 Nan Wu1,2,3,4†
Nan Wu1,2,3,4† Lili Tang1,2,3,4†Xiuxiu Li1,2,3,4Yuwei Dai1,2,3,4Xiaodong Zheng1,2,3,4Min Gao1,2,3,4*Peiguang Wang1,2,3,4*
Lili Tang1,2,3,4†Xiuxiu Li1,2,3,4Yuwei Dai1,2,3,4Xiaodong Zheng1,2,3,4Min Gao1,2,3,4*Peiguang Wang1,2,3,4*- 1Department of Dermatology, The First Affiliated Hospital, Anhui Medical University, Hefei, China
- 2Institute of Dermatology, Anhui Medical University, Hefei, China
- 3Key Laboratory of Dermatology, Anhui Medical University, Ministry of Education, Hefei, China
- 4Provincial Laboratory of Inflammatory and Immune Mediated Diseases, Hefei, China
Dyschromatosis universalis hereditaria (DUH) is a rare genodermatosis characterized by mottled hyperpigmented and hypopigmented macules. SASH1 and ABCB6 have been identified as the causative genes for this disorder. We performed whole exome sequencing on a Chinese family with DUH and genotype-phenotype correlation analysis in DUH and lentiginous phenotype patients. A novel heterozygous missense mutation p.Q518P in SASH1 gene was detected in this family. A majority of patients with SASH1 mutations presented as a distinct clinical phenotype clearly different from that in patients with ABCB6 mutations. Our findings further enrich the reservoir of SASH1 mutations in DUH. The clinical phenotypic difference between SASH1 and ABCB6 variants is suggestive of a close phenotype-genotype link in DUH.
Introduction
Dyschromatosis universalis hereditaria (DUH) is a rare genodermatosis which is characterized by generalized hyperpigmented macules mixed with hypopigmented macules in a reticular pattern, which usually appears from infancy or early childhood. It was firstly described in 1933 by Ichikawa and Hiraga. DUH shares a typical phenotypic feature with dyschromatosis symmetrica hereditaria (DSH), that is reticulate hyperpigmentation and hypopigmentation on extremities. The mode of autosomal dominant inheritance was observed in approximately 50% of all cases. In addition, DUH was rarely accompanied with other abnormalities, such as neurosensory hearing loss, adermatoglyphia, photosensitivity, primary ovarian failure, insulin-dependent diabetes, renal failure, and ocular abnormalities, Dowling-Degos disease (DDD) (Yang and Wong, 1991; Sandhu et al., 2004; Bhoyar et al., 2015; Rojhirunsakool and Vachiramon, 2015; Gupta, 2016; Jayanthi et al., 2016).
DUH consists of three major types, namely DUH1 (OMIM 127500), DUH2 (OMIM 612715), and DUH3 (OMIM 615402) according to the chromosomal mapping of 6q24.2-q25.2, 12q21-q23, and 2q35 region respectively (Xing et al., 2003; Stuhrmann et al., 2008; Zhang et al., 2013). In 2013, Zhou et al. (2013) first identified three heterozygous missense mutations of the SAM (sterile alpha motif) and SH3 (Src homology domain 3) domain containing one (SASH1) gene in three unrelated families with DUH1, whereas, Zhang et al. (2013) identified three heterozygous missense mutations in ABCB6 gene in a large family and six sporadic patients with DUH3. Thereafter, a number of mutations in SASH1 and ABCB6 were successively detected in other affected individuals with DUH (Cui et al., 2013; Liu et al., 2014; Lu et al., 2014; Zhong et al., 2019a, b). We herein described a case of Chinese DUH family with a novel missense mutation in SASH1 gene, and performed genotype-phenotype correlation analysis in the DUH and lentiginous type patients from the published literatures.
Case Presentation
We recruited a four-generation Chinese family with generalized dyschromatosis from Anhui Province in China. The pedigree is suggestive of an autosomal dominant inheritance (Figure 1). The proband was a 6-year-old girl who presented as asymptomatic hyperpigmented macules for 4 years. The pigmented lesions initially appeared on her cheek, and gradually spread to her entire body over a period of 3 years. On dermatological examination, light brown to dark brown macules in 2–4 mm of diameter were diffusely distributed on her face, dorsa of hands and buttocks, while only a small number of similar lesions scattered on her neck, trunk and limbs. Moreover, some irregular hypopigmented macules were also observed on her abdomen and back. Her palms, soles and oral mucosa were spared (Figure 2). Her general health seemed like to be normal. Her father and grandfather showed similar hyperpigmented macules on their whole body. However, obviously reticular hypopigmented macules or patches were also present on their buttocks as well as dorsa of their hands and feet. The hyperpigmentation, hypopigmentation and normal color of skin mingled together and constituted a characteristically variegated appearance on their extremities (Figure 3). The diagnosis of DUH was established by virtue of clinical features of all affected individuals in the family. Her grandmother, mother and younger brother were all normal.
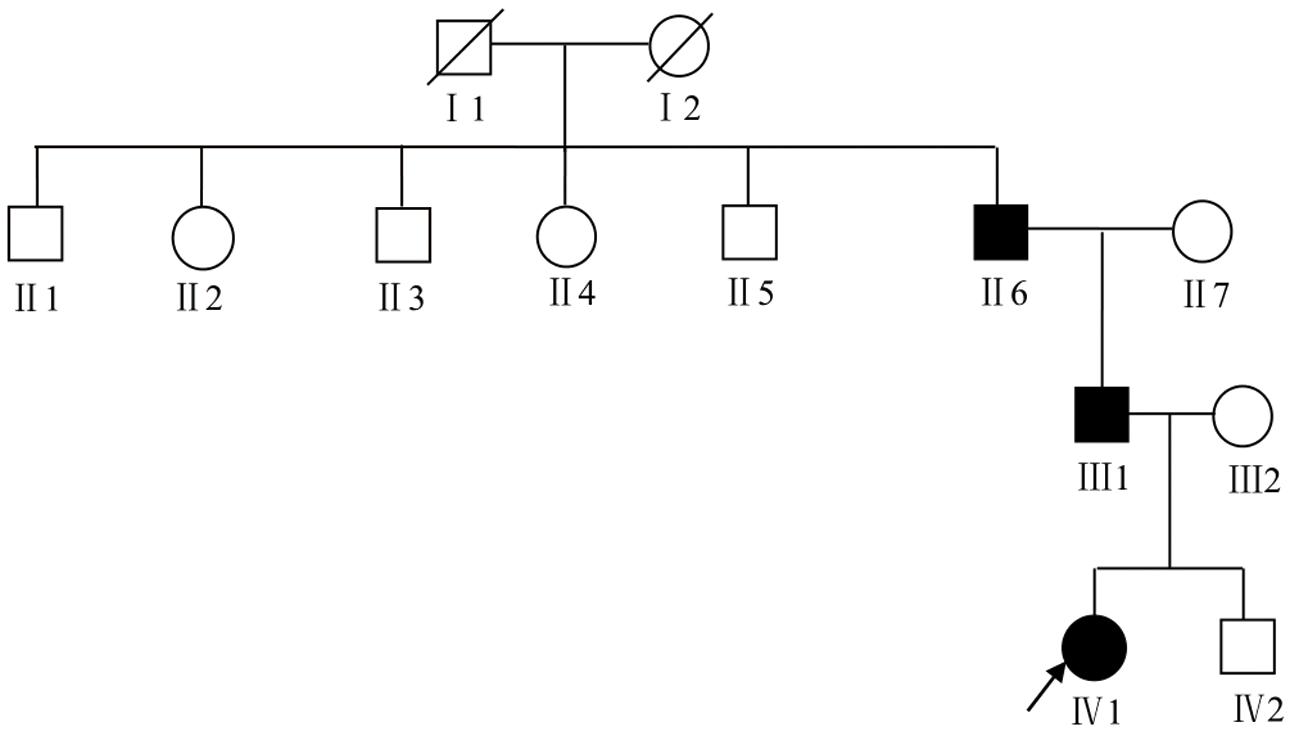
Figure 1. The pedigree chart of DUH family. The proband was marked with an arrow. Females were indicated by circles while males were indicated by squares. Blackened symbols represented patients who were carried the mutation through mutation sequencing.
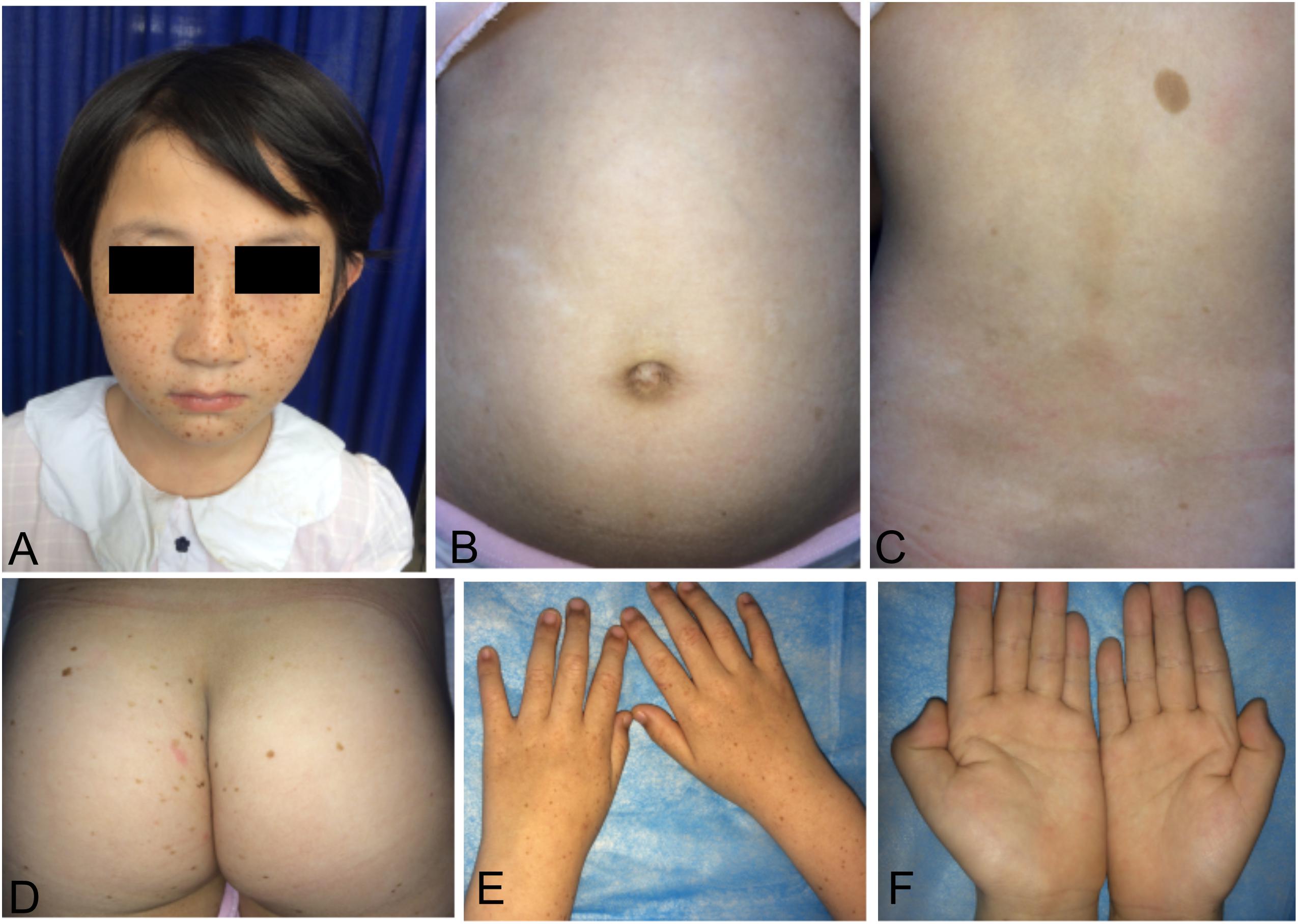
Figure 2. Cutaneous manifestations of the proband. (A,D,E) Light brown to dark brown macules on the her face, buttocks, and dorsa of hands. (B,C) Irregular hypopigmented macules on the her abdomen and back. (F) None of involvement on her palms.
