 Lourdes Hontecillas-Prieto1*†
Lourdes Hontecillas-Prieto1*† Rocío Flores-Campos1†Andrew Silver2Enrique de Álava1,3
Rocío Flores-Campos1†Andrew Silver2Enrique de Álava1,3 Nabil Hajji4‡
Nabil Hajji4‡ Daniel J. García-Domínguez1‡
Daniel J. García-Domínguez1‡- 1Institute of Biomedicine of Seville, Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla/CIBERONC, Seville, Spain
- 2Centre for Genomics and Child Health, Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, United Kingdom
- 3Department of Normal and Pathological Cytology and Histology, School of Medicine, University of Seville, Seville, Spain
- 4Division of Brain Sciences, Imperial College London, London, United Kingdom
Chemotherapy is one of the most established and effective treatments for almost all types of cancer. However, the elevated toxicity due to the non-tumor-associated effects, development of secondary malignancies, infertility, radiation-induced fibrosis and resistance to treatment limit the effectiveness and safety of treatment. In addition, these multiple factors significantly impact quality of life. Over the last decades, our increased understanding of cancer epigenetics has led to new therapeutic approaches and the promise of improved patient outcomes. Epigenetic alterations are commonly found in cancer, especially the increased expression and activity of histone deacetylases (HDACs). Dysregulation of HDACs are critical to the development and progression of the majority of tumors. Hence, HDACs inhibitors (HDACis) were developed and now represent a very promising treatment strategy. The use of HDACis as monotherapy has shown very positive pre-clinical results, but clinical trials have had only limited success. However, combinatorial regimens with other cancer drugs have shown synergistic effects both in pre-clinical and clinical studies. At the same time, these combinations have enhanced the efficacy, reduced the toxicity and tumor resistance to therapy. In this review, we will examine examples of HDACis used in combination with other cancer drugs and highlight the synergistic effects observed in recent preclinical and clinical studies.
Introduction
The role of epigenetics was first described as an essential mechanism for normal cell function. Later, epigenetic disruptions were found to promote malignant cellular transformation leading to cancer (Choi et al., 2001; Ashraf et al., 2006; Adams et al., 2010). Traditionally, cancer is considered a multistep process in which transformational events are mainly associated with genetic changes in oncogenes and tumor suppressor genes (Hanahan and Weinberg, 2011). We now know that cancer initiation and progression involves substantial epigenetic abnormalities along with other genetic alterations (Baylin and Jones, 2011; Sandoval and Esteller, 2012). Changes in the mechanisms of DNA methylation, histone modifications or small non-coding microRNAs (miRNA) are considered now as hallmarks of cancer. Understanding the epigenetic landscape of tumors constitutes a promising research area both in terms of understanding the molecular mechanism involved in tumor development and the identification of novel targets for new and combinational therapies.
Unlike genetic alterations, epigenetic changes in cancer are reversible. Such changes can be exploited as cancer epigenetic biomarkers for use in the development and evaluation of epigenetic drugs for cancer therapy. One of the promising epigenetic drugs is histone deacetylases inhibitors (HDACis). Currently, there are many preclinical studies and clinical trials testing the efficacy, toxicity and utility of different HDACis both as monotherapies and in combination with other therapies.
Histone deacetylases inhibitors, as single agents, have been shown to be effective in preclinical studies involving a wide range of molecular and biological responses in both hematological and solid tumors. In general, HDACis as monotherapy are well-tolerated and are not toxic to normal tissues in preclinical models. However, HDACis as monotherapy have had limited success in clinical trials showing only modest anti-tumor effects, especially in solid tumors, and have caused secondary effects (Munster et al., 2009). These disadvantages could be resolved, at least in part, by combining HDACis with other anticancer drugs. This strategy appears to substantially improve the conventional treatment effect in many cancer studies because of synergistic or additive antitumor effects. HDACis in combination enhance the therapeutic efficacy with respect to their effect as a monotherapy both in preclinical and clinical trials. Additionally, different combinations have shown fewer adverse effects especially in selective HDACis.
Histone Deacetylases: An Overview
Histone deacetylases (HDACs) are a family of enzymes grouped into four classes in humans based on their homology to yeast HDACs analogs. The four classes are different in cellular localization, structure, mechanism of catalysis and expression patterns (Haberland et al., 2009) (Table 1). Class I HDACs include HDAC1, HDAC2, HDAC3, and HDAC8 enzymes located in the nucleus. Class II HDACs include HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10 enzymes, which are located both in the nucleus and cytoplasm. Class II HDACs comprises HDACs 4, 5, 6, 7, 9, and 10 and are subdivided into Class IIA (HDAC4, HDAC5, HDAC7, and HDAC9) and Class IIB (HDAC6 and HDAC10) (Haberland et al., 2009). Class III, also called Sirtuins, include seven members of the Sirtuin HDACs from Sirtuins 1 to 7 that are located in nucleus, cytoplasm and mitochondria. Class IV is represented by HDAC11 (Haberland et al., 2009). HDACs class I, II and IV require Zinc-dependent catalysis as cofactors for their enzymatic activity and HDACs class III or Sirtuins required nicotinamide adenine dinucleotide (NAD)-dependent (Park and Kim, 2020).

Table 1. HDACs family members.
The main physiological function of HDACs is to maintain the steady-state level of lysine acetylation level of histone and non-histone proteins. HDACs are considered as the erasers of the acetyl group, while the histone acetyltransferases (HATs) are the writers. Balanced acetylation and deacetylation levels are controlled by the opposite activity of HDACs and HATs: HDACs are capable of removing acetyl groups from histone tail and induce a chromatin compaction; HATs induce relaxed chromatin by transferring an acetyl group from acetyl CoA to form ε-N-acetyl lysine. Chromatin condensation and relaxation equilibrium occurs across the whole genome, although control of the expression of a small subset of genes can occur in specific areas of the genome when a particular HDAC and HAT balance is modified. HDACs play several important roles in aspects of cancer development including tumor cell proliferation, metastasis, angiogenesis, resistance to apoptosis and alteration of the cell cycle (Figure 1). However, further studies are required to identify the specific HDACs substrates associated with these functions.
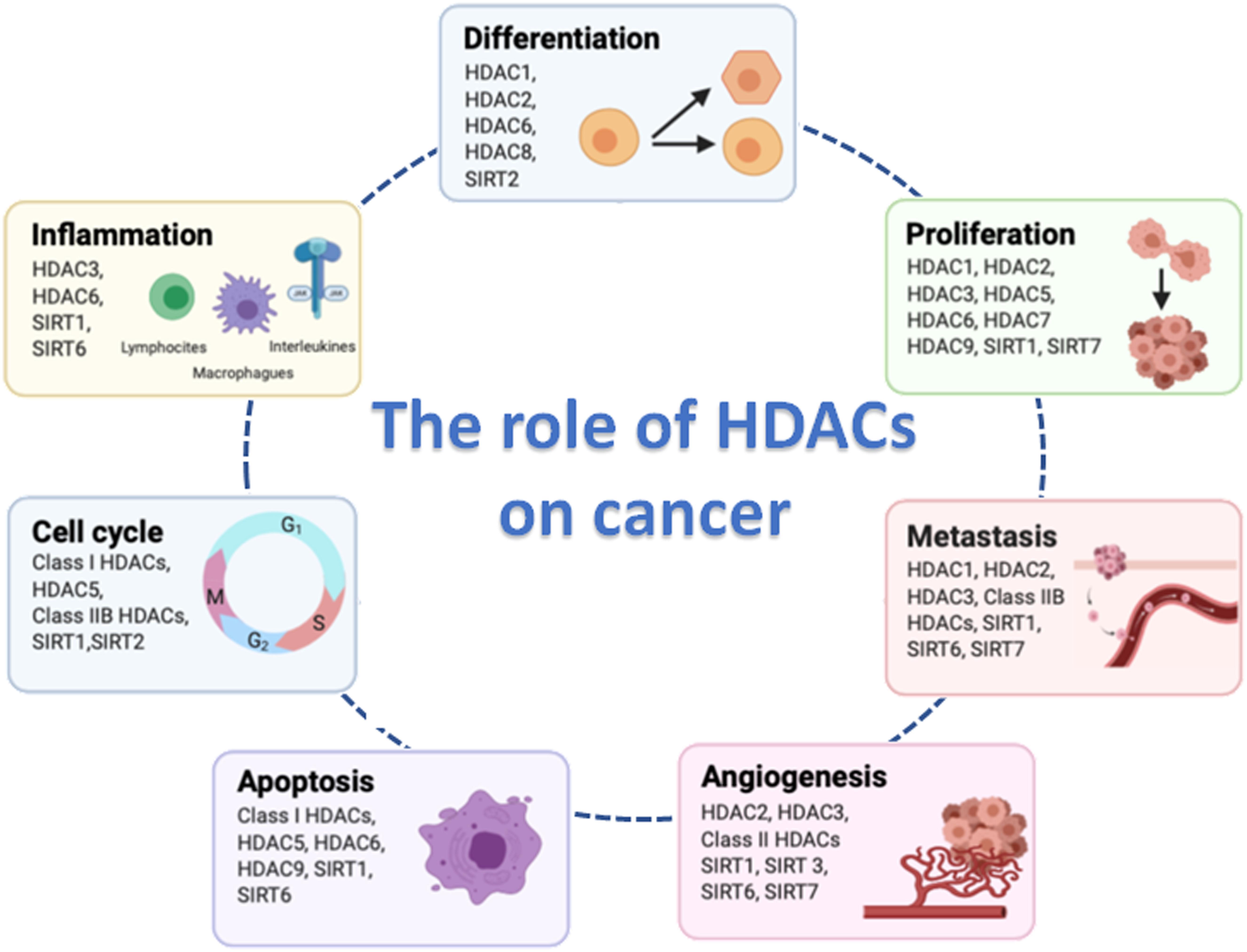
Figure 1. Hallmarks of cancer cell biology in which histone deacetylases (HDACs) are involved. Figure created with BioRender.com.
Histone deacetylases maintain steady-state acetylation levels, which allows them to play an important role in various physiological cellular functions, including differentiation, angiogenesis and metabolism (Kim et al., 2001; Fajas et al., 2002; Knutson et al., 2008). Consequently, HDACs imbalance will promote molecular changes that can influence health. Abnormal activity of HDACs has been implicated in different human diseases such as nervous system (McFarland et al., 2012), cardiovascular (Thal et al., 2012; Xu et al., 2013) and inflammatory diseases (Kim et al., 2012; Zhang et al., 2017b), and in cancer disease where their role has been explored extensively.
Alteration of HDACs activity has been associated with many types of solid tumors and hematological malignancies (Choi et al., 2001; Ashraf et al., 2006; Adams et al., 2010). The mechanisms by which HDACs contribute to cancer are diverse. In most tumors, aberrant expression of HDACs promotes oncogenic signaling by silencing tumor suppressor genes transcription or by the alteration of key target genes expression that regulate oncogenic pathways (Luo et al., 2000; Siddiqui et al., 2003; Cras et al., 2007; Godman et al., 2008; Feng et al., 2013), which are more often associated with poor outcomes in patients. For example, overexpressed of HDAC8 in neuroblastoma, cervical cancer and esophageal squamous cell carcinoma has been significantly correlated with poor prognosis in patients (Oehme et al., 2009; Vanaja et al., 2018). Many cancer cells lines, such as colon, prostate, ovarian and breast, strongly express HDAC11, which control viability and metabolic activity (Deubzer et al., 2013). Moreover, HDAC11 regulates cell cycle, apoptosis and survival in neuroblastoma cells lines (Thole et al., 2017). It has been also described that HDAC8 overexpression promotes proliferation in lung, colon, cervical cancer cells (Vannini et al., 2004) and in hepatocellular carcinoma (Wu et al., 2013). In human breast cancer cell lines overexpression of HDAC1, HDAC6, or HDAC8 contributes to increased invasion via increased metalloproteinase-9 (MMP-9) expression (Park et al., 2011). Furthermore, HDAC6 and HDAC8 was found to regulate cancer cell migration and invasion via α-tubulin acetylation (Wickstrom et al., 2010; Vanaja et al., 2018).
