 James E. Hall
James E. Hall Elijah S. Lawrence1
Elijah S. Lawrence1 Tatum S. Simonson
Tatum S. Simonson- 1Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Medicine, School of Medicine, University of California, San Diego, La Jolla, CA, United States
- 2Department of Anthropology and Global Health, University of California, San Diego, La Jolla, CA, United States
Human populations at high altitude exhibit both unique physiological responses and strong genetic signatures of selection thought to compensate for the decreased availability of oxygen in each breath of air. With the increased availability of genomic information from Tibetans, Andeans, and Ethiopians, much progress has been made to elucidate genetic adaptations to chronic hypoxia that have occurred throughout hundreds of generations in these populations. In this perspectives piece, we discuss specific hypoxia-pathway variants that have been identified in high-altitude populations and methods for functional investigation, which may be used to determine the underlying causal factors that afford adaptation to high altitude.
Introduction
Many of humankind’s smallest but greatest secrets are packaged within the human genome. Recent advancements have accelerated our ability to unravel these mysteries and discover how the genome contributes to shared and distinct variations in human traits, including those that have been essential for survival. Some of the most striking examples of adaptation within our species occurred in populations that migrated to the Tibetan, Andean, and Ethiopian highlands in the past several millennia. Physiologists first noted distinct characteristics among highland populations centuries ago, postulating that specific traits were helpful or harmful to challenges imposed by environmental hypoxia due to decreased oxygen availability at high altitudes (West, 1998). Given that many highland populations have persisted in such environments for hundreds of generations, it was hypothesized that genetic factors provided an adaptive advantage in these groups.
Within the past decade, it has become increasingly feasible to obtain insight into the evolutionary past of our species through genome-wide scans in search of adaptive signatures that highlight outlier patterns within the genome (Simonson, 2015). While many of the original studies of highland Tibetan, Andean, and Ethiopian populations were based on analysis of “tagging” single nucleotide changes scattered throughout the genome, whole genome sequencing (WGS) tools have afforded an opportunity to cultivate multiple large-scale genome datasets, which provide greater resolution for cross-population comparisons. Further technological and molecular advances have provided additional multi-omics insights (e.g., transcriptomics, epigenomics, proteomics, metabolomics), and gene-editing techniques are now available to manipulate and assess the functional consequences of specific genetic variants and their impact on molecular and physiological pathways. This wealth of information regarding genomic, -omics, and functional variation provides an opportunity to more comprehensively examine aspects of evolutionary change in highlanders and place this information into the context of other large-scale, publicly available data sets.
Genomics and Association Studies
Bioinformatic screens of human genetic variation are increasingly used by geneticists and physiologists to better understand features of the human genome, including variable sites, chromosomal arrangements, and population-level variation. Genome-wide tests of selection have been successfully employed across continental populations, yielding candidate genes that are putatively associated with adaptation to high altitude (Beall et al., 2010; Bigham et al., 2010; Simonson et al., 2010; Yi et al., 2010; Alkorta-Aranburu et al., 2012; Scheinfeldt et al., 2012; Huerta-Sánchez et al., 2013). With regard to the specific challenge of hypoxia at high altitude, key players of the hypoxia inducible factor (HIF) pathway that sense and respond to changes in oxygen availability have been highlighted in hundreds of studies focused on genetic adaptation to high altitude (Bigham and Lee, 2014), and other non-HIF genes have also been reported in more than one study (Simonson, 2015). Given the HIF pathway has been implicated as a master transcriptional regulator of hypoxic response on an evolutionary time scale as far back as metazoans (Semenza, 1999; Giaccia et al., 2003; Wenger et al., 2005; Lendahl et al., 2009), these findings have been a major focus of high-altitude research. Last year, William Kaelin Jr, Sir Peter Ratcliffe, and Gregg Semenza were awarded the Nobel Prize in Physiology and Medicine for their ground-breaking research that provided the first insights into HIF signaling, highlighting the immense impact of this work on health and disease (Prabhakar, 2020).
Perhaps the most celebrated genes identified as targets of adaptation to high altitude are those involved in the HIF pathway, e.g., EGLN1 and EPAS1. Both genes are among the top targets of adaptation in Himalayan populations (Beall et al., 2010; Bigham et al., 2010; Simonson et al., 2010; Yi et al., 2010), and EGLN1 was identified among candidate genes initially described in Andeans as well (Bigham et al., 2009, 2010). EPAS1 was recently detected in a test for early stage selection in Andean populations (Eichstaedt et al., 2017) and has been reported as a selection candidate gene in other highland populations in Central Asia (Pagani et al., 2012; Xing et al., 2013; Hackinger et al., 2016).
WGS analyses of DNA from Neanderthal and Denisovan genomes has also provided evidence that genetic material from these archaic human populations is present in modern humans today. Genetic variants in the EPAS1 gene region in Tibetans, noted as one of the strongest adaptive signatures in Tibetans, is most similar to Denisovan DNA compared to DNA of other human populations (Huerta-Sánchez et al., 2014; Hu et al., 2017). While the functional variants have yet to be determined, these studies suggest archaic genetic admixture provided variation that helped Tibetans adapt to the high-altitude environment. This finding highlights the importance of understanding distinct population histories, and unique genetic backgrounds, in studies of genetic adaptation to high altitude.
