 Silvia Murillo-Cuesta1,2,3*
Silvia Murillo-Cuesta1,2,3* Rafael Artuch1,4
Rafael Artuch1,4 Fernando Asensio5
Fernando Asensio5 Pedro de la Villa6
Pedro de la Villa6 Mara Dierssen1,7,8
Mara Dierssen1,7,8 Jose Antonio Enríquez9,10Cristina Fillat1,11
Jose Antonio Enríquez9,10Cristina Fillat1,11 Stéphane Fourcade1,12Borja Ibáñez9,13,14
Stéphane Fourcade1,12Borja Ibáñez9,13,14 Lluis Montoliu1,15
Lluis Montoliu1,15 Eduardo Oliver9,13
Eduardo Oliver9,13 Aurora Pujol1,12,16Eduardo Salido1,17
Aurora Pujol1,12,16Eduardo Salido1,17 Mario Vallejo2,18
Mario Vallejo2,18 Isabel Varela-Nieto1,2,3* for/on behalf of the Working Group on Animal Models of Rare Diseases Biomedical Research and Networking Center for Rare Diseases (CIBERER) Institute of Health Carlos III
Isabel Varela-Nieto1,2,3* for/on behalf of the Working Group on Animal Models of Rare Diseases Biomedical Research and Networking Center for Rare Diseases (CIBERER) Institute of Health Carlos III- 1Biomedical Research Networking Center on Rare Diseases (CIBERER), Institute of Health Carlos III, Madrid, Spain
- 2Instituto de Investigaciones Biomédicas Alberto Sols (IIBM), Consejo Superior de Investigaciones Científicas/Universidad Autónoma de Madrid, Madrid, Spain
- 3Hospital La Paz Institute for Health Research (IdiPAZ), Madrid, Spain
- 4Institut de Recerca Sant Joan de Déu (IRSJD), Barcelona, Spain
- 5Gregorio Marañón Institute for Health Research (IISGM), Madrid, Spain
- 6Faculty of Medicine, University of Alcalá (UAH), Alcalá de Henares, Spain
- 7Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology (BIST), Barcelona, Spain
- 8Universitat Pompeu Fabra (UPF), Barcelona, Spain
- 9Spanish National Center for Cardiovascular Research (CNIC), Institute of Health Carlos III, Madrid, Spain
- 10Biomedical Research Networking Center on Frailty and Healthy Ageing (CIBERFES), Institute of Health Carlos III, Madrid, Spain
- 11Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 12Bellvitge Biomedical Research Institute (IDIBELL), L’Hospitalet de Llobregat, Barcelona, Spain
- 13Biomedical Research Networking Center on Cardiovascular Diseases (CIBERCV), Institute of Health Carlos III, Madrid, Spain
- 14Cardiology Department, Fundación Jiménez Díaz University Hospital Health Research Institute (IIS-FJD), Madrid, Spain
- 15National Center for Biotechnology (CNB), Spanish National Research Council, Madrid, Spain
- 16Catalan Institution of Research and Advanced Studies (ICREA), Barcelona, Spain
- 17Unidad de Investigación, Hospital Universitario de Canarias, Instituto de Tecnologías Biomédicas (ITB), La Laguna, Spain
- 18Biomedical Research Networking Center on Diabetes and Metabolic Diseases (CIBERDEM), Institute of Health Carlos III, Madrid, Spain
Animal models are invaluable for biomedical research, especially in the context of rare diseases, which have a very low prevalence and are often complex. Concretely mouse models provide key information on rare disease mechanisms and therapeutic strategies that cannot be obtained by using only alternative methods, and greatly contribute to accelerate the development of new therapeutic options for rare diseases. Despite this, the use of experimental animals remains controversial. The combination of respectful management, ethical laws and transparency regarding animal experimentation contributes to improve society’s opinion about biomedical research and positively impacts on research quality, which eventually also benefits patients. Here we present examples of current advances in preclinical research in rare diseases using mouse models, together with our perspective on future directions and challenges.
Introduction
Animal research has contributed greatly to advance human health and quality of life. The use of laboratory animals increased exponentially in the 20th century and they are currently employed in almost every field of biomedical research. Animal models reproduce many aspects of human biological and pathological processes, and provide key information on the molecular pathophysiology of human diseases. Non-animal approaches based mainly on cell or tissue/organ culture, and computational methods like data mining/generation, may help to predict clinical outcomes and reduce animal use (Cronin, 2016), but they cannot mimic the complexity of human biology. Animals remain the best model–however imperfect–to predict and characterize disease activity in patients (Garattini and Grignaschi, 2017).
Choosing a suitable animal model is a critical step in basic and preclinical research, and is usually based on a number of criteria, including species similarities to humans (the greater the phylogenetic closeness, the more similar is the genetic composition, anatomy, and physiology), genetic homogeneity, previous knowledge of the model, cost, availability, translatability of the results, ease of manipulation, and ethical implications, among others (National Research Council, 1998). Thus, the laboratory mouse is the most widely used mammalian animal in biomedical research, representing more than 60% of the total number of animals used in the EU (European Commission, 2019).
Genome manipulation and phenotype characterization is a common strategy for studying human pathology in animals, and particularly, in mice. In the last few years, CRISPR-Cas9-based genome editing has transformed the field and greatly expanded the repertoire of animal/cell systems available for disease modeling (Ahmad and Amiji, 2018). Gene homology between mouse and man is an essential prerequisite for pursuing this genotype-to-phenotype approach. Homogenization of the genetic background by inbreeding is also an important factor to reduce experimental variability. In this context, the International Mouse Phenotyping Consortium has generated, phenotyped and archived more than 6000 knockout mice on the C57BL/6 background, the most well-known and widely used inbred mouse strain (Cacheiro et al., 2019). Complete catalogs of genetically modified mouse models are available online at the International Phenotype Mouse Consortium and The Jackson Laboratory webpages (Table 1).

