 Anja Mösch
Anja Mösch Silke Raffegerst2
Silke Raffegerst2- 1Department of Bioinformatics, Wissenschaftszentrum Weihenstephan, Technische Universität München, Freising, Germany
- 2Medigene Immunotherapies GmbH, a subsidiary of Medigene AG, Planegg, Germany
In the last years, immunotherapies have shown tremendous success as treatments for multiple types of cancer. However, there are still many obstacles to overcome in order to increase response rates and identify effective therapies for every individual patient. Since there are many possibilities to boost a patient’s immune response against a tumor and not all can be covered, this review is focused on T cell receptor-mediated therapies. CD8+ T cells can detect and destroy malignant cells by binding to peptides presented on cell surfaces by MHC (major histocompatibility complex) class I molecules. CD4+ T cells can also mediate powerful immune responses but their peptide recognition by MHC class II molecules is more complex, which is why the attention has been focused on CD8+ T cells. Therapies based on the power of T cells can, on the one hand, enhance T cell recognition by introducing TCRs that preferentially direct T cells to tumor sites (so called TCR-T therapy) or through vaccination to induce T cells in vivo. On the other hand, T cell activity can be improved by immune checkpoint inhibition or other means that help create a microenvironment favorable for cytotoxic T cell activity. The manifold ways in which the immune system and cancer interact with each other require not only the use of large omics datasets from gene, to transcript, to protein, and to peptide but also make the application of machine learning methods inevitable. Currently, discovering and selecting suitable TCRs is a very costly and work intensive in vitro process. To facilitate this process and to additionally allow for highly personalized therapies that can simultaneously target multiple patient-specific antigens, especially neoepitopes, breakthrough computational methods for predicting antigen presentation and TCR binding are urgently required. Particularly, potential cross-reactivity is a major consideration since off-target toxicity can pose a major threat to patient safety. The current speed at which not only datasets grow and are made available to the public, but also at which new machine learning methods evolve, is assuring that computational approaches will be able to help to solve problems that immunotherapies are still facing.
Introduction
Immunotherapies have gained more and more importance over the last decades. Checkpoint inhibitors mainly targeting PD1/PDL1 and CTLA4 and personalized cancer vaccines (Gubin et al., 2014; Ott et al., 2017; Sahin et al., 2017) have been and still are heavily investigated in clinical trials. Both depend on patient individual tumor-specific mutations enabling the boost of a cancer-specific T cell-mediated immune response (Snyder et al., 2014; Rizvi et al., 2015; Łuksza et al., 2017). A more direct approach utilizes the adoptive transfer of a patient’s autologous T cells, either genetically modified with a transgenic chimeric antigen receptor (CAR) or T cell receptor (TCR). For CAR-T cell as well as TCR-T cell therapy a defined target, the epitope, needs to be identified. CARs, carrying the functional antigen-binding domain of an antibody, recognize three-dimensional peptide structures on the surface of a cell (Sadelain et al., 2013). By contrast, TCRs recognize predominantly linear peptides presented by the major histocompatibility complex (MHC) called human leucocyte antigen (HLA) in humans. For MHC class I presentation and thus CD8+ T cell detection, these peptides come from proteins that are intracellularly processed by either the constitutive proteasome or the IFNγ induced immunoproteasome (Griffin et al., 1998; Neefjes et al., 2011). After cleavage, the peptides are transported to the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP) complex, where they are loaded onto MHC class I molecules. The peptide-MHCs (pMHCs) are shuttled to the cell surface where they can potentially be recognized by CD8+ cytotoxic T cells, either naturally carrying or engineered to bear a pMHC-specific TCR (see Figure 1). However, there are more than 16,000 different alleles for HLA-A, -B, and -C genes, which bind and present different epitopes (Robinson et al., 2015). Besides MHC class I mediated CD8+ cytotoxic T cell responses, MHC class II bound peptides can induce CD4+ T cell responses that are also reported to play an important role in tumor detection and elimination (Nielsen et al., 2010; Linnemann et al., 2014; Kreiter et al., 2015; Andreatta et al., 2017; Veatch et al., 2018).
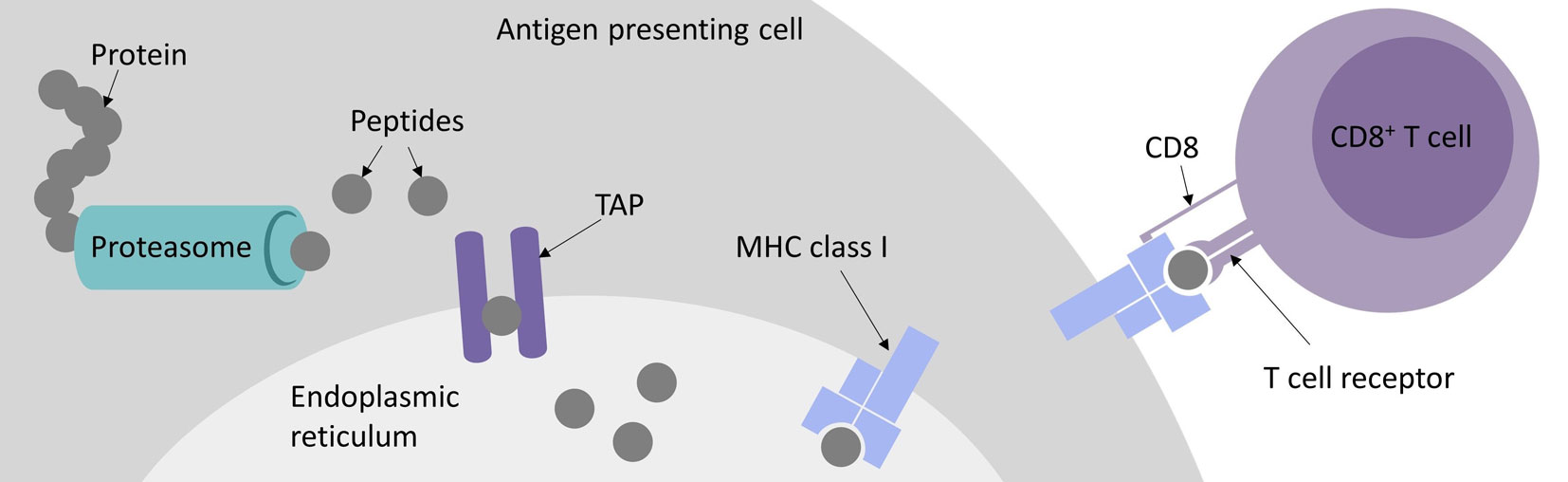
Figure 1 Major histocompatibility complex (MHC) class I antigen presentation pathway for peptides recognized by CD8+ cytotoxic T cells.
A wide spectrum of bioinformatics tools exists for modeling all steps of the MHC class I antigen presentation pathway, including proteasomal cleavage, translocation of the peptides to the ER by TAP, peptide binding to the MHC molecules, and TCR recognition. The overarching goal of these efforts is to enhance our understanding of how T cell epitopes are selected from a virtually unlimited number of short peptides that can be proteolytically generated from the human proteome. The origin of these T cell epitopes can be naturally occurring proteins or peptides derived from somatic mutations. For personalized cancer immunotherapy, these patient- and tumor-specific mutations are usually separately assessed for each patient by exome sequencing, mutation detection and peptide binding prediction (Robbins et al., 2013; Blankenstein et al., 2015; Schumacher and Schreiber, 2015). Predicting these so called neoepitopes or neoantigens is a prevailing challenge for computational methods for immunotherapy and essential for a high-throughput approach to narrow down mutations to be included in vaccines or to be evaluated in vitro for T cell recognition, since only very few mutations are truly immunogenic (Yadav et al., 2014; Strønen et al., 2016; Bjerregaard et al., 2017a).
It is also of utmost importance to evaluate potential cross-reactivity of target-candidate epitopes based on various omics data such as proteomics and peptidomics (Haase et al., 2015; Jaravine et al., 2017a; 2017b). However, all existing approaches based on epitope presentation are only a surrogate for T cell recognition, for which no universal and computationally viable approach exists so far, although the first promising results have been published (Jurtz et al., 2018; Ogishi and Yotsuyanagi, 2019). By now, datasets have been generated that allow sequence-based prediction approaches using deep learning (Shugay et al., 2018; Vita et al., 2018).
In this review, we summarize the current state at the development of prediction algorithms and methods for all steps of antigen presentation, evaluate neoepitope prediction approaches, and discuss progress toward sequence-based TCR binding prediction.
Prediction of T Cell Epitopes
Proteasomal Cleavage Prediction
In order to develop an accurate prediction algorithm for proteosomal cleavages, a thorough mechanistic understanding of the cutting process is required. The PAProC algorithm by Kuttler et al. (Kuttler et al., 2000) relies on a biologically motivated model, which postulates that proteolytic sites are mostly determined by the local sequence context, generally not further away in the sequence than six amino acid residues. The two residues immediately adjacent to the cut make the greatest contribution to the affinity to the active subunits of the proteasome, while the influence of the other surrounding residues is lower. The recognition model is additive in that the total affinity, which ultimately determines the probability of the cut, is considered to be the sum of all individual contributions. Bioinformatics analyses revealed that the amino acids in the six positions preceding the cut and four positions downstream contain sufficient information to reproduce a training dataset of experimentally determined cleavage motifs of 20S proteasomes by a network-based technique. Keşmir et al. (Keşmir et al., 2002) demonstrated that good results in detecting proteasomal cleavage motifs can be achieved by combining experimental data on degradation by the constitutive proteasome with the sequences of peptides bound by the MHC class I molecules, which may be generated either by the constitutive or by the immunoproteasomes. A neural network trained on such a composite dataset, called NetChop, and an updated version NetChop 3.0 (Nielsen et al., 2005), achieved a reasonable accuracy and also yielded useful insights into cleavage-promoting and inhibiting residues as well as into N-terminal extension of peptides after proteasomal cleavage. A recurrent difficulty in predicting proteasomal cleavage is the lack of experimentally verified noncleavage sites. However, such negative data can be artificially generated by considering internal positions of confirmed MHC ligands or randomly generated sites.
TAP Binding Prediction
An early study of Daniel et al. (1998), in which the TAP binding affinity for a large number of peptides of length nine was measured by a peptide binding assay, revealed that positions one to three and nine of the 9-mers make the largest contribution to the selectivity of TAP to peptides. An artificial neural network trained on these data was able to predict the IC50 values with high accuracy. The study also found that HLA class I molecules differed significantly with respect to TAP affinities of their ligands. The predictive scope was later extended to peptides of arbitrary length using a stabilized matrix approach and a scoring scheme that only considers the first three N-terminal residues and the last C-terminal residue (Peters et al., 2003). Since it has been established that the selectivity of peptide transport by TAP is entirely determined by the peptide-binding step (Gubler et al., 1998), affinity predictions can be equated with translocation likelihood predictions. A number of further machine learning methods for predicting peptide binding to TAP were trained on 9-mer data, which is the typical length of the peptides that will subsequently bind to the MHC complex (Bhasin, 2004; Zhang et al., 2006; Diez-Rivero et al., 2010; Lam et al., 2010).
Peptide-MHC Binding Prediction
Sequencing of peptides eluted from MHC class I molecules (Falk et al., 1991) as well as mass-spectrometric (MS) (Hunt et al., 1992) and crystallographic (Madden, 1995) evidence revealed common properties of the epitopes, in particular the typical length range of 8–12 residues. Additionally, it showed the existence of MHC allele-specific anchor residues, usually in positions two and nine of the core nonameric segments, as well as auxiliary anchors, where amino acid preferences are less strict (Rammensee et al., 1993).
Starting from the early nineties, efforts were made to collect available information on MHC class I ligands (Brusic et al., 1994; Rammensee et al., 1995; Rammensee et al.,1999) and to predict them using simple motif- and profile-based techniques (Rothbard and Taylor, 1988; Parker et al., 1994; Reche et al., 2002), based on the notion that peptides highly similar in sequence to experimentally characterized ligands will have a higher binding potential than more distantly related peptides and that individual amino acid side chains make independent contributions to the overall binding energy. Machine learning techniques, such as neural networks and hidden Markov models (Bisset and Fierz, 1993; Mamitsuka, 1998; Nielsen et al., 2003) outperform matrix-based methods in predicting peptide binding affinity (Peters et al., 2006; Lin et al., 2008). They are able to deal with peptides of variable length (Lundegaard et al., 2008) and to take into account nonadditive effects, which may arise, e.g., when two amino acids compete for the same site in the peptide-binding groove of the MHC heterodimer. The latest version of the widely used NetMHC algorithm 4.0 (Andreatta and Nielsen, 2016) was trained on many thousands of quantitative affinity measurements for peptides of length 8–11 and the total of 118 MHC class I alleles from human, other primates, and mouse. Neural networks trained on all peptides (allmer networks) significantly outperformed the networks trained on peptides of each individual length separately. The study also suggested specific binding modes for 10- and 11-mers, which are predicted to bulge out of the MHC grove in contrast to 8- and 9-mers, which are strictly linear epitopes. MHCflurry, which relies on affinity measurement and peptide elution MS data, also uses neural networks trained individually for each HLA allele (O’Donnell et al., 2018b). Additionally, it allows users to train networks locally on data of their choice. This can be important especially for cancer immunotherapy applications, since peptide-binding affinity predictions are traditionally focused on viral epitopes.
