 Chiara Papulino
Chiara Papulino Ugo Chianese
Ugo Chianese Maria Maddalena Nicoletti
Maria Maddalena Nicoletti Rosaria Benedetti
Rosaria Benedetti Lucia Altucci
Lucia Altucci- Department of Precision Medicine, Università degli Studi della Campania “Luigi Vanvitelli”, Naples, Italy
Epigenetics has achieved a profound impact in the biomedical field, providing new experimental opportunities and innovative therapeutic strategies to face a plethora of diseases. In the rare diseases scenario, Beckwith-Wiedemann syndrome (BWS) is a pediatric pathological condition characterized by a complex molecular basis, showing alterations in the expression of different growth-regulating genes. The molecular origin of BWS is associated with impairments in the genomic imprinting of two domains at the 11p15.5 chromosomal region. The first domain contains three different regions: insulin growth like factor gene (IGF2), H19, and abnormally methylated DMR1 region. The second domain consists of cell proliferation and regulating-genes such as CDKN1C gene encoding for cyclin kinase inhibitor its role is to block cell proliferation. Although most cases are sporadic, about 5–10% of BWS patients have inheritance characteristics. In the 11p15.5 region, some of the patients have maternal chromosomal rearrangements while others have Uniparental Paternal Disomy UPD(11)pat. Defects in DNA methylation cause alteration of genes and the genomic structure equilibrium leading uncontrolled cell proliferation, which is a typical tumorigenesis event. Indeed, in BWS patients an increased childhood tumor predisposition is observed. Here, we summarize the latest knowledge on BWS and focus on the impact of epigenetic alterations to an increased cancer risk development and to metabolic disorders. Moreover, we highlight the correlation between assisted reproductive technologies and this rare disease. We also discuss intriguing aspects of BWS in twinning. Epigenetic therapies in clinical trials have already demonstrated effectiveness in oncological and non-oncological diseases. In this review, we propose a potential “epigenetic-based” approaches may unveil new therapeutic options for BWS patients. Although the complexity of the syndrome is high, patients can be able to lead a normal life but tumor predispositions might impair life expectancy. In this sense epigenetic therapies should have a supporting role in order to guarantee a good prognosis.
Introduction
Epigenetic alterations play a crucial role in both cancer and non-oncological diseases (Mau and Yung, 2014) regulating DNA methylation, histone modifications (Nowacka-Zawisza and Wisnik, 2017) and micro-RNA expression that ultimately determines gene expression (Abi Khalil, 2014).
Some rare congenital diseases present alterations in epigenetically regulated genes (Niculescu and Lupu, 2011), providing the rationale to interfere via an epigenetic rebalance to mitigate/overcome these conditions (Nguyen, 2019). BWS is an example of a complex disease characterized by genetic and epigenetic aberrations on chromosomal region 11p15.5 comprising a telomeric and a centromeric domain, both regulated by genomic imprinting (Gomes et al., 2009). BWS is characterized by juvenile abnormal overgrowth (Choufani et al., 2010; Eggermann et al., 2015), affecting 1 child in 10,340–13,700 live births worldwide (Mussa et al., 2013). The associated phenotype may have a range of manifestations (Maas et al., 2016; Dawkins et al., 2018), although the most common clinical features are macroglossia, present in half of the patients with a molecular defect at 11p15.5 (Gaston et al., 2001; Brioude et al., 2018), visceromegalia and abdominal wall defects. Minor features include ear pits, hypoglycemia, nephromegaly, and isolated lateralized overgrowth (Mussa et al., 2016b). BWS is equally incident in males and females, but in monozygotic twins, there is observed an excess in females (Weksberg et al., 2010), and the discordant disease presentation suggests an important epigenetic role (Bliek et al., 2009b).
BWS is considered an imprinting disorder (IDs) affecting growth, development, and metabolism (Netchine et al., 2013) often caused by alterations in imprinting control regions (ICRs) in the parental 11p15.5 region (Abramowitz and Bartolomei, 2012; Begemann et al., 2012; Abi Khalil, 2014). ICRs impairment leads to abnormal methylation state deregulating genes as CDNK1C, H19, IGF2, and KCNQ1OT1 involved in growth, so provoking the onset of BWS features (Wang et al., 2020). The expression/inactivation of a gene is related to differentially methylated regions (DMRs).
The majority of IDs such as BWS present the same four classes of molecular alterations lead to imbalanced gene expression: uniparental disomy (UPD), epimutation (aberrant methylation marks), copy number variations (CNVs) and point mutations in imprinted genes. Since DNA methylation marks are transmitted through generations, an interesting approach for BWS resides in the epigenetic-regulated inheritance study, which is still in an early phase. In that case, the major limitation for it to be carried on is due to the low patients’ number (Nguyen, 2019). For this reason, multi-institutional collaborations are required in order to reach a statistically significant number of patients for the constitution of an observational cohort or a clinical trial (Griggs et al., 2009). Moreover, the low BWS prevalence requires special combined efforts to improve diagnosis, care, and prevention (Ayme and Schmidtke, 2007). In order to overcome this limitation and trying to boost patient care, the Coordination of Rare Diseases at Sanford (CoRDS) started in 2013 a program (NCT01793168) providing a centralized- international patients registry for all rare diseases. This program helps in the identification, advance treatments, and therapies on a large cohort of patients.
Role of Epigenetic Alterations in BWS
Epigenetic alterations associated with BWS are present on two different domains, independently methylated in the 11p15.5 gene cluster (Krzyzewska et al., 2019): H19/IGF2:IG-DMR (intergenic differentially methylated region) and KCNQ1OT1:TSS-DMR (transcription start site differentially methylated region) (Figure 1). The first region is also known as ICR1 consisting of two main imprinted genes, IGF2 and H19. IGF2 is paternally expressed and it encodes s for a fetal growth factor implicated in BWS pathogenesis. H19 is a maternally expressed allele, non-coding RNA, and its function is still unclear, although it has been postulated a tumor suppressor role (Cai and Cullen, 2007; Raveh et al., 2015). IGF2 deregulation determines overgrowth and region-specific tumor development in BWS (Schofield et al., 2001). IGF2 and H19 genes are oppositely imprinted with an enhanced competition (Maher and Reik, 2000). The H19/IGF2:IG-DMR domain works as a chromatin insulator regulating the expression of IGF2 and H19 genes. The chromatin insulator is located 2 Kb upstream of the H19 gene and displays CTCF binding sites (Hark et al., 2000). Once CTCF binding is prevented, the enhancers can interact with IGF2 promoters. Oppositely the unmethylated maternal allele allows CTCF binding to the insulator elements blocking the downstream enhancers of H19 to access the IGF2 promoter (Soejima and Higashimoto, 2013) (Figure 2A). H19/IGF2:IG-DMR-specific histone marks have also been reported: the methylated region presents H3K9me3 and H4K20me3, whereas the unmethylated area carries H3K4me2/3 and H3/H4 acetylated (Nativio et al., 2011). In 5-10% of BWS patients observed with a gain of methylation (GOM) of the H19/IGF2: IG-DMR on the unmethylated maternal allele (Figure 2B) (Eggermann et al., 2014; Brioude et al., 2018). The aberrant methylation is also characterized by variations in histone signatures. Accessible modification of H3K9ac and bivalent H3K4me2/H3K27me3 are converted to the repressive H3K9me3 and H4K20me3 (Nativio et al., 2011). These chromatin alterations prevent CTCF binding to the maternal H19/IGF2:IG-DMR, thus enabling the enhancers to access the IGF2 promoter and leading both to a biallelic expression of IGF2 and reduced expression of H19. In detail, H19/IGF2:IG-DMR is characterized by 59-bp elements hD1 and hD2, composed mainly of two Sox-Oct motif-like sequences (SO motifs) and a single Oct motif (rO) and are accompanied by triple-repeat sequences containing CBS1-3 or CBS4-6 (also known as CBSs). Recently, by using transplantation in mice, it was reported that H19/IGF2:IG-DMR methylation status is regulated by the CBSs and SO motifs (Hori et al., 2012). Interestingly in BWS patients, SO motifs are mutated or deleted. Three Oct motifs variants found in BWS patients disrupt hD1-dependent DNA demethylation and cause the stack of methylation (Kubo et al., 2020). Single nucleotide substitutions in hD1 Oct motifs present in BWS patients show hypermethylation in H19/IGF2:IG-DMR coupled with histone modifications: maternal aberrant DNA methylation is connected with reduction of H3K4me2 and H3K9ac and increase of H3K9me3 and H3K27me3 (Kubo et al., 2020). KCNQ1OT1:TSS-DMR contains six different imprinted genes: KCNQ1, KCNQ1OT1, CDKN1C, SLC22A18, TSSC3, and PHLDA2. CDKN1C is a maternally expressed gene encoding for a cell cycle regulator, a cyclin-dependent kinase inhibitor. It regulates in a negative manner cell proliferation. KCNQ1OT1 is a paternally expressed long non-coding RNA capable to inhibit the expression of genes in the domain in cis. The KCNQ1OT1:TSS-DMR of this domain is localized in the intron 10 of the KCNQ1 gene, and it is methylated on the maternal but not on the paternal allele; thus the unmethylated paternal one allows the transcription of KCNQ1OT1:TSS-DMR, preventing the CDKN1C and KCNQ1 gene expression. In mice, this DMR interacts with G9a and the PRC2 complex leading to repressive histone modifications such as H3K9me3 and H3K27me3. The methylated KCNQ1OT1:TSS-DMR form present on the maternal allele prevents the transcription of the long non-coding RNA, KCNQ1OT1, allowing the expression of several genes present in the domain such as CDKN1C. Loss of methylation (LOM) on the maternal allele occurs in 50% of BWS patients (Eggermann et al., 2014; Brioude et al., 2018). This molecular defect is accompanied by changes in histone modifications, such as the loss of H3K9me2 (Soejima and Higashimoto, 2013). LOM is responsible for the biallelic expression of the KCNQ1OT1 transcript, which reduces the CDNK1C expression provoking BWS phenotype. CDNK1C gene acquires biallelic methylation in some tumors and in 5% of BWS cases, this gene presents point mutations (Du et al., 2003). Another molecular alteration is UPD(11)p present in 10-20% of BWS patients (Eggermann et al., 2014; Brioude et al., 2018) (Figure 2C). UPD condition results when both chromosomes -or part of them- is/are inherited from one parent. In BWS, the UPD is paternal and involves the 11p15 region implying no maternal contribution for it. UPD is related to mitotic recombination during embryonic development. BWS patients with UPD(11)pat show mosaicism suggesting to be a post-zygotic event. The mosaicism level of UPD in different tissues is strongly associated with the pathological phenotype. The size of the UPD region or the level of mosaicism are not correlated with the severity of the disorder (Cooper et al., 2007). Patients with a mosaic UPD(11)pat for the entire chromosome 11 present clinical features comparable to UPD(11)pat cases restricted to a small part of 11p. BWS patients present UPD(11)pat isodisomic, thus determining increased IGF2 expression and reduced H19 expression (Wang et al., 2020). In some BWS patients, it is reported that β2SP gene, a TGF-β/Smad3/4 adaptor protein, a potent tumor suppressor is epigenetically silenced, thus observing a loss or markedly decreased expression of β2SP related to aberrant DNA methylation of CpG islands around its promoter region (Yao et al., 2010).
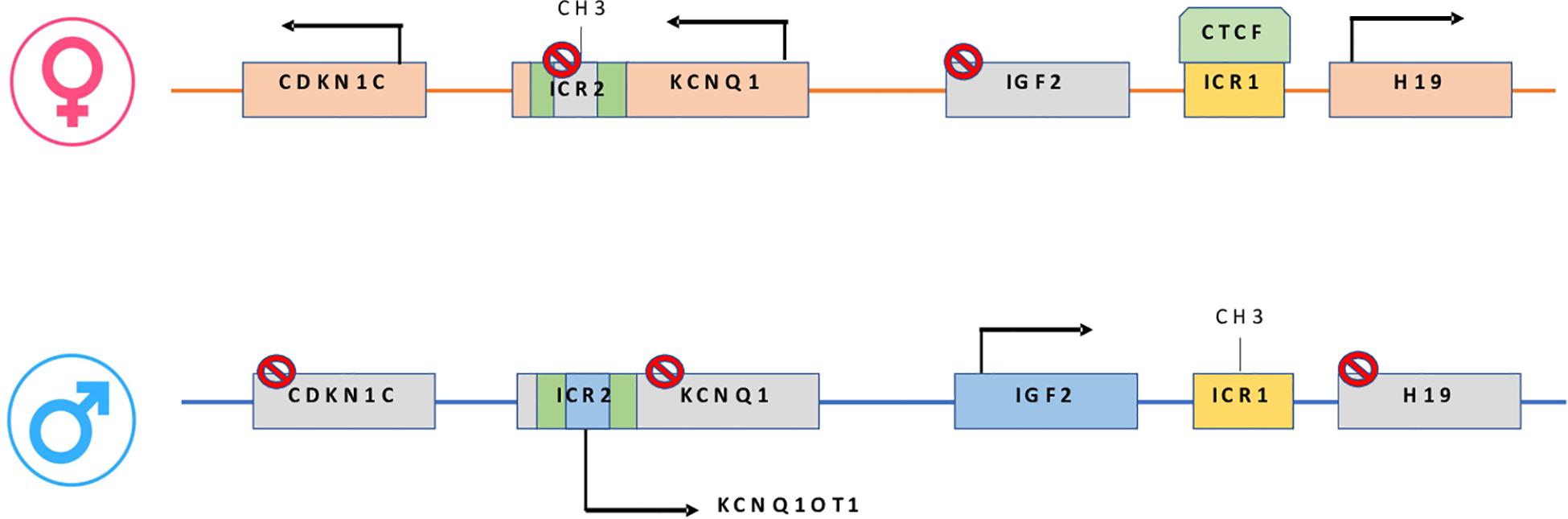
Figure 1. On the maternal allele: CTCF binding H19/IGF2:IG-DMR, prevents IGF2 from accessing the shared enhancers located on the H19 side. On the paternal allele: H19/IGF2:IG-DMR methylation prevents CTCF from binding, allowing IGF2 to engage the enhancers and silencing the H19 promoter. In maternal KCNQ1OT1:TSS-DMR, methylation induces KCNQ1-CDKN1C expression while paternally unmethylated state represses KCNQ1-CDKN1C expression and increase ncKCNQ1OT1 transcription.
