 Osman Sharifi
Osman Sharifi Dag H. Yasui
Dag H. Yasui- LaSalle Laboratory, Department of Medical Microbiology and Immunology, UC Davis School of Medicine, Davis, CA, United States
MeCP2 protein, encoded by the MECP2 gene, binds to DNA and affects transcription. Outside of this activity the true range of MeCP2 function is still not entirely clear. As MECP2 gene mutations cause the neurodevelopmental disorder Rett syndrome in 1 in 10,000 female births, much of what is known about the biologic function of MeCP2 comes from studying human cell culture models and rodent models with Mecp2 gene mutations. In this review, the full scope of MeCP2 research available in the NIH Pubmed (https://pubmed.ncbi.nlm.nih.gov/) data base to date is considered. While not all original research can be mentioned due to space limitations, the main aspects of MeCP2 and Rett syndrome research are discussed while highlighting the work of individual researchers and research groups. First, the primary functions of MeCP2 relevant to Rett syndrome are summarized and explored. Second, the conflicting evidence and controversies surrounding emerging aspects of MeCP2 biology are examined. Next, the most obvious gaps in MeCP2 research studies are noted. Finally, the most recent discoveries in MeCP2 and Rett syndrome research are explored with a focus on the potential and pitfalls of novel treatments and therapies.
Introduction
MeCP2 research efforts have been extensive and have therefore greatly advanced the fields of epigenetics, neuroscience and chromatin research. MeCP2, encoded by the MECP2 gene in humans and the Mecp2 gene in rodents, was first characterized as a methyl CpG DNA binding protein in 1992, thereby establishing it as an epigenetic reader of DNA methylation (Meehan et al., 1992). It was not until the 1999 discovery that mutations in MECP2 contribute to the pathology of the rare disease, Rett syndrome (RTT, OMIM #312750), that the relevance of MeCP2 to normal human development was established (Amir et al., 1999). Virtually all Rett patients are female (Reichow et al., 2015) and mosaic for cells with MECP2 expression as point mutations on the X chromosome are almost always paternally inherited (Trappe et al., 2001). Despite this fact, the function of MeCP2 has been predominately studied in the nervous system of Mecp2 null male mice as ablation of MeCP2 in all brain cells accounts for the most severe disease phenotypes (Guy et al., 2001). Since the initial characterization of MeCP2 as a DNA binding protein in 1992 (Meehan et al., 1992) a whole field of biochemical research has emerged, culminating in the discovery that MeCP2 can organize chromatin by liquid phase separation (Fan et al., 2020; Li et al., 2020; Wang et al., 2020). Since 1999 and the discovery that mutations in MECP2 contribute to Rett syndrome, at least 3,194 papers mentioning MeCP2 have accumulated in the NIH Pubmed database. In this review we will examine the biologic function of MeCP2 in mammals, highlight controversial aspects of MeCP2 research, point out significant gaps in knowledge, and report on paradigm shifting advances in the Rett syndrome field.
Fundamental Aspects of MeCP2 Function in Mammals
MECP2/Mecp2 Mutations and Resulting MeCP2 Deficiencies in Brain Underlie the Majority of Rett Syndrome Like Phenotypes
The key to defining the functions of MeCP2 is to understand the effects that hypomorphic MECP2 has at the whole organism level. Prior to the identification of MECP2 mutations in Rett patients, a general model of MECP2/MeCP2 function had been developed from early studies on the Mecp2 gene and MeCP2 protein in mice (Lewis et al., 1992). Evidence from in vitro studies concluded that MeCP2 acted as a transcriptional repressor of genes in cis (Nan et al., 1997). In 1999 a team led by Huda Zoghbi at Baylor College of Medicine and Uta Francke at Stanford University identified the common mutations for Rett syndrome and mapped them to the MECP2 gene located on the X chromosome (Amir et al., 1999). A Rett syndrome model with a germline Mecp2 exon 3 and 4 deletion allele produced healthy mice at birth but male pups (Mecp2–/y null) soon displayed impaired motor skills and premature lethality, while female mice (Mecp2–/+ deficient heterozygotes) became hypoactive and exhibited motor and breathing defects after 3 months of age Post mortem examination of male Mecp2–/y null mice revealed reduced brain and neuronal size (Guy et al., 2001). These Mecp2 exon 3 and 4 deletion or “Bird Mecp2 deletion” mice have been the primary animal model of Rett syndrome research. A simultaneous germline Mecp2 exon 3 deletion mouse which will be henceforth referred to as the “Jaenisch Mecp2 deletion” model displayed very similar motor defects and premature death as the Bird Mecp2 null mice in males and motor, hypoactivity and respiratory phenotypes in females (Chen et al., 2001). To test the hypothesis that Mecp2 expression is necessary for normal brain function, deletion of Mecp2 in nestin expressing cells, encompassing neurons and glia, also displayed premature death and motor defects in Bird Mecp2–/y null deletion males and delayed motor defects in Bird Mecp2–/+ deficient heterozygous female mice (Guy et al., 2001). Simultaneous histological examination of Jaenisch Mecp2–/y null deletion mice revealed reduced neuronal size along with reduced brain weight, suggestive of brain restricted defects underlying Rett like phenotypes (Chen et al., 2001). To better examine the requirement for Mecp2 expression during brain development, the Jaenisch team also generated mice conditional for Mecp2 deletion in nestin expressing neurons and glia. These mice exhibited the same phenotypes as seen in Mecp2 germline deletion mice as did mice conditional for Mecp2 deletion in CamK expressing, post-mitotic neurons that presented with delayed neurological defects in both Mecp2–/y null and Mecp2–/+ deficient female mice and premature death in male mice (Chen et al., 2001). Together, these Bird and Jaenisch germline and conditional Mecp2 deletion mouse models led to the conclusion that defects in Mecp2 expression of MeCP2 protein during development is necessary for normal central nervous system function and a normal lifespan. Interestingly, subsequent deletion of Mecp2 in specific neuronal subtypes revealed specific network defects but not reduced life span (Fyffe et al., 2008; Samaco et al., 2009; Chao et al., 2010) except for male mice with Mecp2 deletion in somatostatin and parvalbumin neurons (Ito-Ishida et al., 2015). However, MECP2 exon deletions homologous to Mecp2 exon deletion model mice are rarely found in Rett patients and thus represent somewhat artificial genetic constructs of the disease.