Table 1. Reference online resources on RDs, mouse models, legislation, and recommendations on animal experimentation.
Animal Models for Rare Disease Research
The definition of a rare disease (RD) in Europe is a disease with a prevalence of <1 in 2000, whereas ultra-RDs affect <1 in 50,000. RDs comprise more than 7000 different conditions (Orphadata, Table 1), usually severe, clinically complex and chronic, affecting 3.5–5.9% of the world’s population (Nguengang Wakap et al., 2020), most of whom are children. The fact that each RD affects a relatively small number of patients has resulted in limited knowledge of RDs at the clinical level, which often delays an early and accurate diagnosis–patients can wait 8 years before receiving a diagnosis–and a potential therapy. Alarmingly, 95% of RDs have no approved or effective treatments, in part because RDs are underserved by pharmaceutical companies. Accordingly, RDs are considered a public health priority and specific research programs as the International Rare Diseases Research Consortium (IRDiRC) (Table 1) have been established to foster knowledge development (Gahl et al., 2016; European Commission, 2017).
Animal models are indispensable to identify the genetic bases and molecular mechanisms of RDs, as well as to understand their physiopathology, clinical heterogeneity and genotype-phenotype correlations. Indeed, RDs are excellent candidates for animal models, particularly in the context of genetically modified mice, as most RDs involve mutations in a single gene (Institute of Medicine, 2010). Due to the scarcity of available information on RD models, however, one of the major issues hindering translational research is the (incorrect) choice of model in preclinical studies. To address this and other issues some initiatives have been recently launched to generate and register RD mouse models. For instance, the Jackson Laboratory Rare and Orphan Disease Center (Table 1) has generated animal models for Friedreich’s ataxia, Rett syndrome and spinal muscular atrophy. Likewise, the Infrafrontier platform provides access to 670 mouse strains that are related to nearly 1200 distinct RDs. Information about RD mouse models can also be obtained from the governmental agencies responsible for the evaluation of orphan medicinal product designation applications from pharmaceutical companies. In this context, Vaquer et al. (2013) compiled a list of 57 mammalian animal models for metabolic, neuromuscular, and ophthalmological orphan-designated conditions, based on information gathered by the European Medicines Agency (EMA). Additionally, some countries have developed specific national plans to prioritize RD research. For example, in Spain, the Biomedical Research Center Network for RDs (CIBERER) of the Carlos III Health Institute has contributed to the advancement of RD research by (i) developing new animal models, (ii) performing preclinical assays of novel therapeutics, and (iii) creating a mouse model phenotyping unit and a working group to register model information.
Here, we discuss some representative examples of RD mouse models under investigation at CIBERER (Table 2), which serve to illustrate the phenotypic variability of RDs and the possibilities offered by animal modeling to fill the knowledge gaps regarding in this area, and to contribute to the IRDiRC’s goal of accelerating diagnosis and approving 1000 new therapies for RDs by 2027.
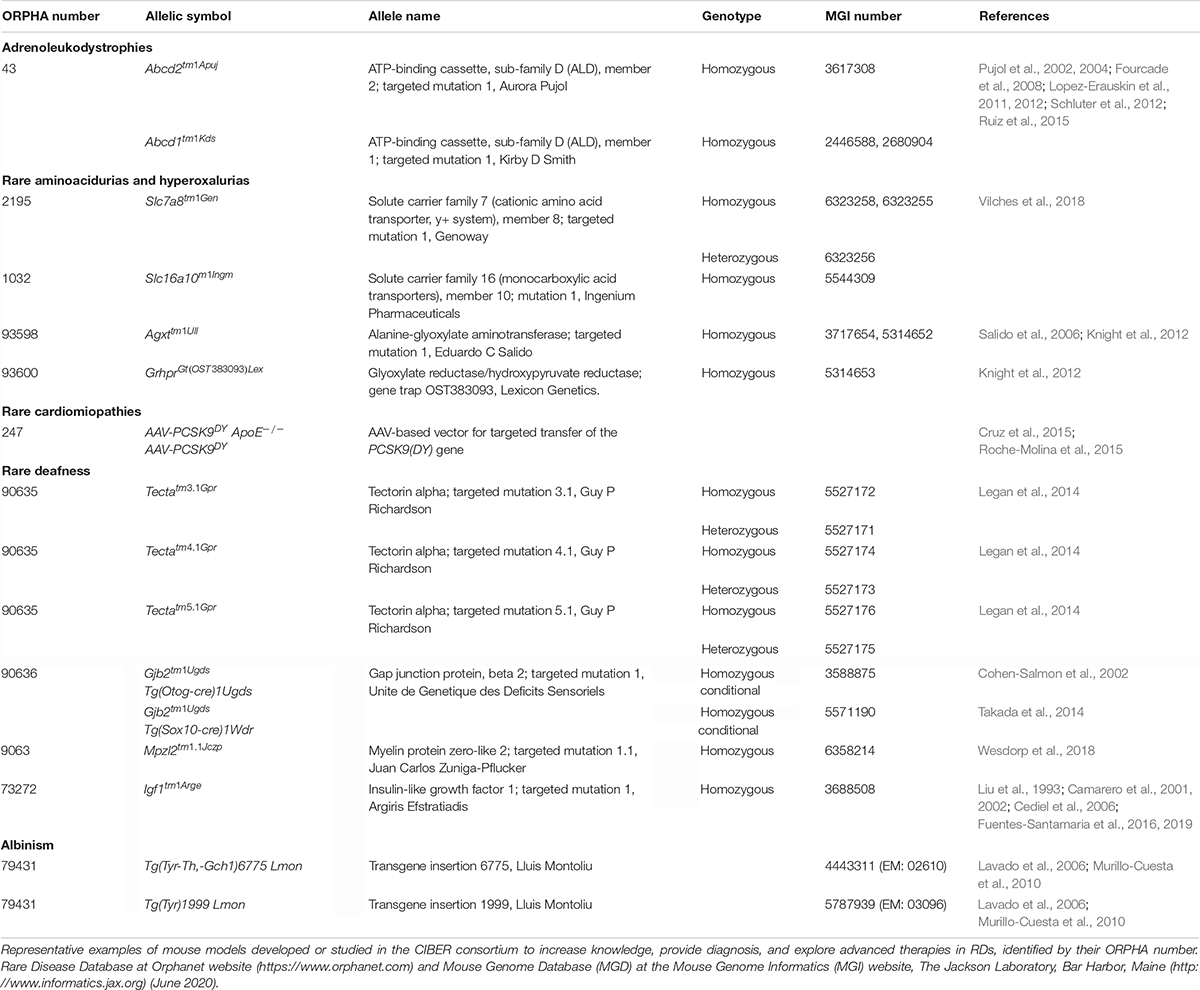
Table 2. Rare disease mouse models.