There is also a growing group of pan-specific methods, including PickPocket (Zhang et al., 2009), NetMHCpan 4.0 (Jurtz et al., 2017), PSSMHCpan (Liu et al., 2017), and ACME (Hu et al., 2019), which take as input both the peptide and the HLA sequence and are able to predict the binding of any peptide to any allele. Most predictions are focused on MHC class I, but there are also methods available for MHC class II, such as NetMHCII 2.3 and NetMHCIIpan 3.2 (Jensen et al., 2018), ProPred (Singh and Raghava, 2001), SMM-align (Nielsen et al., 2007), and NNAlign (Nielsen and Andreatta, 2017), of which the latter also allows to train and use own models, as Garde et al. did for MHC class II prediction using both affinity measurement and MS data (Garde et al., 2019). Many of the aforementioned prediction methods for both MHC class I and II and consensus methods, such as NetMHCcons (Karosiene et al., 2012) and the consensus method by Moutaftsi et al. (Moutaftsi et al., 2006), are integrated into the IEDB epitope analysis resource and can be accessed online (Wang et al., 2010; Fleri et al., 2017; Vita et al., 2018; Dhanda et al., 2019). In addition, combinatory pipelines and frameworks have been published, namely, EpiJen (Doytchinova et al., 2006), NetCTL (Larsen et al., 2007), NetCTLpan (Stranzl et al., 2010), and FRED2 (Schubert et al., 2016), modeling the complete antigen presentation pathway by including proteasomal cleavage and TAP transport predictions.
Epitope presentation, however, is only one step toward T cell recognition. NetMHCstab (Jørgensen et al., 2014) and NetMHCstabpan (Rasmussen et al., 2016) are methods to predict the stability of pMHC complexes, presuming that epitope presentation lasting longer increases the likelihood of T cell recognition and thus immunogenicity. Calis et al. proposed a scoring model to predict true immunogenicity of T cell epitopes (Calis et al., 2013). Despite these efforts, however, true immunogenicity remains far more difficult to predict than mere MHC-binding affinity.
Beyond sequence-based approaches, significant methodological progress has been made in modeling peptide binding to MHC class I molecules on structure level. The diversity of the cognate peptide repertoire and the experimental binding profiles for a particular MHC protein can be accurately captured using both general purpose modeling packages, such as Rosetta (Yanover and Bradley, 2011), and faster specialized methods, such as GradDock (Kyeong et al., 2018), DockTope (Menegatti Rigo et al., 2015), and LYRA (Klausen et al., 2015), of which the latter two are also integrated in the IEDB. Docking experiments are becoming increasingly successful in reproducing crystallographically known peptide-MHC binding geometry (Bordner and Abagyan, 2006; Antunes et al., 2018).
Immunopeptidomics Data
The recent availability of large-scale immunopeptidomics data allowed to explicitly model peptide length distributions and the interdependence between individual sequence positions, leading to more accurate predictions of naturally presented MHC class I ligands (Gfeller et al., 2018). MS profiling provides novel insights into the antigen processing rules, including the discovery of binding motifs, improved description of proteasomal cleavage signatures, cellular localization and sequence features of peptide source proteins, and better understanding of the role of gene expression, protein abundance and degradation (Bassani-Sternberg et al., 2015; Bassani-Sternberg et al., 2017; Abelin et al., 2017). In particular, Abelin et al. (2017) reported that neural networks trained on MS-derived peptides bound to 16 different HLA alleles outperformed affinity-trained predictors.
For immunogenicity, T cell epitope verification by TCRs or TCR-like antibodies would constitute an ideal dataset to train prediction algorithms (Dolan, 2019), but both approaches are highly dependent on specificity and affinity of TCRs and antibodies used and do not reach the high-throughput efficiency of immunopeptidomics. HLA-peptidomics, which is the MS analysis of MHC-eluted peptides, is the most sophisticated method for high-throughput qualitative and quantitative detection of MHC ligands and thereby of potential T cell epitopes (Hunt et al., 1992; Caron et al., 2011; Hassan et al., 2014; Álvaro-Benito et al.,2018; Freudenmann et al., 2018).
The isolation of pMHC complexes from cell surfaces (Sugawara et al., 1987; Storkus et al., 1993; Bassani-Sternberg et al., 2015; Marino et al., 2019) or out of serum (Ritz et al., 2016; 2017) is the first critical step for a high-quality MS HLA-peptidome analysis. After elution from pMHC complexes, peptides are purified, separated by high pressure liquid chromatography (HPLC), and directly injected and analyzed in a mass spectrometer followed by computational processing of MS spectra data (see Figure 2). Successful peptide detection is determined by various factors, such as HLA enrichment, which is dependent on HLA-antibody quality, efficient elution, and physicochemical characteristics of a peptide defined by its amino acid composition. Relevant peptide properties can be mass, hydrophilicity, and hydrophobicity, its ability to be ionized, as well as cysteine content (Gfeller and Bassani-Sternberg, 2018). Therefore, not all peptides are equally likely to be detected by MS but it is difficult to assess how many peptides are missed. Peptide sequences are often determined by tandem MS: a precursor mass spectrum called MS1 spectrum of the eluted peptides is generated and only peptides with high intensities are isolated for fragmentation and analyzed, resulting in a MS2 or MS/MS spectrum. Observed mass spectra are then compared with theoretical mass spectra in general reference databases. Proteogenomic computational pipelines using customized reference datasets also allow the identification of peptides originating from noncanonical and allegedly noncoding reading frames (Laumont and Perreault, 2017; Laumont et al., 2018), unconventional, genomic coding-sequences (Erhard et al., 2018) as well as neoepitopes from somatic alterations (Yadav et al., 2014; Carreno et al., 2015) or intron retentions (Smart et al., 2018). In addition, the generation of customized spectral library databases of high confidence peptides can be used for data-independent acquisition approaches (Ritz et al., 2017), resulting in increased reproducibility and sensitivity.
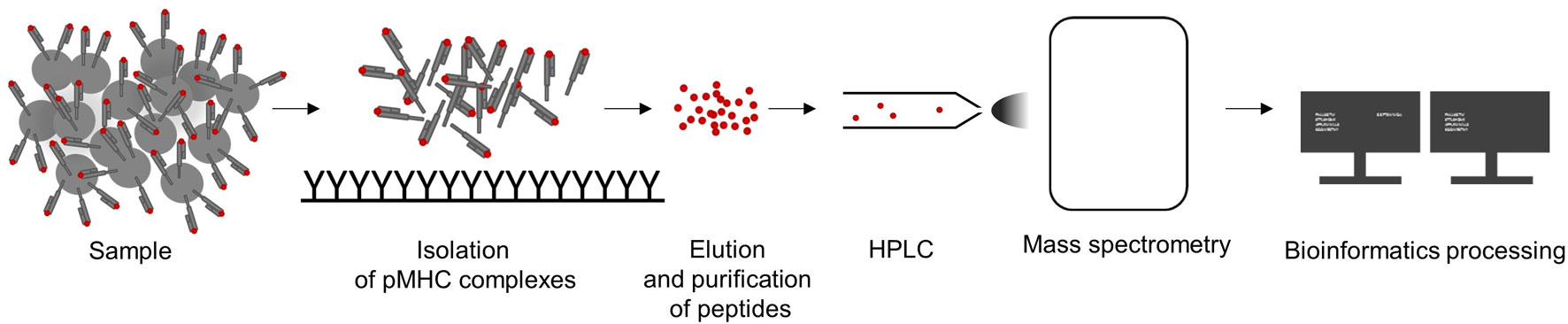
Figure 2 Workflow to analyze of major histocompatibility complex (MHC)-eluted peptides by mass-spectrometric (MS). A sample is lysed, pMHC complexes are captured and peptides are purified by immunoaffinity purification using MHC-specific immobilized antibodies. Eluted peptides are separated by high pressure liquid chromatography (HPLC), analyzed by MS, and the resulting data are computationally processed.
Peptides are often assigned to the HLA molecule from which they were originally eluted by predicting the binding affinity (Freudenmann et al., 2018; Bilich et al., 2019). For common HLA alleles, usually a sufficient number of peptides are identified as binders, resulting in datasets large enough to train prediction algorithms. However, for less frequent HLA alleles, the pool of identified and correctly assigned peptides is more limited, which leads to variability in performance of prediction techniques depending on the rarity of each HLA allele (O’Donnell et al., 2018b). If MS datasets annotated by binding affinity predictions are used to train machine learning algorithms, a self-amplifying bias is introduced. MS profiling of mono-allelic cells (Giam et al., 2015; Abelin et al., 2017) as well as deconvolution approaches (Bassani-Sternberg and Gfeller, 2016) can circumvent this problem and improve the quality of available training data and prediction performance.
Immunotherapy-Specific Applications of Epitope Prediction
Neoepitope Identification
Cancer-specific mutations have been demonstrated to be viable targets for tumor-infiltrating lymphocytes (TILs) enabled by checkpoint inhibitors that block CTLA4 or PD1/PDL1 or by vaccine-induced immune responses (van Rooij et al., 2013; Carreno et al., 2015; Cohen et al., 2015; Gros et al., 2016; McGranahan et al., 2016; Ott et al., 2017; Zacharakis et al., 2018; Hilf et al., 2019). These mutations alter amino acid sequences of proteins and are recognized as so called neoepitopes or neoantigens, with both terms used ambiguously and oftentimes synonymously in the literature. Here, we use the term neoepitopes for epitopes predicted to be presented by a certain MHC and the term neoantigens for confirmed immunogenic mutations. By definition, neoantigens are tumor-specific, which makes them ideal immunotherapy targets, but they are also to a large degree patient-specific. Despite many efforts, only very few shared neoantigens such as KRASG12D/V or BRAFV600E, could be identified, making an off-the-shelf therapy approach hardly feasible (Warren and Holt, 2010; Angelova et al., 2015; Tran et al., 2015; Thorsson et al., 2018). Furthermore, a high individual tumor mutation burden and the ambition to provide personalized medicine for more patients do not allow for testing the immunogenicity of every mutation in vitro. Therefore, the current standard procedure for individual patients relies on exome sequencing followed by mutation calling and machine learning-based neoepitope prediction, which represents the main application of pMHC-binding prediction algorithms in the field of cancer immunotherapy. Here, we reviewed more than 70 publications using binding prediction algorithms to identify neoepitopes of which 49, that provided quantifiable data, are shown in Table 1. Not all studies stated all steps of their neoepitope selection process, including which algorithm parameters were used, how many neoepitopes were found when applying a threshold or how many and what types of mutation were used for predicting neoepitopes, which makes quantitative evaluation and reproducibility difficult. This is aggravated by the large variance in ratio of predicted neoepitopes per mutation, which is caused by thresholds of varying strictness, the number of features used for filtering, and the approach to counting neoepitopes or neoantigens, i.e., if a mutation was counted only once even if presented by more than one HLA allele or contained in multiple epitopes predicted to be immunogenic. Furthermore, some studies could only experimentally validate a subset of predicted neoepitopes and experimental validation was determined by biological assays of varying sensitivity from MHC-ligand confirmation to ELISPOT assays using patient-specific TILs.