The requirement for MeCP2 activity during murine development was examined by leveraging a system in which a stop codon in Mecp2 could be removed using a systemically administered, tamoxifen induced, cre-lox deletion. Using this tool, a landmark series of experiments performed by teams from the Bird and Jaenisch labs indicated that the expression of Mecp2 in neurons during early adult development was able to rescue motor defects, hypo-activity and premature death in Mecp2–/y null male mice bearing Bird and Jaenisch Mecp2 deletion alleles (Giacometti et al., 2007; Guy et al., 2007). To further test the hypothesis that Mecp2 expression is necessary for neurologic function after development in a whole animal, a team led by Huda Zoghbi examined the effects of the Bird Mecp2 deletion allele in adult mice. The Mecp2–/y null male mice acquired motor defects, learning defects, apraxia and lethality within 15 weeks after Mecp2 gene deletion (McGraw et al., 2011). From these collective Mecp2 deletion mouse model studies, it is clear that MeCP2 is necessary for normal neuronal function and overall health throughout the lifespan. One caveat of these and other studies is that Bird Mecp2–/y null deletion males were the primary focus of these studies as they have a robust disease endpoint (death) by 20 weeks of age and present with motor defects at 6 weeks of age (Guy et al., 2001). However, the vast majority of Rett patients are female and are heterozygous for MECP2 mutations as spontaneous MECP2 point mutations are passed from the paternal X chromosome in sperm (Trappe et al., 2001). Female Mecp2–/+ deficient heterozygous deletion mice are more difficult to study as life span is normal and the motor, altered anxiety and respiratory symptoms common to Mecp2–/y null deletion males arise after 6 months of age and are often subtle in presentation (Chen et al., 2001; Guy et al., 2001). It should be noted that maternally inherited MECP2 mutations are extremely rare, tend to be gene duplications and present as a different disease in male patients (Del Gaudio et al., 2006).
Since the generation of the initial Mecp2 deletion mouse models, multiple Mecp2 gene “knock in” models that are based on actual MECP2 mutations identified in Rett patients have been developed. The model that may have the greatest relevance to Rett is the T158A model developed by Zhou (Goffin et al., 2012) as mutation of threonine 158 is the most common MECP2 missense mutation according to the RettBASE database (mecp2.chw.edu.au) and affects the ability of MeCP2 to bind to DNA. The non-sense MECP2 mutation R168X correlates with severe disease symptoms in Rett. This mutation when recapitulated in Mecp2 resulted in motor, cognitive and anxiety defects in Mecp2R168X/y males and females, although the time of onset was delayed in females (Schaevitz et al., 2013). In fact, male T158A mice (Mecp2T158A/y) recapitulated the motor and learning defects as well as premature mortality observed in male Mecp2 null mice (Goffin et al., 2012) while female Mecp2T158A/+ exhibit breathing abnormalities similar to those observed in Rett females (Bissonnette et al., 2014). In 2014 a mouse model based on a human MECP2 exon 1 mutation showed motor defects similar to Bird Mecp2 null mice along with altered anxiety and stereotypic behavior in Mecp2-e1–/y males (Yasui et al., 2014). Additional Mecp2 knock in mouse models based on Rett MECP2 mutations have been developed and exhibit a range of motor and behavioral phenotypes as reviewed in Schmidt and Cardoso (Schmidt et al., 2020). Rat Mecp2 knock in models may have advantages over mouse models as a Mecp2 truncation model exhibits neurologic regression in Mecp2–/+ females and thus more accurately models Rett syndrome than some existing mouse models (Veeraragavan et al., 2015).
Mutations in MECP2/Mecp2 Affect Virtually All Organs and Tissues
Although the most severe disease phenotypes were observed in Mecp2–/y null deletion male mice with deletion engineered in brain (Chen et al., 2001; Guy et al., 2001), defects in MECP2 expression in humans has the potential to affect almost all organs and cell types, as transcripts are detected at significant levels in virtually all adult human tissues (Figure 1). At the systems biology level, male mice engineered to have only peripheral deletion of Mecp2 outside of the nervous system were hypoactive, had reduced exercise capacity and bone defects, but survived beyond 50 weeks after birth (Ross et al., 2016). This study, led by Stuart Cobb, established that Mecp2 expression in multiple organs and cell types likely accounts for these broad phenotypes thereby underlining the importance of MeCP2 function outside of the nervous system.