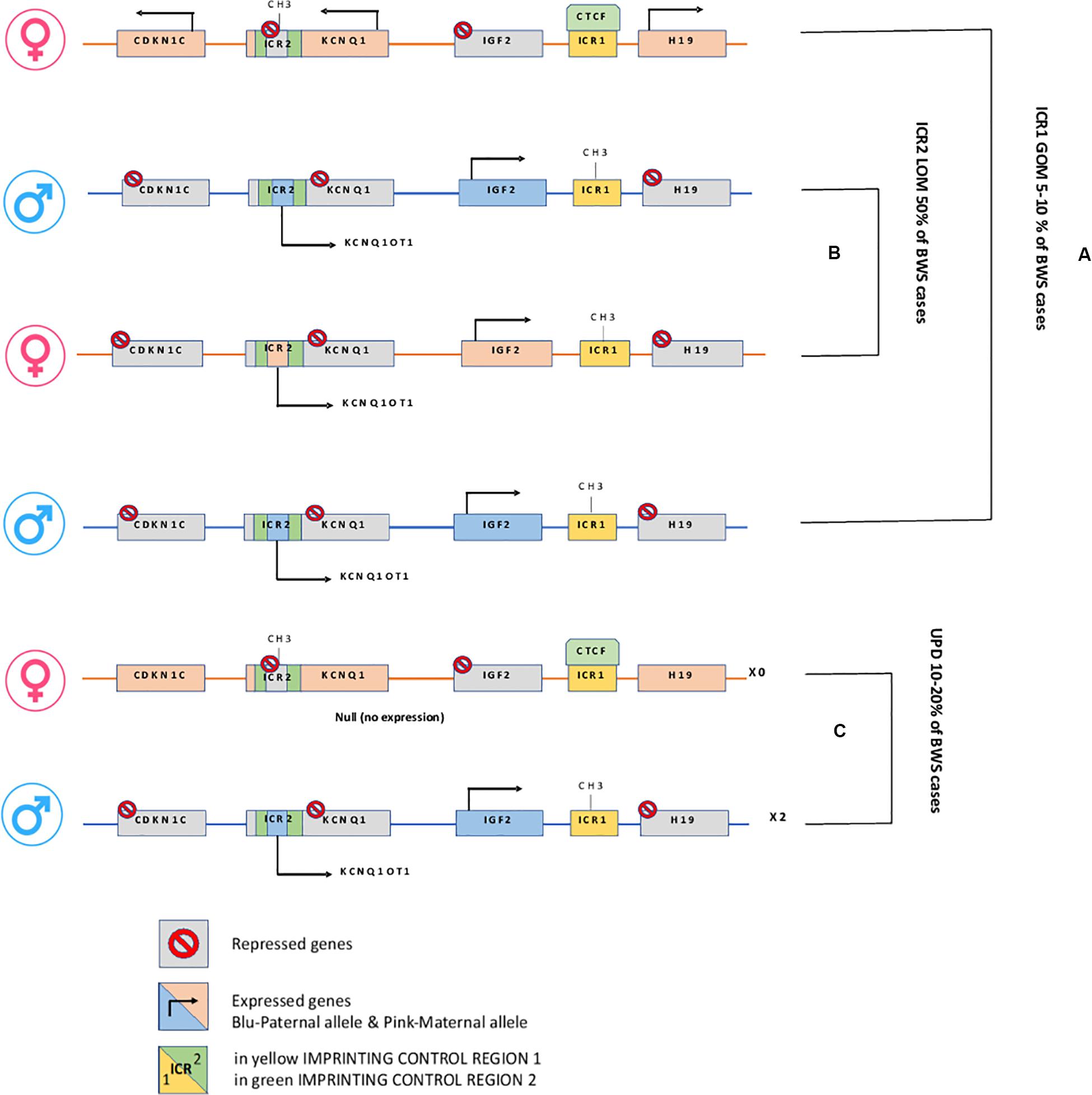
Figure 2. H19/IGF2:IG-DMR and KCNQ1OT1:TSS-DMR. (A) In Maternal allele H19/IGF2:IG-DMR GOM leads to overexpression of the growth factor IGF2. (B) In Maternal KCNQ1OT1:TSS-DMR LOM, the ncRNA KCNQ1OT1 is expressed while cis-linked genes are repressed. (C) Paternal uniparental disomy.
BWS as Multi-Locus Imprinting Disorder
Some BWS patients but also other imprinting disorder cases present additional methylation defects at different imprinted loci determining a pathological condition known as multi-locus imprinting disturbance (MLID) that potentially alters the expression of multiple imprinted gene clusters (Bens et al., 2016; Sanchez-Delgado et al., 2016). The MLID frequency is related to the type of imprinting disorder and appears to vary depending on the technology sensitivity used to analyze and on Imprinted Differentially Methylated Regions (IDMRs) investigated (Poole et al., 2013). Currently, BWS shows at frequencies around 50% in cases with LOM at KCNQ1OT1:TSS-DMR (Court et al., 2013), while MLID condition is less observed among patients with GOM at H19/IGF2:IG-DMR (Maeda et al., 2014). Some of MLID BWS patients present LOM or GOM at both maternal and paternal IDMRs (Court et al., 2013; Poole et al., 2013) while others have a hypomethylation syndrome restricted to maternally imprinted genes (Boonen et al., 2008; Baple et al., 2011). MLID condition usually presents BWS clinical features but in some cases in patients shows complex or atypical phenotypes, conceivably reflecting the loci and tissues mosaicism (Docherty et al., 2014; Begemann et al., 2018) and probably as a consequence of the dominance of one locus on other (Zhang et al., 1997; Azzi et al., 2009). In a cohort study, MLID was identified only in BWS patients with KCNQ1OT1:TSS-DMR LOM and hypomethylation was found only at maternally iDMRs, a condition described as multiple maternal hypomethylation syndrome (MMHS) (Bliek et al., 2009b; Sano et al., 2016; Fontana et al., 2018). A small number of manuscripts on MLID reported as GOM at paternally methylated iDMRs in BWS patients (Court et al., 2013; Maeda et al., 2014). The most frequently altered iDMRs found in BWS patients with MLID are PLAGL1, GRB10, MEST, GNAS, IGF2R, and ZNF331. From a pathological point of view, usually, MLID BWS patients have a decreased level of body weight compared to the one who characterized by a single molecular defect in the 11p15.5 region. Specifically, MLID patients show features not typical of BWS, such as speech retardation, apnea, and feeding difficulties (Bliek et al., 2009b). MLID-associated clinical signs may also only manifest as patients grow up; therefore MLID analysis after molecular confirmation of a specific ID, could guide a patient-tailored follow-up to track subclinical signs before their manifestation (Bakker et al., 2016). In 2005 for the first time there were reported two patients with MLID presented transient neonatal diabetes mellitus (TNDM) and hypomethylation of both iDMRs of KCNQ1OT1 gene (Arima et al., 2005) and of PLAGL1, an antiapoptotic (Shuman et al., 2006) gene located at chromosome 6q24 (Bliek et al., 2009b). A further broad analysis revealed that the MLID prevalence is higher in BWS cases than in other imprinting disorders (Brioude et al., 2018). However, despite the growing number of studies, the etiology of MLID is still unclear as well as mechanisms underlying the co-regulation of imprinting marks across the genome. However, MLID causative mutations have been identified in members of the NLRP and zinc-finger protein families in few BWS patients (Docherty et al., 2014). Their role in the imprinting process and the pattern of inheritance has yet to be fully elucidated. Molecular characterization of MLID is fundamental not only to define the clinical diagnosis of IDs better but also to evidence common functional networks at the basis of the imprinting genome-wide deregulation.
Genes Candidates in Overgrowth Feature in BWS
BWS is a childhood cancer predisposition disorder with increased risk of embryonic tumors, predominately Wilms tumor, and hepatoblastoma (Duffy et al., 2018). The chromosome 11p15.5 region contains imprinted genes that are fetal growth regulators (Brioude et al., 2018). The molecular analysis of domain 1 and 2 has identified two candidates related to tumor development in BWS: CDKN1C (p57Kip2) and IGF2. Their aberrations are associated with growth and development disturbances. IGF2 is a cell cycle regulator gene encoding for a growth factor with different functions in promoting cell growth and proliferation during fetal development (Park et al., 2017). IGF2 binds three receptors, IGF1 receptor (IGF1R), insulin receptor isoform A (IR-A), and the IGF1R–IR-A hybrid receptor, resulting in a cascade of intracellular events and promoting cell survival and mitogenesis (Alvino et al., 2011). IGF2 loss of imprinting from DMR dysregulation on the maternal allele increases its signals, thus promoting growth and anti-apoptosis processes (Gallagher and LeRoith, 2010). In addition, the dysregulation of IGF2 expression has been recently observed also in several tumor onsets such as for breast, ovarian, esophageal, and colorectal cancer, and its presence has been reported and associated with poor prognosis (Livingstone, 2013). IGF2 is involved in prenatal skeletal muscle growth and in muscle regeneration in adults. It has been studied using in vitro differentiation models and in vivo loss of function in mouse models (Park et al., 2017). In mice, the IGF2 overexpression determines BWS clinical features leading to overgrowth, polyhydramnios, fetal and neonatal lethality, disproportionate organ overgrowth, and macroglossia (Murrell et al., 2004). The CDKN1C encodes for a negative regulator of the cell cycle, and its function is to inhibit several Cyclin/CdK complexes. This gene maps in the centromeric region of 11p15, and it is paternally imprinted, thus implying its expression on the maternal allele. CDKN1C is considered a tumor suppressor gene. It blocks cell proliferation by inhibiting cell cycle progression, tissue invasion, metastasis, angiogenesis, and promotes apoptosis and cell differentiation (Kavanagh and Joseph, 2011). CDKN1C protein is composed of 316 amino acids assembled in three different domains: the N-terminal domain is fundamental for CdK inhibition, a central repeating sequence, and a C-terminal domain working as the PCNA binding domain homolog to p27Kip1 it interacts with PCNA protein (Stampone et al., 2018).
In BWS patients, the missense variants of CDNK1C involve highly conserved amino acids of the N-terminal domain with the CdK inhibitory role, impairing its function or its cellular localization. Until 2014 there were described 33 different point mutations in coding and non-coding regions of the gene (Eggermann et al., 2014). Most of them were missense mutations in frame-shift, leading to the production of a truncated form of the protein (Brioude et al., 2015). 5–10% of BWS patients show pathogenic variants of CDKN1C gene on the maternal allele, thus determining its loss of function (Eggermann et al., 2014; Brioude et al., 2018). Several CDKN1C gene point mutations have been identified, 40% with a BWS family history (Romanelli et al., 2010; Eggermann et al., 2014; Brioude et al., 2018). In the Human Gene Mutation Database, 65 different variants are reported in BWS patients exhibiting different clinical features including polydactyly, extra nipple, genital anomalies, and cleft palate, strongly dependent on epigenetic/chromosomal abnormalities at chromosome 11p15.5. BWS patients carrying CDKN1C mutations show a higher frequency of abdominal wall defects and omphalocele (Brioude et al., 2015), whereas mice lacking of a maternal copy of the CDKN1C gene present a phenotype close to BWS with gastrointestinal tract abnormalities or exomphalos (Yan et al., 1997; Zhang et al., 1997). CDKN1C mutations have also been correlated to several types of cancer, such as colorectal, lymph-hematologic, breast cancer (Larson et al., 2005), and it emphasizes the presence of commonalities features between BWS and cancer predisposition.
The Clinical Spectrum of BWS
Genetic and epigenetic changes frequently lead to different clinical phenotypes reported in the clinical criteria applied for the BWS definition. In Figure 3 are reported genetic and epigenetic abnormalities in association with BWS patients’ recurrence.
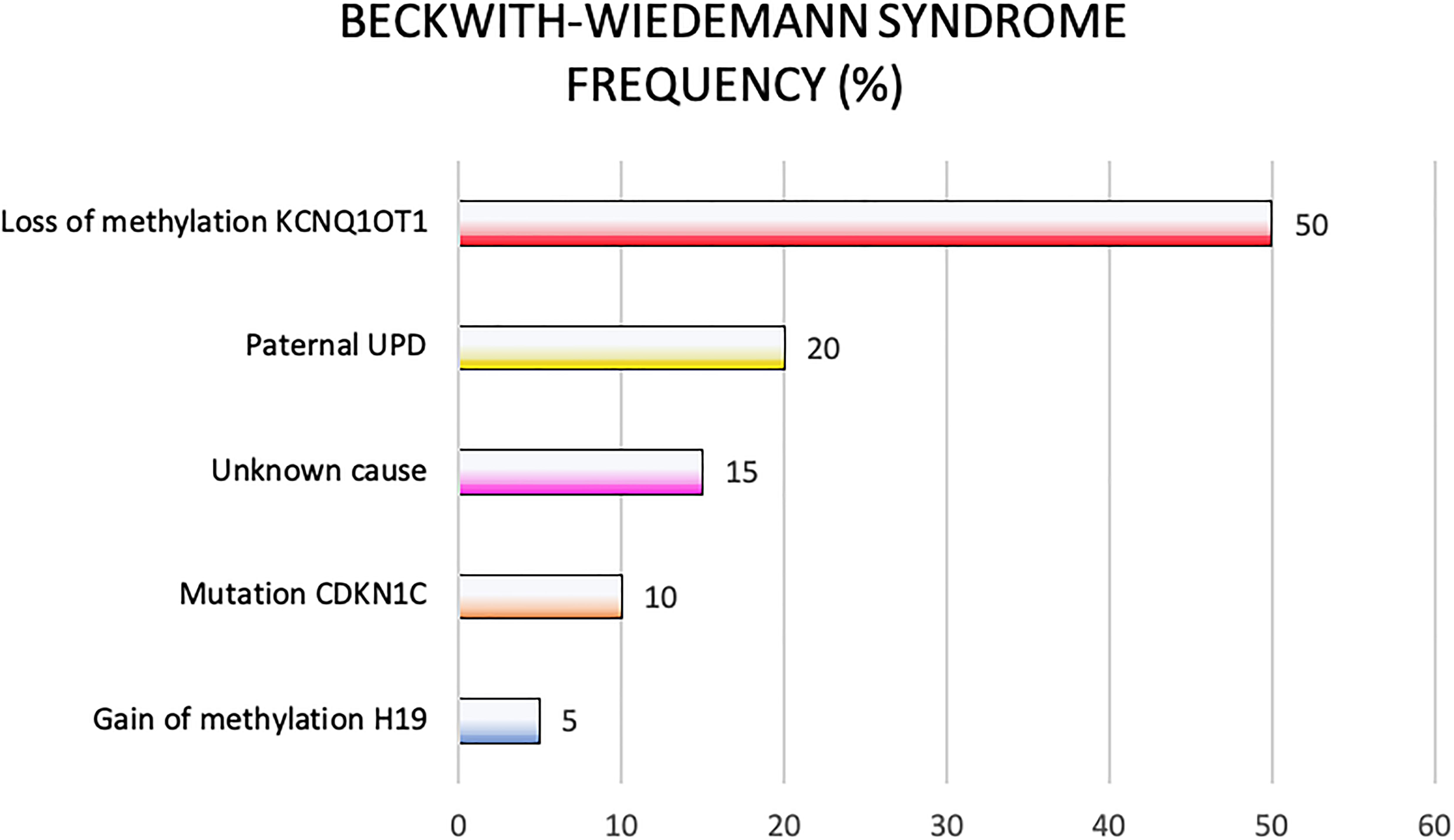
Figure 3. Beckwith-Wiedemann syndrome frequency of molecular defects.