Metabolic RDs
They encompass a large and heterogeneous group of RDs caused by mutations affecting the function of enzymes, transporters, receptors, or hormones involved in metabolizing and transporting small (e.g., amino acids or neurotransmitters) or complex (i.e., glycogens or lipids) molecules, and defects in mitochondrial energy metabolism. One of the most extensively investigated is phenylketonuria, which severely affects the brain by interfering with dopamine and serotonin metabolism (Winn et al., 2018). Pahenu2/enu2 and Pahenu3/enu3 mice mimic human phenylketonuria pathophysiology and have aided in discovering mechanisms and therapies based on phenylalanine-restricted diets (Winn et al., 2018). Similarly, aromatic amino acid decarboxylase deficiency is a defect in dopamine and serotonin synthesis that also causes devastating central nervous system degeneration. DdcTM 1.1Nwlh mutant mice have been used to study the disease (Lee et al., 2013) and to evaluate adeno-associated viral gene therapy, which improved both survival and brain levels of dopamine and serotonin (Lee et al., 2016). A clinical trial using this approach is ongoing with encouraging results (Chien et al., 2017).
X-linked adrenoleukodystrophy (X-ALD) is another severe neurometabolic disease characterized by progressive central demyelination, adrenal insufficiency and accumulation of saturated very long-chain fatty acids, and caused by loss of function of the ABCD1 peroxisomal transporter (Ferrer et al., 2010). To date, no pharmacological treatment has been proven to be beneficial and current therapeutic options are unsatisfactory and restricted to bone marrow transplants and hematopoietic stem cell gene therapy, but most patients remain untreated. Mouse models uncovered the factors that account for genotype-phenotype correlation in human disease variants. The Abcd1– mutant mouse exhibits late-onset axonal degeneration of the spinal cord corticospinal tracts and microglial and astroglial activation, compatible with chronic low-level stimulation of the innate immune response, and constitutes a good model for ALD (Pujol et al., 2002; Ruiz et al., 2015). The Abcd2 gene product shares physiological and biochemical functions with that of Abcd1 (Pujol et al., 2004), and the Abcd1–/Abcd2–/– double mutant presents with an earlier and more severe axonal degenerative phenotype, constituting a more useful model for preclinical evaluation (Pujol et al., 2004). These mouse models revealed that X-ALD shares pathogenic processes with other neurodegenerative disorders (Galea et al., 2012), including redox dyshomeostasis, mitochondrial dysfunction, and proteostasis malfunction (Fourcade et al., 2015). Encouraging preclinical results with neurotrophic factors and antioxidants (Pujol, 2016) have paved the way for the launch of three phase II/III clinical trials for ALD (Casasnovas et al., 2019), and the approval of two orphan drug designations.
Defects in glyoxylate and hydroxyproline hepatic metabolism result in the hepatic overproduction of oxalate and primary hyperoxaluria (PH) – an ultra-RD with a prevalence of 1–3 in 106 individuals (Cochat and Rumsby, 2013). PH1, the most common and severe form, is caused by AGXT mutations (Milliner et al., 1993), whereas PH2 and PH3 are caused by mutations in GRHPR and HOGA1, respectively. Loss of function mutations in any of these genes results in impaired detoxification of glyoxylate, which is converted into oxalate. PH patients present elevated oxalate concentrations in plasma and urine, oxalate deposition in multiple organs, recurrent kidney stone episodes and chronic renal failure, which results in end-stage renal disease. Current therapies include large daily fluid intake and medications to reduce oxalate production (Cochat et al., 2012), but they do not eliminate recurring stones and renal disease. Combined liver and kidney transplantation is the only curative treatment available, but is associated with significant morbi-mortality and problems related to donor organ shortage and life-long immunosuppressive treatment.