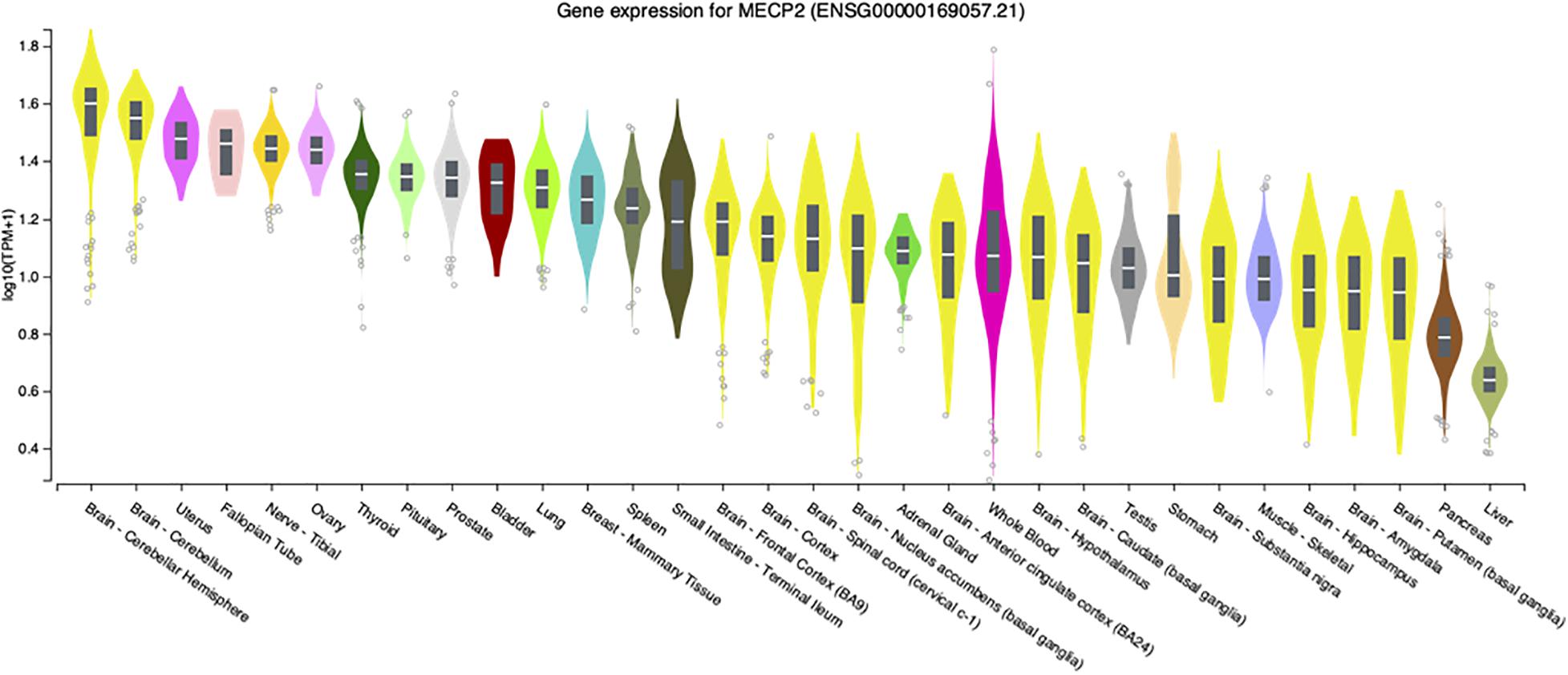
Figure 1. MECP2 transcript levels in select human tissues. Violin plots from the Genome-Tissue expression (GTEx) project are shown for MECP2 with log2 values ranked from high to low levels.
These limited studies indicate that while MeCP2 is necessary for normal neurologic function in mice, disease is still present in other tissues. Similarly, loss or diminished MeCP2 activity contributes to Rett syndrome pathology outside of the nervous system. For example, an early report in 1994 described extended QTc intervals in Rett patients (Sekul et al., 1994). Analysis of Mecp2 deletion mice revealed similar cardiac abnormalities as the patients as well as ventricular tachycardia (McCauley et al., 2011). To further investigate this finding, conditional deletion of Mecp2 from cholinergic neurons was performed by a team led by Jeff Neul. They found that loss of Mecp2 from cholinergic, parasympathetic neurons recapitulated the previous cardiac findings in Bird Mecp2 null mice and symptoms in Rett patients (Herrera et al., 2016). Interestingly, while MECP2 transcripts have relatively low abundance in the human liver (Figure 1) an ENU mutagenesis screen in Bird Mecp2–/y null male mice revealed that MeCP2 function is required for normal lipid metabolism as these animals have dysregulated cholesterol synthesis in brain and elevated cholesterol in liver and serum (Buchovecky et al., 2013; Kyle et al., 2016; Kyle et al., 2018). MECP2 is also expressed at high level in human intestine (Figure 1) and 66% of Rett patients report gastrointestinal pain (Symons et al., 2013). A 2016 study found that Mecp2 null mice had gut hypomotility and reduced nitric oxide synthase expression in enteric neurons (Wahba et al., 2015). To examine the hypothesis that microbial alterations underlie intestinal defects, the overall gut microbiome diversity was examined and appeared to be reduced in Rett patients (Strati et al., 2016; Borghi et al., 2017). As the diets were similar between the Rett and control subjects, these results suggest that altered MECP2 levels in the digestive tract may alter the intestinal environment and thus microbe growth. In fact, it was recently shown that mice with loss of Mecp2 expression solely in the intestine have severe colonic epithelial defects (Millar-Büchner et al., 2016). As there is direct signaling from the gut to the brain, extension of these gut/microbiome studies should be extended to Bird Mecp2–/+ deficient female deletion mice and would thus provide critical insights for female Rett patients. While studies on the role of MeCP2 function in tissues outside of the brain, a recent study found evidence that MeCP2 represses LINE-1 activity in brain, but not in peripheral tissues such as heart and eye (Zhao et al., 2019).
MeCP2 Is a DNA Binding Protein
MeCP2 was first described in the lab of Adrian Bird as a DNA binding protein with a high affinity for CpG methylated DNA (Meehan et al., 1992) via the methyl DNA binding domain (MBD) (Free et al., 2001). Although this report is often overlooked, MeCP2 was later found to be homologous to the nuclear matrix binding protein ARBP, cloned from chicken (Weitzel et al., 1997). This finding is intriguing as the nuclear matrix structures the nucleus in eukaryotic cells and organizes chromatin for replication, DNA repair and transcription among other functions (Wasag and Lenartowski, 2016). These in vitro binding results from the Stratling lab suggested that ARBP/MeCP2 was involved in chromatin loop organization and heterochromatin structure (Weitzel et al., 1997). Basic biochemical analyses of MeCP2 by Jeff Hansen and colleagues revealed that MeCP2 is a highly disordered protein outside of the MBD domain (Adams et al., 2007). Significantly, another study by the Hansen and Woodcock team demonstrated that a common Rett MECP2 mutation, R106W in the MBD altered MeCP2 interaction with nucleosomal DNA (Nikitina et al., 2007). Consistent with these results, MeCP2 lacking the MBD domain was found to bind with low affinity to chromatin in vivo (Stuss et al., 2013). The Hansen group later determined that MeCP2 contains at least two distinct DNA binding domains, the MBD and sequences in the carboxyl terminus (Ghosh et al., 2010).