Phenotype Associations and Coding Variation
Despite tremendous progress on the genomics front, the precise variants that provide functional benefits for high-altitude adaptation remain largely unknown. Scientists are at the early stages of understanding which specific variants in and around the adaptive gene regions identified in highlanders provide functional benefits afforded by natural selection. Phenotype associations made thus far provide a means for prioritizing genes for further investigation. The first three genomic studies to examine relationships between putatively adaptive genes and phenotypes identified relationships between hemoglobin concentration and a combination of candidate genes that exhibited signals of selection, including EGLN1 and EPAS1 (Beall et al., 2010; Simonson et al., 2010; Yi et al., 2010). EGLN1 encodes PHD2, one of three key prolyl hydroxylase oxygen sensors that target the α subunits of the HIF transcription factor for degradation under normoxic conditions, and EPAS1 encodes HIF-2α, which serves to activate the expression of a few hundred genes in response to hypoxia. The downstream effects of these adaptive targets are extensive and relevant to multiple disease states, ranging from cardiopulmonary disease, metabolic dysfunction, to cancer (Semenza, 2020).
Two amino acid variations in PHD2 (EGLN1) were identified in Tibetans: p.Asp4Glu (p.D4E) and p.Cys127Ser (p.C127S) (Lorenzo et al., 2014). While these are located outside of the known functional domains (McDonough et al., 2006), effects on oxygen Km and protein interactions have been observed, with different models for p.D4E/C127S mutations resulting in gain- or loss-of function (Lorenzo et al., 2014; Song et al., 2014). Due to the likely effects of these two mutations on protein-protein interactions and PHD2’s catalytic activity, further molecular characterization will be required to determine their specific role(s). The variants underlying adaptation at the EGLN1 locus appear to be different in Andeans and remain to be determined (Bigham and Lee, 2014; Heinrich et al., 2019).
A single missense EPAS1 variant p.H194R is reported as a target of selection in Argentinian highlanders (Eichstaedt et al., 2017), yet little is known about how this variation affects protein stability and protein-protein interactions with binding partners (e.g., ARNT and PHD2) in humans. However, several variants in non-human species adapted to altitude have been identified and modeled as gain- (Song et al., 2016; Liu et al., 2019) and loss- (Gou et al., 2014) of-function. Given the highly conserved nature of EPAS1 among species, each of these variants provide critical insight into the convergent adaptation to life at high altitude.
While these proteins are involved in hypoxic signaling, many others are also involved, making it difficult to discern the effects of single mutations in a complex signaling network. Reverse engineering these precise mutations, whereby site-specific point mutations are introduced into cells of a standard background in controlled environments, provides an opportunity to functionally investigate impacts on protein structure, protein-protein interactions, and signal transduction as well as gene expression and downstream effects due to alterations in regulatory variants. Experiments in an “adapted” genome are also valuable if multiple genetic targets of selection in that background are carefully considered.
Genome Editing and Functional Investigation of Adaptation
While WGS and association studies are great tools for identifying and prioritizing genetic variants that might be involved in both natural selection and disease phenotypes, these correlative findings do not provide information regarding causality. Gene-editing tools offer physiology and population genetics communities opportunities to investigate the roles of candidate variants and identify precise sites underlying adaptation to hypoxia inherent to high altitude.
In addition to using existing tools, which allow for the reverse engineering of individual single nucleotide variants (SNVs) in isogenic cell lines, multiple SNVs may be examined simultaneously (i.e., multiplex introduction of population specific haplotypes) (Hoehe et al., 2019). This new approach allows for functional investigation of multiple epistatic mutations that are under selection, even if they are located on different chromosomes. These tools could be used to endogenously introduce population-specific high-altitude-adapted haplotypes in physiologically relevant cells lines that recapitulate hypoxic signaling, oxygen metabolism, and posttranslational modification of hypoxia-inducible transcription factors (e.g., A549 Human Lung Adenocarcinoma cells, Sw.71 human trophoblast cells, and ASC52 adipose derived mesenchymal stem cells).
Other publicly available tools may be used to assist in understanding the relationship between genetic variation and tissue-specific expression, including the Genotype-Tissue Expression (GTEx) database (based on data from 17,000 samples, representing 54 tissues from 948 different donors). This resource, in tandem with epigenetic information gathered from resources such as The Encyclopedia of DNA Elements (ENCODE) Consortium, provide the potential to uncover complex gene regulation networks within a wide array of human tissues (Bernstein et al., 2010; Dunham et al., 2012). Such information may be applied to better understand data from various human studies, including those at high altitude, where tissue- and cell-specific sampling is limited. A brief table of techniques and tools for investigating genetic variants in this context are provided in Table 1.

