 Thanh-Huong Truong1,2*†Doan-Loi Do1,2*†
Thanh-Huong Truong1,2*†Doan-Loi Do1,2*† Ngoc-Thanh Kim1,2*Mai-Ngoc Thi Nguyen1,2Thanh-Tung Le1Hong-An Le3
Ngoc-Thanh Kim1,2*Mai-Ngoc Thi Nguyen1,2Thanh-Tung Le1Hong-An Le3- 1Vietnam National Heart Institute, Bach Mai Hospital, Hanoi, Vietnam
- 2Department of Cardiology, Hanoi Medical University, Hanoi, Vietnam
- 3School of Medicine and Pharmacy, Vietnam National University, Hanoi, Vietnam
Familial hypercholesterolemia (FH) is underdiagnosed and undertreated in a majority of the low- and middle-income countries. FH registries could prove useful in bridging the knowledge gaps, supporting genetic and clinical research, and improving health-care planning and patient care. Here, we report the first usage experience of the Vietnam FH (VINAFH) Registry. The VINAFH Registry was established in 2016 as a long-term database for prospective cohorts. FH patients were detected based on the opportunistic and cascade screening. Diagnosis of FH was assessed using the Dutch Lipid Clinic Network criteria, plasma levels of low-density lipoprotein (LDL) cholesterol, and genetic testing. To date, a total of 130 patients with FH have been registered, with 48 index cases and 82 relatives. Of the 130 patients, 8 were homozygous FH patients and 38 were children. Of FH individuals, 46.7% was confirmed by genetic testing: 61 patients (96.8%) carried the LDLR mutation (c.681C > G, c.1427C > G, c.1187-?_2140 ± ?del, c.2529_2530delinsA), and two patients (3.2%) carried the PCSK9 (protein convertase subtilisin/kexin type 9) mutation (c.42_43insTG). The c.2529_2530delinsA mutation detected in this study is novel and reported only in the Vietnamese population. However, only 53.8% of FH patients were followed up post diagnosis, and only 15.3% of these were approved for lipid-lowering therapy and specialized care. Notably, factors such as knowledge about FH in patients and/or guardians of FH children and support of primary care physicians affected patient participation with respect to treatment strategies and follow-up. Genetic identification, screening, and treatment of FH were feasible in Vietnam. The VINAFH Registry significantly contributed to the formation of the government agencies legislative acts that established the importance of FH as a socially and medically important disease requiring appropriate management strategies. Other low- and middle-income countries could, thus, use the VINAFH Registry model as a reference to establish programs for FH management according to the current status.
Introduction
Familial hypercholesterolemia (FH) is a common inherited disorder, affecting one in 250 individuals (de Ferranti et al., 2016; Akioyamen et al., 2017). This disease is characterized by elevated plasma levels of low-density lipoprotein cholesterol (LDL-C) that facilitates the development of atherosclerosis, premature coronary artery disease (CAD), and mortality (Neil et al., 2004; Cuchel et al., 2014). FH involves mutations in the gene encoding LDL receptor (LDLR; 90% of reported FH–causing variants), gene encoding apo-lipoprotein B (APOB; 5–10%), and, rarely, gene encoding protein convertase subtilisin/kexin type 9 (PCSK9; 1%) (Sturm et al., 2018).
Early identification and lipid-lowering therapy play important roles in reducing the health burden for FH. Screening programs such as cascade screening, universal screening, and opportunistic screening are effective for early detection of FH (Kirke et al., 2012). Previously, the Dutch Lipid Clinic Network (DLCN) score was widely accepted for FH diagnosis (Vallejo-Vaz et al., 2018). The European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) recommend that lipid-lowering therapy should be started early and used optimally to achieve LDL-C goals (Mach et al., 2020). However, in reality, majority of FH patients remain underdiagnosed or undertreated. In most countries, less than 1% of FH patients are diagnosed (Nordestgaard et al., 2013). Furthermore, treatment for FH is still limited; for example, the CASCADE-FH Registry reported that only 48% of FH patients achieved LDL-C < 100 mg/dl and 22% achieved LDL-C < 70 mg/dl (Duell et al., 2019).
Because of these challenges, the EAS Familial Hypercholesterolemia Studies Collaboration (EAS-FHSC), the Familial Hypercholesterolemia Foundation, and the World Heart Federation initiated a global call to action for reducing the burden of disease and death due to FH (Vallejo-Vaz et al., 2015; Wilemon et al., 2020). Special attention was given to data registry as an important tool for providing knowledge and support management for FH. In fact, many developed countries have established FH registry. However, only a few low- and middle-income countries have established the same (Bamimore et al., 2015; Mehta et al., 2016; Vallejo-Vaz et al., 2018; Chen et al., 2019). Notably, Vietnam, a large and densely populated country in the Southeast Asia, has spurred rapid economic growth, greater than that of other low- and middle-income countries in the past years. With economic development, Vietnam has also achieved improvement in public health. However, changing lifestyles accompanying the economic growth lead to a double burden of disease; the burden of communicable disease remains, while the burden of non-communicable diseases, such as cardiovascular disease, is increasing (Nguyen and Hoang, 2018). Additionally, there exists a gap in the knowledge about cardiovascular disease, particularly FH. With a population at 97 million, we estimated 500,000 Vietnamese patients with FH, but most of them are underdiagnosed and undertreated (Vallejo-Vaz et al., 2018). This report describes the initial genetic characteristics, clinical characteristics, screening, diagnosis, and treatment of FH patients in Vietnam. We report the experiences gained from the implementation of the Vietnam Familial Hypercholesterolemia (VINAFH) Registry.
Materials and Methods
On the basis of the success of our previous preliminary small-scope research on FH (Truong et al., 2018), we extended the study and established the VINAFH Registry as a long-term prospective cohort promoted by the Vietnam National Heart Institute (VNHI), Bach Mai Hospital – the largest hospital for cardiovascular disease in North Vietnam – and the national referral cardiovascular hospital. Implementation of the VINAFH Registry is presented in Figure 1. The VINAFH Registry was approved by the Council for Science of the VNHI, Bach Mai Hospital (No. 183/VTM-BVBM) and the Council for Science of the Ministry of Science and Technology of Vietnam (No. 828/GXNDGTD-BKHCN). The VINAFH Registry has the following three specific objectives: (1) to identify and enroll heterozygous and homozygous FH individuals in Vietnam; (2) to understand the clinical and genetic characteristics of FH individuals in Vietnam; and (3) to improve the management strategies for FH in Vietnam. Recruitment of FH individuals began in 2016 and is still ongoing. All the FH individuals included in this study provided informed consent. FH individuals younger than 18 years were enrolled only with the explicit consent of a parent or legal guardian.