Several studies have reported a tumor suppressor role for some HDACs. For example, HDAC10 expression was found to suppresses cervical cancer cells metastasis through inhibition of matrix metalloproteinase 2 and 9 (Song et al., 2013). In contrast, low HDAC10 expression is associated with poor prognosis in lung and gastric cancer patients (Osada et al., 2004; Jin et al., 2014). Moreover, low expression of HDAC1 has been correlated with poor clinical outcomes such as metastasis and advanced stages (Chaiyawat et al., 2018) in osteosarcoma. Likewise, low HDAC3, HDAC7 and HDAC9 expressions has been associated with poor prognosis and survival in childhood acute lymphoblastic leukemia (ALL) (Moreno et al., 2010). In this study, it was shown that all the three HDACs are a promising therapeutic target for the treatment of refractory childhood ALL, although it is not clear whether an individual HDAC is more important over the others in the development of malignancy and in response to treatment.
Some studies have reported that the loss of expression and activity of certain HDACs is due somatic mutations associated with tumorigenic effects. For instance, Ropero et al. (2006), found somatic mutations of the HDAC2 gene in carcinomas with microsatellite instability caused a loss of HDAC2 protein expression which made the cells more resistance to HDACis. Additionally, Stark and Hayward described that homozygous deletions of HDAC4 in melanomas (Stark and Hayward, 2007) and Taylor et al. (2011) identified somatic mutations in HDAC1 in 8.3% of dedifferentiated liposarcoma.
The activity of HDACs often requires the formation of a complex among with different HDACs incorporated with Sin3, NuRD (nucleosome remodeling and deacetylation), CoREST (co-repressor for element-1- silencing transcription factor), SMRT (silencing mediator of retinoid and thyroid receptors) and NCoR (nuclear receptor co-repressor) co-repressor complexes (Haberland et al., 2009). Usually in cancer, HDACs are characterized by an aberrant recruitment to these complexes. Furthermore, these co-repressor complexes can be recruited by oncogenic fusion proteins to drive tumorigenesis. For instance, in acute myeloid leukemia PML-RARα, PLZF-RARα, or AML1-ETO oncogenic fusion proteins recruit and bind to HDAC complexes increasing co-repressor activity (Gelmetti et al., 1998; Grignani et al., 1998). In Ewing sarcoma, the oncogenic fusion EWSR1-FLI1 binds to NuRD complex containing HDAC2 and HDAC3 proteins which are considered a chromatin remodeling complex that leads to the repression of gene expression (Sankar et al., 2016). The EWSR1-FLI1 fusion which regulates gene repression was reverted by HDACis leading to inhibition of histone demethylase LSD1, another NuRD complex protein. The inhibition of the formation of this complex was found to affect numerous genes including well-known target genes like CAV1, NKX2.2 (Sankar et al., 2014) and subsequently enhance the anticancer efficacy.
HDACis Treatment to Enhance the Anticancer Efficacy
The development of HDACis has improved our understanding of the molecular events that sustain the biological function of HDACs. The increased sensitivity of cancer cells to HDACis is due to the overexpression of specific HDAC isoform or group of HDACs in cancer cells. Accordingly, the altered activity or expression of the HDACs render them more sensitive to the inhibition of HDAC and subsequent induction of growth arrest, differentiation inhibition and cell death (Kim and Bae, 2011), leaving normal tissue cells unaltered. This ability to modulate gene expression via changes in acetylation status in a reversible manner place HDACs as an attractive target for the treatment of numerous cancers. Knowing which HDAC is expressed in which cancer and at what defined histopathological stage is therefore essential.
Many types of HDACis have been developed, which can be divided into different groups according to their chemical structures. HDACis can be structurally grouped into at least five classes: hydroxamates, cyclic peptides, short chain fatty (aliphatic) acids, benzamides, and sirtuin inhibitors (Kim and Bae, 2011; Eckschlager et al., 2017) (Table 2). Hydroxamates compounds are the most widely explored HDACi in preclinical and clinical studies. These molecules have been developed with a distinct chemical structure consisting of a zinc chelating group, a spacer group and an enzyme binding group that confers specificity, efficiency, and the compound’s pharmacokinetic properties (Finnin et al., 1999; Mottamal et al., 2015). Depending on its ability to inhibit HDAC classes, HDACis are classified as pan-HDAC inhibitors (pan-HDACis) that act on all HDAC classes (not including sirtuins), and selective HDAC inhibitors, that target a specific HDAC (Ceccacci and Minucci, 2016).
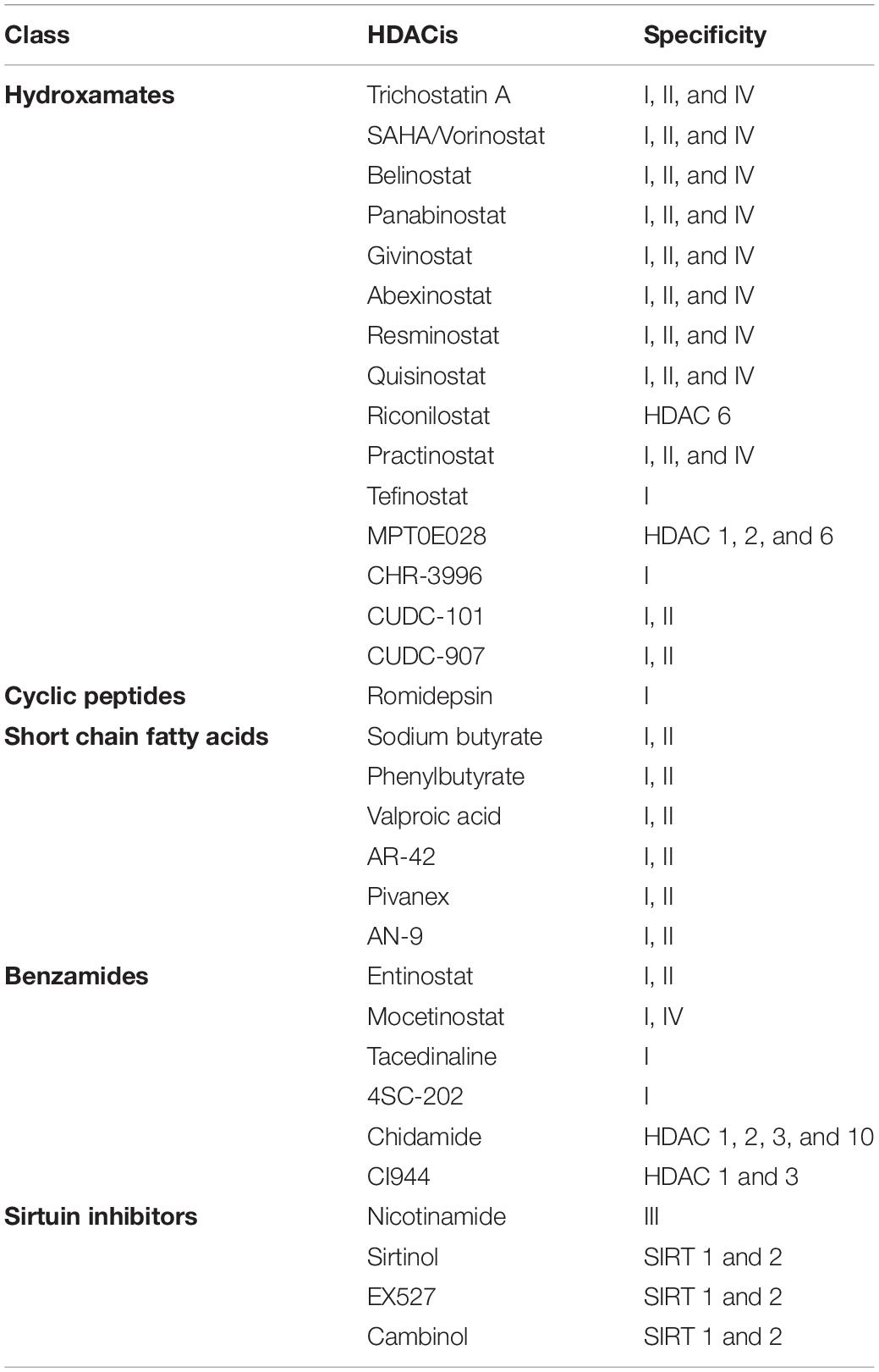
Table 2. Summary of chemical structures and selectivity profiles of HDAC inhibitors.
To date, pan-HDACis have been more widely studied and used rather than selective HDACis. pan-HDACi usage started in 2006 for the treatment of different types of cancers like cutaneous T-cell lymphoma (CTCL), peripheral T-cell lymphoma or multiple myeloma (Halsall and Turner, 2016). Suberoylanilide hydroxamic acid (SAHA; Vorinostat) and Trichostatin A were first generation of HDACis. In fact, SAHA was the first approved pan-HDACis by Food and Drug Administration (FDA) for the treatment of relapsed and refractory cutaneous T-cell lymphoma (CTCL) (Gryder et al., 2012; Eckschlager et al., 2017). Following the successful results with SAHA, many other HDACis have been approved for the clinical treatment in various hematological tumors such as romidepsin or belinostat. Unfortunately, hematological tumor cells have developed drug resistance to HDACis, which promoted the regeneration and maintenance of the malignant phenotype (Wang et al., 2020). The molecular basis for drug resistance by HDACis is still unclear, although drug efflux, chromatin alteration, upregulation of oxidative stress response mechanism, defects or upregulation in apoptotic pathways have been implicated (Wang et al., 2020). Although pan-HDACis are currently approved by FDA, only limited success was achieved when used as single agents against solid tumors in clinical trials compared to the hematological malignancies. In addition, pan-HDACis cause secondary effects such as thrombocytopenia, nausea, vomiting, anorexia, and fatigue (Munster et al., 2009).
Whilst the use of pan-HDACis is relatively successful in clinical applications, the combining of HDACis with other anti-tumor drugs is now considered a major breakthrough in the treatment of both hematological and solid tumors. Recent evidence shows that combinations of HDACis with other antitumor agents increase therapeutic efficacy and decrease toxicity (Munster et al., 2009). Moreover, many studies have described their synergistic effect with different types of drugs such as topoisomerase inhibitors, PARP inhibitors, proteasome inhibitors, radiotherapy, antimetabolites, mTOR inhibitors or monoclonal antibodies amongst others. This has enabled HDACis combinational therapy to be considered as a new therapeutic strategy against solid cancers and/or drug-resistant cancers.
Combining HDACis with other anti-tumor agents may thus be a strategy to achieve their high therapeutic potential. However, side effects and toxicities from pan-HDACis still exist, which is hindering their progress in the clinic. Hence, current research efforts are focused on developing selective HDACis to reduce toxicity and thereby to overcome any adverse consequences caused by off-target effects. Although there are currently few clinical trials with selective HDACis, the results to date have been positive.
Pan-HDACis in Combination
The use of pan-HDACis in cancer has increased considerably in the last few years. A large number of HDACis have been synthetized and tested as antitumor agents in preclinical research or in clinical trials. The following sections describe the studies performed with HDACis, focusing mainly on those synergistic combinations that have improved cancer treatment in preclinical and clinical trials over the last 5 years (Table 3).
