 Nur Shuhadah Mohd Saad
Nur Shuhadah Mohd Saad Anita A. Severn-Ellis
Anita A. Severn-Ellis Aneeta Pradhan
Aneeta Pradhan Jacqueline Batley
Jacqueline Batley- School of Biological Sciences Western Australia and UWA Institute of Agriculture, University of Western Australia, Perth, WA, Australia
Meeting the needs of a growing world population in the face of imminent climate change is a challenge; breeding of vegetable and oilseed Brassica crops is part of the race in meeting these demands. Available genetic diversity constituting the foundation of breeding is essential in plant improvement. Elite varieties, land races, and crop wild species are important resources of useful variation and are available from existing genepools or genebanks. Conservation of diversity in genepools, genebanks, and even the wild is crucial in preventing the loss of variation for future breeding efforts. In addition, the identification of suitable parental lines and alleles is critical in ensuring the development of resilient Brassica crops. During the past two decades, an increasing number of high-quality nuclear and organellar Brassica genomes have been assembled. Whole-genome re-sequencing and the development of pan-genomes are overcoming the limitations of the single reference genome and provide the basis for further exploration. Genomic and complementary omic tools such as microarrays, transcriptomics, epigenetics, and reverse genetics facilitate the study of crop evolution, breeding histories, and the discovery of loci associated with highly sought-after agronomic traits. Furthermore, in genomic selection, predicted breeding values based on phenotype and genome-wide marker scores allow the preselection of promising genotypes, enhancing genetic gains and substantially quickening the breeding cycle. It is clear that genomics, armed with diversity, is set to lead the way in Brassica improvement; however, a multidisciplinary plant breeding approach that includes phenotype = genotype × environment × management interaction will ultimately ensure the selection of resilient Brassica varieties ready for climate change.
Introduction
Predictions of exponential increases in the world population and climate change are forcing re-evaluation of efforts in addressing the demand for global food security. Plant crops account for more than 80% of the diet consumed by the human population, and the production of edible crops has thereby dominated almost half of the world's available land mass since the beginning of the twenty-first century (Leff et al., 2004; Herrera and Garcia-Bertrand, 2018). The Industrial and Green Revolutions have furthermore shaped the ways that these commodities are managed for efficiency and commercialization. The next revolution will require crop improvement not just to curb world hunger but also to address sustainability in the face of biotic and abiotic stresses triggered by the impending climate change. It is estimated that staple crop yield must improve by 70–110% to feed the predicted 10 billion population by 2050 (Saini et al., 2020). Crop projections and modeling studies have suggested that climate change may have already been responsible for a small yearly decrease in yield and calories in certain geographic regions (Ray et al., 2019). More recently, the coronavirus disease 2019 (COVID-19) pandemic has emphasized food security in terms of short-term and local supply (Cappelli and Cini, 2020). While food shortages are not a wide set concern as yet, a prolonged crisis could interfere with the current complex food supply network (de Paulo Farias and dos Santos Gomes, 2020; Siche, 2020). Globalized food distribution, though highly profitable, highlighted the critical gap in local production, closer to consumers and less likely affected by international restrictions (Cappelli and Cini, 2020). Meeting local population demand, while remaining sustainable in a shorter food supply chain, may yet have introduced another facet to food security in the post-COVID-19 era. Predicted environmental variation including rising temperatures and increases in carbon dioxide emissions could result in a drier atmosphere and an increase in evapotranspiration (Ficklin and Novick, 2017). In addition to water, most biological processes are temperature sensitive, and climate change is therefore undoubtedly going to affect all crop performance (Dusenge et al., 2019). As a result, resilience to abiotic stresses, such as heat, drought, and salinity, will become traits that are highly desirable in future crop improvement strategies. Reactive nitrogen plays an important role in plant growth, crop yield, and subsequently human nutrition (Dreccer et al., 2000). Alongside their benefits, agricultural practices such as nitrogen fertilizer application and nitrogen fixing crops have the potential to disturb the global nitrogen cycle and adversely affect human health (Townsend et al., 2003; Bodirsky et al., 2012). These practices, if poorly managed, can contribute significantly toward the release of nitrous oxide into atmosphere, which negatively affects the protective ozone layer and advances climate change (Crutzen and Ehhalt, 1977). In an effort to solve the global nitrogen challenge, Houlton et al. (2019) propose, amongst other things, the improvement of nitrogen-use efficiency in crop production. This could be achieved by altering fertilizers and fertilizer application practices, boosting soil health to promote nitrogen uptake, and developing improved crop varieties that efficiently utilize nitrogen (Houlton et al., 2019).
The genus Brassica consists of extensively agronomically diverse species. Oilseed canola include Brassica rapa, Brassica napus, and Brassica juncea varieties with internationally defined erucic acid and glucosinolate contents (Sharafi et al., 2015). Vegetable Brassicas include B. rapa ssp. rapa (turnip), ssp. oleifera (turnip rape), ssp. chinensis (pak choi/bok choy), and ssp. pekinensis (Chinese cabbage); Brassica oleracea ssp. capitata (cabbages), var. italica (broccoli), var. botrytis (cauliflower), ssp. gemmifera (Brussels sprouts), and ssp. alboglara (Chinese kale); and B. napus var. napobrassica (swede/rutabaga) (Cheng et al., 2016). Mustard Brassica include Brassica nigra (black mustard), Brassica carinata (Ethiopian mustard), and B. juncea (Indian mustard). The U triangle summarized the interspecific hybridization events between diploid progenitors B. rapa (AA), B. nigra (BB), and B. oleracea (CC) resulting in polyploidy, B. juncea (AABB), B. napus (AACC), and B. carinata (BBCC) (Nagaharu et al., 1935; Snowdon et al., 2002).
Brassicas are cultivated on a worldwide scale, and it is therefore almost a certainty that forecasted environmental changes would affect the crop (Francisco et al., 2017). Combined with the abiotic stresses, biotic stress is another challenge facing global Brassica production. Bebber et al. (2013) combined observation data and mathematical equation to project pest distribution and proposed that pathogen and pests affecting global crops are moving polewards as the temperature rises. Pathogens of Brassica oilseed and vegetable crops, such as Leptosphaeria maculans (blackleg or stem canker), Alternaria brassicae (Alternaria blight), Albugo candida (white rust), Pseudocercosporella capsellae (white leaf spot), Plasmodiophora brassicae (club root), and Sclerotinia sclerotiorum (Sclerotinia stem rot), extensively affect yield, seed quality, and crop development (Murray and Brennan, 2012). Breeding Brassica varieties that can withstand the pressures of a changing environment is perhaps the best strategy in ensuring sustainability.
Decreasing nutrient content of modern fruit and vegetable cultivars has raised concern in recent studies conducted in the USA and UK, predicting the future need for agricultural bio-fortification (Mayer, 1997; Davis et al., 2005; White and Broadley, 2005; Davis, 2009). Davis coined the term “genetic dilution effect” in 2005 after observing broccoli hybrids (B. oleracea var. italica) accumulating denser heads without the obvious proportional increase in nutrients (Davis et al., 2005). This is a result of breeding and selection of varieties based on yield and productivity, while overseeing the importance of nutrient content. The World Health Organization considers micronutrient deficiency as a major health challenge especially in developing and poor countries (Khush et al., 2012). Bio-fortification is a cost-effective and sustainable strategy in addressing malnutrition; however, this requires suitable genetic diversity within the genepool to be valuable in breeding (Garg et al., 2018; Kumar et al., 2020).
Available genetic diversity still constitutes the foundation of all breeding efforts. Elite varieties, land races, and crop wild species are important resources of useful variation that can be introgressed, re-introduced, or manipulated to obtain the required biotic and abiotic resilience in Brassica crops (Dwivedi et al., 2017). The identification and exploitation of suitable variation are crucial for crop improvement (Hu et al., 2018) and can be elucidated at the genome scale (Varshney et al., 2018). Genomics can, in addition, contribute toward unraveling the genetic origin and molecular pathways involved in biotic and abiotic stress tolerance traits. Complete and accurate understanding of the ancestry of the Brassica species will assist in the tracking and exploitation of genetic inheritance of useful traits (Bancroft et al., 2011).
Crop improvement has always been a co-evolutionary process between humans and edible plants (Harlan, 1992); changes in plants brought about by cultivation allowed changes in human populations to take place. Plant breeding has largely relied on conventional breeding methods based on phenotypic selection. However, it is in doubt whether conventional breeding approaches alone would be adequate in addressing the impending challenges. During the course of the last three decades, genomics has become an integral part of all life sciences. Rapid advances in sequencing tools followed by cost reductions, as well as the development of high-throughput genotyping techniques, have led to advances in trait mapping, functional characterization, and ultimately crop improvement through genomic selection (GS) (Nepolean et al., 2018).
Without available genetic diversity, the introduction of genes may present a suitable solution. Weed and insect control using genetically modified (GM) crops has assisted farmers worldwide in attaining higher yields with fewer resources (Zhang et al., 2016). Cauliflower (Lu et al., 2006), mustard (Hong et al., 2002), and canola (Garg et al., 2018) have been subjected to transgenic bio-fortification efforts for beta-carotene, gamma-linoleic acid, and phytate degradation, respectively. Despite these efforts, so far, only Phytaseed canola has been released by BASF in the USA. Health concerns, consumer skepticism, and long and expensive regulatory processes are restricting the release of transgenic crops (Watanabe et al., 2005). Recent developments in genome editing (GE) provide an option to alter or introduce specific genes in order to obtain the desired trait expression. Oligo-directed mutagenesis (ODM), programmable sequence-specific nucleases (SSNs), and base-editing tools allow the precise creation of insertions/deletions (indels) or even the introduction of a complete sequence at a predetermined target location within the genome (Scheben et al., 2017).
To further address the pressing demands on crop improvement, an accelerated rate of genetic gain is required. The implementation of GS can fast-track the progress in crop breeding (Wang X. et al., 2018). In GS, predicted breeding values based on phenotype and genome-wide marker scores allow the preselection of promising genotypes, thereby substantially quickening the breeding cycle and enhancing genetic gains (Heffner et al., 2009). Further optimization of mating strategies is essential to prevent inbreeding and ensure long-term genetic gain (Allier et al., 2019).
Genomics armed with diversity is currently leading the way in crop improvement. Here, we review aspects of the Brassica crop improvement cycle (Figure 1) illustrating the importance of genetic diversity, creation of genomic resources, its exploitation in aid of trait discovery, and GE and GS of Brassica.
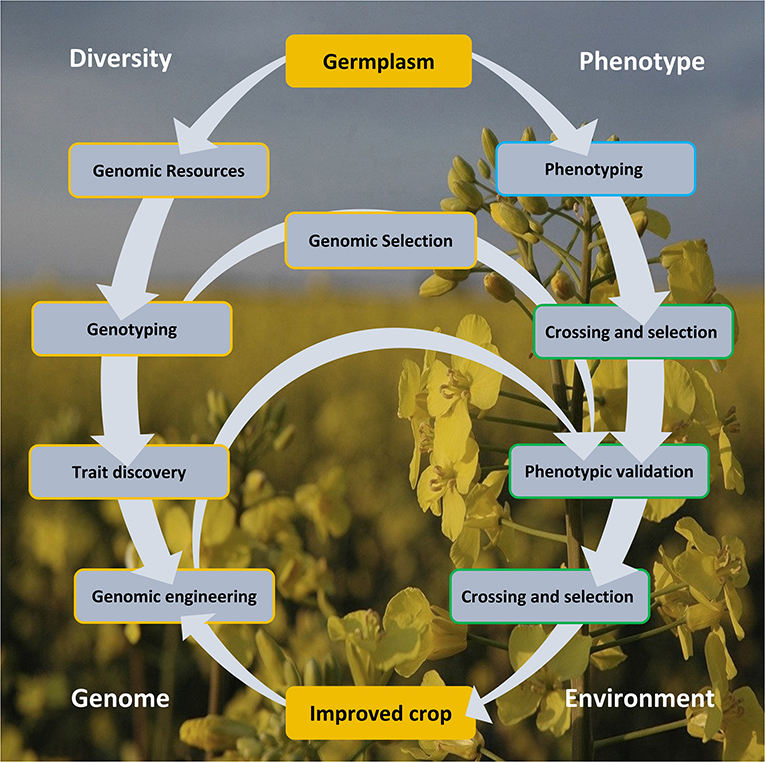
Figure 1. The crop improvement cycle involves the exploitation of genomic diversity and phenotypic variability. These approaches offered a rapid and cost-effective lab-to-field and back loop, centered on arming the genomic arsenal by capitalizing on the variation in the germplasm resource, as defined in the genotype and from trait association. Genomic selection and engineering accelerated crop improvement, circumventing a more laborious and time-consuming conventional crossing and selection approach. Genomic engineering strategies also offer a more precise manipulation for improved crop development. The improved crop must be climate resilient and adaptive to the extreme environmental changes.
Estimation and Conservation of Diversity in Germplasm
Crop breeding is required to be sustainable and to adapt promptly in the face of abiotic and biotic environmental changes (Zhang and Batley, 2020). Most modern crops were developed through repeated cycles of selection, “filtering out” varieties with desirable agronomic traits from ancestral wild species. However, potentially valuable genetic variation is often lost in the process, resulting in lowered trait heritability and increased genetic homogeneity (Rahman, 2013). Comparative population genomics can be used to identify these selective sweeps, or bottlenecks, and potential loci under selection (Slatkin, 2008). An example is the allotetraploid Brassica napus (AACC, 2n = 38), which originated from spontaneous interspecific hybridization events between Brassica rapa (AA, 2n = 20) and Brassica oleracea (CC, 2n = 18) (Chalhoub et al., 2014). Cultivated B. napus have been under intense selection over the past decades, which has led to a severe genetic bottleneck in the species (Becker et al., 1995). This is reflected by the extent of highly conserved regions found between the genomes of B. napus accessions found in almost all major genepools (Werner et al., 2018). Accordingly, the decline of allelic variation and genetic diversity was reported in Canadian (Fu and Gugel, 2010) and Australian spring canola breeding initiatives (Cowling, 2007).
The key to sustainable crop improvement in the face of climate change, and increased pressure from pests and diseases, is maintaining this diversity. Diversity within germplasm provides breeders with valuable material to enable the selection of parental lines, exploitation of heterosis, and the expansion of breeding pools (Yousef et al., 2018). Heritable variation present within the crop germplasm is therefore essential for efficient breeding programs. Characterizing the genetic diversity allows breeders to select alleles at loci of interest and identify trait-associated markers suitable for introgression into new varieties. The assessment of germplasm diversity furthermore assists in the optimization of conservation strategies of germplasm collections (Rao and Hodgkin, 2002). This was highlighted in the study conducted by Yousef et al. (2018), in which cauliflower (B. oleracea var. botrytis) accessions originating from 26 countries were grouped into two major groups representing the two genebanks from which the accessions were obtained and not the country of origin. In this case, composition and accession type influenced the level of diversity and contributed toward the differentiation between the genebanks. Routine monitoring of genetic diversity in ex situ germplasm collections might therefore be essential to prevent potential loss of genetic diversity.
Wild species related to agricultural crops [crop wild relatives or (CWRs)] and landraces offer an attractive alternative source of variation (Dempewolf et al., 2017; Khan et al., 2020). Many of these unique resources are currently available from genebanks and seed repositories (Tanksley and McCouch, 1997). Introgression from CWRs or landraces can broaden the genetic base for modern breeding programs and can contribute to sought-after characteristics associated with relative tolerance to extreme environments and disease resistance. Despite the vast genetic potential locked up in these resources, their utilization is hampered by inconsistent documentation, unintentional duplications, and a lack of available genetic information (Singh N. et al., 2019; Zhang and Batley, 2020). Combining available phenotypic and geographical descriptors with genomic sequence information, perhaps in the form of a universal molecular passport, could facilitate the selection of useful genetic variation and its use in breeding programs (Mascher et al., 2019; Singh N. et al., 2019).
Plant genetic resources, including CWRs in the wild, are considered under threat and in need of conservation (Dempewolf et al., 2017). Climate change, along with habitat fragmentation due to human activity, is predicted to result in drastic plant population declines and even result in extinction (Jump and Peñuelas, 2005). The creation of protected areas, efforts to reduce pollution and legal frameworks to protect endangered species, such as the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES), may aid in the prevention of the decline or loss of biodiversity. Genetic approaches over time have contributed toward the estimation of biodiversity and prioritization of conservation efforts. However, genome-scale data and associated high-density markers can improve estimations of genetic diversity and population structure. Transitioning from a genetics to conservation genomics approach is expected to have a positive impact on future conservation recommendations and policies (Supple and Shapiro, 2018).
Creation of Genomic Resources in Brassica Species
The smaller Brassicaceae genome from Arabidopsis thaliana was the first to be sequenced and benefited progress in Brassica sequencing given the high degree of genomic conservation between A. thaliana and Brassica species. In addition, DNA sequencing technology becomes more affordable with longer reads and higher throughput available at a fraction of the cost (Marri et al., 2018). These developments provided the opportunity to improve de novo genome assembly, increase mapping certainty, and identify structural variants (SVs) (Amarasinghe et al., 2020). Furthermore, re-sequencing efforts have increased over time to resolve challenges and gaps in current studies, especially in complex and agronomically significant crops like Brassica napus.
As technology progresses, more advanced genomic tools become available, expanding the range of analysis and exploitation possibilities of germplasm resources. The PlabiPD database (https://www.plabipd.de/) maintains an updated list of sequenced plant species, including Brassicas, which can be visualized phylogenetically or temporally. Available genomic resources have played an important role in the advancement of breeding programs throughout the world in cereals, legumes, oilseeds, and even ornamental crops. Hence, here we examine the development and availability of nuclear and organellar genome resources generated in Brassicas.
Nuclear Genome
Draft genomes and pangenome assemblies have been created for five domesticated and a wild Brassica species (Table 1). More broadly, 27 members of the Brassicaceae family have been sequenced, including three Arabidopsis (The Arabidopsis Genome Initiative, 2000; Hu T. T. et al., 2011; Akama et al., 2014; Briskine et al., 2017; Michael et al., 2018), three Capsella (Slotte et al., 2013; Kasianov et al., 2017), three Eutrema (Yang et al., 2013; Guo et al., 2018), and two Raphanus species (Kitashiba et al., 2014; Moghe et al., 2014; Shirasawa et al., 2020). The earlier Brassica genome assemblies focused on elite cultivars and combined Illumina paired-end reads with bacterial artificial chromosome (BAC)-end sequences to construct scaffolds and build high-quality assemblies. Markers from genetic maps were used, in addition, to merge assemblies and anchor scaffolds to pseudo-chromosomes. The genomes were either assembled de novo or based on a reference genome of the closest relative. The assembly of B. napus followed an earlier approach for allopolyploids by sequencing the diploid progenitor genome, similar to the methods used in the assembly of the strawberry and cotton genomes (Tennessen et al., 2014; Ming and Man Wai, 2015; Zhang et al., 2015).
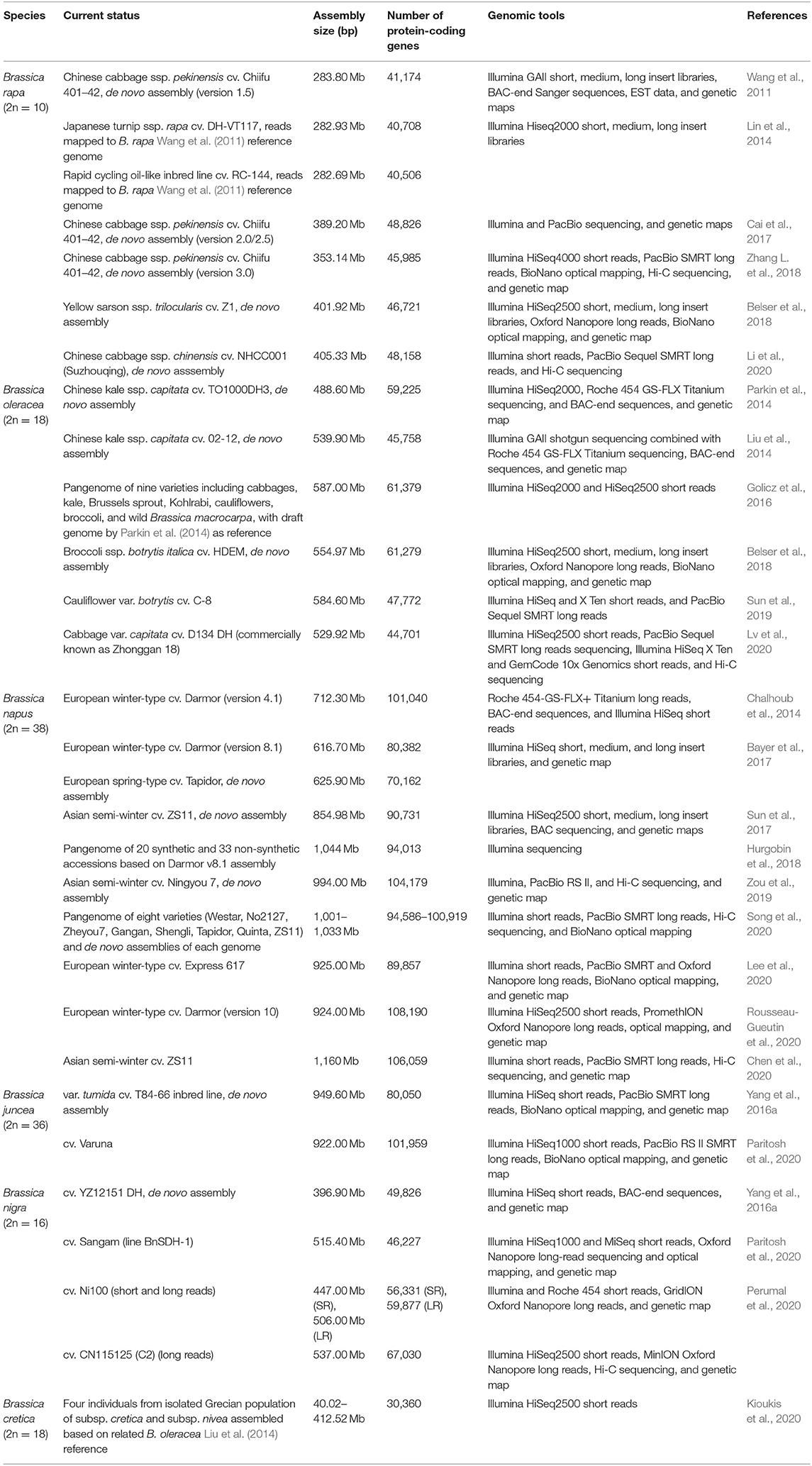
Table 1. List of domesticated and wild Brassica species nuclear genome assemblies.