An abnormal epigenetic variability may cause the onset of pathological mosaic states, one with a normal genotype (Zhang et al., 1997) and the other one carrying modified epigenetic information (Sazhenova and Lebedev, 2008). Thus in 2018, due to this high complexity, the Consensus Group introduced the concept of BWS spectrum (BWSp) including patients with a clinical diagnosis of BWS with or without an epigenetic alteration at the 11p15 locus, patients with “atypical BWS” (a condition defined by fewer cardinal and suggestive features than those needed for a BWS clinical diagnosis) and an epigenetic change at the BWS locus and patients with “isolated lateralized overgrowth.” The accurate identification of clinical aspects is crucial for the diagnosis: cardinal features include macroglossia, exomphalos, lateralized overgrowth, multifocal Wilms tumor, prolonged hyperinsulinism, and distinct pathological findings are unique to BWS. Suggestive features are less specific but may help the clinical diagnosis and the indication for molecular testing. These features are birth weight (>2SDS above the mean), facial nevus simplex, polyhydramnios and/or placentomegaly, ear creases and/or pits, transient hypoglycemia (lasting < 1 week), typical BWSp tumors (neuroblastoma, rhabdomyosarcoma, unilateral Wilms tumor, hepatoblastoma, adrenocortical carcinoma or pheochromocytoma), nephromegaly and/or hepatomegaly, umbilical hernia and/or diastasis recti (Brioude et al., 2018). The consensus criteria for clinical diagnosis apply a scoring system: each cardinal feature gets two points while each suggestive feature gets one point. For the clinical diagnosis, a score ≥ 4 is required, and the molecular confirmation of 11p15.5 anomalies needs to be applied. A genetic test is also required with a score ≥ 2 with further evaluation of a BWS expert (Brioude et al., 2018).
BWS Dedicated Molecular Investigations and Prenatal Test
BWS has genetic and epigenetic abnormalities, and it is phenotypically associated with different clinical aspects; therefore, the analysis of the molecular features of patients is essential for their management and treatment. First molecular testing procedure is usually applied evaluating mosaicism using DNA from blood leukocytes; then, the analysis may be implemented on additional samples such as buccal swabs, skin fibroblasts, or cells of mesenchymal origins. The procedure evaluates H19/IGF2:IG-DMR and KCNQ1OT1:TSS-DMR methylation levels as well as DMR copy number variations (CNVs) (Brioude et al., 2018). The most common diagnostic test for this purpose is Methylation-Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA) able to detect the percent of methylation and DMR copy number status simultaneously (Priolo et al., 2008; Scott et al., 2008; Wang et al., 2020). Low-level mosaicism patients require more sensitive methylation-specific techniques such as MS-PCR, MS quantitative PCR and in particular chromosomal microarray analysis (CMA) can determine the length of the UPD(11)pat region (Coffee et al., 2006; Azzi et al., 2011; Russo et al., 2016). Single gene sequencing of CDKN1C mutations can clarify the recurrence risk in family members (Brioude et al., 2018). Chromosome microarray might be required to define the size and the nature of duplication or deletion in the case of CNV (Baskin et al., 2014; Liu et al., 2015), whereas fluorescence in situ hybridization (FISH) or karyotyping may also provide the identification of chromosomal translocations (Bi et al., 2013). Prenatal molecular tests may allow handling the disease before the birth despite results reliability, and the ethical issues need to be taken into account (Brioude et al., 2018). This type of analysis requires samples such as chorionic villus (CVS) cells, amniotic fluid (AF) cells or fetal blood cells (native and cultured) (Brioude et al., 2018) but it is essential to consider that CVS cells could have different methylation patterns of the 11p15.5 region from embryonic tissues (Paganini et al., 2015) showing false-positive results due to tissue mosaicism (Brioude et al., 2018). For this reason, it is highly recommended that a multicenter audit of cases in order to implement different methods and get a correct diagnosis (Brioude et al., 2018). Different methods and diagnostic rates might be applied (Eggermann et al., 2016; Brioude et al., 2018). Quantitative PCR from amniocytes and cord blood leucocytes in 15 weeks pregnant women (see also NCT01842659) might result useful to evaluate the imprinting region 11p15.5, the Methylation Index (MI), the Interclass Correlation Coefficient (ICC) and the agreement between these two samples. Biochemical screening results such as elevated levels of free β-human chorionic gonadotropin (hCG) in the first trimester and/or increased α-fetoprotein (αFP) levels in the second trimester (associated with exomphalos) can be associated with BWS in the fetus (Gocmen et al., 2005; Kagan et al., 2015). BWS diagnosis can also result from an ultrasonographic detection of an anterior abdominal wall defect, macroglossia, or, less accurately, from macrosomia, visceromegaly, polyhydramnios, placentomegaly, or pancreatic overgrowth. BWS diagnosis can also be proposed by prenatal ultrasound scan (USS) identifying placental mesenchymal dysplasia, urinary tract abnormalities, cardiac defects, adrenal cysts and masses (Kagan et al., 2015; Brioude et al., 2018). Williams et al., 2005 reported diagnostic scheme based on the identification by ultrasound examination of different findings: at least two main (i.e., an abdominal wall defect, macroglossia, or macrosomia), or one main and two minor findings are required to lead BWS diagnosis (i.e., nephromegaly/dysgenesis, adrenal cytomegaly, aneuploidy/abnormal loci, or polyhydramnios) (Williams et al., 2005).
Predisposition in Tumor Development: Estimated Cancer Risk in BWS
Epigenetic aberrations effects in BWS phenotype may determine different tumor predisposition (Segers et al., 2012). Numerous pieces of evidence suggest that cancer is strongly related to the patient’s age (Kalish et al., 2016; MacFarland et al., 2018). There is a high risk during the first 4 years of life (11%) reduced to 3% in the 4–10 years-old patients (DeBaun and Tucker, 1998). Screening and diagnostic procedures should be applied for early diagnosis. The screening phase consists of repeated abdominal ultrasound (every 3–4 months during the first 8-10 years of life) and serum α-fetoprotein (aFP) measurement (every 3 months during the first 30 months of life) (Mussa et al., 2019). These procedures are applied to all patients regardless of the molecular diagnosis and the genotype (Zhang et al., 1997).
BWS molecular subtypes are related to tumor predisposition, and each subtype is associated with the development of one (or more) type of cancer (Mussa et al., 2016a). In Table 1 are reported the molecular subgroups and cancer types occurrence.
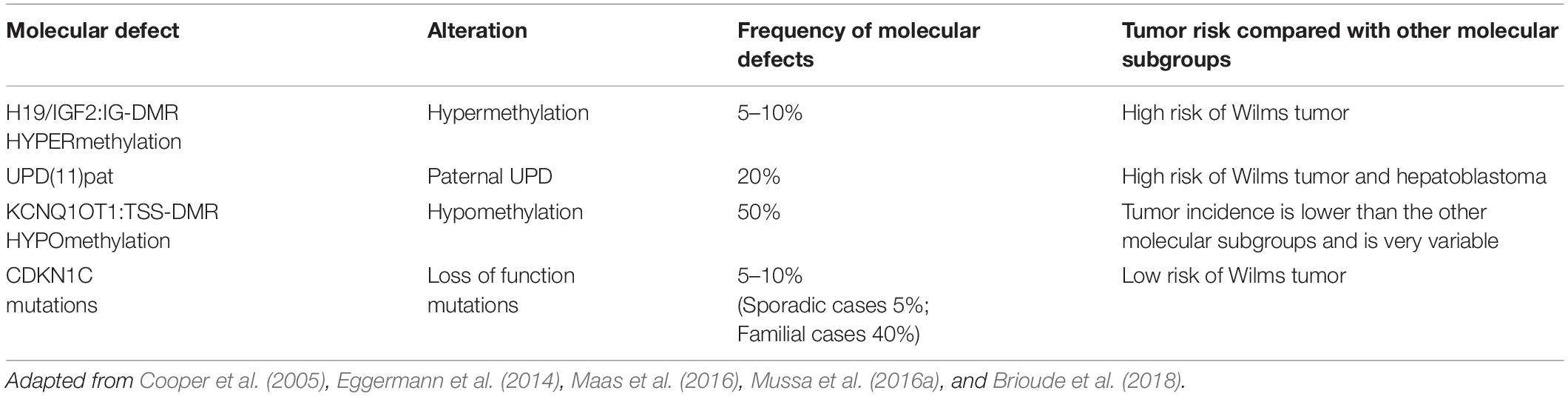
Table 1. Association of BWS molecular defects in subgroups and tumor risk frequency.
The four main molecular subtypes of BWS (KCNQ1OT1:TSS-DMR-LOM, H19/IGF2:IG-DMR -GOM, UPD, and CDKN1C mutations) are characterized by specific genotype-phenotype correlation to tumor development risk (Ibrahim et al., 2014; Mussa et al., 2016a). Patients with 11p15 telomeric domain defects (H19/IGF2:IG-DMR -GOM and UPD) have a higher risk of developing cancer compared to cases presenting centromeric aberrations (KCNQ1OT1:TSS-DMR-LOM mutation and CDKN1C) (Soejima and Higashimoto, 2013; Maas et al., 2016). The molecular subtype characterization of each BWS case increases those patients at the highest cancer risk focusing on a specific cancer. Subtypes analysis and embryonic tumor incidence of BWS cases may guarantee a more efficient surveillance optimizing tumor screening. As previously reported, a meta-analysis collecting results from seven studies, including 1370 genotyped BWS patients identified 102 cases with BWS-related malignancies (Zhang et al., 1997; Mussa et al., 2016a). It is interesting to note how different was the prevalence among the molecular subtypes: 2.5% (21/836) in KCNQ1OT1:TSS-DMR-LOM, 13.8% (47/341) in UPD, 22.8% (28/123) in H19/IGF2:IG-DMR -GOM and 8.6% (6/70) in patients with CDKN1C mutations. Wilms tumor represents the most common cancer in BWS patients (Rump et al., 2005) with a high prevalence in combination with telomeric defects in H19/IGF2:IG-DMR -GOM and UPD subgroups (21.1% vs. 6.2%, respectively, P < 0.001); subjects with centromeric defects display a lower rate. The tumor development in H19/IGF2:IG-DMR -GOM cases is significantly higher compared to UPD cases. Adrenal carcinoma has only been observed in UPD (1.5%, P < 0.001). Hepatoblastoma development has been associated with UPD (4.7%, P < 0.001) (Mussa et al., 2016a) although it is also observed in patients with KCNQ1OT1:TSS-DMR-LOM (0.7%) and H19/IGF2:IG-DMR -GOM (0.8%, P < 0.001). Neuroblastic tumors have been correlated with CDKN1C mutations (4.3%, P = 0.003) despite also observed in KCNQ1OT1:TSS-DMR-LOM (0.5%) and UPD cases (0.9%) with lower prevalence (Brioude et al., 2018). Additional studies are required to implement the available data since some associations seem to be clinically relevant, although not statistically significant (Mussa et al., 2016a). In general, this meta-analysis confirmed previous studies reporting the most common histotypes associated with BWS, such as Wilms tumor, hepatoblastoma, neuroblastic tumors, adrenal carcinoma, and rhabdomyosarcoma (Lapunzina, 2005; Shuman et al., 2006). The overall tumor risk of H19/IGF2:IG-DMR GOM is ∼23%, specifically with a 21% risk of developing Wilms tumor (Maas et al., 2016). The lowest tumor risk regards KCNQ1OT1:TSS-DMR hypomethylated subgroup, although there is a remarkable tumor variability. This subgroup shows different cancer types: hepatoblastoma, rhabdomyosarcoma, and gonadoblastoma but not in Wilms tumor. Patients with H19/IGF2:IG-DMR hypermethylated present Wilms tumor and hepatoblastoma their recurrence is related to IGF2 overexpression during cancer development (Akmal et al., 1995; Rump et al., 2005; Maas et al., 2016; Mussa et al., 2017; Brioude et al., 2019). Several studies observing different cohorts of BWS patients confirmed higher tumor risk associated with the H19/IGF2:IG-DMR hypermethylated and UPD(11)pat subgroup and high frequency for Wilms tumor and hepatoblastoma (Maas et al., 2016; Ounap, 2016; Brioude et al., 2018; Kamien et al., 2018; MacFarland et al., 2018; Wang et al., 2020). Wilms tumor rate is more frequent in the H19/IGF2:IG-DMR subgroup than in the cases observed for the UPD (Wang et al., 2020). The UPD subgroup is associated with a high prevalence of hemihyperplasia and hepatoblastoma (Maas et al., 2016; Mussa et al., 2017). Only four cases of BWS children belonging to KCNQ1OT1:TSS-DMR hypomethylated subgroup had Wilms tumor or nephrogenic remnants. Adrenocortical tumors have a percentage of 3% in BWS cases, and few of them are associated with the LOM KCNQ1OT1 gene (Alsultan et al., 2008; Mama et al., 2014). Two cases of KCNQ1OT1 LOM with adrenocortical tumors also recently observed (Wijnen et al., 2012), and an additional one recently reported, although neither of these patients presented typical phenotypic features of BWS (Wijnen et al., 2012; Eltan et al., 2020). Guidelines are discordant because the European consensus does not recommend this screening (Brioude et al., 2018) that is required in the USA consensus (Kalish et al., 2017).