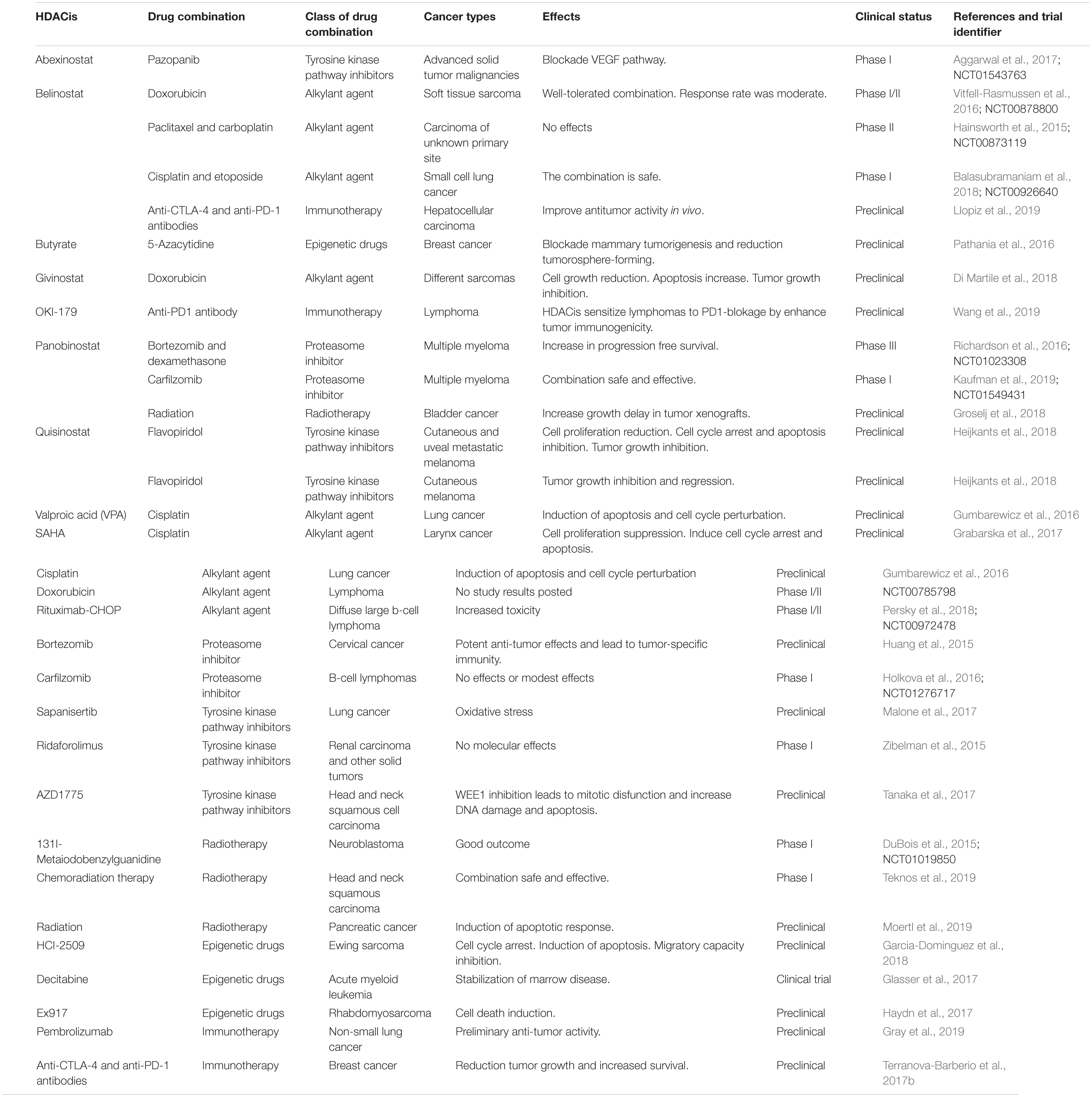
Table 3. Summary of studies evaluating pan-HDAC inhibitors in combination with other antitumor drugs.
Pan-HDACis in Combination With Alkylating Agents
Alkylating agents are used in standard cancer treatments that result in elevated toxicity to normal tissues due to the high used doses. For this reason, several preclinical studies have been conducted over the past 5 years in which these anticancer agents have used lower doses combined with HDACis to reduce toxicity. Currently, preclinical studies have shown greater efficacy when cells were pretreated with HDACis prior to exposure to DNA damaging agents (Suraweera et al., 2018). Based on this treatment strategy, combination therapies with HDACis and alkylating agents as topoisomerase II inhibitors have led to higher nuclear topoisomerase II inhibition accumulation, increased DNA damage, growth inhibition and cell death (Suraweera et al., 2018). In general, the molecular mechanism of HDACis sensitizing cancer cell to DNA damaging agents includes both a mechanistic action by inducing chromatin relaxation and increased accessibility to the exposed DNA and increased binding of transcription factor to reactivate the transcription of proapoptotic genes.
Pan-HDACis reduce tumor growth by inhibiting cancer cell proliferation. For instance, SAHA, combined with cisplatin, was considered as a potential treatment for larynx cancer cells due to the synergistic effect on cell proliferation inhibition (Grabarska et al., 2017). Also, SAHA and TSA was found to synergize with cisplatin in different tumor types, including cholangiocarcinoma cells, inducing cell growth inhibition (Asgar et al., 2016). Furthermore, the effect of cisplatin combined with SAHA or VPA on different cell lines of lung cancer results in synergistic response (Gumbarewicz et al., 2016). For example, SAHA was shown to suppressed cell growth of small-cell lung cancer (SCLC) by causing cell cycle arrest, and, in combination with cisplatin, significantly reduce the expression of Notch1. This is likely beneficial for this particular cancer type where oncogenic Notch1 signaling clearly plays an important role (Wawruszak et al., 2019).
More recently, a specific class I and II HDACi ITF2357 (Givinostat) was shown to induce mitochondrial apoptosis by increasing pro-apoptotic BH3 proteins and a caspases-dependent mechanism (Di Martile et al., 2018). Givinostat combined with doxorubicin (a dual topoisomerase inhibitor I and II and DNA damage agent) has shown significant potential in improving sensitization in different preclinical models of sarcoma such as osteosarcoma, liposarcoma, synovial sarcoma, rhabdomyosarcoma, leiomyosarcoma, or fibrosarcoma (Di Martile et al., 2018).
Histone deacetylases inhibitors can act as potentiators of the cytotoxicity generated by anthracycline-type topoisomerase (topo) II inhibitors (Munster et al., 2009). Following successful preclinical studies, a number of trials have been conducted in patients. For instance, promising results were obtained by combining SAHA and doxorubicin in patients with relapsed or refractory multiple myeloma (NCT00744354) or advanced/refractory lymphoma (NCT00785798) (Suraweera et al., 2018). Another alkylating agent used in solid cancers (e.g., small cell lung cancer) is etoposide; both etoposide or cisplatin have been used in combination with belinostat (PXD101), a hydroxamic acid- type HDACi approved by the FDA, to enhance their efficacy in clinical trial phase I (NCT00926640) (Balasubramaniam et al., 2018). Belinostat is well-tolerated and its combination with conventional cancer therapies has identified no further bone marrow toxicity. This has enabled the use of belinostat in several Phase I/II clinical trials in both hematological cancers and solid tumors such as ovarian cancer, multiple myeloma, adult acute myeloid leukemia and bladder cancer.
Some clinical studies have not had the same outcome. For example, a phase I/II study in patients with advanced stage diffuse large B-cell lymphoma (NCT00972478), in which SAHA was combined with Rituximab-CHOP (treatment based in vincristine and prednisone, a DNA damage inductor and a DNA synthesis inhibitor respectively) showed increased toxicity, particularly neutropenia and sepsis (Persky et al., 2018). A phase II study in patients with carcinoma of unknown primary origin (NCT00873119) showed that the addition of belinostat to paclitaxel/carboplatin did not improve the progression-free survival (PFS) (Hainsworth et al., 2015). Furthermore, a phase I/II trial using a combination of belinostat and doxorubicin in soft tissue sarcoma (NCT00878800) did not provide clear evidence of synergy (Vitfell-Rasmussen et al., 2016). These contrary results might be explained by mechanisms of anticancer effects of HDACis not being identical, and it is also possible that in combinational therapy one drug may interfere or modulate the mode of action of the other (Santoro et al., 2016). Although HDACis in combination with DNA damaging agents have shown promising results in certain types of cancer, only very limited positive outcomes have been identified in others. This depends on the cancer type and the HDACi-drug combinatorial regimes. The understanding of the combination mechanism using relevant preclinical models should improve the selection of the most appropriate combination for specific type of cancer. Also, understanding the mechanistic reasons for unexpected trial results is necessary in order to inform improved rationales for combining HDACis with cytotoxic drugs.
Pan-HDACis in Combination With Proteasome Inhibitors
The high proliferation of cancer cells requires an elevated protein synthesis rate; this makes them highly dependent on the ubiquitin-proteasome system and, therefore, more susceptible to proteasome inhibitors (Goldberg, 2007). Following this reasoning, preclinical and clinical studies have been performed. In fact, the combined treatment SAHA and a proteasome inhibitor, bortezomib, enhances the antitumor effects and immune properties of cervical cancer cells, both in vitro and in vivo (Huang et al., 2015). Furthermore, synergistic effect was observed when bortezomib and dexamethasone were combined with panobinostat in a Phase III (NCT01023308) trial for multiple myeloma patients. This combination worked due to the panobinostat action on different epigenetic and protein metabolism pathways reducing the resistance of tumors in these patients to the treatment of proteasome inhibitors (Richardson et al., 2016). These results paved the way for other studies such as a phase I study (NCT01549431) in patients with relapsed or refractory multiple myeloma (RRMM) which has responded positively to the panobinostat as pan-HDACi that exerts activity on class I, II and IV HDACs combined with carfilzomib, a proteasome inhibitor (Kaufman et al., 2019). In contrast, a modest response was obtained in phase I study (NCT01276717) conducted with a combination of proteasome inhibitor carfilzomib and vorinostat in patients with relapsed or refractory B-cell lymphomas (Holkova et al., 2016). While, carfilzomib with Panobinostat combination is safe and effective in RRMM patients, future trials should explore the molecular mechanism of this combination with different doses in order to optimize treatment tolerability and enhance efficacy.
Pan-HDACis in Combination With Tyrosine Kinase Pathway Inhibitors
Tyrosine kinases are involved in multiple signaling pathways regulating a multitude of biological cell functions. Deregulation of tyrosine kinases are closely associated with cancer development and progression, driving the development of many tyrosine kinase inhibitors.
The multityrosine kinase inhibitor of vascular endothelial growth factor receptor (VEGFR), and other growth factor receptors drugs, have shown significant clinical efficacy in multiple tumor types, although different cancers will readily develop resistance against it. To reduce this resistance, a study in phase I focused on patients with advanced solid tumor malignancies, and used a combination of pazopanib with pan-HDACi abexinostat (PCI24781) (NCT01543763) which resulted in a synergistic effect (Aggarwal et al., 2017). Several mechanisms can be associated with this positive effect, although abexinostat inducing the downregulation of HIF-1α protein expression is one of the more plausible mechanisms that can directly regulate VEGF expression. In this clinical trial is was not clear which HDAC or class of HDACs are involved in regulating VEGF-driven expression in tumors (Aggarwal et al., 2017).
Preclinical studies also reported a synergistic effect between HDACis and tyrosine kinase inhibitors. Both mTOR and its pathway are relevant in cancer studies. It has been shown that mTOR inhibitors exert only cytostatic effects in some NF1-associated malignancies (Malone et al., 2017). The Vorinostat/Sapanisertib combination was identified as a promising drug combination that kills NF1-mutant nervous system malignancies as well as NF1- and KRAS-mutant lung cancers. This combination triggered irresolvable oxidative stress in NF1-mutant malignancies in vitro and in vivo, but did not kill normal cells and was not toxic to mice in vivo (Malone et al., 2017). Moreover, another inhibitor of mTOR pathway, ridaforolimus, has been used combined with vorinostat in a phase I study of advanced renal cell carcinoma and other solid tumors (Zibelman et al., 2015). The combination was tolerated by patients in phase II and the possible mechanism shown to involve the downregulation of various cell cycle regulators such as pAkt and HIF-1α expression. Furthermore, tyrosine kinases such as WEE1 and CDKs are implicated in many signaling pathways. WEE1 is a kinase that has been linked to G2/M arrest and drugs cytotoxic action (Mackintosh et al., 2013). A preclinical study showed a synergistic effect between vorinostat and AZD1775 (WEE1 inhibitor) in vitro and in vivo in head and neck squamous tumor cells expressing high-risk mutant p53 (Tanaka et al., 2017). The combined effect of quisinostat (pan-HDACi) and flavopiridol (CDKi) was a promising therapeutic strategy for both cutaneous and uveal metastatic melanoma (Heijkants et al., 2018). In vitro results showed a synergistic reduction of cell proliferation and a cell death increase in uveal melanoma cell lines. Moreover, quisinostat and flavopiridol effectively reduced tumor growth in vivo, in a patient-derived xenograft model of cutaneous melanoma (Heijkants et al., 2018). This study suggested further investigation of the quisinostat and flavopiridol combination in the clinic was warranted since melanoma patients generally have limited therapeutic options and both drugs are already in clinical trials.