Table 1. Techniques and tools to investigate genetic variation and their applications.
Future Perspectives on Functional Investigation
As seen in Figure 1, the endogenous introduction of putatively adaptive alleles using a precision genome editing (i.e., base-editing signatures of natural selection into isogenic cell lines) offers an effective proof-of-concept for the functional investigation of locally adapted SNVs (Komor et al., 2016; Gaudelli et al., 2018; Tong et al., 2018). However, reverse engineering individual SNVs associated with high-altitude adaptation does not represent the functional investigation of the full spectrum of human genetic variation (i.e., haplotypes, structural variation (SV), and epigenetic inheritance, etc.). Such heterogeneity can be functionally investigated using a number of techniques, and multiple avenues for growth are anticipated to investigate signatures of natural selection in both in vivo and in vitro model systems. The development of multiplexed base-editing systems, which introduce point mutations in aggregate (i.e., introduce haplotypes under selection), will allow for functional investigation of not only epistatic interactions under selection, but also the functional impact of the inheritance of entire haplotype blocks in model systems (Hoehe et al., 2019).
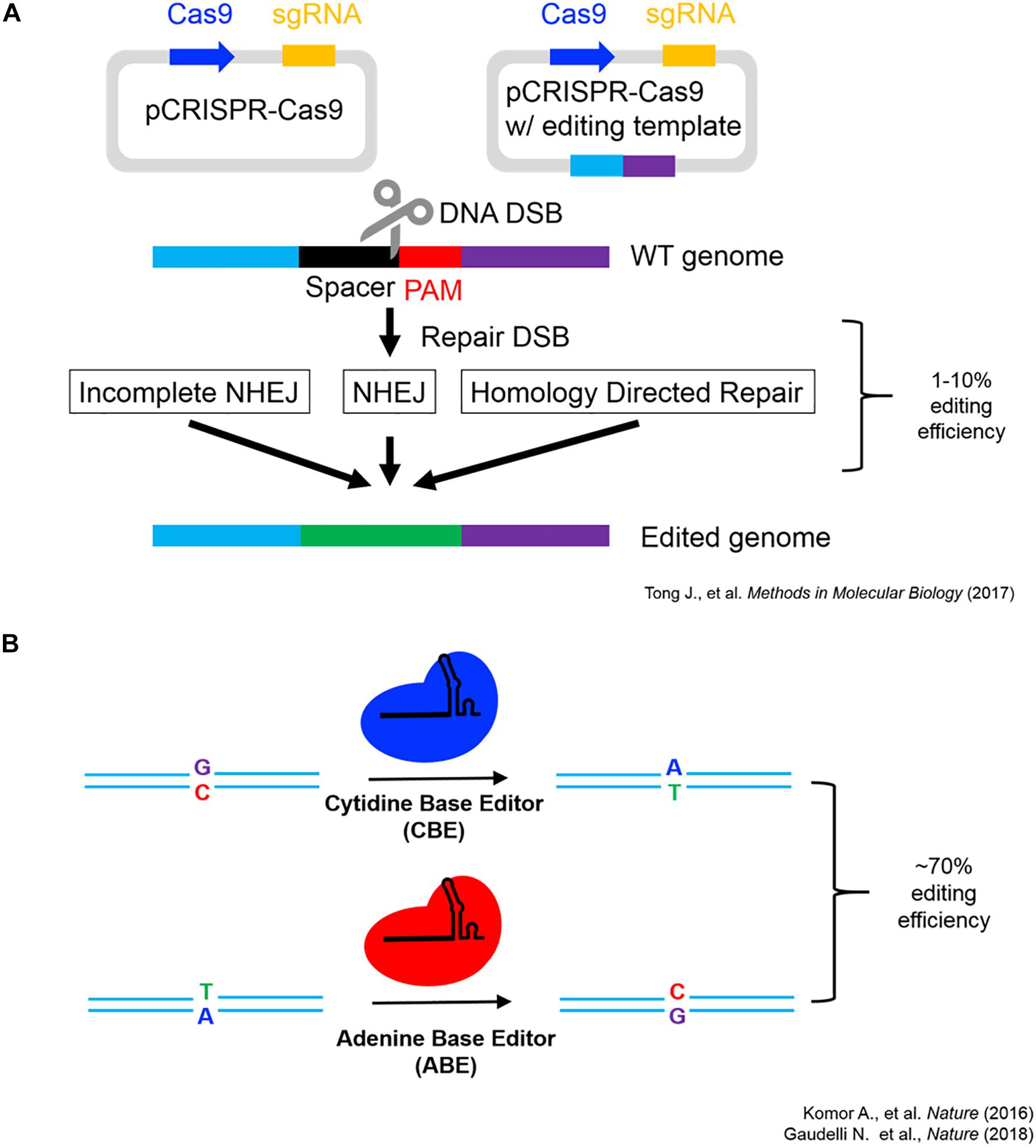
Figure 1. CRISPR-Cas9 schematics and editing efficiency. (A) CRISPR-Cas9 genome editing working model modified from Tong et al. (2018) showcasing the two major routes genome editing via DNA double-strand break repair utilizing non-homologous end joining (NHEJ) or homology-directed repair (HDR) (Tong et al., 2018). (B) Single-base editing utilizing modified Cas9 fusion proteins (Komor et al., 2016; Gaudelli et al., 2018).
