 Leonid Adonin
Leonid Adonin Anatoliy Drozdov
Anatoliy Drozdov Nickolai A. Barlev
Nickolai A. Barlev- 1Moscow Institute of Physics and Technology, Dolgoprudny, Russia
- 2Institute of Environmental and Agricultural Biology (X-BIO), Tyumen State University, Tyumen, Russia
- 3Orekhovich Institute of Biomedical Chemistry, Moscow, Russia
- 4Zhirmunsky National Scientific Center of Marine Biology, Far Eastern Branch of the Russian Academy of Sciences, Vladivostok, Russia
- 5Institute of Cytology, Russian Academy of Sciences, Saint-Petersburg, Russia
The purple sea urchin Strongylocentrotus purpuratus has been used for over 150 years as a model organism in developmental biology. Using this model species, scientists have been able to describe, in detail, the mechanisms of cell cycle control and cell adhesion, fertilization, calcium signaling, cell differentiation, and death. Massive parallel sequencing of the sea urchin genome enabled the deciphering of the main components of gene regulatory networks during the activation of embryonic signaling pathways. This knowledge helped to extrapolate aberrations in somatic cells that may lead to diseases, including cancer in humans. Furthermore, since many, if not all, developmental signaling pathways were shown to be controlled by non-coding RNAs (ncRNAs), the sea urchin organism represents an attractive experimental model. In this review, we discuss the main discoveries in the genetics, genomics, and transcriptomics of sea urchins during embryogenesis with the main focus on the role of ncRNAs. This information may be useful for comparative studies between different organisms, and may help identify new regulatory networks controlled by ncRNAs.
Introduction
The first use of animals for experimental purposes dates back to Ancient Greece. “Generation of Animals” of Aristotle (1942) describes the first systematic study of embryonic development as a phenomenon, which recognizes the key questions about the emergence and relations between hierarchically organized parts of an organism.
Model organisms help in the testing of novel biological hypotheses, which come from in cellulo observations and need to be tested at the whole organism level. Hence, model organisms represent a very important tool in modern biology. Currently, the list of model organisms includes over 100 species of animals, plants, protozoa, and viruses. The most popular model species include the frog, zebrafish, сhicken, mouse, fruit fly, and nematode (Rzepnikowska et al., 2017; Kuo et al., 2018; Chatterjee and Deng, 2019; Marques et al., 2019; Nielsen, 2019; Tang et al., 2019). All these species are used by researchers in a wide range of molecular biological applications, but, unfortunately, none of them are versatile enough to satisfy various experimental needs.
In this review, we focus on the main discoveries in genetics and genomics that were made using a popular model object – the purple sea urchin Strongylocentrotus purpuratus, which has been used in biology for over 150 years (Stimpson, 1857). The species was chosen as a model object for several objective reasons: sea urchins are easy to propagate in the laboratory; it is easy to get synchronous embryo cultures and induce rapid embryogenesis; the embryo is transparent and has a simple structure. Genome sequencing and the description of complex gene regulatory networks during the sea urchin embryogenesis made this model object indispensable for the study of gene expression regulation.
Echinoderms are a sister group of the Chordate phylum. This group has branched out from Chordate before the Cambrian period (more than 500 million years ago, Figure 1; McClay, 2011). Other studies, based on the multigene and multiprotein studies, indicate a more accurate time of divergence (Hedges, 2002; Zaidel-Bar, 2009).
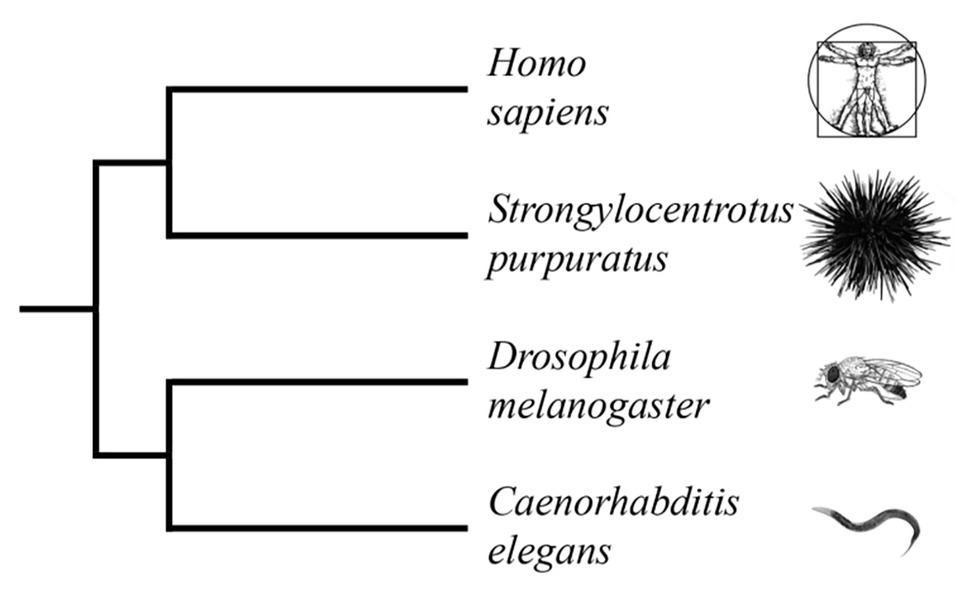
Figure 1. Truncated phylogenetic tree of popular model organisms based on combined analyses of morphology and molecular data (Laumer et al., 2015; Telford et al., 2015; Torruella et al., 2015; Cannon et al., 2016). The tree illustrates the evolutionary relationship between Homo sapiens and Strongylocentrotus purpuratus as members of the deuterostome branch of the animal kingdom. Caenorhabditis elegans and Drosophila melanogaster are members of the protostome branch. (Branch lengths are not proportional to time).
The first studies, which describe the normal embryogenesis of a sea urchin, date back to the middle of the 19th century. Since then, sea urchin embryos have become a popular model in developmental biology. The normal life cycle of sea urchins is shown in Figure 2. At present, certain echinoid species (e.g., S. purpuratus, Strongylocentrotus droebachiensis, Strongylocentrotus intermedius, Hemicentrotus pulcherrimus, Lytechinus variegatus, Paracentrotus lividus, and Mesocentrotus franciscanus) are widely used as experimental models in developmental biology (McClay, 2011). The early stage of embryogenesis of the purple sea urchin was used for studying intercellular communication and cell adhesion (Horstadius, 1939; Giuduce, 1962; McClay and Fink, 1982), cell cycle control mechanisms (Evans et al., 1983), calcium signaling (Whitaker, 2006), fertilization (Briggs and Wessel, 2006), cell differentiation (Giudice, 1973), and cell survival and death (Chiarelli et al., 2016).
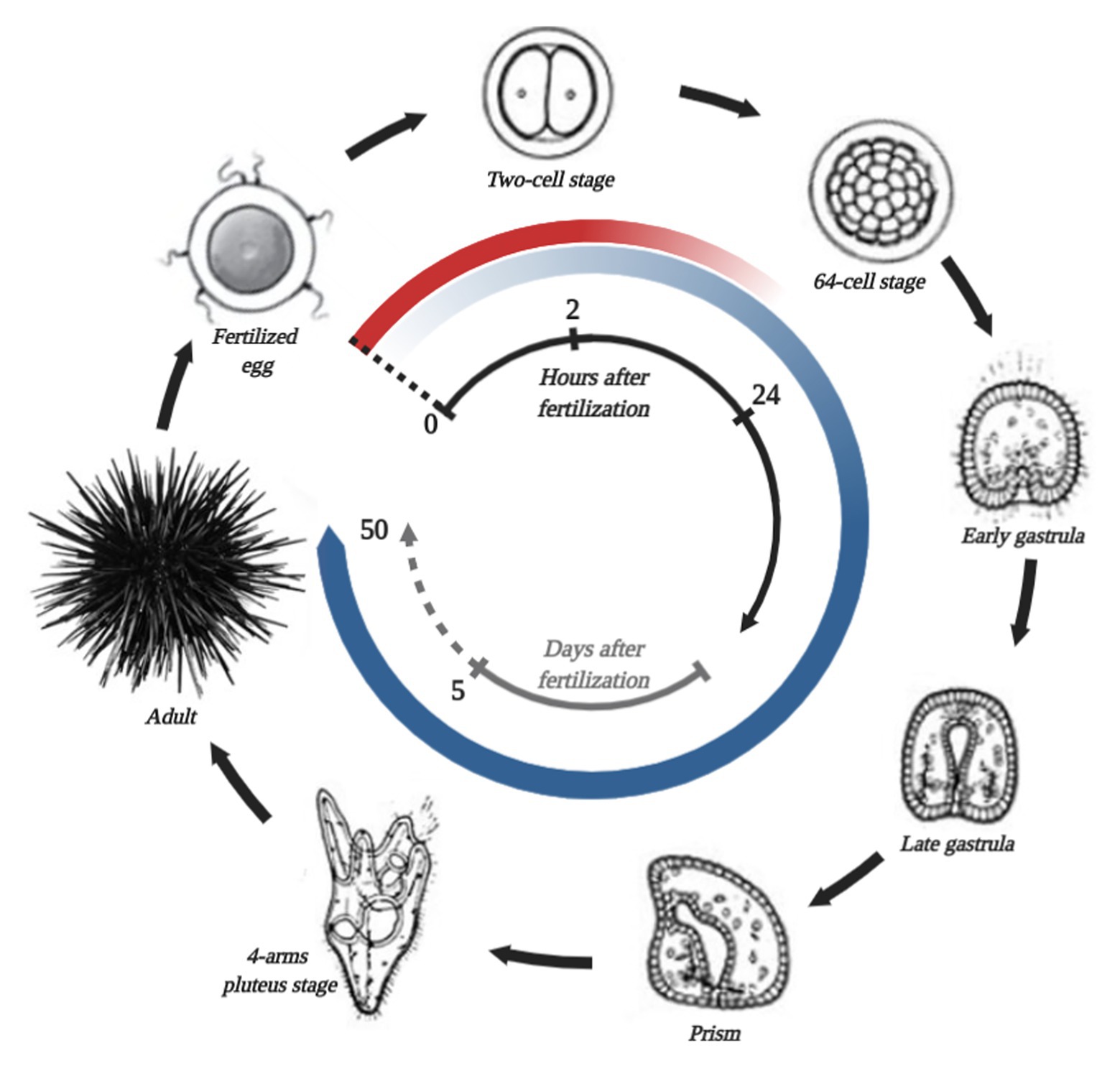
Figure 2. Simplified S. purpuratus life cycle, stages of which are connected by black arrows. The name of each stage is shown in the picture. The beginning of the life cycle is fertilization, which is marked by a black dotted line. In the center, the black and gray time scale represents hours and days after fertilization, correspondingly. The red circular gradient line represents the degradation of the general maternal transcripts. The circular blue gradient shows the beginning of zygotic genome activation and increases in transcribed gene numbers.