Pan-HDACis in Combination With Radiotherapy
Radiotherapy remains one of the most common treatments for cancer. The therapy causes double-stranded DNA breaks that induce cell death. The capacity of tumor cells to repair radiation-induced DNA damage can be decreased by HDACis, by affecting DNA damage signaling and repair pathways (NHEJ and HR) (Groselj et al., 2013). Therefore, combining HDACis and radiotherapy has gained traction in cancer clinical trials. A study in phase I in patients with relapsed or refractory neuroblastoma showed that vorinostat combined with a radiosensitizer such as 131I-Metaiodobenzylguanidine (131I-MIBG) was well-tolerated by patients (NCT01019850) (DuBois et al., 2015). Similarly, vorinostat in combination with chemoradiation therapy in phase I trial of neck squamous cell carcinoma patients was found to be safe and highly effective (Teknos et al., 2019). Preclinical studies using SAHA combined with radiation have demonstrated an increase in tumor radiosensitivity in vitro and in vivo and a subsequent induction of apoptotic response in, for example, pancreatic adenocarcinoma cancer cell lines (Moertl et al., 2019). Another pan-HDACi, panobinostat were tested in a bladder cancer xenograft model and shown to be an efficient tumor radiosensitizer with no increased toxicity (Groselj et al., 2018). In this preclinical study, HDACi induced radiosensitization effects were associated with the DNA repair MRE11-RAD50-NBS1 (MRN) complex, which is known to be regulated primarily by HDAC class I enzymes.
Pan-HDACis in Combination With Other Epigenetic Drugs
Epigenetic mechanisms are somewhat interlinked and inter-dependent. For example, HDACs and LSD1/KDM1A play important roles in regulating this interaction and, as a consequence, have emerged as promising therapeutic targets (Ning et al., 2016). Here, a combination of different epigenetic drugs may enhance efficacy. For example, the combination of a LSD1 inhibitor (HCI-2509) and SAHA exhibited a synergistic inhibition on tumor growth and enhanced apoptosis in Ewing sarcoma cell lines and in patient-derived xenograft mouse models (Garcia-Dominguez et al., 2018). Similarly, another combination of Ex917 (LSD1 inhibitor) and SAHA reported a synergistic induction on cell death in rhabdomyosarcoma cells (Haydn et al., 2017).
An alternative approach under examination is the use of DNA methyltransferase inhibitors (DNMTis) and HDACis combinations. Both DNMTs and HDACs silence gene expression, so their inhibition can be used to enhance tumor suppressor gene expression in various malignancies. Combination of 5-azacytidine (DNMTi) and butyrate (HDACi) efficiently blocked mammary tumorigenesis and reduced the tumorosphere-forming potential of tumor-propagating cells in vitro by reactivating the relevant tumor suppressor genes (Pathania et al., 2016). In addition, a very small scale clinical trial with two patients for relapsed or elderly secondary myelodysplastic syndrome and acute myeloid leukemia, investigated treatment with decitabine (DNMTi) and vorinostat (HDACi). This combination therapy achieved stabilization of marrow disease, outpatient palliation, and family-reported reasonable quality of life (Glasser et al., 2017). A combination of different HDACis/DNMTis could be beneficial in high-risk multiple myeloma patients. The authors developed a score for the prediction of primary multiple myeloma cell sensitivity to HDACi/DNMTi (TSA/decitabine and vorinostat/5-azacitidine) (Bruyer et al., 2018), which could be used in follow-up studies.
Pan-HDACis in Combination With Immunotherapy
In the last few years, immune checkpoint inhibitors (ICIs) such as anti–programmed cell death protein 1 (anti-PD-1) and anti-programmed cell death ligand 1 (anti-PD-L1) have been developed. Due to the relevance of HDAC/HAT pathways in regulating the immune system, HDACis have been considered as immunomodulatory agents (Banik et al., 2019), thus they have been combined with ICIs. Pembrolizumab (an anti-PD1 inhibitor) with vorinostat was well-tolerated in advanced/metastatic non-small cell lung cancer and anti-tumor activity was demonstrated (Gray et al., 2019). Another combination strategy is the use of antibodies, anti-PD-1 or anti-CTLA-4. The novel HDACi, OKI-179 which inhibits class I, IIb and IV HDACs, sensitized resistant lymphoma cells to anti-PD1 in vitro (Wang et al., 2019). The results from combined OKI-179/anti-PD1 antibody treatment in lymphomas showed a synergistic effect compared with more limited effects in monotherapy (Wang et al., 2019). Moreover, the results obtained previously with anti-CTLA-4 antibody use in patients with advanced hepatocellular carcinoma (Duffy et al., 2017) encouraged the use of a combination of anti-CTLA-4 and anti-PD-1 antibodies with the pan-HDACi, belinostat (Llopiz et al., 2019). These combinations were shown to potentiate the already known efficacy of these antibodies by reducing tumor volume and improving their immune functions in a murine hepatocellular carcinoma model (Llopiz et al., 2019). A similar study used a triple combination with vorinostat/anti-CTLA-4/anti-PD-1 in the triple-negative 4T1 breast cancer mouse mode (Terranova-Barberio et al., 2017b). The synergistic interaction of the three drugs resulted in a significant increase in survival and anti-tumor activity in established mouse breast cancer allografts (Terranova-Barberio et al., 2017b).
Selective HDACi in Combination
The intention behind the relatively recent development of specific-HDACis is to improve efficiency and broaden the therapeutic window whilst reducing the adverse effects such as thrombopenia, neutropenia, diarrhea, nausea, vomiting, and fatigue that are associated with pan-HDAC inhibition (Munster et al., 2009; Parbin et al., 2014). This section describes the studies that have used selective HDACis over the last 5 years in both preclinical and clinical trials (Table 4).
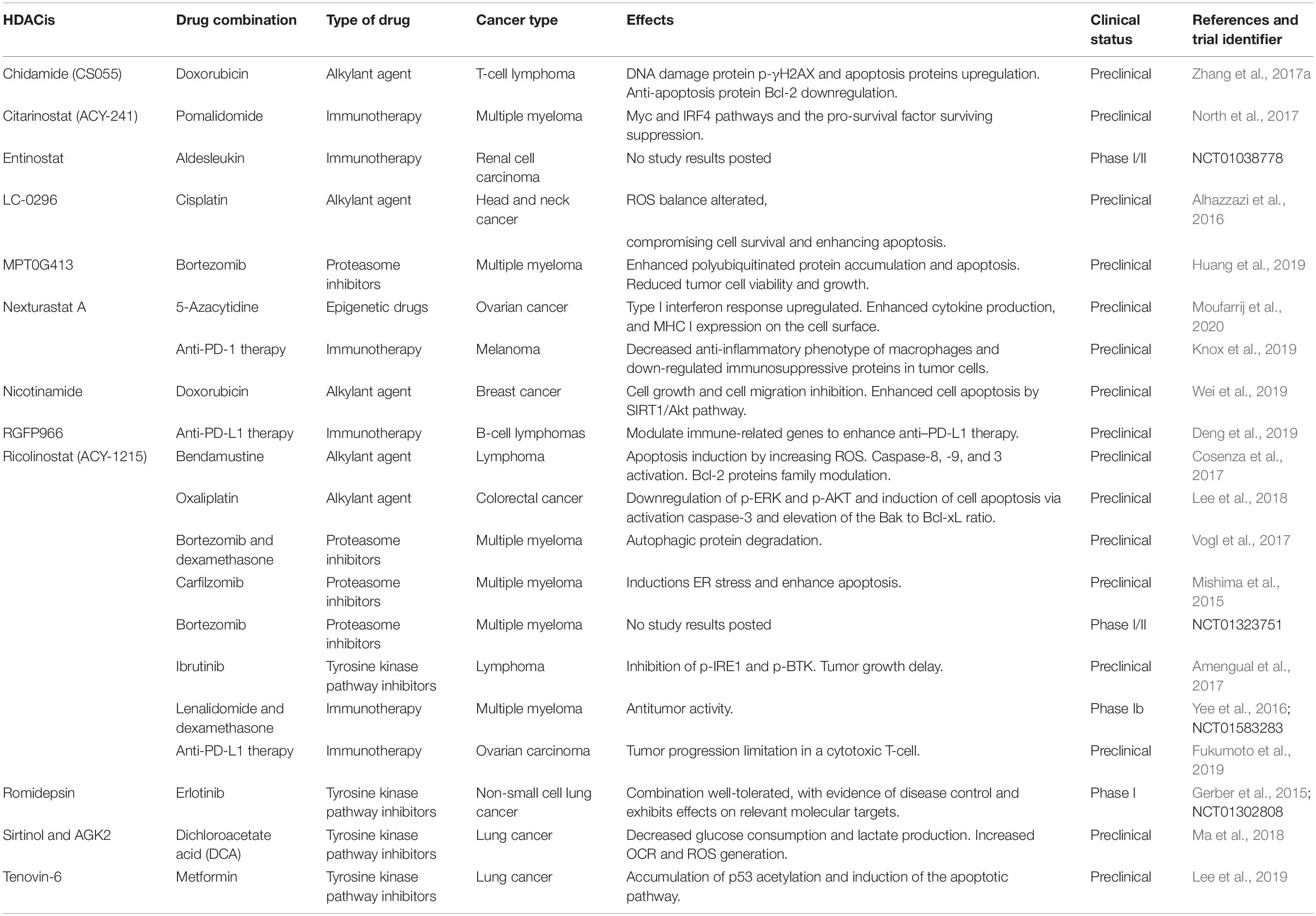
Table 4. Summary of studies evaluating selective HDACs inhibitors in combination with other antitumor drugs.
Selective HDACis in Combination With Alkylating Agents
Combinations of selective HDACis with different alkylating agents have shown synergistic antitumor effects. A selective HDAC6 inhibitor, ricolinostat (ACY-1215) has been combined in different tumors in preclinical studies. Ricolinostat and Bendamustine combination promoted a higher apoptosis induction than when drugs were used as single agents in lymphoma cell lines (Cosenza et al., 2017). The same effects were observed in colorectal cell lines combining ricolinostat with oxaliplatin (Lee et al., 2018). A novel selective HDACi, CS055 (Chidamide) was combined with doxorubicin in peripheral T-cell lymphoma cell lines (PTCL) in a preclinical study that identified a synergistic antitumor effect when this combination was used to treat PTCL cell lines (Zhang et al., 2017a).
Nicotinamide and LC-0296 are selective class III HDAC inhibitors or Sirtuins. The combination of nicotinamide (SIRT1 inhibitor) and doxorubicin increased the inhibition of cell proliferation and apoptosis and reduced resistance to treatment in breast cancer cells (Wei et al., 2019). A novel Sirtuin-3 inhibitor, LC-0296, was developed and its effect studied in head and neck cancer cells. This preclinical study showed how this HDACi inhibited cell survival and proliferation and promoted apoptosis both in monotherapy and in combination with cisplatin despite the resistance of this cancer cells to the alkylating agent described (Alhazzazi et al., 2016). Furthermore, the selective Sirt1 inhibitor the indole EX-527 shows a significant effect in distinct types of cancer, as monotherapy or combined to cancer drugs. However, the efficacy of this Sirt1 inhibitor requires further investigation and subsequent review (Rifai et al., 2018).
Selective HDACis in Combination With Proteasome Inhibitors
Preclinical studies in multiple myeloma conducted prior to the last 5 years (Hideshima et al., 2005) have encouraged exploration of different combinations based on selective HDACis and proteasome inhibitors.