Structural Variation
Assessments of structural variation (SV), also known as copy number variation (CNVs), include identification of insertions, deletions, inversions, and translocations that may help explain some unaccounted heritability (Girirajan and Eichler, 2010). Early analyses from SNV-based microarrays identified a 3.4 kb CNV (deletion) ∼80 kb downstream from EPAS1 in Tibetans that is found at low frequency in other populations (Lou et al., 2015). Newly developed population-specific genome assemblies (Hsieh et al., 2019) allow for the identification of novel SVs as well as the discovery of previously unknown human genes. However, with exception of a recent long-read de novo assembly of a Tibetan individual, population-specific SV has been largely unexplored with respect to high-altitude adaptation (Ouzhuluobu et al., 2019). Ouzhuluobu et al. (2019) identified a 163-bp deletion within an intergenic region of the MKL1 gene (megakaryoblastic leukemia translocation 1), which has been associated with lower systolic pulmonary arterial pressure in Tibetans (Ouzhuluobu et al., 2019).
High-resolution population-specific de novo assemblies may also be compared to archaic hominid assemblies (e.g., Neanderthal and Denisovan) to illuminate population-specific evolutionary histories. Recent comparisons of high-altitude adapted populations from the Himalayas and archaic hominids suggest a higher frequency of genome-wide archaic hominid introgression in Himalayan populations relative to East Asian genomes (Huerta-Sánchez et al., 2014). In the de novo assembly study, a potentially introgressed archaic-hominid sequence (i.e., 662-bp intronic insertion) was identified in the SCUBE2 gene, which has been linked with lung function (i.e., FEV1/FVC ratio) in Tibetans (He et al., 2019; Ouzhuluobu et al., 2019). Many high-altitude adapted SVs will require functional investigation in the appropriate in vitro/vivo systems as new genome-editing tools emerge.
Epigenetics and Transgenerational Inheritance
Nuclear genome heterogeneity does not explain the full spectrum of high-altitude adapted phenotypes. Characterizing environmental exposure (past or present) has the potential to offer key insights into epigenetic and transgenerational inheritance. Previous research indicates that altitude-induced epigenetic changes occur (Childebayeva et al., 2019), and DNA methylation in the promoter region of EPAS1 was lower in individuals living at high versus low altitude in Peru (Childebayeva et al., 2019). Another key factor is the number of years lived at high altitude (i.e., there is a positive association within a known methylated locus dependent on the number of years subsisting at high altitude). Overall, these data suggest that epigenetic modifications are potentially involved in high-altitude adaptation both at birth and over the course of a lifetime (Julian, 2019).
Many genomic studies suggest regulatory variation underlies responses to hypoxia (Frisancho, 2009; Nanduri et al., 2012, 2017; Hartley et al., 2013; Brown and Rupert, 2014; Azad et al., 2017; Hu et al., 2017). Multiple studies in other fields have used next-generation sequencing to reveal genome-wide associations between epigenetic modifications and transcriptional regulatory elements and chromatin accessibility (Fukushima et al., 2019). However, there is a dearth of research regarding such data at altitude. Nonetheless, we anticipate the reverse engineering of specific epigenetic modifications and transcriptional states associated with high-altitude adaptation using targeted in vivo/in vitro epigenome editing systems will provide an opportunity to tease apart direct causal/mechanistic relationships.
Organoids and Assembloids
While isogenic cellular models offer a relevant system to functionally evaluate mutations that are putatively under selection, two dimensional (2D) in vitro cellular systems have a number of limitations. Three dimensional (3D) cellular models (i.e., tissue-specific organoids) offer the opportunity to functionally investigate the effects of cellular development, self-organization, and tissue-specific, organ-specific function (Sterneckert et al., 2014; Hubert et al., 2016; McCauley and Wells, 2017). 3D cellular organoids are collections or aggregations of organ-specific cells developed from pluripotent stem cells or organ progenitor cells. The cellular aggregates are self-organizing and may be used to determine spatially restricted lineage commitment similar to in vivo cellular models. When differentiated, 3D organoids are capable of recapitulating some of the functions of an organ of physiological interest (e.g., contraction, neural activity, endocrine secretion, filtration, excretion, etc.).