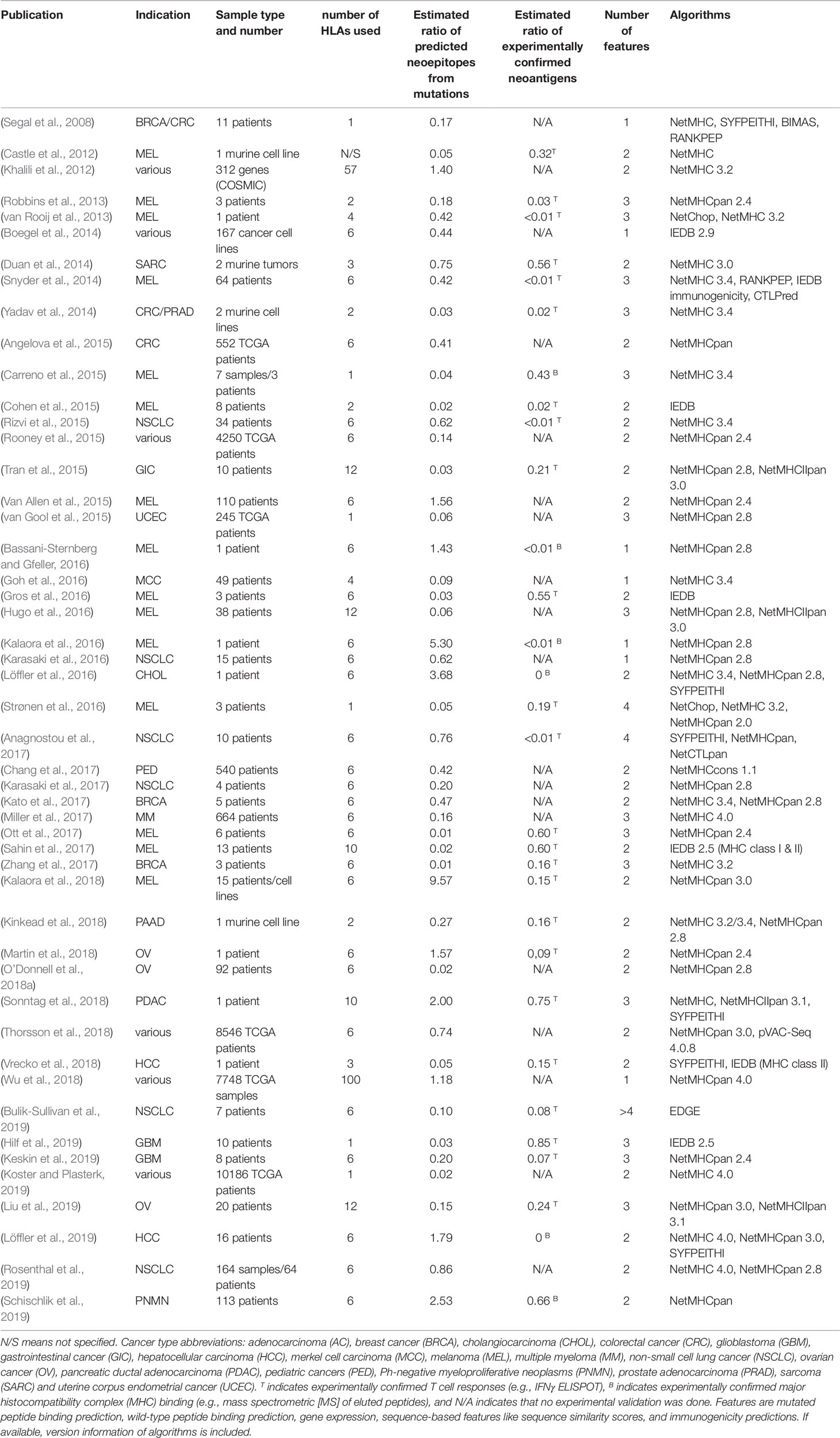
Table 1 Publications describing the application of machine learning approaches to neoepitope prediction.
Not surprisingly, most publications investigated cancer types known for high mutation loads, such as non-small cell lung carcinoma and melanoma, but glioblastoma and chronic lymphocytic leukemia were also shown to harbor neoantigens identified by neoepitope prediction (Rajasagi et al., 2014; Hilf et al., 2019; Keskin et al., 2019). Regarding mutation types, the focus clearly lies on single nucleotide variants (SNVs) considering their abundance in tumors above all other types of mutation, their comparatively easy detection by mutation calling software and easier computational generation of mutated and wild-type peptide sequences (Bailey et al., 2018; Ellrott et al., 2018). However, larger indels, frameshifts, and other more complex mutation types can be the source of more neoepitopes that are also less similar to self and thus highly interesting immunotherapeutic targets. More recent studies from Kahles et al., Koster et al., and Schischlik et al. investigated these types of mutation, benefitting from improvements on sequencing and mutation calling techniques (Kahles et al., 2018; Koster and Plasterk, 2019; Schischlik et al., 2019). Nevertheless, identification of cancer-specific mutation remains a critical step in every neoepitope identification pipeline and the number of mutations obtained varies greatly dependent on the software and thresholds employed (Tran et al., 2015; Karasaki et al., 2017).
The focus of most publications lies on MHC class I presented neoepitopes that can be detected by CD8+ T cells. MHC class I prediction algorithms are more commonly used but there is clear evidence that MHC class II mediated CD4+ T cell responses play a major role in neoantigen immune responses and thus should also be considered for neoepitope detection. (Linnemann et al., 2014; Kreiter et al., 2015; Tran et al., 2015; Hugo et al., 2016; Ott et al., 2017; Reuben et al., 2017; Sahin et al., 2017; Sonntag et al., 2018; Vrecko et al., 2018).
All studies, except Koster et al., who investigated 10-mers only, looked at peptides with a length of 8–10 or 8–11 amino acids or just at 9-mers alone, which are the majority of peptides presented by MHC class I (Trolle et al., 2016). Most studies also relied on matching HLA types for the samples used, often determined by one of the following HLA typing algorithms: ATHLATES, HLAminer, OptiType, PHLAT, POLYSOLVER, and seq2HLA (Boegel et al., 2012; Warren et al., 2012; Liu et al., 2013; Szolek et al., 2014; Shukla et al., 2015; Bai et al., 2018). In contrast, Wu et al. made predictions based on the 100 most frequent HLA alleles in their dataset and Wood et al. based on the general 145 most frequent alleles (Wood et al., 2018; Wu et al., 2018). Whether or not such approaches yield substantial information gain is a debatable issue since most immunogenic mutations are highly individual and restricted by a patient’s individual HLA type (Marty et al., 2017; McGranahan et al., 2017; Rosenthal et al., 2019). HLA-A*02:01 has been extensively studied since it is the most common allele in Caucasian populations and therefore was exclusively used by Segal et al. for their analysis (Segal et al., 2008). Since predictions for A*02:01 still belong to the best performing group and can be more easily validated compared to other alleles due to established in vitro protocols and reagents, Carreno et al., Spranger et al., Strønen et al., van Gool et al., and Hilf et al. also only used A*02:01 for their predictions and the studies that carried out experimental validation accomplished high confirmation of predicted neoepitopes (Carreno et al., 2015; van Gool et al., 2015; Spranger et al., 2016; Strønen et al., 2016; Hilf et al., 2019). Similarly, Koster et al. only used A*02:01 for an unfiltered TCGA dataset although they did not perform experimental validation. Similar to Wood et al., they did not use HLA typing information for TCGA samples, which has been generated but can only be obtained by applying for access to restricted data (Shukla et al., 2015; Charoentong et al., 2017; Marty et al., 2017).
For most studies, algorithms from the NetMHC family were chosen as they are widely known and represent the state-of-the-art prediction methods for binding of a peptide to a given MHC molecule. Van Allen et al. showed that out of 17 validated neoantigens, 14 passed the 500 nM standard threshold, indicating high sensitivity (van Buuren et al., 2014). However, only a handful of the predicted binders will also be recognized by T cells, which requires additional filtering or prediction improvement (Anonymous, 2017). Indeed, using more filtering criteria leads to fewer predicted neoepitopes per mutation, as seen in Figure 3A, although the false negative rate remains unknown. Only a few publications rely on predicting the binding affinity of mutated peptides alone and most use at least one additional threshold criterion, of which gene expression as a premise for antigen recognition is the most common. As RNA-Seq data was not available for Anagnostou et al., Le et al. and Reuben et al., they used TCGA expression data as a proxy to further filter the mutations to test for immunogenicity. Binding of the wild-type peptide was also considered by some studies, but not always used for filtering. Duan et al. proposed a “differential agretopicity index” (DAI), which is the difference between the predicted mutated and wild-type binding affinity, to use as a filtering criterion for neoepitope prediction. Although it yielded promising results based on their mouse data, it seemed less reliable in further investigations by Bjerregaard et al. and Koşaloğlu-Yalçın et al. using human data (Duan et al., 2014; Bjerregaard et al., 2017b; Koşaloğlu-Yalçın et al., 2018). In another study by Ghorani et al., DAI was more predictive for immune infiltration in melanoma and lung cancer compared to neoantigen or mutation load, suggesting that while some neoepitope responses might be enhanced by a reduced cross-reactivity potential, there are also many validated neoantigens whose wild-type counterparts are predicted to bind comparably strong (Ghorani et al., 2018; Koşaloğlu-Yalçın et al., 2018).
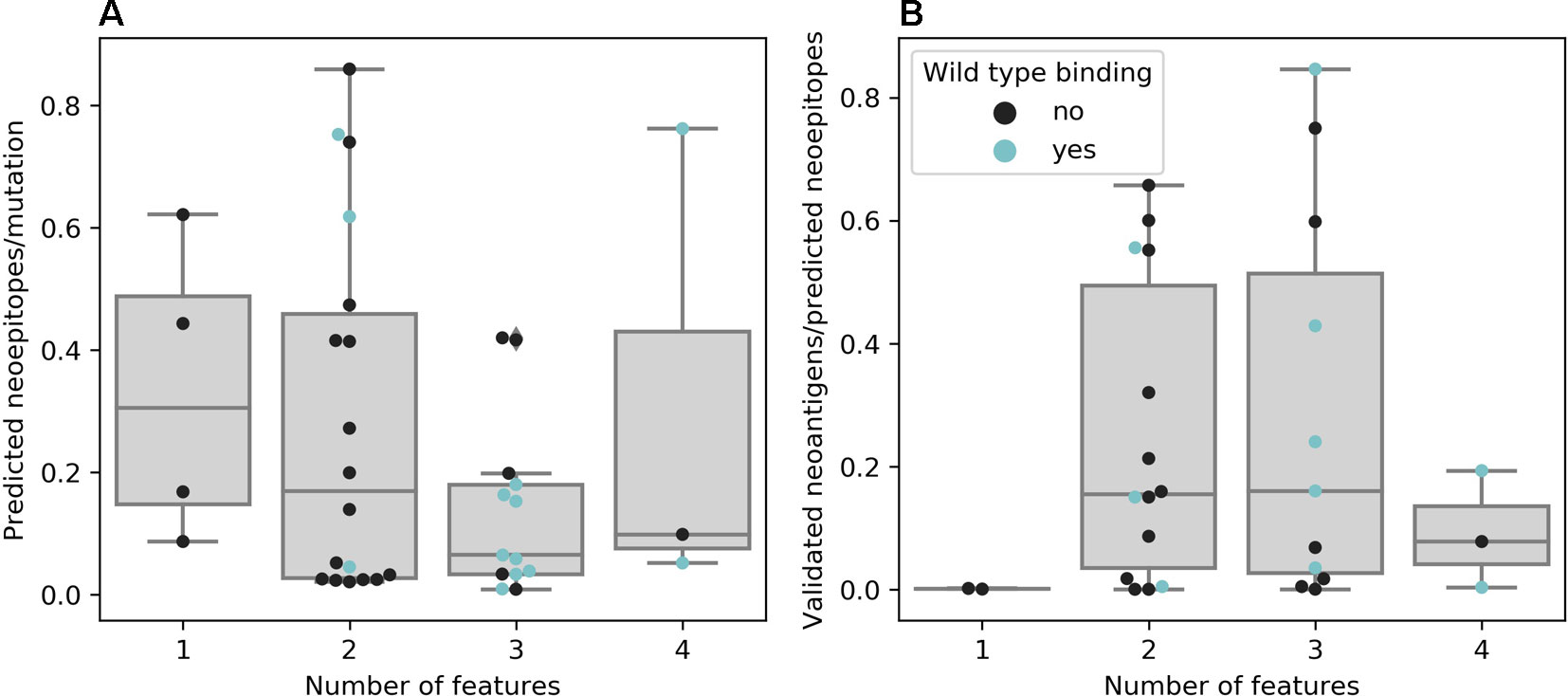
Figure 3 (A) Neoepitopes per mutation grouped by the number of features used for neoepitope selection. Data based on publications that offered comparable data, e.g., not obviously counting a neoepitope predicted to be presented by multiple major histocompatibility complexes (MHCs) multiple times (n = 38). (B) Ratio of confirmed to predicted neoepitopes grouped by the number of features used for neoepitope selection. Data based on publications that experimentally validated all predicted neoepitopes (n = 30)
There is evidence that taking more than one feature into account promises greater success for experimentally validating predicted neoepitopes (see Figure 3B). However, the results of experimental validation are dependent on the sensitivity of the technique used and the reactivity of neoantigen-specific TILs can additionally be hampered by other factors, such as tumor immune suppression or T cell exhaustion (Anonymous, 2017; Bulik-Sullivan et al., 2019).
Some studies chose a quantitative approach, mostly linking neoepitope load and survival (Brown et al., 2014; Rizvi et al., 2015; Miller et al., 2017; Ghorani et al., 2018). It has to be mentioned that neoepitope load and mutational burden are usually highly correlated (Pearson r = 0.89 based on 38 publications with less than 1 neoepitope per mutation from Table 1) and although it can be assumed that an increased survival is linked to the immunogenicity of mutations, quantifying predicted neoepitopes does not necessarily transport more information than mutation burden alone (Nathanson et al., 2017). There are, however, also studies that correlated survival with neoepitopes but not mutational burden or found contradictory results depending on patient cohorts (Snyder et al., 2014; Ghorani et al., 2018).
Among well-described approaches for neoepitope identification based on affinity binding prediction algorithms, there are also pipelines available that automate all analytic steps and rank potential neoepitopes based on peptide affinity prediction and other features (see Table 2). They differ greatly as to their properties and outputs, thus offering choices depending on research questions and dataset sizes. Their availability demonstrates how important neoepitope prediction has become as an application for binding affinity prediction algorithms.

Table 2 Neoepitope prediction pipelines based on mutation data input. Additional features are cancer driver status of the mutated gene used by MuPeXI; differential agretopicity index (DAI), sequence-based immunogenicity score, and more used by Neopepsee; DAI, cleavage, and stability prediction used by pVACtools.