Ricolinostat was combined with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. The selective inhibition of HDAC6 combined with this proteasome inhibitor triggered dual blockade of aggresomal and proteasomal degradation of protein, and therefore synergistic multiple myeloma cell death (Vogl et al., 2017). Similar results were obtained with the same combination in vitro and in vivo, which caused a significant delay in tumor growth and prolonged overall survival in a xenograft mice model (Amengual et al., 2015). Ricolinostat had also been combined with carfilzomib promoting synergistic anti−multiple myeloma effects, even in bortezomib−resistant cells. A decrease in tumor volume associated with apoptosis in an in vivo mouse xenograft was also demonstrated (Mishima et al., 2015). In addition, a novel selective-HDAC6 inhibitor, MPT0G413, was evaluated, in vitro and in vivo, in combination with bortezomib. This showed a synergistic inhibition of multiple myeloma tumor cell viability (Huang et al., 2019). All available preclinical studies have supported clinical trials in multiple myeloma patients with the ACY-1215 and bortezomib combination (NCT01323751).
Selective HDACis in Combination With Tyrosine Kinase Pathway Inhibitors
Sirtinol and AGK2 are two selective inhibitors of SIRT2. These inhibitors were combined with dichloroacetate acid (DCA), a pyruvate dehydrogenase kinase inhibitor in a preclinical study in lung cancer cells (Ma et al., 2018). Both combinations showed a synergistic effect in proliferation inhibition and apoptosis induction in vitro, and a reduction of tumor volume in mice (Ma et al., 2018). In addition, the combination Sirtinol/AGK2/DCA enhanced synergistically all the effects describe previously in vitro and in vivo (Ma et al., 2018).
Tenovin-6, an inhibitor of both SIRT1 and SIRT2, in combination with metformin, an antidiabetic drug and mTOR signaling pathway inhibitor, caused cell growth inhibition and induced apoptosis in lung cancer cells (Lee et al., 2019). Additionally, a preclinical study identified the synergistic interaction of ricolinostat and ibrutinib the first-in-class BTK (Bruton’s tyrosin kinase) inhibitor in a large panel of lymphoma cell lines and in a xenograft model of lymphoma. This combination led to a marked tumor growth delay and prolonged overall survival (Amengual et al., 2017).
A Phase I study in patients with non-small cell lung cancer studied the combination of an EGFR inhibitor, erlotinib, and a selective HDACi, romidepsin (NCT01302808). This combination was based on the involvement of an HDACi to counter erlotinib resistance, although the addition of romidepsin did not appear to alter the frequency and severity of characteristic erlotinib toxicities, such as rash and diarrhea (Gerber et al., 2015).
Selective HDACis in Combination With Other Epigenetic Drugs
Combining nexturastat A (selective HDAC6 inhibitor) with 5-azacytidine (DNMTi) has been described recently as a novel approach for ovarian cancer. Results showed a significant increase of the type I interferon response and cytokine expression in 5/6 ovarian cancer cell lines in vitro compared to each individual treatment. Moreover, a synergistic decrease of PD-L1 protein in ovarian cells was found, which suggested that nexturastat A/5-azacytidine increased tumor immunity. However, this combination only showed a synergistic decrease in tumor burden at week 7 in vivo (Moufarrij et al., 2020).
Selective HDACis in Combination With Immunotherapy
RGFP966, a novel selective HDAC3 inhibitor, enhanced the efficacy of anti-PD-L1 therapy in the treatment of B-cell lymphomas. In fact, tumor regression in syngeneic murine B-cell lymphoma model was observed in the combinatorial therapy (Deng et al., 2019). In a similar way, a HDAC6 inhibitor (riconilostat) blocked the immune checkpoints when it was combined with anti-PD-L1 therapy in ovarian carcinoma cell lines and in in vivo models (Fukumoto et al., 2019).
HDAC6 inhibitors are frequently used in preclinical studies in cancer because of their immunomodulatory properties. Ricolinostat potentiated the effects of lenalidomide (immunomodulatory drug) and dexamethasone in a phase Ib in patients with multiple myeloma (NCT01583283) (Yee et al., 2016). ACY-241 (citarinostat) suppressed proliferation and viability of tumor cells derived from multiple myeloma in combination with pomalidomide. This combination also improved cytotoxicity by decreasing apoptosis and cell cycle arrest (North et al., 2017). A recent study showed that nexturastat A in combination with anti-PD-1 antibodies significantly decreased tumor growth in vivo compared to each agent alone when treating melanoma cells (Knox et al., 2019).
A phase I/II clinical trial (NCT01038778) has studied the combination of entinostat (a selective HDAC1 and HDAC3 inhibitor) and aldesleukin (interleukin 2) in renal cell carcinoma patients. The results showed a greater median progression-free survival without increased toxicity.
HDACis and Multitherapy
Although most of the combinations described evaluation of two drugs, multitherapy evaluate three or more different compounds. This approach is based on the fact that tumors are heterogeneous in their mutational status and involve multiple pathways in their oncogenesis and progression. Also, three-way or more therapeutic combinations may reduce the possibility of resistance, which so often limits single drug usage, and improve treatment efficacy. SAHA was part of a combination with two antimetabolites, cladribine and Gemcitabine and an alkylating agent, busulfan. This preclinical study in lymphoma cell lines demonstrated that any increase in cytotoxicity could be attributed to stable chromatin relaxation mediated by the antimetabolites and SAHA, thereby increasing the susceptibility of genomic DNA to busulfan alkylation (Ji et al., 2016). SAHA was also used in a multitherapy approach in clinical trials. The results of SAHA in combination with radiotherapy led to further trials by adding an alkylating agent to the combination. This strategy was used in the temozolomide phase I/II studies in patients with glioblastoma (NCT00731731) (Galanis et al., 2018) or included antimetabolites such as capecitabine in patients with non-metastatic pancreatic cancer (NCT00983268) (Chan et al., 2016). Like SAHA, other HDACis have been used: belinostat has also been combined with radiation therapy and temozolomide to treat glioblastoma (NCT02137759) (Gurbani et al., 2019); and valproic acid (HDACi) combined with capecitabine and radiotherapy in colorectal cancer where a synergistic antitumor interaction in vitro was shown (Terranova-Barberio et al., 2017a).
The development of a drug that acts on multiple targets is a new therapeutic approach that currently is being evaluated with positive results. Romidepsin and its analogs, FK-A5 and FK-A11, have showed a dual inhibition on HDACs and PI3K resulting in stronger cytotoxic effects in a panel of human cancer cell lines (Saijo et al., 2015). CUDC-101 is a strong inhibitor against HDACs, EGFR and HER2. A Phase I Study combined CUDC-101 with chemoradiation in head and neck squamous cell carcinoma patients at biologically efficacious doses was tolerated (Galloway et al., 2015). Other studies demonstrated the antitumor activity of CUDC-101 in EGFR-overexpressing glioblastoma, anaplastic thyroid cancer (Zhang et al., 2015; Liffers et al., 2016) and in pancreatic cancer when CUDC-101 was combined with gemcitabine (Ji et al., 2018).
Conclusion
The overexpression of HDACs in hematological and solid tumors places HDACis as a promising strategy for the treatment of cancer patients. The early success of HDACis in the treatment of cancer were due to the use of pan-HDACis in hematologic malignancies. The effectiveness of pan-HDACis for cancer treatment relies on its broad-spectrum inhibition of various HDACs. This has triggered numerous studies which have investigated the antitumor effects of these epigenetic drugs. However, toxicities and unintended effects were also observed. All of which has contributed to the development of new strategies.
The use of pan-HDACis in combination with other antitumor agents was the strategy pursued first. The diverse combinations of pan-HDACis with proteasome inhibitors, tyrosine kinase inhibitors, alkylating agents, radiotherapy, immunomodulators have revealed synergistic antitumor effects in many types of cancers both in the preclinical and clinical settings. However, such combinations often trigger various side effects such as fatigue, thrombocytopenia and gastrointestinal issues. Hence, selective HDACis have been developed with higher selectivity and specificity. The difference in increased expression and activity of specific HDAC isoform in tumors but not in normal tissues, have led to the hypothesis that selective HDACis may possess a better therapeutic index, fewer adverse effects, and better patient outcomes with cancer treatment. The current results using selective HDACis in combination with antitumor treatment have had positive results in preclinical and in early clinical studies. In addition, it has been shown that selective HDACis not only reduce the toxicity but also replicate the same effects obtained by those pan-HDACis that target the specific HDAC of selective HDACis (Figure 2).
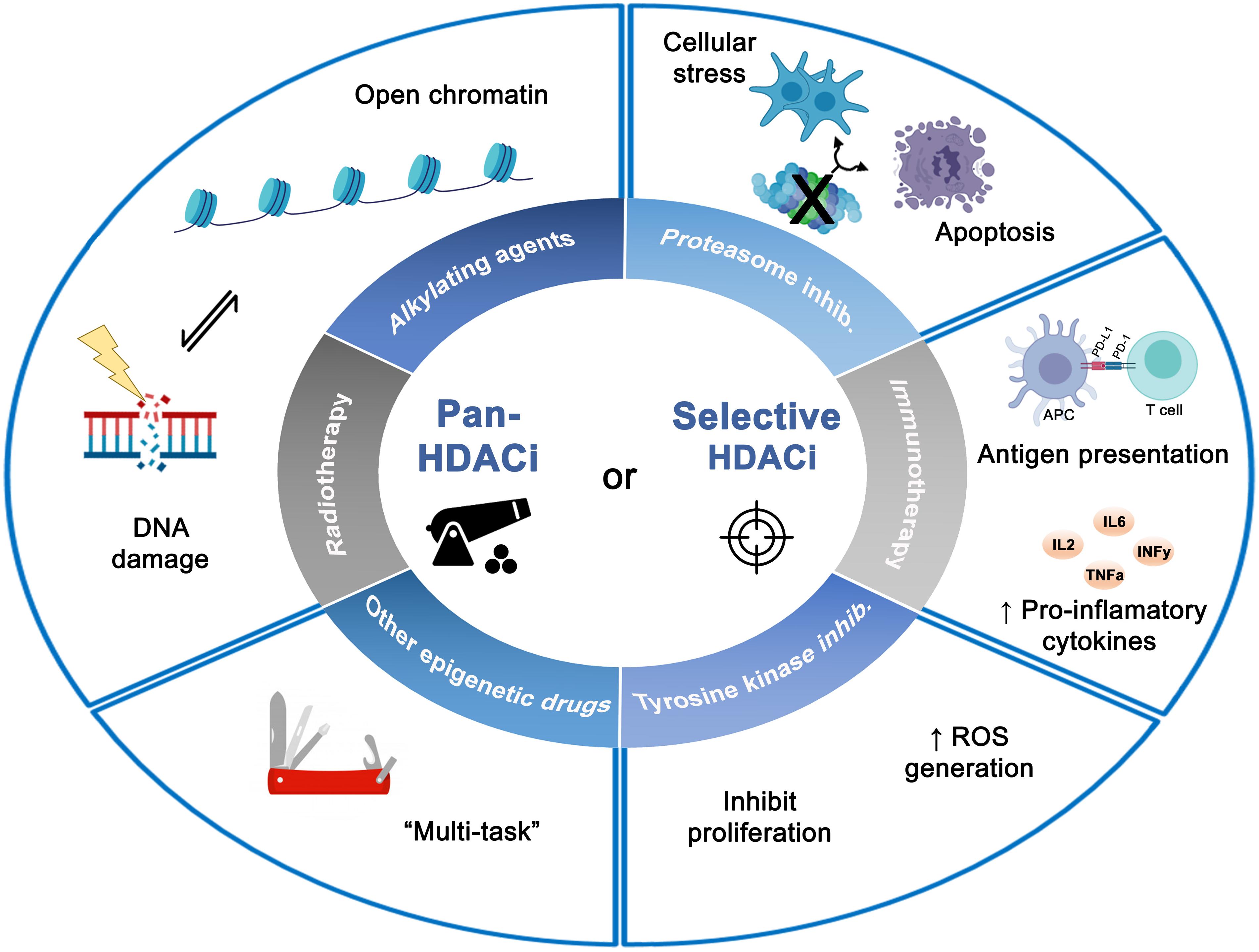
Figure 2. Effects of combined histone deacetylases inhibitors (pan- or selective HDACis) with different antitumor therapies on tumor cells. The diverse combinations with HDACis showed synergistic antitumor effects leading to a wide spectrum of biologic effects such as DNA damage, apoptosis induction, inhibition of proliferation and cellular stress. Figure created with BioRender.com.