Precision genome-editing systems could be used to introduce high-altitude specific mutations (e.g., the SNVs reported in EPAS1 or EGLN1) in pluripotent stem cell tissue systems that may be differentiation into physiologically relevant organoid systems. For example, human blood vessel organoids engineered as models of diabetic vasculopathy could also be genome-edited to create isogenic cellular models to functionally investigate the role of specific mutations in the development and arborization of vascular structures/networks putatively implicated in high-altitude adaptation (Wimmer et al., 2019; Wörsdörfer et al., 2019). Utilization of genome-edited 3D organoid models is not limited to vascular structures (For example, through recent emergence of dynamic “assembloid” systems, investigators are able to engineer and self-assemble multiple compartmentalized brain regions from human pluripotent stem cells and asses their connectivity (Trujillo and Muotri, 2018; Yoon et al., 2019). These technological advancements not only allow for the functional investigation of layered tissue-specific development but the functional investigation of the interplay between sub-tissue systems. They offer a glimpse into the future of functional investigation of high-altitude adapted populations, their physiology, and the dynamic relationships between organs and physiology under conditions of hypoxia.
Single-Cell Genome Sequencing
Another avenue for growth regarding the functional investigation of high-altitude adaptation is the emergence of single-cell DNA and RNA analyses. Single-cell genome sequencing analyses allow for analysis of the entire transcriptome that may be integrated with genome, proteome, and metabolome information. By reverse engineering SNVs via precision genome-editing systems into the appropriate physiological cellular model, it is possible to provide complementary high-resolution views of -omics pathways with in single cells adapted to hypoxia.
Reverse Engineering Hypoxic Environments
While physiologically relevant isogenic cell lines combined with genome-editing technology offer great insight into some of the biological mechanisms involved in high-altitude adaptive phenotypes, we recognize that evaluations of function and mechanism require consideration of environmental pressure(s) at high altitude (Fox et al., 2020). During hypoxia, HIF transcription factors are stabilized and regulate various genes such as those involved in oxygen transport, and there are multiple methods used in laboratory settings to induce hypoxia in physiologically relevant cell cultures (Wu and Yotnda, 2011). We anticipate researchers will introduce putatively adapted mutations in physiologically relevant cell lines to simulate conditions of tissue-appropriate levels of hypoxia in control incubation environments. Other species that evolved to subsist at high altitude may be used to identify mutations associated with hypoxia (Storz and Scott, 2019), and these mutations could be edited in the embryotic state to either remove or introduce mutations associated with hypoxia adaptation.
Seeking Higher Ground With Indigenous Peoples
The potential to engineer new tools to functionally investigate genetic variation is an exciting prospect as it establishes greater accountability in research (e.g., in population genetics, reducing the tendency toward evolutionary adoptionism narratives (Gould et al., 1979). While some researchers overlook the importance of oral history, ethnography, linguistic, and archeological data when drawing conclusions about phenotypic observations and evolutionary analyses (e.g., GWAS and polygenic risk scores) (Nielsen, 2009), it is important to recognize that Indigenous populations should be included in the co-development of evolutionary and medically actionable narratives surrounding human genetic variation collected in their community (Jackson et al., 2019; Fox et al., 2020). Additionally, echoing previous observations by Lewinton and Gould, both ‘empirical evidence and detailed theoretical considerations’ should be used for evolutionary explanations of phenotypic variation observed in various Indigenous populations (Claw et al., 2018).
With the generation of data presented thus far and the ability to manipulate and test functional relevance, the scientific community will continue to gain meaningful insights into hypoxia adaptation. The mechanistic characterization of these observations, through the identification of adaptive gene targets like EGLN1 and EPAS1, would not be made possible without partnerships in Indigenous communities thriving at high altitude. Understanding the function of specific variants in these groups contributes to a more complete picture of the human story and some of the most notable examples of adaptation within our species.
Author Contributions
JH, KF, and TS designed the article. JH and KF designed the figures. JH, EL, TS, and KF drafted and contributed comments to the final manuscript.
Funding
Funding for this work was provided by the University of California San Diego Center for Physiological Genomics of Low Oxygen (CPLGO) and National Geographic Explorer Awards to both TS and KF. JH is supported by NIH 5T32HL134632-04, EL is supported by the UC San Diego Ledell Family Undergraduate Research, Chancellor’s Research Excellence, and Eureka! Undergraduate Research Scholarships, KF is supported by the National Geographic Society Research and Exploration Grant 898GGA, and TS is supported by NIH R01HL145470.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alkorta-Aranburu, G., Beall, C. M., Witonsky, D. B., Gebremedhin, A., Pritchard, J. K., and Di Rienzo, A. (2012). The genetic architecture of adaptations to high altitude in ethiopia. PLoS Genet. 8:e1003110. doi: 10.1371/journal.pgen.1003110
Azad, P., Stobdan, T., Zhou, D., Hartley, I., Akbari, A., Bafna, V., et al. (2017). High-altitude adaptation in humans: from genomics to integrative physiology. J. Mol. Med. 95, 1269–1282. doi: 10.1007/s00109-017-1584-7
Beall, C. M., Cavalleri, G. L., Deng, L., Elston, R. C., Gao, Y., Knight, J., et al. (2010). Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc. Natl. Acad. Sci. U.S.A. 107, 11459–11464. doi: 10.1073/pnas.1002443107
Bernstein, B. E., Stamatoyannopoulos, J. A., Costello, J. F., Ren, B., Milosavljevic, A., Meissner, A., et al. (2010). The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 28, 1045–1048. doi: 10.1038/nbt1010-1045
Bigham, A., Bauchet, M., Pinto, D., Mao, X., Akey, J. M., Mei, R., et al. (2010). Identifying signatures of natural selection in tibetan and andean populations using dense genome scan data. PLoS Genet. 6:e1001116. doi: 10.1371/journal.pgen.1001116
Bigham, A. W., and Lee, F. S. (2014). Human high-altitude adaptation: forward genetics meets the HIF pathway. Genes Dev. 28, 2189–2204. doi: 10.1101/gad.250167.114
Bigham, A. W., Mao, X., Mei, R., Brutsaert, T., Wilson, M. J., Julian, C. G., et al. (2009). Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum. Genomics 4, 79–90. doi: 10.1186/1479-7364-4-2-79
Brown, C. J., and Rupert, J. L. (2014). Hypoxia and environmental epigenetics. High Altitude Med. Biol. 15, 323–330.
Childebayeva, A., Jones, T. R., Goodrich, J. M., Leon-Velarde, F., Rivera-Chira, M., Kiyamu, M., et al. (2019). LINE-1 and EPAS1 DNA methylation associations with high-altitude exposure. Epigenetics 14, 1–15. doi: 10.1080/15592294.2018.1561117
Claw, K. G., Anderson, M. Z., Begay, R. L., Tsosie, K. S., Fox, K., Garrison, N. A., et al. (2018). A framework for enhancing ethical genomic research with Indigenous communities. Nat. Commun. 9:2957. doi: 10.1038/s41467-018-05188-3
Dunham, I., Kundaje, A., Aldred, S. F., Collins, P. J., Davis, C. A., Doyle, F., et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Eichstaedt, C. A., Pagani, L., Antao, T., Inchley, C. E., Cardona, A., Mörseburg, A., et al. (2017). Evidence of early-stage selection on EPAS1 and GPR126 genes in andean high altitude populations. Sci. Rep. 7:13042. doi: 10.1038/s41598-017-13382-4
Fox, K., Rallapalli, K. L., and Komor, A. C. (2020). Rewriting human history and empowering indigenous communities with genome editing tools. Genes 11:88. doi: 10.3390/genes11010088
Frisancho, A. R. (2009). Developmental adaptation: where we go from here. Am. J. Hum. Biol. 21, 694–703. doi: 10.1002/ajhb.20891
Fukushima, H. S., Takeda, H., and Nakamura, R. (2019). Targeted in vivo epigenome editing of H3K27me3. Epigenet. Chromatin 12:17. doi: 10.1186/s13072-019-0263-z
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., et al. (2018). Publisher correction: programmable base editing of AT to GC in genomic DNA without DNA cleavage. Nature 559:E8. doi: 10.1038/s41586-018-0070-x
Giaccia, A., Siim, B. G., and Johnson, R. S. (2003). HIF-1 as a target for drug development. Nat. Rev. Drug Discov. 2, 803–811.
Girirajan, S., and Eichler, E. E. (2010). Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 19, R176–R187. doi: 10.1093/hmg/ddq366
Gou, X., Wang, Z., Li, N., Qiu, F., Xu, Z., Yan, D., et al. (2014). Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Res. 24, 1308–1315. doi: 10.1101/gr.171876.113
Gould, S. J., Lewontin, R. C., Maynard Smith, J., and Holliday, R. (1979). The spandrels of San Marco and the panglossian paradigm: a critique of the adaptationist programme. Proc. R. Soc. Lond. B. Biol. Sci. 205, 581–598. doi: 10.1098/rspb.1979.0086
Hackinger, S., Kraaijenbrink, T., Xue, Y., Mezzavilla, M., Asan, Van Driem, G., et al. (2016). Wide distribution and altitude correlation of an archaic high-altitude-adaptive EPAS1 haplotype in the Himalayas. Hum. Genet. 135, 393–402. doi: 10.1007/s00439-016-1641-2
Hartley, I., Elkhoury, F. F., Heon Shin, J., Xie, B., Gu, X., Gao, Y., et al. (2013). Long-lasting changes in DNA methylation following short-term hypoxic exposure in primary hippocampal neuronal cultures. PLoS One 8:e77859. doi: 10.1371/journal.pone.0077859
He, Y., Lou, H., Cui, C., Deng, L., Gao, Y., Zheng, W., et al. (2019). De novo assembly of a Tibetan genome and identification of novel structural variants associated with high altitude adaptation. bioRxiv [preprint]. doi: 10.1101/753186
Heinrich, E. C., Wu, L., Lawrence, E. S., Cole, A. M., Anza-Ramirez, C., Villafuerte, F. C., et al. (2019). Genetic variants at the EGLN1 locus associated with high-altitude adaptation in Tibetans are absent or found at low frequency in highland Andeans. Ann. Hum. Genet. 83, 171–176. doi: 10.1111/ahg.12299
Hoehe, M. R., Herwig, R., Mao, Q., Peters, B. A., Drmanac, R., Church, G. M., et al. (2019). Significant abundance of cis configurations of coding variants in diploid human genomes. Nucleic Acids Res. 47, 2981–2995. doi: 10.1093/nar/gkz031