The genome size of the purple sea urchin is only a quarter of the human genome, despite it having about the same number of genes. Genome analysis has shown that most of its genes are common to representatives of deuterostomes, which has in turn uncovered an unexpectedly close relation to humans among all used invertebrate model species (Davidson, 2006; Sea Urchin Genome Sequencing Consortium et al., 2006; Cameron, 2014). For example, the sea urchin genome contains orthologs of human disease-associated genes, which are expressed in sea urchin embryogenesis.
The sea urchin genome was shown to contain more than 400 genes, whose products are involved in the regulation of cell homeostasis. Most of these genes display a remarkable conservation of their sequences during the evolution (Goldstone et al., 2006; Rast et al., 2006). The sea urchin genome contains 65 genes of the ATP-binding cassette transporter superfamily (Hamdoun et al., 2004; Goldstone et al., 2006), while, in humans, only 48 members of this family are known to date (Dean and Annilo, 2005). Mutations in these genes lead to several pathologies in humans, including degeneration of the retina, cystic fibrosis, neurological diseases, cholesterol transport disorders, anemia, and many others (Dean et al., 2001). The sea urchin as an experimental model is also frequently used in toxicology and in environmental human health science since it allows an accurate estimate of cancer risk before any epidemiologic evidence is available (Bellé et al., 2007).
Furthermore, sea urchin embryos are used by scientists as a convenient object for elucidating common cellular molecular mechanisms involved in human health and disease. In particular, the sea urchin is used as a model system for studying neurodegenerative disorders that can cause dementia and memory loss (Nakajima et al., 2004).
In the past, it was hypothesized that certain signaling pathways involved in the embryo’s morphogenesis could be aberrantly activated during tumorigenesis. Unfortunately, to develop this idea further, scientists did not have an appropriate human experimental model. This is due to various ethical aspects that restricted human embryo studies (De Wert et al., 2002; De Wert and Mummery, 2003; Holm, 2003; Lo and Parham, 2009). Therefore, an early embryogenesis of sea urchins could be a good model for cancer research.
The normal processes of cell proliferation and differentiation are controlled by several developmental gene regulatory networks. Disunity in these networks leads to the initiation and progression of tumors (Hanahan and Weinberg, 2000; Reya et al., 2001; Pires-daSilva and Sommer, 2003).
Notch, Wnt, and Hedgehog (Hh) signaling pathways are highly conserved from sea urchins to humans. The current model of these pathways, including general components shown on Figure 3 and their main roles in embryogenesis and cancer, is described on Table 1.
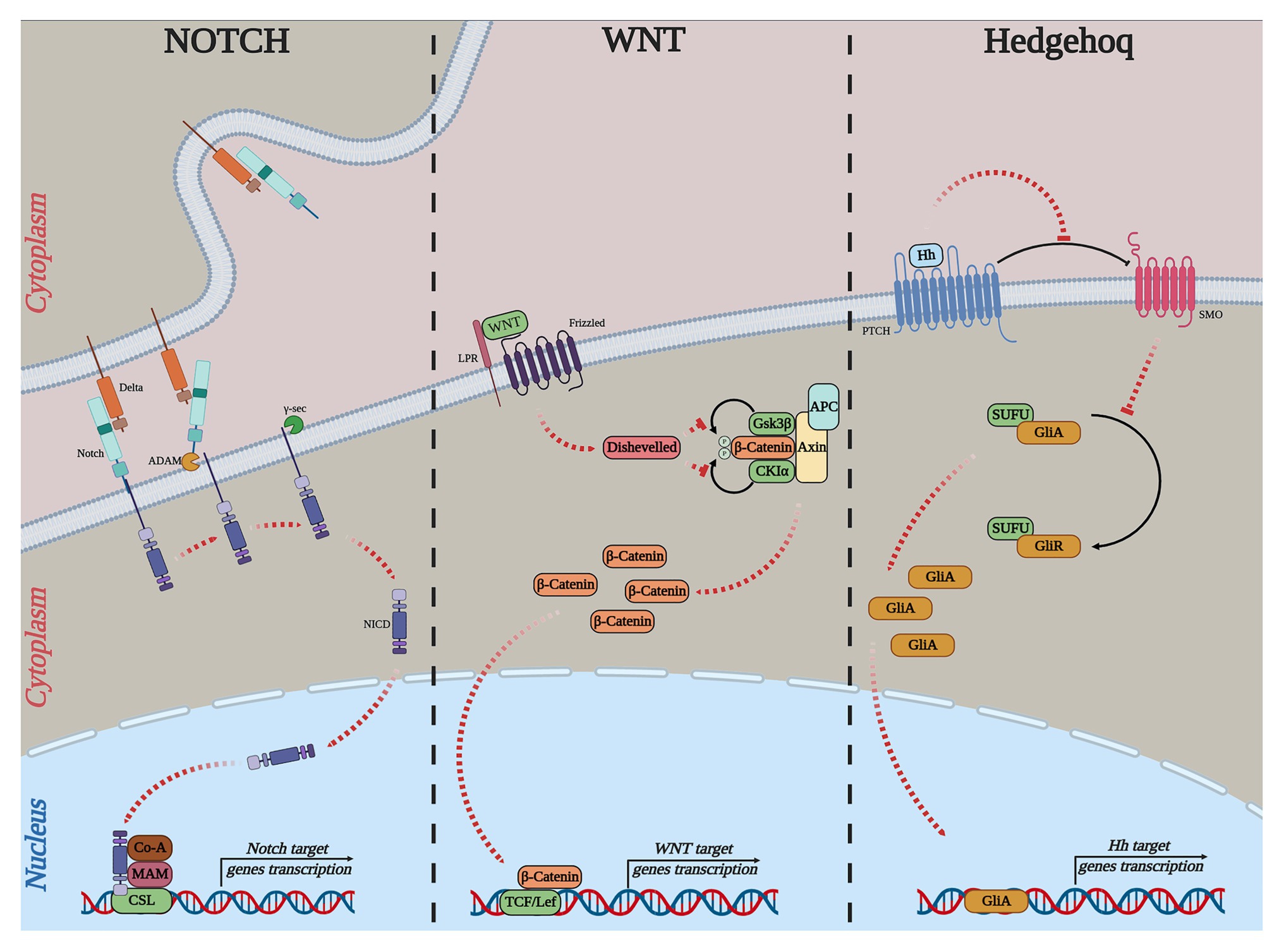
Figure 3. Simple outline of the current models of the canonical Notch, WNT, and Hh pathways. NOTCH: Delta, delta-like ligand, Notch, ADAM, ADAM-family metalloprotease; γ-sec, γ-secretase; NICD, notch intracellular domain; Co-A, transcription coactivator; MAM, conserved and essential nuclear factor mastermind; CSL, DNA-binding transcription factor. WNT: Frizzled; WNT, wingless-type MMTV integration site; LRP, low-density lipoprotein receptor-related protein; APC, adenomatous polyposis coli; Disheveled, cytoplasmic phosphoprotein; GSK3ß, glycogen synthase kinase-3; CK1α, casein kinase 1 alpha; TCF, T-cell-specific transcription factor; LEF, lymphoid enhancer-binding factor. Hedgehog: Hh, hedgehog; PTCH, patched; SMO, smoothened; SUFU, suppressor of fused; GLI, GLI-family zinc finger.

Table 1. Major Notch, WNT, and Hedgehog (Hh) pathways roles in multicellular organisms’ embryo development and cancer.
The Hh pathway plays a crucial role in many fundamental processes of metazoan organisms, including tissue homeostasis and their embryonic development. In the development of the sea urchin, this pathway controls the establishment of the left–right asymmetry in embryos (Warner et al., 2016). According to the previously proposed model, this signaling pathway, similar to vertebrates, provides an asymmetrical expression of Nodal, which is an important cytokine of the TGF beta superfamily. Moreover, in humans, the aberrant activation of this signaling pathway is increasingly associated with various cancers. For example, Hh was shown to control proliferation, malignancy, and metastasis (Sari et al., 2018). In particular, while the Hh signaling pathway is mainly repressed during mammary embryonic development, overexpression of some components (PTCH1, GLI1/2) of the Hh are up-regulated in tumor stem cells of human breast cancer (Liu et al., 2006).