Apart from the advantages of combined HDACis improving cancer treatment, these strategies have increased our knowledge about HDACs mechanisms and their inhibitory effect on tumors. This is particularly important in developing rationales for selecting HDACis for combination therapy.
Although preclinical studies showed that the combination of HDACis with anticancer agents might prove more effective than current therapies, the results obtained in clinical trials have not always been completely successful. This might be explained because many studies are not based on a deep understanding of the molecular events that underpin the synergistic effect of the combinations or are at present ill-defined. It also remains possible that one chemotherapy drug may interfere, or modulate the mode of action of the other (Santoro et al., 2016). A further possibility is that most preclinical research does not test different doses to select the best for use in combinations. This is essential if we are to increase the efficacy in clinical trials and reduce side effects. Importantly, we need to endeavor to understand the mechanistic reasons for reduced drug efficacy in combination trials, as investigating unexpected results will help inform the development improved rationales for selecting HDACis/cytotoxic drug combinations. Improving novel drug combinations with existing therapies need to be based on a thorough understanding of the molecular mechanism involve in cancer cell killing. Also, the study of pharmacodynamics biomarkers as indicators of drugs effects will be of significant use in evaluating the link between drugs regimen, target effects, and biological tumor response. Informative biomarkers means better evaluation of efficacy through improved treatment monitoring. Unfortunately, biomarkers remain a largely unmet clinical need and for those that are potentially available, our knowledge of their pharmacodynamics is limited.
HDACi as single agents have a limited utility in clinical and its combination with anticancer agent’s triggers adverse effects related to toxicity, safety, and efficacy. Accordingly, the use of single HDACis with multiple targets is another attractive and new alternative against solid and drug-resistant tumors, which have been gaining increasing attention. In fact, HDACis are being modified and equipped with additional biochemical activities. This might imply a potent antitumor activity with a better toxicological profile. However, it is a challenge to develop and design these multi-target HDACis.
Epigenetic therapy appears to be a promising and beneficial strategy given the successful results obtained in the treatment of several tumors with HDACis (especially in combination) and with multi-target HDACis. These attractive strategies could be useful for treating those tumors with a low rate of somatic mutations in which epigenetic plays a more central role in oncogenesis and tumor progression (e.g., rhabdoid tumor, Ewing sarcoma, or acute myeloid).
Further studies are needed to understand the mechanisms of action of HDACis in order to increase the efficacy in clinical trials and to reduced side effects. Further intensive investigations are required to provide a firm basis for the successful use of HDACis as anti-cancer agents in the clinic.
Author Contributions
LHP and DGD conceived and wrote the manuscript, and contributed to the figure and table design. RFC wrote the manuscript and contributed to the figure and table design. AS, EDÁ, and NH wrote the manuscript. All authors revised and approved the final version for publication.
Funding
This work was supported by Instituto de Salud Carlos III_FEDER (PI17/00464 and CB16/12/00361), by the Consejería de Salud, Junta de Andalucía (PI-0013-2018), and by Fundación María García Estrada.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, H., Fritzsche, F. R., Dirnhofer, S., Kristiansen, G., and Tzankov, A. (2010). Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin. Ther. Targets 14, 577–584. doi: 10.1517/14728221003796609
Aggarwal, R., Thomas, S., Pawlowska, N., Bartelink, I., Grabowsky, J., Jahan, T., et al. (2017). Inhibiting histone deacetylase as a means to reverse resistance to angiogenesis inhibitors: phase I study of abexinostat plus pazopanib in advanced solid tumor malignancies. J. Clin. Oncol. 35, 1231–1239. doi: 10.1200/jco.2016.70.5350
Alhazzazi, T. Y., Kamarajan, P., Xu, Y., Ai, T., Chen, L., Verdin, E., et al. (2016). A novel Sirtuin-3 inhibitor, LC-0296, inhibits cell survival and proliferation, and promotes apoptosis of head and neck cancer cells. Anticancer Res. 36, 49–60.
Amengual, J. E., Johannet, P., Lombardo, M., Zullo, K., Hoehn, D., Bhagat, G., et al. (2015). Dual targeting of protein degradation pathways with the selective HDAC6 inhibitor ACY-1215 and bortezomib is synergistic in lymphoma. Clin. Cancer Res. 21, 4663–4675. doi: 10.1158/1078-0432.ccr-14-3068
Amengual, J. E., Prabhu, S. A., Lombardo, M., Zullo, K., Johannet, P. M., Gonzalez, Y., et al. (2017). Mechanisms of acquired drug resistance to the HDAC6 selective inhibitor ricolinostat reveals rational drug-drug combination with ibrutinib. Clin. Cancer Res. 23, 3084–3096. doi: 10.1158/1078-0432.ccr-16-2022
Asgar, M. A., Senawong, G., Sripa, B., and Senawong, T. (2016). Synergistic anticancer effects of cisplatin and histone deacetylase inhibitors (SAHA and TSA) on cholangiocarcinoma cell lines. Int. J. Oncol. 48, 409–420. doi: 10.3892/ijo.2015.3240
Ashraf, N., Zino, S., Macintyre, A., Kingsmore, D., Payne, A. P., George, W. D., et al. (2006). Altered sirtuin expression is associated with node-positive breast cancer. Br. J. Cancer 95, 1056–1061. doi: 10.1038/sj.bjc.6603384
Balasubramaniam, S., Redon, C. E., Peer, C. J., Bryla, C., Lee, M. J., Trepel, J. B., et al. (2018). Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anticancer Drugs 29, 457–465.
Banik, D., Moufarrij, S., and Villagra, A. (2019). Immunoepigenetics combination therapies: an overview of the role of HDACs in cancer immunotherapy. Int. J. Mol. Sci. 20:2241. doi: 10.3390/ijms20092241
Baylin, S. B., and Jones, P. A. (2011). A decade of exploring the cancer epigenome – biological and translational implications. Nat. Rev. Cancer 11, 726–734. doi: 10.1038/nrc3130
Bruyer, A., Maes, K., Herviou, L., Kassambara, A., Seckinger, A., Cartron, G., et al. (2018). DNMTi/HDACi combined epigenetic targeted treatment induces reprogramming of myeloma cells in the direction of normal plasma cells. Br. J. Cancer 118, 1062–1073. doi: 10.1038/s41416-018-0025-x
Ceccacci, E., and Minucci, S. (2016). Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Br. J. Cancer 114, 605–611. doi: 10.1038/bjc.2016.36
Chaiyawat, P., Pruksakorn, D., Phanphaisarn, A., Teeyakasem, P., Klangjorhor, J., and Settakorn, J. (2018). Expression patterns of class I histone deacetylases in osteosarcoma: a novel prognostic marker with potential therapeutic implications. Mod. Pathol. 31, 264–274. doi: 10.1038/modpathol.2017.125
Chan, E., Arlinghaus, L. R., Cardin, D. B., Goff, L., Berlin, J. D., Parikh, A., et al. (2016). Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother. Oncol. 119, 312–318.
Choi, J. H., Kwon, H. J., Yoon, B. I., Kim, J. H., Han, S. U., Joo, H. J., et al. (2001). Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 92, 1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x
Cosenza, M., Civallero, M., Marcheselli, L., Sacchi, S., and Pozzi, S. (2017). Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 22, 827–840. doi: 10.1007/s10495-017-1364-4
Cras, A., Darsin-Bettinger, D., Balitrand, N., Cassinat, B., Soulie, A., Toubert, M. E., et al. (2007). Epigenetic patterns of the retinoic acid receptor beta2 promoter in retinoic acid-resistant thyroid cancer cells. Oncogene 26, 4018–4024. doi: 10.1038/sj.onc.1210178
Deng, S., Hu, Q., Zhang, H., Yang, F., Peng, C., and Huang, C. (2019). HDAC3 inhibition upregulates PD-L1 expression in B-cell lymphomas and augments the efficacy of anti-PD-L1 therapy. Mol. Cancer Ther. 18, 900–908. doi: 10.1158/1535-7163.mct-18-1068
Deubzer, H. E., Schier, M. C., Oehme, I., Lodrini, M., Haendler, B., Sommer, A., et al. (2013). HDAC11 is a novel drug target in carcinomas. Int. J. Cancer 132, 2200–2208. doi: 10.1002/ijc.27876
Di Martile, M., Desideri, M., Tupone, M. G., Buglioni, S., Antoniani, B., Mastroiorio, C., et al. (2018). Histone deacetylase inhibitor ITF2357 leads to apoptosis and enhances doxorubicin cytotoxicity in preclinical models of human sarcoma. Oncogenesis 7:20.
DuBois, S. G., Groshen, S., Park, J. R., Haas-Kogan, D. A., Yang, X., Geier, E., et al. (2015). Phase I study of vorinostat as a radiation sensitizer with 131I-Metaiodobenzylguanidine (131I-MIBG) for patients with relapsed or refractory neuroblastoma. Clin. Cancer Res. 21, 2715–2721. doi: 10.1158/1078-0432.ccr-14-3240
Duffy, A. G., Ulahannan, S. V., Makorova-Rusher, O., Rahma, O., Wedemeyer, H., Pratt, D., et al. (2017). Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 66, 545–551. doi: 10.1016/j.jhep.2016.10.029
Eckschlager, T., Plch, J., Stiborova, M., and Hrabeta, J. (2017). Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 18:1414. doi: 10.3390/ijms18071414
Fajas, L., Egler, V., Reiter, R., Hansen, J., Kristiansen, K., Debril, M. B., et al. (2002). The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev. Cell 3, 903–910. doi: 10.1016/s1534-5807(02)00360-x
Feng, D., Wu, J., Tian, Y., Zhou, H., Zhou, Y., Hu, W., et al. (2013). Targeting of histone deacetylases to reactivate tumour suppressor genes and its therapeutic potential in a human cervical cancer xenograft model. PLoS One 8:e80657. doi: 10.1371/journal.pone.0080657
Finnin, M. S., Donigian, J. R., Cohen, A., Richon, V. M., Rifkind, R. A., Marks, P. A., et al. (1999). Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193. doi: 10.1038/43710
Fukumoto, T., Fatkhutdinov, N., Zundell, J. A., Tcyganov, E. N., Nacarelli, T., Karakashev, S., et al. (2019). HDAC6 inhibition synergizes with anti-PD-L1 therapy in ARID1A-inactivated ovarian cancer. Cancer Res. 79, 5482– 5489.