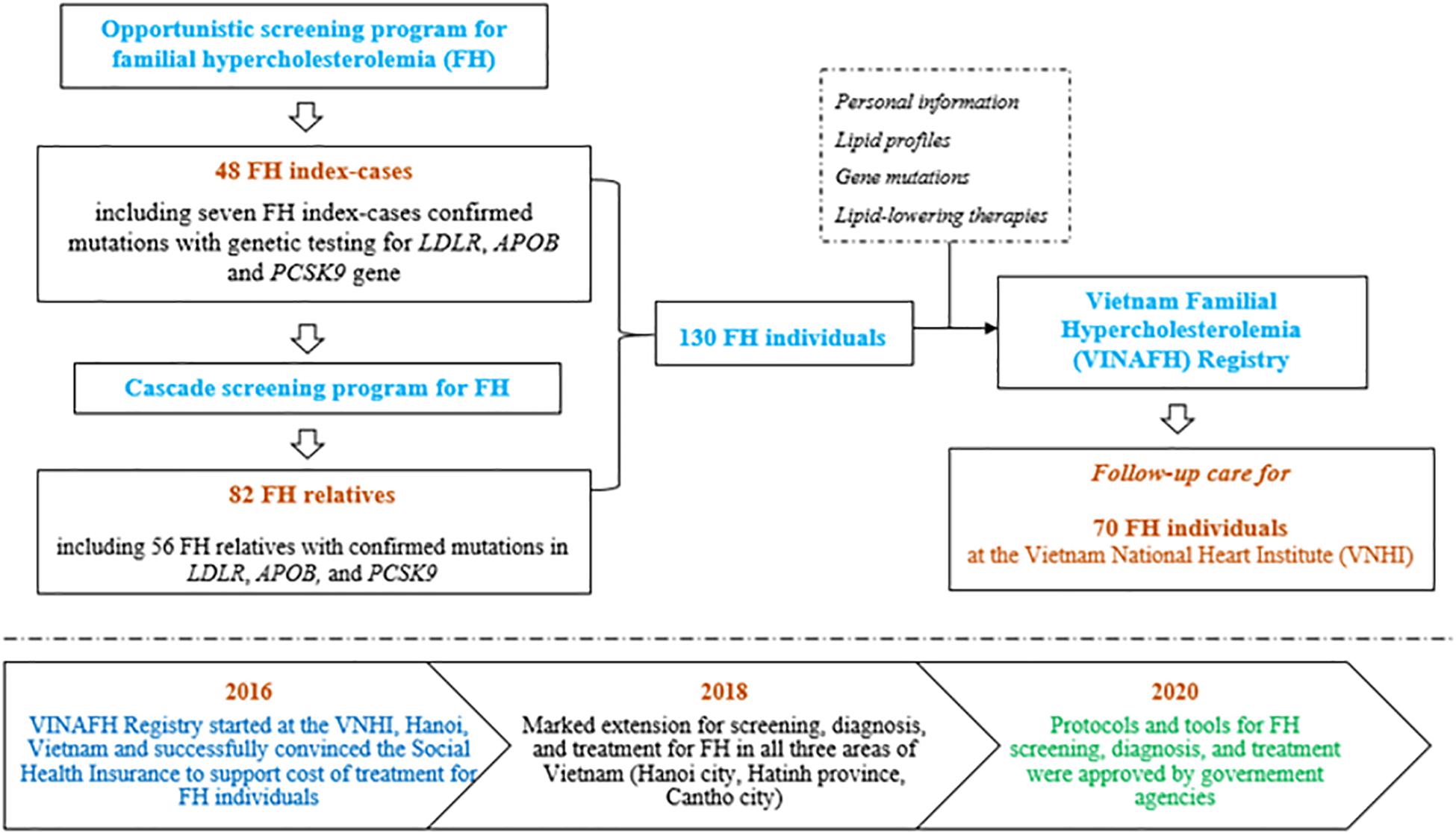
Figure 1. Flow diagram of implementation strategy of the Vietnam Familial Hypercholesterolemia (VINAFH) Registry.
Inclusion Criteria for Familial Hypercholesterolemia Individuals in the Vietnam Familial Hypercholesterolemia Registry
We enrolled phenotypic and/or genotypic FH individuals, including index cases and their relatives in the Registry. Index case was detected by opportunistic screening in patients with premature CAD and/or hypercholesterolemia. Cascade screening was performed to detect FH relatives of index case, as previously described (Truong et al., 2018).
Phenotypic Familial Hypercholesterolemia Individuals
In adults, phenotypic FH was confirmed based on the DLCN criteria (WHO Human Genetics Programme, 1998). Patients with DLCN scores of 3–5 (possible FH), 6–8 (probable FH), and >8 (definite FH) were enrolled. In case of children < 18 years of age, phenotypic FH was diagnosed based on the presence of an LDL-C level consistent with FH in addition to a family history of premature CAD, and/or baseline high cholesterol in one parent, and/or presence of an FH-causing mutation (Wiegman et al., 2015). We also enrolled the likely FH phenotype relatives based on age- and gender-specific LDL-C cutoffs as described by Starr et al. (2008).
Genotypic Familial Hypercholesterolemia Individuals
Mutations in index cases were detected by massively parallel sequencing of FH genes (LDLR, APOB, and PCSK9) and multiplex ligation-dependent probe amplification (MLPA) of LDLR, as previously described (Hooper et al., 2012). Depending on the mutation present in the index case, genetic testing for relatives was performed by Sanger sequencing of the exon containing the family mutation, or by MLPA of LDLR. The ClinVar database, PolyPhen2, and MutationTaster were used to confirm the mutations. Genotypic FH individuals were classified as homozygous for FH (HoFH), carrying the same mutations in both alleles of FH genes, and heterozygous for FH (HeFH), carrying only one mutation in the alleles of FH genes.
Exclusion Criteria for Familial Hypercholesterolemia Individuals in the Vietnam Familial Hypercholesterolemia Registry
Individuals with known medical conditions other than FH that contribute to hyperlipidemia, such as hypothyroidism, nephrotic syndrome, cholestasis, and hypopituitarism, were excluded from the VINAFH Registry. Medical, family, and clinical history were recorded for all registered individuals: characteristics based on the DLCN criteria (WHO Human Genetics Programme, 1998), lipid profiles, and mutation characteristics. Furthermore, information regarding risk factors for cardiovascular disease, including age, sex, smoking, diabetes, hypertension, obesity, premature CAD, and ongoing/past lipid-lowering therapies, were also collected in this Registry.