The key components of Notch signaling are present in all metazoan organisms (Gordon et al., 2008). The canonical Notch pathway begins when a ligand of the Delta/Serrate/LAG-2 (DSL) family binds to the transmembrane receptor protein, Notch (Fehon et al., 1990). The schema of all stages of the process is shown in Figure 3 (part NOTCH). It is well established that the Delta/Notch signaling pathway is intimately involved in mesoderm formation. Subsequently, deregulation of this pathway leads to the elimination of mesoderm derivatives during the embryogenesis of sea urchins (Sherwood and McClay, 1999, 2001; Sweet et al., 2002; Croce and McClay, 2010). In humans, members of the Notch signaling pathway play a key role in embryonic vasculature development (Patel et al., 2005). Notch can in fact be either oncogenic or tumor suppressive depending on the tissue and cellular context. In addition, this pathway is one of the most activated in cancer cells and contributes to metastasis (Venkatesh et al., 2018). For example, the development of squamous cell carcinomas in various epithelial tissues is directly related to mutations in members of the Notch family. These mutations represent the most common cause of misregulation of this signaling pathway (Nowell and Radtke, 2017).
The Wnt signaling pathway regulates the embryogenesis and homeostasis of multicellular organisms. In sea urchin embryos, the Wnt signaling pathway contributes to the activation of the endomesodermal gene regulatory network, whose genes start their expression on the 16-cell stage of embryos (Kumburegama and Wikramanayake, 2008). Also, this pathway regulates the formation of the animal–vegetal (A–V) axis in sea urchin and sea anemone embryos.
In humans, cancer, obesity, and diabetes are the result of the Wnt pathway dysregulation (Langton et al., 2016; Mzoughi et al., 2017). It is shown that the pathway is involved in the regulation of the metabolism of cancer cells, which facilitates tumor progression (Lu et al., 2010; Wang and Kunze, 2015; Poli et al., 2018; Sun et al., 2018).
In the comparison analysis, it was shown that the sea urchin genome contains about 90% of the described homologous components of Wnt signal transduction pathways (Croce et al., 2006; Robert et al., 2014). However, from 13 known Wnt subfamilies, S. purpuratus has only 11: it is missing only Wnt2 and Wnt11 homologs. Meanwhile, last year Croces’ group identified a gene encoding Wnt2 ortholog in the genome of a related sea urchin P. lividus. However, they found no evidence of a bona fide wnt2 gene in S. purpuratus when they reanalyzed its genome (Robert et al., 2019). A consortium of scientists was able to find only about half of the Wnt transcriptional target genes that were reported in the literature.
The purple sea urchin can also be considered as a model of gene expression in the normal developmental processes and is used now as an in vivo model to evaluate the Epithelial/Mesenchymal Transition (EMT; Romancino et al., 2017). In humans, reactivated EMT drives organ fibrosis and tumor progression (Lim and Thiery, 2012; Wu et al., 2012; Nieto et al., 2016). The process of EMT is regulated by a cohort of specific transcription factors that includes Zeb1/Zeb2, Snail, Slug, and Twist (Tulchinsky et al., 2019). Together with chromatin-modifying enzymes, these factors exert both repressive and activating functions. For example, Zeb1, by binding to the E-box consensus site in the DNA, inhibits the transcription of the CDH1 gene, whose product plays a critical role in forming cell-cell junctions. On the other hand, when Zeb1 complexes with Yap1, a member of the Hyppo pathway, it becomes a transcriptional activator to control the transcription of CTGF and AXL genes (Lehmann et al., 2016).
The Postgenomic ERA in Sea Urchin-Related Research
Sea Urchin Genome Sequencing and Analysis
As mentioned before, the sea urchin genome reveals striking similarities to humans and shares with the latter a lot of common gene regulatory pathways.
The first assembly and annotation of the sea urchin genome results were published in 2006 (Sea Urchin Genome Sequencing Consortium et al., 2006) and initiated an active exploration of its genomics and transcriptomics. Over the past 14 years, researchers refined the assembly and annotation of the sea urchin genome. Additional genomic and transcriptomic resources were created, for example, EchinoBase.1
The primary assessment of the purple sea urchin genome was estimated as ∼800 Mb in size (Hinegardner, 1971). After the deep-sequencing refinement, the purple sea urchin genome was predicted to contain 23,300 genes (Sea Urchin Genome Sequencing Consortium et al., 2006). The current genome analysis revealed 33,491 genes (and 556 pseudogenes) that encode 38,439 proteins.2
A comparative analysis of the sea urchin genome with vertebrates revealed an unprecedented complexity relative to other animals in terms of their innate immune recognition receptors (Rast et al., 2006). The SUGSC research team assumed that about 4–5% of all the sea urchin genes identified are involved directly in the immune functions (Sea Urchin Genome Sequencing Consortium et al., 2006).
Around 222 members of the Toll-like receptor family and 203 genes of the NACHT domain–LRR family were described in the sea urchin genome in addition to genes from a large family of cysteine-rich receptor proteins (Rast et al., 2006). The sea urchin immune system showed the presence of a complement system similar to the chordate (Hibino et al., 2006).
Since transcriptional networks are regulated by transcription factors, it is important to mention the work of Howard-Ashby’s team that described the main families of genes coding for transcription factors in the S. purpuratus genome (bHLH, Nuclear Receptor, Basic Leucine Zipper, T-box, Smad, Sox, and other smaller families). The number of genes encoding transcription factors of each family in the sea urchin is comparable to that found in the Drosophila genome, but it is almost twice less than the number of such genes found in the human genome. A similar result was obtained when analyzing the situation with HomeoBox genes (Howard-Ashby et al., 2006). The evolvement of new genes during the evolution since branching Echinodermata from the common Deuterostome branch is associated with the adaptation process, increasing the level of complexity and/or changing the key cellular mechanisms.
During the course of genome analysis, the SUGSC team identified more than 1,200 genes involved in signal transduction. The S. purpuratus genome contains 353 protein kinases (Sea Urchin Genome Sequencing Consortium et al., 2006) and 14 lipid kinases (Bradham et al., 2006). The number of sea urchin protein kinases is higher than that described in the Drosophila genome (about 230 members) but less than in the human kinome (518). Although most of the sea urchin kinase subfamilies are often represented by only a single member, their diversity is surprisingly high and corresponds to approximately 97% of the whole human kinome. Only four subfamilies of kinases are missing (Axl, FastK, H11, and NKF3) in the sea urchin compared to the human kinome, whereas the fruit fly kinome lacks 20 of those, and the kinome of worms misses 32 subfamilies (Bradham et al., 2006). Importantly, it has been shown that approximately 88% of described kinases are expressed during embryogenesis (Bradham et al., 2006; Byrum et al., 2006).
To follow the compilation of similarities in the gene ontology of the sea urchin and of humans, it is important to note that they share common mechanisms of cell cycle control. Perhaps, not surprisingly, a number of genes involved in cell cycle control and DNA metabolism have been described for the sea urchin, although its number is lower compared to the human genome. In addition, a few cases of echinoderm-specific gene diversifications have been described (Fernandez-Guerra et al., 2006). Notably, the sea urchin genome contains orthologs of almost all cyclin-dependent kinases, except CDK3. Members of the NIMA-related kinases family (NEK proteins) are, judging by their complexity, close to vertebrates, whereas the complexity of Polo and Aurora mitotic kinase families are close to those found in the worm (Fernandez-Guerra et al., 2006).
Furthermore, a number of known genes involved in DNA replication, repair, and the mitotic checkpoint were also found in the sea urchin. Interestingly, the sea urchin has a single p63/p73 hybrid homologous to the p53, p63, and p73 members of the p53 family of tumor suppressors (Belyi et al., 2010). In addition, the sea urchin contains two homologs of the pRB tumor suppressor and also one homolog of the p21/p27 family of CDK inhibitors (Fernandez-Guerra et al., 2006). Furthermore, the sea urchin genome shares four families of RAS GTPases with humans: Ras, Rho, Rab, and Afr, although 90% of all small GTPases are expressed during embryogenesis (Beane et al., 2006).
Gene Regulatory Networks
As early as in the pre-genome era, common features, and concepts, of the gene regulatory network (GRN) were described by researchers who used the S. purpuratus sea urchin as a model organism. Genome sequencing and annotation made it possible to structure the information, which led to the creation of one of the most complete networks for the regulation of genes during the early embryogenesis (Davidson et al., 2002; Sea Urchin Genome Sequencing Consortium et al., 2006; Oliveri et al., 2008; Peter and Davidson, 2010, 2017; Peter et al., 2012; Martik et al., 2016).
The key step toward the understanding of basic mechanisms of embryogenesis and global GRN was the deep sequencing of RNA. This allowed the accumulation of data on gene expression networking during the embryogenesis of S. purpuratus (Rafiq et al., 2014; Tu et al., 2014; Barsi et al., 2015; Gildor et al., 2016; Israel et al., 2016; Janies et al., 2016; Pérez-Portela et al., 2016). The described schemes of the GRN became the best tool for the analysis of the development of the genetic control (Peter and Davidson, 2017).
In the study by Tu et al. (2014), the expression profiles of more than 16,000 genes were measured during embryogenesis. For a clearer presentation of the expression profiles of embryonic genes, the authors performed a cluster analysis. The clusters were grouped into four main groups according to their overall dynamics: “off,” “on,” “transient,” and “other” (Tu et al., 2014). They showed that complex expression patterns of many genes underlie embryonic development, especially in the early stages preceding gastrulation.
A study of Rafiq (2014) sets a basis for understanding the genomic regulatory control of a major morphogenetic process – skeletal morphogenesis for embryogenesis. The authors have identified 420 transcripts whose expression levels in primary mesenchymal cells (PMC) were significantly different from other samples. Most of these genes are transcribed at relatively low levels at the stage of mesenchymal blastula. They were targeted at Ets1 and Alx1, key transcription factors that provide regulatory inputs at the top of the PMC regulatory differentiation network.
It was shown that more than half of the identified transcripts received essential inputs from Ets1 and/or Alx1, most of which were positive. All these data point to their key role in the cell-specific identity of PMCs (Rafiq et al., 2014). Additionally, the authors described about 200 transcripts that were not significantly affected by Ets1 or Alx1 knockdown.
The name of the GRN concept implies that exons play a major role in gene cascades. However, The Human Genome Project and the subsequent deciphering of a large number of genomes made it obvious that the bulk of the genome consists of sets of repetitive DNA (Lander et al., 2001). Coding DNA fragments (exons) occupy no more than 2–3% of the genome (Carey, 2017). The major components of the genome are represented by two groups of repetitive DNA sequences: tandem repeats and dispersed repeats or transposons [transposable elements, (TE); Paço et al., 2019]. Almost all eukaryotic genomes contain TE, for example, it occupies about half of the human genome (Wang and Kunze, 2015).
Non-coding RNA in the Sea Urchin
It is well established that almost all of the human genome is transcribed into RNA. Surprisingly, most of the transcribed RNA does not code for proteins and is called non-coding RNA (ncRNA; Lander et al., 2001; Carninci et al., 2005; ENCODE Project Consortium et al., 2007; Djebali et al., 2012; Laurent et al., 2015; Carey, 2017), including microRNA (miRNA, 22–25 bp) and long non-coding RNA (lncRNA, <200 bp). NcRNAs are involved in the transcription regulation of genes and other ncRNA (Li and Liu, 2019). The primary source of all kinds of ncRNA in the genome is transposons (Hadjiargyrou and Delihas, 2013).
MicroRNAs control gene expression via multiple modes (Cai et al., 2009; Steitz and Vasudevan, 2009). In general, the 5' proximal “seed” region (nucleotide 2–8) of miRNAs exhibits imperfect complementarity to the 3’UTR of the target mRNA (Lewis et al., 2005). Subsequently, this newly formed double-stranded RNA is destroyed by dsRNAse, RISC. However, a few cases have been reported when miRNAs regulated the expression by binding the 5'UTR of mRNAs, thereby interfering with the binding of translation initiation factor, eIF4 (Lee et al., 2009; Brümmer and Hausser, 2014).
To date, several models have been proposed describing the consequences of the interaction between the miRNA complex and their targets. The miRNA-dependent gene silencing can be achieved at three stages, including pre-translational, co-translational, and post-translational steps (Finnegan and Matzke, 2003; Garneau et al., 2007; Grewal and Elgin, 2007; Fabian et al., 2010; Carroll et al., 2014).
The lncRNA-driven transcriptional regulation is more complex and includes several mechanisms: (1) lncRNA can recruit a regulatory protein complex to a gene or an entire chromosome; (2) the binding of a transcriptional factor is inhibited by lncRNA; (3) transcription of lncRNAs regulates the transcription of adjacent protein-coding genes; and (4) the heterochromatic or euchromatic organization of regions in close proximity stabilizes these territories and controls the spreading of post-translational modifications to nearby chromatin (Long et al., 2017). Furthermore, lncRNAs play a key role in stem cell differentiation, immune response, epigenetic regulation, inflammation-related diseases, and tumor development (Briggs et al., 2015; Huarte, 2015; Boon et al., 2016; Chen et al., 2017).
Description and analysis of ncRNA regulatory networks could provide new insights into gene transcription regulation not only during the embryonic development, but also in cancer, when specific developmental programs are aberrantly reactivated. Thus, understanding the complexity of these regulatory networks will make it possible to determine the consequences of their disruption in the course of various diseases.
Regulatory Networks Based on Non-Coding RNAs
After the publication of the sea urchin genome annotation, miRNAs were identified (Peterson et al., 2009; Wheeler et al., 2009; Campo-Paysaa et al., 2011). These studies revealed that a few, very conserved, miRNAs are also present in humans (Song et al., 2012). The authors had cloned and sequenced small RNAs (18–40 nucleotides) from different embryo stages, ranging from unfertilized eggs to larva pluteus. Around 49 miRNAs were identified, in which three of these were novel miRNAs (not annotated in miRBase previously). Most of the miRNAs are present in the egg and have dynamic accumulation profiles, with the majority of these being upregulated during gastrulation.
To test the function of miRNA during the embryonic development, authors decided to suppress the dsRNA processing enzyme, Dicer, with Dicer morpholino antisense oligonucleotides (MASO). The majority of injected embryos successfully developed to the stage of blastula. However, at the stage of 48 h p.f., embryos that expressed Dicer MASO failed to enter the gastrulation stage (Song et al., 2012). Developmental defects varied from general retardation to cell death.
One interesting example of opposing functions exerted by one micrRNA is miR-31. Importantly, it is expressed during the embryogenesis of S. purpuratus and suppresses several components of PMC GRN (Pmar1, Alx1, Snail, and VegfR7). Knockdown of miR-31 causes a disturbance of the function of PMC that forms the embryonic skeleton (Stepicheva and Song, 2015). Meanwhile, in humans, miR-31 is considered as a tumor suppressor. Yet, it can affect several signaling pathways that have opposite effects on the proliferation: RAS/MARK and PI3K/AKT stimulate growth, whereas RB/E2F inhibits it. Unfortunately, specific molecular mechanisms that regulate miR-31 in the sea urchin are not known at the moment.
The latest analysis of the sea urchin genome showed that known transposons3 occupy about 15% of the genome, including the major class DNA transposons (Figure 4; Lebedev et al., 2019). The percentage of genome occupied by transposons is higher than that of the worm Caenorhabditis elegans but less than that of fruit fly or human (Kazazian and Moran, 2017).
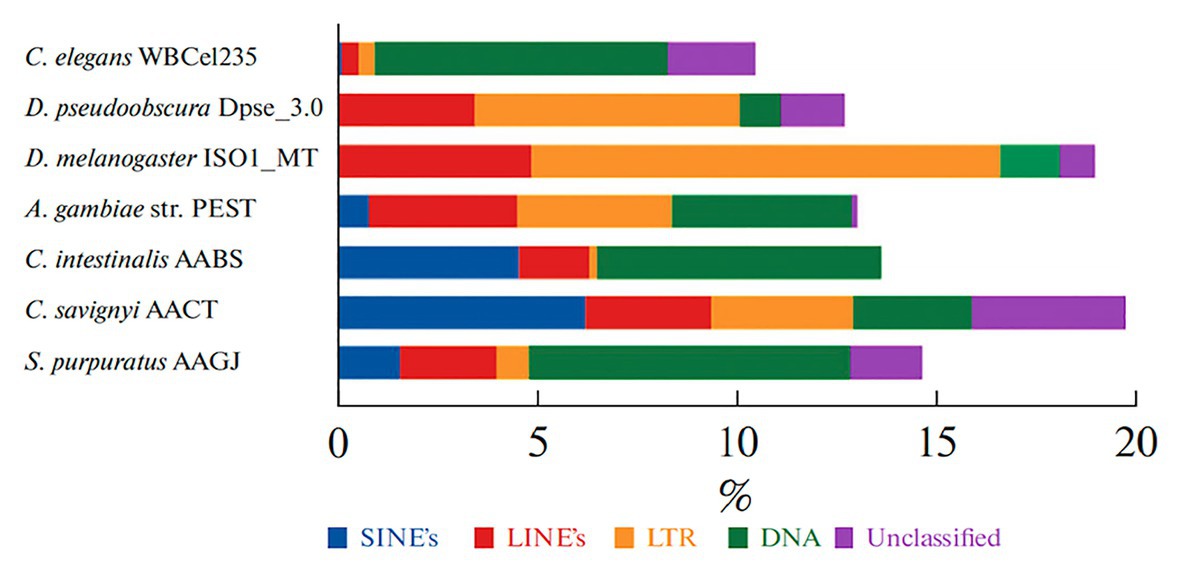
Figure 4. The percent content of transposable elements (TE) in genomes of the sea urchin S. purpuratus and other invertebrates. TE classes are marked with a color: nonLTR SINE – blue; LINE – red; LTR TE – yellow; DNA transposons – green; non-annotated repeats – lilac (from Lebedev et al., 2019).
The development of new sequencing techniques, such as RNA-Seq, has greatly advanced our understanding and knowledge of new RNAs. In one of the latest studies on this, Hezroni and co-authors, using a PLAR-algorithm, predicted more than 5,000 new sequences of lncRNA in sea urchin transcriptomes (Hezroni et al., 2015).
Genes that code for lincRNAs are more species-specific and less conserved than the gene encoding proteins. In the genome of the sea urchin, synthenic (homologous genes situated on the same chromosomes but in different species) lncRNA genes were identified for more than 2,000 human lincRNA genes. This suggests that the sea urchin likely contains a lot of conserved functional vertebrate lincRNA homologs (Hezroni et al., 2015). Of all detected syntenic lincRNAs, only 18 were found in other amniotes.
One example is LINC00261, located downstream of the Foxa2 gene, which codes for a transcription factor. In all vertebrates, this lincRNA is expressed in endodermal tissues, and in sea urchins, it is expressed in the gut. LINC00261 plays the tumor suppressive role being involved in the regulation of DNA damage (Shahabi et al., 2019).
Another syntenic lincRNA (partially annotated as LINC01122 and LOC101927285 in humans) is expressed in the brain and reproductive tissues across vertebrates. In sea urchins, it is expressed in the adult ovary. Unfortunately, the specific functions of both orthologs are unknown. However, this region is located between the Fancl and Bcl11a loci and one can assume that ncRNA expressed from this locus, at least in humans, may participate in the regulation of DNA damage response and/or apoptosis possibly through the p53 regulatory network.
Well-known embryogenesis signaling pathways conserved between different species can serve as the starting point for understanding the complex network of development organization. In recent years, ncRNAs have rapidly emerged as crucial regulators of main signaling pathways in embryo development and cancer (Fu et al., 2019). Regulation takes place at different levels: from the transcriptional to the post-transcriptional and translational levels, for example, LncRNA is involved in the regulation of WNT, Notch, and other signaling pathways in cancer (Li and Kang, 2014; Trimarchi et al., 2014; Yuan et al., 2014; Ong et al., 2017; Peng et al., 2017; Shen et al., 2017).
Future of This Model Organism for Gene Expression Studies
Embryogenesis is regulated by complicated mechanisms to ensure that different types of cells and tissues develop from one cell. All processes of embryogenesis are strictly coordinated by signaling pathways. Next-generation sequencing and bioinformatics methods have made it possible to describe the main components of these pathways (Figure 3) and the branched gene regulatory networks.
Studying the regulatory networks of development and its organization on all levels requires experimental models. It seems to us that this model object – the purple sea urchin S. purpuratus – is the most suitable system for studying the regulation system based on ncRNAs. Further deep bioinformatic analysis of the genome, and the transcriptomic profiling of embryogenesis stages of this model object, will promote important discoveries in gene networking.
Spontaneous alterations in these coordinated gene expression programs can lead to the development of an unhealthy embryo. Furthermore, reactivation of these pathways in somatic cells can cause many diseases, including cancer in humans (Table 1).
Interestingly, the history of modern cancer research begins with the sea urchin: in the first decade of the 20th century, the German biologist Boveri discovered that unproper fertilization of sea-urchin eggs with two sperm rather than one led to chromosomal aberrations and to the failure of proper development (Laubichler and Davidson, 2008). Furthermore, purple sea urchins and some other urchin species (L. variegatus, M. franciscanus) retain the ability to regenerate lost or damaged tissues with age (Bodnar and Coffman, 2016).
However, cancers have not been detected in sea urchins. In fact, the life span of some sea urchin species reaches 100 years (Ebert, 2010; Kober and Bernardi, 2013). How such genomic stability is achieved and what the regulatory transcription mechanisms involved in the longevity of these organisms are require further investigation. Thus, sea urchins can provide insights into the processes in cases of serious human diseases associated with the regulation of transcription.
Author Contributions
LA drafted the manuscript. AD and NB supervised the preparation of the draft. NB helped with writing, proofreading, and editing the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
LA and NB appreciate the support of RSF grant # 20-15-00189. NB appreciates the support of the grant from the Russian Government Program for the Recruitment of the leading scientists into the Russian Institutions of Higher Education 14.W03.31.0029. LA appreciates the Ministry of Science and Higher Education of the Russian Federation (agreement # 075-00337-20-03, project FSMG-2020-0004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Aristotle,. (1942). Generation of animals.
Artavanis-Tsakonas, S., and Muskavitch, M. A. (2010). Notch: the past, the present, and the future. Curr. Top. Dev. Biol. 92, 1–29. doi: 10.1016/S0070-2153(10)92001-2
Bailey, J. M., Mohr, A. M., and Hollingsworth, M. A. (2009). Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 28, 3513–3525. doi: 10.1038/onc.2009.220
Barsi, J. C., Li, E., and Davidson, E. H. (2015). Geometric control of ciliated band regulatory states in the sea urchin embryo. Development 142, 953–961. doi: 10.1242/dev.117986
Beachy, P. A., Karhadkar, S. S., and Berman, D. M. (2004). Tissue repair and stem cell renewal in carcinogenesis. Nature 432, 324–331. doi: 10.1038/nature03100
Beane, W. S., Gross, J. M., and McClay, D. R. (2006). RhoA regulates initiation of invagination, but not convergent extension, during sea urchin gastrulation. Dev. Biol. 292, 213–225. doi: 10.1016/j.ydbio.2005.12.031
Bellé, R., Le Bouffant, R., Morales, J., Cosson, B., Cormier, P., and Mulner-Lorillon, O. (2007). L’embryon d’oursin, le point de surveillance de l'ADN endommagé de la division cellulaire et les mécanismes à l'origine de la cancérisation [Sea urchin embryo, DNA-damaged cell cycle checkpoint and the mechanisms initiating cancer development]. J. Soc. Biol. 201, 317–327. doi: 10.1051/jbio:2007030
Belyi, V. A., Ak, P., Markert, E., Wang, H., Hu, W., Puzio-Kuter, A., et al. (2010). The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2:a001198. doi: 10.1101/cshperspect.a001198
Bodnar, A. G., and Coffman, J. A. (2016). Maintenance of somatic tissue regeneration with age in short- and long-lived species of sea urchins. Aging Cell 15, 778–787. doi: 10.1111/acel.12487
Boon, R. A., Jaé, N., Holdt, L., and Dimmeler, S. (2016). Long noncoding RNAs: from clinical genetics to therapeutic targets? J. Am. Coll. Cardiol. 67, 1214–1226. doi: 10.1016/j.jacc.2015.12.051
Bradham, C. A., Foltz, K. R., Beane, W. S., Arnone, M. I., Rizzo, F., Coffman, J. A., et al. (2006). The sea urchin kinome: a first look. Dev. Biol. 300, 180–193. doi: 10.1016/j.ydbio.2006.08.074
Bray, S. J. (2016). Notch signalling in context. Nat. Rev. Mol. Cell Biol. 17, 722–735. doi: 10.1038/nrm.2016.94
Briggs, E., and Wessel, G. M. (2006). In the beginning...animal fertilization and sea urchin development. Dev. Biol. 300, 15–26. doi: 10.1016/j.ydbio.2006.07.014
Briggs, J. A., Wolvetang, E. J., Mattick, J. S., Rinn, J. L., and Barry, G. (2015). Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron 88, 861–877. doi: 10.1016/j.neuron.2015.09.045
Brümmer, A., and Hausser, J. (2014). MicroRNA binding sites in the coding region of mRNAs: extending the repertoire of post-transcriptional gene regulation. Bioessays 36, 617–626. doi: 10.1002/bies.201300104
Byrum, C. A., Walton, K. D., Robertson, A. J., Carbonneau, S., Thomason, R. T., Coffman, J. A., et al. (2006). Protein tyrosine and serine-threonine phosphatases in the sea urchin, Strongylocentrotus purpuratus: identification and potential functions. Dev. Biol. 300, 194–218. doi: 10.1016/j.ydbio.2006.08.050
Cai, Y., Yu, X., Hu, S., and Yu, J. (2009). A brief review on the mechanisms of miRNA regulation. Genomics Proteomics Bioinformatics 7, 147–154. doi: 10.1016/S1672-0229(08)60044-3
Cameron, R. A. (2014). Tools for sea urchin genomic analysis. Methods Mol. Biol. 1128, 295–310. doi: 10.1007/978-1-62703-974-1_20
Campo-Paysaa, F., Sémon, M., Cameron, R. A., Peterson, K. J., and Schubert, M. (2011). microRNA complements in deuterostomes: origin and evolution of microRNAs. Evol. Dev. 13, 15–27. doi: 10.1111/j.1525-142X.2010.00452.x
Cannon, J. T., Vellutini, B. C., Smith, J. 3rd, Ronquist, F., Jondelius, U., and Hejnol, A. (2016). Xenacoelomorpha is the sister group to Nephrozoa. Nature 530, 89–93. doi: 10.1038/nature16520
Carey, N. (2017). Junk DNA: A journey through the dark matter of the genome. Columbia University Press.
Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., et al. (2005). The transcriptional landscape of the mammalian genome. Science 309, 1559–1563. doi: 10.1126/science.1112014
Carroll, A. P., Goodall, G. J., and Liu, B. (2014). Understanding principles of miRNA target recognition and function through integrated biological and bioinformatics approaches. Wiley Interdiscip. Rev. RNA 5, 361–379. doi: 10.1002/wrna.1217
Chatterjee, D., and Deng, W. -M. (2019). Drosophila model in cancer: an introduction. Adv. Exp. Med. Biol. 1167, 1–14. doi: 10.1007/978-3-030-23629-8_1
Chen, X., Yan, C. C., Zhang, X., and You, Z. H. (2017). Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 18, 558–576. doi: 10.1093/bib/bbw060
Chiarelli, R., Martino, C., Agnello, M., Bosco, L., and Roccheri, M. C. (2016). Autophagy as a defense strategy against stress: focus on Paracentrotus lividus sea urchin embryos exposed to cadmium. Cell Stress Chaperones 21, 19–27. doi: 10.1007/s12192-015-0639-3
Clements, W. M., Wang, J., Sarnaik, A., Kim, O. J., MacDonald, J., Fenoglio-Preiser, C., et al. (2002). Beta-catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 62, 3503–3506.
Croce, J. C., and McClay, D. R. (2010). Dynamics of delta/notch signaling on endomesoderm segregation in the sea urchin embryo. Development 137, 83–91. doi: 10.1242/dev.044149
Croce, J. C., Wu, S. Y., Byrum, C., Xu, R., Duloquin, L., Wikramanayake, A. H., et al. (2006). A genome-wide survey of the evolutionarily conserved Wnt pathways in the sea urchin Strongylocentrotus purpuratus. Dev. Biol. 300, 121–131. doi: 10.1016/j.ydbio.2006.08.045
Davidson, E. H. (2006). The sea urchin genome: where will it lead us? Science 314, 939–940. doi: 10.1126/science.1136252
Davidson, E. H., Rast, J. P., Oliveri, P., Ransick, A., Calestani, C., Yuh, C. -H., et al. (2002). A genomic regulatory network for development. Science 295, 1669–1678. doi: 10.1126/science.1069883
De Wert, G., Berghmans, R. L., Boer, G. J., Andersen, S., Brambati, B., Carvalho, A. S., et al. (2002). Ethical guidance on human embryonic and fetal tissue transplantation: a European overview. Med. Health Care Philos. 5, 79–90. doi: 10.1023/A:1014213125573
De Wert, G., and Mummery, C. (2003). Human embryonic stem cells: research, ethics and policy. Hum. Reprod. 18, 672–682. doi: 10.1093/humrep/deg143
Dean, M., and Annilo, T. (2005). Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genomics Hum. Genet. 6, 123–142. doi: 10.1146/annurev.genom.6.080604.162122
Dean, M., Rzhetsky, A., and Allikmets, R. (2001). The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166. doi: 10.1101/gr.184901
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. doi: 10.1038/nature11233
Ebert, T. A. (2010). Demographic patterns of the purple sea urchin Strongylocentrotus purpuratus along a latitudinal gradient, 1985–1987. Mar. Ecol. Prog. Ser. 406, 105–120. doi: 10.3354/meps08547
Ely, K. A., Bischoff, L. A., and Weiss, V. L. (2018). Wnt signaling in thyroid homeostasis and carcinogenesis. Genes 9:204. doi: 10.3390/genes9040204
ENCODE Project ConsortiumBirney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigó, R., Gingeras, T. R., et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. doi: 10.1038/nature05874
Evans, T., Rosenthal, E. T., Youngblom, J., Distel, D., and Hunt, T. (1983). Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33, 389–396. doi: 10.1016/0092-8674(83)90420-8
Fabian, M. R., Sonenberg, N., and Filipowicz, W. (2010). Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 79, 351–379. doi: 10.1146/annurev-biochem-060308-103103
Fehon, R. G., Kooh, P. J., Rebay, I., Regan, C. L., Xu, T., Muskavitch, M. A., et al. (1990). Molecular interactions between the protein products of the neurogenic loci notch and delta, two EGF-homologous genes in Drosophila. Cell 61, 523–534. doi: 10.1016/0092-8674(90)90534-l
Fernandez-Guerra, A., Aze, A., Morales, J., Mulner-Lorillon, O., Cosson, B., Cormier, P., et al. (2006). The genomic repertoire for cell cycle control and DNA metabolism in S. purpuratus. Dev. Biol. 300, 238–251. doi: 10.1016/j.ydbio.2006.09.012
Finnegan, E. J., and Matzke, M. A. (2003). The small RNA world. J. Cell Sci. 116, 4689–4693. doi: 10.1242/jcs.00838
Flanagan, D. J., Phesse, T. J., Barker, N., Schwab, R. H., Amin, N., Malaterre, J., et al. (2015). Frizzled7 functions as a Wnt receptor in intestinal epithelial Lgr5(+) stem cells. Stem Cell Reports 4, 759–767. doi: 10.1016/j.stemcr.2015.03.003
Flanagan, D. J., Vincan, E., and Phesse, T. J. (2017). Winding back Wnt signalling: potential therapeutic targets for treating gastric cancers. Br. J. Pharmacol. 174, 4666–4683. doi: 10.1111/bph.13890
Fu, P. F., Zheng, X., Fan, X., and Lin, A. F. (2019). Role of cytoplasmic lncRNAs in regulating cancer signaling pathways. J. Zhejiang Univ. Sci. B 20, 1–8. doi: 10.1631/jzus.B1800254
Garneau, N. L., Wilusz, J., and Wilusz, C. J. (2007). The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8, 113–126. doi: 10.1038/nrm2104
Gildor, T., Malik, A., Sher, N., Avraham, L., and De-Leon, S. B. -T. (2016). Quantitative developmental transcriptomes of the Mediterranean sea urchin Paracentrotus lividus. Mar. Genomics 25, 89–94. doi: 10.1016/j.margen.2015.11.013
Giuduce, G. (1962). Restitution of whole larvae from disaggregated cells of sea urchin embryos. Dev. Biol. 5, 402–411. doi: 10.1016/0012-1606(62)90021-0
Goldstone, J. V., Hamdoun, A., Cole, B. J., Howard-Ashby, M., Nebert, D. W., Scally, M., et al. (2006). The chemical defensome: environmental sensing and response genes in the Strongylocentrotus purpuratus genome. Dev. Biol. 300, 366–384. doi: 10.1016/j.ydbio.2006.08.066
Gordon, W. R., Arnett, K. L., and Blacklow, S. C. (2008). The molecular logic of notch signaling--a structural and biochemical perspective. J. Cell Sci. 121, 3109–3119. doi: 10.1242/jcs.035683
Gordon, R. E., Zhang, L., Peri, S., Kuo, Y. M., Du, F., Egleston, B. L., et al. (2018). Statins synergize with hedgehog pathway inhibitors for treatment of medulloblastoma. Clin. Cancer Res. 24, 1375–1388. doi: 10.1158/1078-0432.CCR-17-2923
Grewal, S. I., and Elgin, S. C. (2007). Transcription and RNA interference in the formation of heterochromatin. Nature 447, 399–406. doi: 10.1038/nature05914
Gutzmer, R., and Solomon, J. A. (2019). Hedgehog pathway inhibition for the treatment of basal cell carcinoma. Target. Oncol. 14, 253–267. doi: 10.1007/s11523-019-00648-2
Hadjiargyrou, M., and Delihas, N. (2013). The intertwining of transposable elements and non-coding RNAs. Int. J. Mol. Sci. 14, 13307–13328. doi: 10.3390/ijms140713307
Hamdoun, A. M., Cherr, G. N., Roepke, T. A., and Epel, D. (2004). Activation of multidrug efflux transporter activity at fertilization in sea urchin embryos (Strongylocentrotus purpuratus). Dev. Biol. 276, 452–462. doi: 10.1016/j.ydbio.2004.09.013
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57–70. doi: 10.1016/S0092-8674(00)81683-9
Hedges, S. B. (2002). The origin and evolution of model organisms. Nat. Rev. Genet. 3, 838–849. doi: 10.1038/nrg929
Henrique, D., and Schweisguth, F. (2019). Mechanisms of notch signaling: a simple logic deployed in time and space. Development 146:dev172148. doi: 10.1242/dev.172148
Hezroni, H., Koppstein, D., Schwartz, M. G., Avrutin, A., Bartel, D. P., and Ulitsky, I. (2015). Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 11, 1110–1122. doi: 10.1016/j.celrep.2015.04.023
Hibino, T., Loza-Coll, M., Messier, C., Majeske, A. J., Cohen, A. H., Terwilliger, D. P., et al. (2006). The immune gene repertoire encoded in the purple sea urchin genome. Dev. Biol. 300, 349–365. doi: 10.1016/j.ydbio.2006.08.065
Hinegardner, R. T. (1971). An improved fluorometric assay for DNA. Anal. Biochem. 39, 197–201. doi: 10.1016/0003-2697(71)90476-3
Holm, S. (2003). The ethical case against stem cell research. Camb. Q. Healthc. Ethics 12, 372–383. doi: 10.1017/S0963180103124061
Horstadius, S. (1939). The mechanics of sea urchin development, studied by operative methods. Biol. Rev. 14, 132–179. doi: 10.1111/j.1469-185X.1939.tb00929.x
Howard-Ashby, M., Materna, S. C., Brown, C. T., Chen, L., Cameron, R. A., and Davidson, E. H. (2006). Gene families encoding transcription factors expressed in early development of Strongylocentrotus purpuratus. Dev. Biol. 300, 90–107. doi: 10.1016/j.ydbio.2006.08.033
Huarte, M. (2015). The emerging role of lncRNAs in cancer. Nat. Med. 21, 1253–1261. doi: 10.1038/nm.3981
Israel, J. W., Martik, M. L., Byrne, M., Raff, E. C., Raff, R. A., McClay, D. R., et al. (2016). Comparative developmental transcriptomics reveals rewiring of a highly conserved gene regulatory network during a major life history switch in the sea urchin genus heliocidaris. PLoS Biol. 14:e1002391. doi: 10.1371/journal.pbio.1002391
Janies, D. A., Witter, Z., Linchangco, G. V., Foltz, D. W., Miller, A. K., Kerr, A. M., et al. (2016). EchinoDB, an application for comparative transcriptomics of deeply-sampled clades of echinoderms. BMC Bioinformatics 17:48. doi: 10.1186/s12859-016-0883-2
Karhadkar, S. S., Bova, G. S., Abdallah, N., Dhara, S., Gardner, D., Maitra, A., et al. (2004). Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 431, 707–712. doi: 10.1038/nature02962
Kazazian, H. H. Jr., and Moran, J. V. (2017). Mobile DNA in health and disease. N. Engl. J. Med. 377, 361–370. doi: 10.1056/NEJMra1510092
Kober, K. M., and Bernardi, G. (2013). Phylogenomics of strongylocentrotid sea urchins. BMC Evol. Biol. 13:88. doi: 10.1186/1471-2148-13-88
Konishi, J., Yi, F., Chen, X., Vo, H., Carbone, D. P., and Dang, T. P. (2010). Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene 29, 589–596. doi: 10.1038/onc.2009.366
Kontomanolis, E. N., Kalagasidou, S., Pouliliou, S., Anthoulaki, X., Georgiou, N., Papamanolis, V., et al. (2018). The notch pathway in breast cancer progression. ScientificWorldJournal 2018:2415489. doi: 10.1155/2018/2415489
Kumburegama, S., and Wikramanayake, A. H. (2008). Wnt signaling in the early sea urchin embryo. Methods Mol. Biol. 469, 187–199. doi: 10.1007/978-1-60327-469-2_14
Kuo, C. -J., Hansen, M., and Troemel, E. (2018). Autophagy and innate immunity: insights from invertebrate model organisms. Autophagy 14, 233–242. doi: 10.1080/15548627.2017.1389824
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi: 10.1038/35057062
Langton, P. F., Kakugawa, S., and Vincent, J. P. (2016). Making, exporting, and modulating Wnts. Trends Cell Biol. 26, 756–765. doi: 10.1016/j.tcb.2016.05.011
Laubichler, M. D., and Davidson, E. H. (2008). Boveri’s long experiment: sea urchin merogones and the establishment of the role of nuclear chromosomes in development. Dev. Biol. 314, 1–11. doi: 10.1016/j.ydbio.2007.11.024
Laumer, C. E., Bekkouche, N., Kerbl, A., Goetz, F., Neves, R. C., Sørensen, M. V., et al. (2015). Spiralian phylogeny informs the evolution of microscopic lineages. Curr. Biol. 25, 2000–2006. doi: 10.1016/j.cub.2015.06.068
Laurent, G., Wahlestedt, C., and Kapranov, P. (2015). The landscape of long noncoding RNA classification. Trends Genet. 31, 239–251. doi: 10.1016/j.tig.2015.03.007
Lebedev, E. E., Ostromyshenskii, D. I., Solovyeva, A. I., Turenkod, A. S., Drozdov, A. L., Podgornaya, O. I., et al. (2019). The transposons of the sea urchin strongylocentrotus intermedius agassiz, 1863: in silico versus in vitro. Russ. J. Mar. Biol. 45, 418–424. doi: 10.1134/S1063074019060051
Lee, I., Ajay, S. S., Yook, J. I., Kim, H. S., Hong, S. H., Kim, N. H., et al. (2009). New class of microRNA targets containing simultaneous 5'-UTR and 3’-UTR interaction sites. Genome Res. 19, 1175–1183. doi: 10.1101/gr.089367.108
Lefort, K., Mandinova, A., Ostano, P., Kolev, V., Calpini, V., Kolfschoten, I., et al. (2007). Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 21, 562–577. doi: 10.1101/gad.1484707
Lehmann, W., Mossmann, D., Kleemann, J., Mock, K., Meisinger, C., Brummer, T., et al. (2016). ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 7:10498. doi: 10.1038/ncomms10498
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Li, W., and Kang, Y. (2014). A new Lnc in metastasis: long noncoding RNA mediates the prometastatic functions of TGF-β. Cancer Cell 25, 557–559. doi: 10.1016/j.ccr.2014.04.014
Li, J., and Liu, C. (2019). Coding or noncoding, the converging concepts of RNAs. Front. Genet. 10:496. doi: 10.3389/fgene.2019.00496
Lim, J., and Thiery, J. P. (2012). Epithelial-mesenchymal transitions: insights from development. Development 139, 3471–3486. doi: 10.1242/dev.071209
Liu, S., Dontu, G., Mantle, I. D., Patel, S., Ahn, N. S., Jackson, K. W., et al. (2006). Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 66, 6063–6071. doi: 10.1158/0008-5472.CAN-06-0054
Lloyd-Lewis, B., Mourikis, P., and Fre, S. (2019). Notch signalling: sensor and instructor of the microenvironment to coordinate cell fate and organ morphogenesis. Curr. Opin. Cell Biol. 61, 16–23. doi: 10.1016/j.ceb.2019.06.003
Lo, B., and Parham, L. (2009). Ethical issues in stem cell research. Endocr. Rev. 30, 204–213. doi: 10.1210/er.2008-0031
Long, Y., Wang, X., Youmans, D. T., and Cech, T. R. (2017). How do lncRNAs regulate transcription? Sci. Adv. 3:eaao2110. doi: 10.1126/sciadv.aao2110
Lowry, W. E., Richter, L., Yachechko, R., Pyle, A. D., Tchieu, J., Sridharan, R., et al. (2008). Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 105, 2883–2888. doi: 10.1073/pnas.0711983105
Lu, W., Liu, C. C., Thottassery, J. V., Bu, G., and Li, Y. (2010). Mesd is a universal inhibitor of Wnt coreceptors LRP5 and LRP6 and blocks Wnt/beta-catenin signaling in cancer cells. Biochemistry 49, 4635–4643. doi: 10.1021/bi1001486
Ma, J., Xia, J., Miele, L., Sarkar, F. H., and Wang, Z. (2013). Notch signaling pathway in pancreatic cancer progression. Pancreat. Disord. Ther. 3:1000114.
Marques, I. J., Lupi, E., and Mercader, N. (2019). Model systems for regeneration: zebrafish. Development 146:dev167692. doi: 10.1242/dev.167692
Martik, M. L., Lyons, D. C., and McClay, D. R. (2016). Developmental gene regulatory networks in sea urchins and what we can learn from them. F1000Res. 5:203. doi: 10.12688/f1000research.7381.1
McClay, D. R. (2011). Evolutionary crossroads in developmental biology: sea urchins. Development 138, 2639–2648. doi: 10.1242/dev.048967
McClay, D. R., and Fink, R. D. (1982). Sea urchin hyalin: appearance and function in development. Dev. Biol. 92, 285–293. doi: 10.1016/0012-1606(82)90175-0
Mzoughi, S., Zhang, J., Hequet, D., Teo, S. X., Fang, H., Xing, Q. R., et al. (2017). PRDM15 safeguards naive pluripotency by transcriptionally regulating WNT and MAPK-ERK signaling. Nat. Genet. 49, 1354–1363. doi: 10.1038/ng.3922
Nakajima, Y., Kaneko, H., Murray, G., and Burke, R. D. (2004). Divergent patterns of neural development in larval echinoids and asteroids. Evol. Dev. 6, 95–104. doi: 10.1111/j.1525-142x.2004.04011.x
Nguyen, V., Hough, R., Bernaudo, S., and Peng, C. (2019). Wnt/β-catenin signalling in ovarian cancer: insights into its hyperactivation and function in tumorigenesis. J. Ovarian Res. 12:122. doi: 10.1186/s13048-019-0596-z
Nielsen, J. (2019). Yeast systems biology: model organism and cell factory. Biotechnol. J. 14:e1800421. doi: 10.1002/biot.201800421
Nieto, M. A., Huang, R. Y., Jackson, R. A., and Thiery, J. P. (2016). EMT: 2016. Cell 166, 21–45. doi: 10.1016/j.cell.2016.06.028
Nowell, C. S., and Radtke, F. (2017). Notch as a tumour suppressor. Nat. Rev. Cancer 17, 145–159. doi: 10.1038/nrc.2016.145
Nusse, R., and Clevers, H. (2017). Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999. doi: 10.1016/j.cell.2017.05.016
Oliveri, P., Tu, Q., and Davidson, E. H. (2008). Global regulatory logic for specification of an embryonic cell lineage. Proc. Natl. Acad. Sci. U. S. A. 105, 5955–5962. doi: 10.1073/pnas.0711220105
Ong, M. S., Cai, W., Yuan, Y., Leong, H. C., Tan, T. Z., Mohammad, A., et al. (2017). ‘Lnc’-ing Wnt in female reproductive cancers: therapeutic potential of long non-coding RNAs in Wnt signalling. Br. J. Pharmacol. 174, 4684–4700. doi: 10.1111/bph.13958
Paço, A., Freitas, R., and Vieira-da-Silva, A. (2019). Conversion of DNA sequences: from a transposable element to a tandem repeat or to a gene. Gene 10:1014. doi: 10.3390/genes10121014
Parr, C., Watkins, G., and Jiang, W. G. (2004). The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int. J. Mol. Med. 14, 779–786. doi: 10.3892/ijmm.14.5.779
Patel, N. S., Li, J. L., Generali, D., Poulsom, R., Cranston, D. W., and Harris, A. L. (2005). Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 65, 8690–8697. doi: 10.1158/0008-5472.CAN-05-1208
Peng, W. X., Koirala, P., and Mo, Y. Y. (2017). LncRNA-mediated regulation of cell signaling in cancer. Oncogene 36, 5661–5667. doi: 10.1038/onc.2017.184
Pérez-Portela, R., Turon, X., and Riesgo, A. (2016). Characterization of the transcriptome and gene expression of four different tissues in the ecologically relevant sea urchin Arbacia lixula using RNA-seq. Mol. Ecol. Resour. 16, 794–808. doi: 10.1111/1755-0998.12500
Peter, I. S., and Davidson, E. H. (2010). The endoderm gene regulatory network in sea urchin embryos up to mid-blastula stage. Dev. Biol. 340, 188–199. doi: 10.1016/j.ydbio.2009.10.037
Peter, I. S., and Davidson, E. H. (2017). Assessing regulatory information in developmental gene regulatory networks. Proc. Natl. Acad. Sci. U. S. A. 114, 5862–5869. doi: 10.1073/pnas.1610616114
Peter, I. S., Faure, E., and Davidson, E. H. (2012). Predictive computation of genomic logic processing functions in embryonic development. Proc. Natl. Acad. Sci. U. S. A. 109, 16434–16442. doi: 10.1073/pnas.1207852109
Peterson, K. J., Dietrich, M. R., and McPeek, M. A. (2009). MicroRNAs and metazoan macroevolution: insights into canalization, complexity, and the Cambrian explosion. Bioessays 31, 736–747. doi: 10.1002/bies.200900033
Pires-daSilva, A., and Sommer, R. J. (2003). The evolution of signaling pathways in animal development. Nat. Rev. Genet. 4, 39–49. doi: 10.1038/nrg977
Poli, V., Fagnocchi, L., and Zippo, A. (2018). Tumorigenic cell reprogramming and cancer plasticity: interplay between signaling, microenvironment, and epigenetics. Stem Cells Int. 2018:4598195. doi: 10.1155/2018/4598195
Rafiq, K., Shashikant, T., McManus, C. J., and Ettensohn, C. A. (2014). Genome-wide analysis of the skeletogenic gene regulatory network of sea urchins. Development 141, 950–961. doi: 10.1242/dev.105585
Rast, J. P., Smith, L. C., Loza-Coll, M., Hibino, T., and Litman, G. W. (2006). Genomic insights into the immune system of the sea urchin. Science 314, 952–956. doi: 10.1126/science.1134301
Reya, T., Morrison, S. J., Clarke, M. F., and Weissman, I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. doi: 10.1038/35102167
Robert, N., Hammami, F., Lhomond, G., Dru, P., Lepage, T., Schubert, M., et al. (2019). A wnt2 ortholog in the sea urchin Paracentrotus lividus. Genesis 57:e23331. doi: 10.1002/dvg.23331
Robert, N., Lhomond, G., Schubert, M., and Croce, J. C. (2014). A comprehensive survey of wnt and frizzled expression in the sea urchin Paracentrotus lividus. Genesis 52, 235–250. doi: 10.1002/dvg.22754
Romancino, D. P., Anello, L., Lavanco, A., Buffa, V., Di Bernardo, M., and Bongiovanni, A. (2017). A sea urchin in vivo model to evaluate epithelial-mesenchymal transition. Dev. Growth Differ. 59, 141–151. doi: 10.1111/dgd.12353
Rzepnikowska, W., Flis, K., Muñoz-Braceras, S., Menezes, R., Escalante, R., and Zoladek, T. (2017). Yeast and other lower eukaryotic organisms for studies of Vps13 proteins in health and disease. Traffic 18, 711–719. doi: 10.1111/tra.12523
Sari, I. N., Phi, L., Jun, N., Wijaya, Y. T., Lee, S., and Kwon, H. Y. (2018). Hedgehog signaling in cancer: a prospective therapeutic target for eradicating cancer stem cells. Cells 7:208. doi: 10.3390/cells7110208
Sea Urchin Genome Sequencing ConsortiumSodergren, E., Weinstock, G. M., Davidson, E. H., Cameron, R. A., Gibbs, R. A., et al. (2006). The genome of the sea urchin Strongylocentrotus purpuratus. Science 314, 941–952. doi: 10.1126/science.1133609
Segditsas, S., and Tomlinson, I. (2006). Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 25, 7531–7537. doi: 10.1038/sj.onc.1210059
Shahabi, S., Kumaran, V., Castillo, J., Cong, Z., Nandagopal, G., Mullen, D. J., et al. (2019). LINC00261 is an epigenetically regulated tumor suppressor essential for activation of the DNA damage response. Cancer Res. 79, 3050–3062. doi: 10.1158/0008-5472.CAN-18-2034
Shen, P., Pichler, M., Chen, M., Calin, G. A., and Ling, H. (2017). To Wnt or lose: the missing non-coding linc in colorectal cancer. Int. J. Mol. Sci. 18:2003. doi: 10.3390/ijms18092003
Sheng, T., Li, C., Zhang, X., Chi, S., He, N., Chen, K., et al. (2004). Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer 3:29. doi: 10.1186/1476-4598-3-29
Sherwood, D. R., and McClay, D. R. (1999). LvNotch signaling mediates secondary mesenchyme specification in the sea urchin embryo. Development 126, 1703–1713.
Sherwood, D. R., and McClay, D. R. (2001). LvNotch signaling plays a dual role in regulating the position of the ectoderm-endoderm boundary in the sea urchin embryo. Development 128, 2221–2232.
Skoda, A. M., Simovic, D., Karin, V., Kardum, V., Vranic, S., and Serman, L. (2018). The role of the hedgehog signaling pathway in cancer: a comprehensive review. Bosn. J. Basic Med. Sci. 18, 8–20. doi: 10.17305/bjbms.2018.2756
Song, J. L., Stoeckius, M., Maaskola, J., Friedländer, M., Stepicheva, N., Juliano, C., et al. (2012). Select microRNAs are essential for early development in the sea urchin. Dev. Biol. 362, 104–113. doi: 10.1016/j.ydbio.2011.11.015
Stecca, B., Mas, C., Clement, V., Zbinden, M., Correa, R., Piguet, V., et al. (2007). Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. U. S. A. 104, 5895–5900. doi: 10.1073/pnas.0700776104
Steitz, J. A., and Vasudevan, S. (2009). miRNPs: versatile regulators of gene expression in vertebrate cells. Biochem. Soc. Trans. 37, 931–935. doi: 10.1042/BST0370931
Stepicheva, N. A., and Song, J. L. (2015). microRNA-31 modulates skeletal patterning in the sea urchin embryo. Development 142, 3769–3780. doi: 10.1242/dev.127969
Stimpson, W. (1857). On the Crustacea and Echinodermata of the Pacific shores of North America. Boston J. Nat. Hist. 6, 444–532.
Sun, T., Jiao, L., Wang, Y., Yu, Y., and Ming, L. (2018). SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells. Cell Death Dis. 9:136. doi: 10.1038/s41419-017-0167-4
Sweet, H. C., Gehring, M., and Ettensohn, C. A. (2002). LvDelta is a mesoderm-inducing signal in the sea urchin embryo and can endow blastomeres with organizer-like properties. Development 129, 1945–1955.
Tang, J., Xu, Z., Huang, L., Luo, H., and Zhu, X. (2019). Transcriptional regulation in model organisms: recent progress and clinical implications. Open Biol. 9:190183. doi: 10.1098/rsob.190183
Telford, M. J., Budd, G. E., and Philippe, H. (2015). Phylogenomic insights into animal evolution. Curr. Biol. 25, R876–R887. doi: 10.1016/j.cub.2015.07.060
Torruella, G., de Mendoza, A., Grau-Bové, X., Antó, M., Chaplin, M. A., del Campo, J., et al. (2015). Phylogenomics reveals convergent evolution of lifestyles in close relatives of animals and fungi. Curr. Biol. 25, 2404–2410. doi: 10.1016/j.cub.2015.07.053
Trimarchi, T., Bilal, E., Ntziachristos, P., Fabbri, G., Dalla-Favera, R., Tsirigos, A., et al. (2014). Genome-wide mapping and characterization of notch-regulated long noncoding RNAs in acute leukemia. Cell 158, 593–606. doi: 10.1016/j.cell.2014.05.049
Tu, Q., Cameron, R. A., and Davidson, E. H. (2014). Quantitative developmental transcriptomes of the sea urchin Strongylocentrotus purpuratus. Dev. Biol. 385, 160–167. doi: 10.1016/j.ydbio.2013.11.019
Tulchinsky, E., Demidov, O., Kriajevska, M., Barlev, N. A., and Imyanitov, E. (2019). EMT: a mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta Rev. Cancer 1871, 29–39. doi: 10.1016/j.bbcan.2018.10.003
Venkatesh, V., Nataraj, R., Thangaraj, G. S., Karthikeyan, M., Gnanasekaran, A., Kaginelli, S. B., et al. (2018). Targeting notch signalling pathway of cancer stem cells. Stem Cell Investig. 5:5. doi: 10.21037/sci.2018.02.02
Viatour, P., Ehmer, U., Saddic, L. A., Dorrell, C., Andersen, J. B., Lin, C., et al. (2011). Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J. Exp. Med. 208, 1963–1976. doi: 10.1084/jem.20110198
Vinson, K. E., George, D. C., Fender, A. W., Bertrand, F. E., and Sigounas, G. (2016). The notch pathway in colorectal cancer. Int. J. Cancer 138, 1835–1842. doi: 10.1002/ijc.29800
Wang, Z., and Kunze, R. (2015). “Transposonsin eukaryotes: structures, mechanisms and applications” in eLS. Chichester: John Wiley & Sons, Ltd.
Wang, Q., Zhou, Y., Rychahou, P., Harris, J. W., Zaytseva, Y. Y., Liu, J., et al. (2018). Deptor is a novel target of Wnt/β-catenin/c-Myc and contributes to colorectal cancer cell growth. Cancer Res. 78, 3163–3175. doi: 10.1158/0008-5472.CAN-17-3107
Warner, J. F., Miranda, E. L., and McClay, D. R. (2016). Contribution of hedgehog signaling to the establishment of left-right asymmetry in the sea urchin. Dev. Biol. 411, 314–324. doi: 10.1016/j.ydbio.2016.02.008
Wheeler, B. M., Heimberg, A. M., Moy, V. N., Sperling, E. A., Holstein, T. W., Heber, S., et al. (2009). The deep evolution of metazoan microRNAs. Evol. Dev. 11, 50–68. doi: 10.1111/j.1525-142X.2008.00302.x
Whitaker, M. (2006). Calcium microdomains and cell cycle control. Cell Calcium 40, 585–592. doi: 10.1016/j.ceca.2006.08.018
Wu, C. Y., Tsai, Y. P., Wu, M. Z., Teng, S. C., and Wu, K. J. (2012). Epigenetic reprogramming and post-transcriptional regulation during the epithelial-mesenchymal transition. Trends Genet. 28, 454–463. doi: 10.1016/j.tig.2012.05.005
Yuan, J. H., Yang, F., Wang, F., Ma, J. Z., Guo, Y. J., Tao, Q. F., et al. (2014). A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25, 666–681. doi: 10.1016/j.ccr.2014.03.010
Zaidel-Bar, R. (2009). Evolution of complexity in the integrin adhesome. J. Cell Biol. 186, 17–21. doi: 10.1083/jcb.200811067
Zhou, W., Fu, X. Q., Zhang, L. L., Zhang, J., Huang, X., Lu, X. H., et al. (2013). The AKT1/NF-kappaB/Notch1/PTEN axis has an important role in chemoresistance of gastric cancer cells. Cell Death Dis. 4:e847. doi: 10.1038/cddis.2013.375
Keywords: sea urchin, gene expression, cell signaling, long non-coding RNA, genomics
Citation: Adonin L, Drozdov A and Barlev NA (2021) Sea Urchin as a Universal Model for Studies of Gene Networks. Front. Genet. 11:627259. doi: 10.3389/fgene.2020.627259
Edited by:
Yuriy L. Orlov, I.M. Sechenov First Moscow State Medical University, RussiaReviewed by:
Anna Kudryavtseva, Engelhardt Institute of Molecular Biology (RAS), RussiaAlexander M. Ishov, University of Florida, United States
Copyright © 2021 Adonin, Drozdov and Barlev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonid Adonin, bGVvLmFkb25pbkBnbWFpbC5jb20=; Nickolai A. Barlev, bmljay5hLmJhcmxldkBnbWFpbC5jb20=