Galanis, E., Anderson, S. K., Miller, C. R., Sarkaria, J. N., Jaeckle, K., Buckner, J. C., et al. (2018). Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: results of Alliance N0874/ABTC 02. Neuro Oncol. 20, 546–556. doi: 10.1093/neuonc/nox161
Galloway, T. J., Wirth, L. J., Colevas, A. D., Gilbert, J., Bauman, J. E., Saba, N. F., et al. (2015). A phase I study of CUDC-101, a multitarget inhibitor of HDACs, EGFR, and HER2, in combination with chemoradiation in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 21, 1566–1573. doi: 10.1158/1078-0432.ccr-14-2820
Garcia-Dominguez, D. J., Hontecillas-Prieto, L., Rodriguez-Nunez, P., Pascual-Pasto, G., Vila-Ubach, M., Garcia-Mejias, R., et al. (2018). The combination of epigenetic drugs SAHA and HCI-2509 synergistically inhibits EWS-FLI1 and tumor growth in Ewing sarcoma. Oncotarget 9, 31397–31410. doi: 10.18632/oncotarget.25829
Gelmetti, V., Zhang, J., Fanelli, M., Minucci, S., Pelicci, P. G., and Lazar, M. A. (1998). Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol. Cell Biol. 18, 7185–7191. doi: 10.1128/mcb.18.12.7185
Gerber, D. E., Boothman, D. A., Fattah, F. J., Dong, Y., Zhu, H., Skelton, R. A., et al. (2015). Phase 1 study of romidepsin plus erlotinib in advanced non-small cell lung cancer. Lung Cancer 90, 534–541. doi: 10.1016/j.lungcan.2015.10.008
Glasser, C. L., Lee, A., Eslin, D., Marks, L., Modak, S., and Glade Bender, J. L. (2017). Epigenetic combination therapy for children with secondary myelodysplastic syndrome (MDS)/Acute Myeloid Leukemia (AML) and concurrent solid tumor relapse. J. Pediatr. Hematol. Oncol. 39, 560–564. doi: 10.1097/mph.0000000000000868
Godman, C. A., Joshi, R., Tierney, B. R., Greenspan, E., Rasmussen, T. P., Wang, H. W., et al. (2008). HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol. Ther. 7, 1570–1580. doi: 10.4161/cbt.7.10.6561
Goldberg, A. L. (2007). Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem. Soc. Trans. 35, 12–17. doi: 10.1042/bst0350012
Grabarska, A., Luszczki, J. J., Nowosadzka, E., Gumbarewicz, E., Jeleniewicz, W., Dmoszynska-Graniczka, M., et al. (2017). Histone deacetylase inhibitor SAHA as potential targeted therapy agent for larynx cancer cells. J. Cancer 8, 19–28. doi: 10.7150/jca.16655
Gray, J. E., Saltos, A., Tanvetyanon, T., Haura, E. B., Creelan, B., Antonia, S. J., et al. (2019). Phase I/Ib study of pembrolizumab plus vorinostat in advanced/metastatic non-small cell lung cancer. Clin. Cancer Res. 25, 6623–6632. doi: 10.1158/1078-0432.ccr-19-1305
Grignani, F., De Matteis, S., Nervi, C., Tomassoni, L., Gelmetti, V., Cioce, M., et al. (1998). Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature 391, 815–818. doi: 10.1038/35901
Groselj, B., Ruan, J. L., Scott, H., Gorrill, J., Nicholson, J., Kelly, J., et al. (2018). Radiosensitization in vivo by histone deacetylase inhibition with no increase in early normal tissue radiation toxicity. Mol. Cancer Ther. 17, 381–392. doi: 10.1158/1535-7163.mct-17-0011
Groselj, B., Sharma, N. L., Hamdy, F. C., Kerr, M., and Kiltie, A. E. (2013). Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br. J. Cancer 108, 748–754. doi: 10.1038/bjc.2013.21
Gryder, B. E., Sodji, Q. H., and Oyelere, A. K. (2012). Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 4, 505–524. doi: 10.4155/fmc.12.3
Gumbarewicz, E., Luszczki, J. J., Wawruszak, A., Dmoszynska-Graniczka, M., Grabarska, A. J., Jarzab, A. M., et al. (2016). Isobolographic analysis demonstrates additive effect of cisplatin and HDIs combined treatment augmenting their anti-cancer activity in lung cancer cell lines. Am. J. Cancer Res. 6, 2831–2845.
Gurbani, S. S., Yoon, Y., Weinberg, B. D., Salgado, E., Press, R. H., Cordova, J. S., et al. (2019). Assessing treatment response of glioblastoma to an HDAC inhibitor using whole-brain spectroscopic MRI. Tomography 5, 53–60. doi: 10.18383/j.tom.2018.00031
Haberland, M., Montgomery, R. L., and Olson, E. N. (2009). The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42. doi: 10.1038/nrg2485
Hainsworth, J. D., Daugaard, G., Lesimple, T., Hubner, G., Greco, F. A., Stahl, M. J., et al. (2015). Paclitaxel/carboplatin with or without belinostat as empiric first-line treatment for patients with carcinoma of unknown primary site: a randomized, phase 2 trial. Cancer 121, 1654–1661. doi: 10.1002/cncr.29229
Halsall, J. A., and Turner, B. M. (2016). Histone deacetylase inhibitors for cancer therapy: an evolutionarily ancient resistance response may explain their limited success. Bioessays 38, 1102–1110. doi: 10.1002/bies.201600070
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Haydn, T., Metzger, E., Schuele, R., and Fulda, S. (2017). Concomitant epigenetic targeting of LSD1 and HDAC synergistically induces mitochondrial apoptosis in rhabdomyosarcoma cells. Cell Death Dis. 8:e2879. doi: 10.1038/cddis.2017.239
Heijkants, R., Willekens, K., Schoonderwoerd, M., Teunisse, A., Nieveen, M., Radaelli, E., et al. (2018). Combined inhibition of CDK and HDAC as a promising therapeutic strategy for both cutaneous and uveal metastatic melanoma. Oncotarget 9, 6174–6187. doi: 10.18632/oncotarget.23485
Hideshima, T., Bradner, J. E., Wong, J., Chauhan, D., Richardson, P., Schreiber, S. L., et al. (2005). Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 102, 8567–8572. doi: 10.1073/pnas.0503221102
Holkova, B., Kmieciak, M., Bose, P., Yazbeck, V. Y., Barr, P. M., Tombes, M. B., et al. (2016). Phase 1 trial of carfilzomib (PR-171) in combination with vorinostat (SAHA) in patients with relapsed or refractory B-cell lymphomas. Leuk. Lymphoma 57, 635–643. doi: 10.3109/10428194.2015.1075019
Huang, F. I., Wu, Y. W., Sung, T. Y., Liou, J. P., Lin, M. H., Pan, S. L., et al. (2019). MPT0G413, a novel HDAC6-selective inhibitor, and bortezomib synergistically exert anti-tumor activity in multiple myeloma cells. Front. Oncol. 9:249. doi: 10.3389/fonc.2019.00249
Huang, Z., Peng, S., Knoff, J., Lee, S. Y., Yang, B., Wu, T. C., et al. (2015). Combination of proteasome and HDAC inhibitor enhances HPV16 E7-specific CD8+ T cell immune response and antitumor effects in a preclinical cervical cancer model. J. Biomed. Sci. 22:7. doi: 10.1186/s12929-014-0111-1
Ji, J., Valdez, B. C., Li, Y., Liu, Y., Teo, E. C., Nieto, Y., et al. (2016). Cladribine, gemcitabine, busulfan, and SAHA combination as a potential pretransplant conditioning regimen for lymphomas: a preclinical study. Exp. Hematol. 44, 458–465. doi: 10.1016/j.exphem.2016.03.001
Ji, M., Li, Z., Lin, Z., and Chen, L. (2018). Antitumor activity of the novel HDAC inhibitor CUDC-101 combined with gemcitabine in pancreatic cancer. Am. J. Cancer Res. 8, 2402–2418.
Jin, Z., Jiang, W., Jiao, F., Guo, Z., Hu, H., Wang, L., et al. (2014). Decreased expression of histone deacetylase 10 predicts poor prognosis of gastric cancer patients. Int. J. Clin. Exp. Pathol. 7, 5872–5879.
Kaufman, J. L., Mina, R., Jakubowiak, A. J., Zimmerman, T. L., Wolf, J. J., Lewis, C., et al. (2019). Combining carfilzomib and panobinostat to treat relapsed/refractory multiple myeloma: results of a multiple myeloma research consortium phase I study. Blood Cancer J. 9:3.
Kim, H. J., and Bae, S. C. (2011). Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 3, 166–179.
Kim, M. S., Kwon, H. J., Lee, Y. M., Baek, J. H., Jang, J. E., Lee, S. W., et al. (2001). Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 7, 437–443. doi: 10.1038/86507
Kim, Y., Kim, K., Park, D., Lee, E., Lee, H., Lee, Y. S., et al. (2012). Histone deacetylase 3 mediates allergic skin inflammation by regulating expression of MCP1 protein. J. Biol. Chem. 287, 25844–25859. doi: 10.1074/jbc.m112.348284
Knox, T., Sahakian, E., Banik, D., Hadley, M., Palmer, E., Noonepalle, S., et al. (2019). Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 9:6136.
Knutson, S. K., Chyla, B. J., Amann, J. M., Bhaskara, S., Huppert, S. S., and Hiebert, S. W. (2008). Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 27, 1017–1028. doi: 10.1038/emboj.2008.51
Lee, B. B., Kim, Y., Kim, D., Cho, E. Y., Han, J., Kim, H. K., et al. (2019). Metformin and tenovin-6 synergistically induces apoptosis through LKB1-independent SIRT1 down-regulation in non-small cell lung cancer cells. J. Cell Mol. Med. 23, 2872–2889. doi: 10.1111/jcmm.14194
Lee, D. H., Won, H. R., Ryu, H. W., Han, J. M., and Kwon, S. H. (2018). The HDAC6 inhibitor ACY1215 enhances the anticancer activity of oxaliplatin in colorectal cancer cells. Int. J. Oncol. 53, 844–854.
Liffers, K., Kolbe, K., Westphal, M., Lamszus, K., and Schulte, A. (2016). Histone deacetylase inhibitors resensitize EGFR/EGFRvIII-overexpressing, erlotinib-resistant glioblastoma cells to tyrosine kinase inhibition. Target Oncol. 11, 29–40. doi: 10.1007/s11523-015-0372-y
Llopiz, D., Ruiz, M., Villanueva, L., Iglesias, T., Silva, L., Egea, J., et al. (2019). Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 68, 379–393. doi: 10.1007/s00262-018-2283-0
Luo, J., Su, F., Chen, D., Shiloh, A., and Gu, W. (2000). Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377–381. doi: 10.1038/35042612
Ma, W., Zhao, X., Wang, K., Liu, J., and Huang, G. (2018). Dichloroacetic acid (DCA) synergizes with the SIRT2 inhibitor Sirtinol and AGK2 to enhance anti-tumor efficacy in non-small cell lung cancer. Cancer Biol. Ther. 19, 835–846. doi: 10.1080/15384047.2018.1480281
Mackintosh, C., Garcia-Dominguez, D. J., Ordonez, J. L., Ginel-Picardo, A., Smith, P. G., Sacristan, M. P., et al. (2013). WEE1 accumulation and deregulation of S-phase proteins mediate MLN4924 potent inhibitory effect on Ewing sarcoma cells. Oncogene 32, 1441–1451. doi: 10.1038/onc.2012.153
Malone, C. F., Emerson, C., Ingraham, R., Barbosa, W., Guerra, S., Yoon, H., et al. (2017). mTOR and HDAC inhibitors converge on the TXNIP/Thioredoxin pathway to cause catastrophic oxidative stress and regression of RAS-driven tumors. Cancer Discov. 7, 1450–1463. doi: 10.1158/2159-8290.cd-17-0177
McFarland, K. N., Das, S., Sun, T. T., Leyfer, D., Xia, E., Sangrey, G. R., et al. (2012). Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington’s disease. PLoS One 7:e41423. doi: 10.1371/journal.pone.0041423
Mishima, Y., Santo, L., Eda, H., Cirstea, D., Nemani, N., Yee, A. J., et al. (2015). Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 169, 423–434. doi: 10.1111/bjh.13315
Moertl, S., Payer, S., Kell, R., Winkler, K., Anastasov, N., and Atkinson, M. J. (2019). Comparison of radiosensitization by HDAC inhibitors CUDC-101 and SAHA in pancreatic cancer cells. Int. J. Mol. Sci. 20:3259. doi: 10.3390/ijms20133259
Moreno, D. A., Scrideli, C. A., Cortez, M. A., De Paula Queiroz, R., Valera, E. T., Da Silva Silveira, V., et al. (2010). Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 150, 665–673. doi: 10.1111/j.1365-2141.2010.08301.x
Mottamal, M., Zheng, S., Huang, T. L., and Wang, G. (2015). Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 20, 3898–3941. doi: 10.3390/molecules20033898
Moufarrij, S., Srivastava, A., Gomez, S., Hadley, M., Palmer, E., Austin, P. T., et al. (2020). Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases tumor burden in ovarian cancer. Sci. Rep. 10:3470.