The AgxtTM 1Ull mouse reproduces the main PH1 features (Salido et al., 2006) and has been used to evaluate promising experimental therapies (Martin-Higueras et al., 2017). Regulation of oxalate transepithelial flux in the gut following intestinal colonization with Oxalobacter (Hatch et al., 2011) has received an innovative new drug designation by the United States Food and Drug Administration and is in clinical trials. Similarly, gene therapy with adeno-associated vectors carrying human AGXT under the control of a liver-specific promoter achieved a long-term metabolic correction (Salido et al., 2011), and was granted an EMA orphan drug designation. Deletion of the glycolate oxidase gene, inhibition of its enzymatic product or suppressing its expression with short-interfering RNA (siRNA) resulted in a substantial reversal of the hyperoxaluric phenotype (Martin-Higueras et al., 2016), the latter is currently being evaluated in a clinical trial with encouraging preliminary results. Therapies based on in vivo CRISPR-Cas9 technology are also a potential strategy for curing PH1 by substrate reduction with the administration of AAV-mediated glycolate oxidase-targeted guide RNAs (Zabaleta et al., 2018). We have generated a Grhpr knockout mouse for PH2 (Knight et al., 2012), and both Agxt and Grhpr mutant mice have been used to test the potential of inhibiting hepatic lactate dehydrogenase with siRNA to treat PH (Lai et al., 2018), which has moved to a clinical trial. In contrast to the models for PH1 and PH2, the mouse model for PH3 generated by the International Knockout Mouse Consortium (Hoga1TM 2a(KOMP)Wtsi, MGI:4419886) does not have the expected phenotype and it is currently being used to investigate differences in mouse and human glyoxylate metabolism. This example highlights a key point, which is that the understanding of the differences in the metabolic interactome between species is fundamental for the efficient transfer of the knowledge from experimental models to clinical practice.
Rare aminoacidurias caused by defects in amino acid transporters are being studied with murine models, which emerge as a promising tool to design evidence-based therapies to halt the progression of the disease. Using the Slc16a10–/– Slc7a8–/– mouse and a targeted metabolomics approach, it was confirmed that both transporters functionally cooperate in vivo. This approach also uncovered compensation mechanisms that explain the lack of human basolateral neutral aminoacidurias (Vilches et al., 2018). Similarly, the Slc7a7–/– model of lysinuric protein intolerance resembles the human phenotype, including malabsorption and impaired reabsorption of cationic amino acids, hypoargininemia, and hyperammonemia, and importantly, responses to citrulline treatment, which improved the metabolic derangement and survival (Bodoy et al., 2019).
Rare Cardiac Diseases
Arrhythmogenic right ventricular cardiomyopathy is a severe disease characterized by ventricular fibrofatty replacement of cardiomyocytes, contractile defects, and high risk for developing malignant arrhythmias, which can ultimately lead to sudden cardiac death especially in young athletes (Gandjbakhch et al., 2018). More than 50% of the 380 mutations identified lie within PKP2, which encodes the desmosomal protein plakoglobin-2, a major component of cell-to-cell junctions (van Tintelen et al., 2006). Given the complexity of developing multiple transgenic animals, a novel approach was developed by delivering genes encoding mutated proteins into wild-type mice using adeno-associated viruses (Roche-Molina et al., 2015). Using this strategy, C57BL6/J mice stably expressed the R735X version of PKP2, a dominant-negative mutant, driven by a cardiac-specific promoter, resulting in development of an arrhythmogenic right ventricular cardiomyopathy phenotype following exercise (Cruz et al., 2015). Although no evidence of myocardial fibrosis or fibrofatty cardiomyocyte replacement was observed, a miss localization of the gap-junction protein connexion-43 was evident. This model provides a versatile and accessible tool for investigating this devastating disease.
Albinism
Murine models have been central to understand this rare genetic condition primarily associated with severe visual deficits and variable hypopigmentation, and caused by mutations in at least twenty genes (Montoliu and Marks, 2017). Vision and hearing deficits have been characterized in the Tyr mutant mouse, a model for human oculocutaneous albinism 1 (Lavado et al., 2006; Murillo-Cuesta et al., 2010). Additional mouse models have been generated using CRISPR-Cas9 tools, including those addressing the role of non-coding DNA of regulatory elements in Tyr gene expression (Seruggia et al., 2015).
Sensorineural Hearing Loss
Approximately half of all cases of both non-syndromic and syndromic human hearing loss (HL) are due to rare mutations. TECTA-based human deafness is an example of autosomal dominant non-syndromic HL, in which domain-specific alterations in the glycoprotein Tecta, leading to changes in the tectorial membrane of the cochlea, have been studied using Tecta mutant mice (Legan et al., 2014). Autosomal recessive non-syndromic HL, which in the majority of cases is caused by mutations in GJB2 and GJB6, encoding the gap junction proteins connexin 26 and 30, respectively, has been studied using conditional mutant mice. Thus, Gjb2TM 1Ugds mouse shows a decrease in Cx26 expression, extensive loss of cochlear epithelial cells and an increase in hearing thresholds (Cohen-Salmon et al., 2002; Crispino et al., 2017).
Syndromic HL is a common condition in many RDs including insulin-like growth factor I (IGF-1) deficiency, an ultra-RD caused by homozygous mutations in IGF1 and associated with growth retardation, intellectual deficit, and HL (Varela-Nieto et al., 2013). The use of experimental models is practically the only way to investigate the pathology of ultra-RDs. In this respect, the Igf1TM 1Arge/tm1Arge mouse (Liu et al., 1993) recapitulates the human phenotype, and presents with severe deafness, neural loss (Cediel et al., 2006) and alterations in the auditory central pathway (Fuentes-Santamaria et al., 2016; Fuentes-Santamaria et al., 2019), offering a unique window into the role of the IGF-1 in human hearing.
Discussion
Animal experimentation is essential for understanding the pathogenic mechanisms of RDs and developing new, safe and effective treatments (Garattini and Grignaschi, 2017). This is especially true for RDs whose low prevalence is associated with a lack of knowledge, delays in diagnosis, and absence of effective treatments in most cases (European Commission, 2017). Non-animal experimental approaches provide valuable information, but they are far from reproducing the complexities and interactions in a living organism (Cronin, 2016). Rather than an alternative, non-animal methods are a useful complementary approach that helps to reduce the number of specimens used in biomedical research (European Commission, 2018).