While Woodcock and colleagues had identified DNA binding regions in MeCP2 outside of the MBD, these sites were not well characterized at the time (Ghosh et al., 2010). Later however, these non-MBD binding sites were identified as AT hook regions by the Zoghbi lab in 2013 (Baker et al., 2013). The description of AT hook domains in MeCP2 fits well with an 2005 description of high affinity binding sites containing AT runs adjacent to methylated CpG sites (Klose et al., 2005). To summarize these diverse findings, a very recent paper nicely illustrates the in vivo activity of MeCP2 in live cells. In this study Nat Heintz and colleagues leveraged MeCP2 fluorescent tagging and real-time visual mobility analysis to describe the sum effect of stable MBD domain interactions and transient AT hook interactions that slow the diffusion of MeCP2 from DNA in living cells (Piccolo et al., 2019).
Since the early characterization of MeCP2 as a methylated CpG binding protein (Meehan et al., 1992), the range of MeCP2 binding activity has been extended. A discovery by the Heintz lab revealed that 5-hydroxymethylcytosine (5hmc) is highly abundant in mouse brain (Kriaucionis and Heintz, 2009). This finding was confirmed by Anjana Rao and colleagues who also identified TET1 as the enzyme responsible for the conversion of 5-methylcytosine (5mc) to 5hmc in brain (Tahiliani et al., 2009). Later it was determined by the Heintz lab that MeCP2 can bind to 5hmc with similar affinity as 5mc in actively transcribed genes (Mellén et al., 2012). However, in vitro results had previously shown that 5hmc inhibits binding of the MBD domain of MeCP2 to DNA (Valinluck et al., 2004). Although the activity of TET1 and related factors TET2 and TET3 can each lead to active CpG demethylation, potentially altering the transcriptional state of genes (He et al., 2011) the biologic implications of MeCP2 binding to 5hmc is still not clear.
While 5mc and 5hmc in CpG dinucleotides account for the majority of MeCP2 binding sites in the mammalian genome, MeCP2 can bind to other motifs. In 2014 the Hongjun Song lab revealed that surprisingly, 25% of CpA, CpC, and CpT (CH) dinucleotides are methylated in the brain compared with 75% of CpG sites (Guo et al., 2014). The Song lab also showed that MeCP2 was able to bind to methylated CpH in vitro (Guo et al., 2014). The following year, Harrison Gabel and other members of the Greenberg lab determined that MeCP2 binds to methyl CpA sites within gene bodies, although the effect of this binding is controversial as described in a subsequent section of this review (Gabel et al., 2015). More recently the binding of MeCP2 was correlated with the methylation status of CH sites in the brain (Lavery et al., 2020). These results conflict with data indicating that MeCP2 binding to DNA is mostly due to CpG methylation and that MeCP2 binds to promoters with low CpG density (Baubec et al., 2013). While it is clear that MeCP2 binds to dinucleotides with methylated cytosine, an unbiased statistical analysis of genome-wide MeCP2 ChIP-seq data sets indicated that the GC content of a particular genomic region is the most predictive of MeCP2 binding (Rube et al., 2016). In contrast to these results, recent in vitro and in vivo studies indicate that MeCP2 has minimal binding to non-methylated GT rich DNA (Connelly et al., 2020). To further define MeCP2 binding, Buchmuller et al. (2020) found that MeCP2 was able to bind to DNA in vitro with the asymmetric cytosine modifications C/mC, mC/mC, mC/hmC, and mC/fC, where C is cytosine, mC is 5 methyl cytosine, hmC is 5 hydroxymethylcytosine and fC is 5 formylcytosine. These recent findings appear to account for the ability of MeCP2 to bind virtually anywhere in the genome. It is important to note that the level of MeCP2 protein expression is critical for normal brain function as mice with 50% of normal expression (Kerr et al., 2008; Samaco et al., 2008) and twice normal expression (Collins et al., 2004) exhibit neurological defects. Recent studies suggest that altered neuronal MeCP2 levels correlate with heterochromatin changes and behavioral symptoms (Ito-Ishida et al., 2020).
MeCP2 Is Involved in Higher Order Chromatin Organization
Another fundamental aspect of MeCP2 function that is often overlooked is its ability to regulate large chromosomal domains. For example, some of the earliest in vitro structural studies revealed that MeCP2 could bind and condense nucleosomal arrays that incorporate both methylated and unmethylated DNA (Georgel et al., 2003). Interestingly, these studies did not indicate that MeCP2 was able to form dimers, suggesting that one MeCP2 molecule could bridge two independent arrays. A similar chromatin condensation activity was shown in myoblast cell lines by the Cardoso lab (Brero et al., 2005). Co-immunoprecipitation studies by the El-Osta lab found that the SWI/SNF chromatin remodeling factors Brahma and BAF57 can be recruited to DNA by MeCP2 (Harikrishnan et al., 2005). While these studies examined recombinant MeCP2 linking of artificial nucleosomal constructs, a study from the Kohwi-Shigematsu lab identified a functional chromatin looping activity in vivo. In this study Shin-Ichi Horike and colleagues found that MeCP2 mediated long range looping and thus transcriptional activity of the Dlx5 gene (Horike et al., 2005). In a similar study it was found that long range interactions between the Prader-Willi imprinting center (PWS-IC) and CHRNA7 modulated gene expression in neurons (Yasui et al., 2011). These studies in cell lines were also consistent with in vitro studies from the Hansen lab revealing that MeCP2 was able to interact with DNA outside of the MBD domain, thereby accounting for the ability of one MeCP2 molecule to bind to two sites simultaneously in chromatin looping (Nikitina et al., 2007). This 2007 study also found that MeCP2 molecules encoded by frequent Rett MECP2 mutations were deficient in their ability to compact chromatin (Nikitina et al., 2007). Studies from the LaSalle lab extended this chromatin looping aspect of MeCP2 function further by showing that MeCP2 along with CTCF contributes to both inter and intra chromosomal interactions and gene regulation of 15q11-13 (Meguro-Horike et al., 2011; Yasui et al., 2011). Later studies from the Natalie Berube lab showed that MeCP2 recruits ATRX to imprinted genes to regulate CTCF chromatin looping interactions (Kernohan et al., 2014). Recent atomic force microscopy (AFM) studies reveal that MeCP2 forms loops by bridging distant binding sites on continuous DNA strands (Liu et al., 2020).
MeCP2 Regulates RNA Splicing
While MeCP2 can affect gene transcription directly, one relatively unexplored activity of MeCP2 is its effect on RNA splicing. The first genome wide study of MeCP2 effects on splicing was performed in the Huda Zoghbi lab in 2005. These studies revealed significant splicing defects in Mecp2308/y male mice expressing a truncated MeCP2 protein (Young et al., 2005). In vitro studies revealed that MeCP2 bound to RNA along with YB-1 resulting in gene construct splicing events in neuronal cell lines (Young et al., 2005). Following this report, few attempts were made to confirm the role of MeCP2 in RNA splicing regulation. It was not until 2013 that the Keji Zhao lab reported that Mecp2 ablation correlated with alternatively spliced exon skipping (Maunakea et al., 2013). Research from the LaSalle lab in 2014 confirmed MeCP2 association with YB-1 shown by Young et al. (2005) as well as interaction with the RNA splicing factors MATR3, SFPQ, and SFRS1 (Yasui et al., 2014). The mechanism underlying MeCP2 regulation of splicing was further explored by the Rasko lab. They found that MeCP2 depletion reduces splicing factor recruitment to methylated DNA and increases intron retention events in blood cells (Wong et al., 2017). More recently Nurit Ballas and colleagues reported abnormal intron retention exon skipping in activation induced genes in Mecp2 null neurons compared to wild-type neurons (Osenberg et al., 2018). The latest investigation of the role of MeCP2 in RNA splicing regulation using a machine learning approach, however, found subtle changes in RNA splicing in cells with varying levels of MeCP2 (Chhatbar et al., 2020). While the results may be not always be direct, or of great magnitude, MeCP2 binding to DNA has an effect on mRNA splicing events. However, one wonders how much current RNA-seq methodologies bias this sort of approach.
Unresolved Details of MeCP2 Function
MeCP2 Functions as a Transcriptional Repressor and Activator
The model of MeCP2 as a direct transcriptional repressor of single copy genes in cis derived from in vitro studies dating back to 1992. In 1997 this model was formally proposed in a report showing that affinity purified rat MeCP2 showed repressive activity of CpG methylated gene promoter constructs in vitro (Nan et al., 1997). Subsequent studies from the Alan Wolffe lab demonstrated that MeCP2 recruitment of histone deacetylase activity was correlated with transcriptional repression (Jones et al., 1998). An example of MeCP2 functioning as a transcriptional repressor is the regulation of viral elements that exist in the genome as multiple, highly CpG methylated elements. Specifically, LINE-1 elements were found to be transcriptionally repressed by MeCP2 binding (Lorincz et al., 2001; Yu et al., 2001). In 2010, a paradigm shifting report published by the Gage lab found that mammalian neuronal progenitor cells have a massive activation of LINE-1 transcription and transposition that is absent in cells from peripheral tissues (Muotri et al., 2010). More recently the Cardoso lab demonstrated in cell lines that MBD proteins including MeCP2 repress TET1 mediated 5hmc conversion and activation of LINE1 elements in human cells (Zhang et al., 2017). A recent analysis of LINE1 insertions revealed in that Rett tissues had fewer cells with exonic insertion, consistent with selection against neurons with multiple LINE1 insertion events and other cell types (Zhao et al., 2019). Thus, in vitro and in vivo evidence indicates that MeCP2 binds to CpG methylated DNA promoters and represses transcription of genes and viral elements along with co-factors such as HDACs.
The model of MeCP2 as a transcriptional repressor of single copy genes was first challenged by the genome wide correlation of MeCP2 binding with gene transcripts. In 2007, MeCP2 ChIP-seq analysis combined with gene transcript analysis from the LaSalle lab found that MeCP2 bound to active promoters with low levels of DNA methylation (Yasui et al., 2007). This was closely followed by results published in 2008 from the Zoghbi lab showing that loss of MeCP2 correlated with both reduced and elevated levels of gene transcripts in murine hypothalamus (Chahrour et al., 2008). Further analyses of select gene promoters upregulated by loss of MeCP2 activity revealed binding of the transcriptional activator CREB1 along with MeCP2 to expressed genes (Chahrour et al., 2008). In support of these findings, in 2017 it was shown that MECP2 mutant and MECP2 null human embryonic stem cell derived forebrain neurons had reduced CREB levels along with reduced dendritic complexity, neurite growth, and mitochondrial function (Bu et al., 2017). Most recently, results from the Harrison Gabel lab suggest that DNA methylation at enhancers may repress genes in trans (Clemens et al., 2020). Clearly these results show that MeCP2 binding and recruitment of co-factors correlates with both activation and repression of transcription. However, the location of MeCP2 binding and co-factor interactions appear to be critical for how gene expression is affected.
MeCP2e1 and MeCP2e2 Protein Isoforms Have Distinct Functions
Prior to 2004, MeCP2 was believed to be the gene product of three exons comprising one protein isoform. In that year a team from The Hospital for Sick Children in Toronto described a novel exon upstream of the known MECP2 exons that could splice to exons 3 and 4 to produce a novel protein isoform with a distinctly different amino terminus than the originally described MeCP2 isoform (Mnatzakanian et al., 2004). The presence of this novel exon was also shown in mouse Mecp2 by RT-PCR (Mnatzakanian et al., 2004). Around the same time, the homologous upstream exon in murine Mecp2 was also reported by the lab of Adrian Bird (Kriaucionis and Bird, 2004). Therefore, the alternative splicing of MECP2/Mecp2 exon 1 to exons 3 and 4 produces the MeCP2e1 isoform while translation of mRNA with all four exons leads to production of MeCP2e2 due to the use of an alternative translational start site (Figure 2). In humans and rodents this produces MeCP2 isoforms which have almost identical amino acid sequences but with distinctly different amino termini (Figure 2). Furthermore, it was shown that mutations in MECP2 exon 1 are present in Rett patients (Mnatzakanian et al., 2004) and that MECP2 exon 1 mutations disrupt translation of the MeCP2e1 but not MeCP2e2 isoforms (Gianakopoulos et al., 2012).
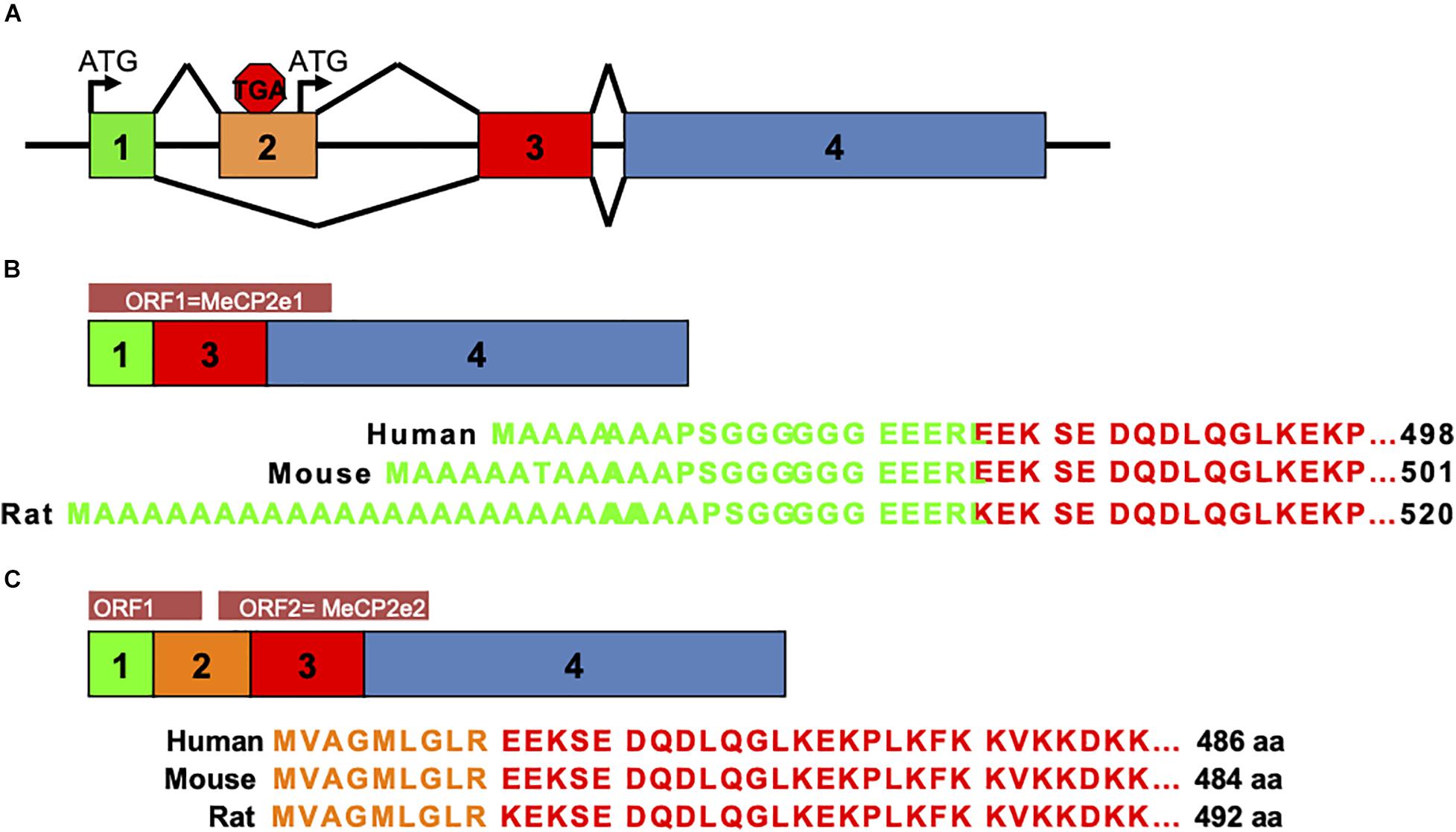
Figure 2. MeCP2 protein isoforms. MeCP2e1 and MeCP2e2 protein isoforms are produced from alternative splicing events. The MeCP2e1 isoform is translated from splicing of exon 1 to exons 3 and 4 with the translational start ATG in exon 1. The MeCP2e2 protein isoform is produced from a transcript from all four exons with a translational start ATG in exon 2. Interestingly, this transcript may encode a small peptide from an ORF in exons 1 and 2. (A) MECP2/Mecp2 exon splicing. (B) MeCP2-e1 isoform. (C) MeCP2-e2 isoform.
Evidence that MeCP2e1 is the isoform underlying Rett syndrome was provided by two key studies. In the first study, knockout of Mecp2 exon 2 which only effects production of MeCP2e2 resulted in mice with normal development and function (Itoh et al., 2012). However, Mecp2 exon 2 deletion did result in placental defects (Itoh et al., 2012). To follow up on these findings the Berge Minassian lab screened a cohort of patients with idiopathic RTT. In one patient they found an MECP2 exon 1 A to T point mutation affecting the translational start site (Saunders et al., 2009). This exon 1 mutation was predicted to prevent translation of only the MeCP2e1 isoform but not the MeCP2e2 isoform, suggesting that it is the loss of MeCP2e1 activity that results in Rett syndrome. Therefore, the LaSalle lab replicated this mutation in a mouse model which resulted in the complete loss of the MeCP2e1 protein isoform while retaining expression of MeCP2e2 (Yasui et al., 2014). Most importantly the results from Yasui and colleagues found that Mecp2e1–/y males had impaired motor function, exhibited apraxia like limb clasping with altered anxiety behavior and premature death (Yasui et al., 2014) similar to Bird Mecp2 deletion male mice (Guy et al., 2001). These MeCP2e1 deficient mice accurately model Rett syndrome as heterozygous Mecp2e1–/+ females exhibited significant motor impairment and altered body composition (Yasui et al., 2014; Vogel Ciernia et al., 2017). Interestingly, both Mecp2e1–/y males and Mecp2e1–/+ females had elevated body fat accumulation correlating with reduced energy expenditure in early adulthood (Vogel Ciernia et al., 2018).