Munster, P. N., Marchion, D., Thomas, S., Egorin, M., Minton, S., Springett, G., et al. (2009). Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 101, 1044–1050. doi: 10.1038/sj.bjc.6605293
Ning, B., Li, W., Zhao, W., and Wang, R. (2016). Targeting epigenetic regulations in cancer. Acta Biochim. Biophys. Sin. (Shanghai) 48, 97–109.
North, B. J., Almeciga-Pinto, I., Tamang, D., Yang, M., Jones, S. S., and Quayle, S. N. (2017). Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS One 12:e0173507. doi: 10.1371/journal.pone.0173507
Oehme, I., Deubzer, H. E., Wegener, D., Pickert, D., Linke, J. P., Hero, B., et al. (2009). Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 15, 91–99. doi: 10.1158/1078-0432.ccr-08-0684
Osada, H., Tatematsu, Y., Saito, H., Yatabe, Y., Mitsudomi, T., and Takahashi, T. (2004). Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer 112, 26–32. doi: 10.1002/ijc.20395
Parbin, S., Kar, S., Shilpi, A., Sengupta, D., Deb, M., Rath, S. K., et al. (2014). Histone deacetylases: a saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 62, 11–33. doi: 10.1369/0022155413506582
Park, S. Y., Jun, J. A., Jeong, K. J., Heo, H. J., Sohn, J. S., Lee, H. Y., et al. (2011). Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol. Rep. 25, 1677–1681.
Park, S. Y., and Kim, J. S. (2020). A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 52, 204–212. doi: 10.1038/s12276-020-0382-4
Pathania, R., Ramachandran, S., Mariappan, G., Thakur, P., Shi, H., Choi, J. H., et al. (2016). Combined inhibition of DNMT and HDAC blocks the tumorigenicity of cancer stem-like cells and attenuates mammary tumor growth. Cancer Res. 76, 3224–3235. doi: 10.1158/0008-5472.can-15-2249
Persky, D. O., Li, H., Rimsza, L. M., Barr, P. M., Popplewell, L. L., Bane, C. L., et al. (2018). A phase I/II trial of vorinostat (SAHA) in combination with rituximab-CHOP in patients with newly diagnosed advanced stage diffuse large B-cell lymphoma (DLBCL): SWOG S0806. Am. J. Hematol. 93, 486–493. doi: 10.1002/ajh.25010
Richardson, P. G., Hungria, V. T., Yoon, S. S., Beksac, M., Dimopoulos, M. A., Elghandour, A., et al. (2016). Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: outcomes by prior treatment. Blood 127, 713–721. doi: 10.1182/blood-2015-09-665018
Rifai, K., Idrissou, M., Penault-Llorca, F., Bignon, Y. J., and Bernard-Gallon, D. (2018). Breaking down the contradictory roles of histone deacetylase SIRT1 in human breast cancer. Cancers (Basel) 10:409. doi: 10.3390/cancers10110409
Ropero, S., Fraga, M. F., Ballestar, E., Hamelin, R., Yamamoto, H., Boix-Chornet, M., et al. (2006). A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 38, 566–569. doi: 10.1038/ng1773
Saijo, K., Imamura, J., Narita, K., Oda, A., Shimodaira, H., Katoh, T., et al. (2015). Biochemical, biological and structural properties of romidepsin (FK228) and its analogs as novel HDAC/PI3K dual inhibitors. Cancer Sci. 106, 208–215. doi: 10.1111/cas.12585
Sandoval, J., and Esteller, M. (2012). Cancer epigenomics: beyond genomics. Curr. Opin. Genet. Dev. 22, 50–55. doi: 10.1016/j.gde.2012.02.008
Sankar, S., Bell, R., Stephens, B., Zhuo, R., Sharma, S., Bearss, D. J., et al. (2016). Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene 35, 6155–6156. doi: 10.1038/onc.2016.142
Sankar, S., Theisen, E. R., Bearss, J., Mulvihill, T., Hoffman, L. M., Sorna, V., et al. (2014). Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin. Cancer Res. 20, 4584–4597. doi: 10.1158/1078-0432.ccr-14-0072
Santoro, V., Jia, R., Thompson, H., Nijhuis, A., Jeffery, R., Kiakos, K., et al. (2016). Role of reactive oxygen species in the abrogation of oxaliplatin activity by cetuximab in colorectal cancer. J. Natl. Cancer Inst. 108:djv394. doi: 10.1093/jnci/djv394
Siddiqui, H., Solomon, D. A., Gunawardena, R. W., Wang, Y., and Knudsen, E. S. (2003). Histone deacetylation of RB-responsive promoters: requisite for specific gene repression but dispensable for cell cycle inhibition. Mol. Cell Biol. 23, 7719–7731. doi: 10.1128/mcb.23.21.7719-7731.2003
Song, C., Zhu, S., Wu, C., and Kang, J. (2013). Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J. Biol. Chem. 288, 28021–28033. doi: 10.1074/jbc.m113.498758
Stark, M., and Hayward, N. (2007). Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 67, 2632–2642. doi: 10.1158/0008-5472.can-06-4152
Suraweera, A., O’byrne, K. J., and Richard, D. J. (2018). Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front. Oncol. 8:92. doi: 10.3389/fonc.2018.00092
Tanaka, N., Patel, A. A., Tang, L., Silver, N. L., Lindemann, A., Takahashi, H., et al. (2017). Replication stress leading to apoptosis within the S-phase contributes to synergism between Vorinostat and AZD1775 in HNSCC harboring high-risk TP53 mutation. Clin. Cancer Res. 23, 6541–6554. doi: 10.1158/1078-0432.ccr-17-0947
Taylor, B. S., Decarolis, P. L., Angeles, C. V., Brenet, F., Schultz, N., Antonescu, C. R., et al. (2011). Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov. 1, 587–597. doi: 10.1158/2159-8290.cd-11-0181
Teknos, T. N., Grecula, J., Agrawal, A., Old, M. O., Ozer, E., Carrau, R., et al. (2019). A phase 1 trial of Vorinostat in combination with concurrent chemoradiation therapy in the treatment of advanced staged head and neck squamous cell carcinoma. Invest. New Drugs 37, 702–710. doi: 10.1007/s10637-018-0696-4
Terranova-Barberio, M., Pecori, B., Roca, M. S., Imbimbo, S., Bruzzese, F., Leone, A., et al. (2017a). Synergistic antitumor interaction between valproic acid, capecitabine and radiotherapy in colorectal cancer: critical role of p53. J. Exp. Clin. Cancer Res. 36:177.
Terranova-Barberio, M., Thomas, S., Ali, N., Pawlowska, N., Park, J., Krings, G., et al. (2017b). HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 8, 114156–114172. doi: 10.18632/oncotarget.23169
Thal, M. A., Krishnamurthy, P., Mackie, A. R., Hoxha, E., Lambers, E., Verma, S., et al. (2012). Enhanced angiogenic and cardiomyocyte differentiation capacity of epigenetically reprogrammed mouse and human endothelial progenitor cells augments their efficacy for ischemic myocardial repair. Circ. Res. 111, 180–190. doi: 10.1161/circresaha.112.270462
Thole, T. M., Lodrini, M., Fabian, J., Wuenschel, J., Pfeil, S., Hielscher, T., et al. (2017). Neuroblastoma cells depend on HDAC11 for mitotic cell cycle progression and survival. Cell Death Dis. 8:e2635. doi: 10.1038/cddis.2017.49
Vanaja, G. R., Ramulu, H. G., and Kalle, A. M. (2018). Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Commun. Signal. 16:20.
Vannini, A., Volpari, C., Filocamo, G., Casavola, E. C., Brunetti, M., Renzoni, D., et al. (2004). Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U.S.A. 101, 15064–15069. doi: 10.1073/pnas.0404603101
Vitfell-Rasmussen, J., Judson, I., Safwat, A., Jones, R. L., Rossen, P. B., Lind-Hansen, M., et al. (2016). A phase I/II clinical trial of belinostat (PXD101) in combination with doxorubicin in patients with soft tissue sarcomas. Sarcoma 2016:2090271.
Vogl, D. T., Raje, N., Jagannath, S., Richardson, P., Hari, P., Orlowski, R., et al. (2017). Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res. 23, 3307–3315. doi: 10.1158/1078-0432.ccr-16-2526
Wang, P., Wang, Z., and Liu, J. (2020). Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 19:5.
Wang, X., Waschke, B. C., Woolaver, R. A., Chen, Z., Zhang, G., Piscopio, A. D., et al. (2019). Histone deacetylase inhibition sensitizes PD1 blockade-resistant B-cell lymphomas. Cancer Immunol. Res. 7, 1318–1331.
Wawruszak, A., Luszczki, J. J., Kalafut, J., Okla, K., Halasa, M., Rivero-Muller, A., et al. (2019). Additive pharmacological interaction between cisplatin (CDDP) and histone deacetylase inhibitors (HDIs) in MDA-MB-231 triple negative breast cancer (TNBC) cells with altered Notch1 activity-an isobolographic analysis. Int. J. Mol. Sci. 20:3663. doi: 10.3390/ijms20153663
Wei, Y., Guo, Y., Zhou, J., Dai, K., Xu, Q., and Jin, X. (2019). Nicotinamide overcomes doxorubicin resistance of breast cancer cells through deregulating SIRT1/Akt pathway. Anticancer Agents Med. Chem. 19, 687–696. doi: 10.2174/1871520619666190114160457
Wickstrom, S. A., Masoumi, K. C., Khochbin, S., Fassler, R., and Massoumi, R. (2010). CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 29, 131–144. doi: 10.1038/emboj.2009.317
Wu, J., Du, C., Lv, Z., Ding, C., Cheng, J., Xie, H., et al. (2013). The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig. Dis. Sci. 58, 3545–3553. doi: 10.1007/s10620-013-2867-7
Xu, Q., Lin, X., Andrews, L., Patel, D., Lampe, P. D., and Veenstra, R. D. (2013). Histone deacetylase inhibition reduces cardiac connexin43 expression and gap junction communication. Front. Pharmacol. 4:44. doi: 10.3389/fphar.2013.00044
Yee, A. J., Bensinger, W. I., Supko, J. G., Voorhees, P. M., Berdeja, J. G., Richardson, P. G., et al. (2016). Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol. 17, 1569–1578. doi: 10.1016/s1470-2045(16)30375-8
Zhang, H., Dong, L., Chen, Q., Kong, L., Meng, B., Wang, H., et al. (2017a). Synergistic antitumor effect of histone deacetylase inhibitor and Doxorubicin in peripheral T-cell lymphoma. Leuk. Res. 56, 29–35. doi: 10.1016/j.leukres.2017.01.025
Zhang, H., Li, X., Zhang, Q., Yang, F., Chu, X., Zhang, D., et al. (2017b). Role of histone deacetylase expression levels and activity in the inflammatory responses of patients with chronic hepatitis B. Mol. Med. Rep. 15, 2744–2752. doi: 10.3892/mmr.2017.6290
Zhang, L., Zhang, Y., Mehta, A., Boufraqech, M., Davis, S., Wang, J., et al. (2015). Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 6, 9073–9085. doi: 10.18632/oncotarget.3268
Zibelman, M., Wong, Y. N., Devarajan, K., Malizzia, L., Corrigan, A., Olszanski, A. J., et al. (2015). Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Invest. New Drugs 33, 1040–1047. doi: 10.1007/s10637-015-0261-3
Keywords: histone deacetylases, HDAC inhibitors (HDACis), combination treatment, cancer, clinical trials, preclinical studies
Citation: Hontecillas-Prieto L, Flores-Campos R, Silver A, de Álava E, Hajji N and García-Domínguez DJ (2020) Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 11:578011. doi: 10.3389/fgene.2020.578011
Received: 30 June 2020; Accepted: 26 August 2020;
Published: 11 September 2020.
Edited by:
Ângela Sousa, University of Beira Interior, PortugalReviewed by:
Yingjie Zhang, Shandong University, ChinaBradley S. Ferguson, University of Nevada, Reno, United States
Copyright © 2020 Hontecillas-Prieto, Flores-Campos, Silver, de Álava, Hajji and García-Domínguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lourdes Hontecillas-Prieto, bGhvbnRlY2lsbGFzLWliaXNAdXMuZXM=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship