 Crescenzio Francesco Minervini†
Crescenzio Francesco Minervini† Cosimo Cumbo†
Cosimo Cumbo† Paola Orsini†
Paola Orsini† Luisa Anelli
Luisa Anelli Antonella Zagaria
Antonella Zagaria Giorgina Specchia
Giorgina Specchia Francesco Albano*
Francesco Albano*- Department of Emergency and Organ Transplantation (D.E.T.O.), Hematology Section, University of Bari, Bari, Italy
The molecular pathogenesis of hematological diseases is often driven by genetic and epigenetic alterations. Next-generation sequencing has considerably increased our genomic knowledge of these disorders becoming ever more widespread in clinical practice. In 2012 Oxford Nanopore Technologies (ONT) released the MinION, the first long-read nanopore-based sequencer, overcoming the main limits of short-reads sequences generation. In the last years, several nanopore sequencing approaches have been performed in various “-omic” sciences; this review focuses on the challenge to introduce ONT devices in the hematological field, showing advantages, disadvantages and future perspectives of this technology in the precision medicine era.
Introduction
The role of genetic and epigenetic alterations in hematopoietic disorders is clear but more and more detailed knowledge is still emerging. Mounting evidence in the last decade has shown that genomic data are crucial to characterize the molecular pathogenesis of hematological diseases, in multiple fields of clinical practice such as diagnosis, prognostic stratification, treatment decision, measurable residual disease detection, hematopoietic stem cell transplantation, and posttransplantation chimerism control (Koutsi and Vervesou, 2018).
The introduction of next-generation sequencing (NGS, more correctly defined as “second generation sequencing”) has considerably improved the throughput of data generated by Sanger sequencing (SS) (first-generation sequencing). NGS approaches, that are becoming ever more widespread in blood diseases research and diagnostic laboratories, have greatly increased our genomic knowledge of hematopoietic disorders (Kuo et al., 2017), but these technologies remain expensive, laborious, time-consuming, and affected by the limits of short-read sequences generation.
In this scenario, in 2012, Oxford Nanopore Technologies (ONT) released the first long-read nanopore-based sequencer, known as MinION: a handheld third generation sequencing device that works connected to a laptop (Loman and Quinlan, 2014). In 2014, ONT launched a community-focused access project: the MinION Access Programme (MAP). The nanopores sit in a membrane that separates two ionic solutions, allowing an electrical current to flow through the nanopores. DNA or RNA molecules are carried through the protein pore, producing an ionic current change which is measured by a sensor with a constant sampling frequency, and later converted into nucleotides thanks to the basecalling algorithms (Goodwin et al., 2016; Magi et al., 2017). In this way, sequencing occurs without the synthesis of new strands as in the second-generation technologies.
In the last six years, the nanopore sequencing (NS) performance has been tested in various “-omic” sciences: genomics, epigenomics and transcriptomics. In this review we summarize the initial attempts to introduce the ONT platforms in the study of hematopoietic disorders, reporting the advantages and disadvantages of long-read sequencing and focusing on the future perspectives of a technology which is still emerging, but offers great promise.
Target Gene Sequencing
NS has been widely applied for target gene sequencing (Table 1) and is currently the most widespread NGS application in the medical field, where the mutational status of a specific set of genes can be decisive for establishing diagnosis, prognosis, and targeted therapy.
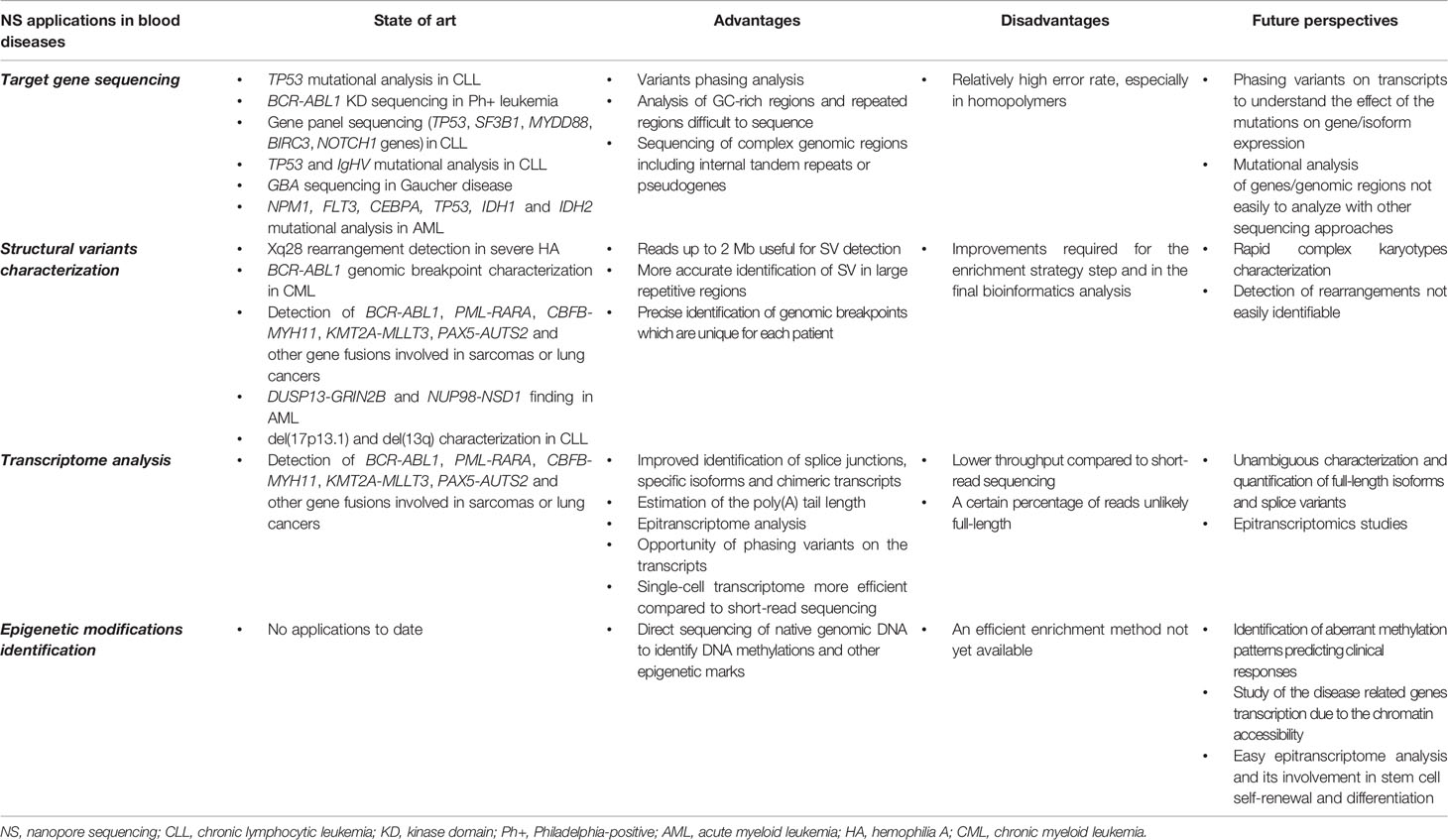
Table 1 State of art and future perspectives of nanopore sequencing applications in blood diseases.
