and in the Il4 promoter in Th2 cells, which are characteristic of each lineage, while consistently increased repressive marks of H3K27me3 have been found in both the Ifng promoter in T-helper Th2 cells and the Il4 promoter in T-helper Th1 cells (Wei et al., 2009). In CD8+ cytotoxic T-cells, the Ifng promoter displayed decreased H3K27me3 levels compared to naive CD8+ T-cells, but H3K4me3 and H3K9ac increased, which are active epigenetic marks (Denton et al., 2011).
In a study performed in 21 autoimmune diseases, genetic and epigenetic data were combined to obtain a more comprehensive view of the etiology of these autoimmune diseases. By using chromatin immunoprecipitation (ChIP) data for H3K27ac, a mark of active enhancers and promoters, the authors elucidated the role of the SNPs detected in the genetic analysis by combining the results with those obtained in the ChIP study, and were able to link disease-associated SNPs with active enhancers (Farh et al., 2015). This example illustrates the utility of histone modification information for clinical research, specifically to know how the study of epigenetic regulation in rare autoimmune-related disorders can help us to completely elucidate the intricate and complex landscape of epigenetic control in the immune system. Thus, we herein describe how epigenetic alterations are linked to SS and RA in an attempt to identify special the relevant epigenetic mechanisms underlying the physiopathology of these autoimmune diseases (Table 2).
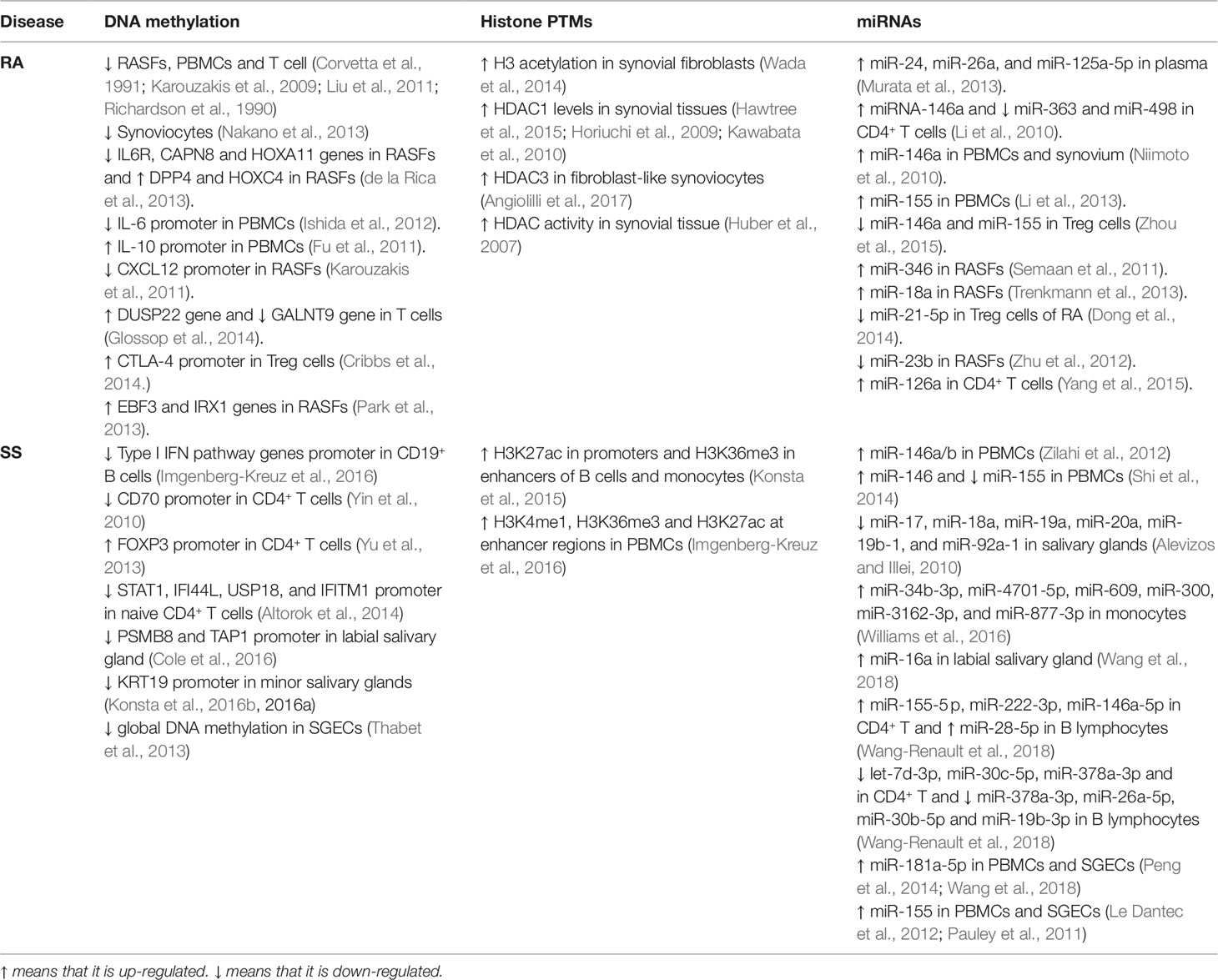
Table 2 Epigenetic Changes in RA and SS.
Sjögren Syndrome
Sjögren syndrome (SS; Disease identification number: OMIM:270150 and ORPHA:289390) is a chronic autoimmune disorder considered a rare disease, characterized by salivary and lacrimal glands being affected, which lead to dry eyes and dry mouth, expose the oral cavity to infections, caries, candidiasis, and imply musculoskeletal pain. SS may occur alone as primary SS or as secondary SS in association with other autoimmune diseases like systemic lupus erythematosus (SLE) or RA. Primary SS prognosis is not favorable and is linked to the onset of respiratory or kidney failure. However, secondary SS is characterized by keratoconjunctivitis and xerostomia, but is always associated with other autoimmune disorders. Therefore, prognosis depends strictly on the primary autoimmune pathology (He et al., 2013a). The problem lies in the presence of the lymphocytic infiltration of exocrine glands and other organs, together with the presence of various autoantibodies [reviewed in (Kapsogeorgou and Tzioufas, 2016); Table 1]. Patients with SS are at higher risk of developing lymphoma and primary liver cirrhosis and can, in some cases, develop non-Hodgkin's lymphoma (Pagano et al., 2013; Ciechomska and O'Reilly, 2016; Vivino, 2017). The exact cause of SS is unclear, but several lines of evidence suggest a wide array of mechanisms including the dysregulation of the immune system which can destroy exocrine glands, as consequence of the production of chemokines by epithelial cells which attract T cells and dendritic cells producing an unappropriated immune response in the acinar and ductal epithelial cells (Nordmark et al., 2006). Moreover, genetic susceptibility and the contribution of epigenetic factors in the SS pathophysiology have been also proposed. Genes IRF5 (interferon regulatory factor 5), CXCR5 (C-X-C chemokine receptor type 5), TNIP1 (TNFAIP3-interacting protein 1), IL12A (interleukin-12 subunit alpha), BLK (B lymphoid tyrosine kinase), and STAT4 (signal transducer and activator of transcription 4) have been proposed in several studies as gene candidates for SS susceptibility (Lessard et al., 2013;Burbelo et al., 2014). Genome-wide and epigenome-wide association studies have been performed to identify the genes and epigenetic marks involved in SS (Low and Witte, 2011; Konsta et al., 2014).
The DNA methylation of immune cells is a key regulatory mechanism that underlies SS. Less global DNA methylation in salivary gland epithelial cells (SGECs) has been observed and linked to decreased DNMT1 and increased GADD45α (Thabet et al., 2013). The DNA methylation levels of SGECs have been inversely correlated with SS severity and B-lymphocyte infiltration. The global DNA methylation levels have been reported to increase in the SGECs of SS patients after administering the anti-CD20 monoclonal antibody rituximab (Devauchelle-Pensec et al., 2007; Thabet et al., 2013). The hypomethylation of the CD70 promoter in CD4+ T-cells brings about CD70 gene expression (Yin et al., 2010). CD70 is a B-cell costimulatory molecule that interacts with CD27 during B- and T-cell contact by promoting plasma cell differentiation and IgG production and, therefore, contributes to the immunological disarrangement and autoimmune response in SS. Otherwise, the hypermethylation of the FOXP3 promoter leads to lower FOXP3 expression, mRNA, and protein levels in the CD4+ T-cells of SS (Yu et al., 2013). Low DNA methylation levels in minor salivary glands generate an increased KRT19 gene expression by producing elevated levels of epithelial protein cytokeratin-19 (CK-19), while hypermethylation has been associated with low protein CK-19 levels. A therapy conducted with DNMT inhibitor 5-azacytidine has increased KRT19 gene expression and CK-19 protein levels (Konsta et al., 2016a), which demonstrates that DNA methylation is a key player in the control of the expression of both genes. A recent genome-wide DNA methylation study conducted in CD19+ B-cells and minor salivary glands has identified prominent hypomethylation sites of interferon (INF)-regulated genes, such as MX1, IFI44L, and PARP9. The hypomethylation of ING-regulated genes in SS B-cells brought about an increase in their gene expression, although an increase in OAS2 gene expression took place in minor salivary glands (Imgenberg-Kreuz et al., 2016). The hypomethylation of some other ING-regulated genes, such as STAT1, IFI44L, USP18, and IFITM1, has been reported (Altorok et al., 2014). The hypomethylation of these genes is congruent with the IFN response activation observed in SS patients. Altorok et al. have also reported that transcription factor gene RUNX1 is hypermethylated in patients with SS and regulates the maturation of hematopoietic stem cells (Okuda et al., 2001; Altorok et al., 2014) (Figure 1). RUNX1 has been associated with cancer predisposition (Asou, 2003; Kundu et al., 2005; Schlegelberger and Heller, 2017), which suggests a possible connection to lymphoma predisposition in SS patients. A recent study by Cole et al. has demonstrated the hypomethylation of PSMB8 and TAP1, which increased the frequency of antigen-presenting cells in labial salivary gland tissues of SS patients (Cole et al., 2016). Moreover, the hypomethylation of SSB gene promoter 1 has been observed in the SGECs of SS patients with SSB/La autoantibodies compared to those from anti-SSB/La-negative patients (Konsta et al., 2016b).