Metabolic Imbalance
Metabolic disorders are one of the major clinical conditions in BWS (Schiff et al., 1973). Among metabolic imbalances, hyperinsulinemia/hypoglycemia are pathological states distressing 50% of BWS patients (Martinez y Martinez et al., 1992). Congenital hyperinsulinism (HI) is associated with a dysregulation of insulin secretion from pancreatic β-cells, and the molecular etiology of HI is due to mutations in ABCC8 and KCNJ11 genes located at the 11p15 region encoding for two subunits of the pancreatic β-cell ATP-sensitive potassium channel (KATP channel), SUR1 and Kir6.2 (Tung et al., 2020). Although only a few cases of BWS show HI, 50% of BWS neonates present transient HI while 5% have persistent HI requiring medical and/or surgical management (Senniappan et al., 2015; Kalish et al., 2016). Hypoglycemia can start in the neonatal period during the first days of life (Sweet et al., 2013). Glycemic disorders are related to aberrations of tumor suppressor genes (IGF2, H19, and p57KIP2) located in the 11p15 region and associated with BWS (Lee et al., 1999). Anomalies in the type 1 sulfonylurea receptor (SUR1) gene on chromosome 11p15 have been reported (de Lonlay-Debeney et al., 1999; Glaser et al., 2000). IGF2 is overexpressed in 20% of BWS individuals (Weksberg et al., 1993), and its loss of imprinting is responsible for hypoglycemia (Lee et al., 1999). Loss of imprinting and UPD on the paternal allele in the 11p15 region (Slavotinek et al., 1997) leads to IGF2 gene overexpression. IGF2 protein binding to the insulin receptor sustains the hyperinsulinemia condition. It is therefore not surprising that hyperinsulinemic hypoglycemia is often associated with BWS diagnosis. Hypoglycemia BWS cases reported hyperinsulinism and inappropriate insulin secretion (Shepherd et al., 2000). Pancreatic β cell dysregulation has been linked to the cause of hyperinsulinism in BWS (Stanley, 1997). In many histological analyses, hypertrophy and hyperplasia (Lteif and Schwenk, 1999) were observed, strengthening the correlation between incorrect pancreatic activity and BWS (Laje et al., 2013). However, the progression toward type 1 diabetes has not been documented (Leibowitz et al., 1995). The high levels of insulin in the blood cause a prolonged lowering of glucose concentrations, although the mechanism of insulin release in the different secretagogues occurs in different ways (Munns and Batch, 2001). In most BWS patients, life expectancy is good (Weng et al., 1995), and the metabolic imbalance tends to improve within time. However, in some cases, prolonged drug treatment is required to control hypoglycemia (Shilyansky et al., 1997). To obtain better results for glycemic control, it depends upon timeliness (Aynsley-Green et al., 2000). Unfortunately, in 20% of BWS cases, hypoglycemia is difficult to control and may cause severe decompensations leading to neurological alterations with severe repercussions in cognitive function development as intellectual impairment (Cresto et al., 1998). In severe cases, partial pancreatectomy may be required (Martinez y Martinez et al., 1992; Laje et al., 2013).
Monozygotic Twins Discordance
In twins, it has been observed a discordant monozygotic phenomenon (Weksberg et al., 2002), whereas BWS afflicts one subject, although the other twin may have some characteristics of the syndrome. Theoretically, monozygotic twins (MZ) resulting from a single zygote should have identical genomes. However, several examples of genetic differences have been reported among MZ, thus suggesting that somatic changes may occur after conception (Bliek et al., 2009a). The mosaicism is leading to a discordance between MZ in BWS, and it is linked to an epigenetic event triggered by the twinning process. Cells involved in this event spread among embryos in a multiple pregnancy creating a mosaic distribution being responsible for the variable phenotypic spectrum observed in BWS cases (Cohen et al., 2019). The timing of epigenetic aberrations influences the twinning, the degree of severity of BWS, and the degree of mosaicism. The theory of “diffuse mosaicism” proposed by Cohen et al. (2019) outlines the time points when the epigenetic event occurs in relation to twinning and the determination of chorionicity. In singleton gestations, the epigenetic aberration has been reported to occur first in non-mosaic patients during embryogenesis and subsequently in mosaic patients. In dichorionic gestations, the zygosity determines the timing of the event. It occurs earlier in the dizygotic dichorionic pregnancies than in the monozygotic dichorionic pregnancies (Cohen et al., 2019). Previous research has shown that an epigenetic event before twinning leads to the formation of two different clonal cell populations (Bell and Spector, 2011). These different cell clones repel each other and trigger the twinning event leading to the formation of separate cell masses (Hall, 1996; Machin, 1996; Weksberg et al., 2002). Therefore it is possible that a cell group carrying an imprinting alteration of KCNQ1OT1 (LOM) could preferably increase its growth rate compared to normal cells, thus generating asymmetry of the entire cell mass and increasing the possibility of cell clones separation genotypically distinct (Weksberg et al., 2002). Most BWS MZ exhibit KCNQ1OT1 imprinting defects (in KCNQ1OT1:TSS-DMR), indicating that monozygotic twinning is mechanically linked to the imprinting error or, conversely, that epigenetic alterations in KvDMR1 (LOM) can increase the possibility of monozygotic twinning. KCNQ1OT1:TSS-DMR hypomethylation is related to a failure in methylation, and it coincides or occurs shortly after the twinning event (Bliek et al., 2009a; Castillo-Fernandez et al., 2014).
Differences in genotype and phenotype can be attributed to various causes, including non-random inactivation X (Machin, 1996). These pieces of evidence agree with the higher incidence observed in monozygotic discordant female twins, suggesting that monozygotic twinning, genomic imprinting, and X inactivation may be mechanically and temporally related events (Lubinsky and Hall, 1991; Weksberg et al., 2002). In most reports, there is a high prevalence of MZ female and very few cases of discordant male twins (Weksberg et al., 2002; Smith et al., 2006; Bliek et al., 2009a; Tierling et al., 2011). In 250 BWS patients, 20 sets of monozygotic and 2 sets of dizygotic twins with high prevalence (16 out of 20) for female MZ were identified (Weksberg et al., 2002). Later in 2009, another study showed a high MZ twinning rate of 2.5% as compared with 0.3–0.4% among normal twins, while dizygotic (DZ) cases reported 0.75% as rate with 0.7–1.1% of prevalence. A female excess among BWS multiple births was observed in this study. In 10 MZ, 9 were females, and all 3 cases of DZ were females (Bliek et al., 2009a). More recently, in 2019, the high incidence of monozygotic female in a cohort of 26 BWS twins was confirmed (Cohen et al., 2019). The significant female preponderance of the MZ discordant for BWS could be associated with a variety of sex-related factors. For example, the developmental error can occur equally in male and female embryos, demonstrating a lethal effect on male MZ or in alternative the delay of early development in female embryos compared to men (Hall, 1996). It is subordinated to the X inactivation process, and it can increase the susceptibility in female MZ embryos to certain errors. The double discrepancy is due to the failure of Dnmt1o (DNMT1oocyte) to maintain methylation in phase S of a cell cycle occurring before or during the twinning event (Bestor, 2003). It has been hypothesized that the excess of twins in BWS patients is secondary to X inactivation with delayed embryogenesis allowing the acquisition of errors such as failure of maintenance methylation (Lubinsky and Hall, 1991; Orstavik et al., 1995; Hall, 1996; Hall and Lopez-Rangel, 1996; Weksberg et al., 2005). This evidence supports the high incidence of female BWS MZ. In two more extensive studies (Gaston et al., 2001; Weksberg et al., 2002) KCNQ1OT1:TSS-DMR hypomethylation from DNA of blood samples has been reported in affected and unaffected twins of discordant couples. It has been proposed that the aberrant methylation in the blood of the healthy twin is caused by vascular connections in the placenta shared by both MZ (monochorionic, diamniotic). This failure occurs in the eight-cell blastocyst stage preceding the moment when MZ (monochorionic, diamniotic) twinning is established. However, not all methylation defects involve twinning, and most BWS patients are singletons (Cohen et al., 2019). An explanation of these assumptions is related to the theory of endangered twins in which twinning occurs in all cases, but the second fetus is reabsorbed in early pregnancy (Landy and Keith, 1998). The twin discordance of BWS patients is observed not only in females but also in males (Smith et al., 2006). A pair of male MZ discordant for BWS was reported, and among these, the affected twin had paternal UPD for chromosome 11p15. The second male twin pair was concordant, and both demonstrated H19/IGF2:IG-DMR hypermethylation, thus suggesting BWS-related molecular heterogeneity in male MZ (Smith et al., 2006).
Assisted Reproduction Techniques (ART) and BWS Epigenome
The association between ART and syndromes related to epigenetic defects has been reported in several cases such as BWS (Dhont et al., 1999; Ferraretti et al., 2012, 2013; Kupka et al., 2014), large offspring syndrome in ruminants (LOF) (de Mouzon et al., 2012) and Angelman syndrome (MIM 105830) (Talaulikar and Arulkumaran, 2012) (MRC Working Party on Children Conceived by In Vitro Fertilization, 1990). In 1995 a BWS patient conceived through ART (Sutcliffe et al., 1995) was reported, Young et al. (1998) described the LOF, etiologically correlated with in vitro fertilization (Kupka et al., 2014). The LOF shows a significant effect on the phenotype connected with ART (Li et al., 2019), and this syndrome is a model for BWS (Chen et al., 2013) presenting similar phenotypic abnormalities (Young et al., 2001). In LOF affected bovines was reported an association between multiple loci imprinted defects and ART. This evidence underlines how ART may cause imprinting disturbances (Mussa et al., 2017). Most of the patients suffering from imprinting disturbances conceived via in vitro fertilization (Kupka et al., 2014) and intracytoplasmic sperm injection (ICSI) showed aberrant imprinted DNA methylation (Hattori et al., 2019). In the paper of DeBaun et al. (2003) were identified seven sporadic cases conceived by ART and epigenetic alterations were present in six of them generally associated with BWS. Several studies have further explored this association (Maher and Reik, 2000; Gicquel et al., 2003; Halliday et al., 2004; Chang et al., 2005; Rossignol et al., 2006; Sutcliffe et al., 2006; Doornbos et al., 2007; Lim et al., 2009; Hiura et al., 2012; Tee et al., 2013). The most recent literature also has corroborated the hypothesis that BWS is related to ART (Vermeiden and Bernardus, 2013; Hattori et al., 2019). Indeed, LOM of KCNQ1OT1:TSS-DMR represents the molecular defect found in BWS patients conceived through ART (Gomes et al., 2009; Mussa et al., 2017). In 2015, the clinical study NCT00773825 on the association between BWS and ART was completed, although the results are not yet reported. The authors investigated the methylation status at nine different loci and other epigenetic marks using Southern blot and methyl-specific quantitative PCR in three groups of patients: children naturally conceived, children conceived after ovarian stimulation, but in vivo fertilization, and a group of children conceived after ovarian stimulation and in vitro fertilization. The aim of this trial was to determine if children born following ART exhibit an increased risk of imprinting defects. Moreover, previous results showed that ART might favor imprinting alterations at the centromeric imprinted 11p15 locus and, consequently, the incidence of BWS. Some of BWS patients reported DNA methylation defects abnormal methylation at loci other than the 11p15 region. This condition was present in both BWS patients naturally, and ART conceived (Gicquel et al., 2008), suggesting that ART procedure could be not specifically involved in the loss of methylation at various imprinting loci (Rossignol et al., 2006).