Outcome Measures
Primary outcomes:
– Number of FH individuals identified.
– Genetic testing for FH-causing mutations (LDLR, PCSK9, and APOB).
Secondary outcomes:
– Number of FH individuals who received dietary and lipid-lowering therapy.
– Number of FH individuals who achieved LDL-C targets.
– Incidence of cardiovascular events (coronary, cerebral, or peripheral vascular diseases) and cardiovascular cause detected during annual follow-up.
– Contribution of the Registry in the formation of national health policies concerning the recognition of FH as an important disease with development of appropriate management strategies.
Statistical Analyses
Continuous variables are expressed as the arithmetic mean and standard deviation if normally distributed and as the median with inter-quartile range for non-normal distribution. Categorical variables are expressed as count and percentage. Normally distributed continuous variables between two groups were compared using the Student t-test for independent samples. Non-normally distributed continuous variables were compared using the Mann–Whitney U test. Proportions were compared by using the chi-squared test with continuity correction or Fisher’s exact test, when appropriate. The analyses were performed with IBM SPSS 22.0 software. The results with p values below 0.05 were considered statistically significant.
Results
Genetic Identification of Familial Hypercholesterolemia Individuals
A total of 130 FH individuals have been registered. Of these, 63 FH individuals (48.5%) underwent genetic testing, confirming the presence of mutations in the genes studied (LDLR, APOB, and PCSK9); eight individuals were HoFH and 55 were HeFH. Four LDLR mutations and one PCSK9 mutation were identified in these individuals, while no pathogenic variants of APOB were identified. Mutations in LDLR were identified in 6/7 index cases (85.7%) in this Registry. We found novel mutations in LDLR, which has not been reported but are annotated in the ClinVar database: c.2529_2530delinsA, (p.Asp843Glufs*86). The variants and their distribution in the study cohort are given in Table 1.
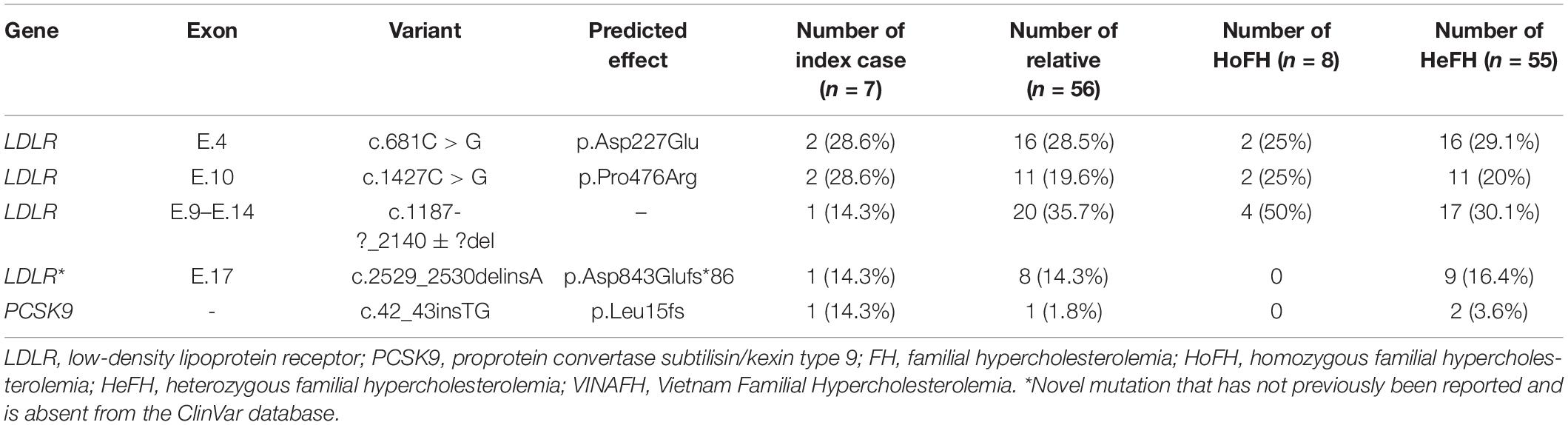
Table 1. LDLR and PCSK9 variants identified in the VINAFH Registry.
Screening of Familial Hypercholesterolemia Individuals
We identified 48 FH index cases and 82 FH relatives. Clinical characteristics of FH individuals are given in Table 2. Notably, the mean ± standard deviation of age for FH diagnosis was 34.8 ± 1.95 years, with an earlier age at diagnosis in the relative group compared with that in the index-case group (28.6 ± 2.43 vs. 45.3 ± 2.66 years, respectively, p < 0.001).
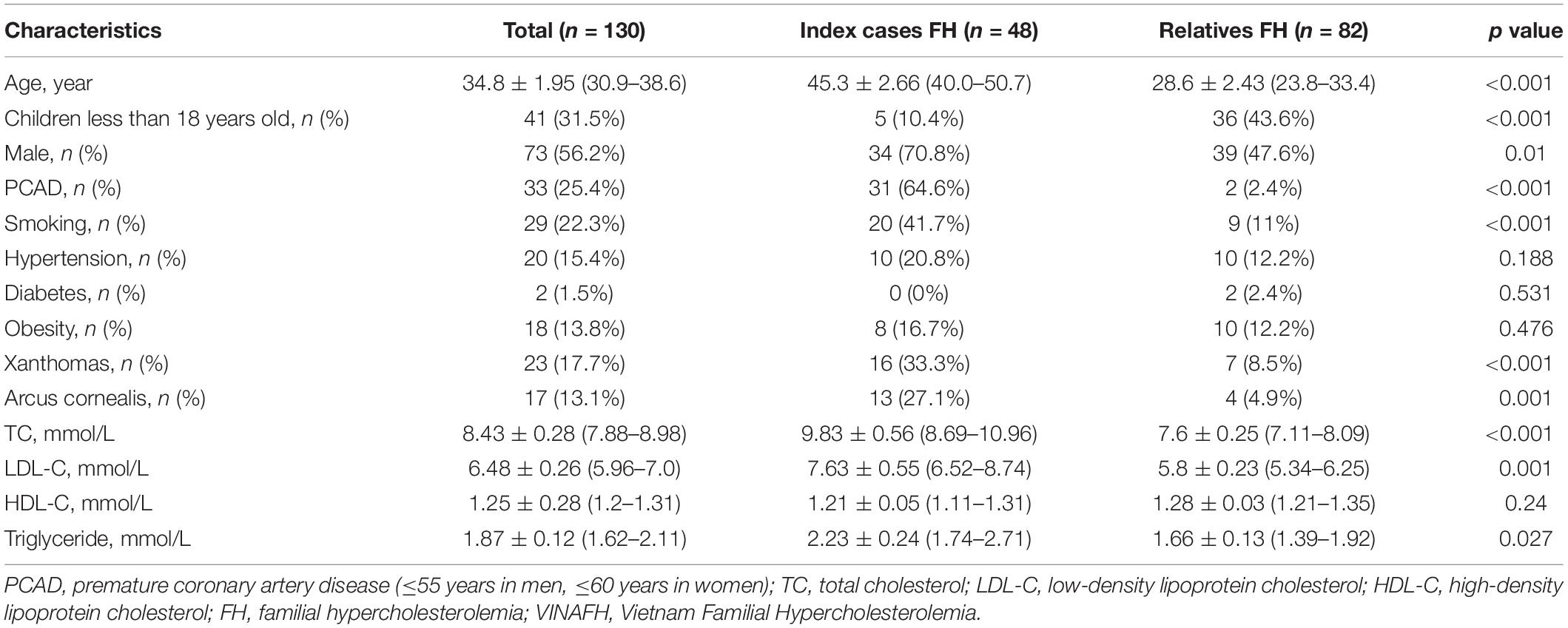
Table 2. Baseline characteristics of FH individuals in the VINAFH Registry.
Treatment of Familial Hypercholesterolemia Individuals
Post diagnosis, 53.8% (n = 70/130) of FH individuals continued to follow-up at the VNHI. All individuals were given standard treatment with dietary supplements containing plant stanols for controlling cardiovascular risks. Only 15.3% (n = 11/70) were given lipid-lowering therapy described in Table 3. Of these 11 individuals, five were HoFH while six were HeFH. After treatment with lipid-lowering therapies for 1 year, 83.3% (n = 5/6) of HeFH patients had LDL-C < 2.5 mmol/L, while the mean of plasma LDL-C of HoFH patients reduced from 17.5 ± 6.0 mmol/L at the time of diagnosis to 10.2 ± 4.1 mmol/L. No new cardiovascular events or mortalities were observed in these individuals. The lower incidence of lipid-lowering therapy was attributed to patient refusal. We recorded two important reasons for patient refusal to lipid-lowering therapy in case of individuals in the VINAFH Registry: lack of knowledge about the effects and side effects of lipid-lowering therapy and barriers due to treatment cost. Besides, FH individuals only agreed to lipid-lowering therapy after we contacted their primary care physicians and successfully convinced these physicians to join as collaborators in our network for FH management.
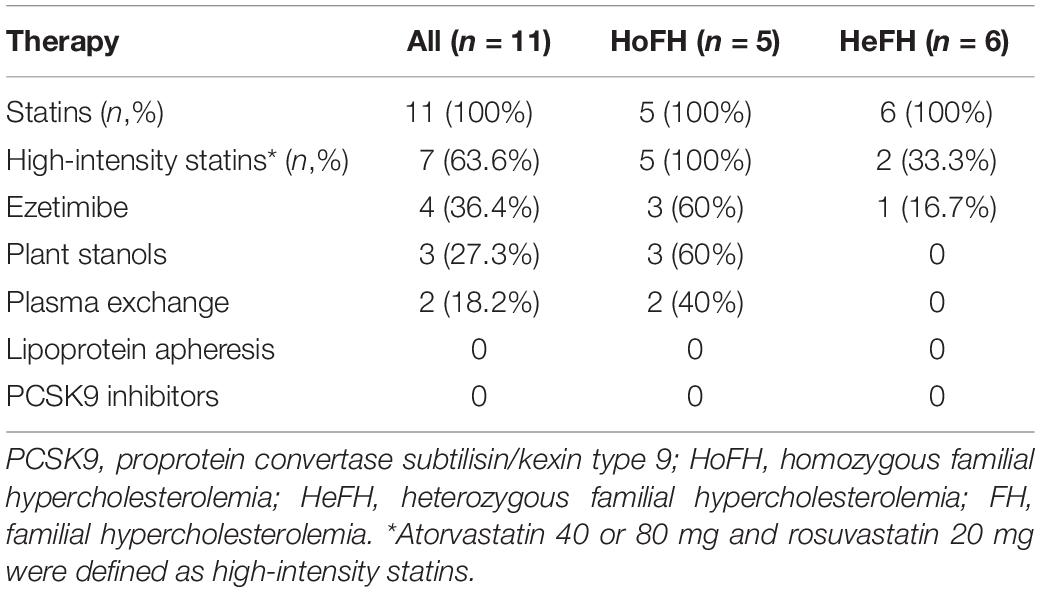
Table 3. Lipid-lowering therapy in 11 FH individuals.
Impact of the Vietnam Familial Hypercholesterolemia Registry on Health Policy
On the basis of experiences gained from the implementation the VINAFH Registry, we created academic documents, including (1) screening and testing (diagnostic and genetic) protocols for FH; (2) guidance on genetic counseling for FH; (3) tools for FH screening, diagnosis, and treatment; and (4) management model for FH. All of them were evaluated, appraised, and approved for clinical practice in Vietnam by the Ministry of Science and Technology of Vietnam. Further, we successfully convinced the Social Health Insurance to cover treatment cost for FH individuals who participated in the VINAFH Registry and were followed up at the VNHI.
Discussion
In our Registry, we registered a higher rate of FH individuals with confirmed mutations (48.5%, n = 63/130) than did few other countries (Nordestgaard and Benn, 2017), based on the support of national genetic experts and colleagues from Australia. We identified four different mutations in LDLR and one in PCSK9. The majority of FH individuals carried mutation in LDLR (85.7% of index cases and 96.8% in total), which is similar to that reported in previous studies (Soutar and Naoumova, 2007; Sjouke et al., 2015; Khera et al., 2016). We found that the exon mutations of LDLR were varied, which was also reported by a previous study in a Korean population (Lee, 2016). LDLR p.Asp227Glu (FH Afrikaner-1, FH Maine) missense variant, which occurs within repeat 5 of the ligand-binding domain of LDLR, has previously been identified (Kotze et al., 1993; Hooper et al., 2012; Sharifi et al., 2016). LDLR c.1187-?_2140 + ?del and PCSK9 c.42_43insTG have also been identified in several cohorts of FH patients (Goldmann et al., 2010; Clinvar database, 2020). Notably, two LDLR mutations (p.Pro476Arg and p.Asp843Glufs*86) that we identified in Vietnamese patients have not been recorded in other ethnicities as confirmed by the Clinvar database (2020). We previously reported the identification of LDLR p.Pro476Arg missense variant, which occurs within the gene encoding the EGF spacer domain of the LDLR, in two Vietnamese families with two HoFH index cases and 11 HeFH relatives (Truong et al., 2018). Pro476 is conserved across species, and prediction algorithms PolyPhen2 and MutationTaster suggest that Pro476Arg is pathogenic. In the VINAFH Registry cohort, we identified the novel mutation, LDLR p.Asp843Glufs*86 frameshift variant, which occurs within the gene encoding the cytoplasmic domain of the LDLR; it was detected in a family with nine HeFH individuals. This frameshift variant results in the loss of 18 amino acids and addition of 86 amino acids at the C-terminal of the LDLR. In general, simultaneous occurrence of both the previously reported variants and the novel variant detected in this study suggest a broad spectrum of mutations and high heterogeneity of FH in the Vietnamese population, which is similar to that observed in other countries (Jiang et al., 2015; Fairoozy et al., 2017).
Massively parallel sequencing detects structural variations with high sensitivity and specificity. However, it is inefficient to detect large deletions/duplications. In contrast, MLPA is highly sensitive to detect these mutations. A previous report showed that in 19/377 (5%) patients with suspected FH, no mutation was found with massively parallel sequencing, whereas MLPA identified large deletions/duplications in LDLR (Taylor et al., 2009). Thus, besides combined massively parallel sequencing, the MLPA method is useful for genetic testing in FH patients. Interestingly, our first index case with HoFH phenotype was also tested using massively parallel sequencing with targeted analysis of FH genes, but no mutations were detected. Therefore, the MLPA method was used to confirm this index case, and homozygotes for a deletion of exons 9 to 14 of LDLR were found. We also detected three HoFH relatives and 17 HeFH relatives carrying this mutation (Truong et al., 2018).
Screening is the first step to improve status of underdiagnosed and undertreated FH cases. In the VINAFH Registry, we combined opportunistic and cascade screening to increase the likelihood of detection. Opportunistic screening was the approach used to detect FH index cases in high-risk individuals such as those with premature CAD, hypercholesterolemia, xanthomas, and arcus cornealis (Nordestgaard et al., 2013). This approach was selected based on a high prevalence of FH in patients with premature CAD (Nanchen et al., 2015; Faggiano et al., 2018; Pirazzi et al., 2019). Elevated plasma total cholesterol and LDL-C levels are important clinical signs of FH. A previous study reported that 1.7% of individuals with LDL-C ≥ 190 mg/dl carried FH mutation (Khera et al., 2016). Moreover, xanthomas and arcus cornealis are also key signs of FH, especially in homozygotes (Bujo et al., 2004). Compared with previous preliminary small-scope report, including the first five FH index cases in Vietnam (Truong et al., 2018), the VINAFH Registry has significantly expanded the number of FH individuals, including 48 index cases detected by opportunistic screening. This provided evidence for the effectiveness of opportunistic screening, especially in developing countries where FH is underdiagnosed.
Because FH is dominantly inherited, each new FH case could become an index case for cascade screening. Indeed, this is a highly effective method for detecting FH in family members and has been approved in many countries (Bell et al., 2015; Jannes et al., 2015; Rubio-Marin et al., 2018). In fact, we undertook cascade screening for FH in close relatives of index cases and detected 82 FH cases. On average, 14 new cases of FH were detected per HoFH index case (Truong et al., 2018). The VINAFH Registry revealed that FH relatives had a younger mean for age than FH index cases. Interestingly, 43.6% of FH relatives were children < 18 years old. Fortunately, only 2.4% of FH relatives had a history of premature CAD, which was lower than the FH index cases (64.6%). Early diagnosis and, thus, prevention or delaying the onset of atherosclerotic cardiovascular disease are the most important factors in the management of FH (Nordestgaard et al., 2013; Knowles et al., 2017; Mach et al., 2020).
The VINAFH Registry has also shown a high prevalence of FH individuals with cardiovascular risk factors including smoking, hypertension, and obesity. Like low- and middle-income countries, Vietnam also faces the increasing burden of non-communicable diseases (Bennett et al., 2018). In a national survey of risk factors for non-communicable diseases in 2015, prevalence of smoking, hypertension, and overweight/obesity was 55.7, 18.5, and 21.1%, respectively, in men and 1.73, 10.2, and 21.2%, respectively, in women (Bui et al., 2016). In FH management, controlling cardiovascular risk factors has been reported to be highly beneficial for reducing cardiovascular events and mortality (Akioyamen et al., 2019). Therefore, the prevention program for FH in Vietnam will focus on lifestyle modification education.
Considerable efforts have been taken for the management of FH in Vietnam, and 53.8% of FH individuals continued to follow-up post diagnosis. FH individuals were also educated in lifestyle modification. However, a large number of Vietnamese FH individuals were undertreated, which is also commonly observed in many countries (Nordestgaard et al., 2013). The VINAFH Registry showed that traditional lipid-lowering therapies, including statin, ezetimibe, plant stanols, and plasma exchange, were effective for reducing plasma LDL-C levels in FH individuals. As per the ESC/EAS guidelines, statin is recommended as the first-line therapy for FH, and high-intensity and maximal potent statin doses are preferred (Nordestgaard et al., 2013; Cuchel et al., 2014; Besseling et al., 2016; Mach et al., 2020). However, in the VINAFH Registry, prevalence of HeFH individuals prescribed with high-intensity statins was limited (33.3%), similar to previous reports in China (Auckle et al., 2017). In Japan, which is a developed country, only 19.2% of FH individuals were treated with high-intensity statins (Teramoto et al., 2018). Lipoprotein apheresis and PCSK9 inhibitors, which are currently presented as efficient methods of treatment of FH (Mach et al., 2020), are still not available in Vietnam. A previous survey by the EAS-FHSC showed that these lipid-lowering therapies are limited in most countries (Vallejo-Vaz et al., 2018).
Limited use of lipid-lowering therapies for FH individuals could be explained by the lack of knowledge and awareness about the disease. Our interviews with FH individuals or legal guardians of FH children noted confusion about the effects and side effects of long-term drug use. It should be noted that in the health-care system, patient-primary care physician relationship is of the utmost importance; everybody highly trusts their physician (O’Malley et al., 2004). Primary care physicians and cardiologists generally advice the patients regarding health and prescribe drugs on the basis of their knowledge. Therefore, updating the physicians’ knowledge about FH is significant to obtain consent for the patient’s treatment. However, Vietnamese physicians had a large deficit in FH knowledge and awareness (Pang et al., 2017). This emphasizes the critical importance of implementing education and awareness programs for both FH individuals and physicians. Ideally, physicians treating FH individual should collaborate for FH management.
As mentioned, many low- and middle-income countries suffer from a double disease burden, the backlog of common infections, and the emerging challenges of non-communicable diseases; moreover, their health resources are also limited. If the relevant information is not available to the government agencies, they may omit important diseases, such as FH, from the national health policy. Therefore, scientists play important roles for providing evidence accumulated though such registries and persuade government agencies to adjust the health policies. In our case, we conducted a preliminary small-scope research for FH, then extended it through the VINAFH Registry, and reported the updated results about FH status in Vietnamese individuals to the government agencies. Simultaneously, we convinced the Social Health Insurance to support cost of treatment for the registered FH individuals. In our experience, initial results of such a registry should provide data and documentation regarding evidence of the existence of the disease pathology, initial results of FH management, and protocols and tools for screening, diagnosis, and treatment of FH. Notably, including genetic information into the registry provides high-value scientific evidence that is a useful factor that increases the persuasiveness of the study for government agencies. Thus, the VINAFH Registry has significantly contributed to the formation of government agencies legislative acts, establishing FH as a socially and medically important disease with appropriate management strategies. It has also led to the deployment of a national screening and disease management program for FH in Vietnam in the future.
Conclusion
In conclusion, the VINAFH Registry is the first database on genetic screening and management of FH in the Vietnamese population. Moreover, we reported a novel variant in LDLR that were identified in our cohort. The likely occurrence of a complex of FH mutations suggests the need for a national FH genetic study. Based on the findings of this study with respect to the treatment strategies for Vietnamese FH patients, we propose the need for awareness and educational programs about FH for patients and doctors, so as to increase the number of diagnosed and treated patients. The VINAFH Registry had an important contribution in the formation of government agencies legislative acts concerning the establishment of FH as a socially and medically important disease and development of appropriate management strategies. Low- and middle-income countries might refer to our Registry to establish similar programs for the management of FH on the basis of genetic testing combined with opportunistic and cascade screening. The management strategies for FH should be implemented in a step-by-step manner on the basis of the personal and financial resources available in these countries.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Council for Science of the VNHI, Bach Mai Hospital (No. 183/VTM-BVBM) and the Council for Science of the Ministry of Science and Technology of Vietnam (No. 828/GXNDGTD-BKHCN). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
T-HT and D-LD initiated the study, designed data collection tools, monitored data collection, cleaned and analyzed the data, and drafted and revised the manuscript. N-TK and M-NN monitored data collection, cleaned and analyzed the data, and revised the draft manuscript. T-TL and H-AL monitored data collection and revised the draft manuscript. All authors read and approved the final manuscript.
Funding
N-TK was supported by the “10 Countries” FH program, funded by the International Atherosclerosis Society (IAS) and Pfizer Independent Grants for Learning and Change (Grant ID: 10839501). Genetic testing was performed in-kind by the Department of Clinical Biochemistry, PathWest Laboratory Medicine (Western Australia) and the Department of Medical Biology and Genetics, Hanoi Medical University, and with the support of the BIMEDTECH Molecular Biology Laboratory.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the team members from the “10 Countries Study” and the EAS-FHSC for their support to establish the VINAFH registry. We thank the medics and nurses from Bach Mai Hospital, medical staff at local health-care facilities, residences, and medical students at the Hanoi Medical University for their support of the FH screening program.
References
Akioyamen, L. E., Genest, J., Chu, A., Inibhunu, H., Ko, D. T., and Tu, J. V. (2019). Risk factors for cardiovascular disease in heterozygous familial hypercholesterolemia: a systematic review and meta-analysis. J. Clin. Lipidol. 13, 15–30. doi: 10.1016/j.jacl.2018.10.012
Akioyamen, L. E., Genest, J., Shan, S. D., Reel, R. L., Albaum, J. M., Chu, A., et al. (2017). Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open 7:e016461. doi: 10.1136/bmjopen-2017-016461
Auckle, R., Su, B., Li, H., Xu, S., Xie, M., Song, Y., et al. (2017). Familial hypercholesterolemia in Chinese patients with premature ST-segment-elevation myocardial infarction: prevalence, lipid management and 1-year follow-up. PLoS One 12:e0186815. doi: 10.1371/journal.pone.0186815
Bamimore, M. A., Zaid, A., Banerjee, Y., Al-Sarraf, A., Abifadel, M., Seidah, N. G., et al. (2015). Familial hypercholesterolemia mutations in the Middle Eastern and North African region: a need for a national registry. J. Clin. Lipidol. 9, 187–194. doi: 10.1016/j.jacl.2014.11.008
Bell, D. A., Pang, J., Burrows, S., Bates, T. R., van Bockxmeer, F. M., Hooper, A. J., et al. (2015). Effectiveness of genetic cascade screening for familial hypercholesterolaemia using a centrally co-ordinated clinical service: an Australian experience. Atherosclerosis 239, 93–100. doi: 10.1016/j.atherosclerosis.2014.12.036
Bennett, J. E., Stevens, G. A., Mathers, C. D., Bonita, R., Rehm, J., Kruk, M. E., et al. (2018). NCD Countdown 2030: worldwide trends in non-communicable disease mortality and progress towards sustainable development goal target 3.4. Lancet 392, 1072–1088. doi: 10.1016/S0140-6736(18)31992-5
Besseling, J., Hovingh, G. K., Huijgen, R., Kastelein, J. J. P., and Hutten, B. A. (2016). Statins in familial hypercholesterolemia: consequences for coronary artery disease and all-cause mortality. J. the Am. Coll. Cardiol. 68, 252–260. doi: 10.1016/j.jacc.2016.04.054
Bui, T. V., Blizzard, C. L., Luong, K. N., Truong Nle, V., Tran, B. Q., Otahal, P., et al. (2016). National survey of risk factors for non-communicable disease in Vietnam: prevalence estimates and an assessment of their validity. BMC Public Health 16:498. doi: 10.1186/s12889-016-3160-4
Bujo, H., Takahashi, K., Saito, Y., Maruyama, T., Yamashita, S., Matsuzawa, Y., et al. (2004). Clinical features of familial hypercholesterolemia in Japan in a database from 1996-1998 by the research committee of the ministry of health, labour and welfare of Japan. J. Atheroscler. Thromb. 11, 146–151. doi: 10.5551/jat.11.146
Chen, P., Chen, X., and Zhang, S. (2019). Current status of familial hypercholesterolemia in China: a need for patient FH registry systems. Front. Physiol. 10:280. doi: 10.3389/fphys.2019.00280
Clinvar database (2020). Available online at: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed March 31, 2020).
Cuchel, M., Bruckert, E., Ginsberg, H. N., Raal, F. J., Santos, R. D., Hegele, R. A., et al. (2014). Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the consensus panel on familial hypercholesterolaemia of the european atherosclerosis society. Eur. Heart J. 35, 2146–2157. doi: 10.1093/eurheartj/ehu274
de Ferranti, S. D., Rodday, A. M., Mendelson, M. M., Wong, J. B., Leslie, L. K., and Sheldrick, R. C. (2016). Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States national health and nutrition examination surveys (NHANES). Circulation 133, 1067–1072. doi: 10.1161/circulationaha.115.018791
Duell, P. B., Gidding, S. S., Andersen, R. L., Knickelbine, T., Anderson, L., Gianos, E., et al. (2019). Longitudinal low density lipoprotein cholesterol goal achievement and cardiovascular outcomes among adult patients with familial hypercholesterolemia: the CASCADE FH registry. Atherosclerosis 289, 85–93. doi: 10.1016/j.atherosclerosis.2019.08.007
Faggiano, P., Pirillo, A., Griffo, R., Ambrosetti, M., Pedretti, R., Scorcu, G., et al. (2018). Prevalence and management of familial hypercholesterolemia in patients with coronary artery disease: the heredity survey. Intern. J. Cardiol. 252, 193–198. doi: 10.1016/j.ijcard.2017.10.105
Fairoozy, R. H., Futema, M., Vakili, R., Abbaszadegan, M. R., Hosseini, S., Aminzadeh, M., et al. (2017). The genetic spectrum of familial hypercholesterolemia (FH) in the Iranian population. Sci. Rep. 7:17087. doi: 10.1038/s41598-017-17181-9
Goldmann, R., Tichy, L., Freiberger, T., Zapletalova, P., Letocha, O., Soska, V., et al. (2010). Genomic characterization of large rearrangements of the LDLR gene in Czech patients with familial hypercholesterolemia. BMC Med. Genet. 11:115. doi: 10.1186/1471-2350-11-115
Hooper, A. J., Nguyen, L. T., Burnett, J. R., Bates, T. R., Bell, D. A., Redgrave, T. G., et al. (2012). Genetic analysis of familial hypercholesterolaemia in Western Australia. Atherosclerosis 224, 430–434. doi: 10.1016/j.atherosclerosis.2012.07.030
Jannes, C. E., Santos, R. D., de Souza Silva, P. R., Turolla, L., Gagliardi, A. C., Marsiglia, J. D., et al. (2015). Familial hypercholesterolemia in Brazil: cascade screening program, clinical and genetic aspects. Atherosclerosis 238, 101–107. doi: 10.1016/j.atherosclerosis.2014.11.009
Jiang, L., Sun, L. Y., Dai, Y. F., Yang, S. W., Zhang, F., and Wang, L. Y. (2015). The distribution and characteristics of LDL receptor mutations in China: a systematic review. Sci. Rep. 5:17272. doi: 10.1038/srep17272
Khera, A. V., Won, H. H., Peloso, G. M., Lawson, K. S., Bartz, T. M., Deng, X., et al. (2016). Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J. Am. Coll. Cardiol. 67, 2578–2589. doi: 10.1016/j.jacc.2016.03.520
Kirke, A., Watts, G. F., and Emery, J. (2012). Detecting familial hypercholesterolaemia in general practice. Austr. Fam. Phys. 41, 965–968.
Knowles, J. W., Rader, D. J., and Khoury, M. J. (2017). Cascade screening for familial hypercholesterolemia and the use of genetic testing. JAMA 318, 381–382. doi: 10.1001/jama.2017.8543
Kotze, M. J., De Villiers, W. J., Steyn, K., Kriek, J. A., Marais, A. D., Langenhoven, E., et al. (1993). Phenotypic variation among familial hypercholesterolemics heterozygous for either one of two Afrikaner founder LDL receptor mutations. Arterioscler. Thromb. 13, 1460–1468. doi: 10.1161/01.atv.13.10.1460
Lee, S. H. (2016). Characteristics and vascular complications of familial hypercholesterolemia in Korea. J. Atheroscler. Thromb. 23, 532–538. doi: 10.5551/jat.34363
Mach, F., Baigent, C., Catapano, A. L., Koskinas, K. C., Casula, M., Badimon, L., et al. (2020). 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur. Heart J. 41, 111–188. doi: 10.1093/eurheartj/ehz455
Mehta, R., Zubiran, R., Martagon, A. J., Vazquez-Cardenas, A., Segura-Kato, Y., Tusie-Luna, M. T., et al. (2016). The panorama of familial hypercholesterolemia in Latin America: a systematic review. J. Lipid Res. 57, 2115–2129. doi: 10.1194/jlr.R072231
Nanchen, D., Gencer, B., Auer, R., Raber, L., Stefanini, G. G., Klingenberg, R., et al. (2015). Prevalence and management of familial hypercholesterolaemia in patients with acute coronary syndromes. Eur. Heart J. 36, 2438–2445. doi: 10.1093/eurheartj/ehv289
Neil, H. A., Seagroatt, V., Betteridge, D. J., Cooper, M. P., Durrington, P. N., Miller, J. P., et al. (2004). Established and emerging coronary risk factors in patients with heterozygous familial hypercholesterolaemia. Heart 90, 1431–1437. doi: 10.1136/hrt.2003.022764
Nguyen, T. T., and Hoang, M. V. (2018). Non-communicable diseases, food and nutrition in Vietnam from 1975 to 2015: the burden and national response. Asia Pac. J. Clin. Nutrit. 27, 19–28. doi: 10.6133/apjcn.032017.13
Nordestgaard, B. G., and Benn, M. (2017). Genetic testing for familial hypercholesterolaemia is essential in individuals with high LDL cholesterol: who does it in the world? Eur. Heart J. 38, 1580–1583. doi: 10.1093/eurheartj/ehx136
Nordestgaard, B. G., Chapman, M. J., Humphries, S. E., Ginsberg, H. N., Masana, L., Descamps, O. S., et al. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the european atherosclerosis society. Eur. Heart J. 34, 3478–3490. doi: 10.1093/eurheartj/eht273
O’Malley, A. S., Sheppard, V. B., Schwartz, M., and Mandelblatt, J. (2004). The role of trust in use of preventive services among low-income African-American women. Prevent. Med. 38, 777–785. doi: 10.1016/j.ypmed.2004.01.018
Pang, J., Hu, M., Lin, J., Miida, T., Nawawi, H. M., Park, J. E., et al. (2017). An enquiry based on a standardised questionnaire into knowledge, awareness and preferences concerning the care of familial hypercholesterolaemia among primary care physicians in the Asia-Pacific region: the ten countries Study. BMJ Open 7:e017817. doi: 10.1136/bmjopen-2017-017817
Pirazzi, C., Håkansson, L., Gustafsson, C., Omerovic, E., Wiklund, O., and Mancina, R. M. (2019). High prevalence of genetic determined familial hypercholesterolemia in premature coronary artery disease. Appl. Clin. Genet. 12, 71–78. doi: 10.2147/TACG.S202942
Rubio-Marin, P., Michan-Dona, A., Maraver-Delgado, J., Arroyo-Olivares, R., Barrado Varea, R., Perez de Isla, L., et al. (2018). Cascade screening program for familial hypercholesterolemia. Endocrinol. Diabetes Nutr. 65, 280–286. doi: 10.1016/j.endinu.2017.12.009
Sharifi, M., Walus-Miarka, M., Idzior-Walus, B., Malecki, M. T., Sanak, M., Whittall, R., et al. (2016). The genetic spectrum of familial hypercholesterolemia in south-eastern Poland. Metab. Clin. Exper. 65, 48–53. doi: 10.1016/j.metabol.2015.10.018
Sjouke, B., Kusters, D. M., Kindt, I., Besseling, J., Defesche, J. C., Sijbrands, E. J., et al. (2015). Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur. Heart J. 36, 560–565. doi: 10.1093/eurheartj/ehu058
Soutar, A. K., and Naoumova, R. P. (2007). Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 4, 214–225. doi: 10.1038/ncpcardio0836
Starr, B., Hadfield, S. G., Hutten, B. A., Lansberg, P. J., Leren, T. P., Damgaard, D., et al. (2008). Development of sensitive and specific age- and gender-specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin. Chem. Lab. Med. 46, 791–803. doi: 10.1515/cclm.2008.135
Sturm, A. C., Knowles, J. W., Gidding, S. S., Ahmad, Z. S., Ahmed, C. D., Ballantyne, C. M., et al. (2018). Clinical genetic testing for familial hypercholesterolemia: JACC scientific expert panel. J. Am. Coll. Cardiol. 72, 662–680. doi: 10.1016/j.jacc.2018.05.044
Taylor, A., Martin, B., Wang, D., Patel, K., Humphries, S. E., and Norbury, G. (2009). Multiplex ligation-dependent probe amplification analysis to screen for deletions and duplications of the LDLR gene in patients with familial hypercholesterolaemia. Clin. Genet. 76, 69–75. doi: 10.1111/j.1399-0004.2009.01168.x
Teramoto, T., Kai, T., Ozaki, A., Crawford, B., Arai, H., and Yamashita, S. (2018). Treatment patterns and lipid profile in patients with familial hypercholesterolemia in Japan. J. Atheroscler. Thromb. 25, 580–592. doi: 10.5551/jat.41483
Truong, T. H., Kim, N. T., Nguyen, M. N. T., Pang, J., Hooper, A. J., Watts, G. F., et al. (2018). Homozygous familial hypercholesterolaemia in Vietnam: case series, genetics and cascade testing of families. Atherosclerosis 277, 392–398. doi: 10.1016/j.atherosclerosis.2018.06.013
Vallejo-Vaz, A. J., De Marco, M., Stevens, C. A. T., Akram, A., Freiberger, T., Hovingh, G. K., et al. (2018). Overview of the current status of familial hypercholesterolaemia care in over 60 countries - the EAS familial hypercholesterolaemia studies collaboration (FHSC). Atherosclerosis 277, 234–255. doi: 10.1016/j.atherosclerosis.2018.08.051
Vallejo-Vaz, A. J., Kondapally Seshasai, S. R., Cole, D., Hovingh, G. K., Kastelein, J. J., Mata, P., et al. (2015). Familial hypercholesterolaemia: a global call to arms. Atherosclerosis 243, 257–259. doi: 10.1016/j.atherosclerosis.2015.09.021
WHO Human Genetics Programme (1998). Familial Hypercholesterolaemia (ıııFH)ııı : Report of a Second WHO Consultation. Geneva: WHO Human Genetics Programme.
Wiegman, A., Gidding, S. S., Watts, G. F., Chapman, M. J., Ginsberg, H. N., Cuchel, M., et al. (2015). Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur. Heart J. 36, 2425–2437. doi: 10.1093/eurheartj/ehv157
Keywords: familial hypercholesterolemia, VINAFH Registry, low- and middle-income country, genetics, screening, treatment
Citation: Truong T-H, Do D-L, Kim N-T, Nguyen M-NT, Le T-T and Le H-A (2020) Genetics, Screening, and Treatment of Familial Hypercholesterolemia: Experience Gained From the Implementation of the Vietnam Familial Hypercholesterolemia Registry. Front. Genet. 11:914. doi: 10.3389/fgene.2020.00914
Received: 02 April 2020; Accepted: 23 July 2020;
Published: 14 August 2020.
Edited by:
Alpo Juhani Vuorio, University of Helsinki, FinlandReviewed by:
Marco Savarese, University of Helsinki, FinlandAnjana Munshi, Central University of Punjab, India
Copyright © 2020 Truong, Do, Kim, Nguyen, Le and Le. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thanh-Huong Truong, bWR0cnRoaHVvbmdAeWFob28uY29t; Doan-Loi Do, ZG9kb2FubG9pQGdtYWlsLmNvbQ==; Ngoc-Thanh Kim, a2ltbmdvY3RoYW5oQGhtdS5lZHUudm4=
†These authors have contributed equally to this work