The mouse is currently the most commonly used species due to its genetic tractability, relative ease of genome editing and cost-efficient management (European Commission, 2019). During the last 20 years, public and private initiatives have made a strong effort to generate and phenotype many hundreds of genetically modified strains (Cacheiro et al., 2019). However, it has been only recently that special attention has been paid to RDs (Institute of Medicine, 2010; Gahl et al., 2016). Information on RD mouse models is limited and scattered across different databases, which could hamper the preclinical testing of new therapeutic approaches. It would be useful to gather all the data from already existing mutant mouse databases with those from the orphan drug evaluation committees in international agencies (Vaquer et al., 2013) and from national initiatives for RDs research. As an example, the Spanish CIBERER consortium has generated mouse models for some RDs that have been shown to be effective for preclinical testing of new drugs (Table 2).
The usefulness of mouse models to advance RD research should not make us forget the importance of the ethical aspects and transparency in animal research. The use of animals in biomedical research remains a contentious issue in society (Matthews, 2008). Citizens demand treatments that require preclinical safety and efficacy testing, but they are increasingly concerned by animal welfare and demand the elimination of pain, and ultimately, of animal experimentation. Authorities and the scientific community are devoted to protect public health and the environment, and require the testing of new medicines, chemicals, and food products in animal models. But they are also fully committed to animal welfare and to the progressive reduction of animal testing (European Commission, 2018), as stated in the current legislation. There is a large body of laws and regulations regarding the use of animals for scientific research and educational purposes. The 2010/63/EU directive (Table 1) states that: (i) animal experimentation can be carried out only after a number of independent evaluations, and authorization from the competent authority; (ii) researchers must reasonably justify the use of experimental animals over alternative methods; (iii) experiments involving animals can only be conducted by competent and experienced professionals in authorized facilities; (iv) the 3Rs principle (reduction, refinement, and replacement) has always to be considered (Mocho, 2020). However, it is critical to improve communication with the general public to convey the fact that animal experimentation is necessary not only to protect human health, but also to protect animals and the environment.
Society also demands transparency regarding animal experimentation. Modern science is now so complex that citizens are often unaware of the gaps in knowledge still existing and wrongly assume that the use of animals is no longer necessary. It is essential that researchers take a stand and clearly explain their position with regard to the use of animals (Van Zutphen, 2002). To fill these gaps, some initiatives have arisen from scientific organizations addressing the requirement for transparency (Montoliu, 2018). The scientific community hopes that society will soon better understand the benefits of the use of animals in research and will provide greater support for animal experimentation, resolving the current controversies. In addition, initiatives like the Animal Research Reporting of In Vivo Experiments (ARRIVE) guidelines and the European Quality in Preclinical Data (EQIPD) have arose to solve challenges with regard to the robustness, rigor, and validity of research data, which often impact the transition from preclinical to clinical testing.
Ethics and transparency in this context will undoubtedly enhance the quality of biomedical research and societal engagement (Van Zutphen, 2002; Montoliu, 2018).
Author Contributions
SM-C, IV-N, RA, LM, SF, AP, BI, EO, and ES wrote the manuscript. All the co-authors revised and approved the manuscript.
Funding
This work was supported by a grant from the Center for Biomedical Research on Rare Diseases (CIBERER) (ER18GDT761) to SM-C. Additional support came from: Multi Target and View FEDER/CM-B2017/BMD-3688 and MINECO/FEDER SAF2017-86107-R grants to SM-C and IV-N; CERCA Program/Generalitat de Catalunya, Autonomous Government of Catalonia (2017SGR1206), and CIBERER (ACCI18-27) to AP; Miguel Servet program CPII16/00016 (Instituto de Salud Carlos III co-funded by European Social Fund ESF) to SF; CNIC (ISCIII, Ministerio de Ciencia, Innovación y Universidades), Pro CNIC Foundation, Severo Ochoa Center of Excellence (SEV-2015-0505) to BI and EO; Generalitat de Catalunya (Grups consolidats 2017 SGR 926) to MD. EO was beneficiary of a grant from “Programa de Atracción de Talento” of Comunidad de Madrid (2017-T1/BMD-5185). SM-C holds a ISCiii CIBER postdoctoral researcher contract. The work at CRG was supported by grants from the Spanish Ministry of Science and Innovation to the EMBL partnership, the Centro de Excelencia Severo Ochoa and the CERCA Programme/Generalitat de Catalunya.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the helpful discussion and support by our research groups and CIBER colleagues. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
References
Ahmad, G., and Amiji, M. (2018). Use of CRISPR/Cas9 gene-editing tools for developing models in drug discovery. Drug Discov. Today 23, 519–533. doi: 10.1016/j.drudis.2018.01.014