Since a variety of different neoepitope identification approaches exist and it is not clear which features are predictive for immunogenicity, Koşaloğlu-Yalçın et al. and Kim et al. integrated and compared features additional to the standard MHC binding affinity by either comparing areas under the curve of receiver operating characteristics or evaluating feature importance derived from trained classifiers (Kim et al., 2018; Koşaloğlu-Yalçın et al., 2018). Both studies found that binding affinity prediction performs best or is the most informative feature. This is not surprising for viral epitopes constituting a major part of data on which most prediction algorithms are trained nor for neoantigens from literature mainly selected by predicted binding affinity, which introduces a bias toward this feature. It still remains unclear how many potential neoantigens are not detected because their binding affinity is predicted to lie beyond thresholds. An approach avoiding this bias has been proposed by Bulik-Sullivan et al. (Bulik-Sullivan et al., 2019). Like the most recent generation of neural network binding prediction algorithms, they developed a deep learning neural network trained on MS data, but apart from improved peptide sequence modeling, they also included features unrelated to the pMHC interaction, namely, quantified gene expression, flanking sequence, and protein family. Although their model is currently limited to HLA alleles of the training data, the approach demonstrated an increased performance of neoepitope discovery over peptide binding prediction and can also be expanded to MHC class II presented antigens.
Cross-Reactivity Assessment
A major challenge for immunotherapies introducing TCRs into patient recipient T cells is the choice of safe target antigens. If an engineered TCR-T cell cross-reacts with self-antigens in healthy tissue, the side-effects can be devastating. Possible TCR toxicity scenarios can be generally divided into on-target and off-target toxicities. On-target toxicities include all aspects of a specific target antigen or epitope expression that lead to an unintentional TCR-mediated destruction of healthy tissues. An example of on-target toxicity is melanocyte destruction, hearing loss, and retina infiltration mediated by MART1-targeting TCR-T cells relating to the same epitope in all cases (Johnson et al., 2009).
Off-target toxicities, in contrast, can appear by unexpected recognition of alternative epitopes that contain amino acid exchanges (mismatches) compared to the known epitope sequence. In rare cases, these mismatched peptides are presented identically on corresponding MHC molecules and are recognized equally well by deployed TCRs.
Targeting epitope sequences of proteins originating from highly homologous family members can cause unforeseen tissue damage as exemplified by the study performed by Morgan et al. (Morgan et al., 2013). Using autologous anti-MAGEA3 TCR-T cells, adoptive transfer led to severe neurotoxicity in several patients. The MAGEA3-specific TCR used in this clinical trial also recognized a MAGEA12, which was retrospectively found to be expressed in the brain. In the Linette et al. study, clinicians adoptively transferred MAGEA3-TCR-modified lymphocytes that also recognized an alternative epitope derived from the protein titin, causing fatal heart failure in two patients (Linette et al., 2013). Each of these examples underline the importance and need of comprehensive preclinical target and TCR analysis to prevent potential adverse events at later stages of clinical development.
With Expitope, we presented the first web server for assessing epitope sharing when evaluating new potential target candidates (Haase et al., 2015). Based on predictions for proteasomal cleavage, TAP transport, and MHC class I binding affinity, Expitope lists peptides with a given number of mismatches including the original target peptide. For these peptides, which are linked to genes by transcripts, the expression values in various healthy tissues, representing all vital human organs, are extracted from RNA-Seq data. However, transcript abundance only indirectly indicates protein expression. Meanwhile, proteome-wide human protein abundance data has become available and now facilitates a more direct approach for the prediction of potential cross-reactivity. The development of a new version 2.0 of Expitope, which computes all possible, naturally occurring epitopes of a peptide sequence and the corresponding cross-reactivity indices using both protein and transcript abundance levels weighted by a proposed hierarchy of importance of various human tissues, should help addressing this issue (Jaravine et al., 2017a). Cross-reactivity potential can also be assessed by calculating structural similarities between pMHC complexes obtained by molecular docking (Antunes et al., 2010) and by clustering pMHC complexes based on their electrostatic properties and the accessible surface area (Mendes et al., 2015). A comprehensive review by Baker et al. (2012) is covering these aspects in great detail.
TCR Binding Prediction
The final piece of the epitope recognition puzzle is the interaction of the pMHC complex with the TCR, which represents a very difficult problem for modeling studies and sequence-based predictions. One reason for that is the complex and noncontiguous nature of the interaction interface, with the CDR1 and CDR2 regions of the TCR α and β chains making contacts with the MHC class I molecule and the CDR3 regions directly interacting with the bound peptide (see Figure 4). Another major hurdle in predicting TCR recognition is the scarcity of experimentally confirmed TCR complementarity determining regions and the sequences of their respective binding partners on the pMHC complex. For example, one of the first feasibility studies of CDR3 sequence patterns was only based on two immunogenic HIV peptides (De Neuter et al., 2018). An additional complication is posed by the fact that repertoire sequencing combined with immune assays determines antigen-specific clonotypes, but does not yield negative controls, i.e., validated pairs of CDRs and pMHC complexes that do not bind each other.
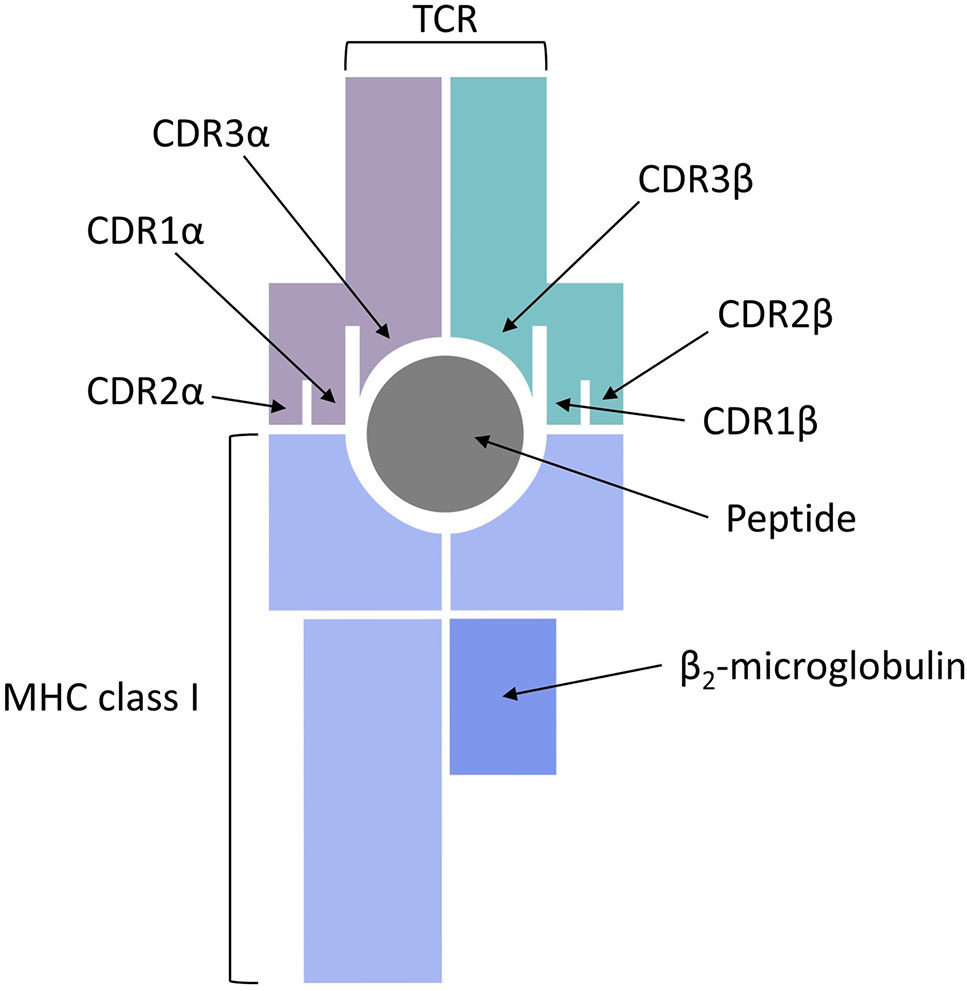
Figure 4 T cell receptor (TCR) binding to a peptide presented by major histocompatibility complex (MHC) class I.
CDR3β chains appear to always be in contact with the antigen bound to the MHC class I molecule, whereas the direct contact of CDR3α chains to the peptide is not always required (Glanville et al., 2017). The involvement of short linear stretches of CDR3β sequence in peptide-TCR interactions creates the opportunity to cluster TCRs in groups of common specificity (Dash et al., 2017; Glanville et al., 2017) and also serves as the basis for developing specialized algorithms for sequence-based prediction of pMHC/TCR binding. Two recent publications addressed this problem from two completely different perspectives. Jurtz et al. presented a proof of concept study, in which they predicted TCR interactions with their cognate HLA-A*02:01-presented peptide targets (Jurtz et al., 2018). A machine learning approach, called NetTCR, was trained on 8,920 TCRβ CDR3 sequences and 91 cognate peptide targets obtained from IEDB and from the immune assay data published by Klinger et al. (2015). A dataset of negative interactions was assembled by randomly matching TCR and peptide pairs. The NetTCR project in its current form is limited to a small number of peptides and it does not consider CDR1/CDR2 interactions with the MHC molecules or CDR3α sequences, but it is an important step forward because it demonstrates that TCR recognition of pMHCs is specific enough to be captured by sequence-level prediction tools.
Ogishi and Yotsuyanagi exploited the existence of immunodominant epitopes, which are targeted by the adaptive immune system in different individuals and would therefore be expected to exhibit some prominent features that make them especially prone to be recognized by T cells (Ogishi and Yotsuyanagi, 2019). The idea behind their repertoire-wide TCR-epitope contact potential profiling is that intermolecular contacts between relevant portions of the epitope and the TCR CDR3β region that closely resemble the contact structure of the interactions involving immunodominant peptides would be more likely to be immunogenic. To quantitatively assess the interaction affinity, they used physicochemical properties of amino acids and an energetic potential, calculated as the sum of all pairwise contact potentials for individual amino acids. The latter were obtained from several previously published amino acid contact potential scales, available from the AAINDEX database (Kawashima et al., 2007). These features were converted to immunogenicity scores using machine learning. It should be noted that the knowledge-based potentials, derived from crystal structures of proteins and protein complexes, reflect either intramolecular interactions driving protein folding and stability or contacts at protein interfaces and may only be a coarse approximation of peptide-TCR interactions. Yet, Ogishi and Yotsuyanagi demonstrated that the most informative contact-based and property-based features strongly correlate with experimentally measured TCR-peptide affinities.
Both approaches by Jurtz et al. and Ogishi and Yotsuyanagi are solely based on CDR3β chains and do not incorporate CDR3α sequence information. This is due to the fact that most datasets and databases such as IEDB and VDJdb did, until recently, consist mainly of CDR3β sequences (Figure 5) derived from bulk sequencing (Shugay et al., 2018; Vita et al., 2018), since identifying functional TCR pairing in repertoire data is technically challenging (Holec et al., 2018). Single cell sequencing eliminates this problem and a large dataset has just been added to VDJdb, which is, however, dominated by only few epitopes and HLA alleles. Another problem regarding TCR-epitope data is the lack of true negative datasets and the inclusion of cross-reactivity information, since many TCRs are able to recognize more than one epitope, which has been elaborated in section “Cross-reactivity assessment.” For this reason, pMHC/TCR binding prediction would also add valuable information to the detection of potential cross-reactivity for clinical candidate TCRs.
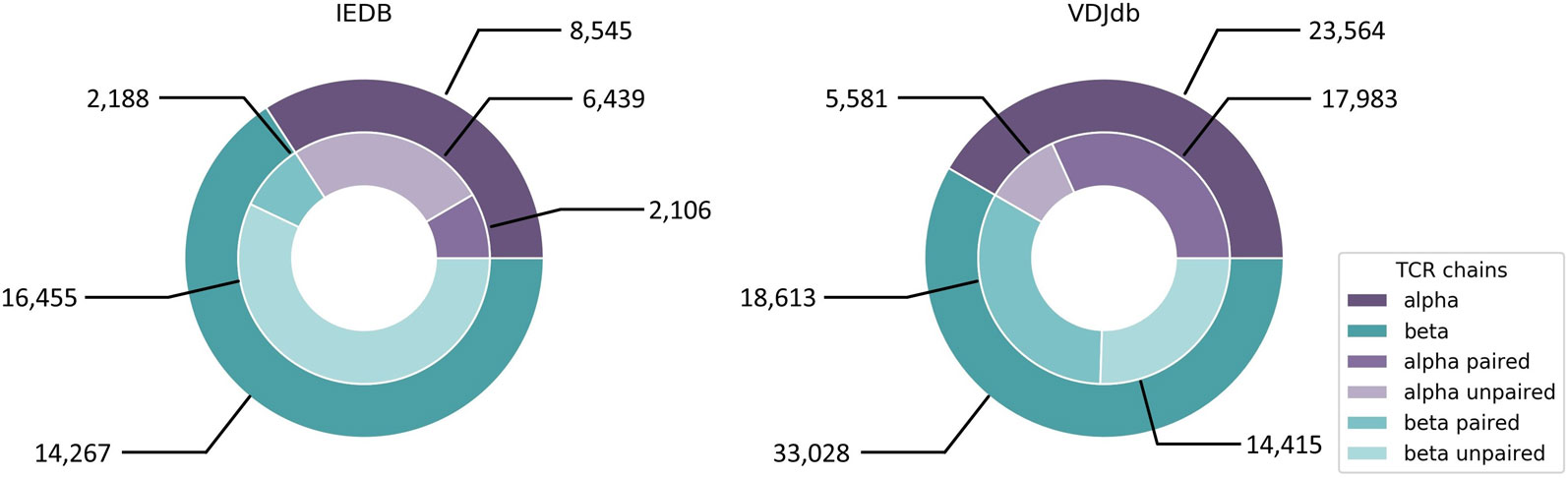
Figure 5 IEDB and VDJdb contents of CDR3α and CDR3β sequences of human origin. IEDB contains 386 unique epitopes linked to CDR3α sequences and 426 unique epitopes linked to CDR3β sequences. For VDJdb there are 93 and 177 unique epitopes, respectively. IEDB data was downloaded from https://www.iedb.org on September 30th, 2019 with the following query parameters: Current Filters: No B cell assays, No major histocompatibility complex (MHC) ligand assays, Restriction Type: Class I, Host: Homo sapiens (human). VDJdb data was taken from https://vdjdb.cdr3.net/overview (last updated on August 7th, 2019).
Further light on the details of pMHC/TCR interactions can be shed by molecular dynamics simulations. This entails understanding the role of hydrogen bonds, hydrophobic contacts, and interactions with the solvent in determining the specificity and cross-reactivity of each individual complex and proposing specific models of TCR engagement with the CDR1, CDR2, and CDR3 regions (Cuendet et al., 2011). Moreover, molecular modeling can help to compare the surface morphology between the complexed wild-type and mutated peptides and their relationship with immunogenicity (Park et al., 2013) and can also help to predict affinity-enhancing TCR mutations (Malecek et al., 2014). In cases where three-dimensional structures are not yet available, accurate models of pMHC/TCR complexes can be obtained by homology modeling (Zoete et al., 2013; Lanzarotti et al., 2019). Finally, a number of both rigid and flexible pMHC/TCR docking protocols have been proposed, which, in many cases, are able to produce accurate complex models starting from unbound structures (Pierce and Weng, 2013).
Conclusion and Outlook
Machine learning has become an indispensable tool for immunotherapeutic applications over the last decades. The established core method is peptide binding affinity prediction and thus target identification for TCR-T therapy or personalized neoantigen vaccination. The constant evolution of available training data as well as machine learning techniques, building on growing computational power, has improved the quality of binding affinity predictions. Focus has been on CD8+ cytotoxic T cells, but the substantial role of CD4+ T cells is increasingly gaining attention and efforts are made to also improve predictions for MHC class II presented epitopes, which poses a more challenging task compared to MHC class I binding due to the larger variety in peptide length and the open binding groove (Brown et al., 1993).
Additional challenges which can be tackled by machine learning remain. Immunogenicity is still an elusive aim for prediction tools, especially when it comes to personalized therapies relying on neoepitope identification. This is owed to the fact that patient immune systems and tumors undergo a process of mutual influence and therefore are highly individual and heterogeneous. The identification of features derived from the immune system that affect T cell recognition of individual epitopes within a tumor could be the key toward more reliable personalized immunotherapy predictions, thereby opening the process to a broader number of patients. Although neoantigens are currently in the focus of cancer immunotherapy, the detection of shared tumor antigens beyond coding DNA regions remains necessary since not all tumors harbor enough immunogenic mutations and the creation of potent TCRs for individual patients is currently impossible. Another challenge, which can be tackled with the help of ongoing data acquisition, is TCR binding prediction. Being able to reliably predict which TCR will recognize which epitope is extremely valuable not only for target epitope identification for TCR-T therapies, but also especially for TCR safety assessment, since it can speed up the process of selecting TCRs for the clinic by reducing in vitro screening of TCR candidates.
As the TCR-T adoptive immunotherapy community grows and data on the impact of sequence variations in both TCR alpha and beta chains on peptide fine specificity, sensitivity of peptide-MHC recognition and TCR cross-reactivity for partially mismatched epitopes emerge, artificial intelligence in the form of machine learning will be critical to advance understanding of pMHC/TCR interactions for many types of antigen and many different HLA allotypes. In particular, these issues will become additionally relevant as this form of immunotherapy is developed for patient populations worldwide. High-throughput TCR discovery platforms, yielding TCR sequence information from natural repertoires of T cells or through TCR mutational analyses, coupled with functional assessment of peptide variants as a means to assess cross-reactivity, offer many opportunities to continually improve understanding of pMHC/TCR interactions that will not only advance the cause of basic science but also help to meet medical needs for patients with cancer, infectious diseases or autoimmunity, where it is envisioned that TCR-Ts have the potential to provide improved therapies worldwide.
In particular, the push to couple TCR sequence data with neoantigen recognition for single patients through analysis of individual tumor samples in order to develop more potent cancer vaccines or TCR-T immunotherapies has already fostered strong collaborations and commercial endeavors to advance the interplay of machine learning and TCR recognition. While it currently seems daunting to imagine how the enormous and fast flow of information now emerging from many sources can be accessed and assembled to rapidly support the broader needs for personalized patient-individualized TCR-based immunotherapies, this review summarizes the challenges as well as the substantial progress that has already been achieved in defining some of the most relevant parameters in the complex cell biology of antigen processing and presentation and pMHC interactions with TCRs that lead to successful immune recognition. Important gaps have also been defined, alerting the community to the types of control data that may already exist in many laboratories, or could be collected, that would help in the refinement of prediction tools to achieve better results in the future. Increased interest and collaborative efforts of machine learning and HLA and TCR specialists will certainly foster further developments to support the rapidly expanding field of T cell-based immunotherapy of high medical relevance.
With the support of bioinformatic tools and improved prediction algorithms, immunotherapy holds the potential to become more precise, more personalized, and more effective than current cancer treatments—and potentially with fewer side effects.
Author Contributions
AM, SR, MW, DS, and DF all contributed to the writing and all approved the content of this review article.
Conflict of Interest
AM, SR, and MW are employees and DS is a Managing Director of Medigene Immunotherapies GmbH, a subsidiary of Medigene AG, Planegg, Germany.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abelin, J. G., Keskin, D. B., Sarkizova, S., Hartigan, C. R., Zhang, W., Sidney, J., et al. (2017). Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 46, 315–326. doi: 10.1016/j.immuni.2017.02.007
Álvaro-Benito, M., Morrison, E., Abualrous, E. T., Kuropka, B., Freund, C. (2018). Quantification of HLA-DM-dependent major histocompatibility complex of class II immunopeptidomes by the peptide landscape antigenic epitope alignment utility. Front. Immunol. 9, 872. doi: 10.3389/fimmu.2018.00872
Anagnostou, V., Smith, K. N., Forde, P. M., Niknafs, N., Bhattacharya, R., White, J., et al. (2017). Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discovery 7, 264–276. doi: 10.1158/2159-8290.CD-16-0828
Andreatta, M., Nielsen, M. (2016). Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517. doi: 10.1093/bioinformatics/btv639
Andreatta, M., Jurtz, V. I., Kaever, T., Sette, A., Peters, B., Nielsen, M. (2017). Machine learning reveals a non-canonical mode of peptide binding to MHC class II molecules. Immunology 152, 255–264. doi: 10.1111/imm.12763
Angelova, M., Charoentong, P., Hackl, H., Fischer, M. L., Snajder, R., Krogsdam, A. M., et al. (2015). Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 16, 64. doi: 10.1186/s13059-015-0620-6
Anonymous (2017) The problem with neoantigen prediction. Nat. Biotechnol. 35, 97–97. doi: 10.1038/nbt.3800
Antunes, D. A., Devaurs, D., Moll, M., Lizée, G., Kavraki, L. E. (2018). General Prediction of peptide-mhc binding modes using incremental docking: a proof of concept. Sci. Rep. 8, 4327. doi: 10.1038/s41598-018-22173-4
Antunes, D. A., Vieira, G. F., Rigo, M. M., Cibulski, S. P., Sinigaglia, M., Chies, J. A. B. (2010). Structural allele-specific patterns adopted by epitopes in the MHC-I cleft and reconstruction of mhc:peptide complexes to cross-reactivity assessment. PloS One 5, e10353. doi: 10.1371/journal.pone.0010353
Bai, Y., Wang, D., Fury, W., (2018). “PHLAT: Inference of high-resolution HLA types from RNA and whole exome sequencing,” in in HLA Typing. Ed. Boegel, S. ((New York, NY: Springer New York), 193–201. doi: 10.1007/978-1-4939-8546-3_13
Bailey, M. H., Tokheim, C., Porta-Pardo, E., Sengupta, S., Bertrand, D., Weerasinghe, A., et al. (2018). Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385.e18. doi: 10.1016/j.cell.2018.02.060
Bais, P., Namburi, S., Gatti, D. M., Zhang, X., Chuang, J. H. (2017). CloudNeo: a cloud pipeline for identifying patient-specific tumor neoantigens. Bioinformatics 33, 3110–3112. doi: 10.1093/bioinformatics/btx375
Baker, B. M., Scott, D. R., Blevins, S. J., Hawse, W. F. (2012). Structural and dynamic control of T-cell receptor specificity, cross-reactivity, and binding mechanism. Immunol Rev. 250, 10–31. doi: 10.1111/j.1600-065X.2012.01165.x
Bassani-Sternberg, M., Gfeller, D. (2016). Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide–HLA interactions. J. Immunol. 197, 2492–2499. doi: 10.4049/jimmunol.1600808
Bassani-Sternberg, M., Chong, C., Guillaume, P., Solleder, M., Pak, H., Gannon, P. O., et al. (2017). Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PloS Comput. Biol. 13, e1005725. doi: 10.1371/journal.pcbi.1005725
Bassani-Sternberg, M., Pletscher-Frankild, S., Jensen, L. J., Mann, M. (2015). Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics 14, 658–673. doi: 10.1074/mcp.M114.042812
Bhasin, M. (2004). Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci. 13, 596–607. doi: 10.1110/ps.03373104
Bilich, T., Nelde, A., Bichmann, L., Roerden, M., Salih, H. R., Kowalewski, D. J., et al. (2019). The HLA ligandome landscape of chronic myeloid leukemia delineates novel T-cell epitopes for immunotherapy. Blood 133, 550–565. doi: 10.1182/blood-2018-07-866830
Bisset, L. R., Fierz, W. (1993). Using a neural network to identify potential HLA-DR1 binding sites within proteins. J. Mol. Recognit. 6, 41–48. doi: 10.1002/jmr.300060105
Bjerregaard, A.-M., Nielsen, M., Hadrup, S. R., Szallasi, Z., Eklund, A. C. (2017a). MuPeXI: prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. 66 (9), 1123–1130 doi: 10.1007/s00262-017-2001-3
Bjerregaard, A.-M., Nielsen, M., Jurtz, V., Barra, C. M., Hadrup, S. R., Szallasi, Z., et al. (2017b). An analysis of natural t cell responses to predicted tumor neoepitopes. Front. Immunol. 8, 1566. doi: 10.3389/fimmu.2017.01566
Blankenstein, T., Leisegang, M., Uckert, W., Schreiber, H. (2015). Targeting cancer-specific mutations by T cell receptor gene therapy. Curr. Opin. Immunol. 33, 112–119. doi: 10.1016/j.coi.2015.02.005
Boegel, S., Löwer, M., Bukur, T., Sahin, U., Castle, J. C. (2014). A catalog of HLA type, HLA expression, and neo-epitope candidates in human cancer cell lines. OncoImmunology 3, e954893. doi: 10.4161/21624011.2014.954893
Boegel, S., Löwer, M., Schäfer, M., Bukur, T., de Graaf, J., Boisguérin, V., et al. (2012). HLA typing from RNA-Seq sequence reads. Genome Med. 4, 102. doi: 10.1186/gm403
Bordner, A. J., Abagyan, R. (2006). Ab initio prediction of peptide-MHC binding geometry for diverse class I MHC allotypes. Proteins: Struct Funct. Bioinf. 63, 512–526. doi: 10.1002/prot.20831
Brown, J. H., Jardetzky, T. S., Gorga, J. C., Stern, L. J., Urban, R. G., Strominger, J. L., et al. (1993). Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1 . Nature 364, 33–39. doi: 10.1038/364033a0
Brown, S. D., Warren, R. L., Gibb, E. A., Martin, S. D., Spinelli, J. J., Nelson, B. H., et al. (2014). Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 24, 743–750. doi: 10.1101/gr.165985.113
Brusic, V., Rudy, G., Harrison, L. C. (1994). MHCPEP: a database of MHC-binding peptides. Nucleic Acids Res. 22, 3663–3665. doi: 10.1093/nar/22.17.3663
Bulik-Sullivan, B., Busby, J., Palmer, C. D., Davis, M. J., Murphy, T., Clark, A., et al. (2019). Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 37, 55–63. doi: 10.1038/nbt.4313
Calis, J. J. A., Maybeno, M., Greenbaum, J. A., Weiskopf, D., De Silva, A. D., Sette, A., et al. (2013). Properties of MHC class I presented peptides that enhance immunogenicity. PloS Comput. Biol. 9, e1003266. doi: 10.1371/journal.pcbi.1003266
Caron, E., Vincent, K., Fortier, M.-H., Laverdure, J.-P., Bramoulle, A., Hardy, M.-P., et al. (2011). The MHC I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Mol. Syst. Biol. 7, 533–533. doi: 10.1038/msb.2011.68
Carreno, B. M., Magrini, V., Becker-Hapak, M., Kaabinejadian, S., Hundal, J., Petti, A. A., et al. (2015). A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808. doi: 10.1126/science.aaa3828
Castle, J. C., Kreiter, S., Diekmann, J., Lower, M., de Roemer, N., de Graaf, J., et al. (2012). Exploiting the mutanome for tumor vaccination. Cancer Res. 72, 1081–1091. doi: 10.1158/0008-5472.CAN-11-3722
Chang, T.-C., Carter, R. A., Li, Y., Li, Y., Wang, H., Edmonson, M. N., et al. (2017). The neoepitope landscape in pediatric cancers. Genome Med. 9, 78. doi: 10.1186/s13073-017-0468-3
Charoentong, P., Finotello, F., Angelova, M., Mayer, C., Efremova, M., Rieder, D., et al. (2017). Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262. doi: 10.1016/j.celrep.2016.12.019
Cohen, C. J., Gartner, J. J., Horovitz-Fried, M., Shamalov, K., Trebska-McGowan, K., Bliskovsky, V. V., et al. (2015). Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Invest. 125, 3981–3991. doi: 10.1172/JCI82416
Cuendet, M. A., Zoete, V., Michielin, O. (2011). How T cell receptors interact with peptide-MHCs: A multiple steered molecular dynamics study. Proteins: Struct Funct. Bioinf. 79, 3007–3024. doi: 10.1002/prot.23104
Daniel, S., Brusic, V., Caillat-Zucman, S., Petrovsky, N., Harrison, L., Riganelli, D., et al. (1998). Relationship between peptide selectivities of human transporters associated with antigen processing and HLA class I molecules. J. Immunol. 161, 617–624.
Dash, P., Fiore-Gartland, A. J., Hertz, T., Wang, G. C., Sharma, S., Souquette, A., et al. (2017). Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93. doi: 10.1038/nature22383
De Neuter, N., Bittremieux, W., Beirnaert, C., Cuypers, B., Mrzic, A., Moris, P., et al. (2018). On the feasibility of mining CD8+ T cell receptor patterns underlying immunogenic peptide recognition. Immunogenetics. 70 (3), 159–168 doi: 10.1007/s00251-017-1023-5
Dhanda, S. K., Mahajan, S., Paul, S., Yan, Z., Kim, H., Jespersen, M. C., et al. (2019). IEDB-AR: immune epitope database—analysis resource in 2019 . Nucleic Acids Res. 47, W502–W506. doi: 10.1093/nar/gkz452
Diez-Rivero, C. M., Chenlo, B., Zuluaga, P., Reche, P. A. (2010). Quantitative modeling of peptide binding to TAP using support vector machine. Proteins: Struct Funct. Bioinf. 78, 63–72. doi: 10.1002/prot.22535
Dolan, B. P. (2019). “Quantitating MHC Class I ligand production and presentation using TCR-like antibodies,” in in Antigen Processing. Ed. van Endert, P. ((New York, NY: Springer New York), 149–157. doi: 10.1007/978-1-4939-9450-2_12
Doytchinova, I. A., Guan, P., Flower, D. R. (2006). EpiJen: a server for multistep T cell epitope prediction. BMC Bioinf. 7, 131. doi: 10.1186/1471-2105-7-131
Duan, F., Duitama, J., Al Seesi, S., Ayres, C. M., Corcelli, S. A., Pawashe, A. P., et al. (2014). Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med. 211, 2231–2248. doi: 10.1084/jem.20141308
Ellrott, K., Bailey, M. H., Saksena, G., Covington, K. R., Kandoth, C., Stewart, C., et al. (2018). Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e7. doi: 10.1016/j.cels.2018.03.002
Erhard, F., Halenius, A., Zimmermann, C., L’Hernault, A., Kowalewski, D. J., Weekes, M. P., et al. (2018). Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods 15, 363–366. doi: 10.1038/nmeth.4631
Falk, K., Rötzschke, O., Stevanovié, S., Jung, G., Rammensee, H.-G. (1991). Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351, 290–296. doi: 10.1038/351290a0
Fleri, W., Paul, S., Dhanda, S. K., Mahajan, S., Xu, X., Peters, B., et al. (2017). The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 8, 278. doi: 10.3389/fimmu.2017.00278
Freudenmann, L. K., Marcu, A., Stevanović, S. (2018). Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology 154, 331–345. doi: 10.1111/imm.12936
Garde, C., Ramarathinam, S. H., Jappe, E. C., Nielsen, M., Kringelum, J. V., Trolle, T., et al. (2019). Improved peptide-MHC class II interaction prediction through integration of eluted ligand and peptide affinity data. Immunogenetics. 71 (7), 445–454. doi: 10.1007/s00251-019-01122-z
Gfeller, D., Bassani-Sternberg, M. (2018). Predicting antigen presentation—what could we learn from a million peptides? Front. Immunol. 9, 1716. doi: 10.3389/fimmu.2018.01716
Gfeller, D., Guillaume, P., Michaux, J., Pak, H.-S., Daniel, R. T., Racle, J., et al. (2018). The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol. 201, 3705–3716. doi: 10.4049/jimmunol.1800914
Ghorani, E., Rosenthal, R., McGranahan, N., Reading, J. L., Lynch, M., Peggs, K. S., et al. (2018). Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Ann. Oncol. 29, 271–279. doi: 10.1093/annonc/mdx687
Giam, K., Ayala-Perez, R., Illing, P. T., Schittenhelm, R. B., Croft, N. P., Purcell, A. W., et al. (2015). A comprehensive analysis of peptides presented by HLA-A1: A comprehensive analysis of peptides. Tissue Antigens 85, 492–496. doi: 10.1111/tan.12565
Glanville, J., Huang, H., Nau, A., Hatton, O., Wagar, L. E., Rubelt, F., et al. (2017). Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98. doi: 10.1038/nature22976
Goh, G., Walradt, T., Markarov, V., Blom, A., Riaz, N., Doumani, R., et al. (2016). Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 7, 3403–3415. doi: 10.18632/oncotarget.6494
Griffin, T. A., Nandi, D., Cruz, M., Fehling, H. J., Kaer, L. V., Monaco, J. J., et al. (1998). Immunoproteasome assembly: cooperative incorporation of interferon γ (IFN-γ)-inducible subunits. J. Exp. Med. 187, 97–104. doi: 10.1084/jem.187.1.97
Gros, A., Parkhurst, M. R., Tran, E., Pasetto, A., Robbins, P. F., Ilyas, S., et al. (2016). Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 22, 433–438. doi: 10.1038/nm.4051
Gubin, M. M., Zhang, X., Schuster, H., Caron, E., Ward, J. P., Noguchi, T., et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581. doi: 10.1038/nature13988
Gubler, B., Daniel, S., Armandola, E. A., Hammer, J., Caillat-Zucman, S., van Endert, P. M. (1998). Substrate selection by transporters associated with antigen processing occurs during peptide binding to TAP. Mol. Immunol. 35, 427–433. doi: 10.1016/s0161-5890(98)00059-5
Haase, K., Raffegerst, S., Schendel, D. J., Frishman, D. (2015). Expitope: a web server for epitope expression. Bioinformatics 31, 1854–1856. doi: 10.1093/bioinformatics/btv068
Hassan, C., Kester, M. G. D., Oudgenoeg, G., de Ru, A. H., Janssen, G. M. C., Drijfhout, J. W., et al. (2014). Accurate quantitation of MHC-bound peptides by application of isotopically labeled peptide MHC complexes. J. Proteomics 109, 240–244. doi: 10.1016/j.jprot.2014.07.009
Hilf, N., Kuttruff-Coqui, S., Frenzel, K., Bukur, V., Stevanović, S., Gouttefangeas, C., et al. (2019). Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565, 240–245. doi: 10.1038/s41586-018-0810-y
Holec, P. V., Berleant, J., Bathe, M., Birnbaum, M. E. (2018). A Bayesian framework for high-throughput T cell receptor pairing. Bioinformatics. 35 (8), 1318–1325. doi: 10.1093/bioinformatics/bty801
Hu, Y., Wang, Z., Hu, H., Wan, F., Chen, L., Yuanpeng, X., et al. (2019). ACME: Pan-specific peptide-MHC class I binding prediction through attention-based deep neural networks. Bioinformatics. doi: 10.1093/bioinformatics/btz427
Hugo, W., Zaretsky, J. M., Sun, L., Song, C., Moreno, B. H., Hu-Lieskovan, S., et al. (2016). Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44. doi: 10.1016/j.cell.2016.02.065
Hundal, J., Kiwala, S., McMichael, J., Miller, C. A., Wollam, A. T., Xia, H., et al. (2019). pVACtools: a computational toolkit to identify and visualize cancer neoantigens. bioRxiv. doi: 10.1101/501817
Hunt, D. F., Henderson, R. A., Shabanowitz, J., Sakaguchi, K., Michel, H., Sevilir, N., et al. (1992). Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 255, 1261–1263. doi: 10.1126/science.1546328
Jaravine, V., Mösch, A., Raffegerst, S., Schendel, D. J., Frishman, D. (2017a). Expitope 2.0: a tool to assess immunotherapeutic antigens for their potential cross-reactivity against naturally expressed proteins in human tissues. BMC Cancer 17, 892. doi: 10.1186/s12885-017-3854-8
Jaravine, V., Raffegerst, S., Schendel, D. J., Frishman, D. (2017b). Assessment of cancer and virus antigens for cross-reactivity in human tissues. Bioinformatics 33, 104–111. doi: 10.1093/bioinformatics/btw567
Jensen, K. K., Andreatta, M., Marcatili, P., Buus, S., Greenbaum, J. A., Yan, Z., et al. (2018). Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154, 394–406. doi: 10.1111/imm.12889
Johnson, L. A., Morgan, R. A., Dudley, M. E., Cassard, L., Yang, J. C., Hughes, M. S., et al. (2009). Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546. doi: 10.1182/blood-2009-03-211714
Jørgensen, K. W., Rasmussen, M., Buus, S., Nielsen, M. (2014). NetMHCstab - predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology 141, 18–26. doi: 10.1111/imm.12160
Jurtz, V. I., Jessen, L. E., Bentzen, A. K., Jespersen, M. C., Mahajan, S., Vita, R., et al. (2018). NetTCR: sequence-based prediction of TCR binding to peptide-MHC complexes using convolutional neural networks. doi:10.1101/433706
Jurtz, V., Paul, S., Andreatta, M., Marcatili, P., Peters, B., Nielsen, M. (2017). NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368. doi: 10.4049/jimmunol.1700893
Kahles, A., Lehmann, K.-V., Toussaint, N. C., Hüser, M., Stark, S. G., Sachsenberg, T., et al. (2018). Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 34, 211–224.e6. doi: 10.1016/j.ccell.2018.07.001
Kalaora, S., Barnea, E., Merhavi-Shoham, E., Qutob, N., Teer, J. K., Shimony, N., et al. (2016). Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 7, 5110–5117. doi: 10.18632/oncotarget.6960
Kalaora, S., Wolf, Y., Feferman, T., Barnea, E., Greenstein, E., Reshef, D., et al. (2018). Combined analysis of antigen presentation and t-cell recognition reveals restricted immune responses in melanoma. Cancer Discovery 8, 1366–1375. doi: 10.1158/2159-8290.CD-17-1418
Karasaki, T., Nagayama, K., Kawashima, M., Hiyama, N., Murayama, T., Kuwano, H., et al. (2016). Identification of individual cancer-specific somatic mutations for neoantigen-based immunotherapy of lung cancer. J. Thoracic Oncol. 11, 324–333. doi: 10.1016/j.jtho.2015.11.006
Karasaki, T., Nagayama, K., Kuwano, H., Nitadori, J., Sato, M., Anraku, M., et al. (2017). Prediction and prioritization of neoantigens: integration of RNA sequencing data with whole-exome sequencing. Cancer Sci. 108, 170–177. doi: 10.1111/cas.13131
Karosiene, E., Lundegaard, C., Lund, O., Nielsen, M. (2012). NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64, 177–186. doi: 10.1007/s00251-011-0579-8
Kato, T., Park, J.-H., Kiyotani, K., Ikeda, Y., Miyoshi, Y., Nakamura, Y. (2017). Integrated analysis of somatic mutations and immune microenvironment of multiple regions in breast cancers. Oncotarget 8, 62029–62038. doi: 10.18632/oncotarget.18790
Kawashima, S., Pokarowski, P., Pokarowska, M., Kolinski, A., Katayama, T., Kanehisa, M. (2007). AAindex: amino acid index database, progress report 2008 . Nucleic Acids Res. 36, D202–D205. doi: 10.1093/nar/gkm998
Keskin, D. B., Anandappa, A. J., Sun, J., Tirosh, I., Mathewson, N. D., Li, S., et al. (2019). Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239. doi: 10.1038/s41586-018-0792-9
Keşmir, C., Nussbaum, A. K., Schild, H., Detours, V., Brunak, S. (2002). Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 15, 287–296. doi: 10.1093/protein/15.4.287
Khalili, J. S., Hanson, R. W., Szallasi, Z. (2012). In silico prediction of tumor antigens derived from functional missense mutations of the cancer gene census. OncoImmunology 1, 1281–1289. doi: 10.4161/onci.21511
Kim, S., Kim, H. S., Kim, E., Lee, M. G., Shin, E., Paik, S., et al. (2018). Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 29 (4), 1030–1036. doi: 10.1093/annonc/mdy022
Kinkead, H. L., Hopkins, A., Lutz, E., Wu, A. A., Yarchoan, M., Cruz, K., et al. (2018). Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 3, e122857. doi: 10.1172/jci.insight.122857
Klausen, M. S., Anderson, M. V., Jespersen, M. C., Nielsen, M., Marcatili, P. (2015). LYRA, a webserver for lymphocyte receptor structural modeling. Nucleic Acids Res. 43, W349–W355. doi: 10.1093/nar/gkv535
Klinger, M., Pepin, F., Wilkins, J., Asbury, T., Wittkop, T., Zheng, J., et al. (2015). Multiplex identification of antigen-specific T cell receptors using a combination of immune assays and immune receptor sequencing. PloS One 10, e0141561. doi: 10.1371/journal.pone.0141561
Koşaloğlu-Yalçın, Z., Lanka, M., Frentzen, A., Logandha Ramamoorthy Premlal, A., Sidney, J., Vaughan, K., et al. (2018). Predicting T cell recognition of MHC class I restricted neoepitopes. OncoImmunology 7, e1492508. doi: 10.1080/2162402X.2018.1492508
Koster, J., Plasterk, R. H. A. (2019). A library of Neo Open Reading Frame peptides (NOPs) as a sustainable resource of common neoantigens in up to 50% of cancer patients. Sci. Rep. 9, 6577. doi: 10.1038/s41598-019-42729-2
Kreiter, S., Vormehr, M., de Roemer, N., Diken, M., Löwer, M., Diekmann, J., et al. (2015). Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696. doi: 10.1038/nature14426
Kuttler, C., Nussbaum, A. K., Dick, T. P., Rammensee, H.-G., Schild, H., Hadeler, K.-P. (2000). An algorithm for the prediction of proteasomal cleavages. J. Mol. Biol. 298, 417–429. doi: 10.1006/jmbi.2000.3683
Kyeong, H.-H., Choi, Y., Kim, H.-S. (2018). GradDock: rapid simulation and tailored ranking functions for peptide-MHC Class I docking. Bioinformatics 34, 469–476. doi: 10.1093/bioinformatics/btx589
Lam, T., Mamitsuka, H., Ren, E., Tong, J. (2010). TAP Hunter: a SVM-based system for predicting TAP ligands using local description of amino acid sequence. Immunome Res. 6, S6. doi: 10.1186/1745-7580-6-S1-S6
Lanzarotti, E., Marcatili, P., Nielsen, M. (2019). T-cell receptor cognate target prediction based on paired α and β chain sequence and structural CDR loop similarities. Front. Immunol. 10, 2080. doi: 10.3389/fimmu.2019.02080
Larsen, M. V., Lundegaard, C., Lamberth, K., Buus, S., Lund, O., Nielsen, M. (2007). Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinf. 8, 424. doi: 10.1186/1471-2105-8-424
Laumont, C. M., Perreault, C. (2017). Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell. Mol. Life Sci. 75 (4), 607–621. doi: 10.1007/s00018-017-2628-4
Laumont, C. M., Vincent, K., Hesnard, L., Audemard, É., Bonneil, É., Laverdure, J.-P., et al. (2018). Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Trans. Med. 10, eaau5516. doi: 10.1126/scitranslmed.aau5516
Lin, H., Ray, S., Tongchusak, S., Reinherz, E. L., Brusic, V. (2008). Evaluation of MHC class I peptide binding prediction servers: Applications for vaccine research. BMC Immunol. 9, 8. doi: 10.1186/1471-2172-9-8
Linette, G. P., Stadtmauer, E. A., Maus, M. V., Rapoport, A. P., Levine, B. L., Emery, L., et al. (2013). Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871. doi: 10.1182/blood-2013-03-490565
Linnemann, C., van Buuren, M. M., Bies, L., Verdegaal, E. M. E., Schotte, R., Calis, J. J. A., et al. (2014). High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 21, 81–85. doi: 10.1038/nm.3773
Liu, C., Yang, X., Duffy, B., Mohanakumar, T., Mitra, R. D., Zody, M. C., et al. (2013). ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res. 41, e142–e142. doi: 10.1093/nar/gkt481
Liu, G., Li, D., Li, Z., Qiu, S., Li, W., Chao, C., et al. (2017). PSSMHCpan: a novel PSSM-based software for predicting class I peptide-HLA binding affinity. GigaScience 6, 1–11. doi: 10.1093/gigascience/gix017
Liu, S., Matsuzaki, J., Wei, L., Tsuji, T., Battaglia, S., Hu, Q., et al. (2019). Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J. Immuno Ther Cancer 7, 156. doi: 10.1186/s40425-019-0629-6
Löffler, M. W., Chandran, P. A., Laske, K., Schroeder, C., Bonzheim, I., Walzer, M., et al. (2016). Personalized peptide vaccine-induced immune response associated with long-term survival of a metastatic cholangiocarcinoma patient. J. Hepatol 65, 849–855. doi: 10.1016/j.jhep.2016.06.027
Löffler, M. W., HEPVAC Consortium Mohr, C., Bichmann, L., Freudenmann, L. K., Walzer, M., et al. (2019). Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 11, 28. doi: 10.1186/s13073-019-0636-8
Łuksza, M., Riaz, N., Makarov, V., Balachandran, V. P., Hellmann, M. D., Solovyov, A., et al. (2017). A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551 (7681), 517–520. doi: 10.1038/nature24473
Lundegaard, C., Lund, O., Nielsen, M. (2008). Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 24, 1397–1398. doi: 10.1093/bioinformatics/btn128
Madden, D. R. (1995). The three-dimensional structure of peptide-MHC complexes. Annu. Rev. Immunol. 13, 587–622. doi: 10.1146/annurev.iy.13.040195.003103
Malecek, K., Grigoryan, A., Zhong, S., Gu, W. J., Johnson, L. A., Rosenberg, S. A., et al. (2014). Specific Increase in Potency via Structure-Based Design of a TCR. J. Immunol. 193, 2587–2599. doi: 10.4049/jimmunol.1302344
Mamitsuka, H. (1998). Predicting peptides that bind to MHC molecules using supervised learning of hidden Markov models. Proteins 33, 460–474. doi: 10.1002/(sici)1097-0134(19981201)33:4<460::aid-prot2>3.0.co;2-m
Marino, F., Chong, C., Michaux, J., Bassani-Sternberg, M., (2019). “High-throughput, fast, and sensitive immunopeptidomics sample processing for mass spectrometry,” in in Immune Checkpoint Blockade. Ed. Pico de Coaña, Y. (New York, NY: Springer New York), 67–79. doi: 10.1007/978-1-4939-8979-9_5
Martin, S. D., Wick, D. A., Nielsen, J. S., Little, N., Holt, R. A., Nelson, B. H. (2018). A library-based screening method identifies neoantigen-reactive T cells in peripheral blood prior to relapse of ovarian cancer. OncoImmunology 7, e1371895. doi: 10.1080/2162402X.2017.1371895
Marty, R., Kaabinejadian, S., Rossell, D., Slifker, M. J., de Haar, J., Engin, H. B., et al. (2017). MHC-I genotype restricts the oncogenic mutational landscape. Cell 171, 1272–1283.e15. doi: 10.1016/j.cell.2017.09.050
McGranahan, N., Furness, A. J. S., Rosenthal, R., Ramskov, S., Lyngaa, R., Saini, S. K., et al. (2016). Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469. doi: 10.1126/science.aaf1490
McGranahan, N., Rosenthal, R., Hiley, C. T., Rowan, A. J., Watkins, T. B. K., Wilson, G. A., et al. (2017). Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171, 1259–1271.e11. doi: 10.1016/j.cell.2017.10.001
Mendes, M. F. A., Antunes, D. A., Rigo, M. M., Sinigaglia, M., Vieira, G. F. (2015). Improved structural method for T-cell cross-reactivity prediction. Mol. Immunol. 67, 303–310. doi: 10.1016/j.molimm.2015.06.017
Menegatti Rigo, M., Amaral Antunes, D., Vaz de Freitas, M., Fabiano de Almeida Mendes, M., Meira, L., Sinigaglia, M., et al. (2015). DockTope: a web-based tool for automated pMHC-I modelling. Sci. Rep. 5, 18413. doi: 10.1038/srep18413
Miller, A., Asmann, Y., Cattaneo, L., Braggio, E., Keats, J., Auclair, D., et al. (2017). High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 7, e612. doi: 10.1038/bcj.2017.94
Morgan, R. A., Chinnasamy, N., Abate-Daga, D., Gros, A., Robbins, P. F., Zheng, Z., et al. (2013). Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immuno ther 36, 133–151. doi: 10.1097/CJI.0b013e3182829903
Moutaftsi, M., Peters, B., Pasquetto, V., Tscharke, D. C., Sidney, J., Bui, H.-H., et al. (2006). A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to vaccinia virus. Nat. Biotechnol. 24, 817–819. doi: 10.1038/nbt1215
Nathanson, T., Ahuja, A., Rubinsteyn, A., Aksoy, B. A., Hellmann, M. D., Miao, D., et al. (2017). Somatic mutations and neoepitope homology in melanomas treated with CTLA-4 blockade. Cancer Immunol. Res. 5, 84–91. doi: 10.1158/2326-6066.CIR-16-0019
Neefjes, J., Jongsma, M. L. M., Paul, P., Bakke, O. (2011). Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11 (12), 823–36. doi: 10.1038/nri3084
Nielsen, M., Andreatta, M. (2017). NNAlign: a platform to construct and evaluate artificial neural network models of receptor–ligand interactions. Nucleic Acids Res. 45, W344–W349. doi: 10.1093/nar/gkx276
Nielsen, M., Justesen, S., Lund, O., Lundegaard, C., Buus, S. (2010). NetMHCIIpan-2.0 - Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 6, 9. doi: 10.1186/1745-7580-6-9
Nielsen, M., Lundegaard, C., Lund, O. (2007). Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinf. 8, 238 . doi: 10.1186/1471-2105-8-238
Nielsen, M., Lundegaard, C., Lund, O., Keşmir, C. (2005). The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 57, 33–41. doi: 10.1007/s00251-005-0781-7
Nielsen, M., Lundegaard, C., Worning, P., Lauemøller, S. L., Lamberth, K., Buus, S., et al. (2003). Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017. doi: 10.1110/ps.0239403
O’Donnell, T. J., Rubinsteyn, A., Bonsack, M., Riemer, A. B., Laserson, U., Hammerbacher, J. (2018b). MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 7 (1), 129–132.e4. doi: 10.1016/j.cels.2018.05.014
O’Donnell, T., Christie, E. L., Ahuja, A., Buros, J., Aksoy, B. A., Bowtell, D. D. L., et al. (2018a). Chemotherapy weakly contributes to predicted neoantigen expression in ovarian cancer. BMC Cancer 18, 87. doi: 10.1186/s12885-017-3825-0
Ogishi, M., Yotsuyanagi, H. (2019). Quantitative prediction of the landscape of T cell epitope immunogenicity in sequence space. Front. Immunol. 10, 827. doi: 10.3389/fimmu.2019.00827
Ott, P. A., Hu, Z., Keskin, D. B., Shukla, S. A., Sun, J., Bozym, D. J., et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. doi: 10.1038/nature22991
Park, M.-S., Park, S. Y., Miller, K. R., Collins, E. J., Lee, H. Y. (2013). Accurate structure prediction of peptide–MHC complexes for identifying highly immunogenic antigens. Mol. Immunol. 56, 81–90. doi: 10.1016/j.molimm.2013.04.011
Parker, K. C., Bednarek, M. A., Coligan, J. E. (1994). Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 152, 163–175.
Peters, B., Bui, H.-H., Frankild, S., Nielson, M., Lundegaard, C., Kostem, E., et al. (2006). A community resource benchmarking predictions of peptide binding to MHC-I molecules. PloS Comput. Biol. 2, e65. doi: 10.1371/journal.pcbi.0020065
Peters, B., Bulik, S., Tampe, R., van Endert, P. M., Holzhutter, H.-G. (2003). Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. 171, 1741–1749. doi: 10.4049/jimmunol.171.4.1741
Pierce, B. G., Weng, Z. (2013). A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci. 22, 35–46. doi: 10.1002/pro.2181
Rajasagi, M., Shukla, S. A., Fritsch, E. F., Keskin, D. B., DeLuca, D., Carmona, E., et al. (2014). Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 124, 453–462. doi: 10.1182/blood-2014-04-567933
Rammensee, H. G., Falk, K., Rötzschke, O. (1993). Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol. 11, 213–244. doi: 10.1146/annurev.iy.11.040193.001241
Rammensee, H. G., Friede, T., Stevanoviíc, S. (1995). MHC ligands and peptide motifs: first listing. Immunogenetics 41, 178–228. doi: 10.1007/bf00172063
Rammensee, H., Bachmann, J., Emmerich, N. P., Bachor, O. A., Stevanović, S. (1999). SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50, 213–219. doi: 10.1007/s002510050595
Rasmussen, M., Fenoy, E., Harndahl, M., Kristensen, A. B., Nielsen, I. K., Nielsen, M., et al. (2016). Pan-specific prediction of peptide-MHC class I complex stability, a correlate of T cell immunogenicity. J. Immunol. 197, 1517–1524. doi: 10.4049/jimmunol.1600582
Reche, P. A., Glutting, J.-P., Reinherz, E. L. (2002). Prediction of MHC class I binding peptides using profile motifs. Hum. Immunol. 63, 701–709. doi: 10.1016/s0198-8859(02)00432-9
Reuben, A., Gittelman, R., Gao, J., Zhang, J., Yusko, E. C., Wu, C.-J., et al. (2017). TCR Repertoire intratumor heterogeneity in localized lung adenocarcinomas: an association with predicted neoantigen heterogeneity and postsurgical recurrence. Cancer Discovery 7, 1088–1097. doi: 10.1158/2159-8290.CD-17-0256
Ritz, D., Gloger, A., Neri, D., Fugmann, T. (2017). Purification of soluble HLA class I complexes from human serum or plasma deliver high quality immuno peptidomes required for biomarker discovery. PROTEOMICS 17, 1600364. doi: 10.1002/pmic.201600364
Ritz, D., Gloger, A., Weide, B., Garbe, C., Neri, D., Fugmann, T. (2016). High-sensitivity HLA class I peptidome analysis enables a precise definition of peptide motifs and the identification of peptides from cell lines and patients’ sera. PROTEOMICS 16, 1570–1580. doi: 10.1002/pmic.201500445
Rizvi, N. A., Hellmann, M. D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J. J., et al. (2015). Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. doi: 10.1126/science.aaa1348
Robbins, P. F., Lu, Y.-C., El-Gamil, M., Li, Y. F., Gross, C., Gartner, J., et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 19, 747–752. doi: 10.1038/nm.3161
Robinson, J., Halliwell, J. A., Hayhurst, J. D., Flicek, P., Parham, P., Marsh, S. G. E. (2015). The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 43, D423–D431. doi: 10.1093/nar/gku1161
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G., Hacohen, N. (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. doi: 10.1016/j.cell.2014.12.033
Rosenthal, R., Cadieux, E. L., Salgado, R., Moore, D. A., Lund, T., Tanić, M., et al. (2019). Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485. doi: 10.1038/s41586-019-1032-7
Rothbard, J. B., Taylor, W. R. (1988). A sequence pattern common to T cell epitopes. EMBO J. 7, 93–100. doi: 10.1002/j.1460-2075.1988.tb02787.x
Sadelain, M., Brentjens, R., Rivière, I. (2013). The basic principles of chimeric antigen receptor design. Cancer Discovery 3, 388–398. doi: 10.1158/2159-8290.CD-12-0548
Sahin, U., Derhovanessian, E., Miller, M., Kloke, B.-P., Simon, P., Löwer, M., et al. (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226. doi: 10.1038/nature23003
Schischlik, F., Jäger, R., Rosebrock, F., Hug, E., Schuster, M. K., Holly, R., et al. (2019). Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood, blood. 2019000519. doi:10.1182/blood.2019000519
Schubert, B., Walzer, M., Brachvogel, H.-P., Szolek, A., Mohr, C., Kohlbacher, O. (2016). FRED 2: an immunoinformatics framework for Python. Bioinformatics 32, 2044–2046. doi: 10.1093/bioinformatics/btw113
Schumacher, T. N., Schreiber, R. D. (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. doi: 10.1126/science.aaa4971
Segal, N. H., Parsons, D. W., Peggs, K. S., Velculescu, V., Kinzler, K. W., Vogelstein, B., et al. (2008). Epitope landscape in breast and colorectal cancer. Cancer Res. 68, 889–892. doi: 10.1158/0008-5472.CAN-07-3095
Shugay, M., Bagaev, D. V., Zvyagin, I. V., Vroomans, R. M., Crawford, J. C., Dolton, G., et al. (2018). VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 46, D419–D427. doi: 10.1093/nar/gkx760
Shukla, S. A., Rooney, M. S., Rajasagi, M., Tiao, G., Dixon, P. M., Lawrence, M. S., et al. (2015). Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 33, 1152–1158. doi: 10.1038/nbt.3344
Singh, H., Raghava, G. P. S. (2001). ProPred: prediction of HLA-DR binding sites. Bioinformatics 17, 1236–1237. doi: 10.1093/bioinformatics/17.12.1236
Smart, A. C., Margolis, C. A., Pimentel, H., He, M. X., Miao, D., Adeegbe, D., et al. (2018). Intron retention is a source of neoepitopes in cancer. Nat. Biotechnol. 36 (11), 1056–1058. doi: 10.1038/nbt.4239
Snyder, A., Makarov, V., Merghoub, T., Yuan, J., Zaretsky, J. M., Desrichard, A., et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. New Engl. J. Med. 371, 2189–2199. doi: 10.1056/NEJMoa1406498
Sonntag, K., Hashimoto, H., Eyrich, M., Menzel, M., Schubach, M., Döcker, D., et al. (2018). Immune monitoring and TCR sequencing of CD4 T cells in a long term responsive patient with metastasized pancreatic ductal carcinoma treated with individualized, neoepitope-derived multipeptide vaccines: a case report. J. Trans. Med. 16, 23. doi: 10.1186/s12967-018-1382-1
Spranger, S., Luke, J. J., Bao, R., Zha, Y., Hernandez, K. M., Li, Y., et al. (2016). Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. 113, E7759–E7768. doi: 10.1073/pnas.1609376113
Storkus, W. J., Zeh, H. J., Salter, R. D., Lotze, M. T. (1993). Identification of T-cell epitopes: rapid isolation of class I-presented peptides from viable cells by mild acid elution. J. Immuno ther Emphasis Tumor Immunol. 14, 94–103.
Stranzl, T., Larsen, M. V., Lundegaard, C., Nielsen, M. (2010). NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62, 357–368. doi: 10.1007/s00251-010-0441-4
Strønen, E., Toebes, M., Kelderman, S., van Buuren, M. M., Yang, W., van Rooij, N., et al. (2016). Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341. doi: 10.1126/science.aaf2288
Sugawara, S., Abo, T., Kumagai, K. (1987). A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3 . J. Immunol. Methods 100, 83–90. doi: 10.1016/0022-1759(87)90175-x
Szolek, A., Schubert, B., Mohr, C., Sturm, M., Feldhahn, M., Kohlbacher, O. (2014). OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316. doi: 10.1093/bioinformatics/btu548
Thorsson, V., Gibbs, D. L., Brown, S. D., Wolf, D., Bortone, D. S., Ou Yang, T.-H., et al. (2018). The immune landscape of cancer. Immunity 48, 812–830.e14. doi: 10.1016/j.immuni.2018.03.023
Tran, E., Ahmadzadeh, M., Lu, Y.-C., Gros, A., Turcotte, S., Robbins, P. F., et al. (2015). Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390. doi: 10.1126/science.aad1253
Trolle, T., McMurtrey, C. P., Sidney, J., Bardet, W., Osborn, S. C., Kaever, T., et al. (2016). The length distribution of class I-restricted t cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J. Immunol. 196, 1480–1487. doi: 10.4049/jimmunol.1501721
Van Allen, E. M., Miao, D., Schilling, B., Shukla, S. A., Blank, C., Zimmer, L., et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. doi: 10.1126/science.aad0095
van Buuren, M. M., Calis, J. J., Schumacher, T. N. (2014). High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. OncoImmunology 3, e28836. doi: 10.4161/onci.28836
van Gool, I. C., Eggink, F. A., Freeman-Mills, L., Stelloo, E., Marchi, E., de Bruyn, M., et al. (2015). POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin. Cancer Res. 21, 3347–3355. doi: 10.1158/1078-0432.CCR-15-0057
van Rooij, N., van Buuren, M. M., Philips, D., Velds, A., Toebes, M., Heemskerk, B., et al. (2013). Tumor exome analysis reveals neoantigen-specific T-Cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 31, e439–e442. doi: 10.1200/JCO.2012.47.7521
Veatch, J. R., Lee, S. M., Fitzgibbon, M., Chow, I.-T., Jesernig, B., Schmitt, T., et al. (2018). Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J. Clin. Invest. 128, 1563–1568. doi: 10.1172/JCI98689
Vita, R., Mahajan, S., Overton, J. A., Dhanda, S. K., Martini, S., Cantrell, J. R., et al. (2018). The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343. doi: 10.1093/nar/gky1006
Vrecko, S., Guenat, D., Mercier-Letondal, P., Faucheu, H., Dosset, M., Royer, B., et al. (2018). Personalized identification of tumor-associated immunogenic neoepitopes in hepatocellular carcinoma in complete remission after sorafenib treatment. Oncotarget 9, 35394–35407. doi: 10.18632/oncotarget.26247
Wang, P., Sidney, J., Kim, Y., Sette, A., Lund, O., Nielsen, M., et al. (2010). Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinf. 11, 568. doi: 10.1186/1471-2105-11-568
Warren, R. L., Holt, R. A. (2010). A census of predicted mutational epitopes suitable for immunologic cancer control. Hum. Immunol. 71, 245–254. doi: 10.1016/j.humimm.2009.12.007
Warren, R. L., Choe, G., Freeman, D. J., Castellarin, M., Munro, S., Moore, R., et al. (2012). Derivation of HLA types from shotgun sequence datasets. Genome Med. 4, 95. doi: 10.1186/gm396
Wood, M. A., Paralkar, M., Paralkar, M. P., Nguyen, A., Struck, A. J., Ellrott, K., et al. (2018). Population-level distribution and putative immunogenicity of cancer neoepitopes. BMC Cancer 18, 414. doi: 10.1186/s12885-018-4325-6
Wu, J., Zhao, W., Zhou, B., Su, Z., Gu, X., Zhou, Z., et al. (2018). TSNAdb: a database for tumor-specific neoantigens from immunogenomics data analysis. Genomics Proteomics Bioinf. 16, 276–282. doi: 10.1016/j.gpb.2018.06.003
Yadav, M., Jhunjhunwala, S., Phung, Q. T., Lupardus, P., Tanguay, J., Bumbaca, S., et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. doi: 10.1038/nature14001
Yanover, C., Bradley, P. (2011). Large-scale characterization of peptide-MHC binding landscapes with structural simulations. Proc. Natl. Acad. Sci. U.S.A. 108, 6981–6986. doi: 10.1073/pnas.1018165108
Zacharakis, N., Chinnasamy, H., Black, M., Xu, H., Lu, Y.-C., Zheng, Z., et al. (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 24, 724–730. doi: 10.1038/s41591-018-0040-8
Zhang, G. L., Petrovsky, N., Kwoh, C. K., August, J. T., Brusic, V. (2006). PRED(TAP): a system for prediction of peptide binding to the human transporter associated with antigen processing. Immunome Res. 2, 3. doi: 10.1186/1745-7580-2-3
Zhang, H., Lund, O., Nielsen, M. (2009). The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics 25, 1293–1299. doi: 10.1093/bioinformatics/btp137
Zhang, X., Kim, S., Hundal, J., Herndon, J. M., Li, S., Petti, A. A., et al. (2017). Breast cancer neoantigens can induce CD8 + T-cell responses and antitumor immunity. Cancer Immunol. Res. 5, 516–523. doi: 10.1158/2326-6066.CIR-16-0264
Keywords: cancer immunotherapy, T cell receptor, neoepitope, neoantigen, cross-reactivity, MHC binding affinity prediction
Citation: Mösch A, Raffegerst S, Weis M, Schendel DJ and Frishman D (2019) Machine Learning for Cancer Immunotherapies Based on Epitope Recognition by T Cell Receptors. Front. Genet. 10:1141. doi: 10.3389/fgene.2019.01141
Received: 26 July 2019; Accepted: 21 October 2019;
Published: 19 November 2019.
Edited by:
Davide Chicco, Peter Munk Cardiac Centre, CanadaReviewed by:
Dhruv Sethi, Obsidian Therapeutics, United StatesGustavo Fioravanti Vieira, Universidade La Salle Canoas, Brazil
Copyright © 2019 Mösch, Raffegerst, Weis, Schendel and Frishman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmitrij Frishman, ZC5mcmlzaG1hbkB3encudHVtLmRl