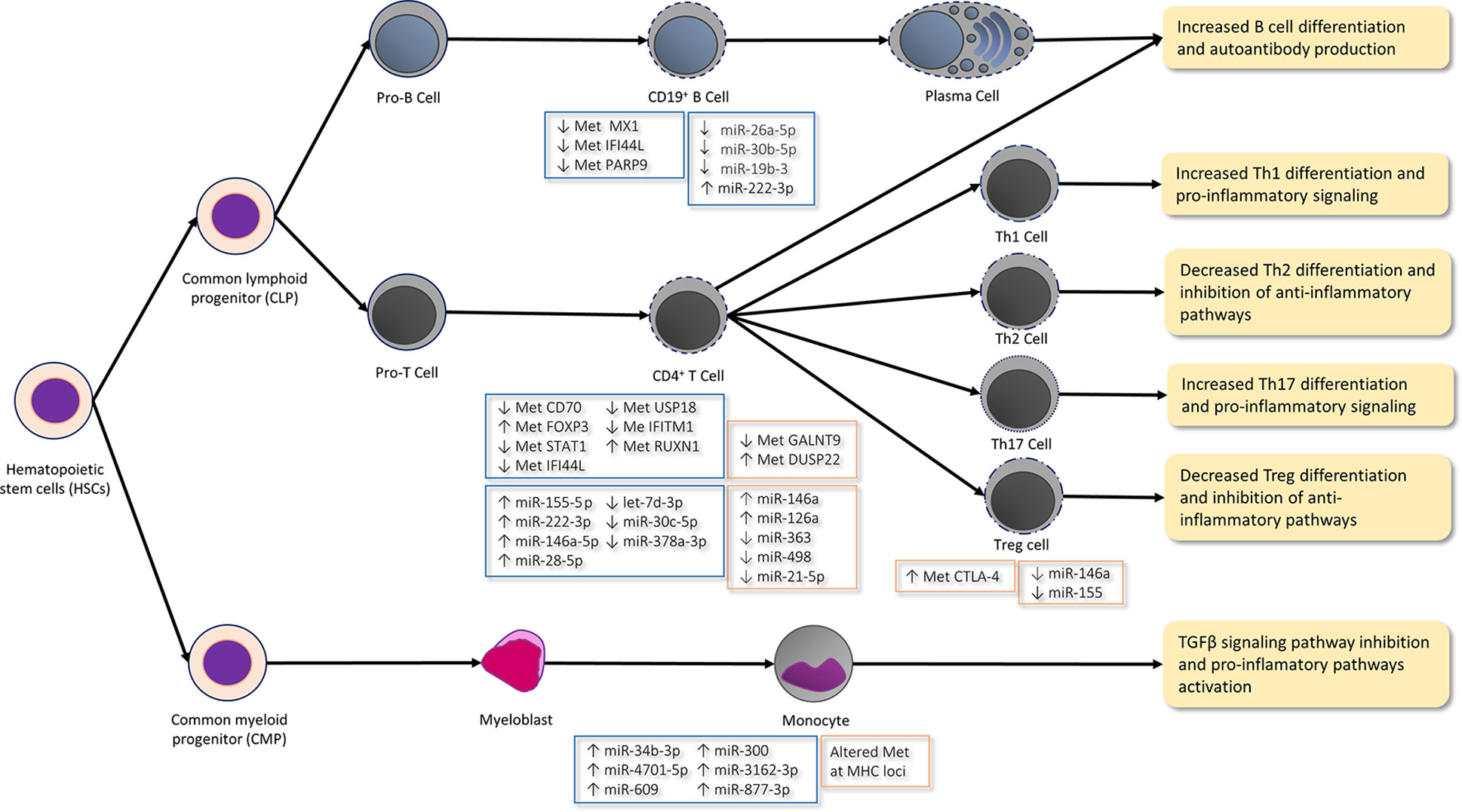
Figure 1 Epigenetic mechanisms in immune cells differentiation and function in arthritis rheumatoid (AR; orange boxes) and Sjögren syndrome (SS; blue boxes). In the CD19+ B-cells of SS patients, type I IFN genes, such as genes MX1, IFI44L, and PARP9, are hypomethylated (↓ Met), which agrees with the hypomethylation of some other IFN-γ-regulated genes (e.g. STAT1, IFI44L, USP18, and IFITM1) in CD4+ T-cells and with the IFN response activation observed in these patients. Moreover, low levels (↓) of miR-26a-5p, miR-30b-5p, and miR-19b-3p and high miR-222-3p levels (↑) are reported in the CD19+ B-cells of SS patients. Low levels miR-30b-5p bring about an increase of the B-cell activating factor (BAFF) and autoantibody production. The hypomethylation of the CD70 promoter in the CD4+ T-cells of SS patients leads to the increased expression of the CD70 gene, which interacts with CD27 during B- and T-cell contact by promoting plasma cell differentiation and IgG production. The hypermethylation (↑ Met) of the RUNX1 gene in SS regulates the maturation of hematopoietic stem cells. The high expression of inflamma-miRs miR-155-5 p, miR-222-3p, and miR-146a-5p and the low expression of let-7d-3p, miR-30c-5p, and miR-378a-3p are described in CD4+ T-cells. ImmflamamiRs miR-155 and mir-146a inhibit Th2 proliferation and promote the Th1 response. Furthermore, miR-155 also promotes the differentiation and function of Th17 and Treg. The hypermethylation of the FOXP3 promoter lowers the FOXP3 expression in the CD4+ T-cells of SS and is characteristic of the differentiated and functional Treg. The hypermethylation of the DUSP22 gene and the hypomethylation of the GALNT9 gene in CD4+ T-cells are implicated in the IL-6/STAT3-mediated signaling pathway by contributing to autoimmunity and promoting the pro-inflammatory Th17 cells differentiation in RA patients. High inflamma-miR miRNA-146a levels and low miR-363, miR-21-5, and miR-498 levels are reported in the CD4+ T-cells of RA patients. Low miR-21-5p levels promote Th17 cell differentiation, while suppressing Treg development. Furthermore, high miR-126a levels inhibit DNM1I which, in turn, produces the hypomethylation of CD11a and CD70 by increasing their expression and promoting the autoimmune response. The methylation of the CTLA4 promoter inhibits Treg activity in RA. In contrast to the miRNA levels observed in the CD4+ T-cells of RA patients, miR-146a and miR-155 are lower in Treg cells after T-cell stimulation. miR-146a regulates NF-kß signaling and targets STAT1 by thus controlling the Treg function. Hence, the low levels of this miRNA bring about the inhibition of Treg differentiation and function. The differential methylated sites located in the MHC region are described in the CD14+ monocytes of RA patients. Moreover, the levels of miR-34b-3p, miR-4701-5p, miR-609, miR-300, miR-3162-3p, and miR-877-3p are higher in the monocytes of SS patients, which may inhibit the TGFβ signaling pathway. Thus, an unbalanced differentiation takes place from the CD4+ T-cells to the Th1 and Th17 cells, which promotes pro-inflammatory pathways and increases autoantibody production in both diseases.
miRNAs are dysregulated in SS. A reduction in cluster miR-17-92 (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a-1) has been observed in the salivary glands of patients with SS (Alevizos and Illei, 2010). This cluster is involved in cell cycle, apoptosis, immune response, and other relevant cellular processes (Mogilyansky and Rigoutsos, 2013). As previously described, this cluster is relevant in lymphoid cell lines as it activates the characteristic expression programs of the Th1 lineage (i.e. T-bet and IFN-γ) and promotes Treg cell differentiation. Another study has observed increased levels of miR-146a, which regulates the innate immune and inflammatory response by producing the repression of IRAK1 and increased TRAF6 gene expression which, in turn, promote the NF-κB target genes expression in the peripheral mononuclear cells of SS patients (Zilahi et al., 2012). Conversely, Williams et al. have reported that miR-34b-3p, miR-4701-5p, miR-609, miR-300, miR-3162-3p, and miR-877-3p increase in SS monocytes (Figure 1). This miRNA profile revealed the broadest coverage of the predicted targets in the canonical TGFβ signaling pathway. Thus, their high levels may suppress this signaling pathway and direct the balance to the pro-inflammatory interleukin-12 and Toll-like receptor/NF-κB pathways (Williams et al., 2016). A recent study has detected the high expression of inflamma-miRs miR-155-5 p, miR-222-3p, miR-146a-5p, and miR-28-5p in CD4+ T-cells and miR-222-3p CD19+ B-cells, and the low expression of let-7d-3p, miR-30c-5p, and miR-378a-3p in CD4+ T-cells, and of miR-26a-5p, miR-30b-5p, and miR-19b-3p in CD19+ B-cells (Wang-Renault et al., 2018). As mentioned above, inflamma-miRs miR-155 and mir-146a inhibit Th2 proliferation and promote the Th1 response (Figure 1). Furthermore, miR-155 also promotes the differentiation and function of Th17 and Treg differentiation (Monticelli et al., 2005;Cobb et al., 2006; Taganov et al., 2006; Rodriguez et al., 2007; Banerjee et al., 2010; Yao et al., 2012). This result agrees with other studies, which have observed how miR-155 increases in the peripheral blood mononuclear cells (PBMCs) and SGECs of SS patients (Pauley et al., 2011; Le Dantec et al., 2012). Conversely, Shi et al. have noted high miR-146a levels and low miR-155 levels in the PBMCs of SS patients (Shi et al., 2014). It is worth mentioning that, as described in the Introduction, miR-146a and miR-155 are important regulators of B- and T-cell differentiation, proliferation and function, and both have been considered two key modulators in the control of the innate and adaptive immune response (Gottfried et al., 2012). Furthermore, low miR-30b-5p levels bring about an increase in the gene expression of the B-cell activating factor (BAFF) in the CD19+ cells of SS patients (Wang-Renault et al., 2018). High BAFF levels have been described in the serum, salivary gland B-cells, T-cells, and epithelial cells of these patients (Lavie et al., 2004;Daridon et al., 2007). In addition, these increased levels correlate with autoantibody production and, accordingly, mice models with high BAFF levels show phenotypic SS features (Mackay et al., 1999; Mariette, 2003).
Remarkably, increased levels of miR-181 and miR-16a in the labial salivary glands and PBMCs of SS patients are associated with the degree of inflammation in SS (Wang et al., 2018), which suggests that both miRNAs are biomarker candidates for monitoring the inflammatory course of SS. miR-181a-5p is associated with antigen sensitivity and the dysfunction of exocrine glands. Peng et al. have observed increased miR-181a-5p levels in the PBMCs of SS (Peng et al., 2014). This miRNA has been described to target the muscarinic receptor 3 gene (CHRM3)(Whisnant et al., 2013), whose variants have been associated with SS (Appel et al., 2011). Accordingly, autoantibodies against CHRM3 have been detected in the serum of SS patients (Gao et al., 2004). Other targets of miR-181a-5p are genes TRIM21 and SSB, which encode for the 52kDa subunit of Ro/SSA ribonucleoprotein and the La/SSB ribonucleoprotein, respectively (Yang et al., 2016). SSA/Ro and SSB/La are regulated by miRNA let-7b, which is repressed in the SGECs of SS (Kapsogeorgou et al., 2011). Autoantibodies against these proteins have been detected in the serum, saliva, salivary gland epithelia, and B-cell infiltrating salivary glands of SS patients (Horsfall et al., 1989; Salomonsson and Wahren-Herlenius, 2003;Routsias and Tzioufas, 2007).
Finally, some authors have studied histone modifications at relevant genomic loci linked to SS risk variants to identify the critical role played by these variants in the disease. Active histone post-translational marks (e.g. H3K4me2, H3K4me3, and H3K9Ac) at the promoters of selected risk variants in B-cells have been observed in contrast to epithelial cells and monocytes. Active marker H3K27Ac and active marker H3K36me3 are enriched in B-cells and monocytes, in contrast to epithelial cells (Konsta et al., 2015). A recent study has reported an increase in the enhancer regions (H3K4me1 and H3K27ac) associated with the CpG sites hypomethylated in the whole blood of SS patients, while hypermethylated sites are underrepresented in these regions and also enriched for active post-translational mark H3K36me3 (Imgenberg-Kreuz et al., 2016). These results reveal that histones PTMs are also key components in the regulation of specific variants, and they also play a critical role in the genetic background and the risk of developing SS. Therefore, further studies should be undertaken to ascertain the role of histone PTMs in the regulation of the genetic risk factors linked to SS.
Rheumatoid Arthritis
RA (Disease Identification Number: OMIM: 180300) is a systemic autoimmune inflammatory disease characterized by chronic joint inflammation and structural damage, with extra-articular manifestation, such as rheumatoid nodules, pulmonary involvement or vasculitis, and systemic comorbidities (Smolen et al., 2016). RA presents a large number of associated autoantibodies [reviewed in (Steiner, 2007) and (Conrad et al., 2010); Table 1]. The autoantibodies produced by the action of B- and T-cells in the synovial membrane promote the destruction of joints, suggesting a particular osteoimmunological interaction in RA (Okamoto and Takayanagi, 2011). Macrophages and granulocytes generate pro-inflammatory cytokines, chemokines, and ROS in synovial fluid by increasing the inflammatory response (Kinne et al., 2007). The origin of RA is unclear, but is considered to be a combination of genetic predisposition and epigenetic factors. Some of the genetic factors of RA are associated with the alleles of HLA-DR4, DR14, and some DR1 beta chains (Smolen et al., 2016), which are polymorphisms found in the gene of protein tyrosine phosphatase and nonreceptor type 22 (PTPN22) that regulate the activity of T- and B-lymphocytes, as well as the polymorphisms of the genes that regulate cytokine production (TNF and STAT4) (Conigliaro et al., 2017).
One of the most important epigenetic factors in RA is DNA methylation, which affects immune-related genes and, therefore, the immune response. Glossop et al. have observed differential DNA methylation in T- and B-cells, and also in synovial fibroblasts in early RA stages (Klein and Gay, 2015;Glossop et al., 2016). Global DNA hypomethylation has been found in rheumatic arthritis synovial fibroblasts (RASFs), PBMCs, and more specifically in the T-cells obtained from RA patients (Richardson et al., 1990;Corvetta et al., 1991; Karouzakis et al., 2009; Liu et al., 2011). The DNA methylation studies performed by Liu et al., by means of Infinium 450K methylation array (Illumina) from sorted CD14+ monocytes of RA patients and controls, found nine differential methylated sites located in the MHC region, which suggests that monocytes are more proximal to the pathogenic cell type (Liu et al., 2013). Here it is necessary to stress that the MHC class II gene expression in the macrophages that came from monocytes has been linked to RA progression (Mueller et al., 2007). Other authors have observed the hypomethylation of the pro-inflammatory cytokine IL6 promoter and the hypermethylation of the anti-inflammatory IL10 gene promoter in the PBMCs of RA patients (Fu et al., 2011; Ishida et al., 2012). Interestingly, reduced IL-10 production should contribute to the balance between CD4+ Th2 cells being lost to favor CD4+ Th1 cell function (Fiorentino et al., 1989; Said et al., 2010) (Figure 1). In line with this, IL-10 generally suppresses the proliferation and cytokine production of all T-cells, as well as the activity of macrophages. Despite the results of Fu et al. (Fu et al., 2011) showing the hypermethylation of the anti-inflammatory IL10 gene in the PBMCs of RA patients, the results of Hernández-Bello et al. found an aberrant overexpression of the IL10 gene in these patients (Hernández-Bello et al., 2017).
An alteration to the methylation of other genes has also been studied. Glossop et al. found the hypermethylation of the DUSP22 gene and the hypomethylation of the GALNT9 gene in the T-cells of RA (Glossop et al., 2014). GALNT9 codifies for an important glycosyltransferase, which is essential in the first step mucin biosynthesis. Mucin is involved in RA (Ishino et al., 2010) and induces IL-6 expression in the PMBC of RA patients (Hamaguchi et al., 2011). DUSP22 codifies for a tyrosine phosphatase that negatively regulates the IL-6/STAT3-mediated signaling pathway. It is noteworthy that IL-6 is a pivotal cytokine in RA. It contributes to autoimmunity (Fonseca et al., 2009) and promotes pro-inflammatory Th17 cells differentiation (Volpe et al., 2008; Yang et al., 2008). The methylation of the CTLA4 promoter inhibits the expression and activation of the indoleamine 2,3-dioxygenase pathway (IDO pathway) in the Treg cells of RA (Cribbs et al., 2014). This pathway is an important regulatory checkpoint that influences Treg activity by stabilizing and augmenting the immunosuppressive phenotype, and by preventing Treg reprogramming into nonsuppressive T helper-like cells (Figure 1). A genome-wide study has shown the hypomethylation of genes IL6R, CAPN8, and HOXA11, and the hypermethylation of DPP4 and HOXC4 in RASFs (de la Rica et al., 2013). The hypomethylation of the chemokine CXCL12 promoter leads to an increase in matrix metalloproteinases which, in turn, cause the destruction of joints in RASFs (Karouzakis et al., 2011). The hypermethylation of genes EBF3 and IRX1 in RASFs has also been observed (Park et al., 2013). Nakano et al. presented a series of hypomethylated genes which clustered in the key pathways related to immune mechanisms, such as cell migration, focal adhesion, cell adhesion, trans-endothelial migration, and extracellular matrix interactions (Nakano et al., 2013). It is worth mentioning that DNA methylation dysregulation in RA could be a consequence of metabolic alterations. For example, increased levels of PMFBP1 (polyamine-modulated factor 1 binding protein 1) and SSAT1 (spermidine synthase) in RASFs promote polyamines metabolism, which may increase S-adenosyl methionine (SAM) consumption and, in turn, decrease the substrate required for DNA methylation in RASFs cells (Karouzakis et al., 2012). Some treatments used in RA patients, such as methotrexate (MTX), have been demonstrated to produce demethylation in FOXP3, which leads to the accumulation of Treg cells (Cribbs et al., 2015). Moreover, due to the changes in the methylation in FOXP3, Wieczorek et al. have suggested analyzing the methylation of FOXP3 as a new method for counting Treg, evaluating disease status and identifying patients' responses to therapy (Wieczorek et al., 2009). Another study has shown that MTX can reverse the hypomethylated status and restore methylation levels once again by reducing the expression of the enzymes involved in demethylation in PBMC (de Andres et al., 2015).
More studies have demonstrated that miRNAs play an important role in RA. MiR-24, miR-26a, and miR-125a-5p are overrepresented in the plasma of RA patients, which suggests their feasible utility as biomarkers (Murata et al., 2013). Li et al. have observed an increase in miRNA-146a that correlated with TNF-α levels and with low levels of miR-363 and miR-498 in CD4+ T-cells (Li et al., 2010). Indeed as we indicate in the Introduction and describe in the SS section, miR-146a has been proposed as a key regulator of the immune system because it can control the secretion of cytokines, B-cell function, and NF-kß signaling, among others. Increased miR-146a levels are associated with IL-17 expression in the PBMCs and synovium of RA patients (Niimoto et al., 2010). In contrast, miR-146a and miR-155 lower in Treg cells after T-cell stimulation in RA patients (Zhou et al., 2015). miR-155 inhibits the transcript of SOCS1 and produces the upregulation of both TNF-α and interleukin IL-1β in the PBMCs of RA (Li et al., 2013). Other miRNAs have also been found to be involved in RA. For example, miR-346 controls TNF-ß synthesis in RASFs by tristetraprolin stabilization (Semaan et al., 2011). Tristetraprolin (also known as the zinc finger protein 36 homolog) is a protein that binds to AU-rich elements in the 3'-untranslated regions of the mRNAs of cytokines and promotes their degradation.
miR-18a plays an important role in the TNF-α-mediated signaling pathway through a feedback loop in the NF-κB signaling in RASFs (Trenkmann et al., 2013). Dong et al. have described a high Th17/Treg cells ratio in the PMBCs of RA patients and they have also found reduced expression levels in miR-21-5p in the CD4+ T-cells of these patients. Low miR-21-5p levels may promote Th17 cell differentiation while suppressing Treg cell development during chronic inflammation in RA (Dong et al., 2014) (Figure 1). Another study has reported that miR-23b show low levels in RASF. It also reported that this miRNA suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3, and IKK-α and that IL-17 is also a negative regulator of miR-23b expression (Zhu et al., 2012). This agrees with the increased IL-17 levels described in RA patients (Zhu et al., 2012), and also with a high ratio of Th17 cells (Dong et al., 2014), which are the main source of this cytokine. In addition, the increase in miR-126a promotes the inhibition of DNMTI, which produces the hypomethylation of the promoters of CD11a and CD70 and, in turn, their expression increases in the CD4+ T-cells of RA (Yang et al., 2015). This mechanism may have similar consequences to those that occur in SS, where the hypomethylation of CD70 in B-cells has been observed and, therefore, contributes to the autoimmune response.
Very few research studies have delved into the role of the PTMs of histones in specific gene or genomic regions in RA. Huber et al. have described an increase in the shifted balance of histone acetylase/histone deacetylase activity to hyperacetylation in the synovial tissue samples of RA patients (Huber et al., 2007). These results agree with increased IL-6 production in the RASFs of RA patients, where elevated H3ac and H3K4me3 levels were found in the proximal IL-6 promoter (Wada et al., 2014). Kawabata et al. have described increased histone deacetylase 1 (HDAC1) levels in the synovial tissues in RA patients, which have been correlated with a high cytoplasmic TNF-α concentration (Horiuchi et al., 2009; Kawabata et al., 2010). In agreement with the previous results, the inhibition of HDAC1 in RASFs and in a mouse collagen-induced arthritis model brought about changes in the expression of the genes related to proliferation, migration, and inflammation by, therefore, improving the RA phenotype (Hawtree et al., 2015). Furthermore, HDAC3 activity is necessary for type I interferon (IFNI) production and the subsequent activation of the signal transducer and activator of transcription 1 (STAT1) in fibroblast-like synoviocytes (FLS) (Angiolilli et al., 2017). In contrast, HDAC5 expression is suppressed in RASFs by inflammatory cytokines, such as IL-1β and TNF-α, which produce interferon regulatory factor (IRF1) nuclear localization and the promotion of IRF1-regulated genes (Angiolilli et al., 2016). Finally, PADI4 is able to deiminate the arginine residues of histones by generating citrulline residues, and an increased PADI4 expression has been observed in RA synovial membranes (Chang et al., 2009).
Conclusions
Recent research demonstrates the importance of epigenetics in the development of the most prevalent human autoimmune diseases because epigenetic dysregulation plays a relevant role in the pathophysiology of several autoimmune/inflammatory disorders, such as SS and RA (Table 2).
The increased expression of IFN-induced genes as a consequence of promoter hypomethylation has been demonstrated in the B-cells of SS patients. The hypomethylated IFNG promoter in pro-inflammatory T-cells (CD4+CD28T− T-cells) has also been found in RA patients (Pieper et al., 2014). Hypomethylation is apparently accompanied by the hyperacetylation of histones, which may contribute to the control of epigenetic programs in enhancer regions. miR-146a and miR-155 appear as relevant epigenetic switches in both autoimmune disorders SS and RA. In particular, miR-146a is elevated in the PBMCs of both SS and RA (Table 2). This is particularly relevant because both miR-146a and miR-155 are recognized as "inflamma-miRs" in aging (Olivieri et al., 2013), a physiological process during which immunosenescence occurs (Fulop et al., 2018). Therefore, it is noteworthy that both miRNAs can be used to monitor not only the inflammation status in SS and RA, but also as markers of dysregulated immunity. Hence, many studies have aimed to elucidate age-dependent changes in chromatin accessibility, DNA methylation, and histone modifications in immune cells, particularly in T-cells (Tserel et al., 2015;Moskowitz et al., 2017; Cheung et al., 2018). The implication of these epigenetic regulation mechanisms in aging will improve our understanding of the phenotypical variations observed in disease progression of both RA and SS patients.
The miR-181 and miR-16a signature measured in the PBMCs of SS patients seems more specific. Both miRNAs are associated with the degree of inflammation (Wang et al., 2018), which indicates the role played by both miRNAs as biomarker candidates to monitor SS progression.
A large number of epigenetic studies in RA and SS have been conducted in PBMCs or affected tissues (RASF or salivary glands). Conversely, accumulating evidence in different subsets of immune cells reveals specific changes in the epigenetic marks that regulate the differentiation and function of each subset of cells (Bock et al., 2012; Sethi et al., 2013; Avgustinova and Benitah, 2016; Kumar Kingsley and Vishnu Bhat, 2017;Wu et al., 2018). Thus, it is reasonable to think that a more specific analysis of the epigenome in specific immune cell subsets should be carried out to unveil which type of immune cells is altered in these autoimmune diseases. This is an important fact because most epigenetic data have been obtained from bulk immune cell populations. However, in order to completely decipher the role of epigenetic mechanisms in autoimmune-related disorders, the whole epigenome needs to be elucidated. Single-cell technologies, such as mass cytometry or single-cell RNA sequencing, have revealed an unprecedented heterogeneity in different immune cell populations (Buenrostro et al., 2015; Cusanovich et al., 2015; Farlik et al., 2015). These strategies have allowed the whole methylome, chromatin regulation to be characterized by the ChIP-seq analysis, as well as the whole miRNome, which helps us to understand the pathogenesis of SS and RA by identifying biomarkers for disease management and predicting the long-term outcomes in these patients. For example, lymphoma is a long-term feature in about 5% of SS patients (Theander et al., 2006).
Furthermore, in order to understand the complexity of immune-related disorders, we must consider not only the complexity of immune cell regulation and epigenetic programs established in immune cell subpopulations, but also the interaction of immune cells with other tissues. The tissue environment can influence genetic programs in immune cells, and it is obvious that environmental factors may affect the inflammatory pathways in both RA and SS. For this reason, another issue to consider is how environmental factors affect the epigenetic regulation in autoimmune diseases. It is known that a narrow range of risk factors has been associated with rheumatic diseases, and also with epigenetic changes (Klareskog et al., 2006;Colafrancesco et al., 2016). In SS, factors such as stress, air pollution, vitamin D, or hormonal factors have been described as risk factors for this autoimmune disease to develop and progress (Colafrancesco et al., 2016). In RA, smoking is considered the commonest environmental risk factor to contribute to RA development and severity (Baka et al., 2009). Air pollution has been widely reported as an epigenetic modulator [see the review in (Li et al., 2017a)] and to increase the probability of developing systemic autoimmune rheumatic diseases such as SS (Bernatsky et al., 2016). Similarly, monocyte-specific smoking-associated DNA methylation patterns have been observed (Reynolds et al., 2017), which well matches an increased risk of RA development (Klareskog et al., 2011;Chang et al., 2014). Although many studies have related environmental factors to the development and/or progression of SS and RA, the actual mechanisms underlying these diseases are still unknown. Hence, epigenetic regulation could provide new pieces in the intricate puzzle of the environmental effects in SS and RA and, thus, more are needed to clarify how external factors can modify the epigenome in the SS and RA physiopathology contexts.
A lot of work is still to be done to improve the efficacy and safety of treatments against autoimmune diseases. However, the identification of the key epigenetic mechanisms and epigenetic switches controlling specific transcriptomic programs can provide new possibilities to elucidate the diagnosis of the wide array of autoimmune disorders. Therefore, research that specifically focuses on elucidating the epigenetic control in specific subpopulations of immune cells will provide new insights into the pathogenesis of autoimmune diseases and will open up new ways to develop novel therapeutic approaches that target epigenetic mechanisms, such as DNA methylation, histone PTMs, and regulating miRNAs expression. HDAC inhibitors (HDACi) are proposed as a therapeutic strategy for RA (Hsieh et al., 2014; Cantley et al., 2015; Oh et al., 2017; Angiolilli et al., 2018; Kim et al., 2018;). HDACi is a good therapeutic approaches because it helps to lower inflammation levels through vascular cell adhesion molecule-1 (VCAM), intercellular adhesion molecule-1 (ICAM), and E-selectin (ESEL) levels, which could contribute to immunosuppression (Inoue et al., 2006). Accordingly, the only clinical trial that has been tested is givinostat for juvenile idiopathic arthritis. Yet despite the benefits of this HDAC inhibitor, after a 12-week safety and efficacy assay (NCT00570661) (Vojinovic et al., 2011), a dose-finding study showed lack of efficacy (NCT0155745), which suggests that it should be used in conjunction with other treatments to improve the effect on patients. Moreover, more efforts should be made to further clarify the special contribution of epigenetics to the etiology of these immune-related disorders and how epigenetic-based drugs can increase the therapeutic options for RA and SS.
Author Contributions
Bibliography compilation and analysis: JG-G, MS-C, JI-C, RO-V, and FP. Manuscript drafting: JG-G, MS-C, JI-C, RO-V, and FP. Review of manuscript content: MS-C and JI-C. Approval of the final version of the manuscript: all the authors.
Funding
This work was supported by FIS (PI12/02263, PI16/01031, and PI16/01036) from ISCIII and was co-financed by European Regional Development Founds (ERDF) and ACCI2014 (CIBERer-ISCIII). JG-G is supported by the Spanish Ministry of Economy and Competitiveness and by the Instituto de Salud Carlos III through CIBERer (Biomedical Network Research Center for Rare Diseases and INGENIO2010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MS-C is a Generalitat Valenciana fellow (APOSTD/2018/A/156) co-financed by European Regional Development Funds (ERDF) at the University of Valencia. JI-C is an "Atracció de Talent" fellow (University of Valencia).
Abbreviations
ChIP, Chromatin Immunoprecipitation; CLP, Common Lymphoid Progenitors; DMRs, Differentially Methylated Regions; DNMTs, DNA Methyltransferases; HSCs, Hematopoietic Stem Cells; HDAC, Histone Deacetylases; IFN, Interferon; MHC, Major histocompatibility complex; PBMCs, Peripheral Blood Mononuclear Cells; PTMs, Post-Translational Modifications; ROS, Reactive Oxygen Species; TREG, Regulatory T-Cells; RA, Rheumatoid Arthritis; RASFs, Rheumatoid Arthritis Synovial Fibroblasts; SGECs, Salivary Gland Epithelial Cells; STAT, Signal Transducer and Activator of Transcription; SNPs, Single Nucleotide Polymorphism; SS, Sjögren Syndrome; miRNA, Small noncoding RNA.
References
Aggarwal, A. (2014). Role of autoantibody testing. Best Pract. Res. Clin. Rheumatol. 28, 907–920. doi: 10.1016/j.berh.2015.04.010
Alarcón, G. S., Williams, G. V., Singer, J. Z., Steen, V. D., Clegg, D. O., Paulus, H. E., et al. (1991). Early undifferentiated connective tissue disease. I. Early clinical manifestation in a large cohort of patients with undifferentiated connective tissue diseases compared with cohorts of well established connective tissue disease. J. Rheumatol. 18, 1332–1339.
Alevizos, I., Illei, G. G. (2010). MicroRNAs in Sjögren’s syndrome as a prototypic autoimmune disease. Autoimmun. Rev. 9, 618–621. doi: 10.1016/j.autrev.2010.05.009
Altorok, N., Coit, P., Hughes, T., Koelsch, K. A., Stone, D. U., Rasmussen, A., et al. (2014). Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary sjögren’s syndrome. Arthritis Rheumatol. 66, 731–739. doi: 10.1002/art.38264
Anaya, J.-M., Rojas-Villarraga, A., Mantilla, R. D., Arcos-Burgos, M., Sarmiento-Monroy, J. C. (2016). Polyautoimmunity in Sjögren Syndrome. Rheum. Dis. Clin. North Am. 42, 457–472. doi: 10.1016/j.rdc.2016.03.005
Angiolilli, C., Grabiec, A. M., Ferguson, B. S., Ospelt, C., Malvar Fernandez, B., van Es, I. E., et al. (2016). Inflammatory cytokines epigenetically regulate rheumatoid arthritis fibroblast-like synoviocyte activation by suppressing HDAC5 expression. Ann. Rheum. Dis. 75, 430–438. doi: 10.1136/annrheumdis-2014-205635
Angiolilli, C., Kabala, P. A., Grabiec, A. M., Rossato, M., Lai, W. S., Fossati, G., et al. (2018). Control of cytokine mRNA degradation by the histone deacetylase inhibitor ITF2357 in rheumatoid arthritis fibroblast-like synoviocytes: beyond transcriptional regulation. Arthritis Res. Ther. 20, 148. doi: 10.1186/s13075-018-1638-4
Angiolilli, C., Kabala, P. A., Grabiec, A. M., Van Baarsen, I. M., Ferguson, B. S., García, S., et al. (2017). Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 76, 277–285. doi: 10.1136/annrheumdis-2015-209064
Appel, S., Le Hellard, S., Bruland, O., Brun, J. G., Omdal, R., Kristjansdottir, G., et al. (2011). Potential association of muscarinic receptor 3 gene variants with primary Sjogren's syndrome. Ann. Rheum. Dis. 70, 1327–1329. doi: 10.1136/ard.2010.138966
Arango Duque, G., Descoteaux, A. (2014). Macrophage cytokines: involvement in immunity and infectious diseases. Front. Immunol. 5, 491. doi: 10.3389/fimmu.2014.00491
Asou, N. (2003). The role of a Runt domain transcription factor AML1/RUNX1 in leukemogenesis and its clinical implications. Crit. Rev. Oncol. Hematol. 45, 129–150. doi: 10.1016/S1040-8428(02)00003-3
Atabaki, M., Hashemi, M., Daneshvar, H., Alijani, E. (2017). Association between interleukin-1 receptor associated kinase 1 rs3027898 A/C gene polymorphism and rheumatoid arthritis. Biomed. Rep. 6, 335–338. doi: 10.3892/br.2017.855
Avgustinova, A., Benitah, S. A. (2016). Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 17, 643–658. doi: 10.1038/nrm.2016.76
Baka, Z., Buzás, E., Nagy, G. (2009). Rheumatoid arthritis and smoking: putting the pieces together. Arthritis Res. Ther. 11, 238. doi: 10.1186/ar2751
Banerjee, A., Schambach, F., DeJong, C. S., Hammond, S. M., Reiner, S. L. (2010). Micro-RNA-155 inhibits IFN-gamma signaling in CD4+ T cells. Eur. J. Immunol. 40, 225–231. doi: 10.1002/eji.200939381
Begum, N. A., Stanlie, A., Nakata, M., Akiyama, H., Honjo, T. (2012). The Histone Chaperone Spt6 is required for activation-induced cytidine deaminase target determination through H3K4me3 regulation. J. Biol. Chem. 287, 32415–32429. doi: 10.1074/jbc.M112.351569
Bernatsky, S., Smargiassi, A., Barnabe, C., Svenson, L. W., Brand, A., Martin, R. V., et al. (2016). Fine particulate air pollution and systemic autoimmune rheumatic disease in two Canadian provinces. Environ. Res. 146, 85–91. doi: 10.1016/j.envres.2015.12.021
Bezman, N. A., Chakraborty, T., Bender, T., Lanier, L. L. (2011). miR-150 regulates the development of NK and iNKT cells. J. Exp. Med. 208, 2717–2731. doi: 10.1084/jem.20111386
Bissels, U., Bosio, A., Wagner, W. (2012). MicroRNAs are shaping the hematopoietic landscape. Haematologica 97, 160–167. doi: 10.3324/haematol.2011.051730
Bock, C., Beerman, I., Lien, W.-H., Smith, Z. D., Gu, H., Boyle, P., et al. (2012). DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 47, 633–647. doi: 10.1016/j.molcel.2012.06.019
Boldin, M. P., Taganov, K. D., Rao, D. S., Yang, L., Zhao, J. L., Kalwani, M., et al. (2011). miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 208, 1189–1201. doi: 10.1084/jem.20101823
Buenrostro, J. D., Wu, B., Litzenburger, U. M., Ruff, D., Gonzales, M. L., Snyder, M. P., et al. (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. doi: 10.1038/nature14590
Burbelo, P. D., Ambatipudi, K., Alevizos, I. (2014). Genome-wide association studies in Sjögren’s syndrome: What do the genes tell us about disease pathogenesis? Autoimmun. Rev. 13, 756–761. doi: 10.1016/j.autrev.2014.02.002
Cabezas-Wallscheid, N., Klimmeck, D., Hansson, J., Lipka, D. B., Reyes, A., Wang, Q., et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA Methylome Analysis. Cell Stem Cell 15, 507–522. doi: 10.1016/j.stem.2014.07.005
Cantley, M. D., Fairlie, D. P., Bartold, P. M., Marino, V., Gupta, P. K., Haynes, D. R. (2015). Inhibiting histone deacetylase 1 suppresses both inflammation and bone loss in arthritis. Rheumatology (Oxford). 54, 1713–1723. doi: 10.1093/rheumatology/kev022
Chang, K., Yang, S., Kim, S., Han, K., Park, S., Shin, J. (2014). Smoking and rheumatoid arthritis. Int. J. Mol. Sci. 15, 22279–22295. doi: 10.3390/ijms151222279
Chang, X., Zhao, Y., Sun, S., Zhang, Y., Zhu, Y. (2009). The expression of PADI4 in synovium of rheumatoid arthritis. Rheumatol. Int. 29, 1411–1416. doi: 10.1007/s00296-009-0870-2
Chatzikyriakidou, A., Voulgari, P. V., Georgiou, I., Drosos, A. A. (2010). A polymorphism in the 3'-UTR of interleukin-1 receptor-associated kinase (IRAK1), a target gene of miR-146a, is associated with rheumatoid arthritis susceptibility. Joint. Bone. Spine 77, 411–413. doi: 10.1016/j.jbspin.2010.05.013
Chen, X., Liu, X., Zhang, Y., Huai, W., Zhou, Q., Xu, S., et al. (2018). Methyltransferase Dot1l preferentially promotes innate IL-6 and IFN-β production by mediating H3K79me2/3 methylation in macrophages. Cell. Mol. Immunol. doi: 10.1038/s41423-018-0170-4
Cheung, P., Vallania, F., Warsinske, H. C., Donato, M., Schaffert, S., Chang, S. E., et al. (2018). Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell 173, 1385–1397.e14. doi: 10.1016/j.cell.2018.03.079
Chowdhury, M., Forouhi, O., Dayal, S., McCloskey, N., Gould, H. J., Felsenfeld, G., et al. (2008). Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proc. Natl. Acad. Sci. U. S. A. 105, 15872–15877. doi: 10.1073/pnas.0808462105
Ciechomska, M., O'Reilly, S. (2016). Epigenetic modulation as a therapeutic prospect for treatment of autoimmune rheumatic diseases. Mediators Inflamm. 2016, 1–11. doi: 10.1155/2016/9607946
Cobb, B. S., Hertweck, A., Smith, J., O’Connor, E., Graf, D., Cook, T., et al. (2006). A role for Dicer in immune regulation. J. Exp. Med. 203, 2519–2527. doi: 10.1084/jem.20061692
Colafrancesco, S., Perricone, C., Shoenfeld, Y. (2016). “Sjögren’s Syndrome and Environmental Factors,” in Sjögren’s Syndrome, eds. Gerli, R., Bartoloni, E., Alunno, A. B. T.-S. S (London: Elsevier), 157–170. doi: 10.1016/B978-0-12-803604-4.00010-1
Cole, M. B., Quach, H., Quach, D., Baker, A., Taylor, K. E., Barcellos, L. F., et al. (2016). Epigenetic Signatures of Salivary Gland Inflammation in Sjögren’s Syndrome. Arthritis Rheumatol. 68, 2936–2944. doi: 10.1002/art.39792
Conigliaro, P., Ciccacci, C., Politi, C., Triggianese, P., Rufini, S., Kroegler, B., et al. (2017). Polymorphisms in STAT4, PTPN2, PSORS1C1 and TRAF3IP2 Genes Are Associated with the Response to TNF Inhibitors in Patients with Rheumatoid Arthritis. PloS One 12, e0169956. doi: 10.1371/journal.pone.0169956
Conrad, K., Roggenbuck, D., Reinhold, D., Dörner, T. (2010). Profiling of rheumatoid arthritis associated autoantibodies. Autoimmun. Rev. 9, 431–435. doi: 10.1016/j.autrev.2009.11.017
Corvetta, A., Della Bitta, R., Luchetti, M. M., Pomponio, G. (1991). 5-Methylcytosine content of DNA in blood, synovial mononuclear cells and synovial tissue from patients affected by autoimmune rheumatic diseases. J. Chromatogr. B Biomed. Sci. Appl. 566, 481–491. doi: 10.1016/0378-4347(91)80265-E
Cribbs, A. P., Kennedy, A., Penn, H., Amjadi, P., Green, P., Read, J. E., et al. (2015). Methotrexate restores regulatory T Cell function through demethylation of the FoxP3 upstream enhancer in patients with rheumatoid arthritis. Arthritis Rheumatol. 67, 1182–1192. doi: 10.1002/art.39031
Cribbs, A. P., Kennedy, A., Penn, H., Read, J. E., Amjadi, P., Green, P., et al. (2014). Treg cell function in rheumatoid arthritis is compromised by CTLA-4 promoter methylation resulting in a failure to activate the indoleamine 2,3-dioxygenase pathway. Arthritis Rheumatol. 66, 2344–2354. doi: 10.1002/art.38715
Cusanovich, D. A., Daza, R., Adey, A., Pliner, H. A., Christiansen, L., Gunderson, K. L., et al. (2015). Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914. doi:.10.1126/science.abb1601.(80-.)
Daridon, C., Devauchelle, V., Hutin, P., Berre, R., Martins-Carvalho, C., et al. (2007). Aberrant expression of BAFF by B lymphocytes infiltrating the salivary glands of patients with primary Sjögren’s syndrome. Arthritis Rheum. 56, 1134–1144. doi: 10.1002/art.22458
de Andres, M. C., Perez-Pampin, E., Calaza, M., Santaclara, F. J., Ortea, I., Gomez-Reino, J. J., et al. (2015). Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res. Ther. 17, 233. doi: 10.1186/s13075-015-0748-5
de la Rica, L., Urquiza, J. M., Gómez-Cabrero, D., Islam, A. B. M. M. K., López-Bigas, N., Tegnér, J., et al. (2013). Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J. Autoimmun. 41, 6–16. doi: 10.1016/j.jaut.2012.12.005
Delacher, M., Imbusch, C. D., Weichenhan, D., Breiling, A., Hotz-Wagenblatt, A., Träger, U., et al. (2017). Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 18, 1160–1172. doi: 10.1038/ni.3799
Denton, A. E., Russ, B. E., Doherty, P. C., Rao, S., Turner, S. J. (2011). Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. 108, 15306–15311. doi: 10.1073/pnas.1112520108
Devauchelle-Pensec, V., Pennec, Y., Morvan, J., Pers, J.-O., Daridon, C., Jousse-Joulin, S., et al. (2007). Improvement of Sjögren’s syndrome after two infusions of rituximab (anti-CD20). Arthritis Rheum. 57, 310–317. doi: 10.1002/art.22536
Di Carlo, V., Mocavini, I., Di Croce, L. (2018). Polycomb complexes in normal and malignant hematopoiesis. J. Cell Biol. 218, 55–69. doi: 10.1083/jcb.201808028
Dong, L., Wang, X., Tan, J., Li, H., Qian, W., Chen, J., et al. (2014). Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J. Cell. Mol. Med. 18, 2213–2224. doi: 10.1111/jcmm.12353
Dooley, J., Linterman, M. A., Liston, A. (2013). MicroRNA regulation of T-cell development. Immunol. Rev. 253, 53–64. doi: 10.1111/imr.12049
Emmrich, S., Rasche, M., Schöning, J., Reimer, C., Keihani, S., Maroz, A., et al. (2014). miR-99a/100∼125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGFβ and Wnt signaling. Genes Dev. 28, 858–874. doi: 10.1101/gad.233791.113
Farh, K. K.-H., Marson, A., Zhu, J., Kleinewietfeld, M., Housley, W. J., Beik, S., et al. (2015). Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343. doi: 10.1038/nature13835
Farlik, M., Sheffield, N. C., Nuzzo, A., Datlinger, P., Schönegger, A., Klughammer, J., et al. (2015). Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 10, 1386–1397. doi: 10.1016/j.celrep.2015.02.001
Fazi, F., Rosa, A., Fatica, A., Gelmetti, V., De Marchis, M. L., Nervi, C., et al. (2005). A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 123, 819–831. doi: 10.1016/j.cell.2005.09.023
Fiorentino, D. F., Bond, M. W., Mosmann, T. R. (1989). Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 170, 2081–2095. doi: 10.1084/jem.170.6.2081
Fonseca, J. E., Santos, M. J., Canhão, H., Choy, E. (2009). Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun. Rev. 8, 538–542. doi: 10.1016/j.autrev.2009.01.012
Fu, L., Ma, C., Cong, B., Li, S., Chen, H., Zhang, J. (2011). Hypomethylation of proximal CpG motif of interleukin-10 promoter regulates its expression in human rheumatoid arthritis. Acta Pharmacol. Sin. 32, 1373–1380. doi: 10.1038/aps.2011.98