Treatment Approaches and Perspectives for Epi-Based Therapies
Currently, there is no dedicated therapy for BWS, and all available treatments are mainly addressing clinical features for ameliorating the quality of life. The current on-going clinical trial NCT01916148 aims to determine the ability F-DOPA PET, a PET radiotracer, to detect focal lesions prior to surgical intervention in BWS patients with hyperinsulinemic hypoglycemia. This study is useful to guarantee an early diagnosis of the pathology and to manage available treatments for BWS patients. Macroglossia occurs in 90% of BWS patients and may regress spontaneously in some children, but 40% of them undergo surgery to reduce tongue size (Brioude et al., 2018). The regional overgrowth in BWS can be progressive or non-progressive (Burkardt et al., 2019). It occurs in 43-60% of patients, and the management is related to the affected limbs (Wang et al., 2020). Leg length discrepancy (LLD) can influence negative life quality and may require shoe lifts or, in some cases, surgical correction (Ghanem et al., 2011; Brioude et al., 2018). The asymmetric overgrowth of the upper limbs generally does not require surgery (Brioude et al., 2018). New strategies are necessary to improve the possible and available treatment for BWS patients (Swinney and Xia, 2014). Many studies have demonstrated that by reprogramming the epigenetic landscape, it is possible to modulate the defects present in the genome leading to the treatment of different diseases (Miranda Furtado et al., 2019). Epigenetic markers can be targeted by activators or inhibitors of epigenetic-modifying proteins (Lauschke et al., 2018), the so-called “epidrugs” currently used mainly in tumor treatments, such as hematological malignancies (Woods et al., 2015; Mazzone et al., 2017). The effectiveness of these treatments is slowly widening toward new fields of application. Epidrugs are demonstrating their potential in other pathologies, such as infectious diseases, metabolic and cardiovascular disorders (Das et al., 2009; Dunn and Rao, 2017). There are promising clinical advances in epigenetics toward new drug discovery (Crea et al., 2011; Glasgow et al., 2015; Lundstrom, 2017) and biomarkers (Shao et al., 2018) in order to limit epigenetic mutation effects (Vitiello et al., 2015; Inamura, 2017). Epigenetic (i.e., hypermethylation of the tumor suppressor gene promoters) and genetic mutations of epigenetic enzymes (loss or gain of function) can be used as predictors of therapy response in different types of diseases (Welch and Clegg, 2010). Prolonged re-expression of epigenetically silenced genes has been demonstrated for various genes, including tumor suppressor genes (Gaur et al., 2015). BWS might represent one of the many new challenges for epigenetic treatment-based applications. For example, it is possible to mitigate CpG island methylation on maternal and/or paternal allele to restore the normal transcriptional activity in the imprinting control regions (Bartolomei and Ferguson-Smith, 2011). Epigenetic treatments might become a valid opportunity for aberrations in 11p15.5 imprinted region (Smith et al., 2012). The Food and Drug Administration (FDA) in the United States has approved several DNA methylation inhibitors, including cytidine analogs 5-azacitidine and zebularine and nucleoside analogs. Histone deacetylase inhibitors (Cheng et al., 2019) such as suberoylanilide hydroxamic acid (SAHA, trade name Vorinostat), romidepsin (trade name Istodax), Valproic acid (VPA) and trichostatin A (TSA) (Kelly et al., 2010; Heerboth et al., 2014) disrupt deacetylation process. New therapeutic programs and technique advancements might be applied to reprogram the epigenetic circuit and to counteract chronic symptoms. Since the main BWS targets are determined by deregulation on epigenetic processes, it should be interesting to evaluate potential targets by using drugs against the activity of DNA methyltransferases and or histone deacetylases and histone acetylation. These drugs should be potential treatments against BWS. Ideally, the first application may be a combination therapy to control disease progression. For example, surgical techniques for phenotypic abnormalities control (Wang et al., 2020) might be supported by the use of epi-based treatments for the metabolic imbalances in young BWS patients. The epigenetic sensitization to radiotherapy might provide promising results in BWS affected by Wilms tumor. Moreover, given the potential therapeutic role of epigenetic modulating agents in metabolic disorders (Crispo et al., 2019), it is tempting to hypothesize that in the near future, by targeting epi-modifiers and remodelers might prove beneficial also in BWS. Clearly, given the genome and epigenome heterogeneity and complexity of this disease, patient’s stratification may represent a ‘conditio sine qua non’ for future epi-based applications.
Conclusion
The progress in epigenetic drugs discovery (Morera et al., 2016; Esteller, 2017; Velasco and Francastel, 2019), the involvement of epigenetic mutations in a wide range of diseases (Dirks et al., 2016; Graca et al., 2016; Jones et al., 2016; Berdasco and Esteller, 2019) and the newly chromatin-based identified disease biomarkers (Dirks et al., 2016) have suggested epi-based approaches as a promising tool for clinical applications. Evidently, potential new therapeutic options require better clinical knowledge. Indeed, the target identification and characterization are at the basis for a correct therapy. In the case of BWS there are some levels of complexity to be decrypted. Being a rare disease, the generally low number of patients is a bottleneck and a hindrance to the development of dedicated therapeutic approaches nor the causal identification of “druggable” targets proven beneficial for the restoration of the health status or, at least, for symptoms defeat. The lesson learned from BWS homozygotic twins not only suggests the existence of a phenotypic link between epigenome deregulation and the complexity of BWS disease, but it also indicates the potential of targeting the epigenome pharmacologically in BWS to at least obtain beneficial effects ameliorating the quality of life. Indeed, on one side, the different disease phenotypes in homozygotic BWS twins have consolidated the idea that the disease penetrance is epigenome-regulated, despite somatic changes may occur after conception. On the other side, this proves in humans the potential of a therapeutic approach targeting BWS epigenome. Recently we have just started to understand the pivotal role of epigenome deregulation in BWS (and other rare diseases) supporting the development of diagnostic, prognostic, and therapeutic approaches also based on this notion. In this perspective, therapeutic approaches might also be applied to epimutations related to tumor predisposition in BWS, which might gain benefit from the use of treatment schemes, including or based on chromatin acting drugs.
Author Contributions
CP, UC, RB, and LA: conceptualization. LA and RB: funding acquisition. CP, UC, MN, and RB: writing – original draft preparation. RB, LA, and MN: writing – review and editing. MN: writing and editing. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by “Epigenetic Hallmarks of Multiple Sclerosis” (acronym Epi-MS) (id:415, Merit Ranking Area ERC LS) in VALERE 2019 Program; V:ALERE 2020 – Progetto competitivo “CIRCE” in risposta al bando D.R. n. 138 del 17/02/2020 Program; Blueprint 282510; MIUR20152TE5PK; EPICHEMBIO CM1406; EPIGEN-MIUR-CNR; AIRC-17217; VALERE: Vanvitelli per la Ricerca; Campania Regional Government Technology Platform Lotta alle Patologie Oncologiche: iCURE; Campania Regional Government FASE2: IDEAL. MIUR, Proof of Concept POC01_00043. POR Campania FSE 2014-2020 ASSE III.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ART, assisted reproductive technologies; BWS, Beckwith-Wiedemann syndrome; BWSp, Beckwith-Wiedemann syndrome spectrum; CNVs, copy number variations; CTCF, CCCTC-binding factor; DMR, differentially methylated region; DZ, dizygotic twins; GOM, gain of methylation; IG-DMR, intergenic differentially methylated region; ICR1, imprinting control region 1; ICR2, imprinting control region 2; LLD, leg length discrepancy; LOF, large offspring syndrome; LOM, loss of methylation; MS-MLPA, methylation-specific multiplex ligation-dependent probe amplification; MZ, monozygotic twins; ncRNA, non-coding RNA; TSS-DMR, transcription start site differentially methylated region; UPD, uniparental disomy.
References
Abi Khalil, C. (2014). The emerging role of epigenetics in cardiovascular disease. Ther. Adv. Chronic. Dis. 5, 178–187. doi: 10.1177/2040622314529325
Abramowitz, L. K., and Bartolomei, M. S. (2012). Genomic imprinting: recognition and marking of imprinted loci. Curr. Opin. Genet. Dev. 22, 72–78. doi: 10.1016/j.gde.2011.12.001
Alsultan, A., Lovell, M. A., Hayes, K. L., Allshouse, M. J., and Garrington, T. P. (2008). Simultaneous occurrence of right adrenocortical tumor and left adrenal neuroblastoma in an infant with Beckwith-Wiedemann syndrome. Pediatr. Blood Cancer 51, 695–698. doi: 10.1002/pbc.21694
Akmal, S. N., Yun, K., MacLay, J., Higami, Y., and Ikeda, T. (1995). Insulin-like growth factor 2 and insulin-like growth factor binding protein 2 expression in hepatoblastoma. Hum. Pathol. 26, 846–851. doi: 10.1016/0046-8177(95)90005-5
Alvino, C. L., Ong, S. C., McNeil, K. A., Delaine, C., Booker, G. W., Wallace, J. C., et al. (2011). Understanding the mechanism of insulin and insulin-like growth factor (IGF) receptor activation by IGF-II. PLoS One 6:e27488. doi: 10.1371/journal.pone.0027488
Arima, T., Kamikihara, T., Hayashida, T., Kato, K., Inoue, T., Shirayoshi, Y., et al. (2005). ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 33, 2650–2660. doi: 10.1093/nar/gki555
Ayme, S., and Schmidtke, J. (2007). Networking for rare diseases: a necessity for Europe. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 50, 1477–1483. doi: 10.1007/s00103-007-0381-9
Aynsley-Green, A., Hussain, K., Hall, J., Saudubray, J. M., Nihoul-Fekete, C., De Lonlay-Debeney, P., et al. (2000). Practical management of hyperinsulinism in infancy. Arch. Dis. Child Fetal Neonatal Ed. 82, F98–F107. doi: 10.1136/fn.82.2.f98
Azzi, S., Rossignol, S., Steunou, V., Sas, T., Thibaud, N., Danton, F., et al. (2009). Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum. Mol. Genet. 18, 4724–4733. doi: 10.1093/hmg/ddp435
Azzi, S., Steunou, V., Rousseau, A., Rossignol, S., Thibaud, N., Danton, F., et al. (2011). Allele-specific methylated multiplex real-time quantitative PCR (ASMM RTQ-PCR), a powerful method for diagnosing loss of imprinting of the 11p15 region in Russell Silver and Beckwith Wiedemann syndromes. Hum. Mutat. 32, 249–258. doi: 10.1002/humu.21403
Bakker, J. P., Wang, R., Weng, J., Aloia, M. S., Toth, C., Morrical, M. G., et al. (2016). Motivational Enhancement for Increasing Adherence to CPAP: a randomized controlled trial. Chest 150, 337–345. doi: 10.1016/j.chest.2016.03.019
Baple, E. L., Poole, R. L., Mansour, S., Willoughby, C., Temple, I. K., Docherty, L. E., et al. (2011). An atypical case of hypomethylation at multiple imprinted loci. Eur. J. Hum. Genet. 19, 360–362. doi: 10.1038/ejhg.2010.218
Bartolomei, M. S., and Ferguson-Smith, A. C. (2011). Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 3:a002592. doi: 10.1101/cshperspect.a002592
Baskin, B., Choufani, S., Chen, Y. A., Shuman, C., Parkinson, N., Lemyre, E., et al. (2014). High frequency of copy number variations (CNVs) in the chromosome 11p15 region in patients with Beckwith-Wiedemann syndrome. Hum. Genet. 133, 321–330. doi: 10.1007/s00439-013-1379-z
Begemann, M., Rezwan, F. I., Beygo, J., Docherty, L. E., Kolarova, J., Schroeder, C., et al. (2018). Maternal variants in NLRP and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. J. Med. Genet. 55, 497–504. doi: 10.1136/jmedgenet-2017-105190
Begemann, M., Spengler, S., Gogiel, M., Grasshoff, U., Bonin, M., Betz, R. C., et al. (2012). Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J. Med. Genet. 49, 547–553. doi: 10.1136/jmedgenet-2012-100967
Bell, J. T., and Spector, T. D. (2011). A twin approach to unraveling epigenetics. Trends Genet. 27, 116–125. doi: 10.1016/j.tig.2010.12.005
Bens, S., Kolarova, J., Beygo, J., Buiting, K., Caliebe, A., Eggermann, T., et al. (2016). Phenotypic spectrum and extent of DNA methylation defects associated with multilocus imprinting disturbances. Epigenomics 8, 801–816. doi: 10.2217/epi-2016-0007
Berdasco, M., and Esteller, M. (2019). Clinical epigenetics: seizing opportunities for translation. Nat. Rev. Genet. 20, 109–127. doi: 10.1038/s41576-018-0074-2
Bestor, T. H. (2003). Imprinting errors and developmental asymmetry. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 1411–1415. doi: 10.1098/rstb.2003.1323
Bi, W., Borgan, C., Pursley, A. N., Hixson, P., Shaw, C. A., Bacino, C. A., et al. (2013). Comparison of chromosome analysis and chromosomal microarray analysis: What is the value of chromosome analysis in today’s genomic array era? Genet. Med. 15, 450–457. doi: 10.1038/gim.2012.152
Bliek, J., Alders, M., Maas, S. M., Oostra, R. J., Mackay, D. M., van der Lip, K., et al. (2009a). Lessons from BWS twins: complex maternal and paternal hypomethylation and a common source of haematopoietic stem cells. Eur. J. Hum. Genet. 17, 1625–1634. doi: 10.1038/ejhg.2009.77
Bliek, J., Verde, G., Callaway, J., Maas, S. M., De Crescenzo, A., Sparago, A., et al. (2009b). Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 17, 611–619. doi: 10.1038/ejhg.2008.233
Boonen, S. E., Porksen, S., Mackay, D. J., Oestergaard, E., Olsen, B., Brondum-Nielsen, K., et al. (2008). Clinical characterisation of the multiple maternal hypomethylation syndrome in siblings. Eur. J. Hum. Genet. 16, 453–461. doi: 10.1038/sj.ejhg.5201993
Brioude, F., Kalish, J. M., Mussa, A., Foster, A. C., Bliek, J., Ferrero, G. B., et al. (2018). Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat. Rev. Endocrinol. 14, 229–249. doi: 10.1038/nrendo.2017.166
Brioude, F., Netchine, I., Praz, F., Le Jule, M., Calmel, C., Lacombe, D., et al. (2015). Mutations of the Imprinted CDKN1C Gene as a Cause of the Overgrowth Beckwith-Wiedemann Syndrome: clinical Spectrum and Functional Characterization. Hum. Mutat. 36, 894–902. doi: 10.1002/humu.22824
Brioude, F., Toutain, A., Giabicani, E., Cottereau, E., Cormier-Daire, V., and Netchine, I. (2019). Overgrowth syndromes - clinical and molecular aspects and tumour risk. Nat. Rev. Endocrinol. 15, 299–311. doi: 10.1038/s41574-019-0180-z
Burkardt, D. D., Tatton-Brown, K., Dobyns, W., and Graham, J. M. Jr. (2019). Approach to overgrowth syndromes in the genome era. Am J. Med. Genet. C Semin. Med. Genet. 181, 483–490. doi: 10.1002/ajmg.c.31757
Cai, X., and Cullen, B. R. (2007). The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 13, 313–316. doi: 10.1261/rna.351707
Castillo-Fernandez, J. E., Spector, T. D., and Bell, J. T. (2014). Epigenetics of discordant monozygotic twins: implications for disease. Genome Med. 6:60. doi: 10.1186/s13073-014-0060-z
Chang, A. S., Moley, K. H., Wangler, M., Feinberg, A. P., and Debaun, M. R. (2005). Association between Beckwith-Wiedemann syndrome and assisted reproductive technology: a case series of 19 patients. Fertil. Steril. 83, 349–354. doi: 10.1016/j.fertnstert.2004.07.964
Chen, Z., Robbins, K. M., Wells, K. D., and Rivera, R. M. (2013). Large offspring syndrome: a bovine model for the human loss-of-imprinting overgrowth syndrome Beckwith-Wiedemann. Epigenetics 8, 591–601. doi: 10.4161/epi.24655
Cheng, Y., He, C., Wang, M., Ma, X., Mo, F., Yang, S., et al. (2019). Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 4:62. doi: 10.1038/s41392-019-0095-0
Choufani, S., Shuman, C., and Weksberg, R. (2010). Beckwith-Wiedemann syndrome. Am J. Med. Genet. C Semin. Med. Genet. 154C, 343–354. doi: 10.1002/ajmg.c.30267
Coffee, B., Muralidharan, K., Highsmith, W. E. Jr., Lapunzina, P., and Warren, S. T. (2006). Molecular diagnosis of Beckwith-Wiedemann syndrome using quantitative methylation-sensitive polymerase chain reaction. Genet. Med. 8, 628–634. doi: 10.1097/01.gim.0000237770.42442.cc
Cohen, J. L., Duffy, K. A., Sajorda, B. J., Hathaway, E. R., Gonzalez-Gandolfi, C. X., Richards-Yutz, J., et al. (2019). Diagnosis and management of the phenotypic spectrum of twins with Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A 179, 1139–1147. doi: 10.1002/ajmg.a.61164
Cooper, W. N., Curley, R., Macdonald, F., and Maher, E. R. (2007). Mitotic recombination and uniparental disomy in Beckwith-Wiedemann syndrome. Genomics 89, 613–617. doi: 10.1016/j.ygeno.2007.01.005
Cooper, W. N., Luharia, A., Evans, G. A., Raza, H., Haire, A. C., Grundy, R., et al. (2005). Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 13, 1025–1032. doi: 10.1038/sj.ejhg.5201463
Court, F., Martin-Trujillo, A., Romanelli, V., Garin, I., Iglesias-Platas, I., Salafsky, I., et al. (2013). Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum. Mutat. 34, 595–602. doi: 10.1002/humu.22276
Crea, F., Nobili, S., Paolicchi, E., Perrone, G., Napoli, C., Landini, I., et al. (2011). Epigenetics and chemoresistance in colorectal cancer: an opportunity for treatment tailoring and novel therapeutic strategies. Drug Resist. Updat. 14, 280–296. doi: 10.1016/j.drup.2011.08.001
Cresto, J. C., Abdenur, J. P., Bergada, I., and Martino, R. (1998). Long-term follow up of persistent hyperinsulinaemic hypoglycaemia of infancy. Arch. Dis. Child 79, 440–444. doi: 10.1136/adc.79.5.440
Crispo, F., Condelli, V., Lepore, S., Notarangelo, T., Sgambato, A., Esposito, F., et al. (2019). Metabolic dysregulations and epigenetics: a bidirectional interplay that drives tumor progression. Cells 8:798. doi: 10.3390/cells8080798
Das, R., Hampton, D. D., and Jirtle, R. L. (2009). Imprinting evolution and human health. Mamm. Genome 20, 563–572. doi: 10.1007/s00335-009-9229-y
Dawkins, H. J. S., Draghia-Akli, R., Lasko, P., Lau, L. P. L., Jonker, A. H., Cutillo, C. M., et al. (2018). Progress in rare diseases research 2010-2016: an IRDiRC perspective. Clin. Transl. Sci. 11, 11–20. doi: 10.1111/cts.12501
de Lonlay-Debeney, P., Poggi-Travert, F., Fournet, J. C., Sempoux, C., Dionisi Vici, C., Brunelle, F., et al. (1999). Clinical features of 52 neonates with hyperinsulinism. N. Engl. J. Med. 340, 1169–1175. doi: 10.1056/NEJM199904153401505
de Mouzon, J., Goossens, V., Bhattacharya, S., Castilla, J. A., Ferraretti, A. P., Korsak, V., et al. (2012). Assisted reproductive technology in Europe, 2007: results generated from European registers by ESHRE. Hum. Reprod. 27, 954–966. doi: 10.1093/humrep/des023
DeBaun, M. R., Niemitz, E. L., and Feinberg, A. P. (2003). Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 72, 156–160. doi: 10.1086/346031
DeBaun, M. R., and Tucker, M. A. (1998). Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J. Pediatr. 132(3 Pt 1), 398–400. doi: 10.1016/s0022-3476(98)70008-3
Dhont, M., De Sutter, P., Ruyssinck, G., Martens, G., and Bekaert, A. (1999). Perinatal outcome of pregnancies after assisted reproduction: a case-control study. Am. J. Obstet. Gynecol. 181, 688–695. doi: 10.1016/s0002-9378(99)70514-4
Dirks, R. A., Stunnenberg, H. G., and Marks, H. (2016). Genome-wide epigenomic profiling for biomarker discovery. Clin. Epigenetics 8:122. doi: 10.1186/s13148-016-0284-4
Docherty, L. E., Rezwan, F. I., Poole, R. L., Jagoe, H., Lake, H., Lockett, G. A., et al. (2014). Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J. Med. Genet. 51, 229–238. doi: 10.1136/jmedgenet-2013-102116
Doornbos, M. E., Maas, S. M., McDonnell, J., Vermeiden, J. P., and Hennekam, R. C. (2007). Infertility, assisted reproduction technologies and imprinting disturbances: a Dutch study. Hum. Reprod. 22, 2476–2480. doi: 10.1093/humrep/dem172
Du, M., Beatty, L. G., Zhou, W., Lew, J., Schoenherr, C., Weksberg, R., et al. (2003). Insulator and silencer sequences in the imprinted region of human chromosome 11p15.5. Hum. Mol. Genet. 12, 1927–1939. doi: 10.1093/hmg/ddg194
Duffy, K. A., Grand, K. L., Zelley, K., and Kalish, J. M. (2018). Tumor Screening in Beckwith-Wiedemann syndrome: parental perspectives. J. Genet. Couns. 27, 844–853. doi: 10.1007/s10897-017-0182-8
Dunn, J., and Rao, S. (2017). Epigenetics and immunotherapy: the current state of play. Mol. Immunol. 87, 227–239. doi: 10.1016/j.molimm.2017.04.012
Eggermann, T., Binder, G., Brioude, F., Maher, E. R., Lapunzina, P., Cubellis, M. V., et al. (2014). CDKN1C mutations: two sides of the same coin. Trends Mol. Med. 20, 614–622. doi: 10.1016/j.molmed.2014.09.001
Eggermann, T., Brioude, F., Russo, S., Lombardi, M. P., Bliek, J., Maher, E. R., et al. (2016). Prenatal molecular testing for Beckwith-Wiedemann and Silver-Russell syndromes: a challenge for molecular analysis and genetic counseling. Eur. J. Hum. Genet. 24, 784–793. doi: 10.1038/ejhg.2015.224
Eggermann, T., Perez de Nanclares, G., Maher, E. R., Temple, I. K., Tumer, Z., Monk, D., et al. (2015). Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenetics 7:123. doi: 10.1186/s13148-015-0143-8
Eltan, M., Arslan Ates, E., Cerit, K., Menevse, T. S., Kaygusuz, S. B., Eker, N., et al. (2020). Adrenocortical carcinoma in atypical Beckwith-Wiedemann syndrome due to loss of methylation at imprinting control region 2. Pediatr. Blood Cancer 67:e28042. doi: 10.1002/pbc.28042
Esteller, M. (2017). Epigenetic drugs: more than meets the eye. Epigenetics 12:307. doi: 10.1080/15592294.2017.1322881
Ferraretti, A. P., Goossens, V., de Mouzon, J., Bhattacharya, S., Castilla, J. A., Korsak, V., et al. (2012). Assisted reproductive technology in Europe, 2008: results generated from European registers by ESHRE. Hum. Reprod. 27, 2571–2584. doi: 10.1093/humrep/des255
Ferraretti, A. P., Goossens, V., Kupka, M., Bhattacharya, S., de Mouzon, J., Castilla, J. A., et al. (2013). Assisted reproductive technology in Europe, 2009: results generated from European registers by ESHRE. Hum. Reprod. 28, 2318–2331. doi: 10.1093/humrep/det278
Fontana, L., Bedeschi, M. F., Maitz, S., Cereda, A., Fare, C., Motta, S., et al. (2018). Characterization of multi-locus imprinting disturbances and underlying genetic defects in patients with chromosome 11p15.5 related imprinting disorders. Epigenetics 13, 897–909. doi: 10.1080/15592294.2018.1514230
Gallagher, E. J., and LeRoith, D. (2010). The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. 21, 610–618. doi: 10.1016/j.tem.2010.06.007
Gaston, V., Le Bouc, Y., Soupre, V., Burglen, L., Donadieu, J., Oro, H., et al. (2001). Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 9, 409–418. doi: 10.1038/sj.ejhg.5200649
Gaur, S., Wen, Y., Song, J. H., Parikh, N. U., Mangala, L. S., Blessing, A. M., et al. (2015). Chitosan nanoparticle-mediated delivery of miRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. Oncotarget 6, 29161–29177. doi: 10.18632/oncotarget.4971
Ghanem, I., Karam, J. A., and Widmann, R. F. (2011). Surgical epiphysiodesis indications and techniques: update. Curr. Opin. Pediatr. 23, 53–59. doi: 10.1097/MOP.0b013e32834231b3
Gicquel, C., El-Osta, A., and Le Bouc, Y. (2008). Epigenetic regulation and fetal programming. Best Pract. Res. Clin. Endocrinol. Metab. 22, 1–16. doi: 10.1016/j.beem.2007.07.009
Gicquel, C., Gaston, V., Mandelbaum, J., Siffroi, J. P., Flahault, A., and Le Bouc, Y. (2003). In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am. J. Hum. Genet. 72, 1338–1341. doi: 10.1086/374824
Glaser, B., Thornton, P., Otonkoski, T., and Junien, C. (2000). Genetics of neonatal hyperinsulinism. Arch. Dis. Child Fetal Neonatal Ed. 82, F79–F86. doi: 10.1136/fn.82.2.f79
Glasgow, M. D., and Chougule, M. B. (2015). Recent developments in active tumor targeted multifunctional nanoparticles for combination chemotherapy in cancer treatment and imaging. J. Biomed. Nanotechnol. 11, 1859–1898. doi: 10.1166/jbn.2015.2145
Gocmen, R., Basaran, C., Karcaaltincaba, M., Cinar, A., Yurdakok, M., Akata, D., et al. (2005). Bilateral hemorrhagic adrenal cysts in an incomplete form of Beckwith-Wiedemann syndrome: MRI and prenatal US findings. Abdom. Imaging 30, 786–789. doi: 10.1007/s00261-005-0337-1
Gomes, M. V., Huber, J., Ferriani, R. A., Amaral Neto, A. M., and Ramos, E. S. (2009). Abnormal methylation at the KvDMR1 imprinting control region in clinically normal children conceived by assisted reproductive technologies. Mol. Hum. Reprod. 15, 471–477. doi: 10.1093/molehr/gap038
Graca, I., Pereira-Silva, E., Henrique, R., Packham, G., Crabb, S. J., and Jeronimo, C. (2016). Epigenetic modulators as therapeutic targets in prostate cancer. Clin. Epigenetics 8:98. doi: 10.1186/s13148-016-0264-8
Griggs, R. C., Batshaw, M., Dunkle, M., Gopal-Srivastava, R., Kaye, E., Krischer, J., et al. (2009). Clinical research for rare disease: opportunities, challenges, and solutions. Mol. Genet. Metab. 96, 20–26. doi: 10.1016/j.ymgme.2008.10.003
Hall, J. G., and Lopez-Rangel, E. (1996). Embryologic development and monozygotic twinning. Acta Genet. Med. Gemellol. 45, 53–57. doi: 10.1017/s0001566000001094
Halliday, J., Oke, K., Breheny, S., Algar, E., and David, J. A. (2004). Beckwith-Wiedemann syndrome and IVF: a case-control study. Am. J. Hum. Genet. 75, 526–528. doi: 10.1086/423902
Hark, A. T., Schoenherr, C. J., Katz, D. J., Ingram, R. S., Levorse, J. M., and Tilghman, S. M. (2000). CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489. doi: 10.1038/35013106
Hattori, H., Hiura, H., Kitamura, A., Miyauchi, N., Kobayashi, N., Takahashi, S., et al. (2019). Association of four imprinting disorders and ART. Clin. Epigenetics 11:21. doi: 10.1186/s13148-019-0623-3
Heerboth, S., Lapinska, K., Snyder, N., Leary, M., Rollinson, S., and Sarkar, S. (2014). Use of epigenetic drugs in disease: an overview. Genet. Epigenet. 6, 9–19. doi: 10.4137/GEG.S12270
Hiura, H., Okae, H., Miyauchi, N., Sato, F., Sato, A., Van De Pette, M., et al. (2012). Characterization of DNA methylation errors in patients with imprinting disorders conceived by assisted reproduction technologies. Hum. Reprod. 27, 2541–2548. doi: 10.1093/humrep/des197
Hori, N., Yamane, M., Kouno, K., and Sato, K. (2012). Induction of DNA demethylation depending on two sets of Sox2 and adjacent Oct3/4 binding sites (Sox-Oct motifs) within the mouse H19/insulin-like growth factor 2 (Igf2) imprinted control region. J. Biol. Chem. 287, 44006–44016. doi: 10.1074/jbc.M112.424580
Ibrahim, A., Kirby, G., Hardy, C., Dias, R. P., Tee, L., Lim, D., et al. (2014). Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin. Epigenetics 6:11. doi: 10.1186/1868-7083-6-11
Inamura, K. (2017). Major tumor suppressor and oncogenic non-coding RNAs: clinical relevance in lung cancer. Cells 6:12. doi: 10.3390/cells6020012
Jones, P. A., Issa, J. P., and Baylin, S. (2016). Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17, 630–641. doi: 10.1038/nrg.2016.93
Kagan, K. O., Berg, C., Dufke, A., Geipel, A., Hoopmann, M., and Abele, H. (2015). Novel fetal and maternal sonographic findings in confirmed cases of Beckwith-Wiedemann syndrome. Prenat. Diagn. 35, 394–399. doi: 10.1002/pd.4555
Kalish, J. M., Boodhansingh, K. E., Bhatti, T. R., Ganguly, A., Conlin, L. K., Becker, S. A., et al. (2016). Congenital hyperinsulinism in children with paternal 11p uniparental isodisomy and Beckwith-Wiedemann syndrome. J. Med. Genet. 53, 53–61. doi: 10.1136/jmedgenet-2015-103394
Kalish, J. M., Doros, L., Helman, L. J., Hennekam, R. C., Kuiper, R. P., Maas, S. M., et al. (2017). Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin. Cancer Res. 23, e115–e122. doi: 10.1158/1078-0432.CCR-17-0710
Kamien, B., Ronan, A., Poke, G., Sinnerbrink, I., Baynam, G., Ward, M., et al. (2018). A clinical review of generalized overgrowth syndromes in the era of massively parallel sequencing. Mol. Syndromol. 9, 70–82. doi: 10.1159/000484532
Kavanagh, E., and Joseph, B. (2011). The hallmarks of CDKN1C (p57. KIP2) in cancer. Biochim. Biophys. Acta 1816, 50–56. doi: 10.1016/j.bbcan.2011.03.002
Kelly, T. K., De Carvalho, D. D., and Jones, P. A. (2010). Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 28, 1069–1078. doi: 10.1038/nbt.1678
Krzyzewska, I. M., Alders, M., Maas, S. M., Bliek, J., Venema, A., Henneman, P., et al. (2019). Genome-wide methylation profiling of Beckwith-Wiedemann syndrome patients without molecular confirmation after routine diagnostics. Clin. Epigenetics 11:53. doi: 10.1186/s13148-019-0649-6
Kubo, S., Murata, C., Okamura, H., Sakasegawa, T., Sakurai, C., Hatsuzawa, K., et al. (2020). Oct motif variants in Beckwith-Wiedemann syndrome patients disrupt maintenance of the hypomethylated state of the H19/IGF2 imprinting control region. FEBS Lett. 594, 1517–1531. doi: 10.1002/1873-3468.13750
Kupka, M. S., Ferraretti, A. P., de Mouzon, J., Erb, K., D’Hooghe, T., Castilla, J. A., et al. (2014). Assisted reproductive technology in Europe, 2010: results generated from European registers by ESHREdagger. Hum. Reprod. 29, 2099–2113. doi: 10.1093/humrep/deu175
Laje, P., Palladino, A. A., Bhatti, T. R., States, L. J., Stanley, C. A., and Adzick, N. S. (2013). Pancreatic surgery in infants with Beckwith-Wiedemann syndrome and hyperinsulinism. J. Pediatr. Surg. 48, 2511–2516. doi: 10.1016/j.jpedsurg.2013.05.016
Landy, H. J., and Keith, L. G. (1998). The vanishing twin: a review. Hum. Reprod. Update 4, 177–183. doi: 10.1093/humupd/4.2.177
Lapunzina, P. (2005). Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am. J. Med. Genet. C Semin. Med. Genet. 137C, 53–71. doi: 10.1002/ajmg.c.30064
Larson, P. S., Schlechter, B. L., de las Morenas, A., Garber, J. E., Cupples, L. A., and Rosenberg, C. L. (2005). Allele imbalance, or loss of heterozygosity, in normal breast epithelium of sporadic breast cancer cases and BRCA1 gene mutation carriers is increased compared with reduction mammoplasty tissues. J. Clin. Oncol. 23, 8613–8619. doi: 10.1200/JCO.2005.02.1451
Lauschke, V. M., Barragan, I., and Ingelman-Sundberg, M. (2018). Pharmacoepigenetics and toxicoepigenetics: novel mechanistic insights and therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 58, 161–185. doi: 10.1146/annurev-pharmtox-010617-053021
Lee, M. P., DeBaun, M. R., Mitsuya, K., Galonek, H. L., Brandenburg, S., Oshimura, M., et al. (1999). Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. U.S.A. 96, 5203–5208. doi: 10.1073/pnas.96.9.5203
Leibowitz, G., Glaser, B., Higazi, A. A., Salameh, M., Cerasi, E., and Landau, H. (1995). Hyperinsulinemic hypoglycemia of infancy (nesidioblastosis) in clinical remission: high incidence of diabetes mellitus and persistent beta-cell dysfunction at long-term follow-up. J. Clin. Endocrinol. Metab. 80, 386–392. doi: 10.1210/jcem.80.2.7852494
Li, Y., Hagen, D. E., Ji, T., Bakhtiarizadeh, M. R., Frederic, W. M., Traxler, E. M., et al. (2019). Altered microRNA expression profiles in large offspring syndrome and Beckwith-Wiedemann syndrome. Epigenetics 14, 850–876. doi: 10.1080/15592294.2019.1615357
Lim, D., Bowdin, S. C., Tee, L., Kirby, G. A., Blair, E., Fryer, A., et al. (2009). Clinical and molecular genetic features of Beckwith-Wiedemann syndrome associated with assisted reproductive technologies. Hum. Reprod. 24, 741–747. doi: 10.1093/humrep/den406
Liu, W., Zhang, R., Wei, J., Zhang, H., Yu, G., Li, Z., et al. (2015). Rapid diagnosis of imprinting disorders involving copy number variation and uniparental disomy using genome-wide SNP microarrays. Cytogenet. Genome Res. 146, 9–18. doi: 10.1159/000435847
Livingstone, C. (2013). IGF2 and cancer. Endocr. Relat. Cancer 20, R321–R339. doi: 10.1530/ERC-13-0231
Lteif, A. N., and Schwenk, W. F. (1999). Hypoglycemia in infants and children. Endocrinol. Metab. Clin. North Am. 28, 619–646, vii. doi: 10.1016/s0889-8529(05)70091-8
Lubinsky, M. S., and Hall, J. G. (1991). Genomic imprinting, monozygous twinning, and X inactivation. Lancet 337:1288. doi: 10.1016/0140-6736(91)92956-3
Lundstrom, K. (2017). Cell-impedance-based label-free technology for the identification of new drugs. Expert Opin. Drug Discov. 12, 335–343. doi: 10.1080/17460441.2017.1297419
Maas, S. M., Vansenne, F., Kadouch, D. J., Ibrahim, A., Bliek, J., Hopman, S., et al. (2016). Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J. Med. Genet. A 170, 2248–2260. doi: 10.1002/ajmg.a.37801
MacFarland, S. P., Duffy, K. A., Bhatti, T. R., Bagatell, R., Balamuth, N. J., Brodeur, G. M., et al. (2018). Diagnosis of Beckwith-Wiedemann syndrome in children presenting with Wilms tumor. Pediatr. Blood Cancer 65:e27296. doi: 10.1002/pbc.27296
Machin, G. A. (1996). Some causes of genotypic and phenotypic discordance in monozygotic twin pairs. Am. J. Med. Genet. 61, 216–228. doi: 10.1002/(sici)1096-8628(19960122)61:3<216::aid-ajmg5>3.0.co;2-s
Maeda, T., Higashimoto, K., Jozaki, K., Yatsuki, H., Nakabayashi, K., Makita, Y., et al. (2014). Comprehensive and quantitative multilocus methylation analysis reveals the susceptibility of specific imprinted differentially methylated regions to aberrant methylation in Beckwith-Wiedemann syndrome with epimutations. Genet. Med. 16, 903–912. doi: 10.1038/gim.2014.46
Maher, E. R., and Reik, W. (2000). Beckwith-Wiedemann syndrome: imprinting in clusters revisited. J. Clin. Invest. 105, 247–252. doi: 10.1172/JCI9340
Mama, N., H’Mida, D., Lahmar, I., Yacoubi, M. T., and Tlili-Graiess, K. (2014). PHACES syndrome associated with carcinoid endobronchial tumor. Pediatr. Radiol. 44, 621–624. doi: 10.1007/s00247-013-2820-0
Martinez y Martinez, R., Martinez-Carboney, R., Ocampo-Campos, R., Rivera, H., Gomez Plascencia y Castillo, J., Cuevas, A., et al. (1992). Wiedemann-Beckwith syndrome: clinical, cytogenetical and radiological observations in 39 new cases. Genet. Couns. 3, 67–76.
Mau, T., and Yung, R. (2014). Potential of epigenetic therapies in non-cancerous conditions. Front. Genet. 5:438. doi: 10.3389/fgene.2014.00438
Mazzone, R., Zwergel, C., Mai, A., and Valente, S. (2017). Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin. Epigenetics 9:59. doi: 10.1186/s13148-017-0358-y
Miranda Furtado, C. L., Dos Santos Luciano, M. C., Silva Santos, R. D., Furtado, G. P., Moraes, M. O., and Pessoa, C. (2019). Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics 14, 1164–1176. doi: 10.1080/15592294.2019.1640546
Morera, L., Lubbert, M., and Jung, M. (2016). Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 8, 57. doi: 10.1186/s13148-016-0223-4
Munns, C. F., and Batch, J. A. (2001). Hyperinsulinism and Beckwith-Wiedemann syndrome. Arch. Dis. Child Fetal Neonatal Ed. 84, F67–F69. doi: 10.1136/fn.84.1.f67
Murrell, A., Heeson, S., and Reik, W. (2004). Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 36, 889–893. doi: 10.1038/ng1402
Mussa, A., Ciuffreda, V. P., Sauro, P., Pagliardini, V., Pagliardini, S., Carli, D., et al. (2019). Longitudinal Monitoring of Alpha-Fetoprotein by Dried Blood Spot for Hepatoblastoma Screening in Beckwith(-)Wiedemann Syndrome. Cancers 11:86. doi: 10.3390/cancers11010086
Mussa, A., Molinatto, C., Baldassarre, G., Riberi, E., Russo, S., Larizza, L., et al. (2016a). Cancer Risk in Beckwith-Wiedemann syndrome: a systematic review and meta-analysis outlining a novel (Epi)Genotype specific histotype targeted screening protocol. J. Pediatr. 176, 142–149.e1. doi: 10.1016/j.jpeds.2016.05.038
Mussa, A., Molinatto, C., Cerrato, F., Palumbo, O., Carella, M., Baldassarre, G., et al. (2017). Assisted reproductive techniques and risk of beckwith-wiedemann syndrome. Pediatrics 140:e20164311. doi: 10.1542/peds.2016-4311
Mussa, A., Russo, S., De Crescenzo, A., Chiesa, N., Molinatto, C., Selicorni, A., et al. (2013). Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am. J. Med. Genet. A 161A, 2481–2486. doi: 10.1002/ajmg.a.36080
Mussa, A., Russo, S., de Crescenzo, A., Freschi, A., Calzari, L., Maitz, S., et al. (2016b). Fetal growth patterns in Beckwith-Wiedemann syndrome. Clin. Genet. 90, 21–27. doi: 10.1111/cge.12759
Nativio, R., Sparago, A., Ito, Y., Weksberg, R., Riccio, A., and Murrell, A. (2011). Disruption of genomic neighbourhood at the imprinted IGF2-H19 locus in Beckwith-Wiedemann syndrome and Silver-Russell syndrome. Hum. Mol. Genet. 20, 1363–1374. doi: 10.1093/hmg/ddr018
Netchine, I., Rossignol, S., Azzi, S., and Le Bouc, Y. (2013). Epigenetic anomalies in childhood growth disorders. Nestle Nutr. Inst. Workshop Ser. 71, 65–73. doi: 10.1159/000342568
Nguyen, K. V. (2019). Potential epigenomic co-management in rare diseases and epigenetic therapy. Nucleosides Nucleotides Nucleic Acids 38, 752–780. doi: 10.1080/15257770.2019.1594893
Niculescu, M. D., and Lupu, D. S. (2011). Nutritional influence on epigenetics and effects on longevity. Curr. Opin. Clin. Nutr. Metab. Care 14, 35–40. doi: 10.1097/MCO.0b013e328340ff7c
Nowacka-Zawisza, M., and Wisnik, E. (2017). DNA methylation and histone modifications as epigenetic regulation in prostate cancer (Review). Oncol. Rep. 38, 2587–2596. doi: 10.3892/or.2017.5972
Orstavik, R. E., Tommerup, N., Eiklid, K., and Orstavik, K. H. (1995). Non-random X chromosome inactivation in an affected twin in a monozygotic twin pair discordant for Wiedemann-Beckwith syndrome. Am. J. Med. Genet. 56, 210–214. doi: 10.1002/ajmg.1320560219
Ounap, K. (2016). Silver-russell syndrome and Beckwith-Wiedemann syndrome: opposite phenotypes with heterogeneous molecular etiology. Mol. Syndromol. 7, 110–121. doi: 10.1159/000447413
Paganini, L., Carlessi, N., Fontana, L., Silipigni, R., Motta, S., Fiori, S., et al. (2015). Beckwith-Wiedemann syndrome prenatal diagnosis by methylation analysis in chorionic villi. Epigenetics 10, 643–649. doi: 10.1080/15592294.2015.1057383
Park, K. S., Mitra, A., Rahat, B., Kim, K., and Pfeifer, K. (2017). Loss of imprinting mutations define both distinct and overlapping roles for misexpression of IGF2 and of H19 lncRNA. Nucleic Acids Res. 45, 12766–12779. doi: 10.1093/nar/gkx896
Poole, R. L., Docherty, L. E., Al Sayegh, A., Caliebe, A., Turner, C., Baple, E., et al. (2013). Targeted methylation testing of a patient cohort broadens the epigenetic and clinical description of imprinting disorders. Am. J. Med. Genet. A 161A, 2174–2182. doi: 10.1002/ajmg.a.36049
Priolo, M., Sparago, A., Mammi, C., Cerrato, F., Lagana, C., and Riccio, A. (2008). MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur. J. Hum. Genet. 16, 565–571. doi: 10.1038/sj.ejhg.5202001
Raveh, E., Matouk, I. J., Gilon, M., and Hochberg, A. (2015). The H19 Long non-coding RNA in cancer initiation, progression and metastasis - a proposed unifying theory. Mol. Cancer 14:184. doi: 10.1186/s12943-015-0458-2
Romanelli, V., Belinchon, A., Benito-Sanz, S., Martinez-Glez, V., Gracia-Bouthelier, R., Heath, K. E., et al. (2010). CDKN1C (p57(Kip2)) analysis in Beckwith-Wiedemann syndrome (BWS) patients: genotype-phenotype correlations, novel mutations, and polymorphisms. Am. J. Med. Genet. A 152A, 1390–1397. doi: 10.1002/ajmg.a.33453
Rossignol, S., Steunou, V., Chalas, C., Kerjean, A., Rigolet, M., Viegas-Pequignot, E., et al. (2006). The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J. Med. Genet. 43, 902–907. doi: 10.1136/jmg.2006.042135
Rump, P., Zeegers, M. P., and van Essen, A. J. (2005). Tumor risk in Beckwith-Wiedemann syndrome: a review and meta-analysis. Am. J. Med. Genet. A 136, 95–104. doi: 10.1002/ajmg.a.30729
Russo, S., Calzari, L., Mussa, A., Mainini, E., Cassina, M., Di Candia, S., et al. (2016). A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver-Russell and Beckwith-Wiedemann syndromes. Clin. Epigenetics 8:23. doi: 10.1186/s13148-016-0183-8
Sanchez-Delgado, M., Riccio, A., Eggermann, T., Maher, E. R., Lapunzina, P., Mackay, D., et al. (2016). Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet. 32, 444–455. doi: 10.1016/j.tig.2016.05.001
Sano, S., Matsubara, K., Nagasaki, K., Kikuchi, T., Nakabayashi, K., Hata, K., et al. (2016). Beckwith-Wiedemann syndrome and pseudohypoparathyroidism type Ib in a patient with multilocus imprinting disturbance: a female-dominant phenomenon? J. Hum. Genet. 61, 765–769. doi: 10.1038/jhg.2016.45
Sazhenova, E. A., and Lebedev, I. N. (2008). [Epimutations of the KCNQ1OT1 imprinting center of chromosome 11 in early human embryo lethality]. Genetika 44, 1609–1616.