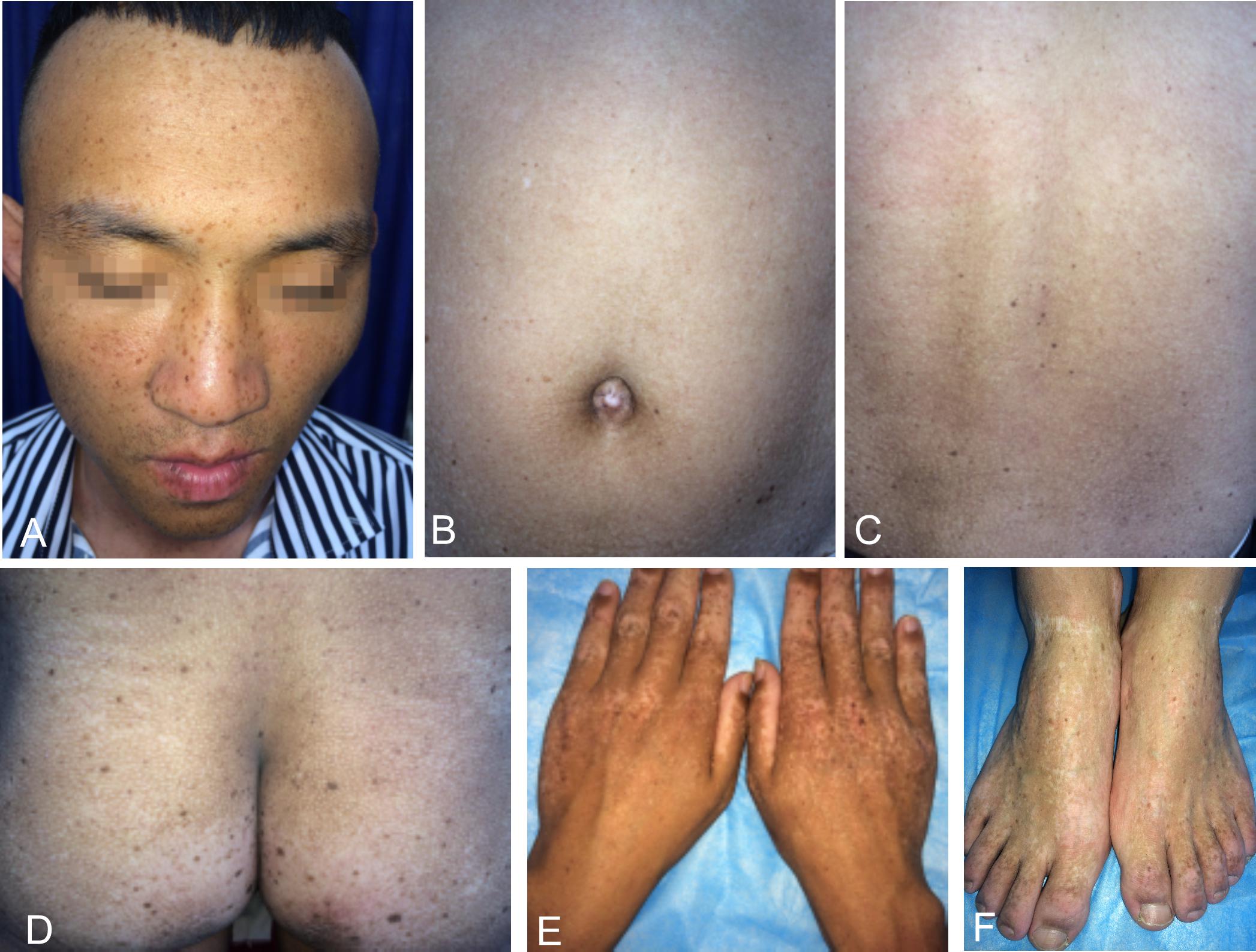
Figure 3. Cutaneous manifestations of the proband’s father. (A–D) Light brown to dark brown macules on his face, abdomen, back and buttocks, more prominent on his face. (B–D) Irregular hypopigmented macules or patches on his abdomen, back and buttocks. (E,F) Reticular dyschromatosis on the dorsa of his hands and feet.
Methods
Peripheral Blood Collection and DNA Extraction
After obtaining informed consent from all participants and approval from Clinical Research Ethics Committee of Anhui Medical University, EDTA anticoagulated venous blood samples were collected from three affected and three unaffected members for DNA analysis. Genomic DNA was extracted using the DNA extraction kit (Promega, Madison, WI, United States) in a standard procedure and stored at −80°C.
Whole Exome Sequencing (WES)
The proband (IV1) and her father (III1) were selected to conduct the WES. Genomic DNA fragments corresponding to all exons in genome were amplified by polymerase chain reaction and subjected to automatic DNA sequencing after purification. Agilent SureSelect XT Library Prep Kit and SureSelect Human All Exon V6 kit were used for the library preparation and capture, followed by 150 bp pair-end sequencing on Illumina Hiseq XTen platform according to the manufacture’s recommendations. After quality control of the raw data, variants comparisons, mutation recognition and annotation, a list of suspected variants were obtained. Screening for disease-associated deleterious mutations was then made with emphases on all the possible pathogenic variations in reported SASH1 and ABCB6 genes, according to the frequency in the control database, variation types, and prediction of mutation function such as the score in SIFT and PolyPhen2 database.
Sanger Sequencing
The possible pathogenic variations identified by WES were confirmed by Sanger sequencing in the proband’s grandparents (II6 and II7), mother (III2), and younger brother (IV2) to detect genotype-phenotype co-segregation. Primers flanking all coding regions of the possible variations in SASH1 or ABCB6 were designed using software Primer Premier 5.0 (Primer Biosystems, Foster City, CA, United States). PCR products from genomic DNA were sequenced using an ABI 3730XL DNA Analyzer (ABI, Foster City, CA, United States). The sequencing results were analyzed using Finch TV (Version 1.5), and the newly discovered mutation was named referring to the principle of the Human Genome Variation Society (HGVs).1
Results
WES Results and Co-segregation Analysis
We generated approximately 14.38 and 12.97 Gb of sequence data in the two affected (III1 and IV1) respectively. A heterozygous missense variation in SASH1 gene (NM_015278.3), c.1553A > C, was identified after integrative analysis. This variation was not present in 700 normal controls. Sanger sequencing revealed that this variation was detected in the proband’s grandfather, but not present in three unaffected family members (Figure 4). The sequences of primers used for validation sequencing were SASH1-Forward primer: AAACCCATGGCAGGACTCGGG, SASH1-Reverse primer: TAGCACCTGAAGGGCAGGACTTG. This variation resulted in an amino acid substitution, p.Gln518Pro (p.Q518P), which co-segregated perfectly with the disease. Moreover, the substitution p.Q518P was predicted to be “deleterious” with a score of 0.001 in SIFT2 and “probably damaging” with a score of 0.999 in PolyPhen2.3 None of possible pathogenic variations was present in ABCB6 gene.
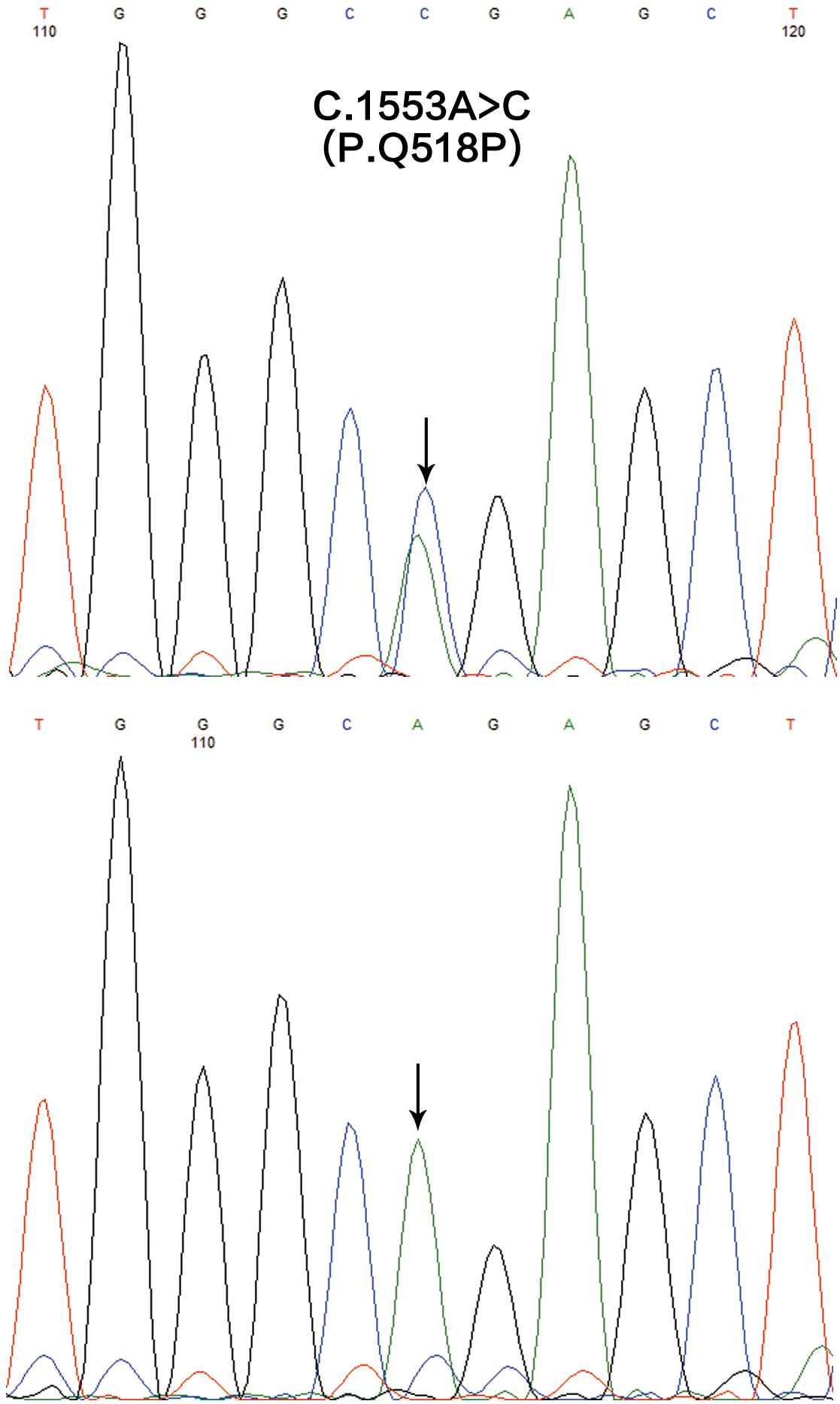
Figure 4. The mutation analysis of SASH1 gene in this family. A heterozygous missense mutation c.1553A > C (p.Q518P) in SASH1 identified in the affected individuals (the upper) and the reference sequencing of SASH1 in the unaffected individuals (the lower).
Genotype-Phenotype Correlation Analysis
So far, totally 11 heterozygous missense mutations in SASH1 have been identified in patients with dyschromatosis (Table 1), in which seven mutations contribute to DUH phenotype and four mutations lead to multiple lentiginous. Comparing the phenotype of SASH1 variants with that of ABCB6 variants in DUH, we found a distinct difference in clinical phenotype between them. The most prominent feature in DUH patients with SASH1 variants was generalized light brown to dark brown macules similar to leopard’s spots, especially on the sun-exposed areas, which may lead to the misdiagnosis as multiple lentigines for those DUH patients with multiple lentigines arising on the background of light-complexioned skin. However, the coexistence of variable retiform or homogenous hypopigmentation helps to differentiate multiple lentigines with DUH. The characteristic manifestation of ABCB6 variants is typically generalized mottled hyperpigmented and hypopigmented macules arranging in a reticular pattern.
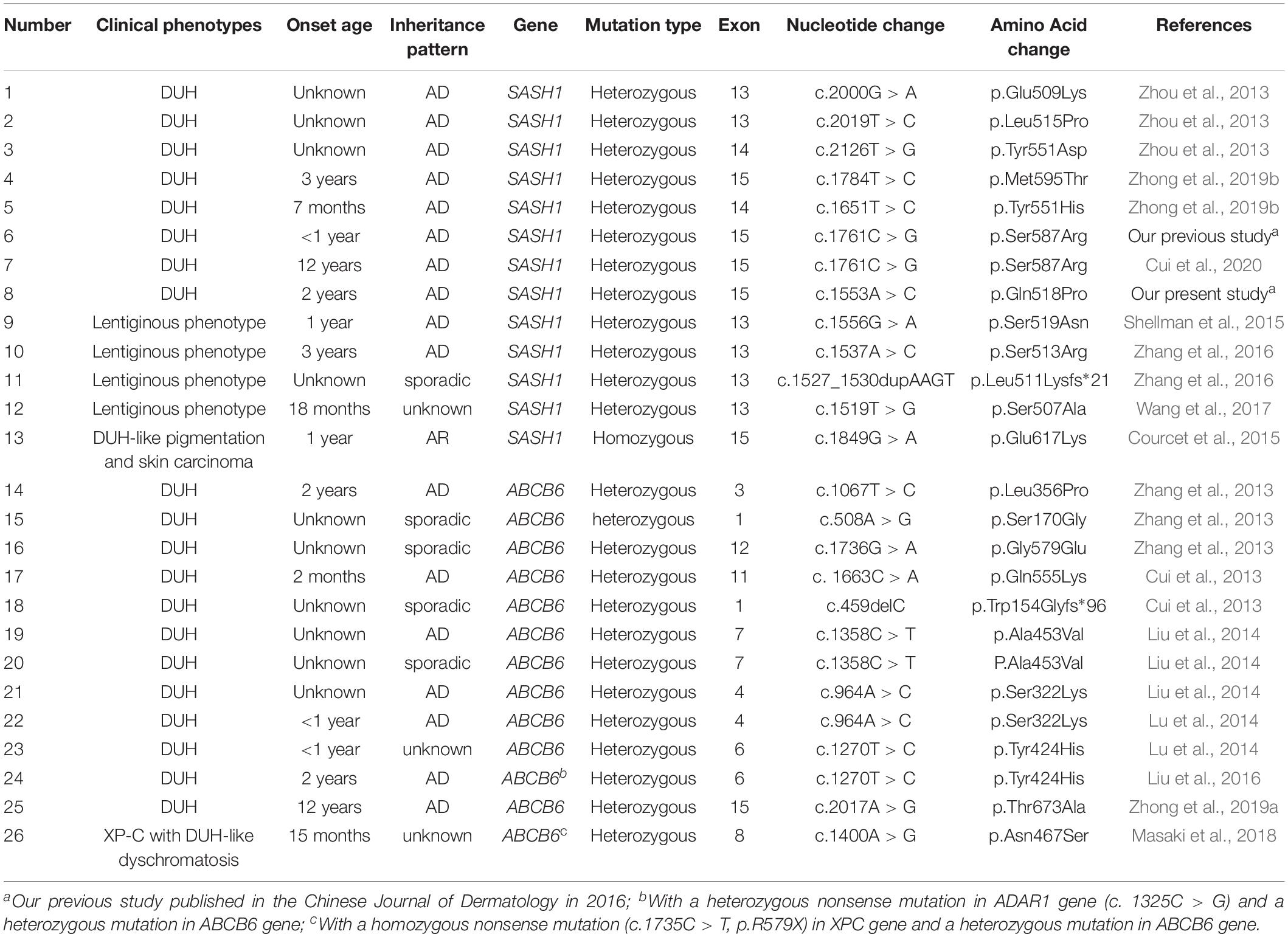
Table 1. Clinical and genetic information in families with DUH or lentiginous phenotype.
Discussion
SASH1 was firstly identified as a potential tumor suppressor gene in breast cancer, which played a crucial role in tumorigenesis, development, invasion and metastasis (Zeller et al., 2003; Lin et al., 2012). In addition, SASH1 was also involved in various complex signaling pathways as a pivotal regulator or mediator (Dauphinee et al., 2013). Zhou et al. (2013) found Gαs-SASH1-IQGAP1-E-Cadherin signaling pathway was responsible for melanocytes transepithelial migration in the progression of DUH. Lately, Zhou et al. (2017) demonstrated that SASH1 mediates skin melanogenesis through a cascade of p53/α-MSH/POMC/Gαs/SASH1. So far, totally 11 heterozygous missense mutations in SASH1 have been identified in some patients with dyschromatosis as illustrated in the Table 1, including nine mutations (p.Y551H, p.S507A, p.S513R, p.L511Kfs∗21, p.S519N, p.Y551D, p.L515P, p.E509K, p.Q518P) in SLY domain and two mutations (p.S587R, p.M595T) in SH3 domain, in which seven mutations contribute to DUH phenotype and four mutations lead to multiple lentiginous phenotype (Shellman et al., 2015; Zhang et al., 2016; Wang et al., 2017). Besides, a homozygous missense mutation p.E617K in SASH1 was responsible for an autosomal recessive syndrome featuring DUH-like pigmentation anomaly, palmoplantar keratoderma and skin carcinoma (Courcet et al., 2015). The protein domains and the amino acid positions of SASH1 mutations responsible for DUH and lentiginous phenotype were showed in a schematic representation (Figure 5). In this study, we detected a missense mutation (p.Q518P) in SASH1 gene, which is located in the highly conserved SLY domain of SASH1. Considering the observed heterogeneity of DUH phenotypes in this family, we wondered whether it was related to SASH1 heterozygous variant coupled with variants in other regions, such as 12q21-q23 which is associated with DUH2. Hence, we explored the variants in this region from the WES data, however, we did not find any pathogenic variation in this region, suggesting that the heterogeneity in this family was related to SASH1 variation.
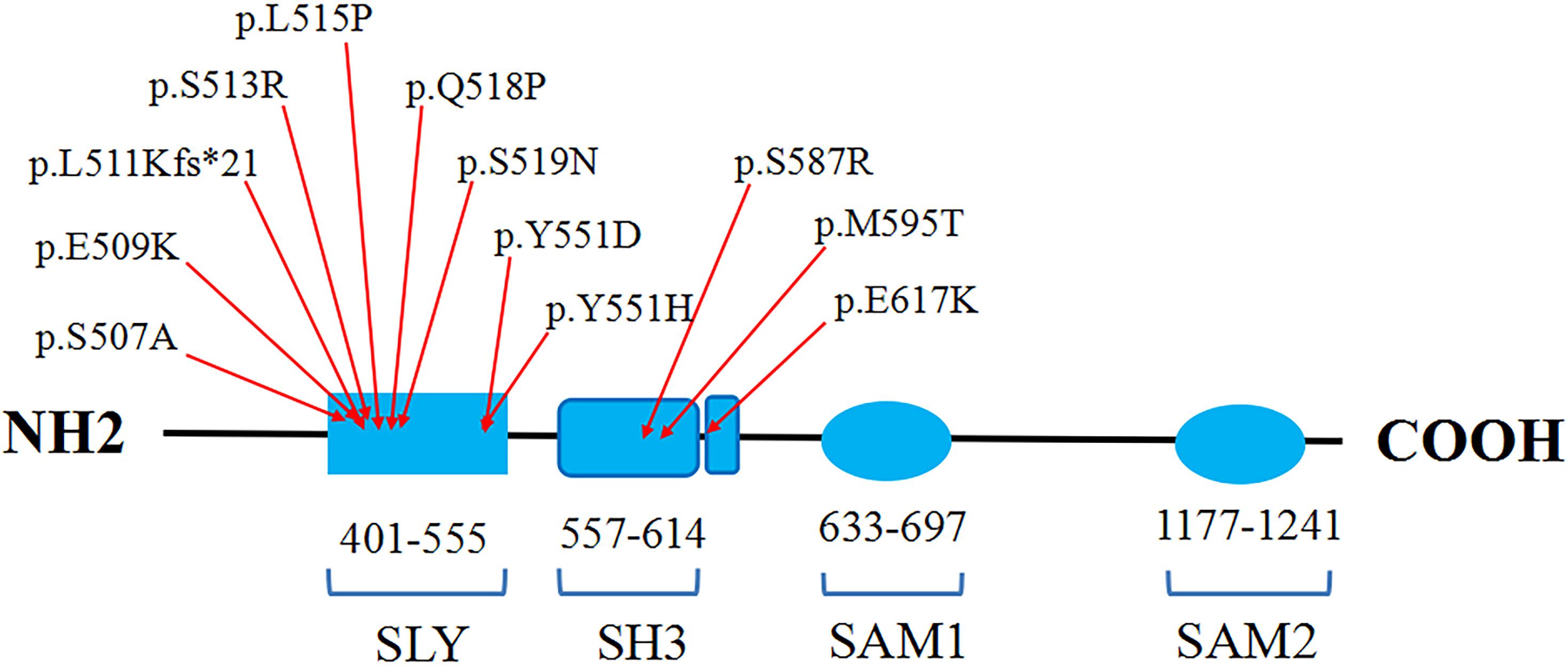
Figure 5. The schematic representation of the protein domains and the amino acid positions of SASH1 mutations responsible for DUH and lentiginous phenotype.
Previously we have identified a heterozygous missense mutation (p.S587R) in SASH1 in a large DUH family from Henan Province, as reported by Yao et al. in Chin J Dermatol in 2016. All seven affected individuals in the family had extensive light brown to dark brown macules with 2–5 mm in diameter on their whole body. Moreover, homogenous hypopigmentation on the rest of skin occurred in six affected individuals. Only one affected individual manifested as generalized reticulate hypopigmented macules. Most recently, this missense mutation (c.1761C > G, p.S587R) has also been identified in a large Chinese family with four generation (Cui et al., 2020). Zhong et al. (2019a) described two unrelated DUH families carrying mutations in SASH1. The probands in these two families both presented with generalized light to dark brown macules on their face, neck, trunk and limbs. In addition, reticular hypopigmentation was also seen on their neck as well as dorsal aspects of hands and feet. Apparently, there is a significant phenotypic heterogeneity among intra-family and inter-family members with DUH in the light of these cases mentioned above.
Besides DUH, Shellman et al. (2015) identified a missense mutation in SASH1 in an autosomal dominant lentiginous phenotype. All affected members exhibited generalized dark brown macules, more prominent in sun-exposed areas. Moreover, some reticular hypopigmented macules were also present on the dorsa of an affected member’s hands. Zhang et al. (2016) detected two mutations in SASH1 in two cases of patients with lentiginous phenotype. These two patients both presented with generalized light brown to dark brown macules, particularly on their face and neck. Furthermore, some scattered hypopigmented spots were also observed on the trunk and face and mild dyschromatosis on the elbow and dorsal area of hands and feet in one patient, while diffuse hypopigmentation occurred on his entire body in the other patient. Later, Wang et al. (2017) reported that a family manifesting as multiple lentigines carried a mutation in SASH1. The proband showed substantially identical phenotype as an affected individual in a DUH family with a mutation (p.S587R) in SASH1. Intriguingly, all affected individuals with lentiginous phenotype shared very similar clinical features with the DUH patients. More importantly, the development of their pigmented defects was associated with a mutation in SASH1. It is well-known that multiple lentigines related to Noonan syndrome or LEOPARD syndrome is caused by mutations in PTPN11 (Tartaglia et al., 2001; Digilio et al., 2002). Hence, we consider that these cases with lentiginous phenotype are actually DUH because of high similarity in their clinical phenotype and molecular genetics.
Three mutations (p.S170G, p.L356P, p.G579E) in ABCB6 were determined in a large Chinese family and six sporadic patients with DUH (Zhang et al., 2013). The proband from the family had generalized hyperpigmented and hypopigmented macules in a reticular arrangement with mottled appearance. Thereafter, six mutations (p.S322K, pA453V, p.Y424H, p.T673A, p.Q555K, c. p.W154Gfs∗96) in ABCB6 were detected in eight families and five sporadic cases with DUH (Cui et al., 2013; Liu et al., 2014; Lu et al., 2014; Zhong et al., 2019a). Besides, a case of xeroderma pigmentosum (XP) complementation group C with DUH-like pigmentation was reported, and a heterozygous missense mutation (p.N467S) was found (Masaki et al., 2018). All affected individuals also exhibited generalized variegated hyperpigmented and hypopigmented macules in a reticulate arrangement over their entire body or abdomen and dorsa of hands. Comparing the phenotype of SASH1 variants with that of ABCB6 variants in DUH, we find a distinct difference in clinical phenotype between them. The most prominent feature of SASH1 variants in DUH is generalized light brown to dark brown macules similar to leopard’s spots, especially on the sun-exposed areas. Therefore, if not carefully examined, they are easily misdiagnosed as multiple lentigines for those DUH patients with multiple lentigines arising on the background of light-complexioned skin. However, the coexistence of variable retiform or homogenous hypopigmentation helps to differentiate multiple lentigines with DUH. The characteristic manifestation of ABCB6 variants is typically generalized mottled hyperpigmented and hypopigmented macules arranging in a reticular pattern. In contrast, it is not difficult to make a correct clinical diagnosis for the DUH cases associated with mutations in ABCB6. Clinically, the DUH case is rare, so herein we only describe a case of DUH family with a novel mutation in SASH1 gene. The analysis of genotype-phenotype correlation in the patients with DUH or lentiginous phenotype were mainly dependent on the information on the published literatures.
In summary, we successfully identified a novel pathogenic mutation in SASH1 in a Chinese family with DUH, thus further extending the mutation spectrum of this gene. The clinical phenotypic difference between SASH1 and ABCB6 variants is suggestive of a close phenotype-genotype link in DUH. Our findings not only help to enhance more comprehensive understanding for DUH, but also provide a useful clue for the correct diagnosis of DUH.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics Statement
Written informed consent was obtained from the individuals or the legal guardian for the publication of any potentially identifiable images or data included in this article.
Author Contributions
NW conducted Sanger sequencing and wrote the manuscript. XL and YD collected clinical data and blood samples, and performed DNA extraction. LT performed whole exome sequencing and wrote the manuscript. XZ performed data analysis of the whole exome sequencing. MG and PW were responsible for the study design and guiding of the study implementation, and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by a grant from the University Natural Science Research Project of Anhui Province (KJ2017A201).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the patients and their family members for participating in this study.
Footnotes
References
Bhoyar, P., Mahajan, B., and Kumar, S. (2015). A case of dyschromatosis universalis hereditaria with adermatoglyphia: a rare association. Indian Dermatol. Online J. 6, 105–109. doi: 10.4103/2229-5178.153013
Courcet, J. B., Elalaoui, S. C., Duplomb, L., Tajir, M., Riviere, J. B., Thevenon, J., et al. (2015). Autosomal-recessive SASH1 variants associated with a new genodermatosis with pigmentation defects, palmoplantar keratoderma and skin carcinoma. Eur. J. Hum. Genet. 23, 957–962. doi: 10.1038/ejhg.2014.213
Cui, H., Guo, S., He, H., Guo, H., Zhang, Y., and Wang, B. (2020). SASH1 promotes melanin synthesis and migration via suppression of TGF-beta1 secretion in melanocytes resulting in pathologic hyperpigmentation. Int. J. Biol. Sci. 16, 1264–1273. doi: 10.7150/ijbs.38415
Cui, Y. X., Xia, X. Y., Zhou, Y., Gao, L., Shang, X. J., Ni, T., et al. (2013). Novel mutations of ABCB6 associated with autosomal dominant dyschromatosis universalis hereditaria. PLoS One 8:e79808. doi: 10.1371/journal.pone.0079808
Dauphinee, S. M., Clayton, A., Hussainkhel, A., Yang, C., Park, Y. J., Fuller, M. E., et al. (2013). SASH1 is a scaffold molecule in endothelial TLR4 signaling. J. Immunol. 191, 892–901. doi: 10.4049/jimmunol.1200583
Digilio, M. C., Conti, E., Sarkozy, A., Mingarelli, R., Dottorini, T., Marino, B., et al. (2002). Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am. J. Hum. Genet. 71, 389–394. doi: 10.1086/341528
Gupta, M. (2016). Dyschromatosis universalis hereditaria with sensorineural hearing loss. Egypt. J. Dermatol. Venerol. 36, 26–27. doi: 10.4103/1110-6530.194160
Jayanthi, N. S., Anandan, V., Jameela, W. A., Kumar, V. S., and Lavanya, P. (2016). A case report of dyschromatosis universalis hereditaria (DUH) with primary ovarian failure (POF). J. Clin. Diagn. Res. 10, WD01–WD02. doi: 10.7860/JCDR/2016/17525.7368
Lin, S., Zhang, J., Xu, J., Wang, H., Sang, Q., Xing, Q., et al. (2012). Effects of SASH1 on melanoma cell proliferation and apoptosis in vitro. Mol. Med. Rep. 6, 1243–1248. doi: 10.3892/mmr.2012.1099
Liu, H., Li, Y., Hung, K. K., Wang, N., Wang, C., Chen, X., et al. (2014). Genome-wide linkage, exome sequencing and functional analyses identify ABCB6 as the pathogenic gene of dyschromatosis universalis hereditaria. PLoS One 9:e87250. doi: 10.1371/journal.pone.0087250
Liu, J. W., Asan, Sun, J., Vano-Galvan, S., Liu, F. X., Wei, X. X., et al. (2016). Differential diagnosis of two chinese families with dyschromatoses by targeted gene sequencing. Chin. Med. J. (Engl.) 129, 33–38. doi: 10.4103/0366-6999.172564
Lu, C., Liu, J., Liu, F., Liu, Y., Ma, D., and Zhang, X. (2014). Novel missense mutations of ABCB6 in two chinese families with dyschromatosis universalis hereditaria. J. Dermatol. Sci. 76, 255–258. doi: 10.1016/j.jdermsci.2014.08.015
Masaki, T., Nakano, E., Okamura, K., Ono, R., Sugasawa, K., Lee, M. H., et al. (2018). A case of xeroderma pigmentosum complementation group C with diverse clinical features. Br. J. Dermatol. 178, 1451–1452. doi: 10.1111/bjd.16339
Rojhirunsakool, S., and Vachiramon, V. (2015). Dyschromatosis universalis hereditaria with renal failure. Case Rep. Dermatol. 7, 51–55. doi: 10.1159/000381174
Sandhu, K., Saraswat, A., and Kanwar, A. J. (2004). Dowling-Degos disease with dyschromatosis universalis hereditaria-like pigmentation in a family. J. Eur. Acad. Dermatol. Venereol. 18, 702–704. doi: 10.1111/j.1468-3083.2004.01028.x
Shellman, Y. G., Lambert, K. A., Brauweiler, A., Fain, P., Spritz, R. A., Martini, M., et al. (2015). SASH1 is involved in an autosomal dominant lentiginous phenotype. J. Invest. Dermatol. 135, 3192–3194. doi: 10.1038/jid.2015.292
Stuhrmann, M., Hennies, H. C., Bukhari, I. A., Brakensiek, K., Nurnberg, G., Becker, C., et al. (2008). Dyschromatosis universalis hereditaria: evidence for autosomal recessive inheritance and identification of a new locus on chromosome 12q21-q23. Clin. Genet. 73, 566–572. doi: 10.1111/j.1399-0004.2008.01000.x
Tartaglia, M., Mehler, E. L., Goldberg, R., Zampino, G., Brunner, H. G., Kremer, H., et al. (2001). Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29, 465–468. doi: 10.1038/ng772
Wang, J., Zhang, J., Li, X., Wang, Z., Lei, D., Wang, G., et al. (2017). A novel de novo mutation of the SASH1 gene in a Chinese family with multiple lentigines. Acta Derm. Venereol. 97, 530–531. doi: 10.2340/00015555-2575
Xing, Q. H., Wang, M. T., Chen, X. D., Feng, G. Y., Ji, H. Y., Yang, J. D., et al. (2003). A gene locus responsible for dyschromatosis symmetrica hereditaria (DSH) maps to chromosome 6q24.2-q25.2. Am. J. Hum. Genet. 73, 377–382. doi: 10.1086/377007
Yang, J. H., and Wong, C. K. (1991). Dyschromatosis universalis with X-linked ocular albinism. Clin. Exp. Dermatol. 16, 436–440. doi: 10.1111/j.1365-2230.1991.tb01230.x
Zeller, C., Hinzmann, B., Seitz, S., Prokoph, H., Burkhard-Goettges, E., Fischer, J., et al. (2003). SASH1: a candidate tumor suppressor gene on chromosome 6q24.3 is downregulated in breast cancer. Oncogene 22, 2972–2983. doi: 10.1038/sj.onc.1206474
Zhang, C., Li, D., Zhang, J., Chen, X., Huang, M., Archacki, S., et al. (2013). Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J. Invest. Dermatol. 133, 2221–2228. doi: 10.1038/jid.2013.145
Zhang, J., Cheng, R., Liang, J., Ni, C., Li, M., and Yao, Z. (2016). Lentiginous phenotypes caused by diverse pathogenic genes (SASH1 and PTPN11): clinical and molecular discrimination. Clin. Genet. 90, 372–377. doi: 10.1111/cge.12728
Zhong, W., Pan, Y., Shao, Y., Yang, Y., Yu, B., and Lin, Z. (2019a). Atypical presentation of dyschromatosis universalis hereditaria with a novel ABCB6 mutation. Clin. Exp. Dermatol. 44, e58–e60. doi: 10.1111/ced.13833
Zhong, W. L., Wang, H. J., Lin, Z. M., and Yang, Y. (2019b). Novel mutations in SASH1 associated with dyschromatosis universalis hereditaria. Indian J. Dermatol. Venereol. Leprol. 85:440. doi: 10.4103/ijdvl.IJDVL_360_17
Zhou, D., Kuang, Z., Zeng, X., Wang, K., Ma, J., Luo, H., et al. (2017). p53 regulates ERK1/2/CREB cascade via a novel SASH1/MAP2K2 crosstalk to induce hyperpigmentation. J. Cell Mol. Med. 21, 2465–2480. doi: 10.1111/jcmm.13168
Keywords: dyschromatosis universalis hereditaria, SASH1 gene, ABCB6 gene, mutation, phenotype
Citation: Wu N, Tang L, Li X, Dai Y, Zheng X, Gao M and Wang P (2020) Identification of a Novel Mutation in SASH1 Gene in a Chinese Family With Dyschromatosis Universalis Hereditaria and Genotype-Phenotype Correlation Analysis. Front. Genet. 11:841. doi: 10.3389/fgene.2020.00841
Received: 28 April 2020; Accepted: 13 July 2020;
Published: 04 August 2020.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
Fan Jin, Zhejiang University, ChinaNicole Ziliotto, University of Milano-Bicocca, Italy
Copyright © 2020 Wu, Tang, Li, Dai, Zheng, Gao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Gao, YWhobmdtQDE2My5jb20=; Peiguang Wang, d3BnMjM3MEAxNjMuY29t
†These authors have contributed equally to this work