Fulop, T., Larbi, A., Dupuis, G., Le Page, A., Frost, E. H., Cohen, A. A., et al. (2018). Immunosenescence and Inflamm-aging as two sides of the same coin: friends or foes? Front. Immunol. 8, 1960. doi: 10.3389/fimmu.2017.01960
Gamper, C. J., Agoston, A. T., Nelson, W. G., Powell, J. D. (2009). Identification of DNA methyltransferase 3a as a T cell receptor-induced regulator of Th1 and Th2 differentiation. J. Immunol. 183, 2267–2276. doi: 10.4049/jimmunol.0802960
Gao, J., Cha, S., Jonsson, R., Opalko, J., Peck, A. B. (2004). Detection of anti-type 3 muscarinic acetylcholine receptor autoantibodies in the sera of Sjögren’s syndrome patients by use of a transfected cell line assay. Arthritis Rheum. 50, 2615–2621. doi: 10.1002/art.20371
Georgantas, R. W., Hildreth, R., Morisot, S., Alder, J., Liu, C., Heimfeld, S., et al. (2007). CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc. Natl. Acad. Sci. U. S. A. 104, 2750–2755. doi: 10.1073/pnas.0610983104
Ghani, S., Riemke, P., Schönheit, J., Lenze, D., Stumm, J., Hoogenkamp, M., et al. (2011). Macrophage development from HSCs requires PU.1-coordinated microRNA expression. Blood 118, 2275–2284. doi: 10.1182/blood-2011-02-335141
Ghisi, M., Corradin, A., Basso, K., Frasson, C., Serafin, V., Mukherjee, S., et al. (2011). Modulation of microRNA expression in human T-cell development: targeting of NOTCH3 by miR-150 . Blood 117, 7053–7062. doi: 10.1182/blood-2010-12-326629
Glossop, J. R., Emes, R. D., Nixon, N. B., Packham, J. C., Fryer, A. A., Mattey, D. L., et al. (2016). Genome-wide profiling in treatment-naive early rheumatoid arthritis reveals DNA methylome changes in T and B lymphocytes. Epigenomics 8, 209–224. doi: 10.2217/epi.15.103
Glossop, J. R., Glossop, J. R., Emes, R. D., Emes, R. D., Nixon, N. B., Nixon, N. B., et al. (2014). Genome-wide DNA methylation profiling in rheumatoid arthritis identifies disease-associated methylation changes that are distinct to individual T- and B-lymphocyte populations. Epigenetics 9, 1228–1237. doi: 10.4161/epi.29718
Gottfried, E., Kreutz, M., Mackensen, A. (2012). Tumor metabolism as modulator of immune response and tumor progression. Semin. Cancer Biol. 22, 335–341. doi: 10.1016/j.semcancer.2012.02.009
Gruber, J. J., Zatechka, D. S., Sabin, L. R., Yong, J., Lum, J. J., Kong, M., et al. (2009). Ars2 links the nuclear cap-binding complex to rna interference and cell proliferation. Cell 138, 328–339. doi: 10.1016/j.cell.2009.04.046
Guerau-de-Arellano, M., Smith, K. M., Godlewski, J., Liu, Y., Winger, R., Lawler, S. E., et al. (2011). Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain 134, 3578–3589. doi: 10.1093/brain/awr262
Guo, S., Lu, J., Schlanger, R., Zhang, H., Wang, J. Y., Fox, M. C., et al. (2010). MicroRNA miR-125a controls hematopoietic stem cell number. Proc. Natl. Acad. Sci. U. S. A. 107, 14229–14234. doi: 10.1073/pnas.0913574107
Hamaguchi, M., Kawahito, Y., Ishino, H., Takeuchi, N., Tokunaga, D., Hojo, T., et al. (2011). Mucin from rheumatoid arthritis synovial fluid enhances interleukin-6 production by human peripheral blood mononuclear cells. Hum. Immunol. 72, 241–248. doi: 10.1016/j.humimm.2010.12.013
Hawtree, S., Muthana, M., Wilkinson, J. M., Akil, M., Wilson, A. G. (2015). Histone deacetylase 1 regulates tissue destruction in rheumatoid arthritis. Hum. Mol. Genet. 24, 5367–5377. doi: 10.1093/hmg/ddv258
He, J., Ding, Y., Feng, M., Guo, J., Sun, X., Zhao, J., et al. (2013a). Characteristics of Sjogren's syndrome in rheumatoid arthritis. Rheumatology 52, 1084–1089. doi: 10.1093/rheumatology/kes374
He, S., Tong, Q., Bishop, D. K., Zhang, Y. (2013b). Histone methyltransferase and histone methylation in inflammatory T-cell responses. Immunotherapy 5, 989–1004. doi: 10.2217/imt.13.101
Hernández-Bello, J., Oregón-Romero, E., Vázquez-Villamar, M., García-Arellano, S., Valle, Y., Padilla-Gutiérrez, J. R., et al. (2017). Aberrant expression of interleukin-10 in rheumatoid arthritis: relationship with IL10 haplotypes and autoantibodies. Cytokine 95, 88–96. doi: 10.1016/j.cyto.2017.02.022
Hori, S., Nomura, T., Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3 . Science 299, 1057–1061. doi: 10.1126/science.1079490
Horiuchi, M., Morinobu, A., Chin, T., Sakai, Y., Kurosaka, M., Kumagai, S. (2009). Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J. Rheumatol. 36, 1580–1589. doi: 10.3899/jrheum.081115
Horsfall, A. C., Rose, L. M., Maini, R. N. (1989). Autoantibody synthesis in salivary glands of Sjögren’s syndrome patients. J. Autoimmun. 2, 559–568. doi: 10.1016/0896-8411(89)90189-3
Hsieh, I.-N., Liou, J.-P., Lee, H.-Y., Lai, M.-J., Li, Y.-H., Yang, C.-R. (2014). Preclinical anti-arthritic study and pharmacokinetic properties of a potent histone deacetylase inhibitor MPT0G009 . Cell Death Dis. 5, e1166. doi: 10.1038/cddis.2014.133
Huber, L. C., Brock, M., Hemmatazad, H., Giger, O. T., Moritz, F., Trenkmann, M., et al. (2007). Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 56, 1087–1093. doi: 10.1002/art.22512
Imgenberg-Kreuz, J., Sandling, J. K., Almlöf, J. C., Nordlund, J., Signér, L., Norheim, K. B., et al. (2016). Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann. Rheum. Dis. 75, 2029–2036. doi: 10.1136/annrheumdis-2015-208659
Inoue, K., Kobayashi, M., Yano, K., Miura, M., Izumi, A., Mataki, C., et al. (2006). Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler. Thromb. Vasc. Biol. 26, 2652–2659. doi: 10.1161/01.ATV.0000247247.89787.e7
Ishida, K., Kobayashi, T., Ito, S., Komatsu, Y., Yokoyama, T., Okada, M., et al. (2012). Interleukin-6 gene promoter methylation in rheumatoid arthritis and chronic periodontitis. J. Periodontol. 83, 917–925. doi: 10.1902/jop.2011.110356
Ishino, H., Kawahito, Y., Hamaguchi, M., Takeuchi, N., Tokunaga, D., Hojo, T., et al. (2010). Expression of Tn and sialyl Tn antigens in synovial tissues in rheumatoid arthritis. Clin. Exp. Rheumatol. 28, 246–249.
Javierre, B. M., Fernandez, A. F., Richter, J., Al-Shahrour, F., Martin-Subero, J. I., Rodriguez-Ubreva, J., et al. (2010). Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 20, 170–179. doi: 10.1101/gr.100289.109
Jensen, K., Brusletto, B. S., Aass, H. C. D., Olstad, O. K., Kierulf, P., Gautvik, K. M. (2013). Transcriptional profiling of mRNAs and microRNAs in human bone marrow precursor B cells identifies subset- and age-specific variations. PloS One 8, e70721. doi: 10.1371/journal.pone.0070721
Jiang, S., Li, C., Olive, V., Lykken, E., Feng, F., Sevilla, J., et al. (2011). Molecular dissection of the miR-17-92 cluster's critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood 118, 5487–5497. doi: 10.1182/blood-2011-05-355644
Kanno, Y., Vahedi, G., Hirahara, K., Singleton, K., O'Shea, J. J. (2012). Transcriptional and epigenetic control of t helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu. Rev. Immunol. 30, 707–731. doi: 10.1146/annurev-immunol-020711-075058
Kapsogeorgou, E. K., Tzioufas, A. G. (2016). “Autoantibodies in Sjögren’s Syndrome and Laboratory Markers,” in, eds. Roccatello, D., Emmi , L. (Cham: Springer International Publishing), 293–308. doi: 10.1007/978-3-319-24535-5_21
Kapsogeorgou, E. K., Gourzi, V. C., Manoussakis, M. N., Moutsopoulos, H. M., Tzioufas, A. G. (2011). Cellular microRNAs (miRNAs) and Sjögren’s syndrome: Candidate regulators of autoimmune response and autoantigen expression. J. Autoimmun. 37, 129–135. doi: 10.1016/j.jaut.2011.05.003
Karouzakis, E., Gay, R. E., Gay, S., Neidhart, M. (2012). Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 64, 1809–1817. doi: 10.1002/art.34340
Karouzakis, E., Gay, R. E., Michel, B. A., Gay, S., Neidhart, M. (2009). DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 60, 3613–3622. doi: 10.1002/art.25018
Karouzakis, E., Rengel, Y., Jüngel, A., Kolling, C., Gay, R. E., Michel, B. A., et al. (2011). DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts. Genes Immun. 12, 643–652. doi: 10.1038/gene.2011.45
Kawabata, T., Nishida, K., Takasugi, K., Ogawa, H., Sada, K., Kadota, Y., et al. (2010). Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-alpha in synovial tissue of rheumatoid arthritis. Arthritis Res. Ther. 12, R133. doi: 10.1186/ar3071
Kim, K.-W., Kim, H. J., Kim, B.-M., Kwon, Y.-R., Kim, H.-R., Kim, Y.-J. (2018). Epigenetic modification of mesenchymal stromal cells enhances their suppressive effects on the Th17 responses of cells from rheumatoid arthritis patients. Stem Cell Res. Ther. 9, 208. doi: 10.1186/s13287-018-0948-4
Kinne, R. W., Stuhlmüller, B., Burmester, G.-R. (2007). Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res. Ther. 9, 224. doi: 10.1186/ar2333
Klareskog, L., Malmström, V., Lundberg, K., Padyukov, L., Alfredsson, L. (2011). Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin. Immunol. 23, 92–98. doi: 10.1016/j.smim.2011.01.014
Klareskog, L., Padyukov, L., Lorentzen, J., Alfredsson, L. (2006). Mechanisms of disease: Genetic susceptibility and environmental triggers in the development of rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 2, 425–433. doi: 10.1038/ncprheum0249
Klein, K., Gay, S. (2015). Epigenetics in rheumatoid arthritis. Curr. Opin. Rheumatol. 27, 76–82. doi: 10.1097/BOR.0000000000000128
Konsta, O. D., Charras, A., Le Dantec, C., Kapsogeorgeou, E., Bordron, A., Brooks, W. H., et al. (2016a). Epigenetic modifications in salivary glands from patients with Sjögren’s syndrome affect cytokeratin 19 expression. Bull. Group. Int. Rech. Sci. Stomatol. Odontol. 53, e01.
Konsta, O. D., Le Dantec, C., Charras, A., Brooks, W. H., Arleevskaya, M. I., Bordron, A., et al. (2015). An in silico approach reveals associations between genetic and epigenetic factors within regulatory elements in B cells from primary sjögren’s syndrome patients. Front. Immunol. 6, 437. doi: 10.3389/fimmu.2015.00437
Konsta, O. D., Le Dantec, C., Charras, A., Cornec, D., Kapsogeorgou, E. K., Tzioufas, A. G., et al. (2016b). Defective DNA methylation in salivary gland epithelial acini from patients with Sjögren’s syndrome is associated with SSB gene expression, anti-SSB/LA detection, and lymphocyte infiltration. J. Autoimmun. 68, 30–38. doi: 10.1016/j.jaut.2015.12.002
Konsta, O. D., Thabet, Y., Le Dantec, C., Brooks, W. H., Tzioufas, A. G., Pers, J.-O., et al. (2014). The contribution of epigenetics in Sjögren’s Syndrome. Front. Genet. 5, 71. doi: 10.3389/fgene.2014.00071
Kumar Kingsley, S. M., Vishnu Bhat, B. (2017). Role of MicroRNAs in the development and function of innate immune cells. Int. Rev. Immunol. 36, 154–175. doi: 10.1080/08830185.2017.1284212
Kundu, M., Compton, S., Garrett-Beal, L., Stacy, T., Starost, M. F., Eckhaus, M., et al. (2005). Runx1 deficiency predisposes mice to T-lymphoblastic lymphoma. Blood 106, 3621–3624. doi: 10.1182/blood-2005-04-1447
Lai, A. Y., Mav, D., Shah, R., Grimm, S. A., Phadke, D., Hatzi, K., et al. (2013). DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res. 23, 2030–2041. doi: 10.1101/gr.155473.113
Lal, G., Zhang, N., van der Touw, W., Ding, Y., Ju, W., Bottinger, E. P., et al. (2009). Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 182, 259–273. doi: 10.4049/jimmunol.182.1.259
Lavie, F., Miceli-Richard, C., Quillard, J., Roux, S., Leclerc, P., Mariette, X. (2004). Expression of BAFF(BLyS) in T cells infiltrating labial salivary glands from patients with Sjögren’s syndrome. J. Pathol. 202, 496–502. doi: 10.1002/path.1533
Le Dantec, C., Varin, M.-M., Brooks, W. H., Pers, J.-O., Youinou, P., Renaudineau, Y. (2012). Epigenetics and Sjögren’s syndrome. Curr. Pharm. Biotechnol. 13, 2046–53. doi: 10.2174/138920112802273326
Lee, P. P., Fitzpatrick, D. R., Beard, C., Jessup, H. K., Lehar, S., Makar, K. W., et al. (2001). A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774. doi: 10.1016/s1074-7613(01)00227-8
Lessard, C. J., Li, H., Adrianto, I., Ice, J. A., Rasmussen, A., Grundahl, K. M., et al. (2013). Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat. Genet. 45, 1284–1292. doi: 10.1038/ng.2792
Li, J., Li, W. X., Bai, C., Song, Y. (2017a). Particulate matter-induced epigenetic changes and lung cancer. Clin. Respir. J. 11, 539–546. doi: 10.1111/crj.12389
Li, J., Wan, Y., Guo, Q., Zou, L., Zhang, J., Fang, Y., et al. (2010). Altered microRNA expression profile with miR-146a upregulation in CD4+ T cells from patients with rheumatoid arthritis. Arthritis Res. Ther. 12, R81. doi: 10.1186/ar3006
Li, X., Tian, F., Wang, F. (2013). Rheumatoid Arthritis-Associated MicroRNA-155 Targets SOCS1 and Upregulates TNF-α and IL-1β in PBMCs. Int. J. Mol. Sci. 14, 23910–23921. doi: 10.3390/ijms141223910
Li, X., Zhang, Q., Shi, Q., Liu, Y., Zhao, K., Shen, Q., et al. (2017b). Demethylase Kdm6a epigenetically promotes IL-6 and IFN-β production in macrophages. J. Autoimmun. 80, 85–94. doi: 10.1016/j.jaut.2017.02.007
Liu, C.-C., Fang, T.-J., Ou, T.-T., Wu, C.-C., Li, R.-N., Lin, Y.-C., et al. (2011). Global DNA methylation, DNMT1, and MBD2 in patients with rheumatoid arthritis. Immunol. Lett. 135, 96–99. doi: 10.1016/j.imlet.2010.10.003
Liu, Y., Aryee, M. J., Padyukov, L., Fallin, M. D., Hesselberg, E., Runarsson, A., et al. (2013). Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 31, 142–147. doi: 10.1038/nbt.2487
Loizou, J. I., Oser, G., Shukla, V., Sawan, C., Murr, R., Wang, Z.-Q., et al. (2009). Histone Acetyltransferase Cofactor Trrap Is Essential for Maintaining the Hematopoietic Stem/Progenitor Cell Pool. J. Immunol. 183, 6422–6431. doi: 10.4049/jimmunol.0901969
Low, H., Witte, T. (2011). Aspects of innate immunity in Sjögren’s syndrome. Arthritis Res. Ther. 13, 218. doi: 10.1186/ar3318
Lu, L.-F., Boldin, M. P., Chaudhry, A., Lin, L.-L., Taganov, K. D., Hanada, T., et al. (2010). Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 142, 914–929. doi: 10.1016/j.cell.2010.08.012
Lu, Y., Wahl, L. M. (2005). Oxidative stress augments the production of matrix metalloproteinase-1, cyclooxygenase-2, and prostaglandin e 2 through enhancement of nf-κb activity in lipopolysaccharide-activated human primary monocytes. J. Immunol. 175, 5423–5429. doi: 10.4049/jimmunol.175.8.5423
MacGregor, A. J., Snieder, H., Rigby, A. S., Koskenvuo, M., Kaprio, J., Aho, K., et al. (2000). Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 43, 30–37. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B
Mackay, F., Woodcock, S. A., Lawton, P., Ambrose, C., Baetscher, M., Schneider, P., et al. (1999). Mice Transgenic for Baff Develop Lymphocytic Disorders along with Autoimmune Manifestations. J. Exp. Med. 190, 1697–1710. doi: 10.1084/jem.190.11.1697
Mariette, X. (2003). The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjogren's syndrome. Ann. Rheum. Dis. 62, 168–171. doi: 10.1136/ard.62.2.168
Marques, S. C., Laursen, M. B., Bødker, J. S., Kjeldsen, M. K., Falgreen, S., Schmitz, A., et al. (2015). MicroRNAs in B-cells: from normal differentiation to treatment of malignancies. Oncotarget 6, 7–25. doi: 10.18632/oncotarget.3057
Mattes, J., Collison, A., Plank, M., Phipps, S., Foster, P. S. (2009). Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc. Natl. Acad. Sci. U. S. A. 106, 18704–18709. doi: 10.1073/pnas.0905063106
Mehta, A., Baltimore, D. (2016). MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 16, 279–294. doi: 10.1038/nri.2016.40
Mogilyansky, E., Rigoutsos, I. (2013). The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 20, 1603–1614. doi: 10.1038/cdd.2013.125
Monticelli, S., Ansel, K. M., Xiao, C., Socci, N. D., Krichevsky, A. M., Thai, T.-H., et al. (2005). MicroRNA profiling of the murine hematopoietic system. Genome Biol. 6, R71. doi: 10.1186/gb-2005-6-8-r71
Morales-Nebreda, L., McLafferty, F. S., Singer, B. D. (2019). DNA methylation as a transcriptional regulator of the immune system. Transl. Res. 204, 1–18. doi: 10.1016/j.trsl.2018.08.001
Moskowitz, D. M., Zhang, D. W., Hu, B., Le Saux, S., Yanes, R. E., Ye, Z., et al. (2017). Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol. 2, eaag0192. doi: 10.1126/sciimmunol.aag0192
Mueller, R. B., Skapenko, A., Wendler, J., Schuch, F., Kalden, J. R., Schulze-Koops, H. (2007). MHC class II expression on myeloid cells inversely correlates with disease progression in early rheumatoid arthritis. Rheumatology 46, 931–933. doi: 10.1093/rheumatology/kem025
Murata, K., Furu, M., Yoshitomi, H., Ishikawa, M., Shibuya, H., Hashimoto, M., et al. (2013). Comprehensive microRNA Analysis Identifies miR-24 and miR-125a-5p as Plasma Biomarkers for Rheumatoid Arthritis. PloS One 8, e69118. doi: 10.1371/journal.pone.0069118
Nakano, K., Whitaker, J. W., Boyle, D. L., Wang, W., Firestein, G. S. (2013). DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 72, 110–117. doi: 10.1136/annrheumdis-2012-201526
Niimoto, T., Nakasa, T., Ishikawa, M., Okuhara, A., Izumi, B., Deie, M., et al. (2010). MicroRNA-146a expresses in interleukin-17 producing T cells in rheumatoid arthritis patients. BMC Musculoskelet. Disord. 11, 209. doi: 10.1186/1471-2474-11-209
Nordmark, G., Alm, G. V., Rönnblom, L. (2006). Mechanisms of Disease: primary Sjögren’s syndrome and the type I interferon system. Nat. Clin. Pract. Rheumatol. 2, 262–269. doi: 10.1038/ncprheum0173
O’Connell, R. M., Taganov, K. D., Boldin, M. P., Cheng, G., Baltimore, D. (2007). MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U. S. A. 104, 1604–1609. doi: 10.1073/pnas.0610731104
Oh, B. R., Suh, D.-H., Bae, D., Ha, N., Choi, Y., Yoo, H. J., et al. (2017). Therapeutic effect of a novel histone deacetylase 6 inhibitor, CKD-L, on collagen-induced arthritis in vivo and regulatory T cells in rheumatoid arthritis in vitro. Arthritis Res. Ther. 19, 154. doi: 10.1186/s13075-017-1357-2
Okamoto, K., Takayanagi, H. (2011). Regulation of bone by the adaptive immune system in arthritis. Arthritis Res. Ther. 13, 219. doi: 10.1186/ar3323
Okuda, T., Nishimura, M., Nakao, M., Fujitaa, Y. (2001). RUNX1/AML1: a central player in hematopoiesis. Int. J. Hematol. 74, 252–257. doi: 10.1007/BF02982057
Olivieri, F., Rippo, M. R., Procopio, A. D., Fazioli, F. (2013). Circulating inflamma-miRs in aging and age-related diseases. Front. Genet. 4, 121. doi: 10.3389/fgene.2013.00121
Pagano, G., Castello, G., Pallardó, F. V. (2013). Sjøgren’s syndrome-associated oxidative stress and mitochondrial dysfunction: Prospects for chemoprevention trials. Free Radic. Res. 47, 71–73. doi: 10.3109/10715762.2012.748904
Park, S.-H., Kim, S.-K., Choe, J.-Y., Moon, Y., An, S., Park, M. J., et al. (2013). Hypermethylation of EBF3 and IRX1 genes in synovial fibroblasts of patients with rheumatoid arthritis. Mol. Cells 35, 298–304. doi: 10.1007/s10059-013-2302-0
Parkes, M., Cortes, A., van Heel, D. A., Brown, M. A. (2013). Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat. Rev. Genet. 14, 661–673. doi: 10.1038/nrg3502
Pauley, K. M., Stewart, C. M., Gauna, A. E., Dupre, L. C., Kuklani, R., Chan, A. L., et al. (2011). Altered miR-146a expression in Sjögren’s syndrome and its functional role in innate immunity. Eur. J. Immunol. 41, 2029–2039. doi: 10.1002/eji.201040757
Peng, L., Ma, W., Yi, F., Yang, Y.-J., Lin, W., Chen, H., et al. (2014). MicroRNA Profiling in Chinese Patients with Primary Sjögren Syndrome Reveals Elevated miRNA-181a in Peripheral Blood Mononuclear Cells. J. Rheumatol. 41, 2208–2213. doi: 10.3899/jrheum.131154
Pieper, J., Johansson, S., Snir, O., Linton, L., Rieck, M., Buckner, J. H., et al. (2014). Peripheral and site-specific CD4(+) CD28(null) T cells from rheumatoid arthritis patients show distinct characteristics. Scand. J. Immunol. 79, 149–155. doi: 10.1111/sji.12139
Ramos-Casals, M., Brito-Zerón, P., Font, J. (2007). The Overlap of Sjögren’s Syndrome with Other Systemic Autoimmune Diseases. Semin. Arthritis Rheum. 36, 246–255. doi: 10.1016/j.semarthrit.2006.08.007
Reynolds, L. M., Lohman, K., Pittman, G. S., Barr, R. G., Chi, G. C., Kaufman, J., et al. (2017). Tobacco exposure-related alterations in DNA methylation and gene expression in human monocytes: the Multi-Ethnic Study of Atherosclerosis (MESA). Epigenetics 12, 1092–1100. doi: 10.1080/15592294.2017.1403692
Richardson, B., Scheinbart, L., Strahler, J., Gross, L., Hanash, S., Johnson, M. (1990). Evidence for impaired t cell dna methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 33, 1665–1673. doi: 10.1002/art.1780331109
Rodriguez, A., Vigorito, E., Clare, S., Warren, M. V., Couttet, P., Soond, D. R., et al. (2007). Requirement of bic/microRNA-155 for normal immune function. Science 316, 608–611. doi: 10.1126/science.1139253
Routsias, J. G., Tzioufas, A. G. (2007). Sjögren’s syndrome–study of autoantigens and autoantibodies. Clin. Rev. Allergy Immunol. 32, 238–251. doi: 10.1007/s12016-007-8003-8
Said, E. A., Dupuy, F. P., Trautmann, L., Zhang, Y., Shi, Y., El-Far, M., et al. (2010). Programmed death-1–induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 16, 452–459. doi: 10.1038/nm.2106
Salomonsson, S., Wahren-Herlenius, M. (2003). Local production of Ro/SSA and La/SSB autoantibodies in the target organ coincides with high levels of circulating antibodies in sera of patients with Sjögren’s syndrome. Scand. J. Rheumatol. 32, 79–82. doi: 10.1080/03009740310000076
Schlegelberger, B., Heller, P. G. (2017). RUNX1 deficiency (familial platelet disorder with predisposition to myeloid leukemia, FPDMM). Semin. Hematol. 54, 75–80. doi: 10.1053/j.seminhematol.2017.04.006
Schoenborn, J. R., Dorschner, M. O., Sekimata, M., Santer, D. M., Shnyreva, M., Fitzpatrick, D. R., et al. (2007). Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat. Immunol. 8, 732–742. doi: 10.1038/ni1474
Sellars, M., Huh, J. R., Day, K., Issuree, P. D., Galan, C., Gobeil, S., et al. (2015). Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat. Immunol. 16, 746–754. doi: 10.1038/ni.3198
Semaan, N., Frenzel, L., Alsaleh, G., Suffert, G., Gottenberg, J.-E., Sibilia, J., et al. (2011). miR-346 Controls Release of TNF-α Protein and Stability of Its mRNA in Rheumatoid Arthritis via Tristetraprolin Stabilization. PloS One 6, e19827. doi: 10.1371/journal.pone.0019827
Sethi, A., Kulkarni, N., Sonar, S., Lal, G. (2013). Role of miRNAs in CD4 T cell plasticity during inflammation and tolerance. Front. Genet. 4, 8. doi: 10.3389/fgene.2013.00008
Shi, H., Zheng, L., Zhang, P., Yu, C. (2014). miR-146a and miR-155 expression in PBMCs from patients with Sjögren’s syndrome. J. Oral Pathol. Med. 43, 792–797. doi: 10.1111/jop.12187
Smolen, J. S., Aletaha, D., McInnes, I. B. (2016). Rheumatoid arthritis. Lancet 388, 2023–2038. doi: 10.1016/S0140-6736(16)30173-8
Steiner, G. (2007). Auto-antibodies and autoreactive T-cells in rheumatoid arthritis. Clin. Rev. Allergy Immunol. 32, 23–35. doi: 10.1007/BF02686079
Taganov, K. D., Boldin, M. P., Chang, K.-J., Baltimore, D. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 103, 12481–12486. doi: 10.1073/pnas.0605298103
Thabet, Y., Le Dantec, C., Ghedira, I., Devauchelle, V., Cornec, D., Pers, J.-O., et al. (2013). Epigenetic dysregulation in salivary glands from patients with primary Sjögren’s syndrome may be ascribed to infiltrating B cells. J. Autoimmun. 41, 175–181. doi: 10.1016/j.jaut.2013.02.002
Theander, E., Henriksson, G., Ljungberg, O., Mandl, T., Manthorpe, R., Jacobsson, L. T. H. (2006). Lymphoma and other malignancies in primary Sjögren’s syndrome: a cohort study on cancer incidence and lymphoma predictors. Ann. Rheum. Dis. 65, 796–803. doi: 10.1136/ard.2005.041186
Trenkmann, M., Brock, M., Gay, R. E., Michel, B. A., Gay, S., Huber, L. C. (2013). Tumor Necrosis Factor α-Induced MicroRNA-18a Activates Rheumatoid Arthritis Synovial Fibroblasts Through a Feedback Loop in NF-κB Signaling. Arthritis Rheum. 65, 916–927. doi: 10.1002/art.37834
Tserel, L., Kolde, R., Limbach, M., Tretyakov, K., Kasela, S., Kisand, K., et al. (2015). Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci. Rep. 5, 13107. doi: 10.1038/srep13107
Vento-Tormo, R., Company, C., Rodríguez-Ubreva, J., de la Rica, L., Urquiza, J. M., Javierre, B. M., et al. (2016). IL-4 orchestrates STAT6-mediated DNA demethylation leading to dendritic cell differentiation. Genome Biol. 17, 4. doi: 10.1186/s13059-015-0863-2
Vidal, M., Starowicz, K. (2017). Polycomb complexes PRC1 and their function in hematopoiesis. Exp. Hematol. 48, 12–31. doi: 10.1016/j.exphem.2016.12.006
Vigorito, E., Kohlhaas, S., Lu, D., Leyland, R. (2013). miR-155: an ancient regulator of the immune system. Immunol. Rev. 253, 146–157. doi: 10.1111/imr.12057
Vivino, F. B. (2017). Sjogren's syndrome: Clinical aspects. Clin. Immunol. 182, 48–54. doi: 10.1016/j.clim.2017.04.005
Vojinovic, J., Damjanov, N., D'Urzo, C., Furlan, A., Susic, G., Pasic, S., et al. (2011). Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 63, 1452–1458. doi: 10.1002/art.30238
Volpe, E., Servant, N., Zollinger, R., Bogiatzi, S. I., Hupé, P., Barillot, E., et al. (2008). A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 9, 650–657. doi: 10.1038/ni.1613
Wada, T. T., Araki, Y., Sato, K., Aizaki, Y., Yokota, K., Kim, Y. T., et al. (2014). Aberrant histone acetylation contributes to elevated interleukin-6 production in rheumatoid arthritis synovial fibroblasts. Biochem. Biophys. Res. Commun. 444, 682–686. doi: 10.1016/j.bbrc.2014.01.195
Wang, Y., Zhang, G., Zhang, L., Zhao, M., Huang, H. (2018). Decreased microRNA-181a and -16 expression levels in the labial salivary glands of Sjögren syndrome patients. Exp. Ther. Med. 15, 426–432. doi: 10.3892/etm.2017.5407
Wang-Renault, S.-F., Boudaoud, S., Nocturne, G., Roche, E., Sigrist, N., Daviaud, C., et al. (2018). Deregulation of microRNA expression in purified T and B lymphocytes from patients with primary Sjögren’s syndrome. Ann. Rheum. Dis. 77, 133–140. doi: 10.1136/annrheumdis-2017-211417
Wei, G., Wei, L., Zhu, J., Zang, C., Hu-Li, J., Yao, Z., et al. (2009). Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167. doi: 10.1016/j.immuni.2008.12.009
Whisnant, A. W., Bogerd, H. P., Flores, O., Ho, P., Powers, J. G., Sharova, N., et al. (2013). In-Depth Analysis of the Interaction of HIV-1 with Cellular microRNA Biogenesis and Effector Mechanisms. MBio. 4, e000193. doi: 10.1128/mBio.00193-13
Wieczorek, G., Asemissen, A., Model, F., Turbachova, I., Floess, S., Liebenberg, V., et al. (2009). Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res. 69, 599–608. doi: 10.1158/0008-5472.CAN-08-2361
Williams, A. E. G., Choi, K., Chan, A. L., Lee, Y. J., Reeves, W. H., Bubb, M. R., et al. (2016). Sjögren’s syndrome-associated microRNAs in CD14+ monocytes unveils targeted TGFβ signaling. Arthritis Res. Ther. 18, 95. doi: 10.1186/s13075-016-0987-0
Wilson, A., Laurenti, E., Trumpp, A. (2009). Balancing dormant and self-renewing hematopoietic stem cells. Curr. Opin. Genet. Dev. 19, 461–468. doi: 10.1016/j.gde.2009.08.005
Wilson, A., Laurenti, E., Oser, G., van der Wath, R. C., Blanco-Bose, W., Jaworski, M., et al. (2008). Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 135, 1118–1129. doi: 10.1016/j.cell.2008.10.048
Wilting, R. H., Yanover, E., Heideman, M. R., Jacobs, H., Horner, J., van der Torre, J., et al. (2010). Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 29, 2586–2597. doi: 10.1038/emboj.2010.136
Wu, H., Deng, Y., Feng, Y., Long, D., Ma, K., Wang, X., et al. (2018). Epigenetic regulation in B-cell maturation and its dysregulation in autoimmunity. Cell. Mol. Immunol. 15, 676–684. doi: 10.1038/cmi.2017.133
Wu, H., Neilson, J. R., Kumar, P., Manocha, M., Shankar, P., Sharp, P. A., et al. (2007). miRNA profiling of naïve, effector and memory CD8 T cells. PloS One 2, e1020. doi: 10.1371/journal.pone.0001020
Xiao, C., Calado, D. P., Galler, G., Thai, T.-H., Patterson, H. C., Wang, J., et al. (2007). MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 131, 146–159. doi: 10.1016/j.cell.2007.07.021
Yang, G., Wu, D., Zeng, G., Jiang, O., Yuan, P., Huang, S., et al. (2015). Correlation between miR-126 expression and DNA hypomethylation of CD4+ T cells in rheumatoid arthritis patients. Int. J. Clin. Exp. Pathol. 8, 8929–8936.
Yang, H., Bian, S., Chen, H., Wang, L., Zhao, L., Zhang, X., et al. (2018). Clinical characteristics and risk factors for overlapping rheumatoid arthritis and Sjögren’s syndrome. Sci. Rep. 8, 6180. doi: 10.1038/s41598-018-24279-1
Yang, L., Anderson, D. E., Baecher-Allan, C., Hastings, W. D., Bettelli, E., Oukka, M., et al. (2008). IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature 454, 350–352. doi: 10.1038/nature07021
Yang, Y., Peng, L., Ma, W., Yi, F., Zhang, Z., Chen, H., et al. (2016). Autoantigen-targeting microRNAs in Sjögren’s syndrome. Clin. Rheumatol. 35, 911–917. doi: 10.1007/s10067-016-3203-3
Yao, R., Ma, Y.-L., Liang, W., Li, H.-H., Ma, Z.-J., Yu, X., et al. (2012). MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1 . PloS One 7, e46082. doi: 10.1371/journal.pone.0046082
Yin, H., Zhao, M., Wu, X., Gao, F., Luo, Y., Ma, L., et al. (2010). Hypomethylation and overexpression of CD70 (TNFSF7) in CD4+ T cells of patients with primary Sjögren’s syndrome. J. Dermatol. Sci. 59, 198–203. doi: 10.1016/j.jdermsci.2010.06.011
Yu, X., Liang, G., Yin, H., Ngalamika, O., Li, F., Zhao, M., et al. (2013). DNA hypermethylation leads to lower FOXP3 expression in CD4+ T cells of patients with primary Sjögren’s syndrome. Clin. Immunol. 148, 254–257. doi: 10.1016/j.clim.2013.05.005
Zhang, Z., Zhang, C., Li, F., Zhang, B., Zhang, Y. (2018). Regulation of Memory CD8+ T Cell Differentiation by MicroRNAs. Cell. Physiol. Biochem. 47, 2187–2198. doi: 10.1159/000491532
Zhao, J. L., Ma, C., O'Connell, R. M., Mehta, A., DiLoreto, R., Heath, J. R., et al. (2014). Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 14, 445–459. doi: 10.1016/j.stem.2014.01.007
Zhou, Q., Haupt, S., Kreuzer, J. T., Hammitzsch, A., Proft, F., Neumann, C., et al. (2015). Decreased expression of miR-146a and miR-155 contributes to an abnormal Treg phenotype in patients with rheumatoid arthritis. Ann. Rheum. Dis. 74, 1265–1274. doi: 10.1136/annrheumdis-2013-204377
Zhu, S., Pan, W., Song, X., Liu, Y., Shao, X., Tang, Y., et al. (2012). The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat. Med. 18, 1077–1086. doi: 10.1038/nm.2815
Keywords: epigenetics, autoimmune diseases, epigenetic pathways, rheumatic diseases, DNA methylation, histone modifications, miRNAs
Citation: Ibáñez-Cabellos JS, Seco-Cervera M, Osca-Verdegal R, Pallardó FV and García-Giménez JL (2019) Epigenetic Regulation in the Pathogenesis of Sjögren Syndrome and Rheumatoid Arthritis. Front. Genet. 10:1104. doi: 10.3389/fgene.2019.01104
Received: 28 March 2019; Accepted: 11 October 2019;
Published: 13 November 2019.
Edited by:
Sung-Chou Li, Kaohsiung Chang Gung Memorial Hospital, TaiwanReviewed by:
Girdhari Lal, National Centre for Cell Science, IndiaJoy Tseng, Chang Gung University College of Medicine, Taiwan
Copyright © 2019 Ibáñez-Cabellos, Seco-Cervera, Osca-Verdegal, Pallardó and García-Giménez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Seco-Cervera, bWFydGEuc2Vjb0B1di5lcw==; José Luis García-Giménez, ai5sdWlzLmdhcmNpYUB1di5lcw==