Additional evidence that MeCP2e1 has non-overlapping function with MeCP2e2 is the finding by the Rastegar lab that MeCP2e1 is expressed in brain prior to MeCPe2 at higher levels and a more consistent distribution pattern between brain regions (Olson et al., 2014). Consistent with the mouse studies, the Ellis lab found that human MeCP2e1 deficient iPS derived neurons recapitulate synaptic current, soma size, membrane potential and maturation defects common to Rett syndrome further establishing the necessity of MeCP2e1 in neuronal development (Djuric et al., 2015). MeCP2e1 and MeCPe2 were also found by the Ausio lab to have differential DNA binding kinetics, distinct co-factor association and preferred different DNA binding motifs (Martínez De Paz et al., 2019). Perhaps the most compelling evidence that MeCP2e1 has an essential function that differs from MeCP2e2 is the finding that MeCP2e1 is the evolutionarily older isoform, as orthologs have been identified in vertebrates back to bony fish (Osteichthyes) and amphibians (Mnatzakanian et al., 2004).
Glial Cells Are Critical for Rett Syndrome Pathology
Another contentious issue in the MeCP2/Rett field is the contribution of non-neuronal cells to the disease process, despite considerable evidence that MECP2 transcripts are present at high levels in virtually all tissues (Figure 1). Although cell sorting experiments show that MeCP2 is present at near octomer levels in neurons, MeCP2 is present in glia, albeit at much lower levels (Skene et al., 2010). Direct evidence for the role of non-neural cells in Rett pathology was provided by research performed by the Jin lab at UC Davis where it was found that astrocytes from Bird Mecp2–/y null male mice were abnormal and produced underdeveloped wild-type neurons compared with wild-type astrocytes in a co-culture system (Maezawa et al., 2009). A subsequent study by the Jin lab revealed that Bird Mecp2–/y null male microglia damaged adjacent neurons in a non-cell autonomous manner by releasing glutamate (Maezawa and Jin, 2010). The relevance of cell autonomous vs. non-cell autonomous effects in the Mecp2 mutant brain is discussed further in the next section.
In support of these studies, a paper by the Mandel lab described that re-expression of Mecp2 solely in astrocytes was able to reduce motor defects, breathing irregularity as well as prolong life in Bird Mecp2–/y null male mice (Lioy et al., 2011). Surprisingly, microglia, which comprise a tiny percentage of brain cells, were found to be important for Rett pathology when it was shown that replacement of Mecp2 null microglia with wild-type microglia in Bird Mecp2–/y null male brain improved life span, normalized breathing and reduced motor impairment (Derecki et al., 2012). Although these results were not independently reproduced, a recent study by the Stevens lab established that microglial engulfment of synapses damages neural circuits in Mecp2–/y null male mice (Schafer et al., 2016). To further address the specific contribution of astrocytes to Rett phenotypes, Qiang Chang from the University of Wisconsin Madison derived wild-type and MECP2 mutant iPS astrocytes and examined their function in culture with neurons. They found that MECP2 mutant astrocytes have excess calcium stores that correlated with elevated NMDA receptor expression in neighboring neurons and hyper neuronal network excitability (Dong et al., 2018). Therefore, the authors conclude that astrocytes act both cell-autonomously and non-cell autonomously to mediate Rett like pathology (Dong et al., 2018). An extensive review on the function of MeCP2 in glia has recently become available (Kahanovitch et al., 2019).
MECP2/Mecp2 Has Both Cell Autonomous and Non-cell Autonomous Effects
The vast majority of Rett patients are female MECP2 heterozygotes and are thus cellular mosaics for wild-type and mutant MECP2 expressing cells. It was first reported by the LaSalle lab that Bird Mecp2 null neurons affected the expression of adjacent wild-type neurons in Mecp2–/+ heterozygous female brains (Braunschweig et al., 2004). This finding led to the “bad neighborhood” or non-cell autonomous effects hypothesis of Mecp2 deficiency where defects in Mecp2 mutant cells affect the function of local wild-type cells in the brain or other tissues (Braunschweig et al., 2004). This “bad neighborhood” becomes important when examining disease processes in female but not male mice where all cells are deficient in Mecp2 expression. Subsequent studies by the Gail Mandel lab showed that MeCP2 deficient glia impair neuronal function in a non-cell autonomous manner in vitro (Ballas et al., 2009). As mentioned in the previous section, a similar in vitro culture system showed that Mecp2–/y null astrocytes were able to impair the growth of wild-type neuronal dendrites through soluble factors (Maezawa et al., 2009). To study these effects in vivo by transplanting either Mecp2 mutant or wild-type neuroblasts into wild-type brain Kishi and Macklis found that MeCP2 functions largely cell autonomously in development of the cortex but that non-cell autonomous effects on neuronal function exist (Kishi and Macklis, 2010). In the most definitive study to date, the Zhou lab examined Mecp2 mutant tagged and wild-type cells from female brains and found that non-cell autonomous effects were greater in magnitude than cell autonomous effects and tended to effect cell to cell signaling and phosphorylation (Johnson et al., 2017). These results were consistent with subsequent single cell RNA-sequencing (sc-RNAseq) analysis of neurons from Mecp2–/+ deficient female brain suggesting that MeCP2 can affect gene expression in a non-cell autonomous manner in different neuronal cell types, Renthal et al. (2018).
MeCP2 Preferentially Regulates Long Genes Over Short Genes
The hypothesis that MeCP2 preferentially regulates long genes was first proposed by Sacha Nelson in 2014 (Sugino et al., 2014). This report was supported by data published from the Greenberg lab in 2015 (Gabel et al., 2015). This hypothesis was further tested in an important study mentioned previously by the Zhou lab which found a trend toward long genes being more severely affected by MeCP2 deficiency than short genes (Johnson et al., 2017). However, these findings were refuted by a re-analysis of the data and transcriptional analysis using alternative methods which together, suggested that traditional PCR methods bias the RNA-seq results toward long genes (Raman et al., 2018). The controversy over regulation of long genes is discussed further in a review by Connolly and Zhou (2019).