Bodoy, S., Sotillo, F., Espino-Guarch, M., Sperandeo, M. P., Ormazabal, A., Zorzano, A., et al. (2019). Inducible Slc7a7 knockout mouse model recapitulates lysinuric protein intolerance disease. Int. J. Mol. Sci. 20:5294. doi: 10.3390/ijms20215294
Cacheiro, P., Haendel, M. A., and Smedley, D., International Mouse Phenotyping Consortium, and The Monarch Initiative (2019). New models for human disease from the International mouse phenotyping consortium. Mamm. Genome 30, 143–150. doi: 10.1007/s00335-019-09804-5
Camarero, G., Avendano, C., Fernandez-Moreno, C., Villar, A., Contreras, J., De Pablo, F., et al. (2001). Delayed inner ear maturation and neuronal loss in postnatal Igf-1-deficient mice. J. Neurosci. 21, 7630–7641. doi: 10.1523/jneurosci.21-19-07630.2001
Camarero, G., Villar, M. A., Contreras, J., Fernandez-Moreno, C., Pichel, J. G., Avendano, C., et al. (2002). Cochlear abnormalities in insulin-like growth factor-1 mouse mutants. Hear. Res. 170, 2–11. doi: 10.1016/s0378-5955(02)00447-1
Casasnovas, C., Ruiz, M., Schluter, A., Naudi, A., Fourcade, S., Veciana, M., et al. (2019). Biomarker identification, safety, and efficacy of high-dose antioxidants for adrenomyeloneuropathy: a phase II pilot study. Neurotherapeutics 16, 1167–1182. doi: 10.1007/s13311-019-00735-2
Cediel, R., Riquelme, R., Contreras, J., Diaz, A., and Varela-Nieto, I. (2006). Sensorineural hearing loss in insulin-like growth factor I-null mice: a new model of human deafness. Eur. J. Neurosci. 23, 587–590. doi: 10.1111/j.1460-9568.2005.04584.x
Chien, Y. H., Lee, N. C., Tseng, S. H., Tai, C. H., Muramatsu, S. I., Byrne, B. J., et al. (2017). Efficacy and safety of AAV2 gene therapy in children with aromatic L-amino acid decarboxylase deficiency: an open-label, phase 1/2 trial. Lancet Child Adolesc. Health 1, 265–273. doi: 10.1016/s2352-4642(17)30125-6
Cochat, P., Hulton, S. A., Acquaviva, C., Danpure, C. J., Daudon, M., and De Marchi, M. (2012). Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol. Dial. Transplant. 27, 1729–1736. doi: 10.1093/ndt/gfs078
Cohen-Salmon, M., Ott, T., Michel, V., Hardelin, J. P., Perfettini, I., Eybalin, M., et al. (2002). Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr. Biol. 12, 1106–1111. doi: 10.1016/s0960-9822(02)00904-1
Crispino, G., Galindo Ramirez, F., Campioni, M., Zorzi, V., Praetorius, M., Di Pasquale, G., et al. (2017). In vivo genetic manipulation of inner ear connexin expression by bovine adeno-associated viral vectors. Sci. Rep. 7:6567.
Cruz, F. M., Sanz-Rosa, D., Roche-Molina, M., Garcia-Prieto, J., Garcia-Ruiz, J. M., Pizarro, G., et al. (2015). Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll. Cardiol. 65, 1438–1450. doi: 10.1016/j.jacc.2015.01.045
European Commission (2017). Rare Diseases: A Major Unmet Medical Need. Brussels: European Commission.
European Commission (2018). Commission replies to “Stop Vivisection” European Citizens’ Initiative. Brussels: European Commission.
European Commission (2019). 2019 Report on The Statistics on the Use of Animals for Scientific Purposes in the Member States of the European Union in 2015-2017, Report From The Commission To The European Parliament And The Council. Brussels: European Commission.
Ferrer, I., Aubourg, P., and Pujol, A. (2010). General aspects and neuropathology of X-linked adrenoleukodystrophy. Brain Pathol. 20, 817–830. doi: 10.1111/j.1750-3639.2010.00390.x
Fourcade, S., Ferrer, I., and Pujol, A. (2015). Oxidative stress, mitochondrial and proteostasis malfunction in adrenoleukodystrophy: a paradigm for axonal degeneration. Free Radic. Biol. Med. 88, 18–29. doi: 10.1016/j.freeradbiomed.2015.05.041
Fourcade, S., Lopez-Erauskin, J., Galino, J., Duval, C., Naudi, A., Jove, M., et al. (2008). Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum. Mol. Genet. 17, 1762–1773. doi: 10.1093/hmg/ddn085
Fuentes-Santamaria, V., Alvarado, J. C., Rodriguez-De La Rosa, L., Juiz, J. M., and Varela-Nieto, I. (2019). Neuroglial Involvement in Abnormal Glutamate Transport in the Cochlear Nuclei of the Igf1 (-/-) Mouse. Front. Cell. Neurosci. 13:67. doi: 10.3389/fncel.2019.00067
Fuentes-Santamaria, V., Alvarado, J. C., Rodriguez-De La Rosa, L., Murillo-Cuesta, S., Contreras, J., Juiz, J. M., et al. (2016). IGF-1 deficiency causes atrophic changes associated with upregulation of VGluT1 and downregulation of MEF2 transcription factors in the mouse cochlear nuclei. Brain Struct. Funct. 221, 709–734. doi: 10.1007/s00429-014-0934-2
Gahl, W. A., Mulvihill, J. J., Toro, C., Markello, T. C., Wise, A. L., and Ramoni, R. B. (2016). The NIH undiagnosed diseases program and network: applications to modern medicine. Mol. Genet. Metab. 117, 393–400. doi: 10.1016/j.ymgme.2016.01.007
Galea, E., Launay, N., Portero-Otin, M., Ruiz, M., Pamplona, R., Aubourg, P., et al. (2012). Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochim. Biophys. Acta 1822, 1475–1488. doi: 10.1016/j.bbadis.2012.02.005
Gandjbakhch, E., Redheuil, A., Pousset, F., Charron, P., and Frank, R. (2018). Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J. Am. Coll. Cardiol. 72, 784–804.
Garattini, S., and Grignaschi, G. (2017). Animal testing is still the best way to find new treatments for patients. Eur. J. Intern. Med. 39, 32–35. doi: 10.1016/j.ejim.2016.11.013
Hatch, M., Gjymishka, A., Salido, E. C., Allison, M. J., and Freel, R. W. (2011). Enteric oxalate elimination is induced and oxalate is normalized in a mouse model of primary hyperoxaluria following intestinal colonization with Oxalobacter. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G461–G469.