Schiff, D., Colle, E., Wells, D., and Stern, L. (1973). Metabolic aspects of the Beckwith-Wiedemann syndrome. J. Pediatr. 82, 258–262. doi: 10.1016/s0022-3476(73)80163-5
Schofield, P. N., Joyce, J. A., Lam, W. K., Grandjean, V., Ferguson-Smith, A., Reik, W., et al. (2001). Genomic imprinting and cancer; new paradigms in the genetics of neoplasia. Toxicol. Lett. 120, 151–160. doi: 10.1016/s0378-4274(01)00294-6
Scott, R. H., Douglas, J., Baskcomb, L., Nygren, A. O., Birch, J. M., Cole, T. R., et al. (2008). Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) robustly detects and distinguishes 11p15 abnormalities associated with overgrowth and growth retardation. J. Med. Genet. 45, 106–113. doi: 10.1136/jmg.2007.053207
Segers, H., Kersseboom, R., Alders, M., Pieters, R., Wagner, A., and van den Heuvel-Eibrink, M. M. (2012). Frequency of WT1 and 11p15 constitutional aberrations and phenotypic correlation in childhood Wilms tumour patients. Eur. J. Cancer 48, 3249–3256. doi: 10.1016/j.ejca.2012.06.008
Senniappan, S., Ismail, D., Shipster, C., Beesley, C., and Hussain, K. (2015). The heterogeneity of hyperinsulinaemic hypoglycaemia in 19 patients with Beckwith-Wiedemann syndrome due to KvDMR1 hypomethylation. J. Pediatr. Endocrinol. Metab 28, 83–86. doi: 10.1515/jpem-2013-0390
Shao, Q., Xu, J., Deng, R., Wei, W., Zhou, B., Yue, C., et al. (2018). Long non-coding RNA-422 acts as a tumor suppressor in colorectal cancer. Biochem. Biophys. Res. Commun. 495, 539–545. doi: 10.1016/j.bbrc.2017.10.076
Shepherd, R. M., Cosgrove, K. E., O’Brien, R. E., Barnes, P. D., Ammala, C., and Dunne, M. J. (2000). Hyperinsulinism of infancy: towards an understanding of unregulated insulin release. European Network for Research into Hyperinsulinism in Infancy. Arch. Dis. Child Fetal Neonatal Ed. 82, F87–F97. doi: 10.1136/fn.82.2.f87
Shilyansky, J., Cutz, E., and Filler, R. M. (1997). Endogenous hyperinsulinism: diagnosis, management, and long-term follow-up. Semin. Pediatr. Surg. 6, 115–120.
Shuman, C., Smith, A. C., Steele, L., Ray, P. N., Clericuzio, C., Zackai, E., et al. (2006). Constitutional UPD for chromosome 11p15 in individuals with isolated hemihyperplasia is associated with high tumor risk and occurs following assisted reproductive technologies. Am. J. Med. Genet. A 140, 1497–1503. doi: 10.1002/ajmg.a.31323
Slavotinek, A., Gaunt, L., and Donnai, D. (1997). Paternally inherited duplications of 11p15.5 and Beckwith-Wiedemann syndrome. J. Med. Genet. 34, 819–826. doi: 10.1136/jmg.34.10.819
Smith, A. C., Rubin, T., Shuman, C., Estabrooks, L., Aylsworth, A. S., McDonald, M. T., et al. (2006). New chromosome 11p15 epigenotypes identified in male monozygotic twins with Beckwith-Wiedemann syndrome. Cytogenet. Genome Res. 113, 313–317. doi: 10.1159/000090847
Smith, A. C., Suzuki, M., Thompson, R., Choufani, S., Higgins, M. J., Chiu, I. W., et al. (2012). Maternal gametic transmission of translocations or inversions of human chromosome 11p15.5 results in regional DNA hypermethylation and downregulation of CDKN1C expression. Genomics 99, 25–35. doi: 10.1016/j.ygeno.2011.10.007
Soejima, H., and Higashimoto, K. (2013). Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders. J. Hum. Genet. 58, 402–409. doi: 10.1038/jhg.2013.51
Stampone, E., Caldarelli, I., Zullo, A., Bencivenga, D., Mancini, F. P., Della Ragione, F., et al. (2018). Genetic and epigenetic control of CDKN1C expression: importance in cell commitment and differentiation, tissue homeostasis and human diseases. Int. J. Mol. Sci. 19:1055. doi: 10.3390/ijms19041055
Stanley, C. A. (1997). Hyperinsulinism in infants and children. Pediatr. Clin. North Am. 44, 363–374. doi: 10.1016/s0031-3955(05)70481-8
Sutcliffe, A. G., D’Souza, S. W., Cadman, J., Richards, B., McKinlay, I. A., and Lieberman, B. (1995). Minor congenital anomalies, major congenital malformations and development in children conceived from cryopreserved embryos. Hum. Reprod. 10, 3332–3337. doi: 10.1093/oxfordjournals.humrep.a135915
Sutcliffe, A. G., Peters, C. J., Bowdin, S., Temple, K., Reardon, W., Wilson, L., et al. (2006). Assisted reproductive therapies and imprinting disorders–a preliminary British survey. Hum. Reprod. 21, 1009–1011. doi: 10.1093/humrep/dei405
Sweet, C. B., Grayson, S., and Polak, M. (2013). Management strategies for neonatal hypoglycemia. J. Pediatr. Pharmacol. Ther. 18, 199–208. doi: 10.5863/1551-6776-18.3.199
Swinney, D. C., and Xia, S. (2014). The discovery of medicines for rare diseases. Future Med. Chem. 6, 987–1002. doi: 10.4155/fmc.14.65
Talaulikar, V. S., and Arulkumaran, S. (2012). Reproductive outcomes after assisted conception. Obstet. Gynecol. Surv. 67, 566–583. doi: 10.1097/OGX.0b013e31826a5d4a
Tee, L., Lim, D. H., Dias, R. P., Baudement, M. O., Slater, A. A., Kirby, G., et al. (2013). Epimutation profiling in Beckwith-Wiedemann syndrome: relationship with assisted reproductive technology. Clin. Epigenetics 5:23. doi: 10.1186/1868-7083-5-23
Tierling, S., Souren, N. Y., Reither, S., Zang, K. D., Meng-Hentschel, J., Leitner, D., et al. (2011). DNA methylation studies on imprinted loci in a male monozygotic twin pair discordant for Beckwith-Wiedemann syndrome. Clin. Genet. 79, 546–553. doi: 10.1111/j.1399-0004.2010.01482.x
Tung, J. Y., Lai, S. H. Y., Au, S. L. K., Yeung, K. S., Kan, A. S. Y., Loong, F., et al. (2020). Coexistence of paternally-inherited ABCC8 mutation and mosaic paternal uniparental disomy 11p hyperinsulinism. Int. J. Pediatr. Endocrinol. 2020:13. doi: 10.1186/s13633-020-00083-5
Velasco, G., and Francastel, C. (2019). Genetics meets DNA methylation in rare diseases. Clin. Genet. 95, 210–220. doi: 10.1111/cge.13480
Vermeiden, J. P., and Bernardus, R. E. (2013). Are imprinting disorders more prevalent after human in vitro fertilization or intracytoplasmic sperm injection? Fertil. Steril. 99, 642–651. doi: 10.1016/j.fertnstert.2013.01.125
Vitiello, M., Tuccoli, A., and Poliseno, L. (2015). Long non-coding RNAs in cancer: implications for personalized therapy. Cell. Oncol. 38, 17–28. doi: 10.1007/s13402-014-0180-x
Wang, R., Xiao, Y., Li, D., Hu, H., Li, X., Ge, T., et al. (2020). Clinical and molecular features of children with Beckwith-Wiedemann syndrome in China: a single-center retrospective cohort study. Ital. J. Pediatr. 46:55. doi: 10.1186/s13052-020-0819-3
Weksberg, R., Shen, D. R., Fei, Y. L., Song, Q. L., and Squire, J. (1993). Disruption of insulin-like growth factor 2 imprinting in Beckwith-Wiedemann syndrome. Nat. Genet. 5, 143–150. doi: 10.1038/ng1093-143
Weksberg, R., Shuman, C., and Beckwith, J. B. (2010). Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 18, 8–14. doi: 10.1038/ejhg.2009.106
Weksberg, R., Shuman, C., Caluseriu, O., Smith, A. C., Fei, Y. L., Nishikawa, J., et al. (2002). Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 11, 1317–1325. doi: 10.1093/hmg/11.11.1317
Weksberg, R., Shuman, C., and Smith, A. C. (2005). Beckwith-Wiedemann syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 137C, 12–23. doi: 10.1002/ajmg.c.30058
Welch, G. R., and Clegg, J. S. (2010). From protoplasmic theory to cellular systems biology: a 150-year reflection. Am. J. Physiol. Cell Physiol. 298, C1280–C1290. doi: 10.1152/ajpcell.00016.2010
Weng, E. Y., Mortier, G. R., and Graham, J. M. Jr. (1995). Beckwith-Wiedemann syndrome. An update and review for the primary pediatrician. Clin. Pediatr. 34, 317–326. doi: 10.1177/000992289503400605
Wijnen, M., Alders, M., Zwaan, C. M., Wagner, A., and van den Heuvel-Eibrink, M. M. (2012). KCNQ1OT1 hypomethylation: a novel disguised genetic predisposition in sporadic pediatric adrenocortical tumors? Pediatr. Blood Cancer 59, 565–566. doi: 10.1002/pbc.23398
Williams, D. H., Gauthier, D. W., and Maizels, M. (2005). Prenatal diagnosis of Beckwith-Wiedemann syndrome. Prenat. Diagn. 25, 879–884. doi: 10.1002/pd.1155
Woods, D. M., Sodre, A. L., Villagra, A., Sarnaik, A., Sotomayor, E. M., and Weber, J. (2015). HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 3, 1375–1385. doi: 10.1158/2326-6066.CIR-15-0077-T
Yan, Y., Frisen, J., Lee, M. H., Massague, J., and Barbacid, M. (1997). Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 11, 973–983. doi: 10.1101/gad.11.8.973
Yao, Z. X., Jogunoori, W., Choufani, S., Rashid, A., Blake, T., Yao, W., et al. (2010). Epigenetic silencing of beta-spectrin, a TGF-beta signaling/scaffolding protein in a human cancer stem cell disorder: Beckwith-Wiedemann syndrome. J. Biol. Chem. 285, 36112–36120. doi: 10.1074/jbc.M110.162347
Young, L. E., Fernandes, K., McEvoy, T. G., Butterwith, S. C., Gutierrez, C. G., Carolan, C., et al. (2001). Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat. Genet. 27, 153–154. doi: 10.1038/84769
Young, L. E., Sinclair, K. D., and Wilmut, I. (1998). Large offspring syndrome in cattle and sheep. Rev. Reprod. 3, 155–163. doi: 10.1530/ror.0.0030155
Keywords: Beckwith-Wiedemann syndrome, rare diseases, cancer predisposition, epigenetics, metabolic disorders, DNA methylation, monozygotic twins
Citation: Papulino C, Chianese U, Nicoletti MM, Benedetti R and Altucci L (2020) Preclinical and Clinical Epigenetic-Based Reconsideration of Beckwith-Wiedemann Syndrome. Front. Genet. 11:563718. doi: 10.3389/fgene.2020.563718
Received: 19 May 2020; Accepted: 26 August 2020;
Published: 15 September 2020.
Edited by:
Mojgan Rastegar, University of Manitoba, CanadaReviewed by:
Alessandro Mussa, University of Turin, ItalySilvia Russo, Istituto Auxologico Italiano (IRCCS), Italy
Lidia Larizza, Italian Auxological Institute (IRCCS), Italy
Copyright © 2020 Papulino, Chianese, Nicoletti, Benedetti and Altucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosaria Benedetti, cm9zYXJpYS5iZW5lZGV0dGlAdW5pY2FtcGFuaWEuaXQ=; Lucia Altucci, bHVjaWEuYWx0dWNjaUB1bmljYW1wYW5pYS5pdA==
†These authors have contributed equally to this work
‡These authors share last authorship