 Luca Cozzuto1
Luca Cozzuto1 Huanle Liu1Leszek P. Pryszcz1,2
Huanle Liu1Leszek P. Pryszcz1,2 Toni Hermoso Pulido1Anna Delgado-Tejedor1,3
Toni Hermoso Pulido1Anna Delgado-Tejedor1,3 Julia Ponomarenko1,3*
Julia Ponomarenko1,3* Eva Maria Novoa1,3,4,5*
Eva Maria Novoa1,3,4,5*- 1Centre for Genomic Regulation, The Barcelona Institute of Science and Technology, Barcelona, Spain
- 2International Institute of Molecular and Cell Biology, Warsaw, Poland
- 3Universitat Pompeu Fabra, Barcelona, Spain
- 4Department of Neuroscience, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia
- 5St Vincent’s Clinical School, UNSW Sydney, Darlinghurst, NSW, Australia
The direct RNA sequencing platform offered by Oxford Nanopore Technologies allows for direct measurement of RNA molecules without the need of conversion to complementary DNA, fragmentation or amplification. As such, it is virtually capable of detecting any given RNA modification present in the molecule that is being sequenced, as well as provide polyA tail length estimations at the level of individual RNA molecules. Although this technology has been publicly available since 2017, the complexity of the raw Nanopore data, together with the lack of systematic and reproducible pipelines, have greatly hindered the access of this technology to the general user. Here we address this problem by providing a fully benchmarked workflow for the analysis of direct RNA sequencing reads, termed MasterOfPores. The pipeline starts with a pre-processing module, which converts raw current intensities into multiple types of processed data including FASTQ and BAM, providing metrics of the quality of the run, quality-filtering, demultiplexing, base-calling and mapping. In a second step, the pipeline performs downstream analyses of the mapped reads, including prediction of RNA modifications and estimation of polyA tail lengths. Four direct RNA MinION sequencing runs can be fully processed and analyzed in 10 h on 100 CPUs. The pipeline can also be executed in GPU locally or in the cloud, decreasing the run time fourfold. The software is written using the NextFlow framework for parallelization and portability, and relies on Linux containers such as Docker and Singularity for achieving better reproducibility. The MasterOfPores workflow can be executed on any Unix-compatible OS on a computer, cluster or cloud without the need of installing any additional software or dependencies, and is freely available in Github (https://github.com/biocorecrg/master_of_pores). This workflow simplifies direct RNA sequencing data analyses, facilitating the study of the (epi)transcriptome at single molecule resolution.
Introduction
Next generation sequencing (NGS) technologies have revolutionized our understanding of the cell and its biology. However, NGS technologies are heavily limited by their inability to sequence long reads, thus requiring complex bioinformatic algorithms to assemble back the DNA pieces into a full genome or transcriptome. Moreover, NGS technologies require a PCR amplification step, and as such, they are typically blind to DNA or RNA modifications (Novoa et al., 2017).
The field of epitranscriptomics, which studies the biological role of RNA modifications, has experienced an exponential growth in the last few years. Systematic efforts coupling antibody immunoprecipitation or chemical treatment with next-generation sequencing (NGS) have revealed that RNA modifications are much more widespread than originally thought, are reversible (Jia et al., 2011), and can play major regulatory roles in determining cellular fate (Batista et al., 2014), differentiation (Lin et al., 2017; Furlan et al., 2019; Lee et al., 2019) and sex determination (Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017), among others. However, the lack of selective antibodies and/or chemical treatments that are specific for a given modification have largely hindered our understanding of this pivotal regulatory layer, limiting our ability to produce genome-wide maps for 95% of the currently known RNA modifications (Jonkhout et al., 2017; Boccaletto et al., 2018).
Third-generation sequencing (TGS) platforms, such as the one offered by Oxford Nanopore Technologies (ONT), allow for direct measurement of both DNA and RNA molecules without prior fragmentation or amplification (Brown and Clarke, 2016), thus putting no limit on the length of DNA or RNA molecule that can be sequenced. In the past few years, ONT technology has revolutionized the fields of genomics and (epi)transcriptomics, by showing its wide range of applications in genome assembly (Jain et al., 2018), study of structural variations within genomes (Cretu Stancu et al., 2017), 3′ poly(A) tail length estimation (Krause et al., 2019; Workman et al., 2019), accurate transcriptome profiling (Bolisetty et al., 2015; Sessegolo et al., 2019), identification of novel isoforms (Byrne et al., 2017; Križanovic et al., 2018) and direct identification of DNA and RNA modifications (Carlsen et al., 2014; Simpson et al., 2017; Garalde et al., 2018; Leger et al., 2019; Liu et al., 2019; Parker et al., 2020). Thus, not only this technology overcomes many of the limitations of short-read sequencing, but importantly, it also can directly measure RNA and DNA modifications in their native molecules. Although ONT can potentially address many problems that NGS technologies cannot, the lack of proper standardized pipelines for the analysis of ONT output has greatly limited its reach to the scientific community.
To overcome these limitations, workflow management systems together with Linux containers offer an efficient solution to analyze large-scale datasets in a highly reproducible, scalable and parallelizable manner. In the last year, several workflows to analyze nanopore data have become available, which are aimed at facilitating genome assembly (e.g., Katuali),1 genome annotation (e.g., Pinfish2) and single nucleotide polymorphism analyses (e.g., NanoPipe3). However, none of the current available pipelines cannot be used for the analysis of direct RNA sequencing datasets.
Here we provide a scalable and parallelizable workflow for the analysis of direct RNA (dRNA) sequencing datasets, termed MasterOfPores,4 which uses as input raw direct RNA sequencing FAST5 reads, which is a flexible HDF5 format used by ONT to store raw sequencing data, which includes current intensity values, metadata of the sequencing run and base-called fasta sequences, among other features. The MasterOfPores workflow performs both data pre-processing (base-calling, quality control, demultiplexing, filtering, mapping, estimation of per-gene or per-transcript abundances) and data analysis (prediction of RNA modifications and estimation of polyA tail lengths) (Figure 1). Thus, the MasterOfPores workflow facilitates the analysis of nanopore (epi)transcriptomics sequencing data.