The main NS drawback is the relatively high error rate compared to standard short-read methods (Table 2). Indeed, variant calling error rates from ONT data still remain relatively higher; however, starting from the first works with MinION sequencer (Ammar et al., 2015; Minervini et al., 2016), significant improvements to base-calling and single nucleotide variant (SNV)–calling steps have been made, making NS a device that may potentially be used in routine molecular tests (Minervini et al., 2016; Minervini et al., 2017; Orsini et al., 2018).
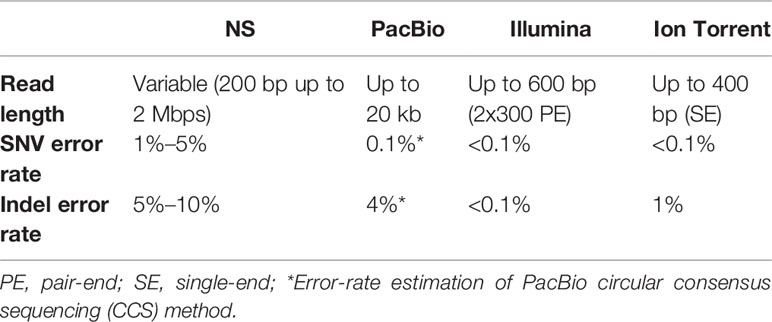
Table 2 Error-rate comparison of NS, PacBio, Ilumina, and Ion Torrent sequencing platforms.
Since the first MinION release, the error rate has considerably improved thanks to changes in sequencing chemistry, the introduction of new base-calling algorithms, postsequencing correction tools and nanopore SNVs/insertions-deletions (INDELs) calling tools (Rang et al., 2018; Makałowski and Shabardina, 2019). Most of the substitution/deletion errors in nanopore reads occur in homopolymers, because their length is not correctly inferred from the electric signal (Rang et al., 2018). This issue has improved over time, reducing the SNVs/INDELs error rate from 40% to 5%–15% on average (Magi et al., 2017; Rang et al., 2018).
In general, NS and other long read sequencing platforms such as Pacific Biosciences (PacBio) suffer of a high error rate and companies are working hard to overcome this drawback. ONT improved data quality by sequencing both template and complementary strands to obtain a final more accurate consensus read; to this aim 2D and subsequently 1D2 library preparation protocols have been developed, although the error rate was still about 5%–10%.
Recently, PacBio has developed the Circular Consensus long-read Sequencing method (Wenger et al., 2019), able to combine the high-accuracy sequencing with long reads generation, thus reducing the mean error rate from 12%–15% to 0.1% (Table 2). Likewise, to improve NS results the Rolling Circle Amplification to Concatemeric Consensus has been proposed (Volden et al., 2018).
In this context, by comparing the two platforms, PacBio currently shows better data quality for the resulting consensus data; on the other hand, NS allows to obtain longer reads, provide a higher throughput, as the nanopores can sequence multiple molecules, and has more advantageous and competitive costs than PacBio sequencing.
More promising data for NS came from a new sequencing method combining unique molecular identifiers (UMI’s) with NS has been proposed (Karst et al., 2019), obtaining consensus sequences with a mean error rate of 0.03%, similar to short-read sequencing, and representing a promising strategy to improve NS data accuracy.
Furthermore, error correction tools have already been tested (Minervini et al., 2016; Orsini et al., 2018; Makałowski and Shabardina, 2019); however, further improvements would be desirable to make the NS performance comparable to standard NGS technologies.
Target enrichment is the other critical aspect of the target sequencing and is closely related to the sequencing technology. To date, the most used target enrichment approach is Polymerase Chain Reaction (PCR) based even if the capture technology is another possible alternative. Noteworthy, a PCR free target enrichment strategy based on Cas9 technology has been recently proposed (Stevensid et al., 2019). In this strategy, sample DNA is firstly 5’- dephosphorylated, to prevent the ligase binding and Cas9 is then used to cleave the DNA at predetermined sites, exposing ligatable ends. These latter are then used to attach sequencing adapters for library preparation. In this way only the region of interest is available for sequencing. Furthermore, a modified Cas9 negative enrichment strategy contemplates digestion with exonuclease after the Cas9 action. In this protocol Cas9, after the cutting step, protects the DNA target region from being digested (Stevensid et al., 2019).
NS in Hematology
A major advantage of NS gene mutational analysis is the possibility of phasing genetic variations, by establishing the allelic context of mutations affecting the same gene but too physically distant to be detected with the short-read methods (Minervini et al., 2017; Cumbo et al., 2019). NS also allows the analysis of GC-rich regions or repeated regions that are difficult to study with conventional NGS sequencing approaches. As regards variant calling tools, starting from the methods used in the first works, many bioinformatic resources have been developed over time (Makałowski and Shabardina, 2019); moreover, in addition to custom procedures for phasing analysis, some specific software are currently available, such as the WhatsHap tool (Martin et al., 2016).
In the blood diseases context, NS has been widely tested to evaluate the mutational status of single or multiple genes involved in a specific disease. For example, it is well known that altered p53 function, due to a 17p deletion (del(17p)) and/or TP53 gene mutation, is associated with poor prognosis in chronic lymphocytic leukemia (CLL) patients, who are candidates for Bruton tyrosine kinase inhibitor treatment (Malcikova et al., 2018). TP53 NS with MinION was initially performed as a single test (Minervini et al., 2016), then as part of a gene panel including the most frequently mutated genes in CLL (Orsini et al., 2018), and was finally included in a screening assay encompassing TP53 mutational analysis, del(17p) detection, and IgHV mutational status evaluation (Burns et al., ). Compared to SS, TP53 analysis by NS can be performed even from one single amplicon, instead of one PCR per exon, obtaining long reads throughout the gene; this approach is less laborious and, in cases when more than one variant is identified, allows their phasing to be established.
A nanopore assay was also developed to identify BCR-ABL1 kinase domain (KD) mutations in Philadelphia-positive (Ph +) leukemias: chronic myeloid leukemia (CML) patients with treatment failure and acute lymphoblastic leukemia (ALL) cases at diagnosis (Minervini et al., 2017). Indeed, about 15%–30% of newly diagnosed chronic phase (CP)-CML patients will not reach an optimal response with first-line tyrosine kinase inhibitors (TKIs) therapy, and in about 25%–50% of them a BCR-ABL1 KD mutation will be identified (Soverini et al., 2011). This frequency increases among accelerated phase (AP) and blast crisis (BC) patients (Jabbour et al., 2006). The frequency of these mutations is much higher in ALL-Ph + patients at the time of relapse than CML (Jones et al., 2008). Compared to SS, that is considered the gold standard method, BCR-ABL1 KD NS has the advantage of potentially identifying low-level variants (< 15%–20% variant frequency, out of SS ability detection) and distinguishing between “compound mutants” (multiple mutations in the same clone) and “polyclonal mutants” (different clones) (Minervini et al., 2017). Indeed, it has been widely documented that the presence of BCR-ABL1 KD “compound mutations” defines a subset of patients with an increased likelihood of disease progression and resistance to second generation TKIs (Parker et al., 2016; Soverini et al., 2016).