Hsieh, P., Vollger, M. R., Dang, V., Porubsky, D., Baker, C., Cantsilieris, S., et al. (2019). Adaptive archaic introgression of copy number variants and the discovery of previously unknown human genes. Science 366:eaax2083. doi: 10.1126/science.aax2083
Hu, H., Petousi, N., Glusman, G., Yu, Y., Bohlender, R., Tashi, T., et al. (2017). Evolutionary history of Tibetans inferred from whole-genome sequencing. PLoS Genet. 13:e1006675. doi: 10.1371/journal.pgen.1006675
Hubert, C. G., Rivera, M., Spangler, L. C., Wu, Q., Mack, S. C., Prager, B. C., et al. (2016). A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found <em>In Vivo</em>. Cancer Res. 76:2465. doi: 10.1158/0008-5472.CAN-15-2402
Huerta-Sánchez, E., Degiorgio, M., Pagani, L., Tarekegn, A., Ekong, R., Antao, T., et al. (2013). Genetic signatures reveal high-altitude adaptation in a set of ethiopian populations. Mol. Biol. Evol. 30, 1877–1888. doi: 10.1093/molbev/mst089
Huerta-Sánchez, E., Jin, X., Asan, Bianba, Z., Peter, B. M., Vinckenbosch, N., et al. (2014). Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197. doi: 10.1038/nature13408
Jackson, L. F., Kuhlman, C., Jackson, F. L., and Fox, K. (2019). Including vulnerable populations in the assessment of data from vulnerable populations. Front. Big Data 2:19. doi: 10.3389/fdata.2019.00019
Julian, C. G. (2019). An aptitude for altitude: are epigenomic processes involved? Front. Physiol. 10:1397. doi: 10.3389/fphys.2019.01397
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi: 10.1038/nature17946
Lendahl, U., Lee, K. L., Yang, H., and Poellinger, L. (2009). Generating specificity and diversity in the transcriptional response to hypoxia. Nat. Rev. Genet. 10, 821–832. doi: 10.1038/nrg2665
Liu, X., Zhang, Y., Li, Y., Pan, J., Wang, D., Chen, W., et al. (2019). EPAS1 gain-of-function mutation contributes to high-altitude adaptation in tibetan horses. Mol. Biol. Evol. 36, 2591–2603. doi: 10.1093/molbev/msz158
Lorenzo, F. R., Huff, C., Myllymäki, M., Olenchock, B., Swierczek, S., Tashi, T., et al. (2014). A genetic mechanism for Tibetan high-altitude adaptation. Nat. Genet. 46, 951–956. doi: 10.1038/ng.3067
Lou, H., Lu, Y., Lu, D., Fu, R., Wang, X., Feng, Q., et al. (2015). A 3.4-kb copy-number deletion near EPAS1 is significantly enriched in high-altitude Tibetans but absent from the Denisovan sequence. Am. J. Hum. Genet. 97, 54–66. doi: 10.1016/j.ajhg.2015.05.005
McCauley, H. A., and Wells, J. M. (2017). Pluripotent stem cell-derived organoids: using principles of developmental biology to grow human tissues in a dish. Development 144:958. doi: 10.1242/dev.140731
McDonough, M. A., Li, V., Flashman, E., Chowdhury, R., Mohr, C., Liénard, B. M. R., et al. (2006). Cellular oxygen sensing: crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. U.S.A. 103, 9814–9819. doi: 10.1073/pnas.0601283103
Nanduri, J., Makarenko, V., Reddy, V. D., Yuan, G., Pawar, A., Wang, N., et al. (2012). Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc. Natl. Acad. Sci. U.S.A. 109:2515. doi: 10.1073/pnas.1120600109
Nanduri, J., Peng, Y.-J., Wang, N., Khan, S. A., Semenza, G. L., Kumar, G. K., et al. (2017). Epigenetic regulation of redox state mediates persistent cardiorespiratory abnormalities after long-term intermittent hypoxia. J. Physiol. 595, 63–77. doi: 10.1113/JP272346
Nielsen, R. (2009). Adaptionism—30 years after gould and lewontin. Evol. Int. J. Org. Evol. 63, 2487–2490. doi: 10.1111/j.1558-5646.2009.00799.x
Ouzhuluobu, He, Y., Lou, H., Cui, C., Deng, L., Gao, Y., et al. (2019). De novo assembly of a Tibetan genome and identification of novel structural variants associated with high altitude adaptation. Natl. Sci. Rev. 7, 391–402.