Knight, J., Holmes, R. P., Cramer, S. D., Takayama, T., and Salido, E. (2012). Hydroxyproline metabolism in mouse models of primary hyperoxaluria. Am. J. Physiol. Renal Physiol. 302, F688–F693.
Lai, C., Pursell, N., Gierut, J., Saxena, U., Zhou, W., Dills, M., et al. (2018). Specific inhibition of hepatic lactate dehydrogenase reduces oxalate production in mouse models of primary hyperoxaluria. Mol. Ther. 26, 1983–1995. doi: 10.1016/j.ymthe.2018.05.016
Lavado, A., Jeffery, G., Tovar, V., De La Villa, P., and Montoliu, L. (2006). Ectopic expression of tyrosine hydroxylase in the pigmented epithelium rescues the retinal abnormalities and visual function common in albinos in the absence of melanin. J. Neurochem. 96, 1201–1211. doi: 10.1111/j.1471-4159.2006.03657.x
Lee, N. C., Lee, Y. M., Chen, P. W., Byrne, B. J., and Hwu, W. L. (2016). Mutation-adapted U1 snRNA corrects a splicing error of the dopa decarboxylase gene. Hum. Mol. Genet. 25, 5142–5147.
Lee, N. C., Shieh, Y. D., Chien, Y. H., Tzen, K. Y., Yu, I. S., Chen, P. W., et al. (2013). Regulation of the dopaminergic system in a murine model of aromatic L-amino acid decarboxylase deficiency. Neurobiol. Dis. 52, 177–190. doi: 10.1016/j.nbd.2012.12.005
Legan, P. K., Goodyear, R. J., Morin, M., Mencia, A., Pollard, H., Olavarrieta, L., et al. (2014). Three deaf mice: mouse models for TECTA-based human hereditary deafness reveal domain-specific structural phenotypes in the tectorial membrane. Hum. Mol. Genet. 23, 2551–2568. doi: 10.1093/hmg/ddt646
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J., and Efstratiadis, A. (1993). Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59–72. doi: 10.1016/s0092-8674(05)80084-4
Lopez-Erauskin, J., Fourcade, S., Galino, J., Ruiz, M., Schluter, A., Naudi, A., et al. (2011). Antioxidants halt axonal degeneration in a mouse model of X-adrenoleukodystrophy. Ann. Neurol. 70, 84–92. doi: 10.1002/ana.22363
Lopez-Erauskin, J., Galino, J., Bianchi, P., Fourcade, S., Andreu, A. L., Ferrer, I., et al. (2012). Oxidative stress modulates mitochondrial failure and cyclophilin D function in X-linked adrenoleukodystrophy. Brain 135, 3584–3598. doi: 10.1093/brain/aws292
Martin-Higueras, C., Luis-Lima, S., and Salido, E. (2016). Glycolate oxidase is a safe and efficient target for substrate reduction therapy in a mouse model of primary hyperoxaluria type I. Mol. Ther. 24, 719–725. doi: 10.1038/mt.2015.224
Martin-Higueras, C., Torres, A., and Salido, E. (2017). Molecular therapy of primary hyperoxaluria. J. Inherit. Metab. Dis. 40, 481–489.
Matthews, R. A. (2008). Medical progress depends on animal models - doesn’t it? J. R. Soc. Med. 101, 95–98. doi: 10.1258/jrsm.2007.070164
Milliner, D. S., Harris, P. C., Cogal, A. G., and Lieske, J. C. (1993). “Primary hyperoxaluria type 1,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: NIH).
Montoliu, L. (2018). Transparency in Animal Experimentation [Online]. Available online at: https://network.febs.org/users/104011-lluis-montoliu/posts/32348-transparency-in-animal-experimentation (accessed May 7, 2018).
Montoliu, L., and Marks, M. S. (2017). A new type of syndromic albinism associated with mutations in AP3D1. Pigment Cell. Melanoma Res. 30, 5–7. doi: 10.1111/pcmr.12543
Murillo-Cuesta, S., Contreras, J., Zurita, E., Cediel, R., Cantero, M., Varela-Nieto, I., et al. (2010). Melanin precursors prevent premature age-related and noise-induced hearing loss in albino mice. Pigment Cell. Melanoma Res. 23, 72–83. doi: 10.1111/j.1755-148x.2009.00646.x
Institute of Medicine (2010). Rare Diseases and Orphan Products: Accelerating Research and Development. Washington, DC: The National Academies Press. doi: 10.17226/12953
National Research Council (1998). Biomedical Models and Resources: Current Needs and Future Opportunities. Washington, DC: National Research Council.
Nguengang Wakap, S., Lambert, D. M., Olry, A., Rodwell, C., Gueydan, C., Lanneau, V., et al. (2020). Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur. J. Hum. Genet. 28, 165–173. doi: 10.1038/s41431-019-0508-0
Pujol, A. (2016). Novel therapeutic targets and drug candidates for modifying disease progression in adrenoleukodystrophy. Endocr. Dev. 30, 147–160. doi: 10.1159/000439340
Pujol, A., Ferrer, I., Camps, C., Metzger, E., Hindelang, C., Callizot, N., et al. (2004). Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum. Mol. Genet. 13, 2997–3006. doi: 10.1093/hmg/ddh323
Pujol, A., Hindelang, C., Callizot, N., Bartsch, U., Schachner, M., and Mandel, J. L. (2002). Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum. Mol. Genet. 11, 499–505. doi: 10.1093/hmg/11.5.499
Roche-Molina, M., Sanz-Rosa, D., Cruz, F. M., Garcia-Prieto, J., Lopez, S., Abia, R., et al. (2015). Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler. Thromb. Vasc. Biol. 35, 50–59. doi: 10.1161/atvbaha.114.303617
Ruiz, M., Jove, M., Schluter, A., Casasnovas, C., Villarroya, F., Guilera, C., et al. (2015). Altered glycolipid and glycerophospholipid signaling drive inflammatory cascades in adrenomyeloneuropathy. Hum. Mol. Genet. 24, 6861–6876.