Moreover, the long-reads performances simplify some hard tasks in acute myeloid leukemia (AML) molecular evaluation.
The presence of biallelic mutations in CEBPA gene, identifies a subgroup of patients which is a separate entity in the recent WHO classification, with distinct biological and clinical features (Amriah, 2006; Cumbo et al., 2019). As already discussed for BCR-ABL1 short-reads methods show difficulties to phase variants and biallelic status can only be inferred. The use of a long-read technology allows a direct detection of simultaneous presence of mutations over the distance in CEBPA gene. As well, conventional NGS approaches show limitations for the detection of longer FLT3 internal tandem duplications (ITDs) (Schranz et al., 2018) whose characterization is crucial in cytogenetically normal AML without NPM1 mutation, where FLT3-ITDs length was recently associated to a worse outcome (Chen et al., 2019). In the near future, targeted gene sequencing approaches may be surpassed by whole-genome sequencing (WGS) that will be used more and more frequently in clinical medicine in diagnostic contexts and to inform treatment choices (Bowden et al., 2019). Further improving base calling, alignment and variant calling methods will be crucial for this purpose. NS has also been applied to GBA gene sequencing (Leija-Salazar et al., 2019). Biallelic GBA mutations are responsible for Gaucher’s disease, the most frequent lysosomal storage disorder. GBA sequencing is complicated by some issues, especially the presence of a nearby pseudogene, GBAP1, a complex surrounding genomic region, and some exons that are difficult to sequence. In this context, long-read sequencing has proved to identify mutations that are otherwise difficult to observe, intronic variants not detectable with the conventional SS approach, and to perform phasing analysis, thus contributing to a deeper knowledge of the biological effect of GBA mutations.
Overall, given the low costs (from around 90€/Gb to 2€/Gb depending of the platform used) and the ease of library preparation protocols, NS could potentially enable more laboratories to perform targeted gene sequencing analysis; the introduction of user-friendly bioinformatic tools specific for NS data will further improve its diffusion in medical field.
Structural Variants Characterization
Structural variants (SVs) of DNA are defined as regions larger than 50 bp characterized by a location shift or a change in copy number in the genome. They include translocations, inversions, insertions, deletions, duplications, expansion of repetitive sequences and combinations of these (Escaramís et al., 2015; Sudmant et al., 2015). These genomic alterations are frequently observed in hematological diseases even if their identification is sometimes difficult.
The techniques generally used in clinical practice for this purpose range from cytogenetics to molecular biology. Conventional and molecular cytogenetic techniques are rather laborious, time consuming and affected by low sensitivity; on the other hand, molecular biology approaches, although easier and more sensitive, can answer few specific diagnostic questions. The advent of NGS approaches currently used in diagnostics has not markedly improved this practice, because of the short length of reads generated. NS allows the processing of fragments of thousands of kb and the generation of long reads up to 2 Mb, a new record for a single contiguous DNA sequence (Payne et al., 2019). Clearly, this ability permits a more accurate characterization of SV especially for the assembly of large repetitive regions (De Coster and Van Broeckhoven, 2019).
Regarding the complete NS workflow for SV characterization, the two critical points to be considered are the choice of the enrichment strategy (during library preparation) and the final bioinformatic analysis. Although the targeted approach is the most used strategy to perform SV analysis, NS allows SV detection also by the WGS approach.
NS in Hematology
Targeted Approach
The superiority of single molecule long-read sequencing was demonstrated in the characterization of a new Xq28 rearrangement disrupting F8, in a case of genetically unresolved severe hemophilia A (HA) (Chatron et al., 2019). The study presented a 3.8‐Mb Xq11.1q12 inserted duplication in the F8 intron 25, producing an F8/Xq12 (noncoding sequence) fusion transcript. Nanopore technology could improve HA molecular diagnosis, particularly in those cases (about 2%) in which conventional molecular technologies fail.
The nanopore ability to generate long-reads was also exploited to characterize the BCR-ABL1 genomic breakpoint in ten patients affected by CML, in order to develop an approach for the “personalized monitoring” of residual disease during follow-up (Cumbo et al., 2018). NS simplifies the identification of genomic junctions that are different in each patient; this procedure is difficult and expensive to accomplish with the short-read sequencing approach and not suitable for clinical practice. The sequence thus obtained can then be used to design and test a patient-specific droplet digital PCR assay to make an absolute quantification of residual leukemic cells in patients’ peripheral blood (Cumbo et al., 2018).
The chance to detect fusion oncogenes was developed in order to develop a rapid assay for cases needing fast results (Jeck et al., 2019). The pipeline proposed was tested on a cell line (K562) and on 16 multiplexed specimens (14 clinical samples and two control analytes) for the identification of BCR-ABL1, PML-RARA, CBFB-MYH11, KMT2A-MLLT3, PAX5-AUTS2 and other gene fusions involved in solid tumors such as sarcomas or lung cancers. Comparing with cytogenetics and molecular techniques, NS offers a more rapid approach leading to a result in two hours and sometimes in 30 minutes thanks to the possibility to analyze data in real time. Moreover, NS allows the detection of all possible fusion partnering and variants. Furthermore, authors reported the advantages of NS compared with Illumina and other NGS platforms: faster results, low initial investment costs, no need to batch samples to reduce costs and workload. The assay showed high sensitivity and specificity, with no false positives (in the high-quality calls) when compared to Illumina sequencing, even if further improvements will be required in the base-call quality step (to reduce the known higher error rates in nanopore reads) and in the development of an ecosystem of tools for real-time processing of nanopore data (to rapidly perform the relevant fusion calls).
WGS Approach
Recently, the rapid detection and breakpoint characterization of two chromosomal translocations in a newly diagnosed AML patient, by WGS has been reported (Au et al., 2019). A 26,194 bp sequencing read revealed the exact translocation breakpoint between DUSP13 (chromosome 10q22.2) and GRIN2B (chromosome 12p13.1): a nonproductive passenger event yet identified by conventional cytogenetics; a 20,709 bp sequencing read, instead, characterizes the breakpoint between NUP98 (chromosome 11p15.4) and NSD1 (chromosome 5q35.3), a known cryptic driver translocation not amendable for detection by routine karyotyping. The case study suggests the potential use of NS for rapid general SV screening, even if a test with such a low depth of coverage (1x) cannot be separated from a validation step. Moreover, WGS performed in 11 CLL cases identified, with an average depth of coverage of 1.6x (range 0.1–3.4x) and an average read length of 6.3 kbp, two important chromosome deletions (Burns et al., ). In fact, in three cases del(17p13.1) were identified, 21.3 Mb, 20.3 Mb, and 22 Mb in length, respectively; instead four cases harbored del(13q), with an 800kb minimally deleted region encompassing MIR16, MIR15A and DLEU1 (Burns et al., ). As regards the sequencing run, the accuracy of the different chemistry developed by ONT in the last years is suitable for this kind of approach. In fact, the SV detection is not affected by the relatively high error rate of the platform (De Coster et al., 2019).
Transcriptome and Epitranscriptome Sequencing
RNA-sequencing (RNA-seq) has been a real revolution, making it possible to analyze gene expression, uncover novel RNA molecules and isoforms and identify mutations in transcripts (Pan et al., 2008; Piskol et al., 2013). Nowadays, RNA-seq methods are routinely applied for differential genes/isoforms expression analysis as well as other specific applications, such as gene fusion detection, targeted sequencing and single-cell analysis.
Up to now, short-read NGS has been used for RNA-seq; however, the main drawback of these technologies remains the prior fragmentation of the messenger RNA (mRNA) molecules, and the later computational reconstruction and assembly of the short-reads. Indeed, the assembled transcripts do not always contain the precise isoform information (for example, due to repetitive sequences or a low expression level with a consequently insufficient number of reads, or for multimapping of the short-reads) and alternative splicing patterns detection may be not accurate.
Short-read RNA-seq is also hampered by systematic issues, such as the PCR bias, and up to now all the RNA-seq preparation methods have required the conversion of RNA into complementary DNA (cDNA), amplification of these molecules and then sequencing. Each of these procedures can potentially introduce artifacts (Reid-Bayliss and Loeb, 2017).
Short-read sequencing can produce reads of up to 350 bp, but the read depth is generally high and the error rate is very low; on the other hand, long-read sequencing produces long reads with a relatively higher error rate, but good performance with splice junctions and repetitive regions resolution (Kukurba and Montgomery, 2015). Moreover, Rolling Circle Amplification to Concatemeric Consensus method (Volden et al., 2018) showed to generate more accurate reads of full-length RNA transcript isoforms than any other available long-read sequencing approaches by generating consensus sequences with increased base accuracy.
Recently, long-read sequencing has demonstrated peculiar advantages even for transcriptome analysis. In fact, this approach eliminates the need for the assembly of short-reads, ensures a more efficient alignment to repetitive regions and allows the detection of unambiguous isoforms (Stark et al., 2019).
The NS approach has been a real revolution for the RNA-seq procedure, because PCR bias can be eliminated by using direct cDNA/RNA sequencing. The main strengths of this strategy are the possibility to obtain full-length transcripts, with unambiguous identification of transcripts and isoforms, and the opportunity to study splice variants and chimeric transcripts and SVs more accurately. As in NS the read length corresponds to the fragment length, the limits typical of short-read sequencing are overcome.
Nanopore transcriptome analysis also allows estimation of the poly(A) tail length (Workman et al., 2018), that is important for RNA stability and translation, and is difficult to study with standard short-read sequencing methods (Seki et al., 2019). Overall, NS currently offers three different approaches for RNA-seq: cDNA-PCR sequencing, direct cDNA sequencing and direct RNA sequencing; the latter is the first direct RNA sequencing method (Garalde et al., 2018). Compared to short-read sequencing, long-read RNA-seq, whether long-read cDNA or direct RNA, is potentially a more appropriate approach for isoform discovery, fusion transcript discovery, de novo transcriptome analysis and analysis of complex loci.
Moreover, direct RNA-seq allows epitranscriptome analysis, through the detection of transcriptional modifications inferred from the signal as the RNA molecule passes through the nanopore (Schwartz and Motorin, 2017; Workman et al., 2018; Wongsurawat et al., 2018; Liu et al., 2019), as subsequently described. However, the main drawback of nanopore transcriptome analysis is a lower throughput compared to short-read sequencing; the throughput further decreases when using the direct RNA approach, that is the method with the highest input requirement, due to a slower transition through the pore compared to the DNA strand. In direct RNA-seq, 500 ng of polyA mRNA are needed as starting material, which may not be available for some critical samples. Moreover, RNA degradation can compromise an efficient detection of splice variants; the application of method for isolating intact transcripts (for instance, by targeting the eukaryotic 5′ cap) may be very effective for this purpose (Garalde et al., 2018).
In some papers, nanopore cDNA and direct RNA sequencing approaches have been compared, illustrating the strengths and limitations of both (Workman et al., 2018; Seki et al., 2019; Soneson et al., 2019). Although direct sequencing of full-length cDNA/RNA molecules is promising, it has been reported that a certain percentage of the raw nanopore reads are unlikely to be full-length reference transcripts, both with direct cDNA and RNA sequencing, thus interfering with the true identification/quantification of transcripts; this aspect needs to be improved (Soneson et al., 2019).
Regarding bioinformatics tools to be used for data analysis, dedicated pipelines/software have recently been developed and are currently available for nanopore RNA-seq analysis (Makałowski and Shabardina, 2019; Seki et al., 2019) (https://long-read-tools.org/). Some applications are more efficiently performed with NS compared to short-read sequencing: the detection of fusion transcripts, the opportunity of identifying allele-specific transcriptional events by phasing variants on the transcripts, and single-cell transcriptome analysis (Seki et al., 2019). Until now, most of the transcriptome studies in human research performed with NS namely were aimed at unambiguously characterizing and quantifying full-length isoforms and splice variants (Clark et al., 2018; Sakamoto et al., 2019; Rahimi et al., 2019; Hardwick et al., 2019), and identifying fusion transcripts and determining variants phasing (Byrne et al., 2017; Suzuki et al., 2017; Jeck et al., 2019), especially in cancer and neurological disorders.
NS in Hematology
In the hematological field, nanopore RNA-seq is still in its infancy, but some applications are currently emerging. By combining the MinION system and Anchored Multiplex PCR (AMP)-based libraries, a nanopore-based sequencing assay was developed to rapidly detect fusion genes in acute leukemia, where fusion detection is time sensitive and clinically relevant to choose the best treatment depending on the leukemia subtype (Jeck et al., 2019). Similarly, a NS assay based on the analysis of full-length cDNA has been developed to analyze the single-cell transcriptome, and has been applied to study the transcriptional heterogeneity of B cells with different surface receptors. Single-cell transcriptome analysis coupled with long-read sequencing makes it possible to study the real cellular transcriptional diversity at both gene and isoform level in these cells, defining and quantifying complex and/or never before reported isoforms (Byrne et al., 2017) and so extending the knowledge of physiological cellular processes and their alterations.
NS was introduced as a “validation tool” as a part of a new method, called Genotyping for Transcriptome (GoT), developed to associate the somatic genotyping of expressed genes with transcriptional profiling of single cells (Nam et al., 2019). This approach was applied to CD34+ cells from patients with myeloproliferative neoplasms (MPNs) to inspect the biological effect of calreticulin gene mutations on the cells transcriptome.
Long-read sequencing applied to transcriptome analysis may help acquire more biological information in various hematological diseases; for example, in Philadelphia chromosome (Ph)-like ALL, a crucial question is how to easily and rapidly identify Ph-like ALL (Tasian et al., 2017); gene/isoform expression analysis and fusion transcripts detection by NS may help to elucidate this aspect. In multiple myeloma (MM), where gene expression profiling has been used to identify defined molecular subgroups (Broyl et al., 2010), long-read sequencing may be useful to further improve the biological knowledge of MM cells transcriptomes.
The possibility of phasing variants on transcripts may also be exploited to understand the effect of the mutations identified in genes involved in leukemic transformation on gene/isoform expression. However, it should be underlined that the potential advantages associated with long-read analysis from blood samples have to do with the difficulty of obtaining good quality RNA from formalin-fixed paraffin-embedded (FFPE) tissue specimens from solid tumors and lymphomas.
In the future, direct RNA-sequencing may elucidate some still unknown biological issues in hematological diseases. It is known that methyladenosine (m6A) is a reversible nucleotide modification catalyzed by the METTL3/METTL14 methyltransferase complex4, that is involved in various normal biological processes, such as stem cell self-renewal and differentiation (Deng et al., 2018), as well as in AML pathogenesis (Vu et al., 2017; Barbieri et al., 2017). Several data support the concept that in AML cells the methylation of specific transcripts is crucial for AML translational regulation, and that the depletion of METTL3 can alter this process, inducing cell apoptosis in malignant cells, thereby suggesting this gene as a new potential target. In this context, direct RNA-seq could provide a direct map of m6A modifications in leukemic cells and evaluate the biological impact of this mechanism on transcriptomes in AML.
Epigenetic Modifications Identification
DNA bases chemical modifications can influence the biological functions. Nowadays, different base modifications are described to play an important role in mammals, in physiological processes such as aging, gene regulation, imprinting, as well as disease. Methylcytosine (5-mC) is the most common among these modified bases in the genome. In addition to methylation of the cytosine (C) residue, other modifications such as oxidation of the 5-mC to 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), 5-carboxylcytosine (5-caC), and methylation of adenine (A) to N6-methyladenine (m6A), are being identified as important epigenetic regulators (Smith and Meissner, 2013; Klungland and Robertson, 2017; Kumar et al., 2018). The cancer cell genome undergoes dramatic shifts in the pattern of genomic methylation, including genome-wide hypomethylation in conjunction with local areas of hypermethylation. Aberrant hypomethylation causes the expression of certain genes, such as oncogenes, while hypermethylation causes the inhibition of tumor suppressor genes (Feinberg and Vogelstein, 1983; Esteller et al., 2001). Additionally, the spatial and temporal regulation of DNA methylation in the hematopoietic developmental hierarchy is critical to hematopoietic homeostasis.
NS in Hematology
Recent studies suggest that the epigenetic factors act in concert with the transcriptional factors to ensure hematopoietic homeostasis (Cullen et al., 2014; Sashida and Iwama, 2012; Smith and Meissner, 2013; Beerman and Rossi, 2014; Gore and Weinstein, 2016; Kumar et al., 2018). An improper orchestration of the epigenetic mechanisms that control this balance can cause an aberrant hematopoietic stem cell (HSC) function and induce several hematological disorders/malignancies, such as leukemias and lymphomas, MPN, myelodysplastic syndromes (MDS), and MM. In the context of hematological malignancies, the methylation and, more generally the epigenetic status, could affect the prognosis (Toyota et al., 2001; Gore and Weinstein, 2016; Timms et al., 2016). Changes in the promoter methylation of TAL1, ERHV-3, CDKN2A, CALCA, p15, IDH1/2, and TP53 genes have been associated with relapse in T-cell acute lymphoblastic leukemia (T-ALL) (Hogan et al., 2011; Nordlund et al., 2013). Moreover, an increasing number of recent studies are unveiling aberrant methylation patterns that predict survival in many blood malignancies, supporting the significance of measuring DNA methylation before treatment in order to predict clinical responses (Toyota et al., 2001). However, to assess the epigenomic alterations, the currently available methodologies are sophisticated and time-consuming, making them difficult to standardize and implement. Short-reads NGS strategies contemplate bisulfite conversion of unmethylated C to uracil (U) before sequencing, and so are affected by the conversion efficiency and by difficulties in repetitive genomic sequences (Barros-Silva et al., 2018). In the field of long-reads technology, a more affordable NS is also able to detect directly modified DNA and RNA bases by measuring the fluctuations in the ionic current signal between modified and unmodified bases (Simpson et al., 2017). Several bioinformatics approaches have been developed to call modified bases from the electric events obtained during the sequencing run (https://long-read-tools.org/). Basically, all such methods are able to call 5-mC but they differ as regards the training data needs and the ability to distinguish other specific modifications. The major advantage in using the nanopore approach for the base modification analysis is the possibility to directly sequence the native genomic DNA. However, when a specific target region needs to be analyzed, this advantage becomes a limit due to the lack of an efficient enrichment methodology. For this purpose, Cas9 enrichment strategy previously described, allows to overcome this issue. Apart from the methylation, other epigenetic marks could be investigated using NGS, such as the nucleosome occupancy or the histone modifications. The degree of packaging of the DNA in the chromatin has an effect on the gene expression because it controls access to the factors that regulate gene transcription. Recently, some authors have published a study on erythroid differentiation highlighting the role of the changes in the chromatin accessibility during various stages of erythropoiesis (Ludwig et al., 2019). Furthermore, other authors showed specific increases of chromatin accessibility in MM as compared to normal B cells. These changes cause enhancer activation and disease-related genes transcription (Jin et al., 2018). For this purpose, some authors applied nanopore detection of the 5-mC, to define the chromatin accessibility using a method already known as NOME-seq (Kelly et al., 2012; Lee et al., 2019). NOME-seq uses GpC methyltransferase (M.CviPI) and NGS to generate a high resolution footprint of the nucleosome position. The technique allows simultaneous detection of the nucleosome occupancy and the DNA methylation pattern. Using long-read NS, the nucleosome occupancy can be observed on the single read. Furthermore, the authors propose the use of different methyltransferases, such as EcoGII which methylates A to m6A in order to obtain a “multicolor” measurement which could allow discrimination between endogenous 5-mC and induced modifications (Lee et al., 2019).
Closing Remarks
The advent of third generation sequencing can be considered a revolution in NGS technologies. What was considered impossible only a few years ago in terms of throughput, potential, ease of use and costs, today is becoming achievable. Genetic and genomic approaches are now a routine part of biological research and are becoming ever more widespread in clinical practice. As regards hematological diseases, and especially in the context of blood malignancies, several NS approaches have been performed with the aim of elucidating some still unknown biological issues and testing the performance of the ONT platforms in diagnostic contexts (Table 1). As previously discussed, many advantages are to be gained from long-read sequencing: efficient variants-phasing, the ability to analyze GC-rich or repeated regions, easy characterization of the genomic SVs and the identification of full-length transcripts and isoforms. Furthermore, the ONT peculiarity of being able to directly sequence the nucleic acids allows a better study of splice variants and chimeric transcripts, and direct detection of the epigenetic modifications of DNA and RNA molecules. The scalability of the technology makes it possible to perform different approaches in terms of throughput (2 Gb–10.5 Tb), ranging from rapid target gene sequencing to WGS or whole transcriptome analysis. ONT released the GridION and PromethION platforms, allowing higher throughput sequencing runs (from 250 Gb to 10.5 Tb, respectively, according to Oxford Nanopore information); on the other hand, MinION can be adapted for smaller rapid experiments using Flongle: a single use flow cell that is ideal for target sequencing (throughput: 2 Gb) (Kumar et al., 2019). These emerging strengths, together with the constant improvements in nanopore technology may, in the near future, pave the way for the introduction of ONT in hematological research and diagnostic laboratories. In the precision medicine era, a platform with this kind of performance will help to increase the understanding of the “-omic” mechanisms in these diseases, providing clinicians with valid support in diagnosis and prognosis, thus promoting the personalized treatment of patients.
Author Contributions
CM, CC, and PO conceived and wrote the review. LA and AZ performed the literature analysis. FA and GS supervised and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by “Associazione Italiana contro le Leucemie (AIL)-BARI”. The authors would like to thank Ms. MVC Pragnell, B.A. for language revision of the manuscript.
References
Ammar, R., Paton, T. A., Torti, D., Shlien, A., Bader, G. D. (2015). Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Research 4. doi: 10.12688/f1000research.6037.1
Au, C. H., Ho, D. N., Ip, B. B. K., Wan, T. S. K., Ng, M. H. L., Chiu, E. K. W., et al. (2019). Rapid detection of chromosomal translocation and precise breakpoint characterization in acute myeloid leukemia by nanopore long-read sequencing. Cancer Genet. 239, 22–25. doi: 10.1016/j.cancergen.2019.08.005
Barbieri, I., Tzelepis, K., Pandolfini, L., Shi, J., Millán-Zambrano, G., Robson, S. C., et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia via m6A-dependent translation control Europe PMC Funders Group. Nature 552, 126–131. doi: 10.1038/nature24678
Barros-Silva, D., Joana Marques, C., Henrique, R., Jerónimo, C. (2018). Profiling DNA Methylation based on next-generation sequencing approaches: new insights and clinical applications. Genes 9 (9), 429. doi: 10.3390/genes9090429
Beerman, I., Rossi, D. J. (2014). Epigenetic regulation of hematopoietic stem cell aging. Exp. Cell Res. 329, 192–199. doi: 10.1016/j.yexcr.2014.09.013
Bowden, R., Davies, R. W., Heger, A., Pagnamenta, A. T., de Cesare, M., Oikkonen, L. E., et al. (2019). Sequencing of human genomes with nanopore technology. Nat. Commun. 10, 1–9. doi: 10.1038/s41467-019-09637-5
Broyl, A., Hose, D., Lokhorst, H., De Knegt, Y., Peeters, J., Jauch, A., et al. (2010). Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 116, 2543–2553. doi: 10.1182/blood-2009-12-261032
Burns, A., Disalvo-Williams, D., Bruce, D., Robbe, P., Timbs, A., Stamatopoulos, B., et al. The diagnostic chronic lymphocytic leukaemia genome by nanopore sequencing. bioRxiv 750059. doi: 10.1101/750059
Byrne, A., Beaudin, A. E., Olsen, H. E., Jain, M., Cole, C., Palmer, T., et al. (2017). Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 8, 1–11. doi: 10.1038/ncomms16027
Chatron, N., Schluth-Bolard, C., Frétigny, M., Labalme, A., Vilchez, G., Castet, S. M., et al. (2019). Severe hemophilia A caused by an unbalanced chromosomal rearrangement identified using nanopore sequencing. J. Thromb. Haemost. 17:1097–1103. doi: 10.1111/jth.14460
Chen, F., Sun, J., Yin, C., Cheng, J., Ni, J., Jiang, L., et al. (2019). Impact of FLT3-ITD allele ratio and ITD length on therapeutic outcome in cytogenetically normal AML patients without NPM1 mutation. Bone Marrow Transplant. doi: 10.1038/s41409-019-0721-z
Clark, M., Wrzesinski, T., Garcia-Bea, A., Kleinman, J., Hyde, T., Weinberger, D., et al. (2018). Long-read sequencing reveals the splicing profile of the calcium channel gene CACNA1C in human brain. bioRxiv, 260562. doi: 10.1101/260562
Cullen, S. M., Mayle, A., Rossi, L., Goodell, M. A. (2014). “Hematopoietic Stem Cell Development,” in Current topics in developmental biology, 107, 39–75. doi: 10.1016/B978-0-12-416022-4.00002-0
Cumbo, C., Impera, L., Minervini, C. F., Orsini, P., Anelli, L., Zagaria, A., et al. (2018). Genomic BCR-ABL1 breakpoint characterization by a multistrategy approach for “personalized monitoring” of residual disease in chronic myeloid leukemia patients. Oncotarget 9, 10978–10986. doi: 10.18632/oncotarget.23971
Cumbo, C., Minervini, C. F., Orsini, P., Anelli, L., Zagaria, A., Minervini, A., et al. (2019). Nanopore targeted sequencing for rapid gene mutations detection in acute myeloid leukemia. Genes 2019 10, 1026. doi: 10.3390/GENES10121026
De Coster, W., Van Broeckhoven, C. (2019). Newest methods for detecting structural variations. Trends Biotechnol. 37 (9), 973–982. doi: 10.1016/j.tibtech.2019.02.003
De Coster, W., De Rijk, P., De Roeck, A., De Pooter, T., D’Hert, S., Strazisar, M., et al. (2019). Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 29, 1178–1187. doi: 10.1101/gr.244939.118
Deng, X., Su, R., Weng, H., Huang, H., Li, Z., Chen, J. (2018). RNA N 6 -methyladenosine modification in cancers: current status and perspectives. Cell Res. 28, 507–517. doi: 10.1038/s41422-018-0034-6
Escaramís, G., Docampo, E., Rabionet, R. (2015). A decade of structural variants: description, history and methods to detect structural variation. Brief Funct. Genomics 14, 305–314. doi: 10.1093/bfgp/elv014
Esteller, M., Corn, P. G., Baylin, S. B., Herman, J. G. (2001). A gene hypermethylation profile of human cancer. Cancer Res. 61, 3225–3229. [Accessed July 5, 2019].
Feinberg, A. P., Vogelstein, B. (1983). Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301, 89–92. doi: 10.1038/301089a0
Garalde, D. R., Snell, E. A., Jachimowicz, D., Sipos, B., Lloyd, J. H., Bruce, M., et al. (2018). Highly parallel direct RN A sequencing on an array of nanopores. Nat. Methods 15, 201–206. doi: 10.1038/nmeth.4577
Goodwin, S., McPherson, J. D., McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Gore, A. V., Weinstein, B. M. (2016). DNA methylation in hematopoietic development and disease. Exp. Hematol. 44, 783–790. doi: 10.1016/j.exphem.2016.04.013
Hardwick, S. A., Bassett, S. D., Kaczorowski, D., Blackburn, J., Barton, K., Bartonicek, N., et al. (2019). Targeted, high-resolution RNA sequencing of non-coding genomic regions associated with neuropsychiatric functions. Front. Genet. 10, 1–17. doi: 10.3389/fgene.2019.00309
Hogan, L. E., Meyer, J. A., Yang, J., Wang, J., Wong, N., Yang, W., et al. (2011). Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 118, 5218–5226. doi: 10.1182/blood-2011-04-345595
Jabbour, E., Kantarjian, H., Jones, D., Talpaz, M., Bekele, N., O’Brien, S., et al. (2006). Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia 20, 1767–1773. doi: 10.1038/sj.leu.2404318
Jeck, W. R., Lee, J., Robinson, H., Le, L. P., Iafrate, A. J., Nardi, V. (2019). A nanopore sequencing–based assay for rapid detection of gene fusions. J. Mol. Diagnostics 21, 58–69. doi: 10.1016/j.jmoldx.2018.08.003
Jin, Y., Chen, K., De Paepe, A., Hellqvist, E., Krstic, A. D., Metang, L., et al. (2018). Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 131, 2138–2150. doi: 10.1182/blood-2017-09-808063
Jones, D., Thomas, D., Yin, C. C., O’Brien, S., Cortes, J. E., Jabbour, E., et al. (2008). Kinase domain point mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia emerge after therapy with BCR-ABL kinase inhibitors. Cancer 113, 985–994. doi: 10.1002/cncr.23666
Karst, S. M., Ziels, R. M., Kirkegaard, R. H., Albertsen, M. (2019). Enabling high-accuracy long-read amplicon sequences using unique molecular identifiers and Nanopore sequencing. bioRxiv, 645903. doi: 10.1101/645903
Kelly, T. K., Liu, Y., Lay, F. D., Liang, G., Berman, B. P., Jones, P. A. (2012). Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22, 2497–2506. doi: 10.1101/gr.143008.112
Klungland, A., Robertson, A. B. (2017). Oxidized C5-methyl cytosine bases in DNA: 5-Hydroxymethylcytosine; 5-formylcytosine; and 5-carboxycytosine. Free Radic. Biol. Med. 107, 62–68. doi: 10.1016/j.freeradbiomed.2016.11.038
Koutsi, A., Vervesou, E.-C. (2018). Diagnostic molecular techniques in haematology: recent advances. Ann. Transl. Med. 6, 242–242. doi: 10.21037/atm.2018.05.30
Kukurba, K. R., Montgomery, S. B. (2015). RNA sequencing and analysis. Cold Spring Harb Protoc. 2015, 951. doi: 10.1101/PDB.TOP084970
Kumar, S., Chinnusamy, V., Mohapatra, T. (2018). Epigenetics of modified DNA Bases: 5-Methylcytosine and beyond. Front. Genet. 9, 640. doi: 10.3389/fgene.2018.00640
Kumar, K., Cowley, M., Davis, R. (2019). Next-generation sequencing and emerging technologies. Semin. Thromb. Hemost 1, 661–673. doi: 10.1055/s-0039-1688446
Kuo, F. C., Mar, B. G., Lindsley, R. C., Lindeman, N. I. (2017). The relative utilities of genome-wide, gene panel, and individual gene sequencing in clinical practice. Blood 130, 433–439. doi: 10.1182/blood-2017-03-734533
Lee, I., Razaghi, R., Gilpatrick, T., Molnar, M., Sadowski, N., Simpson, J. T., et al. (2019). Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing. bioRxiv, 504993. doi: 10.1101/504993
Leija-Salazar, M., Sedlazeck, F. J., Toffoli, M., Mullin, S., Mokretar, K., Athanasopoulou, M., et al. (2019). Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genom. Med. 7. doi: 10.1002/mgg3.564
Liu, H., Begik, O., Lucas, M. C., Christopher, E., Schwartz, S., Mattick, J. S., et al. (2019). Accurate detection of m6A RNA modifications in native RNA sequences. bioRxiv, 525741. doi: 10.1101/525741
Loman, N. J., Quinlan, A. R. (2014). Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30, 3399–3401. doi: 10.1093/bioinformatics/btu555
Ludwig, L. S., Lareau, C. A., Bao, E. L., Nandakumar, S. K., Muus, C., Ulirsch, J. C., et al. (2019). Transcriptional states and chromatin accessibility underlying human erythropoiesis. Cell Rep. 27, 3228–3240.e7. doi: 10.1016/J.CELREP.2019.05.046
Magi, A., Semeraro, R., Mingrino, A., Giusti, B., D’Aurizio, R. (2017). Nanopore sequencing data analysis: state of the art, applications and challenges. Brief Bioinform. 19, 1256–1272. doi: 10.1093/bib/bbx062
Makałowski, W., Shabardina, V. (2019). Bioinformatics of nanopore sequencing. J. Hum. Genet. 65, 1–7. doi: 10.1038/s10038-019-0659-4
Malcikova, J., Tausch, E., Rossi, D., Sutton, L. A., Soussi, T., Zenz, T., et al. (2018). ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia - Update on methodological approaches and results interpretation. Leukemia 32, 1070–1080. doi: 10.1038/s41375-017-0007-7
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Gunnar, W., et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv 085050, 1–18. doi: 10.1101/085050
Minervini, C. F., Cumbo, C., Orsini, P., Brunetti, C., Anelli, L., Zagaria, A., et al. (2016). TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn. Pathol. 11, 1–9. doi: 10.1186/s13000-016-0550-y
Minervini, C. F., Cumbo, C., Orsini, P., Anelli, L., Zagaria, A., Impera, L., et al. (2017). Mutational analysis in BCR-ABL1 positive leukemia by deep sequencing based on nanopore MinION technology. Exp. Mol. Pathol. 103, 33–37. doi: 10.1016/j.yexmp.2017.06.007
Nam, A. S., Kim, K. T., Chaligne, R., Izzo, F., Ang, C., Taylor, J., et al. (2019). Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 571, 355–360. doi: 10.1038/s41586-019-1367-0
Nordlund, J., Bäcklin, C. L., Wahlberg, P., Busche, S., Berglund, E. C., Eloranta, M.-L., et al. (2013). Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 14, r105. doi: 10.1186/GB-2013-14-9-R105
Orsini, P., Minervini, C. F., Cumbo, C., Anelli, L., Zagaria, A., Minervini, A., et al. (2018). Design and MinION testing of a nanopore targeted gene sequencing panel for chronic lymphocytic leukemia. Sci. Rep. 8, 11798. doi: 10.1038/s41598-018-30330-y
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415. doi: 10.1038/ng.259
Parker, W. T., Yeung, D. T. O., Yeoman, A. L., Altamura, H. K., Jamison, B. A., Field, C. R., et al. (2016). The impact of multiple low-level BCR-ABL1 mutations on response to ponatinib. Blood 127, 1870–1880. doi: 10.1182/blood-2015-09-666214
Payne, A., Holmes, N., Rakyan, V., Loose, M. (2019). BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics 35, 2193–2198. doi: 10.1093/bioinformatics/bty841
Piskol, R., Ramaswami, G., Li, J. B. (2013). Reliable Identification of Genomic Variants from RNA-Seq Data. Am. J. Hum. Genet. 93, 641–651. doi: 10.1016/j.ajhg.2013.08.008
Rahimi, K., Venø, M. T., Dupont, D. M., Kjems, J. (2019). Nanopore sequencing of full-length circRNAs in human and mouse brains reveals circRNA-specific exon usage and intron retention. bioRxiv, 567164. doi: 10.1101/567164
Rang, F. J., Kloosterman, W. P., de Ridder, J. (2018). From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 19, 1–11. doi: 10.1186/s13059-018-1462-9
Reid-Bayliss, K. S., Loeb, L. A. (2017). Accurate RNA consensus sequencing for high-fidelity detection of transcriptional mutagenesis-induced epimutations. Proc. Natl. Acad. Sci. 114, 9415–9420. doi: 10.1073/PNAS.1709166114
Sakamoto, Y., Xu, L., Seki, M., Yokoyama, T. T., Kasahara, M., Kashima, Y., et al. (2019). Long read sequencing reveals a novel class of structural aberrations in cancers: identification and characterization of cancerous local amplifications. bioRxiv, 620047. doi: 10.1101/620047
Sashida, G., Iwama, A. (2012). Epigenetic regulation of hematopoiesis. Int. J. Hematol. 96, 405–412. doi: 10.1007/s12185-012-1183-x
Schranz, K., Hubmann, M., Harin, E., Vosberg, S., Herold, T., Metzeler, K. H., et al. (2018). Clonal heterogeneity of FLT3-ITD detected by high-throughput amplicon sequencing correlates with adverse prognosis in acute myeloid leukemia. Oncotarget 9, 30128–30145. doi: 10.18632/oncotarget.25729
Schwartz, S., Motorin, Y. (2017). Next-generation sequencing technologies for detection of modified nucleotides in RNAs. RNA Biol. 14, 1124–1137. doi: 10.1080/15476286.2016.1251543
Seki, M., Katsumata, E., Suzuki, A., Sereewattanawoot, S., Sakamoto, Y., Mizushima-Sugano, J., et al. (2019). Evaluation and application of RNA-Seq by MinION. DNA Res. 26, 55–65. doi: 10.1093/dnares/dsy038
Simpson, J. T., Workman, R. E., Zuzarte, P. C., David, M., Dursi, L. J., Timp, W. (2017). Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410. doi: 10.1038/nmeth.4184
Smith, Z. D., Meissner, A. (2013). DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220. doi: 10.1038/nrg3354
Soneson, C., Yao, Y., Bratus-Neuenschwander, A., Patrignani, A., Robinson, M. D., Hussain, S. (2019). A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun. 10. doi: 10.1038/s41467-019-11272-z
Soverini, S., Hochhaus, A., Nicolini, F. E., Gruber, F., Lange, T., Saglio, G., et al. (2011). BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: Recommendations from an expert panel on behalf of European LeukemiaNet. Blood 118, 1208–1215. doi: 10.1182/blood-2010-12-326405
Soverini, S., De Benedittis, C., Polakova, K. M., Linhartova, J., Castagnetti, F., Gugliotta, G., et al. (2016). Next-generation sequencing for sensitive detection of BCR-ABL1 mutations relevant to tyrosine kinase inhibitor choice in imatinib-resistant patients. Oncotarget 7, 21982–21990. doi: 10.18632/oncotarget.8010
Stark, R., Grzelak, M., Hadfield, J. (2019). RNA sequencing: the teenage years. Nat. Rev. Genet. 20, 631–656. doi: 10.1038/s41576-019-0150-2
Stevensid, R. C., Steele, J. L., Glover, W. R., Sanchez-Garcia, J. F., Simpson, S. D., O’rourkeid, D., et al. (2019). A novel CRISPR/Cas9 associated technology for sequence-specific nucleic acid enrichment. PLoS One 14 (4), e0215441. doi: 10.1371/journal.pone.0215441
Sudmant, P. H., Rausch, T., Gardner, E. J., Handsaker, R. E., Abyzov, A., Huddleston, J., et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. doi: 10.1038/nature15394
Suzuki, A., Suzuki, M., Mizushima-Sugano, J., Frith, M. C., Makałowski, W., Kohno, T., et al. (2017). Sequencing and phasing cancer mutations in lung cancers using a long-read portable sequencer. DNA Res. 24, 585–596. doi: 10.1093/dnares/dsx027
Tasian, S. K., Loh, M. L., Hunger, S. P. (2017). Philadelphia chromosome–like acute lymphoblastic leukemia. Blood 130, 2064–2072. doi: 10.1182/blood-2017-06-743252
Timms, J. A., Relton, C. L., Rankin, J., Strathdee, G., McKay, J. A. (2016). DNA methylation as a potential mediator of environmental risks in the development of childhood acute lymphoblastic leukemia. Epigenomics 8, 519–536. doi: 10.2217/epi-2015-0011
Toyota, M., Kopecky, K. J., Toyota, M. O., Jair, K. W., Willman, C. L., Issa, J. P. (2001). Methylation profiling in acute myeloid leukemia. Blood 97, 2823–2829. doi: 10.1182/blood.v97.9.2823
Volden, R., Palmer, T., Byrne, A., Cole, C., Schmitz, R. J., Green, R. E., et al. (2018). Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA. Proc. Natl. Acad. Sci. U. S. A 115, 9726–9731. doi: 10.1073/pnas.1806447115
Vu, L. P., Pickering, B. F., Cheng, Y., Zaccara, S., Nguyen, D., Minuesa, G., et al. (2017). The N 6 -methyladenosine (m 6 A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376. doi: 10.1038/nm.4416
Wenger, A. M., Peluso, P., Rowell, W. J., Chang, P. C., Hall, R. J., Concepcion, G. T., et al. (2019). Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 37, 1155–1162. doi: 10.1038/s41587-019-0217-9
Wongsurawat, T., Jenjaroenpun, P., Wassenaar, T. M., Wadley, T. D., Wanchai, V., Akel, N. S., et al. (2018). Decoding the epitranscriptional landscape from native rna sequences. bioRxiv 17, 487819. doi: 10.1101/487819
Keywords: nanopore sequencing, blood diseases, target gene sequencing, structural variants, transcriptome, epigenetic modifications
Citation: Minervini CF, Cumbo C, Orsini P, Anelli L, Zagaria A, Specchia G and Albano F (2020) Nanopore Sequencing in Blood Diseases: A Wide Range of Opportunities. Front. Genet. 11:76. doi: 10.3389/fgene.2020.00076
Received: 06 November 2019; Accepted: 23 January 2020;
Published: 19 February 2020.
Edited by:
Youri I. Pavlov, University of Nebraska Medical Center, United StatesReviewed by:
Thidathip Wongsurawat, University of Arkansas for Medical Sciences, United StatesYinbo Zhang, University of Washington, United States
Copyright © 2020 Minervini, Cumbo, Orsini, Anelli, Zagaria, Specchia and Albano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Albano, ZnJhbmNlc2NvLmFsYmFub0B1bmliYS5pdA==
†These authors have contributed equally to this work