Pagani, L., Ayub, Q., Macarthur, D. G., Xue, Y., Baillie, J. K., Chen, Y., et al. (2012). High altitude adaptation in Daghestani populations from the Caucasus. Hum. Genet. 131, 423–433. doi: 10.1007/s00439-011-1084-8
Scheinfeldt, L. B., Soi, S., Thompson, S., Ranciaro, A., Woldemeskel, D., Beggs, W., et al. (2012). Genetic adaptation to high altitude in the Ethiopian highlands. Genome Biol. 13:R1. doi: 10.1186/gb-2012-13-1-r1
Semenza, G. L. (1999). Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15, 551–578. doi: 10.1146/annurev.cellbio.15.1.551
Semenza, G. L. (2020). The genomics and genetics of oxygen homeostasis. Annu. Rev. Genom. Hum. Genet. 21. https://doi.org/10.1146/annurev-genom-111119-073356 [Epub ahead of print].
Simonson, T. S. (2015). Altitude adaptation: a glimpse through various lenses. High Alt. Med. Biol. 16, 125–137. doi: 10.1089/ham.2015.0033
Simonson, T. S., Yang, Y., Huff, C. D., Yun, H., Qin, G., Witherspoon, D. J., et al. (2010). Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75. doi: 10.1126/science.1189406
Song, D., Li, L.-S., Arsenault, P. R., Tan, Q., Bigham, A. W., Heaton-Johnson, K. J., et al. (2014). Defective Tibetan PHD2 binding to p23 links high altitude adaption to altered oxygen sensing. J. Biol. Chem. 289, 14656–14665. doi: 10.1074/jbc.M113.541227
Song, S., Yao, N., Yang, M., Liu, X., Dong, K., Zhao, Q., et al. (2016). Exome sequencing reveals genetic differentiation due to high-altitude adaptation in the Tibetan cashmere goat (Capra hircus). BMC Genomics 17:122. doi: 10.1186/s12864-016-2449-0
Sterneckert, J. L., Reinhardt, P., and Schöler, H. R. (2014). Investigating human disease using stem cell models. Nat. Rev. Genet. 15, 625–639. doi: 10.1038/nrg3764
Storz, J. F., and Scott, G. R. (2019). Life ascending: mechanism and process in physiological adaptation to high-altitude hypoxia. Annu. Rev. Ecol. Evol. Syst. 50, 503–526. doi: 10.1242/jeb.048181
Tong, Y., Robertsen, H. L., Blin, K., Weber, T., and Lee, S. Y. (2018). “CRISPR-Cas9 toolkit for actinomycete genome editing,” in Synthetic Metabolic, Pathways, Methods, and Protocols, eds M. K. Jensen & J. D. Keasling (New York, NY: Springer), 163–184. doi: 10.1007/978-1-4939-7295-1_11
Trujillo, C. A., and Muotri, A. R. (2018). Brain organoids and the study of neurodevelopment. Trends Mol. Med. 24, 982–990.
Wenger, R. H., Stiehl, D. P., and Camenisch, G. (2005). Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005:re12. doi: 10.1126/stke.3062005re12
West, J. B. (ed.) (1998). “Other recent high-altitude studies,” in High Life, (Cham: Springer), 364–400.
Wimmer, R. A., Leopoldi, A., Aichinger, M., Wick, N., Hantusch, B., Novatchkova, M., et al. (2019). Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565, 505–510. doi: 10.1038/s41586-018-0858-8
Wörsdörfer, P., Dalda, N., Kern, A., Krüger, S., Wagner, N., Kwok, C. K., et al. (2019). Generation of complex human organoid models including vascular networks by incorporation of mesodermal progenitor cells. Sci. Rep. 9:15663. doi: 10.1038/s41598-019-52204-7
Wu, D., and Yotnda, P. (2011). Induction and testing of hypoxia in cell culture. J. Vis. Exp. 54:2899. doi: 10.3791/2899
Xing, J., Wuren, T., Simonson, T. S., Watkins, W. S., Witherspoon, D. J., Wu, W., et al. (2013). Genomic analysis of natural selection and phenotypic variation in high-altitude mongolians. PLoS Genet. 9:e1003634. doi: 10.1371/journal.pgen.1003634
Yi, X., Liang, Y., Huerta-Sanchez, E., Jin, X., Cuo, Z. X. P., Pool, J. E., et al. (2010). Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78. doi: 10.1126/science.1190371
Keywords: genetic adaptation, genome editing, high-altitude adaptation, functional investigation, adaptive variation
Citation: Hall JE, Lawrence ES, Simonson TS and Fox K (2020) Seq-ing Higher Ground: Functional Investigation of Adaptive Variation Associated With High-Altitude Adaptation. Front. Genet. 11:471. doi: 10.3389/fgene.2020.00471
Received: 16 February 2020; Accepted: 16 April 2020;
Published: 14 May 2020.
Edited by:
Jacob A. Tennessen, Harvard University, United StatesReviewed by:
Xuebin Qi, Kunming Institute of Zoology, ChinaChristina Alessandra Eichstaedt, Heidelberg University Hospital, Germany
Copyright © 2020 Hall, Lawrence, Simonson and Fox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatum S. Simonson, dHNpbW9uc29uQGhlYWx0aC51Y3NkLmVkdQ==; Keolu Fox, cGtmb3hAdWNzZC5lZHU=