 Qitao Zhan1†
Qitao Zhan1† Liya Wang1†
Liya Wang1† Xiangrong Xu1
Xiangrong Xu1 Yan Sun2Lejun Li1Xuchen Qi3Feng Chen1Xiaoming Wei4Michael L. Raff5Ping Yu6
Yan Sun2Lejun Li1Xuchen Qi3Feng Chen1Xiaoming Wei4Michael L. Raff5Ping Yu6 Fan Jin1*
Fan Jin1*- 1Key Laboratory of Reproductive Genetics (Ministry of Education), Department of Reproductive Endocrinology, Women’s Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Department of Obstetrics and Gynecology, Beijing Tongren Hospital, Capital Medical University, Beijing, China
- 3Department of Neurosurgery, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 4BGI-Shenzhen, Shenzhen, China
- 5Genomics Institute, MultiCare Health System, Tacoma, WA, United States
- 6Department of Cell Biology and Medical Genetics, Zhejiang University School of Medicine, Hangzhou, China
Background: Familial adenomatous polyposis (FAP) is an autosomal dominant disorder characterized primarily by the development of numerous adenomatous polyps in the colon and a high risk for colorectal cancer. FAP is caused by germline mutations of the adenomatous polyposis coli (APC) gene. The proband in this family was a 39-year-old female patient with the pathologic diagnosis of adenomatous polyps, and then a five-generation kindred with FAP was characterized in the following years. This article identified an APC mutation, and demonstrated the practical use of APC-linked STR markers, which could be used to reduce misdiagnosis of prenatal diagnosis or preimplantation genetic diagnosis resulted from contamination or allele drop-out.
Methods: Next-generation sequencing (NGS) was used to identify the possible APC mutations in an affected individual from a family with autosomal dominant colon cancer. Targeted sequencing then used to identify additional related individuals with the mutation. Three short tandem repeat (STR) loci, D5S299, D5S134, and D5S346, were used for PCR-based microsatellite analysis of the APC gene in the extended family.
Results: We identified an APC: p.W553X mutation. The STR haplotype at the APC locus, A1B4C1, was shared by all clinically affected individuals with the APC: p.W553X mutation. In addition, the APC: p.D1822V variant was observed in 40% affected individuals and in two unaffected individuals.
Conclusion: We described a protein truncation mutation, APC: p.W553X; demonstrated the value of APC-linked STR markers (D5S299, D5S134, and D5S346) haplotypes; and suggested the potential role of these haplotypes in detecting loss of heterozygosity of the APC gene.
Introduction
Familial adenomatous polyposis (FAP) is an autosomal dominant disorder characterized primarily by the development of numerous adenomatous polyps in the colon and a high risk for colorectal cancer (Koskenvuo et al., 2016). Without a proper diagnosis to ensure heightened colon surveillance and timely surgical intervention, affected individuals are likely to develop colorectal adenocarcinoma. FAP is a clinically heterogeneous disorder, including variation in the age of onset, presence of extracolonic lesions, and the number, distribution, and histology of colorectal adenomas. Even within a single affected pedigree, there is also significant clinical variability.
Familial adenomatous polyposis is caused by germline mutations of the adenomatous polyposis coli (APC) gene, located on chromosome 5q21-22 (van der Luijt et al., 1995; Grover et al., 2012). The coding region of APC encompasses 8532 bp that was distributed among 15 exons. APC gene mutations have been identified in 60–80% of all FAP families and remain unknown in 20–30% (Friedl et al., 2001). Large deletions that evade detection by conventional DNA screening methods, mutations located in introns, regulatory regions or splice-acceptor site, and mutations in other unidentified genes are possible explanations for this low APC gene mutation detection rate (Stekrova et al., 2007; Zhang et al., 2016, 2017; Wang et al., 2017). The use of next-generation sequencing (NGS) has revolutionized the way of mapping a mutation and identifying its molecular identity, leading to massive improvements in our understanding on the role of nucleic acids functions (Doitsidou et al., 2016; Del Vecchio et al., 2017). Targeted sequencing of candidate cancer genes and whole-exome and whole-genome sequencing, coupled with encouraging clinical results based on the use of targeted therapeutics and biomarker-guided clinical trials, are fueling further technological advancements of NGS technology (Khotskaya et al., 2017). Nevertheless, the use of informative polymorphic markers linked to the APC gene may still be valuable in the diagnoses of FAP family members.
Short-tandem repeats (STR) are highly polymorphic two-to-five base pair CA repeat units interspersed throughout the genome. The information provided by polymorphic markers can be markedly increased by constructing haplotypes-defined patterns of alleles at closely linked polymorphic loci. STR markers D5S318 (Wijnen et al., 1991; Gonzalez et al., 2005), D5S299 (van Leeuwen et al., 1991; Wijnen et al., 1991), D5S82 (Breukel et al., 1991; Wijnen et al., 1991), D5S134 and D5S346 are located within 1–10 cM of the APC gene (Wijnen et al., 1991; Flintoff et al., 2001). Of these, D5S134 is within 1 cM of the 5′ end of the gene, and D5S346 is tightly linked within 30–70 kb of the 3′ end of the gene (Wong et al., 1996; Barth et al., 1997). If a particular haplotype of these markers can be linked to the FAP phenotype in a pedigree, genetic diagnosis of pre-symptomatic individuals is possible without requiring an identified APC gene mutation. Furthermore, if a particular haplotype can be linked to a pathogenic APC gene mutation, misdiagnosis of prenatal diagnosis or preimplantation genetic diagnosis resulted from contamination or allele drop-out could be avoided.
In this large kindred, we firstly confirmed the disease through the pathology results. Then we analyzed the whole data of a five-generation kindred with 100 control individuals to find and verify disease related mutations. We used NGS to identify possible APC mutations, and employed APC-linked STR marker analysis to complete the molecular investigation of the five-generation family with the APC mutations.
Method
Patients
The proband (III-17), a 39-year-old female patient with the pathologic diagnosis of adenomatous polyps by local hospital, went to the First Affiliated Hospital of Medical School of Zhejiang University for further treatment. Colonoscopy revealed that the surface of her colon and rectum segment was covered with polyps. There were about 55 polyps per 5 cm in the ileocecal region and about 25 polyps per 5 cm in the relatively sparse area. Then the patient underwent total colorectal resection and gastroduodenostomy with ileal pouch. Microscopic examination showed that the largest polyp, at a distance of 29 cm from the upper incision, showed tubular adenoma change with grade II-III epithelial dysplasia. The rest of the polyps showed tubular adenoma change with grade I-II epithelial dysplasia. Pathological diagnosis was FAP. After the recovery, this patient came to Women’s Hospital, School of Medicine, Zhejiang University, for genetic counseling of preimplantation genetic diagnosis, and became the first to be characterized with APC: p.W553X mutation and APC: p.D1822V variant in this family. Figure 1 shows the pedigree of a five-generation family with FAP. The diagnosis of FAP in affected individuals was based on colonoscopy or histological verification after colectomy. Affected individuals are shaded black (II-11, III-11, III-15, III-17, III-30, and IV-1). The clinical and molecular characterization of individual family members is shown in Table 1. In all, 30 descendants of the first generation and nine biologically unrelated spouses were studied at the molecular level.
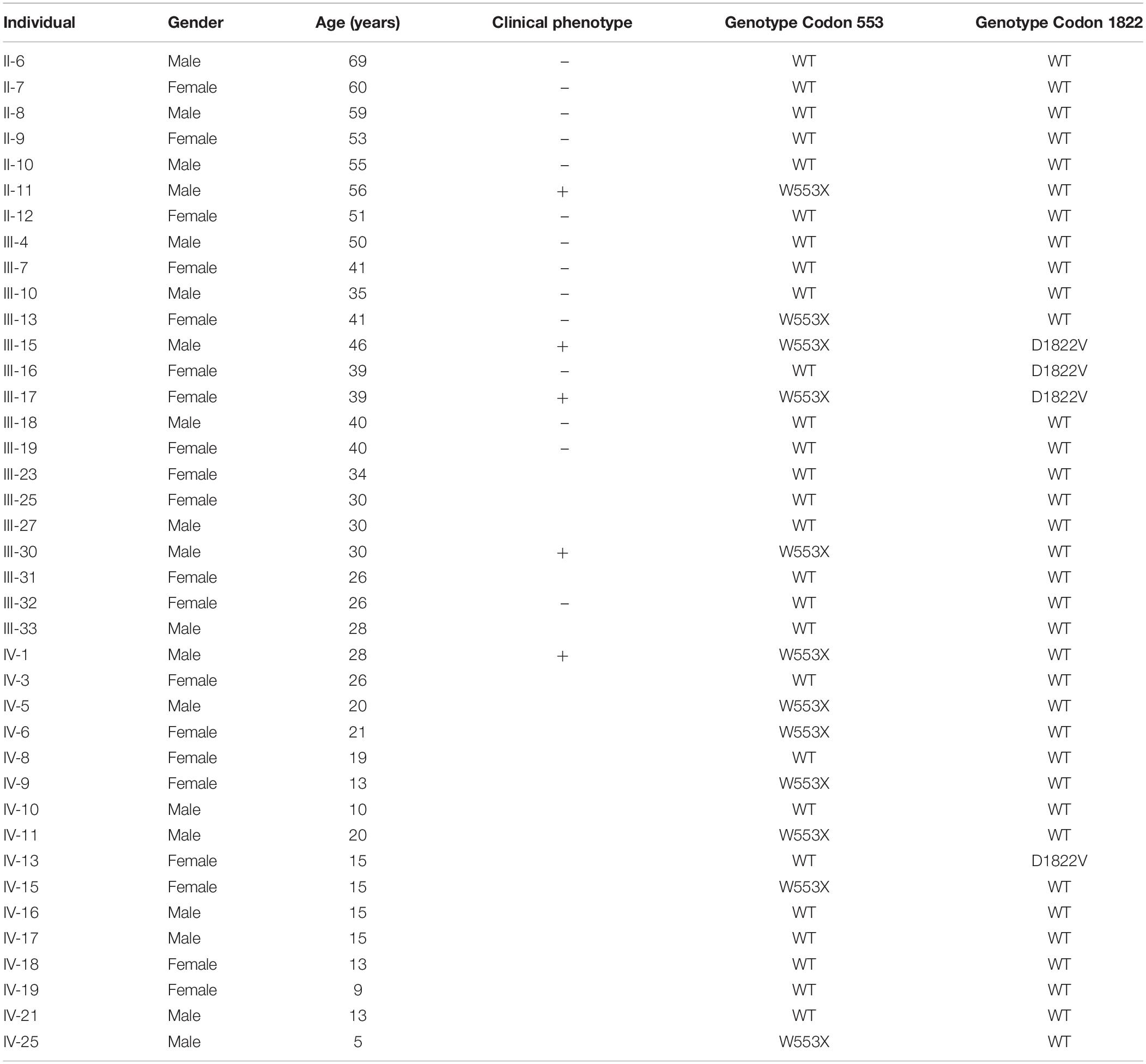
Table 1. Clinical characteristics of individual family members.
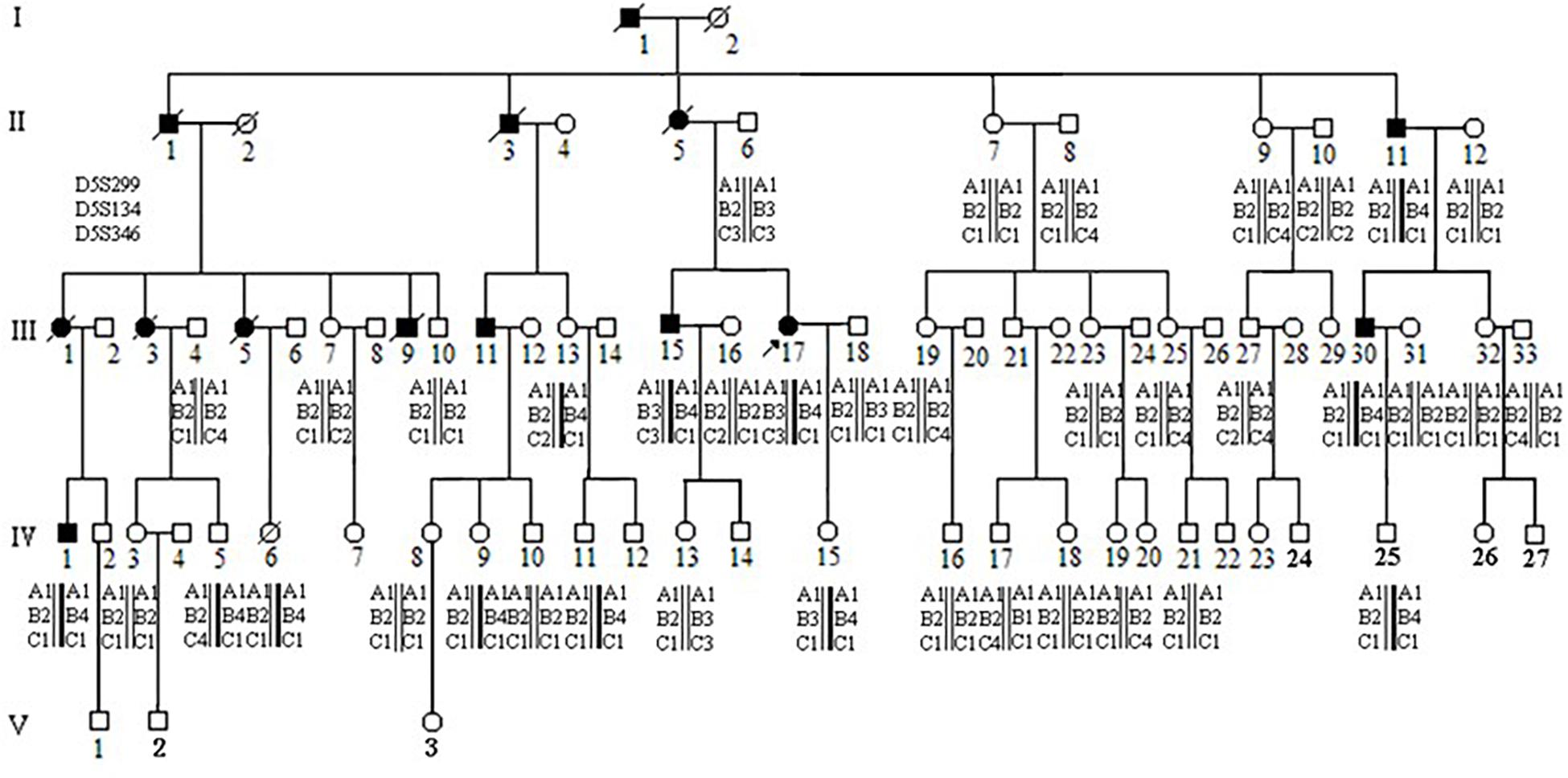
Figure 1. Pedigree and haplotype analysis with STR markers D5S299, D5S134, D5S346. Individual III-17 (arrow) is the proband. By PCR and electrophoresis, D5S299 alleles are designated A1-4; D5S134 alleles, B1-4; and D5S346 alleles, C1-4. Haplotype A1B4C1 (bold) segregates with the disease phenotype.
Morphological Analysis
Individual III-17 underwent a preventive total colectomy in 1998, and the 85 cm resected colon and rectum was studied. Individual III-15 underwent a total colectomy in 1997, and the 65 cm resected colon was studied. The tissues were fixed in 4% paraformaldehyde and embedded in wax. Sections 5 μm thick were stained with hematoxylin and eosin, and observed using an Olympus BX51 microscope.
Detection of APC Gene Mutation
DNA sample was obtained from affected individual III-17 (Figure 1), a 39 year old female with prior colectomy, and sequenced using microarray-based NGS. A custom Sequence Capture 2.1 M Human Array from Roche NimbleGen (Madison, United States) was designed to capture a total of 1.5 Mb of DNA containing 3093 exons (including 100 bp of flanking DNA) of 222 genes associated with common genetic diseases, including APC. The preparation of libraries was consistent with published standard operating protocols (Gnirke et al., 2009). In each pooled batch, 10 to 33 samples were sequenced simultaneously on Illumina HiSeq2000 Analyzers (Illumina, San Diego, United States) for 90 cycles. Image analysis, error estimation, and base calling were performed using Illumina Pipeline software (version 1.3.4). Raw sequence reads were screened following established filtering criteria. Clean reads with a length of 90 bp were aligned to the reference human genome from the NCBI database (GRCh37) using the Burrows Wheeler Aligner (BWA) Multi-Vision software package with output files in BAM format. The BAM data were used for reads coverage in the target region and sequencing depth computation, SNP and INDEL calling, and CNV detection. The APC mutation detected in the proband was not found in 100 control individuals. Direct DNA sequencing was used to determine the FAP mutation status of additional family members.
Sanger Sequencing
Sanger sequencing was then carried out to confirm the identified mutations by exome sequencing. Specific primers for amplifying exon 13 of the APC gene (NM_001127511) were designed using the Primer 3.0 (primer sequencing: 13-Forward CAG CCT CCC AAA GTG ATA GG, and 13-Reverse ATG GCT AAA AGA AGG CAG CA). DNA was amplified in a 20 μl reaction volume: 10 μl of PCR mix (2× HS Taq PCR Mix, Takara, Japan), 0.2 μM of each primer, 100 ng genomic DNA and 7.2 ul of DNase/RNase free water. The PCR cycling profile was as follow: initial denaturation at 94°C for 5 min, followed by 30 cycles at 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 10 min. The PCR products were checked on 2% agarose (w/v) gel. Sequence analysis was run on an ABI 3730XL DNA Analyzer (Applied Biosystems, United States).
Polymorphism in Codon 1822 Confirmed by Restriction Enzyme BpiI
APC: p.D1882V was assayed in all family member and 37 control individuals by restriction enzyme BpiI (5′-GAAGAC(N)2↓-3′, 3′-CTTCTG(N)6↓-5′).
Linkage Analysis
Three short tandem repeat (STR) loci, D5S299 (GDB:180445), D5S134 (GDB:196678), and D5S346 (GDB:181171), were used for PCR-based microsatellite analysis of the APC gene. After amplification, PCR products were separated by 8% native polyacrylamide gel electrophoresis at 800 v for 4–5 h and DNA fragments detected with silver staining. Based on a decreasing migration rate of PCR product, the amplification products of D5S346 were named A1, A2, A3, and A4; D5S134 products were named B1, B2, B3, and B4; D5S299 were named C1, C2, C3, and C4.
Results
Morphological Analysis
III-17
There were approximately 15 small polyps with a diameter range of 0.1–0.6 cm. Histologically, these polyps were typical tubular adenomas or microadenomas with low grade intra-epithelial neoplasia (Figures 2A,B). The characteristic single dysplastic crypts (unicryptal adenomas), the initiation of adenomas, were occasionally found in the colon mucosa (Figures 2C,D).
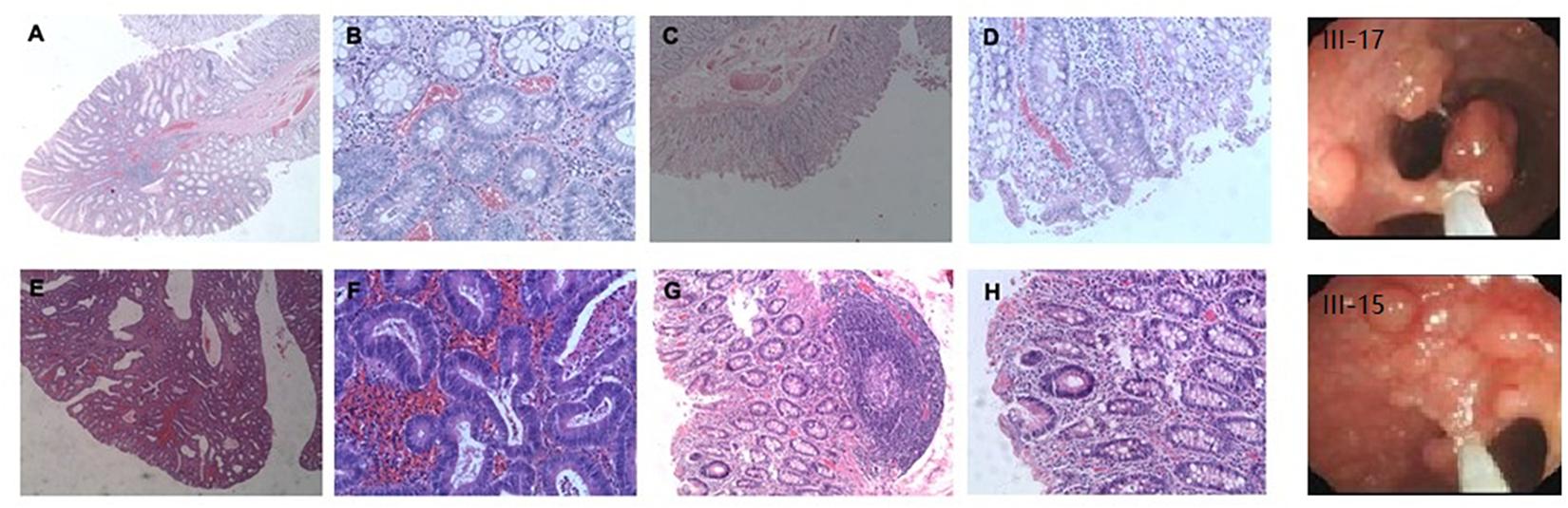
Figure 2. Adenomatous polyps in colorectum of individual III-17 and III-15. (A–D), Polyps in III-17. (E–H) Polyps in III-15. (A,B,E,F) Typical tubular adenomas or microadenomas. (C,D,G,H) Characteristic single dysplastic crypts.
III-15
There were more than 500 polyps with diameters ranging from 0.1 to 2.0 cm in the colon. Histologically, these polyps appeared as tubular adenomas or microadenomas (Figure 2E∗50). Most adenomas showed low grade intra-epithelial neoplasia except for the largest one which was high grade (Figure 2F∗200). The characteristic single dysplastic crypt (unicryptal adenomas) was occasionally found in the colonic mucosa (Figures 2G∗100, H∗200).
Mutation Detection
Microarray-based NGS of the proband (individual III-17 in Figure 1) revealed a novel heterozygous mutation, TGG > TGA (Figure 3). This nonsense mutation substitutes a stop codon for a tryptophan codon (W553X). Direct sequencing identified the same mutation in 11 additional individuals across three generations of the extended family (II-11, III-13, III-15, III-30, IV-1, IV-5, IV-6, IV-9, IV-11, IV-15, and IV-25 in Figure 1), five of whom (II-11, III-15, III-17, III-30, and IV-1) manifest clinical findings of familial adenomatous polyposis, others could not finished the examinations because of the young age or disagreement of their parents.
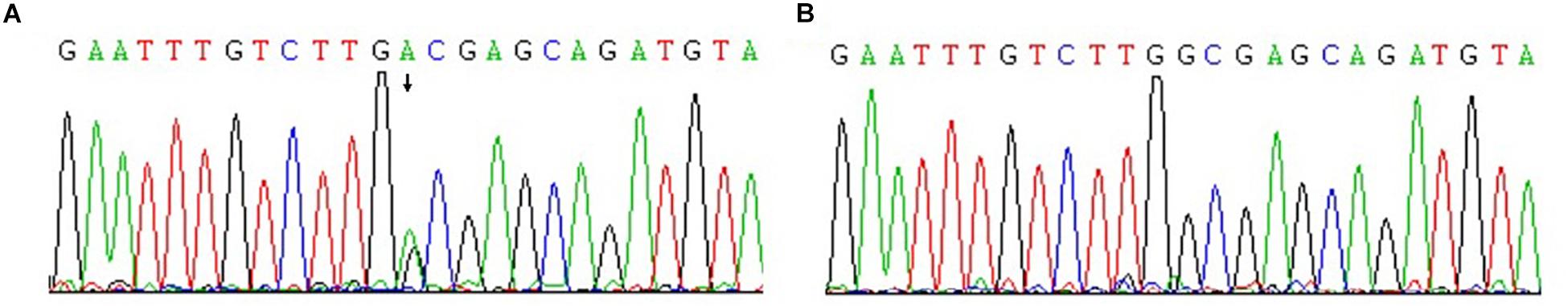
Figure 3. Confirmation of novel G-to-A mutation (arrow) by DNA sequencing. (A) The affected proband (III-17); (B) An unaffected family member.
The proband’s STR haplotype at the APC locus, A1B4C1, was shared by all clinically affected individuals as well as all individuals, affected and unaffected, with the APC: p.W553X mutation (Figure 1).
Interestingly, the APC: p.D1822V variant was observed linked to the A1B3C3 haplotype but was not seen in individual II-6, who passed on this haplotype to his two children. These two children had also inherited clinical findings of FAP from their affected mother (II-5) as well as (by indirect evidence) the A1B3C3 haplotype and the APC: p.W553X mutation. This result implied that the APC: p.D1822V variant had arisen anew in individual II-6 linked to the A1B3C3 haplotype and was present presumably as a gonadal mosaic, which was consistent with previous studies (Iwaizumi et al., 2015; Kahyo et al., 2017).
Discussion
Familial adenomatous polyposis is a hereditary colorectal cancer syndrome that arises from germline mutations in the APC tumor suppressor gene (Lamlum et al., 1999). This study describes a novel c.1659G > A mutation that creates a stop codon at a tryptophan codon–W553X–in the APC protein. The resulting truncated protein product lacks all axin- and β-catenin-binding sites and is therefore presumed defective in the regulation of β-catenin phosphorylation and ubiquitination (Fearnhead et al., 2001). Such a truncated protein might instead function as an activator for Wnt signaling which provides a strong selective advantage by affecting cell proliferation, migration, apoptosis and possibly differentiation of intestinal stem cells (Zhang et al., 2012).
We identified this mutation in 12 individuals among 30 tested in a multi-generational family with familial adenomatous polyposis. Of these twelve, five are clinically affected by the disorder. Among the seven asymptomatic mutation carriers, one (III-13) is 41 years old, while the others are all under 21 years of age. 95% of individuals who have inherited an APC mutation will manifest clinical findings by age 35 years, while only 50% will have manifested findings by age 16 years (Petersen et al., 1991; Aihara et al., 2014). Thus, because of the asymptomatic 41 year old, this family displays incomplete penetrance (Evans et al., 1993). Halling et al. reported two families with truncated mutations at codons 1982–1983 and 1982 with desmoid tumors, which are observed in 10% of FAP families (Halling et al., 1999) but with incomplete penetrance for the development of intestinal polyps 0.16 Even when there is complete penetrance, considerable variation can be seen among FAP patients, both in the spectrum of extracolonic features and in the extent of intestinal polyposis (Fearon, 1997; Koskenvuo et al., 2016). Dinucleotide repeat microsatellite markers D5S299, D5S134, and D5S346, are located immediately adjacent to the APC gene (Koorey et al., 1992; Gray et al., 2011). In this study, all five clinically affected individuals in our FAP extended family shared the same haplotype for these markers: A1B4C1. All were confirmed to carry the APC: p.W553X mutation. Seven additional family members (III-13, IV-5, IV-6, IV-9, IV-11, IV-15, and IV-25) were found to have the same A1B4C1 haplotype. The presence of the APC: p.W553X mutation in these individuals confirmed the accuracy of the haplotyping. These seven individuals are at high risk for developing colorectal cancer and should receive appropriate surveillance, including annual colonoscopy 0.1 This includes the individual who is asymptomatic at 41 years of age (III-13), as non-penetrance cannot be assumed with certainly, and the two adolescent individuals (IV-9 and IV-15). This does not yet include the 5 year old individual (IV-25) as screening does not begin until 10–15 years of age. These STR markers can also be used to detect loss of heterozygosity of the APC locus in tumor tissue from colon cancer as well as from other tumors where loss of the APC gene is implicated, such as gastric cancer, non-small cell lung cancer, renal cell cancer, endometrial cancer, and squamous cell carcinoma.
We observed that the frequency of the APC: p.D1822V variant allele in this family was not higher than in the healthy control population (10% vs. 18.9%), consistent with its reported lack of clinical significance (Ruiz-Ponte et al., 2001; Tranah et al., 2005). This missense variant changes a hydrophilic asparate to a hydrophobic valine residue between the fourth and fifth of the seven 20-amino-acid repeats involved in the binding and down-regulation of β-catenin (Polakis, 1997). Efficient β-catenin down-regulation requires a minimum of three of the seven binding repeats (coinciding with the 3′ limit of the mutation cluster region), suggesting that the APC: p.D1822V polymorphism does not have an appreciable effect on β-catenin degradation (Tranah et al., 2005). This variant is reported in the exome variant server with a frequency of 21%.
Obviously, STR linkages were prominent in this large kindred, but there were no extended evidences for whether these STRs were really useful to search for detecting new APC mutations in other families. They would be useful in LOH (loss of heterozygosity) research, but this would be so when we investigate somatic change in tumors arising in the patients of W553X carriers. Besides, in our study, we were hoping for new discoveries in this FAP family; however, most of these genes were not targeted to FAP and indeed there were no new related findings.
In summary, microarray-based NGS and direct sequencing identified a novel mutation, APC: p.W553X, in a large Chinese kindred with familial adenomatous polyposis. In addition, the value of APC-linked STR marker (D5S299, D5S134, and D5S346) haplotypes was demonstrated in tracking inheritance of APC alleles, and their use in uncovering loss of heterozygosity of the APC gene suggested for future studies.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The studies involving human participants were reviewed and approved by the Key Laboratory of Reproductive Genetics of Women’s Hospital, Zhejiang University School of Medicine. Written informed consent to participate in this study was provided by the participants or their legal guardian/next of kin.
Author Contributions
QZ, LW, and FJ designed the study and performed the major genetic experiments. XX, YS, and PY performed the molecular biological experiments and the analysis of pathological examination. FC was in charge of specimen collection and finished the patients’ follow-ups. XW carried out the next generation sequencing. QZ and XQ wrote the manuscript. YS, LL, and MR revised the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFC1004900) and the National Natural Science Foundation of China (81702974).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SB declared a shared affiliation, though no other collaboration, with one of the authors PY to the handling Editor.
Acknowledgments
We are grateful for the participation of all the patients and their families and for the great effort of Pathologist Bingjian Lv on histopathological analysis.
References
Aihara, H., Kumar, N., and Thompson, C. C. (2014). Diagnosis, surveillance, and treatment strategies for familial adenomatous polyposis: rationale and update. Eur. J. Gastroenterol. Hepatol. 26, 255–262. doi: 10.1097/MEG.0000000000000010
Barth, A. I., Pollack, A. L., Altschuler, Y., Mostov, K. E., and Nelson, W. J. (1997). NH2-terminal deletion of beta-catenin results in stable colocalization of mutant beta-catenin with adenomatous polyposis coli protein and altered MDCK cell adhesion. J. Cell Biol. 136, 693–706. doi: 10.1083/jcb.136.3.693
Breukel, C., Tops, C., van Leeuwen, C., van der Klift, H., Nakamura, Y., Fodde, R., et al. (1991). CA repeat polymorphism at the D5S82 locus, proximal to adenomatous polyposis coli (APC). Nucleic Acids Res. 19:5804. doi: 10.1093/nar/19.20.5804
Del Vecchio, F., Mastroiaco, V., Di Marco, A., Compagnoni, C., Capece, D., Zazzeroni, F., et al. (2017). Next-generation sequencing: recent applications to the analysis of colorectal cancer. J. Transl. Med. 15:246. doi: 10.1186/s12967-017-1353-y
Doitsidou, M., Jarriault, S., and Poole, R. J. (2016). Next-generation sequencing-based approaches for mutation mapping and identification in Caenorhabditis elegans. Genetics 204, 451–474. doi: 10.1534/genetics.115.186197
Evans, D. G., Guy, S. P., Thakker, N., Armstrong, J. G., Dodd, C., Davies, D. R., et al. (1993). Non-penetrance and late appearance of polyps in families with familial adenomatous polyposis. Gut 34, 1389–1393. doi: 10.1136/gut.34.10.1389
Fearnhead, A. S., Britton, M. P., and Bodmer, W. F. (2001). The ABC of APC. Hum. Mol. Genet. 10, 721–733. doi: 10.1093/hmg/10.7.721
Fearon, E. R. (1997). Human cancer syndromes: clues to the origin and nature of cancer. Science 278, 1043–1050. doi: 10.1126/science.278.5340.1043
Flintoff, K. J., Sheridan, E., Turner, G., Chu, C. E., and Taylor, G. R. (2001). Submicroscopic deletions of the APC gene: a frequent cause of familial adenomatous polyposis that may be overlooked by conventional mutation scanning. J. Med. Genet. 38, 129–132. doi: 10.1136/jmg.38.2.129
Friedl, W., Caspari, R., Sengteller, M., Uhlhaas, S., Lamberti, C., Jungck, M., et al. (2001). Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut 48, 515–521. doi: 10.1136/gut.48.4.515
Gnirke, A., Melnikov, A., Maguire, J., Rogov, P., LeProust, E. M., Brockman, W., et al. (2009). Solution hybrid selection with ultra-longoligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189. doi: 10.1038/nbt.1523
Gonzalez, S., Blanco, I., Campos, O., Julia, M., Reyes, J., Llompart, A., et al. (2005). Founder mutation in familial adenomatous polyposis (FAP) in the Balearic Islands. Cancer Genet. Cytogenet. 158, 70–74. doi: 10.1016/j.cancergencyto.2004.07.003
Gray, S. E., Kay, E. W., Leader, M., and Mabruk, M. (2011). Analysis of APC allelic imbalance/loss of heterozygosity and APC protein expression in cutaneous squamous cell carcinomas. Cancer Genomics Proteomics 8, 149–155.
Grover, S., Kastrinos, F., Steyerberg, E. W., Cook, E. F., Dewanwala, A., Burbidge, L. A., et al. (2012). Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA 308, 485–492. doi: 10.1001/jama.2012.8780
Halling, K. C., Lazzaro, C. R., Honchel, R., Bufill, J. A., Powell, S. M., Arndt, C. A., et al. (1999). Hereditary desmoid disease in a family with a germline Alu I repeat mutation of the APC gene. Hum. Hered. 49, 97–102. doi: 10.1159/000022852
Iwaizumi, M., Tao, H., Yamaguchi, K., Yamada, H., Shinmura, K., Kahyo, T., et al. (2015). A novel APC mosaicism in a patient with familial adenomatous polyposis. Hum. Genome Var. 2:15057. doi: 10.1038/hgv.2015.57
Kahyo, T., Iwaizumi, M., Yamada, H., Tao, H., Kurachi, K., and Sugimura, H. (2017). Application of digital PCR with chip-in-a-tube format to analyze Adenomatous polyposis coli (APC) somatic mosaicism. Clin. Chim. Acta 475, 91–96. doi: 10.1016/j.cca.2017.10.015
Khotskaya, Y. B., Mills, G. B., and Mills Shaw, K. R. (2017). Next-generation sequencing and result interpretation in clinical oncology: challenges of personalized cancer therapy. Annu. Rev. Med. 68, 113–125. doi: 10.1146/annurev-med-102115-021556
Koorey, D. J., McCaughan, G. W., Trent, R. J., and Gallagher, N. D. (1992). Dinucleotide repeat polymorphism at the D5S134 locus linked to the adenomatous polyposis coli (APC) gene. Hum. Mol. Genet. 1:655. doi: 10.1093/hmg/1.8.655
Koskenvuo, L., Pitkaniemi, J., Rantanen, M., and Lepisto, A. (2016). Impact of screening on survival in familial adenomatous polyposis. J. Clin. Gastroenterol. 50, 40–44. doi: 10.1097/MCG.0000000000000426
Lamlum, H., Ilyas, M., Rowan, A., Clark, S., Johnson, V., Bell, J., et al. (1999). The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson’s ‘two-hit’ hypothesis. Nat. Med. 5, 1071–1075. doi: 10.1038/12511
Petersen, G. M., Slack, J., and Nakamura, Y. (1991). Screening guidelines and premorbid diagnosis of familial adenomatous polyposis using linkage. Gastroenterology 100, 1658–1664. doi: 10.1016/0016-5085(91)90666-9
Polakis, P. (1997). The adenomatous polyposis coli (APC) tumor suppressor. Biochim. Biophys. Acta 1332, F127–F147.
Ruiz-Ponte, C., Vega, A., Conde, R., Barros, F., and Carracedo, A. (2001). The Asp1822Val variant of the APC gene is a common polymorphism without clinical implications. J. Med. Genet. 38:E33.
Stekrova, J., Sulova, M., Kebrdlova, V., Zidkova, K., Kotlas, J., Ilencikova, D., et al. (2007). Novel APC mutations in Czech and Slovak FAP families: clinical and genetic aspects. BMC Med. Genet. 8:16. doi: 10.1186/1471-2350-8-16
Tranah, G. J., Giovannucci, E., Ma, J., Fuchs, C., and Hunter, D. J. A. P. C. (2005). Asp1822Val and Gly2502Ser polymorphisms and risk of colorectal cancer and adenoma. Cancer Epidemiol. Biomarkers Prev. 14, 863–870. doi: 10.1158/1055-9965.epi-04-0687
van der Luijt, R. B., Vasen, H. F., Tops, C. M., Breukel, C., Fodde, R., and Meera Khan, P. (1995). APC mutation in the alternatively spliced region of exon 9 associated with late onset familial adenomatous polyposis. Hum. Genet. 96, 705–710. doi: 10.1007/bf00210303
van Leeuwen, C., Tops, C., Breukel, C., van der Klift, H., Fodde, R., and Khan, P. M. (1991). CA repeat polymorphism at the D5S299 locus linked to adenomatous polyposis coli (APC). Nucleic Acids Res. 19:5805. doi: 10.1093/nar/19.20.5805-a
Wang, D., Liang, S., Zhang, Z., Zhao, G., Hu, Y., Liang, S., et al. (2017). A novel pathogenic splice acceptor site germline mutation in intron 14 of the APC gene in a Chinese family with familial adenomatous polyposis. Oncotarget 8, 21327–21335. doi: 10.18632/oncotarget.15570
Wijnen, J., Tops, C., Breukel, C., van Leeuwen, C., Goverde, A., van der Klift, H., et al. (1991). CA repeat polymorphism from YAC JW25 at the D5S318 locus, distal to adenomatous polyposis coli (APC). Nucleic Acids Res. 19:6965. doi: 10.1093/nar/19.24.6965-a
Wong, M. H., Hermiston, M. L., Syder, A. J., and Gordon, J. I. (1996). Forced expression of the tumor suppressor adenomatosis polyposis coli protein induces disordered cell migration in the intestinal epithelium. Proc. Natl. Acad. Sci. U.S.A. 93, 9588–9593. doi: 10.1073/pnas.93.18.9588
Zhang, Z., Chen, L., Gao, L., Lin, K., Zhu, L., Lu, Y., et al. (2012). Structural basis for the recognition of Asef by adenomatous polyposis coli. Cell Res. 22, 372–386. doi: 10.1038/cr.2011.119
Zhang, Z., Liang, S., Huang, H., Wang, D., Zhang, X., Wu, J., et al. (2016). A novel pathogenic large germline deletion in adenomatous polyposis coli gene in a Chinese family with familial adenomatous polyposis. Oncotarget 7, 50392–50400. doi: 10.18632/oncotarget.10408
Keywords: familial adenomatous polyposis, APC gene, STR marker, next generation sequencing, mutation
Citation: Zhan Q, Wang L, Xu X, Sun Y, Li L, Qi X, Chen F, Wei X, Raff ML, Yu P and Jin F (2020) An APC Mutation in a Large Chinese Kindred With Familial Adenomatous Polyposis Was Identified Using Both Next Generation Sequencing and Simple STR Marker Haplotypes. Front. Genet. 11:191. doi: 10.3389/fgene.2020.00191
Received: 19 July 2019; Accepted: 18 February 2020;
Published: 04 March 2020.
Edited by:
Ahmed Rebai, Centre of Biotechnology of Sfax, TunisiaReviewed by:
Haruhiko Sugimura, Hamamatsu University School of Medicine, JapanSantasree Banerjee, Zhejiang University, China
Copyright © 2020 Zhan, Wang, Xu, Sun, Li, Qi, Chen, Wei, Raff, Yu and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Jin, amluZmFuQHpqdS5lZHUuY24=
†These authors have contributed equally to this work