Salido, E., Rodriguez-Pena, M., Santana, A., Beattie, S. G., Petry, H., and Torres, A. (2011). Phenotypic correction of a mouse model for primary hyperoxaluria with adeno-associated virus gene transfer. Mol. Ther. 19, 870–875. doi: 10.1038/mt.2010.270
Salido, E. C., Li, X. M., Lu, Y., Wang, X., Santana, A., Roy-Chowdhury, N., et al. (2006). Alanine-glyoxylate aminotransferase-deficient mice, a model for primary hyperoxaluria that responds to adenoviral gene transfer. Proc. Natl. Acad. Sci. U.S.A. 103, 18249–18254. doi: 10.1073/pnas.0607218103
Schluter, A., Espinosa, L., Fourcade, S., Galino, J., Lopez, E., Ilieva, E., et al. (2012). Functional genomic analysis unravels a metabolic-inflammatory interplay in adrenoleukodystrophy. Hum. Mol. Genet. 21, 1062–1077. doi: 10.1093/hmg/ddr536
Seruggia, D., Fernandez, A., Cantero, M., Pelczar, P., and Montoliu, L. (2015). Functional validation of mouse tyrosinase non-coding regulatory DNA elements by CRISPR-Cas9-mediated mutagenesis. Nucleic Acids Res. 43, 4855–4867. doi: 10.1093/nar/gkv375
Takada, Y., Beyer, L. A., Swiderski, D. L., O’neal, A. L., Prieskorn, D. M., Shivatzki, S., et al. (2014). Connexin 26 null mice exhibit spiral ganglion degeneration that can be blocked by BDNF gene therapy. Hear. Res. 309, 124–135. doi: 10.1016/j.heares.2013.11.009
van Tintelen, J. P., Entius, M. M., Bhuiyan, Z. A., Jongbloed, R., Wiesfeld, A. C., Wilde, A. A., et al. (2006). Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 113, 1650–1658. doi: 10.1161/circulationaha.105.609719
Van Zutphen, L. F. (2002). Use of animals in research: a science - society controversy? The European perspective. Altex 19, 140–144.
Vaquer, G., Riviere, F., Mavris, M., Bignami, F., Llinares-Garcia, J., Westermark, K., et al. (2013). Animal models for metabolic, neuromuscular and ophthalmological rare diseases. Nat. Rev. Drug Discov. 12, 287–305. doi: 10.1038/nrd3831
Varela-Nieto, I., Murillo-Cuesta, S., Rodriguez-De La Rosa, L., Lassatetta, L., and Contreras, J. (2013). IGF-I deficiency and hearing loss: molecular clues and clinical implications. Pediatr. Endocrinol. Rev. 10, 460–472.
Vilches, C., Boiadjieva-Knopfel, E., Bodoy, S., Camargo, S., Lopez De Heredia, M., Prat, E., et al. (2018). Cooperation of antiporter LAT2/CD98hc with uniporter TAT1 for renal reabsorption of neutral amino acids. J. Am. Soc. Nephrol. 29, 1624–1635. doi: 10.1681/asn.2017111205
Wesdorp, M., Murillo-Cuesta, S., Peters, T., Celaya, A. M., Oonk, A., Schraders, M., et al. (2018). MPZL2, encoding the epithelial junctional protein myelin protein zero-like 2, is essential for hearing in man and mouse. Am. J. Hum. Genet. 103, 74–88. doi: 10.1016/j.ajhg.2018.05.011
Winn, S. R., Scherer, T., Thony, B., Ying, M., Martinez, A., Weber, S., et al. (2018). Blood phenylalanine reduction corrects CNS dopamine and serotonin deficiencies and partially improves behavioral performance in adult phenylketonuric mice. Mol. Genet. Metab. 123, 6–20. doi: 10.1016/j.ymgme.2017.10.009
Keywords: orphan diseases, animal models, preclinical research, novel therapies, ethics, transparency
Citation: Murillo-Cuesta S, Artuch R, Asensio F, de la Villa P, Dierssen M, Enríquez JA, Fillat C, Fourcade S, Ibáñez B, Montoliu L, Oliver E, Pujol A, Salido E, Vallejo M and Varela-Nieto I (2020) The Value of Mouse Models of Rare Diseases: A Spanish Experience. Front. Genet. 11:583932. doi: 10.3389/fgene.2020.583932
Received: 15 July 2020; Accepted: 14 September 2020;
Published: 14 October 2020.
Edited by:
E. Ann Yeh, Hospital for Sick Children, CanadaReviewed by:
Rameshwar Nath Koul Bamezai, Jawaharlal Nehru University, IndiaNicolina Cristina Sorrentino, Telethon Institute of Genetics and Medicine (TIGEM), Italy
Copyright © 2020 Murillo-Cuesta, Artuch, Asensio, de la Villa, Dierssen, Enríquez, Fillat, Fourcade, Ibáñez, Montoliu, Oliver, Pujol, Salido, Vallejo and Varela-Nieto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Murillo-Cuesta, c211cmlsbG9AaWliLnVhbS5lcw==; Isabel Varela-Nieto, aS52YXJlbGEubmlldG9AY3NpYy5lcw==