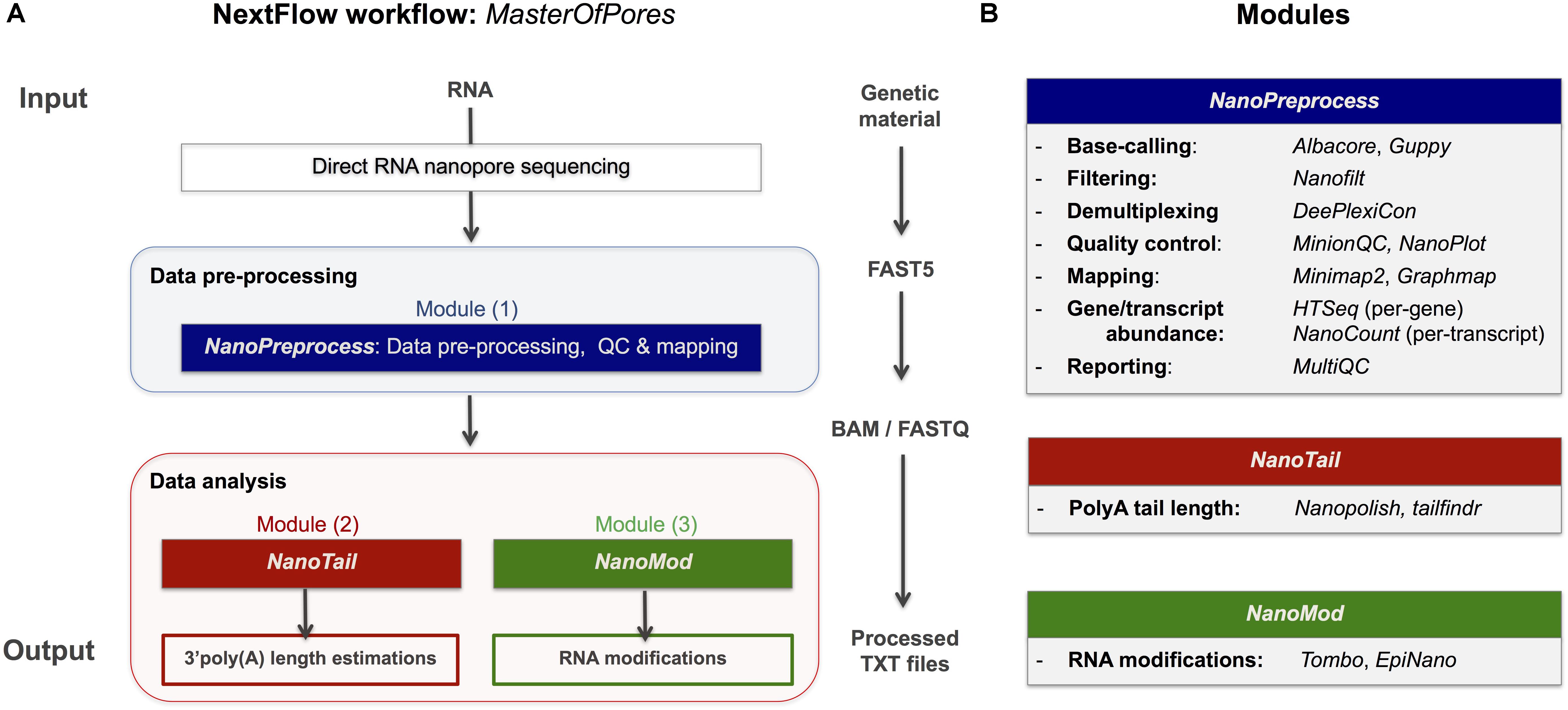
Figure 1. Overview of the MasterOfPores workflow for the processing of direct RNA nanopore sequencing datasets. (A) Overview of the 4 modules included in the MasterOfPores workflow. The pre-processing module (NanoPreprocess) accepts both single FAST5 and multi-FAST5 reads and includes 8 main steps: (i) base-calling, (ii) demultiplexing (iii) filtering, (iv) quality control, (v) mapping and (vi) gene or transcript quantification and (vii) final report building. The outputs generated by NanoPreprocess (BAM, FastQ and base-called Fast5) are used as input by the subsequent MasterOfPores data analysis modules, to predict RNA modifications (NanoMod) and polyA tail length estimations (NanoTail). (B) Detailed description of the individual steps and software used for each of the 4 modules included in MasterOfPores.
For each step, the workflow extracts metrics which are compiled in a final HTML report that can be easily visualized an analyzed by non-expert bioinformaticians. For each sequencing run, the pipeline produces as output a FASTQ file containing the base-called reads, a BAM file containing the mapped reads, and up to three plain text files containing gene or isoform quantifications, polyA tail length estimations and RNA modification predictions. A direct RNA sequencing run produced by MinION or GridION devices, which typically comprises 1-2M reads, takes ∼2 h to process on a CPU cluster using 100 nodes, and ∼1 h or less on a single GPU (see Table 1 for detailed metrics). Moreover, the pipeline can also be run on the cloud (see section “Running on AWS”).
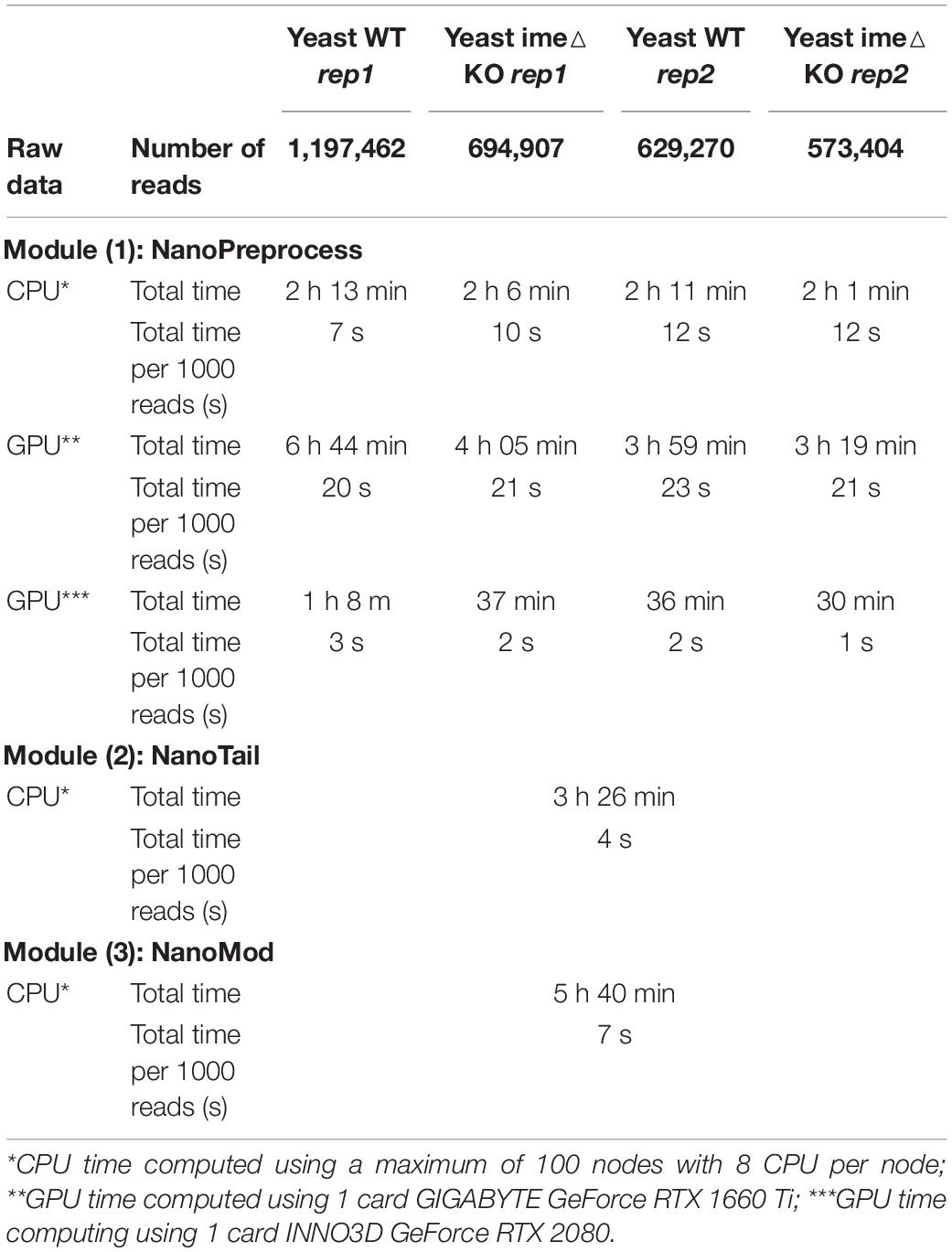
Table 1. Comparison of computing time and RAM used to run the pipeline for the four S. cerevisiae polyA(+) direct RNA sequencing datasets used in this study.
MasterOfPores simplifies the analysis of direct RNA sequencing data by providing a containerized pipeline implemented in the NextFlow framework. It is important to note that this approach avoids the heavy-lifting of installing dependencies by the user, and thus, is simple and accessible to any researcher with little bioinformatics expertise. We expect that our workflow will greatly facilitate the access of Nanopore direct RNA sequencing to the community.
Results
Overview of the MasterOfPores Workflow
Workflow management systems together with Linux containers offer a solution to efficiently analyze large scale datasets in a highly reproducible, scalable and parallelizable manner. During the last decade, an increasing interest in the field has led to the development of different programs such as Snakemake (Köster and Rahmann, 2012), NextFlow (Di Tommaso et al., 2017), Galaxy (Afgan et al., 2018), SciPipe (Lampa et al., 2019) or GenPipes (Bourgey et al., 2019), among others. These tools enable the prototyping and deployment of pipelines by abstracting computational processes and representing pipelines as directed graphs, in which nodes represent tasks to be executed and edges represent either data flow or execution dependencies between different tasks.
Here we chose the workflow framework NextFlow (Di Tommaso et al., 2017) because of its native support of different batch schedulers (SGE, LSF, SLURM, PBS, and HTCondor), cloud platforms (Kubernetes, Amazon AWS, and Google Cloud) and GPU computing, which is crucial for processing huge volumes of data produced by nanopore sequencers. NextFlow has tight integration with lightweight Linux containers, such as Docker and Singularity. Automatic organization of intermediate results produced during the NextFlow pipeline execution allows reducing the complexity of intermediary file names and the possibility of name clashing. Continuous check-pointing with the possibility of resuming failed executions, interoperability and meticulous monitoring and reporting of resource usage are among other thought-after features of NextFlow. The executables of the presented pipeline have been bundled within Docker images accessible at DockerHub that can be converted on the fly into a Singularity image, thus allowing the HPC usage.
The MasterOfPores workflow includes all steps needed to process raw FAST5 files produced by Nanopore direct RNA sequencing and executes the following steps, allowing users a choice among different algorithms (Figure 1). The pipeline consists of 3 modules:
(i) NanoPreprocess: this module takes as input the raw Fast5 reads and produces as output base-called sequences both in FAST5 and FASTQ formats, as well as alignments in BAM format. The pre-processing module performs base-calling, demultiplexing, filtering, quality control, mapping and gene and/or transcript quantification, generating a final report of the performance and results of each of the steps performed.
(ii) NanoTail: this module takes as input the output from the NanoPreprocess module and produces polyA tail length estimations using two different algorithms.
(iii) NanoMod: this module takes as input the files generated during the pre-processing step, and produces flat text files with the predicted RNA modifications using two different algorithms.
Pre-processing Module: NanoPreprocess
The NanoPreprocess module consists of 8 main steps (Figure 2):
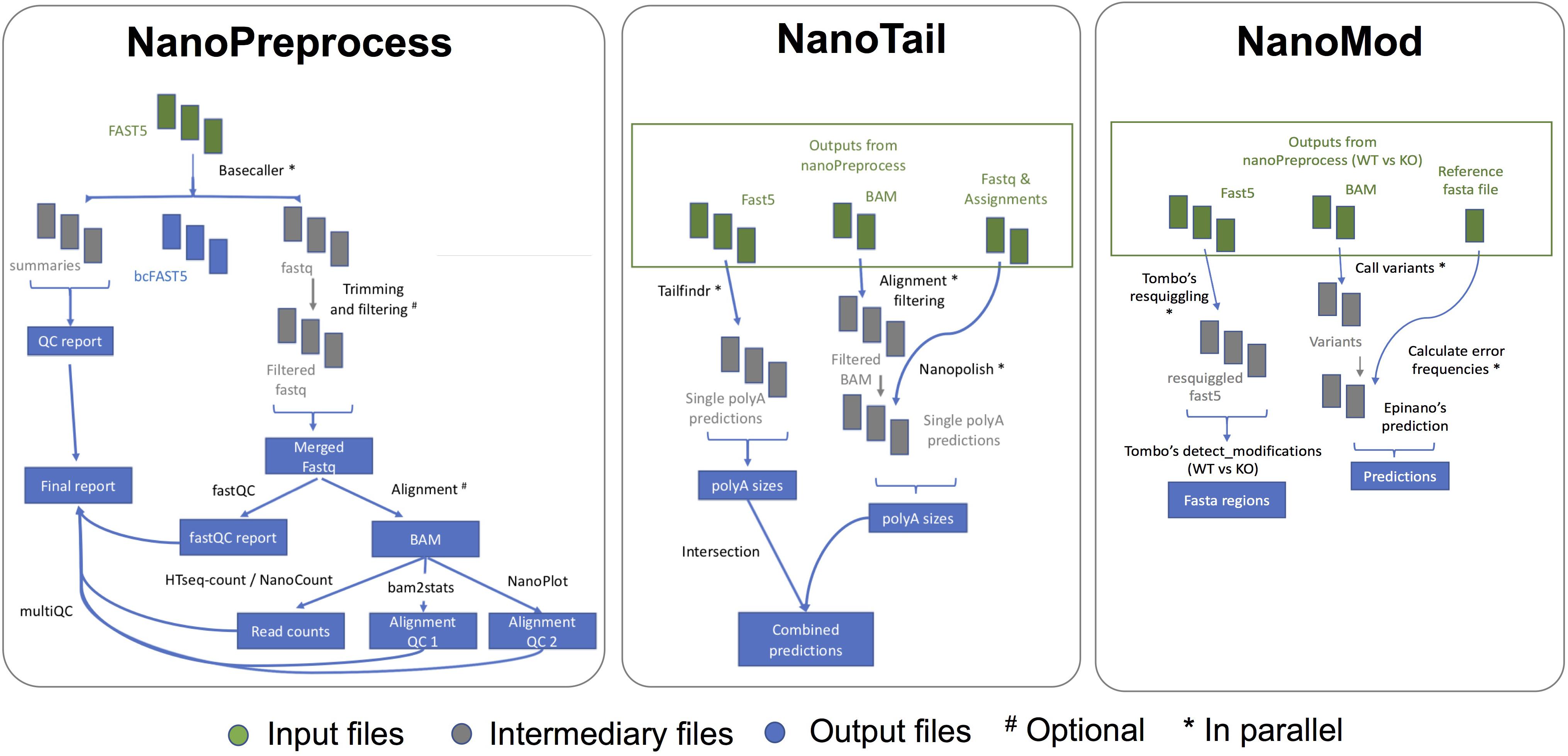
Figure 2. Scheme of the individual steps performed, inputs and outputs of the three modules (NanoPreprocess, NanoTail, and NanoMod) included in MasterOfPores workflow. The inputs required by each module are depicted in green, whereas final outputs generated by each module are shown in blue.
(i) Read base-calling with the algorithm of choice, using Albacore5 or Guppy5. This step can be run in parallel and the user can decide the number of files to be processed in a single job by using the command –granularity.
(ii) Demultiplexing of the reads using DeePlexiCon (Smith et al., 2019). This step is optional, and can only be used if the libraries have been barcoded using the oligonucleotides used to train the deep neural classifier6
(iii) Filtering of the resulting fastq files using Nanofilt (De Coster et al., 2018). This step is optional and can be run in parallel.
(iv) Quality control of the base-called data, using MinIONQC (Lanfear et al., 2019) and FastQC.7
(v) Read mapping to the reference genome or transcriptome, using minimap28 or graphmap2.9
(vi) Quality control on the alignment, using NanoPlot10 and bam2stats.11
(vii) Gene or transcript quantification, using HTSeq (Anders et al., 2015) or NanoCount.12 The latter estimates transcript abundance using an expectation-maximization algorithm. NanoCount will be run if reads have been mapped to the transcriptome, using the flag –reference_type transcriptome, whereas HTSeq will be employed to quantify per-gene counts if the reads have been mapped to the genome.
(viii) Final report of the data processing using multiQC13 that combines the single quality controls done previously, as well as global run statistics (Figure 3).
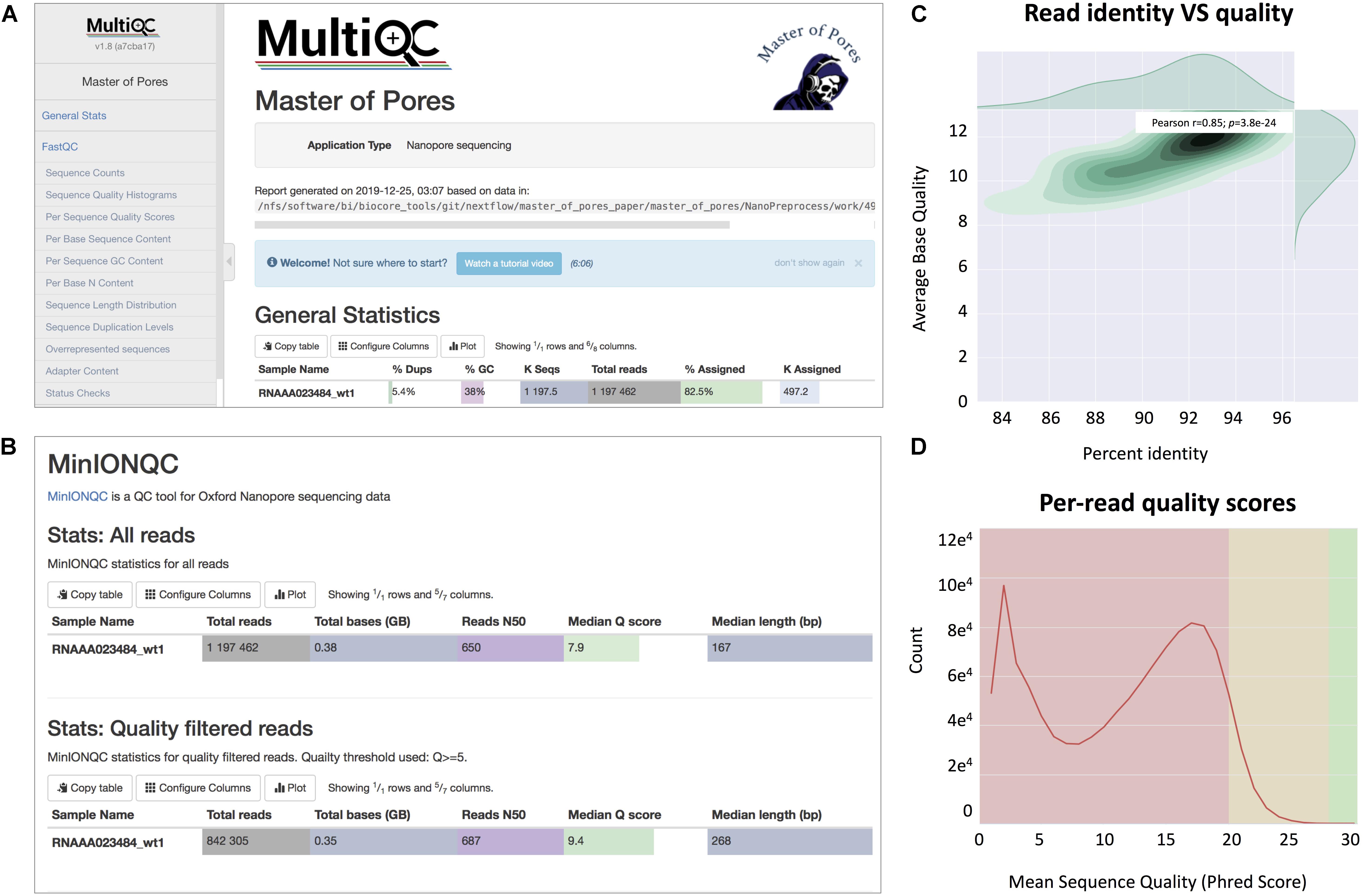
Figure 3. Snapshots of the final report generated by MasterOfPores. (A) Main menu and overview of the final report generated by MasterOfPores. (B) The report includes detailed metrics on the input reads (“MinIONQC”), as well as on the mapped reads (“AlignmentQC”). (C,D) Example of plots that are included as part of the MasterOfPores final report, some of which are generated by integrating Nanoplot (C) and FastQC (D) software.
Data Analysis Modules: NanoTail and NanoMod
The MasterOfPores pipeline contains two additional modules for the downstream analyses of the mapped reads, namely NanoTail and NanoMod, which provide polyA tail length estimations and RNA modification predictions, respectively (Figure 2). The modules can be run using as input the output from the NanoPreprocess module.
The NanoTail module estimates polyA tail lengths using Nanopolish14 and TailfindR,15 producing a plain text file with polyA tail length estimations for each read, computed using both algorithms. The correlation between the two algorithms is also reported as a plot.
The NanoMod module predicts RNA modifications using Tombo16 and EpiNano,17 producing a plain text files with the predicted sites by each algorithm. The NanoMod module is run “paired mode,” i.e., providing two conditions, as both EpiNano and Tombo identify RNA modifications by comparing two conditions.
Running MasterOfPores: Installation, Input, Parameters and Output
To run MasterOfPores, the following steps are required:
(i) Install NextFlow (version 19.10.0):
$ curl -s https://get.nextflow.io | bash
(ii) Clone the MasterOfPores repository:
$ git clone –depth 1 https://github.com/biocorecrg/master_of_pores.git
(iii) Install Docker or Singularity (for Singularity, version 2.6.1 and Docker 19.03 or later are required):
Docker: https://docs.docker.com/install/
Singularity: https://sylabs.io/guides/2.6/user-guide/quick_start.html#quick-installation-steps
(iv) Download Nanopore base-calling algorithms: guppy with or without GPU support and or the albacore Wheel file (a standard built-package format used for Python distributions) and install them inside the bin folder inside the MasterOfPores directory. The users can place their preferred version of guppy and/or albacore in the bin folder (in the example below, albacore version 2.1.7 and guppy 3.1.5).
$ cd master_of_pores/NanoPreprocess/bin
$ tar -zvxf ont-guppy_3.1.5_linux64.tar.gz
$ ln -s ont-guppy_3.1.5_linux64/ont-guppy/bin/guppy_∗.
$ pip3 install –target = ./albacore ont_albacore-2.1.7-cp36-cp36m-manylinux1_x86_64.whl
$ ln -s albacore/bin/multi_to_single_fast5
$ ln -s albacore/bin/read_fast5_basecaller.py
(v) Optional step: install CUDA drivers (only needed for GPU support):
https://docs.nvidia.com/cuda/cuda-installation-guide-linux/index.html
(vi) Run the pre-processing step of the pipeline (using singularity or docker):
$ cd./
$ nextflow run nanopreprocess.nf-with-singularity
or
$ nextflow run nanopreprocess.nf-with-docker
(vii) Run polyA tail estimation module
$ cd./NanoTail
$ nextflow run nanotail.nf-bg-with-singularity –input_folders “.NanoPreprocess/output/RNA∗”
(viii) Run RNA modification prediction module
$ cd./NanoMod
$ nextflow run nanomod.nf -with-singularityinput_path “.NanoPreprocess/output/”
The NanoPreprocess module can handle both single- and multi-FAST5 reads as input. To execute the workflow, several parameters can be defined by the user, including the choice of the basecaller (albacore or guppy), mapper (minimap2 or graphmap2), as well as their command line options. If these are not specified by the user, the workflow will be run with default parameter settings specified in the params.config file (Table 2). The final report includes four different types of metrics: (i) General statistics of the input, including the total number of reads, GC content and number of identical base-called sequences; (ii) Per-read statistics of the input data, including scatterplots of the average read length versus sequence identity, the histogram of read lengths, and the correlation between read quality and identity; (iii) Alignment statistics, including the total number of mapped reads, the total number of mapped bases, the average length of mapped reads, and the mean sequence identity; (iv) Quality filtering statistics, including the number of filtered reads, median Q-score and read length, compared to those observed in all sequenced reads; and (v) Per-read analysis of biases, including information on duplicated reads, over-represented reads and possible adapter sequences (Figure 3). The final outputs of this module include:
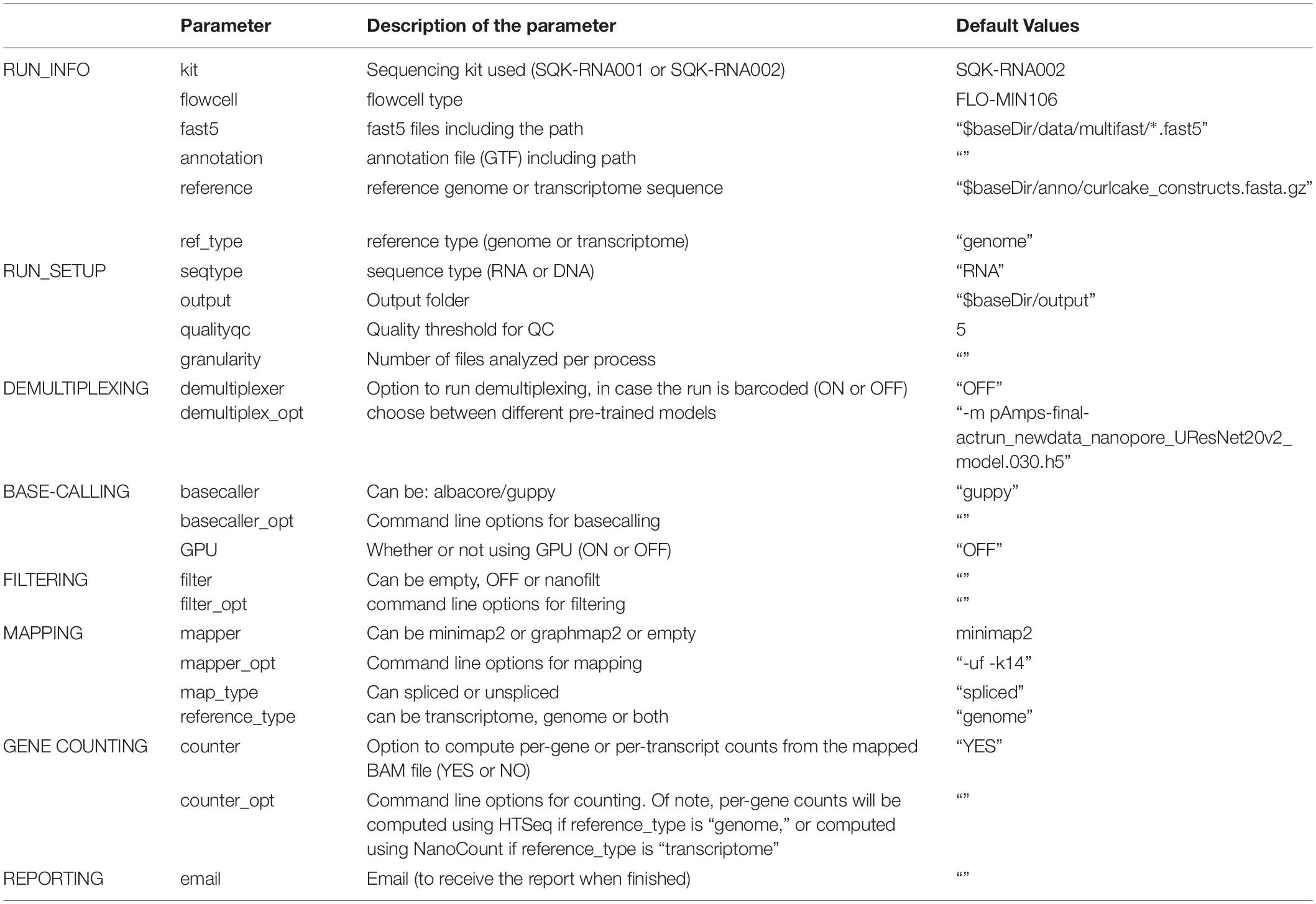
Table 2. Settings and parameters that can be customized to run the NanoPreprocess module of the MasterOfPores workflow.
– Basecalled fast5 files within the “fast5_files” folder.
– Filtered fastq files within “fastq_files” folder.
– QC reports within “QC” folder.
– Final report within “report” folder.
– Aligned reads in sorted BAM files within the “aln” folder.
– Read counts within the “counts” folder.
The NanoMod module requires two samples to detect RNA modifications, typically wild-type and knock-out (or knock-down) matched conditions. The user must provide a tab-delimited file (–comparison “comparison.tsv”) indicating which input file is the wild-type condition and which one is the knock-out or knock-down condition (see, for example18), which is specified in the parameter file. The NanoMod module will output the results into two different folders:
– RNA modification results predicted using Tombo in the “Tombo” folder
– RNA modification results predicted using EpiNano in the “EpiNano” folder
The NanoTail module will output the results into three different folders:
– PolyA tail length estimates predicted using Nanopolish, in the “Nanopolish” folder.
– PolyA tail length estimates predicted using tailfindR, in the “Tailfindr” folder.
– In this module, an additional “NanoMod_final” folder is generated, containing combined Nanopolish and tailfindR estimates of polyA tail lengths, as well as information regarding the geneID or transcriptID where the read is mapped to.
Running MasterOfPores on the Cloud (AWS Batch and AWS EC2)
Nanopore sequencing allows for real-time sequencing of samples. While GridION devices come with built-in GPUs that allows live base-calling, smaller MinION devices do not have built-in CPU or GPU. Thus, the user has to connect the MinION to a computer with sufficient CPU/GPU capabilities, or run base-calling after the sequencing. In all these contexts, the possibility of running the MasterOfPores pipeline on the cloud presents a useful alternative.
The Amazon Web Services (AWS) Batch is a computing service that enables users to submit jobs to a cloud-based user-defined infrastructure, which can be easily set up via either code-based definitions or a web-based interface. Computation nodes can be allocated in advance or according to resource availability. Cloud infrastructure can be also deployed or dismantled on demand using automation tools, such as CloudFormation or Terraform.
Here we show that the MasterOfPores pipeline can be successfully implemented on the cloud, and provide the Terraform script for running MasterOfPores on the AWS Batch CPU environments, available in the GitHub repository.19 To run the pipeline using the AWS Batch, the users needs to change only a few parameters related to their accounts in a configuration file. The pipeline can be run from either a local workstation or an Amazon EC2 entrypoint instance initiated for this purpose (we recommend the latter). Data to be analyzed can be uploaded to an Amazon S3 storage bucket.
Similarly, we also tested whether our pipeline could be run in Amazon Web Services (AWS) Elastic Compute Cloud (EC2), which is one of the most popular cloud services (Supplementary Table S1). Compared to AWS Batch, to run any workflow in AWS EC2, the user must first create an Amazon Machine Image (AMI). The AMI can be created using the same instructions as provided in Supplementary File S1, starting from the official Ubuntu 18.04 LTS AMI, and including both Docker and Singularity software with NVIDIA libraries support. Here we show that the resulting image can be used to run the MasterOfPores workflow with NVIDIA Tesla V100 GPU cards. Automation scripts to run MasterOfPores in AWS EC2 can be found in the GitHub repository.20
Test Case: Analysis of Saccharomyces cerevisiae SK1 PolyA(+) RNA
Running the MasterOfPores Pipeline on S. cerevisiae PolyA(+) RNA
To benchmark the performance of the MasterOfPores workflow, we employed four publicly available direct RNA sequencing runs of polyA(+)-selected S. cerevisiae WT and ime4△ strains, in biological replicates, which had been sequenced using MinION and GridION devices, producing a total of ∼3 million reads (Table 1). We used up to 100 nodes with 8 CPUs for testing the base-calling in CPU mode and 1 node with 1 GPU card for testing the base-calling in GPU mode (Table 1).
The MasterOfPores NanoPreprocess module was ran using guppy version 3.1.5 as the base-caller and minimap2 version 2.17 as the mapping algorithm. Reads were filtered by running nanofilt with the options “-q 0 –headcrop 5 –tailcrop 3 –readtype 1D”. Filtered reads were mapped to the yeast SK1 fasta genome. Specifically, the command that was executed to run the pipeline with these settings was:
$ cd master_of_pores/NanoPreprocess
$ nextflow run nanopreprocess.nf –basecaller guppy –seqtype RNA \
–fast5 “FOLDERNAME/∗.fast5” –demultiplexing “OFF” \
–map_type “spliced” –mapper_opt “-uf -k14” \
–reference genome.fa.gz –mapper minimap2 –ref_type “genome”\
–filter nanofilt –filter_opt “-q 0 –headcrop 5 –tailcrop 3 –readtype 1D”.
Then, the two data analysis modules were executed as follows:
$ nextflow run nanotail.nf –input_folders “./NanoPreprocess/output/∗” \ –nanopolish_opt “” –tailfindr_opt “” –reference “genome.fa.gz”
$ nextflow run nanomod.nf –input_path “./NanoPreprocess/output/” \ –comparison “./comparison.tsv” –reference “genome.fa.gz” \ –tombo_opt “–num-bases 5” –epinano_opt “”
Benchmarking the Time Used for the Analysis of S. cerevisiae PolyA(+) RNA
Here we have tested the pipeline using both CPU and GPU computing. Specifically, we ran the pipeline on the following configurations: (i) a single CPU node (e.g., emulating the computing time on a single laptop); (ii) a CPU cluster with 100 nodes; (iii) a single mid-range GPU card (RTX2080); and (iv) a single high-end GPU card (GTX1080 Ti). We found that the computing time required to run the pipeline on a single GPU card was significantly lower than the running time in parallel on a high performance CPU cluster with 100 nodes, 8 cores per node (Table 1, see also Supplementary Table S1). Moreover, we found that the computing time of the NanoPreprocess module can be significantly reduced depending on the GPU card (base-calling step was ∼2X faster for GTX1080 Ti than for RTX2080).
Reporting Resources Used for the Analysis of S. cerevisiae PolyA(+) RNA
Taking advantage of the NextFlow reporting functions, the pipeline can produce detailed reports on the time and resources consumed by each process (Figure 4), in addition to the output files (bam, fastq) and final report (html), if the workflow is executed with parameters -with-report (formatted report) or-with-trace (plain text report). Running the base-calling on each multi-fast5 file in parallel on our dataset showed that the most memory intensive tasks (about 5 Gbytes) were the mapping step (using minimap2) and the quality control step (using Nanoplot) (Table 3), while the most CPU-intensive and time-consuming step (∼80 min) was the base-calling (using Guppy) (Table 4).
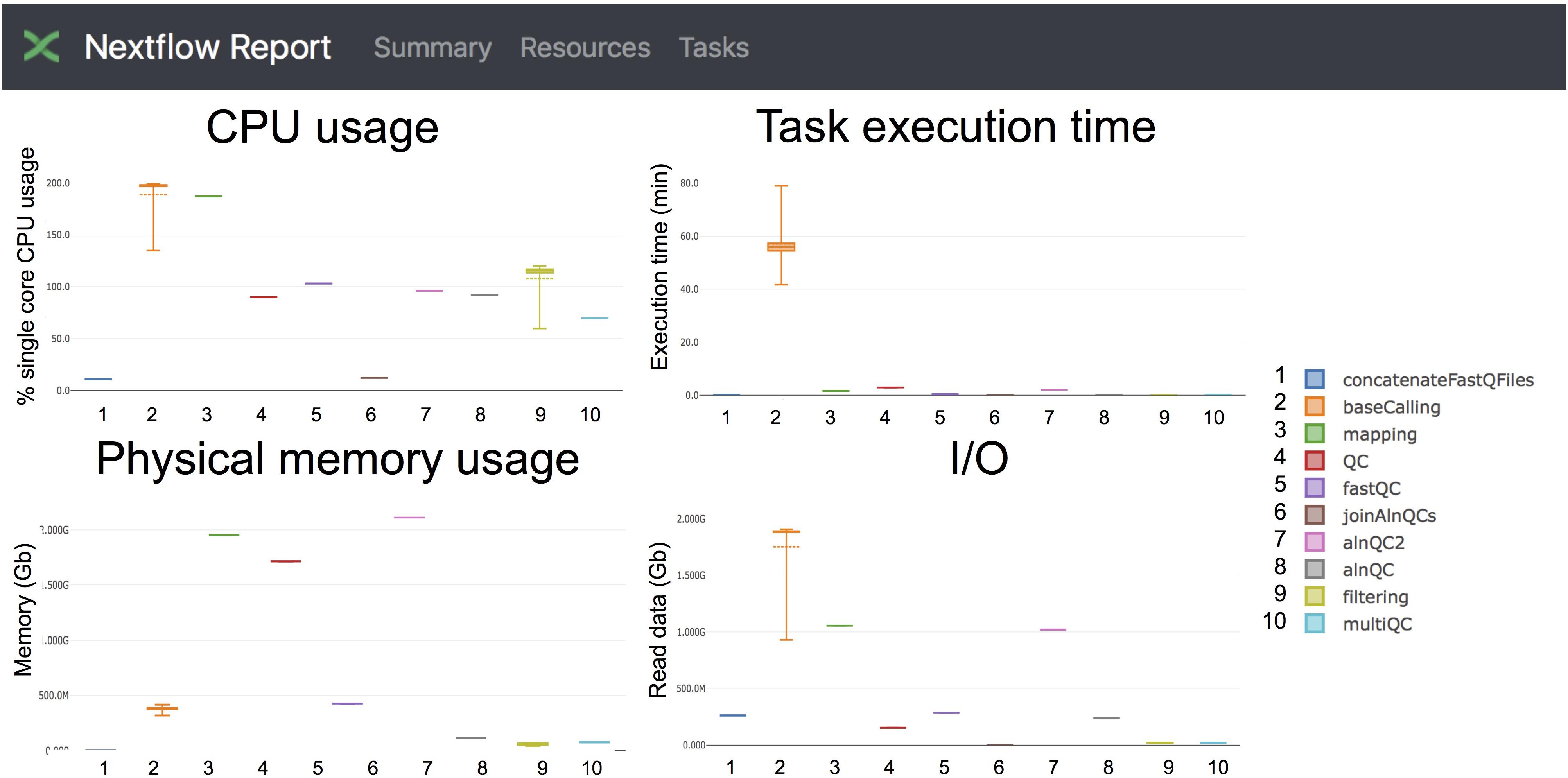
Figure 4. Snapshot of the NextFlow resources report. The report includes detailed information of the computing resources and time needed to execute each of the modules of the pipeline. Base-calling and mapping are the most CPU demanding tasks. The base-calling step is the longest to run, whereas mapping and generation of alignment QC metrics are the most memory-demanding tasks.
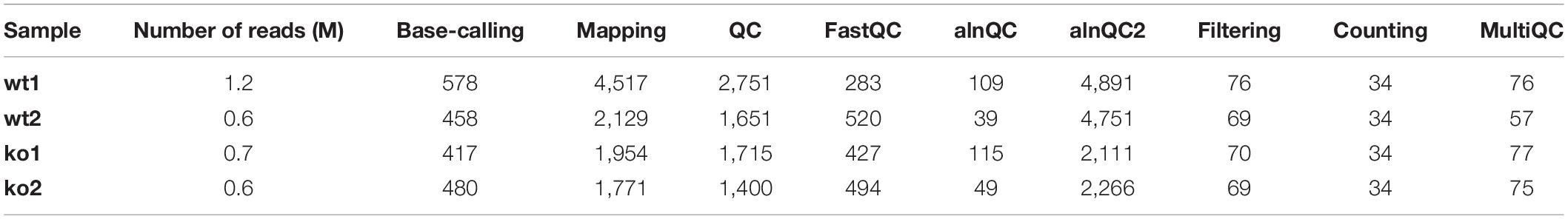
Table 3. RAM peak (Mbytes) used by each of the pre-processing module.
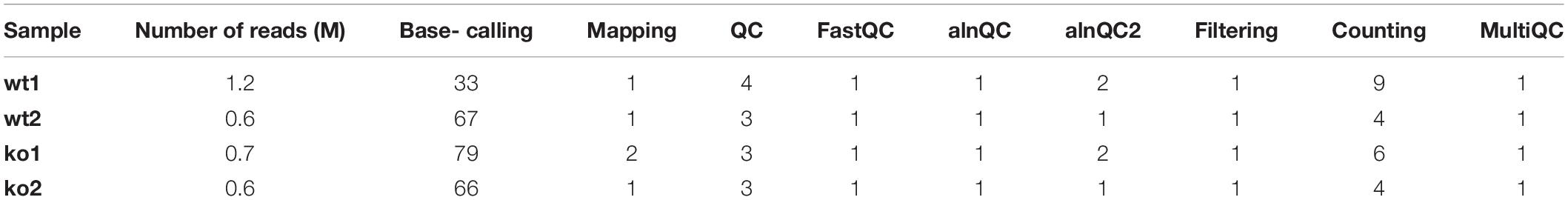
Table 4. CPU time peak (min) used by each of the steps of the pre-processing module.
Finally, we should note that the latest (19.10.0) version of NextFlow allows the user to control the execution of a pipeline remotely. To enable this feature, the user needs to login to the https://tower.nf/website developed by the NextFlow authors and retrieve a token for communicating with the pipeline. For doing that, the user should set this token as an environmental variable and run the pipeline as follows:
$ export TOWER_ACCESS_TOKEN = YOUR_TOKEN
$ cd master_of_pores/NanoPreprocess
$ nextflow run nanopreprocess.nf -with-docker -with-report -bg -with-tower
Discussion
The direct RNA sequencing technology developed by Oxford Nanopore technologies (ONT) offers the possibility of sequencing native RNA molecules, allowing to investigate the (epi)transcriptome at an unprecedented resolution, in full-length RNA molecules and in its native context. Although the direct RNA sequencing library preparation kit was made available in April 2017, only a modest number of researchers have started to adopt this new technology, partly due to the complexity of analyzing the resulting raw FAST5 data. Moreover, even in those cases when specific software and tools have been made available, the users typically experience many difficulties in installing dependencies and running the software. To overcome these issues and facilitate the data analysis of direct RNA sequencing to the general user, we propose the use of NextFlow workflows.
Specifically, we propose the use of MasterOfPores workflow for the analysis of direct RNA sequencing datasets, which is a containerized pipeline implemented in the NextFlow framework. MasterOfPores can handle both single- and multi-FAST5 reads as input, is highly customizable by the user (Table 2) and produces informative detailed reports on both the FAST5 data processing and analysis (MultiQC report, Figure 3) as well as on the computing resources used to perform each step (NextFlow report, see Figure 4). Thus, the current outputs of the MasterOfPores workflow include: (i) base-called FAST5 files, (ii) base-called fastq file, (iii) sorted BAM file containing mapped reads, (iv) per-gene or per-transcript counts (depending on algorithm choice), (v) MultiQC report, (vi) NextFlow report, (vii) per-read polyA tail length estimations, including the correlation of predictions using two distinct algorithms, and (viii) per-site RNA modification predictions, including a final plain text file containing the consensus sites predicted by two distinct algorithms.
The process of Nanopore read base-calling, that is, converting ion current changes into the sequence of RNA/DNA bases, has significantly improved during the last few years, mainly due to the adoption of deep learning approaches, such as the use of convolutional neural networks (CNNs) and recurrent neural networks (RNNs), which are currently the most commonly used strategies for base-calling. The adoption of RNN and CNN-based base-calling algorithms has led to a dramatic improvement in base-calling accuracy. However, this has come at the expense of a higher computational cost: only 5–10 reads can be base-called on 1 CPU core per second using the latest versions of the base-calling algorithms. The use of graphic processing units (GPUs) can greatly accelerate certain CPU-intensive computational tasks, thus allowing to process 50–500 reads per second (Supplementary Table S1). We therefore developed our pipeline for both CPU and GPU computing. Moreover, we provide the GPU-enabled docker image and detailed information on how to setup the GPU computing (see section: “Running MasterOfPores”). We encourage users to adopt the GPU computing for the analysis of Nanopore sequencing data whenever possible, as this option is both more time- and cost-efficient.
Materials and Methods
Code Availability
The pipeline is publicly available at https://github.com/biocorecrg/master_of_pores under an MIT license. The example input data as well as expected outputs are included in the GitHub repository. Detailed information on program versions used can be found in the GitHub repository. EpiNano was modified from its original version (1.0) to decrease the computing time of the pipeline (EpiNano version 1.1, available at https://github.com/enovoa/EpiNano).
Documentation Availability
Detailed documentation on how to install and use the pipeline can be found at: https://biocorecrg.github.io/master_of_pores/
Availability of Docker Files and Docker Images
The pipeline uses software that is embedded within Docker containers. Docker files are available in the GitHub repository.21 The pipeline retrieves a specific Docker image from DockerHub. In particular, the workflow retrieves four distinct images: one for basecalling,22 one for demultiplexing,23 one for pre-processing24 and one for measuring polyA tail lengths and detecting RNA modifications.25
Integration of Base-Calling Algorithms in the Docker Images
Due to the terms and conditions that users agree to when purchasing Nanopore products, we are not allowed to distribute Nanopore software (binaries or in packaged form like docker images). While the original version of the MasterOfPores pipeline includes both guppy and albacore, we are not legally allowed to distribute it with the binaries. Therefore, here we only make available a version where the binaries must be downloaded and placed into a specific folder by the user. We expect future versions of MasterOfPores will include these programs within the docker image once this issue is solved.
CPU and GPU Computing Time and Resources
The MasterOfPores workflow was tested both locally (using either CPU or GPU) as well as in the cloud (AWS). Computing times for each mode are shown in Table 1. CPU time was determined using a maximum of 100 nodes simultaneously with maximum 8 cores CPU per node (2.8–3.5 GHz, 80–130 Watt). GPU time was computed using either GIGABYTE GeForce RTX 1660 Ti (1536 CUDA cores @ 1770 MHz with 6GB of GDDR6 vRAM memory, 120 Watt) or INNO3D GeForce RTX 2080 (2944 CUDA cores @ 1710 MHz with 8 GB of GDDR6 vRAM memory, 225 Watt) or NVIDIA Tesla V100 (5120 CUDA cores + 640 Tensor cores @ 1462 MHz with 16 GB of HBM2 memory). For GPU computing, both system memory (RAM) and GPU memory (vRAM) are used. Base-calling with guppy typically uses 1 or 4.2 Gb of vRAM in fast and high accuracy mode, respectively. As a result, only one base-calling process can be performed on above mentioned cards in high accuracy mode at given time. The execution time in the AWS EC2 p3.2xlarge instance involves reading files already placed in a previously set-up S3 storage bucket but not writing back output results into it.
Data Availability Statement
Direct RNA sequencing datasets for Saccharomyces cerevisiae SK1 PolyA(+) RNA were taken from publicly available GEO datasets (GSE126213).
Author Contributions
LC wrote the pipeline. HL optimized EpiNano code to incorporate it into the pipeline and tested the pipeline. LP tested the pipeline and implemented GPU computing for containers. TP implemented and tested the workflow for AWS cloud computing. AD-T tested the pipeline with MinION, GridION, and PromethION sequencing runs and helped with the optimization of the NanoPreprocess module. JP and EN supervised the work. LC and EN made figures and tables. EN conceived the project. LC, JP, and EN wrote the manuscript, with contributions from all authors.
Funding
This work was partly supported by the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) (PGC2018-098152-A-100 to EN) and by the Australian Research Council (DP180103571 to EN). LP was supported by funding from the European Union’s H2020 Research and Innovation Programme under Marie Skłodowska-Curie grant agreement no. 754422. We acknowledge the support of the MEIC to the EMBL partnership, Centro de Excelencia Severo Ochoa and CERCA Programme/Generalitat de Catalunya. This article is based upon work from COST Action EPITRAN supported by COST (European Cooperation in Science and Technology).
Conflict of Interest
EN has received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the members of the Novoa lab for their valuable insights and discussion. We would also like to thank Benjamin Lang for letting us benchmark our pipeline on his hardware (GTX1080 Ti).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00211/full#supplementary-material
Footnotes
- ^ https://github.com/nanoporetech/katuali
- ^ https://github.com/nanoporetech/pipeline-pinfish-analysis
- ^ https://github.com/IOB-Muenster/nanopipe2
- ^ https://biocorecrg.github.io/master_of_pores/
- ^ https://nanoporetech.com
- ^ https://github.com/Psy-Fer/deeplexicon
- ^ http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- ^ https://github.com/lh3/minimap2
- ^ https://github.com/lbcb-sci/graphmap2
- ^ https://github.com/wdecoster/NanoPlot
- ^ https://github.com/lpryszcz/bin
- ^ https://github.com/a-slide/NanoCount
- ^ https://github.com/ewels/MultiQC
- ^ https://github.com/jts/nanopolish
- ^ https://github.com/adnaniazi/tailfindr
- ^ https://github.com/nanoporetech/tombo
- ^ https://github.com/enovoa/EpiNano
- ^ https://github.com/biocorecrg/master_of_pores/NanoMod/comparison.tsv
- ^ https://biocorecrg.github.io/master_of_pores/
- ^ https://biocorecrg.github.io/master_of_pores/
- ^ https://github.com/biocorecrg/master_of_pores/tree/master/docker/
- ^ https://cloud.docker.com/u/biocorecrg/repository/docker/biocorecrg/mopbasecall
- ^ https://hub.docker.com/repository/docker/biocorecrg/mopdem
- ^ https://cloud.docker.com/u/biocorecrg/repository/docker/biocorecrg/mopprepr
- ^ https://cloud.docker.com/u/biocorecrg/repository/docker/biocorecrg/mopmod
References
Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Cech, M., et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. doi: 10.1093/nar/gky379
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Batista, P. J., Molinie, B., Wang, J., Qu, K., Zhang, J., Li, L., et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. doi: 10.1016/j.stem.2014.09.019
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., Wirecki, T. K., et al. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307. doi: 10.1093/nar/gkx1030
Bolisetty, M. T., Rajadinakaran, G., and Graveley, B. R. (2015). Determining exon connectivity in complex mRNAs by nanopore sequencing. Genome Biol. 16:204. doi: 10.1186/s13059-015-0777-z
Bourgey, M., Dali, R., Eveleigh, R., Chen, K. C., Letourneau, L., Fillon, J., et al. (2019). GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience 8:giz037. doi: 10.1093/gigascience/giz037
Brown, C. G., and Clarke, J. (2016). Nanopore development at Oxford Nanopore. Nat. Biotechnol. 34, 810–811. doi: 10.1038/nbt.3622
Byrne, A., Beaudin, A. E., Olsen, H. E., Jain, M., Cole, C., Palmer, T., et al. (2017). Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 8:16027. doi: 10.1038/ncomms16027
Carlsen, A. T., Zahid, O. K., Ruzicka, J. A., Taylor, E. W., and Hall, A. R. (2014). Selective detection and quantification of modified DNA with solid-state nanopores. Nano Lett. 14, 5488–5492. doi: 10.1021/nl501340d
Cretu Stancu, M., van Roosmalen, M. J., Renkens, I., Nieboer, M. M., Middelkamp, S., de Ligt, J., et al. (2017). Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 8:1326. doi: 10.1038/s41467-017-01343-4
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Di Tommaso, P., Chatzou, M., Floden, E. W., Barja, P. P., Palumbo, E., and Notredame, C. (2017). Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319. doi: 10.1038/nbt.3820
Furlan, M., Galeota, E., de Pretis, S., Caselle, M., and Pelizzola, M. (2019). m6A-Dependent RNA dynamics in T Cell differentiation. Genes 10:28. doi: 10.3390/genes10010028
Garalde, D. R., Snell, E. A., Jachimowicz, D., Sipos, B, Lloyd, J. H., Bruce, M et al. (2018). Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206. doi: 10.1038/nmeth.4577
Haussmann, I. U., Bodi, Z., Sanchez-Moran, E., Mongan, N. P., Archer, N., Fray, R. G., et al. (2016). m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540, 301–304. doi: 10.1038/nature20577
Jain, M., Koren, S., Miga, K. H., Quick, J., Rand, A. C., Sasani, T. A., et al. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345. doi: 10.1038/nbt.4060
Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887. doi: 10.1038/nchembio.687
Jonkhout, N., Tran, J., Smith, M. A., Schonrock, N., Mattick, J. S., and Novoa, E. M. (2017). The RNA modification landscape in human disease. RNA 23, 1754–1769. doi: 10.1261/rna.063503.117
Kan, L., Grozhik, A. V., Vedanayagam, J., Patil, D. P., Pang, N., Lim, K.-S., et al. (2017). The m6A pathway facilitates sex determination in Drosophila. Nat. Commun. 8:15737. doi: 10.1038/ncomms15737
Köster, J., and Rahmann, S. (2012). Snakemake–a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. doi: 10.1093/bioinformatics/bts480
Krause, M., Niazi, A. M., Labun, K., Torres Cleuren, Y. N., Müller, F. S., and Valen, E. (2019). tailfindr: alignment-free poly(A) length measurement for oxford nanopore RNA and DNA sequencing. RNA 25, 1229–1241. doi: 10.1261/rna.071332.119
Križanovic, K., Echchiki, A., Roux, J., and Šikic, M. (2018). Evaluation of tools for long read RNA-seq splice-aware alignment. Bioinformatics 34, 748–754. doi: 10.1093/bioinformatics/btx668
Lampa, S., Dahlö, M., Alvarsson, J., and Spjuth, O. (2019). SciPipe: a workflow library for agile development of complex and dynamic bioinformatics pipelines. Gigascience 8:giz044. doi: 10.1093/gigascience/giz044
Lanfear, R., Schalamun, M., Kainer, D., Wang, W., and Schwessinger, B. (2019). MinIONQC: fast and simple quality control for MinION sequencing data. Bioinformatics 35, 523–525. doi: 10.1093/bioinformatics/bty654
Lee, H., Bao, S., Qian, Y., Geula, S., Leslie, J., Zhang, C., et al. (2019). Stage-specific requirement for Mettl3-dependent m6A mRNA methylation during haematopoietic stem cell differentiation. Nat. Cell Biol. 21, 700–709. doi: 10.1038/s41556-019-0318-1
Leger, A., Amaral, P. P., Pandolfini, L., and Capitanchik, C. (2019). RNA modifications detection by comparative Nanopore direct RNA sequencing. BioRxiv. [preprint]. Available at: https://www.biorxiv.org/content/10.1101/843136v1.abstract
Lence, T., Akhtar, J., Bayer, M., Schmid, K., Spindler, L., Ho, C. H., et al. (2016). m6A modulates neuronal functions and sex determination in Drosophila. Nature 540, 242–247. doi: 10.1038/nature20568
Lin, Z., Hsu, P. J., Xing, X., Fang, J., Lu, Z., Zou, Q., et al. (2017). Mettl3-/Mettl14-mediated mRNA N6-methyladenosine modulates murine spermatogenesis. Cell Res. 27, 1216–1230. doi: 10.1038/cr.2017.117
Liu, H., Begik, O., Lucas, M. C., Mason, C. E., and Schwartz, S. (2019). Accurate detection of m6A RNA modifications in native RNA sequences. bioRxiv [preprint]. doi: 10.1038/s41467-019-11713-9
Novoa, E. M., Mason, C. E., and Mattick, J. S. (2017). Charting the unknown epitranscriptome. Nat. Rev. Mol. Cell Biol. 18, 339–340. doi: 10.1038/nrm.2017.49
Parker, M. T., Knop, K., Sherwood, A. V., Schurch, N. J., Mackinnon, K., Gould, P. D., et al. (2020). Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. eLife 9:e49658. doi: 10.7554/elife.49658
Sessegolo, C., Cruaud, C., Da Silva, C., Cologne, A., Dubarry, M., Derrien, T., et al. (2019). Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci. Rep. 9:14908. doi: 10.1038/s41598-019-51470-9
Simpson, J. T., Workman, R. E., Zuzarte, P. C., David, M., Dursi, L. J., and Timp, W. (2017). Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410. doi: 10.1038/nmeth.4184
Smith, M. A., Ersavas, T., Ferguson, J. M., Liu, H., Lucas, M. C., Begik, O., et al. (2019). Barcoding and demultiplexing Oxford Nanopore native RNA sequencing reads with deep residual learning. bioRxiv [pre print]. doi: 10.1101/864322
Keywords: Nextflow, direct RNA sequencing, nanopore, Docker, singularity
Citation: Cozzuto L, Liu H, Pryszcz LP, Pulido TH, Delgado-Tejedor A, Ponomarenko J and Novoa EM (2020) MasterOfPores: A Workflow for the Analysis of Oxford Nanopore Direct RNA Sequencing Datasets. Front. Genet. 11:211. doi: 10.3389/fgene.2020.00211
Received: 25 October 2019; Accepted: 21 February 2020;
Published: 17 March 2020.
Edited by:
Mattia Pelizzola, Italian Institute of Technology, ItalyReviewed by:
Cuncong Zhong, University of Kansas, United StatesSarath Chandra Janga, Indiana University, Purdue University Indianapolis, United States
Copyright © 2020 Cozzuto, Liu, Pryszcz, Pulido, Delgado-Tejedor, Ponomarenko and Novoa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia Ponomarenko, anVsaWEucG9ub21hcmVua29AY3JnLmV1; Eva Maria Novoa, ZXZhLm5vdm9hQGNyZy5ldQ==; ZXZhbWFyaWEubm92b2FAZ21haWwuY29t