Brozynska et al. (2016) explored the progress of CWR sequencing efforts and noted fewer efforts compared with domesticated relatives, a trend also observed in Brassica. Higher levels of heterozygosity in CWRs, which can result in greater assembly difficulties, might be contributing toward the trend (Brozynska et al., 2016). Recently, the genomes of two wild diploid perennial Brassica C-genome species were sequenced (Golicz et al., 2016; Kioukis et al., 2020). Brassica macrocarpa and Brassica cretica, both native to Greece, are potential wild progenitors of Brassica oleracea, which was thereby used as reference in the assembly of the two CWRs (Branca et al., 2012).
Next-generation sequencing (NGS) technologies, although extensively and successfully used in genome assembly, are limited by their relatively short read lengths. Shortcomings include misassembly and gaps in long repeat regions, difficulties in detecting larger SVs, transcript isoforms, and haplotype phasing (Van Dijk et al., 2018). Long-read third-generation sequencing (TGS) technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) allow less bias and more homologous coverage of the genome, thereby overcoming the challenges such as polyploidy and frequent repetitive elements. TGS and NGS are often combined with long-range mapping technologies (BioNano Genomics) and chromosome conformation capture (Hi-C) (Van Berkum et al., 2010), to enable the assembly of highly contiguous chromosome-level crop genome assemblies (Hu et al., 2018; Schreiber et al., 2018). Song et al. (2020) created eight high-quality B. napus reference genomes by integrating different combinations of Illumina, PacBio, Hi-C, and BioNano data. These high-quality B. napus reference genomes allowed the identification of SVs, including copy number variants (CNVs) and presence and absence variations (PAVs), and improve our understanding of the genome structure and genetic basis behind phenotype differentiation in B. napus.
Genomic comparison between two B. napus reference genomes by Bayer et al. (2017) highlighted the limitations of a single reference assembly, given the wealth of variation between individuals, concluding that the genetic diversity at a species level cannot be sufficiently captured by a single reference genome (Hurgobin and Edwards, 2017). The pangenome was “born” in an effort to capture the totality of diversity in a species or something broader (Vernikos, 2020). B. oleracea (Golicz et al., 2016) and B. napus (Hurgobin et al., 2018) were the first Brassica pangenomes to be published and investigated the diversity within the species. The second B. napus pangenome (Song et al., 2020) combined TGS technologies, including PacBio single-molecule real-time (SMRT) and ultralong nanopore sequencing. These pangenome assemblies demonstrated the prospect of uncovering and mining diversity within secondary crop genepools for crop improvement (Voss-Fels and Snowdon, 2016).
Affordability of sequencing is promoting a combination of de novo assembly and whole-genome re-sequencing (WGRS) efforts of wider genepools, including close relatives and CWRs in an effort to identify and explore useful genetic variation (Brozynska et al., 2016). While the pangenome represents the genomic makeup of a species (Tettelin et al., 2005), the super-pangenome includes the CWR genetic variability that has been lost due to domestication and breeding selection bottlenecks (Khan et al., 2020). This was well-demonstrated by the discovery of an abundance of unique genes upon the inclusion of the wild Brassica macrocarpa, in comparison with the other eight domesticated varieties, in the B. oleracea pangenome (Golicz et al., 2016). These genes form part of the pangenome's dispensable genome, and the findings yet again emphasize the value of the genetic variability captured within the wild Brassica spp. It is predicted that efforts to catalog and include more lines into pangenomes and super-pangenomes would probably never cease, thereby providing a constant contribution of valuable resources for crop improvement efforts in an ever-changing environment. Future exploration in genomic resources in Brassica will likely involve additional WGRS efforts and further pangenome studies to explore breeding histories and identify loci associated with important agronomic traits such as oil content and composition, seed quality, and disease resistance (Wang Y. et al., 2016; Lu et al., 2019; Dolatabadian et al., 2020; Gabur et al., 2020; Yan et al., 2020; Zhang et al., 2020).
Organelle Genomes
In Brassica, the mitochondrial (mt) genome assembly predates that of the whole genome due to its significantly smaller size (~20 kbp) (Palmer and Herbon, 1988; Handa, 2003; Kode et al., 2005) and high copy number per cell (Lima et al., 2016) (Table 2). Due to its small size in comparison with other higher plants, the Brassica mt genome was used as an early model in the understanding the plant mt, structure, function, and content (Grewe et al., 2014). Comparative analysis of the mt genome can be used to study interspecific phylogenetic relationships (Darracq et al., 2011) and uncover the plant's evolutionary history (Xue et al., 2020). For example, the mt genomes of Brassica rapa subspecies with distinct morphologies were found to be highly conserved (Hatono et al., 2017). Furthermore, the rate of mutation for Brassica mt DNA was four times slower than that of the chloroplast (Cp) DNA (Palmer and Herbon, 1988). Xue et al. (2020) compared mt DNA of the six members of the U triangle (Nagaharu et al., 1935) and revealed that B. oleracea was undergoing the most mt genomic change, while the B-genomes containing Brassica carinata and Brassica nigra were identified as maternally more distantly related to the remaining Brassica accessions of the U triangle (Xue et al., 2020). It was also suggested that Sinapis arvensis could have been misclassified based on both phylogenetic and mt genomic organization, placing it within the Brassica species and sister to the B. nigra–B. carinata lineage (Sang et al., 2020; Xue et al., 2020). Sang et al. (2020) also confirmed that the genome structure and evolutionary analysis of the S. arvensis organellar genomes were more similar to those of B. nigra and B. carinata. In addition, mt genes, encoding putative proteins with transmembrane domains, were discovered, which may explain the alloplasmic male sterility of novel cytoplasmic male sterility (CMS) derived from somatic cell hybridization between B. napus and S. arvensis.
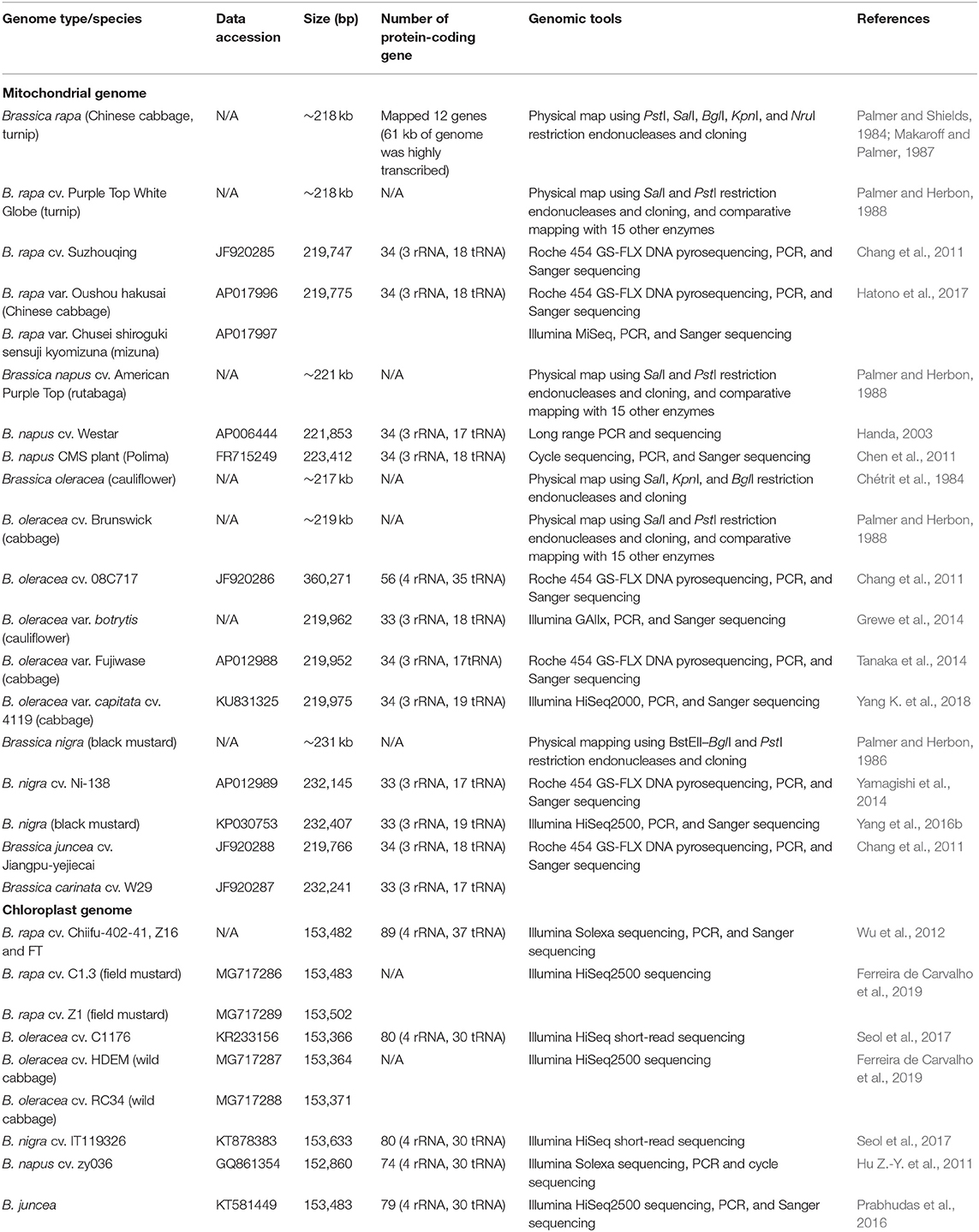
Table 2. List of domesticated Brassica species organelle genome sequences.
Cp genomes of most land plants vary between 120 and 160 kbp in size (Wicke et al., 2011). Xiao-Ming et al. (2017) further established that cp gene lengths were proportionally to cp genome size, based on the analysis of 272 species including B. napus and 14 other members of the Brassicaceae family. Phylogenetic analysis of the cp genomes of B. nigra and B. oleracea with those of 10 reported species in the order Brassicales suggested that B. oleracea is closely related to B. rapa and B. napus while B. nigra was more diverse than the neighbor species Raphanus sativus (Seol et al., 2017). Li et al. (2017) completed the de novo assembly of cp genomes of 60 Brassica genotypes of the six U triangle species. Subsequent phylogenetic analyses divided the Brassica genus into four clades: B. carinata and Brassica juncea, in accordance with the U-triangle model, shared their cp genome with hybridization donors B. nigra and B. rapa, respectively. Two types of cp genomes were discovered in B. rapa, while the presence of both B. rapa cp genomes in B. napus strongly suggests two independent hybridization events. These findings were consistent with mt genome findings (Palmer and Herbon, 1988).
Besides photosynthesis, a number of essential metabolic reactions are catalyzed in the cp. These include the biosynthesis of partial amino acids, lipids and fatty acids, vitamins, and isoprenoids, as well as the reduction of nitrites and sulfates (Chen et al., 2018). The cp is furthermore involved in the synthesis defense-related hormones and signaling molecules metabolites associated with disease response and environment changes including heat and light (Lu and Yao, 2018). Genes present in the cp genomes could potentially be explored in Brassica to improve yield and resistance to biotic and abiotic stresses. It is clear that the composition and structure of the organellar genome not only hold potential in the elucidation of organellar genome evolution and phylogeny but and understanding thereof may be useful in the identification breeding compatible germplasm resources and CMS and provide opportunities for the introducing of new agronomic and horticultural traits into Brassica crops (Daniell et al., 2016). Organellar genome sequences are therefore valuable assets in the future crop improvement efforts.
Functional Trait Discovery and Characterization
The characterization and subsequent exploration of discovered genetic diversity can uncover useful genes linked to adaptation and resistance to abiotic and biotic constraints (Khan et al., 2020). Available Brassica reference genomes have provided the foundation to facilitate the fine-scale mapping and elucidation of functionally significant variations in Brassica accessions. In this section, we review genome-wide approaches utilized in the identification of alleles associated with desirable traits.
Genome-Wide Single-Nucleotide Polymorphism Discovery Through Whole Genome Re-sequencing
Available reference genomes provide a valuable tool in the study and detection of genetic variation, which can be reliably integrated and reproduced between studies (Malmberg et al., 2018). Alignment and comparison of WGRS data to the reference genome allow the simultaneous detection of large numbers of unbiased genomic single-nucleotide polymorphisms (SNPs), indels, and SVs. WGRS therefore permits a more in-depth interrogation of the genome than complexity reduction methods, resulting in a significant increase in the number of SNP markers detected.
Annotated, high-quality SNPs and SVs set the stage for high-resolution genome-wide association studies (GWASs). Quantitative trait locus (QTL) discovery can lead to the identification of genetic loci and subsequently provide the basis for the functional validation of candidate genes controlling important traits (Lu et al., 2019). Subsequently, by identifying these traits of interest, marker-assisted selection (MAS) could be introduced to advance breeding efforts for easily inherited traits (Harper et al., 2012). For example, in gene pyramiding, multiple genes or major QTLs are introgressed into a single genetic background (Pérez-de-Castro et al., 2012). This approach was recently implemented to introgress multiple genes conveying Sclerotinia resistance from wild Brassica oleracea into canola (Mei et al., 2020). Furthermore, as long-read sequencing technology advances and improves in accuracy, both PAVs and CNVs are likely to drive the latest source of rich adaptive variation including in crops subjected to biotic and abiotic stresses (Gabur et al., 2018).
In Brassica species, WGRS has facilitated the identification of intraspecific and interspecific genetic polymorphisms. Comparative analysis between different Brassica rapa morphotypes: Japanese turnip, rapid cycling, and Chinese cabbage cv. Chiifu, revealed, respectively 1,090, 1,118, and 1,464 unique genes in each of the genomes (Lin et al., 2014). Orthologous gene comparison suggested earlier divergence between the three varieties before a more recent domestication event. Gazave et al. (2016) used WGRS to survey the genetic diversity present in a worldwide collection of 782 accessions of Brassica napus. A total of 30,881 high-confidence SNP markers were identified, which upon analysis revealed distinct evolutionary histories for the A and C subgenomes. Wu et al. (2019) re-sequenced 991 spring, winter, and semi-winter B. napus germplasm accessions, originating from 39 countries. By mapping reads to the “Darmor-bzh” and “Tapidor” reference genomes, a total of 5.56 and 5.53 million SNPs, in addition to 1.86 and 1.92 million indels, were respectively identified. Comparison of SNPs using GWAS revealed a global pattern of genetic polymorphisms as well as paths of allelic drift within the main populations of B. napus. Selective sweeps disclosed the genetic basis of divergence between the ecotypes, while SNPs discovered in the promotor regions of FLOWERING LOCUS T and FLOWERING LOCUS C orthologs also corresponded with ecotype groups. Malmberg et al. (2018) furthermore utilized WGRS to develop genomic resources consisting of 4,029,750 high-confidence annotated SNPs with predicted effects, as well as SVs in the form of 10,976 deletions and 2,556 insertions. These valuable genomic resources have the potential to bring together global breeding efforts in the development of locally adapted B. napus varieties.
Single-Nucleotide Polymorphism Arrays
Available sequencing data for Brassica crops allowed researchers to develop and use high-throughput molecular markers, such as SNPs, more efficiently (Clarke et al., 2013). These markers form an integral part of genomic diversity and, due to their abundance across the plant genome, have become an invaluable tool in crop improvement programs (Scheben et al., 2019). SNP screening can be carried out by WGRS, genotyping-by-sequencing (GBS), or alternatively using SNP arrays. High-density SNP arrays provide an alternative and reproducible genotyping platform, which has been widely used in the characterization of germplasm, GWAS, and QTL studies including the analysis of structural variation (You et al., 2018; Scheben et al., 2019).
A community-driven Brassica 60K (AC genomes) Illumina Infinium™ array (Clarke et al., 2016) was developed and more recently expanded to include the B-genome in the Brassica 90K Illumina Infinium™ array (Scheben et al., 2019). The usefulness of Brassica SNP arrays was demonstrated in the genotyping of resistance genes on chromosome A7 in B. napus (Dalton-Morgan et al., 2014), prediction of candidate genes for clubroot disease resistance in B. napus (Li et al., 2016), and the assessment of de novo homologous recombination events in B. napus (Higgins et al., 2018). In addition, several closely linked candidate genes were identified using the 60K Brassica SNP array in the development of functional haplotype markers for the improvement of the oleic acid content in rapeseed (Yao et al., 2020). Several yield associated traits have also been identified using the array platform including branching number (He et al., 2017), ovule numbers (Khan et al., 2019), seed quality (Gajardo et al., 2015), and stem strength (Li H. et al., 2018).
The creation of artificial Brassica allohexaploid could potentially result in the development of new oilseed and vegetable crops types with greater inter-subgenomic heterosis. These synthetic tri-genomic hexaploid Brassica species are potentially more vigorous and adaptable to a wider range of environmental conditions (Yan et al., 2009; Pradhan et al., 2010; Tian et al., 2010; Geng et al., 2013; Malek et al., 2013; Li et al., 2015; Gupta et al., 2016; Zhou et al., 2016; Mwathi et al., 2020). SNP genotyping was carried out by Gaebelein et al. (2019) using the 90K Brassica SNP array and GWAS to determine the relative impact of genome rearrangement events and inherited allelic variants on meiotic stability. A strong correlation between fertility and meiotic behavior in populations of Brassica allohexaploids segregating for alleles from parent allotetraploid species B. napus, Brassica juncea, and Brassica carinata was found. Potential genes of interest were subsequently identified for further investigation into meiotic regulation and future establishment of stable A, B, and C allohexaploids (Gaebelein et al., 2019).
Although SNP arrays can provide vital data to breeders and researchers, public accessibility to genotypes identified can be limited due to the lack of public repositories or databases designed to host crop SNP array data. To address this constraint, Scheben et al. (2019) established the CropSNP database (http://snpdb.appliedbioinformatics.com.au) for SNP array data generated on the Illumina Infinium™ Brassica 60 and 90K array platforms.
Transcriptomics
One of the key challenges in genomics-based breeding remains the complex linking of genotype to phenotype across tissue types, developmental stages, and environmental conditions. Transcriptomics, although part of associated “omics,” have emerged as an exceptional tool in the functional inference of genetic variability (Wang et al., 2019) with technological innovation constantly advancing the field (Lowe et al., 2017; Wang et al., 2019).
RNA-Seq technology has been extensively used in the mapping of exon/intron boundaries, improvement of genome annotations, and the detection of rare transcripts and splicing variants (Pérez-de-Castro et al., 2012). For example, the transcriptional regulation of anthocyanin biosynthesis in B. juncea was studied to identify differentially expressed genes between the purple and green leaves from a backcrossed BC3 segregation population. Genes associated with phenylpropanoid biosynthesis, phenylalanine metabolism, and flavonoid biosynthesis were differentially expressed, while genes involved with anthocyanin biosynthesis (BjTT8 and BjMYC2) were up-regulated in the purple leaves. Understanding anthocyanin biosynthesis and its regulatory network in Brassica is a prerequisite in the development of health-promoting anthocyanin-rich vegetables (Heng et al., 2020).
The complex defense response between a Sclerotinia sclerotiorum resistant and susceptible line of B. napus was analyzed in a study by Wu et al. (2016). Dynamic transcriptome analyses uncovered differences between susceptibility and resistance associated with the magnitude of expression changes in genes involved in pathogen recognition, MAPK signaling cascade, WRKY transcription regulation, jasmonic acid/ethylene signaling pathways, and biosynthesis of defense-related protein and indolic glucosinolate. Valuable insights gained will assist in the development of effective strategies in Sclerotinia-resistance breeding (Lu et al., 2014; Wu et al., 2016).
Limitations such as misassembly, associated with short read RNA-Seq, hampers the full-length assembly of transcripts from highly repetitive regions or analogous gene families. These difficulties are often more pronounced in polyploid plants. TGS technologies such as SMRT by PacBio and Oxford Nanopore single molecule structure sequencing (SMS-seq) provide an opportunity to construct full-length transcripts, with the possibility to capture structural variations, tertiary interactions, and the dynamics of riboswitch ligand binding (Bizuayehu et al., 2020). Full-length transcriptome sequencing (PacBio) was used by Tan et al. (2019) to explore the transcript and splice isoforms expressed during anther development in Chinese cabbage (B. rapa ssp. pekinensis). In addition to predicted fusion transcripts and poly-A sites, 53 key genes active during anther development were detected, of which eight annotated loci had alternatively spliced isoforms. The transcripts generated provided a valuable resource for the characterization of anther-specific gene expression and improved Chinese cabbage genome annotation (Tan et al., 2019).
An et al. (2019) conducted a comprehensive study comparing the genetic diversity of 183 B. napus, 112 B. rapa, and 42 B. oleracea accessions, along with 20 wild relatives (Brassica hilarionis, Brassica villosa, Brassica montana, Brassica macrocarpa, Brassica rupestris, Brassica incana, and Brassica insularis) and five other Brassicaceae species as outgroups in order to improve the understanding in the origin and diversification of B. napus. RNA-Seq reads generated from B. rapa accessions and B. oleracea and other wild C genome species were respectively mapped to the A and C genomes of the B. napus Darmor-bzh reference genome to identify SNPs. Six genetic clusters of B. napus were identified, which were shown to have undergone different selective pressures in accordance with known breeding histories. Although the multi-origin of B. napus remained elusive, the study contributed toward the identification of putative candidate genes to important agronomic traits, which, along with high-quality SNPs identified, have the potential to facilitate rapeseed improvement and germplasm preservation.
In line with the pangenomics approach, He et al. (2015) assembled the first pan-transcriptome resources for the Brassica A and C genomes. The pan transcriptome was established using existing coding DNA sequence (CDS) gene models from the B. oleracea TO1000 and B. napus Darmor-bzh reference genomes in addition to preliminary CDS models from the B. rapa Chiifu genome sequence assembly. The construction of the pan-transcriptome allows, in a similar fashion as the pangenome, the discovery of functional dispensable genes (Jin et al., 2016).
Epi-Genomics
In addition to the identification of novel genes and useful haplotypes, epigenetic variation has the potential to contribute toward crop adaptation and productivity (Dwivedi et al., 2017). These epigenetic variations are plant developmental and adaptation responses to environmental constraints (Gallusci et al., 2017; Tirnaz and Batley, 2019b). Epigenomics encapsulates genotype × environment interactions and their independent influence on the phenotype (Seymour and Becker, 2017) and may contribute as potential phenotypic resources for breeding.
Epigenetic regulation is independent of DNA sequence alteration and stably inherited during mitosis or meiosis (Weigel and Colot, 2012). DNA (de)methylation, histone modification, and chromatin remodeling involves regulatory reprogramming at transcriptional and post-transcriptional levels (Paszkowski and Whitham, 2001; Tirnaz and Batley, 2019a). DNA methylation in plants includes de novo methylation, as well as maintenance and demethylation, as a means of regulatory check and balance in gene expression (Elhamamsy, 2016; Tirnaz and Batley, 2019a). Histone modification refers to the methylation or (de)acetylation of histone proteins at the N-terminal. Mono-, di-, or trimethylation of the lysine residue in the former results in various functional responses, while the histone acetylation and deacetylation are associated with gene activation and repression, respectively (Fuchs et al., 2006). Additionally, chromatin remodeling is influenced by histone octomer movement (Perrella and Kaiserli, 2016), ATP-dependent enzyme affecting nucleosome composition (Tariq and Paszkowski, 2004) or histone variants (Rando and Ahmad, 2007) cause the DNA sequence to become inaccessible to transcriptional mechanisms, resulting in transcriptional silencing.
Epialleles, or epivariants, first coined in mammals by Rakyan et al. (2002), refer to genetically identical and stable alleles that are variably expressed due to epigenetic regulations and result in difference in phenotype (Richards, 2006; Dolinoy et al., 2010). Earlier studies on epialleles lack whole-genome information to decipher association with desirable traits (Seymour and Becker, 2017). While more studies have been conducted recently, epigenomics is still considered in its infancy compared with the characterization of genetic sequence- or structural-caused variation.
Epigenomic studies in Brassica crops have predominantly involved global or targeted methylation profiling for natural or experimentally induced epivariants as defined by Gallusci et al. (2017). Early Brassica methylation studies relied on a chemical demethylation agent treatment using 5-azacytidine (5-AzaC) and on cytological (Solís et al., 2015) as well as targeted allele-specific and methylation-sensitive amplified polymorphism (MSAP) techniques to capture the global DNA methylation pattern (Shiba et al., 2006; Hauben et al., 2009) (refer Table 3). Hypomethylated populations created using 5-AzaC treatment could also be mined for epiallelic variation (Amoah et al., 2012). The forward and reverse screening of epigenetic variation was employed in functional and inheritance studies in B. rapa var. trilocularis and suggested to have potential as an intervention strategy for crop improvement (Amoah et al., 2012).
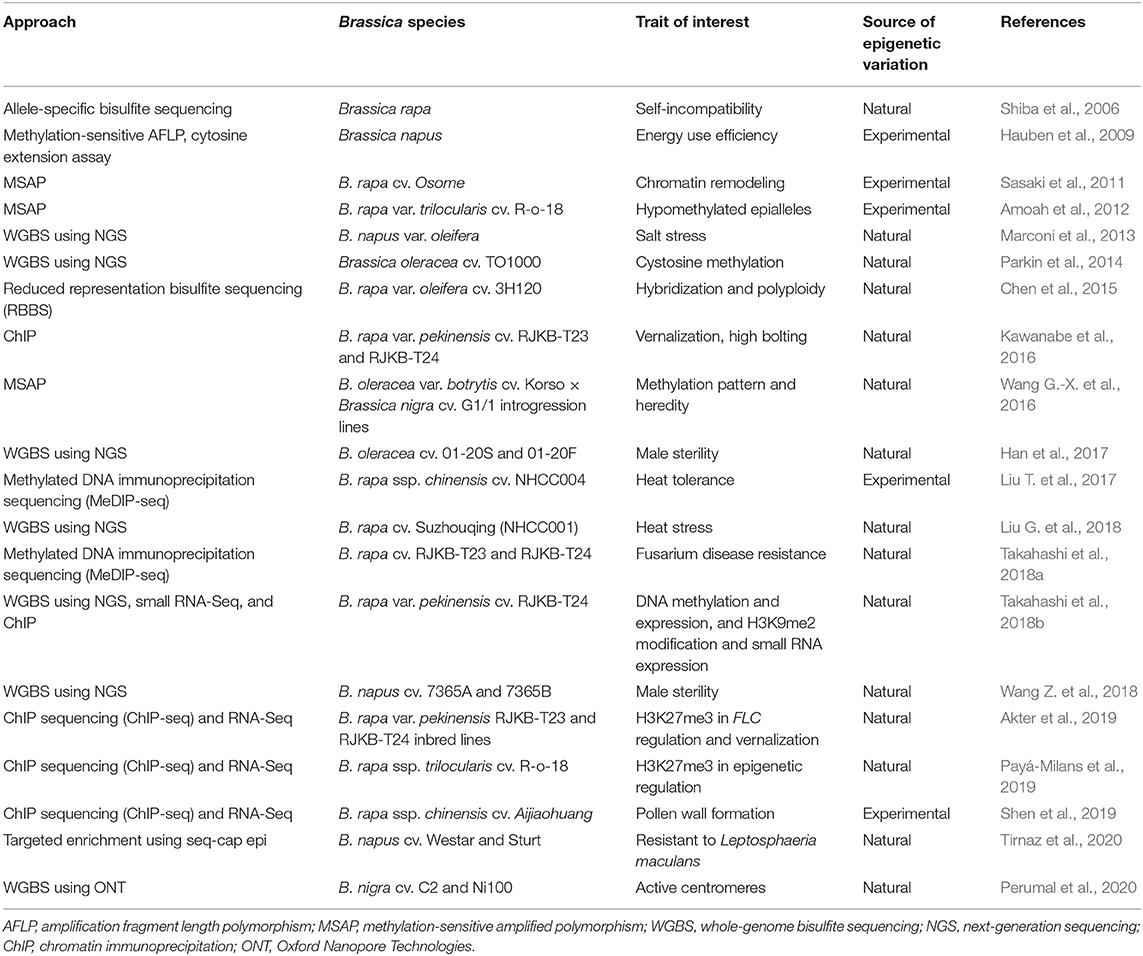
Table 3. Exploration of epigenetic resources in Brassica for natural and experimentally induced epivariants.
These techniques were superseded by whole-genome bisulfite sequencing (WGBS) first described in Arabidopsis (Cokus et al., 2008; Lister et al., 2008). With the use of the WGBS approach, DNA hypomethylation of the multiallelic Bnams4 gene associated with male sterility was detected in young floral buds (Wang Z. et al., 2018). The male sterility trait promotes heterosis and hybrid development and hence is favored in crop improvement and breeding strategies (Saxena and Hingane, 2015).
Chromatin immunoprecipitation (ChIP) is another useful strategy for protein–gene interaction studies and, in the case of methylation, for investigating specific histone modifications (Das et al., 2004; Kawanabe et al., 2016). Kawanabe et al. (2016) developed positive and negative control primers to validate ChIP assays. The primers were targeted at histone modifications at H3K4me3 (trimethylation of the 4th lysine of H3), H3K9me2, H3K27me3, and H3K36me3 and used to study the response of 4 FLC paralogs to vernalization in B. rapa var. pekinensis. ChIP combined with WGBS further expanded the ability to investigate complex interactions between genetic and epigenetic factors (Li and Tollefsbol, 2011). These studies assisted in the identification of epigenetic markers, epigenetic QTL, and genes associated with floral and pollen development, self-incompatibility, salt and heat stress, vernalization, disease resistance, and male sterility and in the assessment of methylation profiles of introgression lines for crop improvement resource (Table 3). Nonetheless, due to the complexity of epigenetic interaction and its involvement in complex regulatory networks, these epigenomic approaches still require extensive investigation before their application in crop improvement can be implemented.
Reverse Genetics
One of the most cost-effective and quick approaches used to identify genetic variation in crop populations is by Targeting Induced Local Lesions in Genomes (TILLING). This method combines chemical mutagenesis, to create lesions on the genome, and molecular techniques such as PCR and DNA pooling, to identify point mutations within the population. TILLING was first demonstrated in Arabidopsis using ethyl methanesulfonate (EMS) (McCallum et al., 2000), which results in base transitions by causing the G residues to alkylate and as a result pair with T instead of C (Rashid et al., 2011). Soon after, the technique was explored in Brassica species for various traits of interest including fatty acid content and shatter tolerance (Table 4). However, this method is less feasible and becomes more time-consuming if multiple genes are targeted, such as those involved in specific biopathways (Sashidhar et al., 2020). Examples of canola varieties developed that benefit from this approach include PodGuard trait for In Vigor R5520P and 1H51 RR varieties, which were commercially marketed by Bayer for their resistance to pod shattering (Raman et al., 2019).
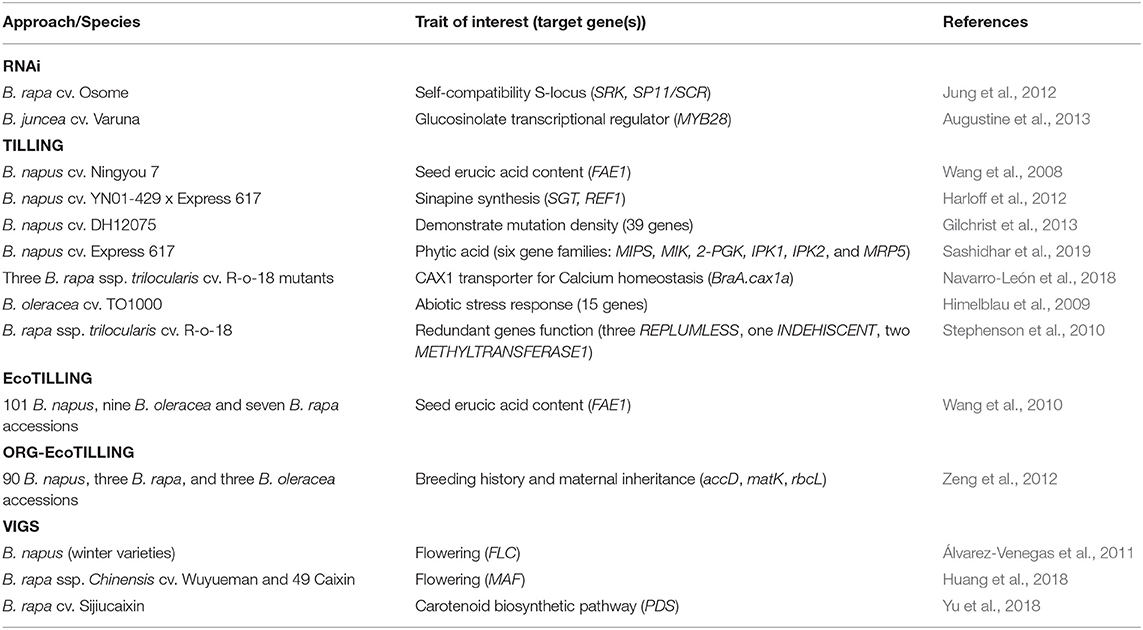
Table 4. Reverse genetic resource in Brassica over the last 10 years.
TILLING has been further developed to include EcoTILLING (Ecotype) and ORG-EcoTILLING. EcoTILLING involves pooling DNA from only two individuals consisting of a reference and queried genotypes (Backes, 2013); therefore, instead of creating mutant populations, this more recent approach involves a study of allelic variation. EcoTILLING requires re-sequencing efforts to characterize and locate the genotypic polymorphism (Wang et al., 2010). Additionally, this approach is high-throughput and can associate natural variants with gene function, trait association, and phylogenetic relationships (Zeng et al., 2012). ORG-EcoTILLING was first utilized in Brassicas to explore the use of TILLING in organelle genomes by combining CEL1 endonuclease, which cuts specific mismatches in heteroplex DNA, and PCR, for three cp genes and one mt gene. ORG-EcoTILLING consistently confirmed B. rapa as B. napus maternal progenitors based on phylogenetic analysis. Additionally, it also uncovered the possibility of multiple origins and evolution throughout B. napus domestication with the identification of three additional divergences in the accessions.
The TILLING approach is considerably more accessible in terms of cost and time than other specialized reverse genetics approaches like RNA interference (RNAi) or gene silencing, and virus-induced gene silencing (VIGS). RNAi gained traction with the ability to knock down gene functions mediated by small interfering RNA (siRNA) or microRNA (miRNA) (Pe'ery et al., 2003; Limera et al., 2017). Traits introduced using RNAi, such as self-compatibility in B. rapa, have been found to be stable even in crosses with commercial variety (Jung et al., 2012). This will help improve likelihood for the seeds to be used in commercial cultivation. Similar to RNAi, VIGS also involves introduction of dsRNA molecules. A viral vector genome such as Cabbage Leaf Curl Virus (CaLCuV) is modified to include the plant target gene fragment (150–800 bp) and to remove the viral inducing host gene, thus forming a recombinant virus (Lu et al., 2003; Ramegowda et al., 2014; Bekele et al., 2019). The recombinant vector introduces infection in the plant, is amplified, and generates dsRNA molecules. These dsRNA molecules triggered post transcriptional gene silencing, once detected by the host plant, causing it to be cleaved into siRNA. The RNAi silencing complex and antisense siRNA strands associates together and begin to target RNAs, which complemented the siRNAs. These target-specific RNAs were screened and destroyed, which subsequently caused the target gene to be silenced. VIGS can also be utilized for tissue-specific gene silencing, helpful in screening stress responses, and induced transcriptional gene silencing by targeting the gene promoter (Kanazawa et al., 2011; Senthil-Kumar and Mysore, 2011; Bekele et al., 2019). VIGS research in Brassica includes interest in understanding the vernalization pathway in Brassicas as a means to improve flowering and reproductive development by silencing genes associated with late flowering (Álvarez-Venegas et al., 2011) or floral organ transition (Huang et al., 2018).
Genome Manipulation
Traditional breeding approaches rely on the diversity found in local land races, mutation panels, or even germplasm from related species to introduce desired traits or elite alleles through costly and time-consuming backcrossing programs (Dwivedi et al., 2017). The absence of natural genetic diversity and potential linkage drag introducing closely linked unwanted agronomic characteristics has plagued crop improvement efforts (Holme et al., 2013). Increased availability of genomic resources, identified and well-characterized genes, as well as a deeper understanding of underlying molecular mechanisms has led the way for the introduction of innovative approaches to overcome these limitations and fast-track crop breeding (Scheben et al., 2016: Hickey et al., 2019).
Gene Transformation
Genetic modification through the introduction of transgenes was developed in an attempt to expand the available genepool (Kamthan et al., 2016). Agrobacterium tumefaciens and biolistic techniques are widely and efficiently employed to mediate the transfer of selected exogenous genes or regulatory elements from an unrelated species or even non-plant organism (Moloney et al., 1989; Altpeter et al., 2016). Numerous transgenic Brassica spp. have been developed in an attempt to introduce traits such as salt tolerance (Kim et al., 2016), disease resistance (Grison et al., 1996; Aghazadeh et al., 2016; Zarinpanjeh et al., 2016), reduced sinapine content (Wolfram et al., 2010; Harloff et al., 2012), and herbicide tolerance (Beversdorf et al., 1988; De Block et al., 1989; Cuthbert et al., 2001). The potential of transgenics in advanced plant metabolic engineering is however best demonstrated in the development of transgenic omega-3 Brassica napus varieties by BASF and Cargill, and Nuseed, CSIRO and GRDC (Napier et al., 2019) respectively, as well as Brassica juncea by Wu et al. (2005). Transgenic B. napus accumulating long-chain polyunsaturated fatty acids (LC-PUFAs) were engineered by each of the initiatives through the introduction of large multi-transgene cassettes. The BASF cassette (~44 kbp) contained 12 genes, while the Nuseed initiative's cassette (~23 kbp) contained six omega-3 LC-PUFA biosynthetic genes. Each gene was under the regulation of a seed-specific promoter. Each cassette also contained a gene for herbicide resistance (Connelly and MacIntosh, 2018; Sottosanto et al., 2018). The LC-PUFA profile produced by the two omega-3 transgenic canola varieties varied and respectively contained ~7% eicosapentaenoic acid (EPA), ~3% docosapentaenoic acid (DPA), and ~1% docosahexaenoic acid (DHA) (LBFLFK); and <0.5% EPA, ~1% DPA, and ~10% DHA (NS-B50027-4) respectively (Napier et al., 2019). Very-long-chain (VLC) PUFA accumulating B. juncea was on the other hand engineered in a stepwise approach through a series of transformations with increasing numbers of transgenes. The resulting transgenic B. juncea yielded up to ~15% EPA, ~4% DPA, and ~1.5% DHA (Wu et al., 2005).
In addition to the transformation of the nuclear genome, modification of the cp genome has also been established. The introduction of foreign genes into the cp can address nuclear transgenic limitations such as low-level transgene expression (Jin and Daniell, 2015) and the potential transgene escape via pollen (Daniell, 2007). The stable integration and expression of more than 40 cp-based transgenes has been reported by Daniell et al. (2016), most of which were aimed at potentially enhancing biotic stress tolerance and consequently yield. An example is the development of a cabbage-plastid transformation system for introduction of the insecticidal cry1Ab gene. With the use of a species-specific vector, the expression of the BT-toxin facilitated the control of the diamond moth, an economically important Brassica pest (Liu C. W. et al., 2008). Introduced insect resistance using cry genes was also reported for rapeseed (Schuler et al., 2004) and collards (Cao et al., 2005).
Improved weed and insect control using GM crops has assisted farmers in attaining higher yields with fewer resources. The analysis by Brookes and Barfoot (2018) estimated that the adoption of commercial herbicide-tolerant GM canola and sugar beet led to a total global income gain of $559 million in 2016 and $6.44 billion cumulatively since 1996. Despite the commercial benefits, growing consumer skepticism (Frewer et al., 2013), as well as potential risks to human health and the environment (Zhang et al., 2016), has encouraged the development of alternative genomic modification technologies.
Cisgenesis and intragenesis, based on the same gene transfer technologies as transgenesis, were consequently developed. Genetic crop modification thereby involved the introduction of target DNA from the same plant species, or a sexually compatible species, for crop improvement. Resulting crop plants are free from any foreign DNA mitigating associated risk to some extent (Espinoza et al., 2013; Holme et al., 2013). Alternative approaches such as reverse breeding exploit the use of transgenes to accelerate initial breeding. The unwanted transgene is eliminated through Mendelian segregation during the later stages of the breeding process (Dirks et al., 2009; Basso et al., 2020).
Genome Editing
Recently, GE technologies have come to the foreground, allowing the precise and permanent modification of specific genes or genomic regions. ODM, programmable SSNs, and base editing provide an opportunity to study gene function and alter crop traits through the mutation of specific genes, reprogramming of epigenetic markers, and the generation of site-specific sequence modifications (Voytas and Gao, 2014; Ran et al., 2017; Jansing et al., 2019).
Oligo-Directed Mutagenesis
Traditional mutation breeding using chemical or irradiation results in random mutations in the genome. The movement toward a more desired and controlled site-specific targeted mutagenesis took shape in the 1970s (Lusser and Davies, 2013). ODM, also known as targeted gene repair, oligonucleotide-directed gene targeting, genoplasty, and chimeraplasty, makes use of 20–100 bp of DNA or RNA oligonucleotides to introduce mutations at the target site. The synthesized oligonucleotides are designed to be homologous to the target site with the exception of 1–4 bp (Lusser et al., 2012). Upon transfection, the oligonucleotides associate with the target site, prompting DNA repair at the sequence mismatch sites resulting in base pair mutations, deletions, or reversal of mutations (Lusser et al., 2012; Lusser and Davies, 2013). With a difference of often just a few nucleotides underlying important traits in plants, the application of ODM held potential as a non-GM organism (GMO) base pair-specific oligonucleotide-directed gene editing platform to augment the genetic diversity of a specific genotype. The use of ODM was furthered by Cibus as part of the commercial Rapid Trait Development System (RTDS™) introducing novel and commercially valuable traits such as herbicide resistance into a variety of crops including oilseed rape (Gocal et al., 2015). Ruiter et al. (2003) found that spontaneous mutation in plants obscured the intended sequence modifications in B. napus mediated through self-complementary RNA–DNA chimeric oligonucleotides or chimeraplasty. Studies by Sauer et al. (2016) confirmed that significant precise gene-editing events in plants could be realized by ODM alone and suggested that ODM efficiency could be further improved in combination with reagents that cause DNA double-stranded breaks (DSBs).
Programmable Sequence-Specific Nuclease
GE using programmable SSNs is generally achieved through the induction of a controlled DSB at a target locus using SSNs. The DBS activates the intracellular DNA-repair pathways and is repaired through either non-homologous end joining (NHEJ) or homology-directed repair (HDR) (Gaj et al., 2013). The imprecise re-joining of the DBS through NHEJ leads to the introduction of indels at the target loci and disruption of gene function. On the contrary, HDR entails the use of an exogenous DNA-repair template to bridge the DSB site. The repair template, a double-stranded DNA vector or a single-stranded DNA oligonucleotide, enables the introduction of a precise mutation or insertion to alter gene function (Zhang et al., 2013). Several engineered nuclease systems have been developed including meganucleases (MN), zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR) coupled with a CRISPR-associated protein (Cas).
Zinc Finger Nucleases
ZFN, first described by Kim et al. (1996), employs novel hybrid site-specific endonucleases created by the linking of two different zinc finger proteins (ZFPs) to the cleavage domain of the bacterial FokI endonuclease. The zinc finger domains are designed to each recognize and bind to a unique 3- to 4-bp DNA sequence adjacent to the target site. The tandem repeats can be constructed to recognize an extended 9- to 18-bp DNA sequence (Lin and Musunuru, 2016). In an attempt to modify seed oil composition in B. napus, Gupta et al. (2012) engineered ZFP transcription factors (TFs) to fuse to a conserved region downstream of the transcription start site of two canola KASII genes. The modification resulted in the escalated expression of the KASII mRNA, increasing C18 and lowering palmitic acid levels well as the overall saturated fatty acid content in the seed. Canola oil with a lower saturated fat content is more desirable and potentially poses health benefits (Hyseni et al., 2017). Despite the progress in technology, the engineering of the desired ZFN-binding domain remains challenging and time-consuming, with further limitations presenting in selection of the target site (Cox et al., 2015).
Transcription Activator-Like Effector Nucleases
TALENs are similar in structure to ZFNs, composed of di-meric DNA-binding proteins fused to the nuclease domain FokI (Cermak et al., 2011). The central domain of the TAL effector consists of a polymorphic repeat of ~34 amino acids with hypervariable di-amino acids at positions 12 and 13. These so-called repeat variable di-residues (RVDs) associate and recognize a corresponding C, T, A, or G nucleotide (Scholze and Boch, 2010). The longer DNA recognition sites promote specificity and reduce potential off-target effects (Li et al., 2012). The TAL effector DNA binding domain is more flexible and can be customized, which broadens its potential application.
Sun et al. (2013) demonstrated the suitability of the TALEN construct to alter the endogenous FRIGIDA (vernalization determinant) gene in Brassica oleracea var. capitata, further suggesting that the method could be applied to related Brassica spp. TALENs with mt localization signals (mitoTALENs) were designed by Kazama et al. (2019) to knock out CMS-associated genes at orf79 and orf125, respectively, of CMS varieties of rice and B. napus (SW18). Induced mt modifications restored fertility without causing noticeable phenotypic changes and were found to be stable and maternally inherited. The successful modifications of the mt genome pose the prospect of “mitochondrial breeding” in plants (Kazama et al., 2019), which can play an important role in the study and future conditioning of plant responses toward climate change (Budar and Roux, 2011; Sweetman et al., 2019; Florez-Sarasa et al., 2020). TALENs similar to ZFNs are time-consuming genome manipulation techniques (Razzaq et al., 2019) and require the extensive screening of large numbers of manipulated individuals.
RNA-Guided Nucleases
The second-generation CRISPR/Cas9 system provides an alternative approach in targeted nucleases. In contrast to the ZFN's and TALEN's engineered protein associated DNA-binding systems, the CRISPR/Cas9 system has a single-guide RNA (sgRNA) bound to the Cas9 endonuclease that directs the complex to a specific site in the genome (Jinek et al., 2012; Cong et al., 2013). Recognition is attained through base pairing between the programmable 20-bp-long spacer region at the 5′-end leading sequence of the gRNA and specific DNA target. The Cas9 nuclease uses the CRISPR gRNA–DNA pairing as guide in combination with adjacent the DNA protospacer-adjacent motif (PAM) to cleave the DNA. This simplicity and flexibility in the programming of the CRISPR/Cas9 system have facilitated its adoption and exploitation in plants including Brassica (Zhang et al., 2018b). Lawrenson et al. (2015) demonstrated the efficiency of the CRISPR/Cas9 GE tool for the first time in the knockout of target genes in B. oleracea. Introduced mutations were stably inherited and transgene-free plants obtained through segregation.
Due to the allotetraploid nature of some Brassica species, the observed effect of single gene modification is often limited by its potential redundant function. Modification of all homologous genes is hence required to obtain a reliable altered genotype and phenotype (Sashidhar et al., 2019). Multiple guide sequences can be encoded into a single CRISPR array, allowing the simultaneous editing of several sites (Cong et al., 2013), making the CRISPR/Cas9 system a valuable tool in the knockout of redundant genes or parallel pathways in polyploids. Sashidhar et al. (2019) illustrate a case in point with the CRISPR/Cas9-mediated knockout of multiple paralogs of the key enzyme inositol tetrakisphosphate kinase (ITPK) involved in the synthesis of phytic acid in B. napus seed. A noticeable change in the phytic acid content of B. napus seed was observed only in triple mutants of the essential BnITPK genes.
The CRISPR/Cas9 GE tool has been widely adopted in the manipulation and study of a variety of genes underlying agronomical important traits in Brassica. These include traits such as pod shatter resistance (Braatz et al., 2017; Zhai et al., 2019), multi-ocular silique (Yang Y. et al., 2018), increase in oleic acid content in seed (Okuzaki et al., 2018), and seed coat color (Zhai et al., 2020) (Table 5). Even though the use of the CRISPR/Cas9 system is popular in gene knockout or knock-in studies, it is limited by the introduction of random indels at the target site in addition to the possibility off-target mutations.
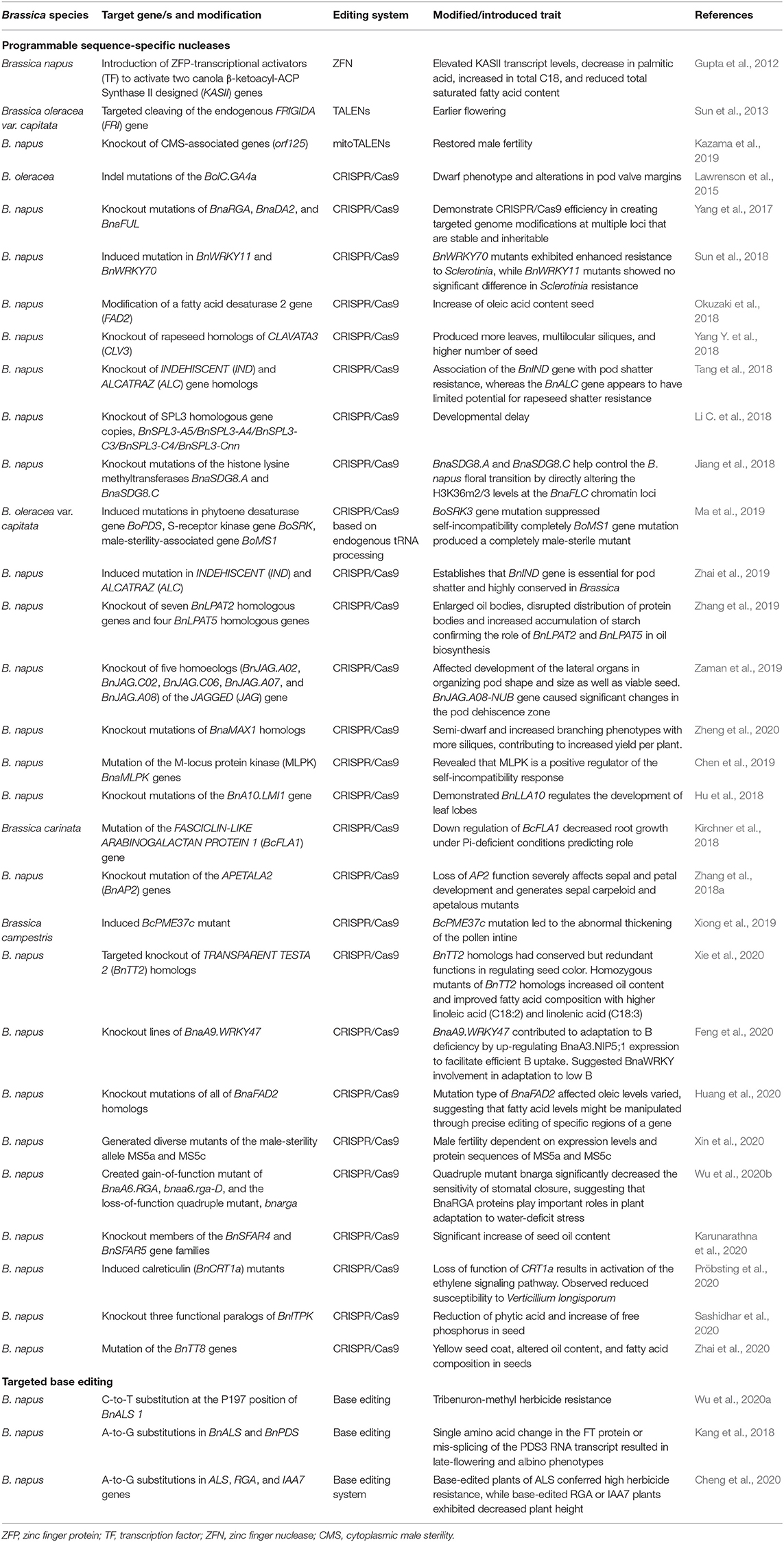
Table 5. Application of programmable sequence-specific nucleases and targeted base editing tools in genome editing of Brassica.
Targeted Base Editing
Targeted base editing is one of the newest additions to GE. The technique is based on the CRISPR/Cas9 system and enables the direct and irremediable conversion of a selected target base without the induction and repair of a DSB (Komor et al., 2016). In the base-editing system, a cytosine or adenosine deaminase domain is fused to the N-terminus of a deactivated Cas9 (dCas9) of Cas9 nickase (nCas9). Although the Cas9 retains the ability to be guided by the gRNA, it instead mediates the direct conversion of cytidine (C) to uridine (U) resulting in a C-to-T or G-to-A substitution resulting in a single controlled point mutation or base correction rather than a random gene disruption. The precise mutations can lead to the introduction of stop codons, changes in amino acids, and regulatory site modification, thereby improving the resolution in the functional analysis of genes and proteins to a single nucleotide or amino acid (Komor et al., 2016). Kang et al. (2018) established an adenosine base editing (ABE) system in B. napus, demonstrating the efficiency of ABE in generating A-to-G substitutions at the target BnALS and BnPDS loci. The substitution resulted in a single amino acid change in the FT protein or mis-splicing of the PDS3 RNA transcript generating germline transmissible transgenic Brassica plants with late-flowering and albino phenotypes. The nCas9 cytosine base-editing system was employed by Wu et al. (2020a) to introduce a C-to-T conversion at the P197 position of the BnALS 1 gene in B. napus. The P197S substitution conferred tribenuron-methyl resistance generating transgene-free homozygous mutants. Herbicide resistance and dwarfed plant architecture, both important traits in the commercial cultivation of oilseed rape, were reportedly introduced by Cheng et al. (2020) using the A3A-PBE base-editing system. The A3A-PBE base-editing system enabled the substitution of C to T with increased efficiency (>20%) and wider editing window.
Modifications brought upon by genome-editing technologies pose a significantly lower risk than those associated with transgenics. In general, only a few selected nucleotides are altered, rendering changes similar to those that occur in natural populations (Voytas and Gao, 2014). After the genomic-editing agents were segregated out, it is not possible to differentiate between a naturally occurring mutation and the gene edit. GE is therefore a valuable tool to establish rapid and precise changes to aid crop improvement (Zhang et al., 2018b).
Genomic Selection in Crop Improvement
Plant breeding is founded on the principles of collection, induction, and rearrangement of genetic diversity followed by phenotypic-driven selection. Conventional breeding success was achieved through the exploitation of natural or mutation-induced variation followed by efficient selection of desirable traits largely based on phenotypic observation (Pérez-de-Castro et al., 2012). This approach has several limitations including long periods required (5–12 years) to develop a crop variety, high environmental noise, and being less effective in the improvement of complex and low heritable traits (Tuberosa, 2012).
Desirable traits often include characteristics such as increased yield, plant architecture, tolerance to environmental stresses, and resistance against pests and diseases. An interlinking network of multiple “minor” genes regulates the expression of these agronomically important features. Phenotypic expression is further shaped by non-genetic factors including genotype–environment interactions (Werner et al., 2018). It is not possible to accurately access these intricate and dynamic interdependencies based on phenotypic observation alone in conventional breeding. Crop development is therefore limited by extended periods of selections of up to 10 years, environmental noise, and low heredity of complex traits in conventional breeding approaches.
With the onset of the genomics era (Nepolean et al., 2018), marker-assisted breeding (MAS) was developed to address the limitations posed by conventional breeding (Collard and Mackill, 2008). In MAS, functional markers linked to QTL are used to detect important traits through linkage mapping or GWAS. As only statistically significant marker–trait associations are retained (Arruda et al., 2016), MAS is restricted to the detection of traits controlled by a limited number of QTL with large contributions to phenotypic variation. MAS has therefore limited value in the selection of traits under complex genetic control and is, as such, outperformed by traditional phenotypic selection (Bernardo, 2001, 2016; Zhao et al., 2014).
Fast-evolving genomic tools and vast amounts of available genomic sources are permitting the establishment of genotype–phenotype relationships, in particular for complex multi-genic traits (Pérez-de-Castro et al., 2012). Genome-wide selection or GS (Meuwissen et al., 2001), contrary to MAS, includes all marker information in the prediction model, reducing marker bias and allowing the potential to explain variance even with small-effect QTL. Predicted marker effects based on phenotype and high-density marker scores are then used to estimate the breeding value of untested genotypes (Zhao et al., 2015). This estimate, applied in the preselection of promising genotypes, can accelerate progress in crop breeding and reduce cost in comparison with conventional breeding (Wang X. et al., 2018).
Hybrid breeding has been widely used in the improvement of crop performance through the exploitation of heterosis (Liu et al., 2020). In heterosis, hybrid offspring created have the potential to outclass agronomic characteristics of the parents. The selection of suitable parental combinations is therefore of essence and can pose a major challenge in the development of hybrids. However, GS has shown the potential to predict hybrid performance (Zhao et al., 2015); resulting hybrid genotypes can be inferred from their inbred parents and potentially reduce genotyping cost and generation interval (Wang X. et al., 2018). GS methods for hybrid canola breeding was evaluated by Jan et al. (2016); genome-wide SNP profiles were used to evaluate the prediction of the best possible parental combination of pollinators crossed with the two tester lines in a testcross performance for a number of important traits in spring canola. Based on genome-wide SNP markers, it was determined that testcross performance prediction in canola breeding could be an effective and efficient method to preselect promising pollinators for combinations with available male-sterile maternal lines, thereby promoting the efficient allocation of breeding resources (Jan et al., 2016).
Würschum et al. (2014) investigated the potential of GS in rapeseed breeding reporting medium-to-high prediction accuracies for several morphological-, quality-, and yield-related traits. Despite lower accuracy in the prediction of some novel families, it was concluded that with increased marker availability, GS will provide a valuable genomic tool in knowledge-based rapeseed breeding. GS has further been applied in winter-type oilseed rape (Werner et al., 2017), spring-sown canola (Jan et al., 2016), and a biparental population based on a cross between a European winter cultivar and a Chinese semi-winter cultivar (Zou et al., 2016; Liu P. et al., 2017). Werner et al. (2018) investigated the value of marker selection approaches in Asian rapeseed and illustrated that high prediction accuracies for polygenic traits are achievable with low marker density, given that the representative markers were selected with regard to the genome-wide linkage disequilibrium (LD) structure in a population.
Increased phenotypic heritability has been shown to have a greater impact on whole-genome prediction accuracies, more so than training set population size and marker density (Zhang et al., 2017). Fikere et al. (2020) reported moderate-to-high genomic prediction accuracies using genomic best linear unbiased prediction (GBLUP) models upon evaluating genetic correlations and genomic prediction accuracies for several agronomic, disease, and seed quality traits in canola. The inclusion of genotype-by-environment interaction in the GBLUP model resulted in further, though slight, improvements in predictions. Koscielny et al. (2020) confirmed these findings, demonstrating higher accuracy in whole-genome predictions within the stress treatment than within the control treatment for the majority of traits evaluated. It is therefore important, even in the genomics era, to link selected phenotypic or demographic models with the underlying processes of genomic variation. As demonstrated in the CWR Brassica cretica, if variation is largely selectively neutral, it is not possible to assume that a diverse population will inescapably display the wide-ranging adaptive diversity required for further crop improvement (Kioukis et al., 2020).
Future Prospects
Mechanisms of interaction for stress responses involve complex interactions and traits and are therefore more difficult to investigate than direct interaction (Werner et al., 2018). While genomic advances have exponentially increased during the past decades, high-throughput phenotyping has not caught up yet. To accelerate plant breeding and improve our understanding of genotype underlying expressed phenotype, dedicated high-throughput phenotyping approaches are required (Singh D. et al., 2019). It is therefore not surprising that high-throughput phenomics has increased in popularity, especially for the management and data collection of Brassica oilseed and vegetable crops. Improvements in sensor, drone, and remote sensing technology, as well as high throughput phenotyping techniques, are simplifying and enabling the quantification of complex phenotypic traits without the necessity of destructive sampling (Parmley et al., 2019). Brassica physiological studies, for example, plant height and biomass data (Moeckel et al., 2018), flower number (Wan et al., 2018), vegetation and flower fraction (Fang et al., 2016), and nitrogen nutrient studies (Graeff et al., 2008; Liu S. et al., 2018), have been generated using unmanned aerial vehicles. Assimilating large amounts of phenotypic data with the capabilities of machine learning will provide breeders with the analytical tools to optimize cultivar development in relation to target environment and accelerate the rate of genetic gain (Parmley et al., 2019).
Besides phenotypic characteristics, crop breeding requirements are also dictated by an assortment of additional major and minor variables such as environment, cultivation, and management practices and fluctuating consumer needs (Araus et al., 2018). Aligning breeding objectives to an increasing number of critical factors will require cross-disciplinary approaches driven by breeding teams, climate specialist, bioinfomaticians, and crop modelers (Beveridge et al., 2019; Stöckle and Kemanian, 2020). Crop modeling can assist breeders in comprehending the influence and interaction of variable factors in the selection of desirable varieties (Stöckle and Kemanian, 2020).
Conclusion
Crop breeding has benefitted from the advancement of genomic tools and associated analysis pipelines. Available genomic resources and lower cost of high-throughput sequencing have contributed toward the increase in WGRS efforts. The vast amount of genomic information created and advances in genomic tools developed will significantly improve capturing the range of genetic diversity estimation and enhance the capturing and exploitation of diversity in Brassica germplasm profiles. The genetic libraries of CWRs should be further explored, as quality of available references and assembly methods has improved. The availability of GE tools has improved in precision and specificity; these systems are highly customizable and can be advantageously exploited to fast-track crop improvement. Although genomics is currently taking the center stage, a multidisciplinary plant breeding approach that includes phenotype = genotype × environment × management interaction backed by big data capabilities will ultimately ensure the selection of future-proof Brassica crops.
Author Contributions
AS-E, NM, and JB conceptualized the manuscript. NM, AS-E, and AP wrote the manuscript, with additions and edits from JB and DE. The tables were prepared by NM and AS-E. AS-E illustrated the figure. All authors read and approved the final manuscript.
Funding
This work was funded by Australian Research Council Projects DP1601004497 and LP160100030.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aghazadeh, R., Zamani, M., Motallebi, M., Moradyar, M., and Moghadassi Jahromi, Z. (2016). Co-transformation of canola by chimeric chitinase and tlp genes towards improving resistance to Sclerotinia sclerotiorum. World J. Microbiol. Biotechnol. 3:144. doi: 10.1007/s11274-016-2104-6
Akama, S., Shimizu-Inatsugi, R., Shimizu, K. K., and Sese, J. (2014). Genome-wide quantification of homeolog expression ratio revealed nonstochastic gene regulation in synthetic allopolyploid Arabidopsis. Nucleic Acids Res. 42:e46. doi: 10.1093/nar/gkt1376
Akter, A., Takahashi, S., Deng, W., Shea, D. J., Itabashi, E., Shimizu, M., et al. (2019). The histone modification H3 lysine 27 tri-methylation has conserved gene regulatory roles in the triplicated genome of Brassica rapa L. DNA Res. 26, 433–443. doi: 10.1093/dnares/dsz021
Allier, A., Lehermeier, C., Charcosset, A., Moreau, L., and Teyssèdre, S. (2019). Improving short- and long-term genetic gain by accounting for within-family variance in optimal cross-selection. Front. Genet. 10:1006. doi: 10.3389/fgene.2019.01006
Altpeter, F., Springer, N. M., Bartley, L. E., Blechl, A. E., Brutnell, T. P., Citovsky, V., et al. (2016). Advancing crop transformation in the era of genome editing. Plant Cell 28, 1510–1520. doi: 10.1105/tpc.16.00196
Álvarez-Venegas, R., Zhang, Y., Kraling, K., and Tulsieram, L. (2011). Flowering without vernalization in winter canola (Brassica napus): use of Virus-Induced Gene Silencing (VIGS) to accelerate genetic gain. Nova Sci. 3, 29–50.
Amarasinghe, S. L., Su, S., Dong, X., Zappia, L., Ritchie, M. E., and Gouil, Q. (2020). Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 21:30. doi: 10.1186/s13059-020-1935-5
Amoah, S., Kurup, S., Rodriguez Lopez, C. M., Welham, S. J., Powers, S. J., Hopkins, C. J., et al. (2012). A hypomethylated population of Brassica rapa for forward and reverse epi-genetics. BMC Plant Biol. 12:193. doi: 10.1186/1471-2229-12-193
An, H., Qi, X., Gaynor, M. L., Hao, Y., Sarah, C., Gebken, S. C., et al. (2019). Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus. Nat. Commun. 10:2878. doi: 10.1038/s41467-019-10757-1
Araus, J. L., Kefauver, S. C., Zaman-Allah, M., Olsen, M. S., and Cairns, J. E. (2018). Translating high-throughput phenotyping into genetic gain. Trends Plant Sci. 23, 451–466. doi: 10.1016/j.tplants.2018.02.001
Arruda, M. P., Lipka, A. E., Brown, P. J., Krill, A. M., Thurber, C., Brown-Guedira, G., et al. (2016). Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat (Triticum aestivum L.). Mol. Breed. 36:84. doi: 10.1007/s11032-016-0508-5
Augustine, R., Mukhopadhyay, A., and Bisht, N. C. (2013). Targeted silencing of BjMYB28 transcription factor gene directs development of low glucosinolate lines in oilseed Brassica juncea. Plant Biotechnol. J. 11, 855–866. doi: 10.1111/pbi.12078
Backes, G. (2013). “TILLING and EcoTILLING,” in Diagnostics in Plant Breeding, eds T. Lübberstedt and R. K. Varshney (Dordrecht: Springer), 145–165.
Bancroft, I., Morgan, C., Fraser, F., Higgins, J., Wells, R., Clissold, L., et al. (2011). Dissecting the genome of the polyploid crop oilseed rape by transcriptome sequencing. Nat. Biotechnol. 29, 762–766. doi: 10.1038/nbt.1926
Basso, M. F., Arraes, F. B. M., Grossi-de-Sa, M., Moreira, V. J. V., Alves-Ferreira, M., and Grossi-de-Sa, M. F. (2020). Insights into genetic and molecular elements for transgenic crop development. Front. Plant Sci. 11:509. doi: 10.3389/fpls.2020.00509
Bayer, P. E., Hurgobin, B., Golicz, A. A., Chan, C.-K. K., Yuan, Y., Lee, H., et al. (2017). Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 15, 1602–1610. doi: 10.1111/pbi.12742
Bebber, D. P., Ramotowski, M. A. T., and Gurr, S. J. (2013). Crop pests and pathogens move polewards in a warming world. Nat. Clim.Change 3, 985–988. doi: 10.1038/nclimate1990
Becker, H. C., Engqvist, G. M., and Karlsson, B. (1995). Comparison of rapeseed cultivars and resynthesized lines based on allozyme and RFLP markers. Theor. Appl. Genet. 91, 62–67. doi: 10.1007/BF00220859
Bekele, D., Kassahun, T., and Fikre, A. (2019). Applications of virus induced gene silencing (VIGS) in plant functional genomics studies. J. Plant Biochem. Physiol. 7:1. doi: 10.4172/2329-9029.1000229
Belser, C., Istace, B., Denis, E., Dubarry, M., Baurens, F.-C., Falentin, C., et al. (2018). Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 4, 879–887. doi: 10.1038/s41477-018-0289-4
Bernardo, R. (2001). What if we knew all the genes for a quantitative trait in hybrid crops? Crop Sci. 41, 1–4. doi: 10.2135/cropsci2001.4111
Bernardo, R. (2016). Bandwagons I, too, have known. Theor. Appl. Genet. 129, 2323–2332. doi: 10.1007/s00122-016-2772-5
Beveridge, L., Whitfield, S., and Challinor, A. (2019). Crop modelling: towards locally relevant and climate-informed adaptation. Clim. Change 147, 475–489. doi: 10.1007/s10584-018-2160-z
Beversdorf, W. D., Hume, D. J., and Daonnelly-Vanderloo, M. J. (1988). Agronomic performance of trianzine-resistant and susceptible reciprocal spring canola hybrids. Crop Sci. 28, 932–934. doi: 10.2135/cropsci1988.0011183X002800060012x
Bizuayehu, T. T., Kornel Labun, K., Jefimov, K., and Valen, E. (2020). Single molecule structure sequencing reveals RNA structural dependencies, breathing and ensembles. bioRxiv. doi: 10.1101/2020.05.18.101402v1
Bodirsky, B. L., Popp, A., Weindl, I., Dietrich, J. P., Rolinski, S., Scheiffele, L., et al. (2012). N2O emissions from the global agricultural nitrogen cycle – current state and future scenarios. Biogeoscience 9, 4169–4197. doi: 10.5194/bg-9-4169-2012
Braatz, J., Harloff, H. J., Mascher, M., Stein, N., Himmelbach, A., and Jung, C. (2017). CRISPR-Cas9 targeted mutagenesis leads to simultaneous modification of different homoeologous gene copies in polyploid oilseed rape (Brassica napus). Plant Physiol. 174, 935–942. doi: 10.1104/pp.17.00426
Branca, F., Argento, S., and Alessandro, T. (2012). “Assessing genetic reserves in Sicily (Italy): The Brassica wild relatives case study,” in Agrobiodiversity Conservation: Securing the Diversity of Crop Wild Relatives and Landraces, eds N. Maxted, M. Ehsan Dulloo, B. V. Ford-Lloyd, L. Frese, J. M. Iriondo, and M. A. A. Pinheiro de Carvalho (Wallingford: Centre for Agriculture and Bioscience International), 52–58.
Briskine, R. V., Paape, T., Shimizu-Inatsugi, R., Nishiyama, T., Akama, S., Sese, J., et al. (2017). Genome assembly and annotation of Arabidopsis halleri, a model for heavy metal hyperaccumulation and evolutionary ecology. Mol. Ecol. Res. 17, 1025–1036. doi: 10.1111/1755-0998.12604
Brookes, G., and Barfoot, P. (2018). Farm income and production impacts of using GM crop technology 1996-2016. GM Crop. Food 9, 59–89. doi: 10.1080/21645698.2018.1464866
Brozynska, M., Furtado, A., and Henry, R. J. (2016). Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotechnol. J. 14, 1070–1085. doi: 10.1111/pbi.12454
Budar, F., and Roux, F. (2011). The role of organelle genomes in plant adaptation: time to get to work! Plant Signal. Behav. 6, 635–639. doi: 10.4161/psb.6.5.14524
Cai, C., Wang, X., Liu, B., Wu, J., Liang, J., Cui, Y., et al. (2017). Brassica rapa genome 2.0: a reference upgrade through sequence re-assembly and gene re-annotation. Mol. Plant. 10, 649–651. doi: 10.1016/j.molp.2016.11.008
Cao, J., Shelton, A. M., and Earle, E. D. (2005). Development of transgenic collards (Brassica oleracea L., var. acephala) expressing a cry1Ac or cry1C Bt gene for control of the diamondback moth. Crop Prot. 24, 804–813. doi: 10.1016/j.cropro.2004.12.014
Cappelli, A., and Cini, E. (2020). Will the COVID-19 pandemic make us reconsider the relevance of short food supply chains and local productions? Trends Food Sci. Tech. 99, 566–567. doi: 10.1016/j.tifs.2020.03.041
Cermak, T., Doyle, E. L., Christian, M., Wang, L., Zhang, Y., Schmidt, C., et al. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 3:e82. doi: 10.1093/nar/gkr218
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A. P., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 34599, 950–953. doi: 10.1126/science.1253435
Chang, S., Yang, T., Du, T., Huang, Y., Chen, J., Yan, J., et al. (2011). Mitochondrial genome sequencing helps show the evolutionary mechanism of mitochondrial genome formation in Brassica. BMC Genome 12:497. doi: 10.1186/1471-2164-12-497
Chen, F., Yang, Y., Li, B., Liu, Z., Khan, F., Zhang, T., et al. (2019). Functional analysis of M-Locus Protein Kinase revealed a novel regulatory mechanism of self-incompatibility in Brassica napus L. Int. J. Mol. Sci. 20:3303. doi: 10.3390/ijms20133303
Chen, J., Guan, R., Chang, S., Du, T., Zhang, H., and Xing, H. (2011). Substoichiometrically different mitotypes coexist in mitochondrial genomes of Brassica napus L. PLoS ONE 6:e17662. doi: 10.1371/journal.pone.0017662
Chen, X., Ge, X., Wang, J., Tan, C., King, G. J., and Liu, K. (2015). Genome-wide DNA methylation profiling by modified reduced representation bisulfite sequencing in Brassica rapa suggests that epigenetic modifications play a key role in polyploid genome evolution. Front. Plant Sci. 6:836. doi: 10.3389/fpls.2015.00836
Chen, X., Tong, C., Zhang, X., Song, A., Hu, M., Dong, W., et al. (2020). A high-quality Brassica napus genome reveals expansion of transposable elements, subgenome evolution and disease resistance. Plant Biotechnol. J. doi: 10.1111/pbi.13493
Chen, Y., Zhou, B., Li, J., Tang, H., Tang, J., and Yang, Z. (2018). Formation and change of chloroplast-located plant metabolites in response to light conditions. Int. J. Mol. Sci. 19:654. doi: 10.3390/ijms19030654
Cheng, F., Wu, J., Cai, C., Fu, L., Liang, J., Borm, T., et al. (2016). Genome resequencing and comparative variome analysis in a Brassica rapa and Brassica oleracea collection. Sci. Data 3:160119. doi: 10.1038/sdata.2016.119
Cheng, H., Hao, M., Ding, B., Mei, D., Wang, W., Wang, H., et al. (2020). Base editing with high efficiency in allotetraploid oilseed rape by A3A-PBE base editing system. Plant Biotechnol. J. 19, 87–97. doi: 10.1111/pbi.13444
Chétrit, P., Mathieu, C., Muller, J. P., and Vedel, F. (1984). Physical and gene mapping of cauliflower (Brassica oleracea) mitochondrial DNA. Curr. Genet. 8, 413–421. doi: 10.1007/BF00433907
Clarke, W. E., Higgins, E. E., Plieske, J., Wieseke, R., Sidebottom, C., Khedikar, Y., et al. (2016). A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor. Appl. Genet. 129, 1887–1899. doi: 10.1007/s00122-016-2746-7
Clarke, W. E., Parkin, I. A., Gajardo, H. A., Gerhardt, D. J., Higgins, E., Sidebottom, C., et al. (2013). Genomic DNA enrichment using sequence capture microarrays: a novel approach to discover sequence nucleotide polymorphisms (SNP) in Brassica napus L. PLoS ONE 8:e081992. doi: 10.1371/journal.pone.0081992
Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., et al. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219. doi: 10.1038/nature06745
Collard, B. C., and Mackill, D. J. (2008). Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 363, 557–572. doi: 10.1098/rstb.2007.2170
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Connelly, M., and MacIntosh, S. (2018). Petition for Determination of Nonregulated Status for DHA Canola. Available online at: https://www.aphis.usda.gov/brs/aphisdocs/17_23601p.pdf
Cowling, W. A. (2007). Genetic diversity in Australian canola and implications for crop breeding for changing future environments. Field Crop Res. 104, 103–111. doi: 10.1016/j.fcr.2006.12.014
Cox, D. B. T., Platt, R. J., and Zhang, F. (2015). Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131. doi: 10.1038/nm.3793
Crutzen, P. J., and Ehhalt, D. H. (1977). Effects of nitrogen fertilizers and combustion on the stratospheric ozone layer. Ambio 6, 112–117.
Cuthbert, J. L., McVetty, P. B. E., Freyssinet, G., and Freyssinet, M. (2001). Comparison of the performance of bromoxynil resistant and susceptible near-isogenic populations of oilseed rape. Can. J. Plant Sci. 81, 367–372. doi: 10.4141/P00-115
Dalton-Morgan, J., Hayward, A., Alamery, S., Tollenaere, R., Mason, A. S., Campbell, E., et al. (2014). A high-throughput SNP array in the amphidiploid species Brassica napus shows diversity in resistance genes. Funct. Integr. Genomics 14, 643–655. doi: 10.1007/s10142-014-0391-2
Daniell, H. (2007). Transgene containment by maternal inheritance: effective or elusive? Proc. Natl. Acad. Sci. U.S.A. 104, 6879–6880. doi: 10.1073/pnas.0702219104
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134–134. doi: 10.1186/s13059-016-1004-2
Darracq, A., Varré, J. S., Maréchal-Drouard, L., Courseaux, A., Castric, V., Saumitou-Laprade, P., et al. (2011). Structural and content diversity of mitochondrial genome in beet: a comparative genomic analysis. Genome Biol. Evol. 3, 723–736. doi: 10.1093/gbe/evr042
Das, P. M., Ramachandran, K., Van Wert, J., and Singal, R. (2004). Chromatin immunoprecipitation assay. Biotech. 37, 961–969. doi: 10.2144/04376RV01
Davis, D. (2009). Declining fruit and vegetable nutrient composition: what is the evidence? Hortic. Sci. 44, 15–19. doi: 10.21273/HORTSCI.44.1.15
Davis, D. R., Epp, M. D., and Riordan, H. D. (2005). Changes in USDA food composition data for 43 garden crops, 1950 to 1999. J. Am. Coll. Nutr. 23, 669–682. doi: 10.1080/07315724.2004.10719409
De Block, M., De Brouwer, D., and Tenning, P. (1989). Transformation of Brassica napus and Brassica oleracea using Agrobacterium tumefaciens and the expression of the bar and neo genes in the transgenic plants. Plant Physiol. 91, 694–701. doi: 10.1104/pp.91.2.694
de Paulo Farias, D., and dos Santos Gomes, M. G. (2020). COVID-19 outbreak: what should be done to avoid food shortages? Trends Food Sci. Technol. 102, 291–292. doi: 10.1016/j.tifs.2020.06.007
Dempewolf, H., Baute, G., Anderson, J., Kilian, B., Smith, C., and Guarino, L. (2017). Past and future use of wild relatives in crop breeding. Crop Sci. 57, 1070–1082. doi: 10.2135/cropsci2016.10.0885
Dirks, R., van Dun, K., de Snoo, C. B., van den Berg, M., Lelivelt, C. L., Voermans, W., et al. (2009). Reverse breeding: a novel breeding approach based on engineered meiosis. Plant Biotechnol. J. 7, 837–845. doi: 10.1111/j.1467-7652.2009.00450.x
Dolatabadian, A., Bayer, P. E., Tirnaz, S., Hurgobin, B., Edwards, D., and Batley, J. (2020). Characterization of disease resistance genes in the Brassica napus pangenome reveals significant structural variation. Plant Biotechnol. J. 18, 969–982. doi: 10.1111/pbi.13262
Dolinoy, D. C., Weinhouse, C., Jones, T. R., Rozek, L. S., and Jirtle, R. L. (2010). Variable histone modifications at the Avy metastable epiallele. Epigenetics. 5, 637–644. doi: 10.4161/epi.5.7.12892
Dreccer, M. F., Schapendonk, A. H. C. M., Slafer, G. A., and Rabbinge, R. (2000). Comparative response of wheat and oilseed rape to nitrogen supply: absorption and utilisation efficiency of radiation and nitrogen during the reproductive stages determining yield. Plant Soil 220, 189–205. doi: 10.1023/A:1004757124939
Dusenge, M. E., Duarte, A. G., and Way, D. A. (2019). Plant carbon metabolism and climate change: elevated CO2 and temperature impacts on photosynthesis, photorespiration and respiration. New Phytol. 221, 32–49. doi: 10.1111/nph.15283
Dwivedi, S. L., Scheben, A., Edwards, D., Spillane, C., and Ortiz, R. (2017). Assessing and exploiting functional diversity in germplasm pools to enhance abiotic stress adaptation and yield in cereals and food legumes. Front. Plant Sci. 8:1461. doi: 10.3389/fpls.2017.01461
Elhamamsy, A. R. (2016). DNA methylation dynamics in plants and mammals: overview of regulation and dysregulation. Cell Biochem. Funct. 34, 289–298. doi: 10.1002/cbf.3183
Espinoza, C., Schlechter, R., Herrera, D., Torres, E., Serrano, A., Medina, C., et al. (2013). Cisgenesis and intragenesis: new tools for improving crops. Biol. Res. 46, 323–331. doi: 10.4067/S0716-97602013000400003
Fang, S., Tang, W., Peng, Y., Gong, Y., Dai, C., Chai, R., et al. (2016). Remote estimation of vegetation fraction and flower fraction in oilseed rape with unmanned aerial vehicle data. Remote Sens. 8:416. doi: 10.3390/rs8050416
Feng, Y., Cui, R., Wang, S., He, M., Hua, Y., Shi, L., et al. (2020). Transcription factor BnaA9.WRKY47 contributes to the adaptation of Brassica napus to low boron stress by up-regulating the boric acid channel gene BnaA3.NIP5;1. Plant Biotechnol. J. 18, 1241–1254. doi: 10.1111/pbi.13288
Ferreira de Carvalho, J., Lucas, J., Deniot, G., Falentin, C., Filangi, O., Gilet, M., et al. (2019). Cytonuclear interactions remain stable during allopolyploid evolution despite repeated whole-genome duplications in Brassica. Plant J. 98, 434–447. doi: 10.1111/tpj.14228
Ficklin, D. L., and Novick, K. A. (2017). Historic and projected changes in vapor pressure deficit suggest a continental-scale drying of the United States atmosphere. J. Geophys. Res. Atmos. 122, 2061–2079. doi: 10.1002/2016jd025855
Fikere, M., Barbulescu, D. M., Malmberg, M. M., Maharjan, P., Salisbury, P. A., Kant, S., et al. (2020). Genomic prediction and genetic correlation of agronomic, blackleg disease, and seed quality traits in canola (Brassica napus L.). Plants 9:719. doi: 10.3390/plants9060719
Florez-Sarasa, I., Fernie, A. R., and Gupta, K. J. (2020). Does the alternative respiratory pathway offer protection against the adverse effects resulting from climate change? J. Exp. Bot. 71, 465–469. doi: 10.1093/jxb/erz428
Francisco, M., Tortosa, M., Martínez-Ballesta, M. d,.C., Velasco, P., García-Viguera, C., et al. (2017). Nutritional and phytochemical value of Brassica crops from the agri-food perspective. Ann. Appl. Biol. 170, 273–285. doi: 10.1111/aab.12318
Frewer, L. J., van der Lans, I., Fischer, A. R. H., Reinders, M. J., Menozzi, D., Zhang, X., et al. (2013). Public perceptions of agri-food applications of genetic modification – A systematic review and meta-analysis. Trends Food Sci. Technol. 30, 142–152. doi: 10.1016/j.tifs.2013.01.003
Fu, Y-B., and Gugel, R. K. (2010). Genetic diversity of Canadian elite summer rape (Brassica napus L.) cultivars from the pre- to post-canola quality era. Can. J. Plant Sci. 90, 23–33. doi: 10.4141/CJPS09073
Fuchs, J., Demidov, D., Houben, A., and Schubert, I. (2006). Chromosomal histone modification patterns – from conservation to diversity. Trends Plant Sci. 11, 199–208. doi: 10.1016/j.tplants.2006.02.008
Gabur, I., Chawla, H. S., Lopisso, D. T., von Tiedemann, A., Snowdon, R. J., and Obermeier, C. (2020). Gene presence-absence variation associates with quantitative Verticillium longisporum disease resistance in Brassica napus. Sci. Rep. 10:4131. doi: 10.1038/s41598-020-61228-3
Gabur, I., Chawla, H. S., Snowdon, R. J., and Parkin, I. A. P. (2018). Connecting genome structural variation with complex traits in crop plants. Theor. Appl. Genet. 132, 733–750. doi: 10.1007/s00122-018-3233-0
Gaebelein, R., Schiessl, S. V., Samans, B., Batley, J., and Mason, A. S. (2019). Inherited allelic variants and novel karyotype changes influence fertility and genome stability in Brassica allohexaploids. New Phytol. 223, 965–978. doi: 10.1111/nph.15804
Gaj, T., Gersbach, C. A., and Barbas, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405. doi: 10.1016/j.tibtech.2013.04.004
Gajardo, H. A., Wittkop, B., Soto-Cerda, B., Higgins, E. E., Parkin, I. A. P., Snowdon, R. J., et al. (2015). Association mapping of seed quality traits in Brassica napus L. using GWAS and candidate QTL approaches. Mol. Breed. 35:143. doi: 10.1007/s11032-015-0340-3
Gallusci, P., Dai, Z., Génard, M., Gauffretau, A., Leblanc-Fournier, N., Richard-Molard, C., et al. (2017). Epigenetics for plant improvement: current knowledge and modeling avenues. Trends Plant Sci. 22, 610–623. doi: 10.1016/j.tplants.2017.04.009
Garg, M., Sharma, N., Sharma, S., Kapoor, P., Kumar, A., Chunduri, V., et al. (2018). Biofortified crops generated by breeding, agronomy, and transgenic approaches are improving lives of millions of people around the world. Front. Nutr. 5:12. doi: 10.3389/fnut.2018.00012
Gazave, E., Tassone, E. E., Ilut, D. C., Wingerson, M., Datema, E., et al. (2016). Population genomic analysis reveals differential evolutionary histories and patterns of diversity across subgenomes and subpopulations of Brassica napus L. Front. Plant Sci. 7:525. doi: 10.3389/fpls.2016.00525
Geng, X. X., Chen, S., Astarini, I. A., Yan, G. J., Tian, E., Meng, J. L., et al. (2013). Doubled haploids of novel trigenomic Brassica derived from various interspecific crosses. Plant Cell Tissue Organ Cult. 113, 501–511. doi: 10.1007/s11240-013-0292-4
Gilchrist, E. J., Sidebottom, C. H. D., Koh, C. S., MacInnes, T., Sharpe, A. G., and Haughn, G. W. (2013). A mutant Brassica napus (canola) population for the identification of new genetic diversity via TILLING and next generation sequencing. plos ONE 8:e84303. doi: 10.1371/journal.pone.0084303
Gocal, G. F. W., Schöpke, C., and Beetham, P. R. (2015). “Oligo-Mediated Targeted Gene Editing,” in Advances in New Technology for Targeted Modification of Plant Genomes, eds F. Zhang, H. Puchta, and J. Thomson (New York, NY: Springer), 73–89.
Golicz, A. A., Bayer, P. E., Barker, G. C., Edger, P. P., Kim, H., Martinez, P. A., et al. (2016). The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 7:13390. doi: 10.1038/ncomms13390
Graeff, S., Pfenning, J., Claupein, W., and Liebig, H.-P. (2008). Evaluation of image analysis to determine the N-fertilizer demand of broccoli plants (Brassica oleracea convar. botrytis var. italica). Adv. Opt. Tech. 2008, 1–8. doi: 10.1155/2008/359760
Grewe, F., Edger, P. P., Keren, I., Sultan, L., Pires, J. C., Ostersetzer-Biran, O., et al. (2014). Comparative analysis of 11 Brassicales mitochondrial genomes and the mitochondrial transcriptome of Brassica oleracea. Mitochondrion 19, 135–143. doi: 10.1016/j.mito.2014.05.008
Grison, R., Grezes-Besset, B., Schneider, M., Lucante, N., Olsen, L., Leguay, J. J., et al. (1996). Field tolerance to fungal pathogens of Brassica napus constitutively expressing a chimeric chitinase gene. Nat. Biotechnol. 14, 643–646. doi: 10.1038/nbt0596-643
Guo, X., Hu, Q., Hao, G., Wang, X., Zhang, D., Ma, T., et al. (2018). The genomes of two Eutrema species provide insight into plant adaptation to high altitudes. DNA Res. 25, 307–315. doi: 10.1093/dnares/dsy003
Gupta, M., Atri, C., Agarwal, N., and Banga, S. S. (2016). Development and molecular-genetic characterization of a stable Brassica allohexaploid. Theor. Appl. Genet. 129, 2085–2100. doi: 10.1007/s00122-016-2759-2
Gupta, M., DeKelver, R. C., Palta, A., Clifford, C., Gopalan, S., Miller, J. C., et al. (2012). Transcriptional activation of Brassica napus β-ketoacyl-ACP synthase II with an engineered zinc finger protein transcription factor. Plant Biotechnol. J. 10, 783–791. doi: 10.1111/j.1467-7652.2012.00695.x
Han, F., Zhang, X., Liu, X., Su, H., Kong, C., Fang, Z., et al. (2017). Comparative analysis of genome wide DNA methylation profiles for the genic male sterile cabbage line 01-20S and its maintainer line. Genes 8:159. doi: 10.3390/genes8060159
Handa, H. (2003). The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): Comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 31, 5907–5916. doi: 10.1093/nar/gkg795
Harlan, J. R. (1992). Crops and Man. 2nd Edn. Madison, WI: American Society of Agronomy and Crop Science Society of America. doi: 10.2135/1992
Harloff, H.-J., Lemcke, S., Mittasch, J., Frolov, A., Wu, J. G., Dreyer, F., et al. (2012). A mutation screening platform for rapeseed (Brassica napus L.) and the detection of sinapine biosynthesis mutants. Theor. Appl. Genet. 124, 957–969. doi: 10.1007/s00122-011-1760-z
Harper, A. L., Trick, M., Higgins, J., Fraser, F., Clissold, L., Wells, R., et al. (2012). Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat. Biotechnol. 30, 798–802. doi: 10.1038/nbt.2302
Hatono, S., Nishimura, K., Murakami, Y., Tsujimura, M., and Yamagishi, H. (2017). Complete mitochondrial genome sequences of Brassica rapa (Chinese cabbage and mizuna), and intraspecific differentiation of cytoplasm in B. rapa and Brassica juncea. Breed. Sci. 67, 357–362. doi: 10.1270/jsbbs.17023
Hauben, M., Haesendonckx, B., Standaert, E., Van Der Kelen, K., Azmi, A., Akpo, H., et al. (2009). Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. U.S.A. 106, 20109–20114. doi: 10.1073/pnas.0908755106
He, Y., Wu, D., Wei, D., Fu, Y., Cui, Y., Dong, H., et al. (2017). GWAS, QTL mapping and gene expression analyses in Brassica napus reveal genetic control of branching morphogenesis. Sci. Rep. 7:15971. doi: 10.1038/s41598-017-15976-4
He, Z., Cheng, F., Li, Y., Wang, X., Parkin, I.A., Chalhoub, B., et al. (2015). Construction of Brassica A and C genome-based ordered pan-transcriptomes for use in rapeseed genomic research. Data Brief 4, 357–362. doi: 10.1016/j.dib.2015.06.016
Heffner, E. L., Sorrells, M. E., and Jannink, J.-L. (2009). Genomic selection for crop improvement. Crop Sci. 49, 1–12. doi: 10.2135/cropsci2008.08.0512
Heng, S., Wang, L., Yang, X., Huang, H., Chen, G., Cui, M., et al. (2020). Genetic and comparative transcriptome analysis revealed degs involved in the purple leaf formation in Brassica juncea. Front. Genet. 11:322. doi: 10.3389/fgene.2020.00322
Herrera, R. J., and Garcia-Bertrand, R. (eds.). (2018). “The agricultural revolutions,” in Ancestral DNA, Human Origins, and Migrations (London: Academic Press), 475–509.
Hickey, L. T., Hafeez, A. N., Robinson, H., Jackson, S. A., Leal-Bertioli, S., Tester, M., et al. (2019). Breeding crops to feed 10 billion. Nat. Biotechnol. 37, 744–754. doi: 10.1038/s41587-019-0152-9
Higgins, E. E., Clarke, W. E., Howell, E. C., Armstrong, S. J., and Parkin, I. A. P. (2018). Detecting de novo homoeologous recombination events in cultivated Brassica napus using a genome-wide SNP array. G3 8, 2673–2683. doi: 10.1534/g3.118.200118
Himelblau, E., Gilchrist, E. J., Buono, K., Bizzell, C., Mentzer, L., Vogelzang, R., et al. (2009). Forward and reverse genetics of rapid-cycling Brassica oleracea. Theor. Appl. Genet. 118, 953–961. doi: 10.1007/s00122-008-0952-7
Holme, I. B. K., Wendt, T., and Holm, P. B. (2013). Intragenesis and cisgenesis as alternatives to transgenic crop development. Plant Biotechnol. J. 11, 395–407. doi: 10.1111/pbi.12055
Hong, H., Datla, N., Reed, D. W., Covello, P. S., MacKenzie, S. L., and Qiu, X. (2002). High-level production of γ-linolenic acid in Brassica juncea using a Δ6 desaturase from Pythium irregulare. Plant Physiol. 129, 354–362. doi: 10.1104/pp.001495
Houlton, B. Z., Almaraz, M., Aneja, V., Austin, A. T., Bai, E., Cassman, K. G., et al. (2019). A world of cobenefits: solving the global nitrogen challenge. Earths Future 7, 865–872. doi: 10.1029/2019EF001222
Hu, H., Scheben, A., and Edwards, D. (2018). Advances in integrating genomics and bioinformatics in the plant breeding pipeline. Agriculture 8:75. doi: 10.3390/agriculture8060075
Hu, T. T., Pattyn, P., Bakker, E. G., Cao, J., Cheng, J.-F., Clark, R. M., et al. (2011). The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–481. doi: 10.1038/ng.807
Hu, Z.-Y., Hua, W., Huang, S.-M., and Wang, H.-Z. (2011). Complete chloroplast genome sequence of rapeseed (Brassica napus L.) and its evolutionary implications. Genet. Resour. Crop Evol. 58, 875–887. doi: 10.1007/s10722-010-9626-9
Huang, F., Liu, T., and Hou, X. (2018). Isolation and functional characterization of a floral repressor, BcMAF1, from Pak-choi (Brassica rapa ssp. chinensis). Front. Plant Sci. 9:290. doi: 10.3389/fpls.2018.00290
Huang, H., Cui, T., Zhang, L., Yang, Q., Yang, Y., Xie, K., et al. (2020). Modifications of fatty acid profile through targeted mutation at BnaFAD2 gene with CRISPR/Cas9-mediated gene editing in Brassica napus. Theor. Appl. Genet. 133, 2401–2411. doi: 10.1007/s00122-020-03607-y
Hurgobin, B., and Edwards, D. (2017). SNP discovery using a pangenome: has the single reference approach become obsolete? Biology 6:21. doi: 10.3390/biology6010021
Hurgobin, B., Golicz, A. A., Bayer, P. E., Chan, C.-K. K., Tirnaz, S., Dolatabadian, A., et al. (2018). Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 16, 1265–1274. doi: 10.1111/pbi.12867
Hyseni, L., Bromley, H., Kypridemos, C., O'Flaherty, M., Lloyd-Williams, F., Guzman-Castillo, M., et al. (2017). Systematic review of dietary trans-fat reduction interventions. Bull. World Health Organ. 95, 821G−830G. doi: 10.2471/BLT.16.189795
Jan, H. U., Abbadi, A., Lücke, S., Nichols, R. A., and Snowdon, R. J. (2016). Genomic prediction of testcross performance in canola (Brassica napus). PLoS ONE 11:e0147769. doi: 10.1371/journal.pone.0147769
Jansing, J., Schiermeyer, A., Schillberg, S., Fischer, R., and Bortesi, L. (2019). Genome editing in agriculture: Technical and practical considerations. Int. J. Mol. Sci. 20:2888. doi: 10.3390/ijms20122888
Jiang, L., Li, D., Jin, L., Ruan, Y., Shen, W.-H., and Liu, C. (2018). Histone lysine methyltransferases BnaSDG 8.A and BnaSDG 8.C are involved in the floral transition in Brassica napus. Plant J. 95, 672–685. doi: 10.1111/tpj.13978
Jin, M., Liu, H., He, C., Fu, J., Xiao, Y., Wanget, Y., et al. (2016). Maize pan-transcriptome provides novel insights into genome complexity and quantitative trait variation. Sci. Rep. 6:18936. doi: 10.1038/srep18936
Jin, S., and Daniell, H. (2015). The engineered chloroplast genome just got smarter. Trends Plant Sci. 20, 622–640. doi: 10.1016/j.tplants.2015.07.004
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Jump, A. S., and Peñuelas, J. (2005), Running to stand still: adaptation the response of plants to rapid climate change. Ecol. Lett. 8, 1010–1020. doi: 10.1111/j.1461-0248.2005.00796.x
Jung, H.-J., Jung, H.-J., Ahmed, N. U., Park, J.-I., Kang, K.-K., Hur, Y., et al. (2012). Development of self-compatible B. rapa by RNAi-mediated S locus gene silencing. PLoS ONE 7:e49497. doi: 10.1371/journal.pone.0049497
Kamthan, A., Chaudhuri, A., Kamthan, M., and Datta, A. (2016). Genetically modified (GM) crops: milestones and new advances in crop improvement. Theor. Appl. Genet. 129, 1639–1655. doi: 10.1007/s00122-016-2747-6
Kanazawa, A., Inaba, J-i., Kasai, M., Shimura, H., and Masuta, C. (2011). RNA-mediated epigenetic modifications of an endogenous gene targeted by a viral vector. Plant Signal. Behav. 6, 1090–1093. doi: 10.4161/psb.6.8.16046
Kang, B., Yun, J., Kim, S., Shin, Y., Ryu, J., Choi, M., et al. (2018). Precision genome engineering through adenine base editing in plants. Nat. Plants 4, 427–431. doi: 10.1038/s41477-018-0178-x
Karunarathna, N. L., Wang, H., Harloff, H. J., Jiang, L., and Jung, C. (2020). Elevating seed oil content in a polyploid crop by induced mutations in SEED FATTY ACID REDUCER genes. Plant Biotechnol. J. 18:11. doi: 10.1111/pbi.13381
Kasianov, A. S., Klepikova, A. V., Kulakovskiy, I. V., Gerasimov, E. S., Fedotova, A. V., Besedina, E. G., et al. (2017). High-quality genome assembly of Capsella bursa-pastoris reveals asymmetry of regulatory elements at early stages of polyploid genome evolution. Plant J. 91, 278–291. doi: 10.1111/tpj.13563
Kawanabe, T., Osabe, K., Itabashi, E., Okazaki, K., Dennis, E. S., and Fujimoto, R. (2016). Development of primer sets that can verify the enrichment of histone modifications, and their application to examining vernalization-mediated chromatin changes in Brassica rapa L. Genes Genet. Syst. 91:1. doi: 10.1266/ggs.15-00058
Kazama, T., Okuno, M., Watari, Y., Yanase, S., Koizuka, C., Tsuruta, Y., et al. (2019). Curing cytoplasmic male sterility via TALEN-mediated mitochondrial genome editing. Nat. Plants 5, 722–730. doi: 10.1038/s41477-019-0459-z
Khan, A. W., Garg, V., Roorkiwal, M., Golicz, A. A., Edwards, D., and Varshney, R. K. (2020). Super-pangenome by integrating the wild side of a species for accelerated crop improvement. Trends Plant Sci. 25, 148–158. doi: 10.1016/j.tplants.2019.10.012
Khan, S. U., Yangmiao, J., Liu, S., Zhang, K., Khan, M. H. U., Zhai, Y., et al. (2019). Genome-wide association studies in the genetic dissection of ovule number, seed number, and seed weight in Brassica napus L. Ind. Crop Prod. 142:111877. doi: 10.1016/j.indcrop.2019.111877
Khush, G. S., Lee, S., Cho, J.-I., and Jeon, J.-S. (2012). Biofortification of crops for reducing malnutrition. Plant Biotechnol. Rep. 6, 195–202. doi: 10.1007/s11816-012-0216-5
Kim, J. A., Jung, H. E., Hong, J. K., Hermand, V., McClung, C. R., Lee, Y. H., et al. (2016). Reduction of GIGANTEA expression in transgenic Brassica rapa enhances salt tolerance. Plant Cell Rep. 35, 1943–1954. doi: 10.1007/s00299-016-2008-9
Kim, Y. G., Cha, J., and Chandrasegaran, S. (1996). Hybrid restriction enzymes: zinc finger fusions to FokI cleavage domain. Proc. Natl Acad. Sci. U.S.A. 93, 1156–1160. doi: 10.1073/pnas.93.3.1156
Kioukis, A., Michalopoulou, V. A., Briers, L., Pirintsos, S., Studholme, D. J., Pavlidis, P., et al. (2020). Intraspecific diversification of the crop wild relative Brassica cretica Lam. using demographic model selection. BMC Genomics 21:48. doi: 10.1186/s12864-019-6439-x
Kirchner, T.W., Niehaus, M., Rössig, K.L., Lauterbach, T., Herde, M., Küster, H., et al. (2018). Molecular background of Pi deficiency-induced root hair growth in Brassica carinata?a fasciclin-like arabinogalactan protein is involved. Front. Plant Sci. 9:1372. doi: 10.3389/fpls.2018.01372
Kitashiba, H., Li, F., Hirakawa, H., Kawanabe, T., Zou, Z., Hasegawa, Y., et al. (2014). Draft sequences of the radish (Raphanus sativus L.) genome. DNA Res. 21, 481–490. doi: 10.1093/dnares/dsu014
Kode, V., Mudd, E. A., Iamtham, S., and Day, A. (2005). The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 44, 237–244. doi: 10.1111/j.1365-313X.2005.02533.x
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi: 10.1038/nature17946
Koscielny, C. B., Gardner, S. W., Technow, F., and Duncan, R. W. (2020). Linkage mapping and whole-genome predictions in canola (Brassica napus) subjected to differing temperature treatments. Crop Pasture Sci. 71, 229–238. doi: 10.1071/CP19387
Kumar, M. S., Mawlong, I., and Rani, R. (2020). “Biofortification of Brassicas for quality improvement,” in Brassica Improvement: Molecular, Genetics and Genomic Perspectives, eds S. H. Wani, A. K. Thakur and Y. Jeshima Khan (Cham: Springer International Publishing), 127–145.
Lawrenson, T., Shorinola, O., Stacey, N., Li, C., Østergaard, L., Patron, N., et al. (2015). Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA-guided Cas9 nuclease. Genome Biol. 16:258. doi: 10.1186/s13059-015-0826-7
Lee, H., Chawla, H. S., Obermeier, C., Dreyer, F., Abbadi, A., and Snowdon, R. (2020). Chromosome-scale assembly of winter oilseed rape Brassica napus. Front. Plant Sci. 11:496. doi: 10.3389/fpls.2020.00496
Leff, B., Ramankutty, N., and Foley, J. A. (2004). Geographic distribution of major crops across the world. Glob. Biogeochem. Cycles 18:1. doi: 10.1029/2003gb002108
Li, C., Hao, M., Wang, W., Wang, H., Chen, F., Chu, W., et al. (2018). An efficient CRISPR/Cas9 platform for rapidly generating simultaneous mutagenesis of multiple gene homoeologs in allotetraploid oilseed rape. Front. Plant Sci. 9:442. doi: 10.3389/fpls.2018.00442
Li, H., Cheng, X., Zhang, L., Hu, J., Zhang, F., Chen, B., et al. (2018). An integration of genome-wide association study and gene co-expression network analysis identifies candidate genes of stem lodging-related traits in Brassica napus. Front. Plant Sci. 9:796. doi: 10.3389/fpls.2018.00796
Li, J., Rao, L., Meng, Q., Ghani, M. A., and Chen, L. (2015). Production of Brassica tri-genomic vegetable germplasm by hybridisation between tuber mustard (Brassica juncea) and red cabbage (B. oleracea). Euphytica 204, 323–333. doi: 10.1007/s10681-014-1336-5
Li, L., Liu, Y., Chen, B., Xu, K., Zhang, F., Li, H., et al. (2016). A genome-wide association study reveals new loci for resistance to clubroot disease in Brassica napus. Front. Plant Sci. 7:1483. doi: 10.3389/fpls.2016.01483
Li, P., Zhang, S., Li, F., Zhang, S., Zhang, H., Wang, X., et al. (2017). A phylogenetic analysis of chloroplast genomes elucidates the relationships of the six economically important Brassica species comprising the Triangle of U. Front. Plant Sci. 8:111. doi: 10.3389/fpls.2017.00111
Li, T., Lui, B., Spalding, M. H., Weeks, D. P., and Yang, B. (2012). High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 30, 390–392. doi: 10.1038/nbt.2199
Li, Y., Liu, G-F., Ma, L-M., Liu, T-K., Zhang, C-W., Xiao, D., et al. (2020). A chromosome-level reference genome of non-heading Chinese cabbage [Brassica campestris (syn. Brassica rapa) ssp. chinensis]. Hortic. Res. 7:212. doi: 10.1038/s41438-020-00449-z
Li, Y., and Tollefsbol, T. O. (2011). Combined chromatin immunoprecipitation and bisulfite methylation sequencing analysis. Methods Mol. Biol. 791, 239–251. doi: 10.1007/978-1-61779-316-5_18
Lima, M. S., Woods, L. C., Cartwright, M. W., and Smith, D. R. (2016). The (in)complete organelle genome: exploring the use and nonuse of available technologies for characterizing mitochondrial and plastid chromosomes. Mol. Ecol. Res. 16, 1279–1286. doi: 10.1111/1755-0998.12585
Limera, C., Sabbadini, S., Sweet, J. B., and Mezzetti, B. (2017). New biotechnological tools for the genetic improvement of major woody fruit species. Front. Plant Sci. 8:1418. doi: 10.3389/fpls.2017.01418
Lin, J., and Musunuru, K. (2016). Genome engineering tools for building cellular models of disease. FEBS J. 283, 3222–3231. doi: 10.1111/febs.13763
Lin, K., Zhang, N., Severing, E. I., Nijveen, H., Cheng, F., Visser, R. G. F., et al. (2014). Beyond genomic variation - comparison and functional annotation of three Brassica rapa genomes: a turnip, a rapid cycling and a Chinese cabbage. BMC Genomics 15:250. doi: 10.1186/1471-2164-15-250
Lister, R., O'Malley, R. C., Tonti-Filippini, J., Gregory, B. D., Berry, C. C., Millar, A. H., et al. (2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536. doi: 10.1016/j.cell.2008.03.029
Liu, C. W., Lin, C. C., Yiu, J. C., Chen, J. J., and Tseng, M. J. (2008). Expression of a Bacillus thuringiensis toxin (cry1Ab) gene in cabbage (Brassica oleracea L. var. capitata L.) chloroplasts confers high insecticidal efficacy against Plutella xylostella. Theor. Appl. Genet. 117, 75–88. doi: 10.1007/s00122-008-0754-y
Liu, G., Xia, Y., Liu, T., Dai, S., and Hou, X. (2018). The DNA methylome and association of differentially methylated regions with differential gene expression during heat stress in Brassica rapa. Int. J. Mol. Sci. 19:1414. doi: 10.3390/ijms19051414
Liu, J., Li, M., Zhang, Q., Wei, X., and Huang, X. (2020). Exploring the molecular basis of heterosis for plant breeding. J. Integr. Plant Biol. 62, 287–298. doi: 10.1111/jipb.12804
Liu, P., Zhao, Y., Liu, G., Wang, M., Hu, D., Hu, J., et al. (2017). Hybrid performance of an immortalized F2 rapeseed population is driven by additive, dominance, and epistatic effects. Front. Plant Sci. 8:815. doi: 10.3389/fpls.2017.00815
Liu, S., Li, L., Gao, W., Zhang, Y., Liu, Y., Wang, S., et al. (2018). Diagnosis of nitrogen status in winter oilseed rape (Brassica napus L.) using in-situ hyperspectral data and unmanned aerial vehicle (UAV) multispectral images. Comput. Electron. Agric. 151, 185–195. doi: 10.1016/j.compag.2018.05.026
Liu, S., Liu, Y., Yang, X., Tong, C., Edwards, D., Parkin, I. A. P., et al. (2014). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5:3930. doi: 10.1038/ncomms4930
Liu, T., Li, Y., Duan, W., Huang, F., and Hou, X. (2017). Cold acclimation alters DNA methylation patterns and confers tolerance to heat and increases growth rate in Brassica rapa. J. Exp. Bot. 68, 1213–1224. doi: 10.1093/jxb/erw496
Lowe, R., Shirley, N., Bleackley, M., Dolan, S., and Shafee, T. (2017). Transcriptomics technologies. PLoS Comput. Biol. 13:e1005457. doi: 10.1371/journal.pcbi.1005457
Lu, G., Harper, A. L., Trick, M., Morgan, C., Fraser, F., O'Neill, C., et al. (2014). Associative transcriptomics study dissects the genetic architecture of seed glucosinolate content in Brassica napus. DNA Res. 21, 613–625. doi: 10.1093/dnares/dsu024
Lu, K., Wei, L., Li, X., Wang, Y., Wu, J., Liu, M., et al. (2019). Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 10:1154. doi: 10.1038/s41467-019-09134-9
Lu, R., Martin-Hernandez, A. M., Peart, J. R., Malcuit, I., and Baulcombe, D. C. (2003). Virus-induced gene silencing in plants. Methods 30, 296–303. doi: 10.1016/S1046-2023(03)00037-9
Lu, S., Van Eck, J., Zhou, X., Lopez, A. B., O'Halloran, D. M., Cosman, K. M., et al. (2006). The cauliflower Or gene encodes a DnaJ cysteine-rich domain-containing protein that mediates high levels of β-carotene accumulation. Plant Cell 18, 3594–3605. doi: 10.1105/tpc.106.046417
Lu, Y., and Yao, J. (2018). Chloroplasts at the crossroad of photosynthesis, pathogen infection and plant defense. Int. J. Mol. Sci. 19:3900. doi: 10.3390/ijms19123900
Lusser, M., and Davies, H. V. (2013). Comparative regulatory approaches for groups of new plant breeding techniques. New Biotechnol. 30, 437–446. doi: 10.1016/j.nbt.2013.02.004
Lusser, M., Parisi, C., Plan, D., and Rodríguez-Cerezo, E. (2012). Deployment of new biotechnologies in plant breeding. Nat. Biotechnol. 30, 231–239. doi: 10.1038/nbt.2142
Lv, H., Wang, Y., Han, F., Ji, J., Fang, Z., Zhuang, M., et al. (2020). A high-quality reference genome for cabbage obtained with SMRT reveals novel genomic features and evolutionary characteristics. Sci. Rep. 10:12394. doi: 10.1038/s41598-020-69389-x
Ma, C., Zhu, C., Zheng, M., Liu, M., Zhang, D., Liu, B., et al. (2019). CRISPR/Cas9-mediated multiple gene editing in Brassica oleracea var. capitata using the endogenous tRNA-processing system. Hortic. Res. 6:20. doi: 10.1038/s41438-018-0107-1
Makaroff, C. A., and Palmer, J. D. (1987). Extensive mitochondrial specific transcription of the Brassica campestris mitochondrial genome. Nucleic Acids Res. 15, 5141–5156. doi: 10.1093/nar/15.13.5141
Malek, M., Rahman, L., Das, M., Hassan, L., and Rafii, M. (2013). Development of hexaploid 'Brassica' (AABBCC) from hybrids (ABC) of 'Brassica carinata' (BBCC) x B. rapa (AA). Aust. J. Crop Sci. 7, 1375–1382.
Malmberg, M. M., Shi, F., Spangenberg, G. C., Daetwyler, H. D., and Cogan, N. O. I. (2018). Diversity and genome analysis of Australian and global oilseed Brassica napus L. germplasm using transcriptomics and whole genome re-sequencing. Front. Plant Sci. 9:508. doi: 10.3389/fpls.2018.00508
Marconi, G., Pace, R., Traini, A., Raggi, L., Lutts, S., Chiusano, M., et al. (2013). Use of MSAP markers to analyse the effects of salt stress on DNA methylation in rapeseed (Brassica napus var. oleifera). PLoS ONE. 8:e75597. doi: 10.1371/journal.pone.0075597
Marri, P. R., Ye, L., Jia, Y., Jiang, K., and Rounsley, S. D. (2018). “Advances in sequencing and resequencing in crop plants,” in Plant Genetics and Molecular Biology, eds R. K. Varshney, M. K. Pandey, and A. Chitikineni (Cham: Springer International Publishing), 11–35.
Mascher, M., Schreiber, M., Scholz, U., Graner, A., Reif, J. C., and Stein, N. (2019). Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat. Genet. 51, 1076–1081. doi: 10.1038/s41588-019-0443-6
Mayer, A. (1997). Historical changes in the mineral content of fruits and vegetables. Br. Food J. 99, 207–211. doi: 10.1108/00070709710181540
McCallum, C. M., Comai, L., Greene, E. A., and Henikoff, S. (2000). Targeting Induced Local Lesions IN Genomes (TILLING) for plant functional genomics. Plant Physiol. 123, 439–442. doi: 10.1104/pp.123.2.439
Mei, J., Shao, C., Yang, R., Feng, Y., Gao, Y., Ding, Y., et al. (2020). Introgression and pyramiding of genetic loci from wild Brassica oleracea into B. napus for improving Sclerotinia resistance of rapeseed. Theor. Appl. Genet. 133, 1313–1319. doi: 10.1007/s00122-020-03552-w
Meuwissen, T. H., Hayes, B. J., and Goddard, M. E. (2001). Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829.
Michael, T. P., Jupe, F., Bemm, F., Motley, S. T., Sandoval, J. P., Lanz, C., et al. (2018). High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 9:541. doi: 10.1038/s41467-018-03016-2
Ming, R., and Man Wai, C. (2015). Assembling allopolyploid genomes: no longer formidable. Genome Biol. 16:27. doi: 10.1186/s13059-015-0585-5
Moeckel, T., Dayananda, S., Nidamanuri, R. R., Nautiyal, S., Hanumaiah, N., Buerkert, A., et al. (2018). Estimation of vegetable crop parameter by multi-temporal UAV-borne images. Remote Sens. 10:805. doi: 10.3390/rs10050805
Moghe, G. D., Hufnagel, D. E., Tang, H., Xiao, Y., Dworkin, I., Town, C. D., et al. (2014). Consequences of whole-genome triplication as revealed by comparative genomic analyses of the wild radish Raphanus raphanistrum and three other Brassicaceae species. Plant Cell 26, 1925–1937. doi: 10.1105/tpc.114.124297
Moloney, M. M., Walker, J. M., and Sharma, K. K. (1989). High efficiency transformation of Brassica napus using Agrobacterium vectors. Plant Cell Rep. 8, 238–242. doi: 10.1007/BF00778542
Murray, G. M., and Brennan, J. P. (2012). The Current and Potential Costs From Diseases of Oilseed Crops in Australia. Grains Research and Development Corporation. Available online at: https://grdc.com.au/__data/assets/pdf_file/0021/82641/grdcreportdiseasecostoilseedspdf.pdf.pdf
Mwathi, M. W., Gupta, M., Quezada-Martinez, D., Pradhan, A., Batley, J., Mason, A. S., et al. (2020). Fertile allohexaploid Brassica hybrids obtained from crosses between B. oleracea and B. juncea via ovule rescue and colchicine treatment of cuttings. Plant Cell Tissue Organ Cult. 140, 301–313. doi: 10.1007/s11240-019-01728-x
Napier, J. A., Olsen, R. E., and Tocher, D. R. (2019). Update on GM canola crops as novel sources of omega-3 fish oils. Plant Biotechnol. J. 17, 703–705. doi: 10.1111/pbi.13045
Navarro-León, E., Ruiz, J. M., Graham, N., and Blasco, B. (2018). Physiological profile of CAX1a TILLING mutants of Brassica rapa exposed to different calcium doses. Plant Sci. 272, 164–172. doi: 10.1016/j.plantsci.2018.04.019
Nepolean, T., Kaul, J., Mukri, G., and Mittal, S. (2018). Genomics-enabled next-generation breeding approaches for developing system-specific drought tolerant hybrids in maize. Front. Plant Sci. 9:361. doi: 10.3389/fpls.2018.00361
Okuzaki, A., Ogawa, T., Koizuka, C., Kaneko, K., Inaba, M., Imamura, J., et al. (2018). CRISPR/Cas9-mediated genome editing of the fatty acid desaturase 2 gene in Brassica napus. Plant Physiol. Biochem. 131, 63–69. doi: 10.1016/j.plaphy.2018.04.025
Palmer, J. D., and Herbon, L. A. (1986). Tricircular mitochondrial genomes of Brassica and Raphanus: Reversal of repeat configurations by inversion. Nucleic Acids Res. 14, 9755–9764. doi: 10.1093/nar/14.24.9755
Palmer, J. D., and Herbon, L. A. (1988). Plant mitochondrial DNA evolved rapidly in structure, but slowly in sequence. J. Mol. Evol. 28, 87–97. doi: 10.1007/BF02143500
Palmer, J. D., and Shields, C. R. (1984). Tripartite structure of the Brassica campestris mitochondrial genome. Nature 307, 437–440. doi: 10.1038/307437a0
Paritosh, K., Yadava, S. K., Singh, P., Bhayana, L., Mukhopadhyay, A., Gupta, V., et al. (2020). A chromosome-scale assembly of allotetraploid Brassica juncea (AABB) elucidates comparative architecture of the A and B genomes. Plant Biotechnol. J. doi: 10.1111/pbi.13492
Parkin, I. A. P., Koh, C., Tang, H., Robinson, S. J., Kagale, S., Clarke, W. E., et al. (2014). Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15:R77. doi: 10.1186/gb-2014-15-6-r77
Parmley, K. A., Higgins, R. H., Ganapathysubramanian, B., Sakar, S., and Singh, A. K. (2019). Machine learning approach for prescriptive plant breeding. Sci. Rep. 9:17132. doi: 10.1038/s41598-019-53451-4
Paszkowski, J., and Whitham, S. A. (2001). Gene silencing and DNA methylation processes. Curr. Opin. Plant Biol. 4, 123–129. doi: 10.1016/S1369-5266(00)00147-3
Payá-Milans, M., Poza-Viejo, L., Martín-Uriz, P. S., Lara-Astiaso, D., Wilkinson, M. D., and Crevillén, P. (2019). Genome-wide analysis of the H3K27me3 epigenome and transcriptome in Brassica rapa. GigaScience 8:12. doi: 10.1093/gigascience/giz147
Pe'ery, T., Mathews, M. B., and Baulcombe, D. (2003). RNA interference. Methods 30, 287–288. doi: 10.1016/S1046-2023(03)00035-5
Pérez-de-Castro, A. M., Vilanova, S., Cañizares, J., Pascual, L., Blanca, J. M., Díez, M. J., et al. (2012). Application of genomic tools in plant breeding. Curr. Genomics 13, 179–195. doi: 10.2174/138920212800543084
Perrella, G., and Kaiserli, E. (2016). Light behind the curtain: Photoregulation of nuclear architecture and chromatin dynamics in plants. New Phytol. 212, 908–919. doi: 10.1111/nph.14269
Perumal, S., Koh, C. S., Jin, L., Buchwaldt, M., Higgins, E. E., Zheng, C., et al. (2020). A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants 6, 929–941. doi: 10.1038/s41477-020-0735-y
Prabhudas, S. K., Raju, B., Kannan Thodi, S., Parani, M., and Natarajan, P. (2016). The complete chloroplast genome sequence of Indian mustard (Brassica juncea L.). Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 27, 4622–4623. doi: 10.3109/19401736.2015.1101586
Pradhan, A., Plummer, J. A., Nelson, M. N., Cowling, W. A., and Yan, G. (2010). Successful induction of trigenomic hexaploid Brassica from a triploid hybrid of B. napus L. and B. nigra (L.) Koch. Euphytica 176:87–98. doi: 10.1007/s10681-010-0218-8
Pröbsting, M., Schenke, D., Hossain, R., Häder, C., Thurau, T., Wighardt, L., et al. (2020). Loss of function of CRT1a (calreticulin) reduces plant susceptibility to Verticillium longisporum in both Arabidopsis thaliana and oilseed rape (Brassica napus). Plant Biotechnol. J. 18, 2328–2344. doi: 10.1111/pbi.13394
Rahman, H. (2013). Review: Breeding spring canola (Brassica napus L.) by the use of exotic germplasm. Can. J. Plant Sci. 93, 363–373. doi: 10.4141/cjps2012-074
Rakyan, V. K., Blewitt, M. E., Druker, R., Preis, J. I., and Whitelaw, E. (2002). Metastable epialleles in mammals. Trends Genet. 18, 348–351. doi: 10.1016/S0168-9525(02)02709-9
Raman, H., Uppal, R. K., and Raman, R. (2019). “Genetic solutions to improve resilience of canola to climate change,” in Genomic Designing of Climate-Smart Oilseed Crops, ed C. Kole (Cham: Springer International Publishing), 75–131.
Ramegowda, V., Mysore, K. S., and Senthil-Kumar, M. (2014). Virus-induced gene silencing is a versatile tool for unraveling the functional relevance of multiple abiotic-stress-responsive genes in crop plants. Front. Plant Sci. 5:323. doi: 10.3389/fpls.2014.00323
Ran, Y., Liang, Z., and Gao, C. (2017). Current and future editing reagent delivery systems for plant genome editing. Sci. China Life Sci. 60, 490–505. doi: 10.1007/s11427-017-9022-1
Rando, O. J., and Ahmad, K. (2007). Rules and regulation in the primary structure of chromatin. Curr. Opin. Cell Biol. 19, 250–256. doi: 10.1016/j.ceb.2007.04.006
Rao, V. R., and Hodgkin, T. (2002). Genetic diversity and conservation and utilization of plant genetic resources. Plant Cell Tissue Organ Cult. 68, 1–19. doi: 10.1023/A:1013359015812
Rashid, M., He, G., Guanxiao, Y., and Ziaf, K. (2011). Relevance of TILLING in plant genomics. Aust. J. Crop Sci. 5, 411–420.
Ray, D. K., West, P. C., Clark, M., Gerber, J. S., Prishchepov, A. V., and Chatterjee, S. (2019). Climate change has likely already affected global food production. PLoS ONE 14:e0217148. doi: 10.1371/journal.pone.0217148
Razzaq, A., Saleem, F., Kanwal, M., Mustafa, G., Yousaf, S., Imran Arshad, H. M., et al. (2019). Modern trends in plant genome editing: an inclusive review of the CRISPR/Cas9 toolbox. Int. J. Mol. Sci. 20:4045. doi: 10.3390/ijms20164045
Richards, E. J. (2006). Inherited epigenetic variation — revisiting soft inheritance. Nat. Rev. Genet. 7, 395–401. doi: 10.1038/nrg1834
Rousseau-Gueutin, M., Belser, C., Da Silva, C., Richard, G., Istace, B., Cruaud, C., et al. (2020). Long-reads assembly of the Brassica napus reference genome, Darmor-bzh. GigaScience 9:12. doi: 10.1093/gigascience/giaa137
Ruiter, R., van den Brande, I., Stals, E., Delauré, S., Cornelissen, M., and D'Halluin, K. (2003). Spontaneous mutation frequency in plants obscures the effect of chimeraplasty. Plant Mol. Biol. 53, 675–689. doi: 10.1023/B:PLAN.0000019111.96107.01
Saini, P., Saini, P., Kaur, J. J., Francies, R. M., Gani, M., Rajendra, A. A., et al. (2020). “Molecular approaches for harvesting natural diversity for crop improvement,” in Rediscovery of Genetic and Genomic Resources for Future Food Security, eds R. K. Salgotra and S. M. Zargar (Singapore: Springer), 67–169.
Sang, S., Cheng, H., Mei, D., Fu, L., Wang, H., Liu, J., et al. (2020). Complete organelle genomes of Sinapis arvensis and their evolutionary implications. Crop J. 8, 505–514. doi: 10.1016/j.cj.2019.12.001
Sasaki, T., Fujimoto, R., Kishitani, S., and Nishio, T. (2011). Analysis of target sequences of DDM1s in Brassica rapa by MSAP. Plant Cell Rep. 30, 81–88. doi: 10.1007/s00299-010-0946-1
Sashidhar, N., Harloff, H.-J., and Jung, C. (2019). Identification of phytic acid mutants in oilseed rape (Brassica napus) by large-scale screening of mutant populations through amplicon sequencing. New Phytol. 225:2022–2034. doi: 10.1111/nph.16281
Sashidhar, N., Harloff, H. J., Potgieter, L., and Jung, C. (2020). Gene editing of three BnITPK genes in tetraploid oilseed rape leads to significant reduction of phytic acid in seeds. Plant Biotechnol. J. 18, 2241–2250. doi: 10.1111/pbi.13380
Sauer, N. J., Narváez-Vásquez, J., Mozoruk, J., Miller, R. B., Warburg, Z. J., Woodward, M. J., et al. (2016). Oligonucleotide-mediated genome editing provides precision and function to engineered nucleases and antibiotics in plants. Plant Physiol. 170, 1917–1928. doi: 10.1104/pp.15.01696
Saxena, K. B., and Hingane, A. J. (2015). “Male sterility systems in major field crops and their potential role in crop improvement,” in Plant Biology and Biotechnology, eds B. Bahadur, M. Venkat Rajam, L. Sahijram, and K. Krishnamurthy (New Delhi: Springer), 639–656.
Scheben, A., Berpaalen, B., Lawley, C. T., Chan, C. K. K., Bayer, P. E., Batley, J., et al. (2019). CropSNPdb: a database of SNP array data for Brassica crops and hexaploid bread wheat. Plant J. 98, 142–152. doi: 10.1111/tpj.14194
Scheben, A., Wolter, F., Batley, J., Puchta, H., and Edwards, D. (2017). Towards CRISPR/Cas crops bringing together genomics and genome editing. New Phytol. 216, 682–698. doi: 10.1111/nph.14702
Scheben, A., Yuan, Y., and Edwards, D. (2016). Advances in genomics for adopting crops to climate change. Curr. Plant Biol. 6, 2–10. doi: 10.1016/j.cpb.2016.09.001
Scholze, H., and Boch, J. (2010). TAL effector-DNA specificity. Virulence 1:428–432. doi: 10.4161/viru.1.5.12863
Schreiber, M., Stein, N., and Mascher, M. (2018). Genomic approaches for studying crop evolution. Genome Biol. 19:140. doi: 10.1186/s13059-018-1528-8
Schuler, T. H., Denholm, I., Clark, S. J., Stewart, C. N., and Poppy, G. M. (2004). Effects of Bt plants on the development and survival of the parasitoid Cotesia plutellae (Hymenoptera: Braconidae) in susceptible and Bt-resistant larvae of the diamondback moth, Plutella xylostella (Lepidoptera: Plutellidae). J. Insect Physiol. 50, 435–443. doi: 10.1016/j.jinsphys.2004.03.001
Senthil-Kumar, M., and Mysore, K. S. (2011). New dimensions for VIGS in plant functional genomics. Trends Plant Sci. 16, 656–665. doi: 10.1016/j.tplants.2011.08.006
Seol, Y.-J., Kim, K., Kang, S.-H., Perumal, S., Lee, J., and Kim, C.-K. (2017). The complete chloroplast genome of two Brassica species, Brassica nigra and B. oleracea. Mitochondrial DNA Part A DNA Mapp. Seq. Appl. 28, 167–168. doi: 10.3109/19401736.2015.1115493
Seymour, D. K., and Becker, C. (2017). The causes and consequences of DNA methylome variation in plants. Curr. Opin. Plant Biol. 36, 56–63. doi: 10.1016/j.pbi.2017.01.005
Sharafi, Y., Majidi, M. M., Goli, S. A. H., and Rashidi, F. (2015). Oil content and fatty acids composition in Brassica species. Int. J. Food Prop. 18, 2145–2154. doi: 10.1080/10942912.2014.968284
Shen, X., Xu, L., Liu, Y., Dong, H., Zhou, D., Zhang, Y., et al. (2019). Comparative transcriptome analysis and ChIP-sequencing reveals stage-specific gene expression and regulation profiles associated with pollen wall formation in Brassica rapa. BMC Genomics 20:264. doi: 10.1186/s12864-019-5637-x
Shiba, H., Kakizaki, T., Iwano, M., Tarutani, Y., Watanabe, M., Isogai, A., et al. (2006). Dominance relationships between self-incompatibility alleles controlled by DNA methylation. Nat. Genet. 38, 297–299. doi: 10.1038/ng1734
Shirasawa, K., Hirakawa, H., Fukino, N., Kitashiba, H., and Isobe, S. (2020). Genome sequence and analysis of a Japanese radish (Raphanus sativus) cultivar named ‘Sakurajima Daikon’ possessing giant root. DNA Res. 27:2. doi: 10.1093/dnares/dsaa010
Siche, R. (2020). What is the impact of COVID-19 disease on agriculture? Sci. Agrop. 11, 3–6. doi: 10.17268/sci.agropecu.2020.01.00
Singh, D., Wang, X., Kumar, U., Gao, L., Noor, M., Imtiaz, M., et al. (2019). High-throughput phenotyping enabled genetic dissection of crop lodging in wheat. Front. Plant Sci. 10:394. doi: 10.3389/fpls.2019.00394
Singh, N., Wu, S., Raupp, W. J., Sehgal, S., Arora, S., Tiwari, V., et al. (2019). Efficient curation of genebanks using next generation sequencing reveals substantial duplication of germplasm accessions. Sci. Rep. 9:650. doi: 10.1038/s41598-018-37269-0
Slatkin, M. (2008). Linkage disequilibrium? understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 9, 477–485. doi: 10.1038/nrg2361
Slotte, T., Hazzouri, K. M., Ågren, J. A., Koenig, D., Maumus, F., Guo, Y.-L., et al. (2013). The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat. Genet. 45, 831–835. doi: 10.1038/ng.2669
Snowdon, R. J., Friedrich, T., Friedt, W., and Köhler, W. (2002). Identifying the chromosomes of the A- and C-genome diploid Brassica species B. rapa (syn. campestris) and B. oleracea in their amphidiploid B. napus. Theor. Appl. Genet. 104, 533–538. doi: 10.1007/s00122-001-0787-y
Solís, M. T., El-Tantawy, A. A., Cano, V., Risueño, M. C., and Testillano, P. S. (2015). 5-azacytidine promotes microspore embryogenesis initiation by decreasing global DNA methylation, but prevents subsequent embryo development in rapeseed and barley. Front. Plant Sci. 6:472. doi: 10.3389/fpls.2015.00472
Song, J.-M., Guan, Z., Hu, J., Guo, C., Yang, Z., Wang, S., et al. (2020). Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 6, 34–45. doi: 10.1038/s41477-019-0577-7
Sottosanto, J., Andre, C., Arias, D. I., Bhatti, M., Breazeale, S., Fu, H., et al. (2018). Petition for the Determination of Non-regulatory Status for EPA+DHA Canola Event LBFLFK. Available online at: https://www.aphis.usda.gov/brs/aphisdocs/17_32101p.pdf
Stephenson, P., Baker, D., Girin, T., Perez, A., Amoah, S., King, G. J., et al. (2010). A rich TILLING resource for studying gene function in Brassica rapa. BMC Plant Biol. 10:62. doi: 10.1186/1471-2229-10-62
Stöckle, C. O., and Kemanian, A. R. (2020). Can crop models identify critical gaps in genetics, environment, and management interactions? Front. Plant Sci. 11:737. doi: 10.3389/fpls.2020.00737
Sun, D., Wang, C., Zhang, X., Zhang, W., Jiang, H., Yao, X., et al. (2019). Draft genome sequence of cauliflower (Brassica oleracea L. var. botrytis) provides new insights into the C genome in Brassica species. Hortic. Res. 6:82. doi: 10.1038/s41438-019-0164-0
Sun, F., Fan, G., Hu, Q., Zhou, Y., Guan, M., Tong, C., et al. (2017). The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 92, 452–468. doi: 10.1111/tpj.13669
Sun, Q., Lin, L., Liu, D., Wu, D., Fang, Y., Wu, J., et al. (2018). CRISPR/Cas9-mediated multiplex genome editing of the BnWRKY11 and BnWRKY70 genes in Brassica napus L. Int. J. Mol. Sci. 19:2716. doi: 10.3390/ijms19092716
Sun, Z., Li, N., Huang, G., Xu, J., Pan, Y., Wang, Z., et al. (2013). Site-specific gene targeting using transcription activator-like effector (TALE)-based nuclease in Brassica oleracea. J. Integr. Plant Biol. 55, 1092–1103. doi: 10.1111/jipb.12091
Supple, M. A., and Shapiro, B. (2018). Conservation of biodiversity in the genomics era. Genome Biol. 19:131. doi: 10.1186/s13059-018-1520-3
Sweetman, C., Waterman, C. D., Rainbird, B. M., Smith, P., Jenkins, C. D., Day, D. A., et al. (2019). AtNDB2 is the main external NADH dehydrogenase in mitochondria and is important for tolerance to environmental stress. Plant Physiol. 181, 774–788. doi: 10.1104/pp.19.00877
Takahashi, S., Fukushima, N., Osabe, K., Itabashi, E., Shimizu, M., Miyaji, N., et al. (2018a). Identification of DNA methylated regions by using methylated DNA immunoprecipitation sequencing in Brassica rapa. Crop Pasture Sci. 69, 107–120. doi: 10.1071/CP17394
Takahashi, S., Osabe, K., Fukushima, N., Takuno, S., Miyaji, N., Shimizu, M., et al. (2018b). Genome-wide characterization of DNA methylation, small RNA expression, and histone H3 lysine nine di-methylation in Brassica rapa L. DNA Res. 25, 511–520. doi: 10.1093/dnares/dsy021
Tan, C., Liu, H., Ren, J., Ye, X., Feng, H., and Liu, Z. (2019). Single-molecule real-time sequencing facilitates the analysis of transcripts and splice isoforms of anthers in Chinese cabbage (Brassica rapa L. ssp. pekinensis). BMC Plant Biol. 1:517. doi: 10.1186/s12870-019-2133-z
Tanaka, Y., Tsuda, M., Yasumoto, K., Terachi, T., and Yamagishi, H. (2014). The complete mitochondrial genome sequence of Brassica oleracea and analysis of coexisting mitotypes. Curr. Genet. 60, 277–284. doi: 10.1007/s00294-014-0433-2
Tang, T., Yu, X., Yang, H., Gao, Q., Ji, H., Wang, Y., et al. (2018). Development and validation of an effective CRISPR/Cas9 vector for efficiently isolating positive transformants and transgene-free mutants in a wide range of plant species. Front. Plant Sci. 9:1533. doi: 10.3389/fpls.2018.01533
Tanksley, S. D., and McCouch, S. R. (1997). Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277, 1063–1066. doi: 10.1126/science.277.5329.1063
Tariq, M., and Paszkowski, J. (2004). DNA and histone methylation in plants. Trends Genet. 20, 244–251. doi: 10.1016/j.tig.2004.04.005
Tennessen, J. A., Govindarajulu, R., Ashman, T.-L., and Liston, A. (2014). Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol. Evol. 6, 3295–3313. doi: 10.1093/gbe/evu261
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
The Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
Tian, E., Jiang, Y., Chen, L., Liu, F., and Menget, J. (2010). Synthesis of a Brassica trigenomic allohexaploid (B. carinata × B. rapa) de novo and its stability in subsequent generations. Theor. Appl. Genet. 121, 1431–1440. doi: 10.1007/s00122-010-1399-1
Tirnaz, S., and Batley, J. (2019a). DNA methylation: toward crop disease resistance improvement. Trends Plant Sci. 24, 1137–1150. doi: 10.1016/j.tplants.2019.08.007
Tirnaz, S., and Batley, J. (2019b). Epigenetics: potentials and challenges in crop breeding. Mol Plant 12, 1309–1311. doi: 10.1016/j.molp.2019.09.006
Tirnaz, S., Merce, C., Bayer, P. E., Severn-Ellis, A. A., Edwards, D., and Batley, J. (2020). Effect of Leptosphaeria maculans infection on promoter DNA methylation of defence genes in Brassica napus. Agronomy 10:1072. doi: 10.3390/agronomy10081072
Townsend, A. R., Howarth, R. W., Bazzaz, F. A., Booth, M. S., Cleveland, C. C., Collinge, S. K., et al. (2003). Human health effects of a changing global nitrogen cycle. Front. Ecol. Environ. 1, 240–246. doi: 10.1890/1540-9295(2003)001[0240:HHEOAC]2.0.CO;2
Tuberosa, R. (2012). Phenotyping for drought tolerance of crops in the genomics era. Front. Physiol. 3:347. doi: 10.3389/fphys.2012.00347
Nagaharu, U., Nagaharu, N., and Nagaharu. (1935). Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 7, 389–452.
Van Berkum, N. L., Lieberman-Aiden, E., Williams, L., Imakaev, M., Gnirke, A., Mirny, L. A., et al. (2010). Hi-C: a method to study the three-dimensional architecture of genomes. J. Vis. Exp. 39:e1869. doi: 10.3791/1869
Van Dijk, E. L., Jaszczyszyn, Y., Naquin, D., and Thermes, C. (2018). The third revolution in sequencing technology. Trends Genet. 34, 666–681. doi: 10.1016/j.tig.2018.05.008
Varshney, R. K., Pandey, M. K., Bohra, A., Singh, V. K., Thudi, M., and Saxena, R. K. (2018). Toward the sequence-based breeding in legumes in the post-genome sequencing era. Theor. Appl. Genet. 132, 797–816. doi: 10.1007/s00122-018-3252-x
Vernikos, G. S. (2020). “A review of pangenome tools and recent studies,” in The Pangenome, eds H. Tettelin, and D. Medini (Cham: Springer), 89–112.
Voss-Fels, K., and Snowdon, R. J. (2016). Understanding and utilizing crop genome diversity via high-resolution genotyping. Plant Biotechnol. J. 14, 1086–1094. doi: 10.1111/pbi.12456
Voytas, D. F., and Gao, C. (2014). Precision genome engineering and agriculture: opportunities and regulatory challenges. PLoS Biol. 12:e1001877. doi: 10.1371/journal.pbio.1001877
Wan, L., Li, Y., Cen, H., Zhu, J., Yin, W., Wu, W., et al. (2018). Combining UAV-based vegetation indices and image classification to estimate flower number in oilseed rape. Remote Sens. 10:1484. doi: 10.3390/rs10091484
Wang, B., Kumar, V., Olson, A., and Ware, D. (2019). Reviving the transcriptome studies: an insight into the emergence of single-molecule transcriptome sequencing. Front. Genet. 1:384. doi: 10.3389/fgene.2019.00384
Wang, G.-X., Lv, J., Zhang, J., Han, S., Zong, M., Guo, N., et al. (2016). Genetic and epigenetic alterations of Brassica nigra introgression lines from somatic hybridization: a resource for cauliflower improvement. Front. Plant Sci. 7:1258. doi: 10.3389/fpls.2016.01258
Wang, N., Shi, L., Tian, F., Ning, H., Wu, X., Long, Y., et al. (2010). Assessment of FAE1 polymorphisms in three Brassica species using EcoTILLING and their association with differences in seed erucic acid contents. BMC Plant Biol. 10:137. doi: 10.1186/1471-2229-10-137
Wang, N., Wang, Y., Tian, F., King, G. J., Zhang, C., Long, Y., et al. (2008). A functional genomics resource for Brassica napus: development of an EMS mutagenized population and discovery of FAE1 point mutations by TILLING. New Phytol. 180, 751–765. doi: 10.1111/j.1469-8137.2008.02619.x
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Wang, X., Xu, Y., Hu, Z., and Xu, C. (2018). Genomic selection methods for crop improvement: current status and prospects. Crop J. 6, 330–340. doi: 10.1016/j.cj.2018.03.001
Wang, Y., Xiao, L., Guo, S., An, F., and Du, D. (2016). Fine mapping and whole-genome resequencing identify the seed coat color gene in Brassica rapa. PLoS ONE 11:e0166464. doi: 10.1371/journal.pone.0166464
Wang, Z., Wu, X., Wu, Z., An, H., Yi, B., Wen, J., et al. (2018). Genome-wide DNA methylation comparison between Brassica napus genic male sterile line and restorer line. Int. J. Mol. Sci. 19:2689. doi: 10.3390/ijms19092689
Watanabe, K., Sassa, Y., Suda, E., Chen, C.-H., Inaba, M., and Kikuchi, A. (2005). Global political, economic, social and technological issues on transgenic crops. Plant Biotechnol. 22, 515–522. doi: 10.5511/plantbiotechnology.22.515
Weigel, D., and Colot, V. (2012). Epialleles in plant evolution. Genome Biol. 13:249. doi: 10.1186/gb-2012-13-10-249
Werner, C. R., Qian, L., Voss-Fels, K. P., Abbadi, A., Leckband, G., Frisch, M., et al. (2017). Genome-ide regression models considering general and specifc combining ability predict hybrid performance in oilseed rape with similar accuracy regardless of trait architecture. Theor. Appl. Genet. 131, 299–317. doi: 10.1007/s00122-017-3002-5
Werner, C. R., Voss-Fels, K. P., Miller, C. N., Qian, W., Hua, W., Guan, et al. (2018). Effective genomic selection in a narrow-genepool crop with low-density markers: Asian rapeseed as an example. Plant Genome 11:170084. doi: 10.3835/plantgenome2017.09.0084
White, P. J., and Broadley, M. R. (2005). Historical variation in the mineral composition of edible horticultural products. J. Hortic. Sci. Biotechnol. 80, 660–667. doi: 10.1080/14620316.2005.11511995
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wolfram, K., Schmidt, J., Wray, V., Milkowski, C., Schliemann, W., and Strack, D. (2010). Profiling of phenylpropanoids in transgenic low-sinapine oilseed rape (Brassica napus). Phytochemistry 71, 1076–1084. doi: 10.1016/j.phytochem.2010.04.00
Wu, D., Liang, Z., Yan, T., Xu, Y., Xuan, L., Tang, J., et al. (2019). Whole-genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Mol. Plant. 12, 30–43. doi: 10.1016/j.molp.2018.11.007
Wu, G., Truksa, M., Datla, N., Vrinten, P., Bauer, J., Zank, T., et al. (2005). Stepwise engineering to produce high yields of very long-chain polyunsaturated fatty acids in plants. Nat. Biotechnol. 23, 1013–1017. doi: 10.1038/nbt1107
Wu, J., Chen, C., Xian, G., Liu, D., Lin, L., Yin, S., et al. (2020a). Engineering herbicide-resistant oilseed rape by CRISPR/Cas9-mediated cytosine base-editing. Plant Biotechnol. J. 18, 1857–1859. doi: 10.1111/pbi.13368
Wu, J., Liu, B., Cheng, F., Ramchiary, N., Choi, S.R., Lim, Y.P., et al. (2012). Sequencing of chloroplast genome using whole cellular DNA and Solexa sequencing technology. Front. Plant Sci. 3:243. doi: 10.3389/fpls.2012.00243
Wu, J., Yan, G., Duan, Z., Wang, Z., Kang, C., Guo, L., et al. (2020b). Roles of the Brassica napus DELLA Protein BnaA6.RGA, in modulating drought tolerance by interacting with the ABA signalling component BnaA10.ABF2. Front. Plant Sci. 11:577. doi: 10.3389/fpls.2020.00577
Wu, J., Zhao, Q., Yang, Q., Liu, H., Li, Q., Yi, X., et al. (2016). Comparative transcriptomic analysis uncovers the complex genetic network for resistance to Sclerotinia sclerotiorum in Brassica napus. Sci. Rep. 6:19007. doi: 10.1038/srep19007
Würschum, T., Abel, S., and Zhao, Y. (2014). Potential of genomic selection in rapeseed (Brassica napus L.) breeding. Plant Breed. 133, 45–51. doi: 10.1111/pbr.12137
Xiao-Ming, Z., Junrui, W., Li, F., Sha, L., Hongbo, P., Lan, Q., et al. (2017). Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 7:1555. doi: 10.1038/s41598-017-01518-5
Xie, T., Chen, X., Guo, T., Rong, H., Chen, Z., Sun, Q., et al. (2020). Targeted knockout of BnTT2 homologues for yellow-seeded Brassica napus with reduced flavonoids and improved fatty acid composition. J. Agric. Food Chem. 68, 5676–5690. doi: 10.1021/acs.jafc.0c01126
Xin, Q., Wang, X., Gao, Y., Xu, D., Xie, Z., Dong, F., et al. (2020). Molecular mechanisms underpinning the multiallelic inheritance of MS5 in Brassica napus. Plant J. Cell Mol. Biol. 103, 1723–1734. doi: 10.1111/tpj.14857
Xiong, X., Zhou, D., Xu, L., Liu, T., Yue, X., Liu, W., et al. (2019). BcPME37c is involved in pollen intine formation in Brassica campestris. Biochem. Biophys. Res. Commun. 517, 63–68. doi: 10.1016/j.bbrc.2019.07.009
Xue, J.-Y., Wang, Y., Chen, M., Dong, S., Shao, Z.-Q., and Liu, Y. (2020). Maternal inheritance of U's triangle and evolutionary process of Brassica mitochondrial genomes. Front. Plant Sci. 11:805. doi: 10.3389/fpls.2020.00805
Yamagishi, H., Tanaka, Y., and Terachi, T. (2014). Complete mitochondrial genome sequence of black mustard (Brassica nigra; BB) and comparison with Brassica oleracea (CC) and Brassica carinata (BBCC). Genome 57, 577–582. doi: 10.1139/gen-2014-0165
Yan, C., Huang, Y., Liu, Z., Guo, F., Jiao, Z., Yang, W., et al. (2020). Rapid identification of yellow-flowered gene Bofc in cauliflower (Brassica oleracea var. botrytis) by bulked segregant analysis and whole-genome resequencing. Euphytica 216:26. doi: 10.1007/s10681-020-2560-9
Yan, G., Nelson, M., Pradhan, A., Mason, A., Weerakoon, S., Si, P., et al. (2009). Progress towards the creation of trigenomic brassica hexaploid populations. SABRAO, J. Breed. Genet. 41:00274.
Yang, H., Wu, J., Tang, T., Liu, K.-D., and Dai, C. (2017). CRISPR/Cas9-mediated genome editing efficiently creates specific mutations at multiple loci using one sgRNA in Brassica napus. Sci. Rep. 7:7489. doi: 10.1038/s41598-017-07871-9
Yang, J., Liu, D., Wang, X., Ji, C., Cheng, F., Liu, B., et al. (2016a). The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232. doi: 10.1038/ng.3657
Yang, J., Liu, G., Zhao, N., Chen, S., Liu, D., Ma, W., et al. (2016b). Comparative mitochondrial genome analysis reveals the evolutionary rearrangement mechanism in Brassica. Plant Biol. 18, 527–536. doi: 10.1111/plb.12414
Yang, K., Nath, U. K., Biswas, M. K., Kayum, M. A., Yi, G-e., Lee, J., et al. (2018). Whole-genome sequencing of Brassica oleracea var. capitata reveals new diversity of the mitogenome. PLoS ONE 13:e0194356. doi: 10.1371/journal.pone.0194356
Yang, R., Jarvis, D. E., Chen, H., Beilstein, M. A., Grimwood, J., Jenkins, J., et al. (2013). The reference genome of the halophytic plant Eutrema salsugineum. Front. Plant Sci. 4:46. doi: 10.3389/fpls.2013.00046
Yang, Y., Zhu, K., Li, H., Han, S., Meng, Q., Khan, S. U., et al. (2018). Precise editing of CLAVATA genes in Brassica napus L. regulates multilocular silique development. Plant Biotechnol. J. 16, 1322–1335. doi: 10.1111/pbi.12872
Yao, M., Guan, M., Zhang, Z., Cui, Y., Chen, H., Liu, W., et al. (2020). GWAS and co-expression network combination uncovers multigenes with close linkage effects on the oleic acid content accumulation in Brassica napus. BMC Genomics 21:320. doi: 10.1186/s12864-020-6711-0
You, Q., Yang, X., Peng, Z., Xu, L., and Wang, J. (2018). Development and applications of a high throughput genotyping tool for polyploid crops: single nucleotide polymorphism (SNP) array. Front. Plant Sci. 9:104. doi: 10.3389/fpls.2018.00104
Yousef, E., Müller, T., Börner, A., and Schmid, K. J. (2018). Comparative analysis of genetic diversity and differentiation of cauliflower (Brassica oleracea var. botrytis) accessions from two ex situ genebanks. PLoS ONE 13:e0192062. doi: 10.1371/journal.pone.0192062
Yu, J., Yang, X. D., Wang, Q., Gao, L. W., Yang, Y., Xiao, D., et al. (2018). Efficient virus-induced gene silencing in Brassica rapa using a turnip yellow mosaic virus vector. Biol. Plant. 62, 826–834. doi: 10.1007/s10535-018-0803-6
Zaman, Q. U., Chu, W., Hao, M., Shi, Y., Sun, M., Sang, S. F., et al. (2019). CRISPR/Cas9-mediated multiplex genome editing of JAGGED gene in Brassica napus L. Biomolecules 9:725. doi: 10.3390/biom9110725
Zarinpanjeh, N., Motallebi, M., Zamani, M. R., and Ziaei, M. (2016). Enhanced resistance to Sclerotinia sclerotiorum in Brassica napus by co-expression of defensin and chimeric chitinase genes. J. Appl. Genet. 57:417–425. doi: 10.1007/s13353-016-0340-y
Zeng, C.-L., Wang, G.-Y., Wang, J.-B., Yan, G.-X., Chen, B.-Y., Xu, K., et al. (2012). High-throughput discovery of chloroplast and mitochondrial DNA polymorphisms in Brassicaceae species by ORG-EcoTILLING. PLoS ONE 7:e47284. doi: 10.1371/journal.pone.0047284
Zhai, Y., Cai, S., Hu, L., Yang, Y., Amoo, O., Fan, C., et al. (2019). CRISPR/Cas9-mediated genome editing reveals differences in the contribution of INDEHISCENT homologues to pod shatter resistance in Brassica napus L. Theor. Appl. Genet. 132, 2111–2123. doi: 10.1007/s00122-019-03341-0
Zhai, Y., Yu, K., Cai, S., Hu, L., Amoo, O., Xu, L., et al. (2020). Targeted mutagenesis of BnTT8 homologs controls yellow seed coat development for effective oil production in Brassica napus L. Plant Biotech J. 18, 1153–1168. doi: 10.1111/pbi.13281
Zhang, A., Wang, H., Beyene, Y., Samagn, K., Liu, Y., Cao, S., et al. (2017). Effect of trait heritability, training population size and marker density on genomic prediction accuracy estimation in 22 bi-parental tropical maize populations. Front. Plant Sci. 8:1916. doi: 10.3389/fpls.2017.01916
Zhang, C., Wohlhueter, R., and Zhang, H. (2016). Genetically modified foods: a critical review of their promise and problems. Food Sci. Hum. Wellness 5, 116–123. doi: 10.1016/j.fshw.2016.04.002
Zhang, F., and Batley, J. (2020). Exploring the application of wild species for crop improvement in a changing climate. Curr. Opin. Plant Biol. 56, 218–222. doi: 10.1016/j.pbi.2019.12.013
Zhang, K., Nie, L., Cheng, Q., Yin, Y., Chen, K., Qi, F., et al. (2019). Effective editing for lysophosphatidic acid acyltransferase 2/5 in allotetraploid rapeseed (Brassica napus L.) using CRISPR-Cas9 system. Biotechnol. Biofuels 12:225. doi: 10.1186/s13068-019-1567-8
Zhang, L., Cai, X., Wu, J., Liu, M., Grob, S., Cheng, F., et al. (2018). Improved Brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic. Res. 5:50. doi: 10.1038/s41438-018-0071-9
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. doi: 10.1038/nbt.3207
Zhang, Y., Huang, S., Wang, X., Liu, J., Guo, X., Mu, J., et al. (2018a). Defective APETALA2 genes lead to sepal modification in Brassica crops. Front. Plant Sci. 9:367. doi: 10.3389/fpls.2018.00367
Zhang, Y., Massel, K., Godwin, I. D., and Gao, G. (2018b). Applications and potential of genome editing in crop improvement. Genome Biol. 19:210. doi: 10.1186/s13059-018-1586-y
Zhang, Y., Thomas, W., Bayer, P. E., Edwards, D., and Batley, J. (2020). Frontiers in dissecting and managing Brassica diseases: from reference-based RGA candidate identification to building pan-RGAomes. Int. J. Mol. Sci. 21:8964. doi: 10.3390/ijms21238964
Zhang, Z., Zhang, S., Huang, X., Orwig, K. E., and Sheng, Y. (2013). Rapid assembly of customized TALENs into multiple delivery systems. PLoS ONE 8:e80281. doi: 10.1371/journal.pone.0080281
Zhao, Y., Mette, M. F., Gowda, M., Longin, C. F. H., and Reif, J. C. (2014). Bridging the gap between marker-assisted and genomic selection of heading time and plant height in hybrid wheat. Heredity 112, 638–645. doi: 10.1038/hdy.2014.1
Zhao, Y. S., Mette, M. F., and Reif, J. C. (2015). Genomic selection in hybrid breeding. Plant Breed. 134, 1–10. doi: 10.1111/pbr.12231
Zheng, M., Zhang, L., Tang, M., Liu, J., Liu, H., Yang, H., et al. (2020). Knockout of two BnaMAX1 homologs by CRISPR/Cas9-targeted mutagenesis improves plant architecture and increases yield in rapeseed (Brassica napus L.). Plant Biotech. J. 18, 644–654. doi: 10.1111/pbi.13228
Zhou, J., Chen, T., Cheng, C., Xianhong, G., and Zaiyun, L. (2016). Distinct subgenome stabilities in synthesized Brassica allohexaploids. Theor. Appl. Genet. 129, 1257–1271. doi: 10.1007/s00122-016-2701-7
Zou, J., Mao, L., Qiu, J., Wang, M., Jia, L., Wu, D., et al. (2019). Genome-wide selection footprints and deleterious variations in young Asian allotetraploid rapeseed. Plant Biotechnol. J. 17, 1998–2010. doi: 10.1111/pbi.13115
Keywords: Brassica, plant genomics, crop improvement, molecular breeding, food security
Citation: Mohd Saad NS, Severn-Ellis AA, Pradhan A, Edwards D and Batley J (2021) Genomics Armed With Diversity Leads the Way in Brassica Improvement in a Changing Global Environment. Front. Genet. 12:600789. doi: 10.3389/fgene.2021.600789
Received: 31 August 2020; Accepted: 15 January 2021;
Published: 18 February 2021.
Edited by:
Reyazul Rouf Mir, Sher-e-Kashmir University of Agricultural Sciences and Technology, IndiaReviewed by:
Raju Datla, Global Institute for Food Security (GIFS), CanadaAndreas Stahl, Julius Kühn-Institut, Germany
Asif Mohammad Iqbal Qureshi, Sher-e-Kashmir University of Agricultural Sciences and Technology, India
Copyright © 2021 Mohd Saad, Severn-Ellis, Pradhan, Edwards and Batley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacqueline Batley, amFjcXVlbGluZS5iYXRsZXlAdXdhLmVkdS5hdQ==