At What Developmental Stage Do MeCP2 Defects Impair Development
A lack of a developmental time course of how MeCP2 defects are manifested impedes Rett research. Although there is an abundance of data about MeCP2 defects in adult mice, the earliest stages of development have not been well characterized, aside from a 2003 study which found MeCP2 positive cells in E14 rat cortex (Jung et al., 2003). To address this gap, the Landsberger lab analyzed neurons from embryonic Bird Mecp2–/y null male cortex (Bedogni et al., 2016). They found that Bird Mecp2–/y null embryonic neurons have altered gene expression, altered morphology, reduced calcium flux and mobility compared to wild-type neurons (Bedogni et al., 2016). Independent evidence suggests that there are deficiencies in Mecp2 transcripts and MeCP2 protein in the mouse cortex as early as E14 (Zachariah et al., 2012). However, one limitation of these molecular studies of early MeCP2 expression in mice is that they employed conventional methods in bulk tissue such as qRT-PCR and Western blot and that the investigators studied Bird Mecp2–/y null male mice which do accurately model Rett syndrome in MECP2 heterozygous females. To date there has been only one study of MeCP2 embryonic function at single-cell resolution in disease relevant Bird Mecp2–/+ deficient mouse brain by the Greenberg group. However only adult Bird Mecp2–/+ deficient deletion female mice and adult human Rett brains were analyzed (Renthal et al., 2018). Yet it is known that Rett syndrome girls experience a postnatal developmental regression at about 6–18 months of age (Zoghbi, 2005). Thus, the molecular changes during this critical early time are still not well defined. While it is clear that MeCP2 function is critical for normal neurological function, the molecular phenotypes in distinct brain cell types have not been investigated over the full-time course of development in mouse models. The Rett field is lacking an extensive, longitudinal study in mosaic Mecp2–/+ deficient mice investigating the mechanisms and molecular pathways that are impacted by perturbed Mecp2/MeCP2 expression throughout disease progression.
MeCP2 Is an RNA Binding Protein
Coding RNAs and non-coding RNA such as long non-coding RNAs, small nucleolar RNAs, and micro RNAs establish a diverse set of functions due to their direct interactions with RNA-binding proteins (RBPs). In one of the few studies on this topic it was found that MeCP2 binds with high affinity to mRNA and siRNA outside of the MBD in vitro (Jeffery and Nakielny, 2004). MeCP2 was also found to associate with long non-coding RNAs and imprinted genes in mouse brain extracts (Maxwell et al., 2013). To examine MeCP2 interaction with RNA in vivo, MeCP2 RNA-immunoprecipitation of small RNAs was performed in the lab of Assam El-Osta who found that MeCP2 bound to specific RNAs including micro RNAs (Khan et al., 2017). These studies investigating MeCP2 interactions with RNA were performed in a targeted manner, therefore an unbiased, genome wide screen of RNA binding would provide a significant advance in understanding this MeCP2 activity.
Recent Developments in the MeCP2 Field
MeCP2 Enables Liquid Phase Separation Events in Nuclear Compartmentalization
In the last 2 years the biologic functions of MeCP2 have expanded appreciably. One concept that has emerged is the effect that liquid-liquid phase separations (LLPS) have on the formation of sub-nuclear compartments (Alberti et al., 2019). In their review Alberti, Gladfelter, and Mittag explain that the nucleus of eukaryotic cells consists of membrane less structures such as nucleoli and heterochromatin that form by LLPS through the activity of proteins and nucleic acids that condense into a dense phase (heterochromatin) and a loose phase depending on conditions such as molecular concentration, salt concentration and pH (Alberti et al., 2019). Thus, the three near simultaneous reports that MeCP2 is involved with LLPS DNA phase separation in the cell nucleus was of great relevance to the field. In the first report, Wang and colleagues found that MeCP2 was able to condense chromatin constructs in vitro and that DNA methylation enhanced this effect (Wang et al., 2020). Furthermore, Wang et al found that mutant MeCP2 proteins from Rett patients were defective in chromatin condensation and LLPS (Wang et al., 2020). Results from Fan and colleagues confirmed that wild-type MeCP2 was able to form condensates with DNA in vitro (Fan et al., 2020). Finally, research published by Richard Young and Rudolph Jaenisch at MIT also showed that MeCP2 condensed chromatin in vitro and MeCP2 mutant forms were defective in LLPS activity (Li et al., 2020). What set apart the Young and Jaenisch study from the other studies is the finding that the intrinsically disordered regions (IDR) of MeCP2 mediate the phase separation activity in the nucleus (Li et al., 2020). The concept of MeCP2 as a nuclear organizer via LLPS is reminiscent of early reports that an MeCP2 homolog, ARBP, functions as a component of the nuclear matrix as mentioned previously (Weitzel et al., 1997).
MeCP2 Functions With DNMT3A/Dnmt3a to Regulate Gene Expression
MeCP2 and DNMT3A, a de novo DNA methyltransferase have long been linked by the fact that the MeCP2 MBD preferentially binds to CpG methylated DNA (mCG). Recently it was found that there is a direct molecular link between the two molecules as the MeCP2 TRD binds to the DNMT3A ADD domain. This interaction inhibits DNMT3A activity and this inhibition is countered by DNMT3A interaction with H3K4 through the ADD domain in vitro (Rajavelu et al., 2018). Similar results were shown by a collaboration between the Joe Ecker and Huda Zoghbi labs that examined the in vivo effects of MeCP2 and DNMT3A interaction. It had been previously established by the Ecker lab that high levels of DNA methylation at CH (mCH) sites (H = Adenosine, Cytosine, and Thymidine) in neurons, is due to the activity of DNMT3A (Lister et al., 2009). Therefore, Zoghbi and Ecker examined the hypothesis that as MeCP2 is the sole reader of methyl mCH and that DNMT3A is the sole writer of methyl CH, deletion of either factor in neurons would have similar effects on gene transcription. Surprisingly, loss of DNMT3A in neurons affected transcription of significantly more genes than the loss of MeCP2, highlighting the importance of mCH to neurologic gene regulation and function (Lavery et al., 2020). The relationship between mCH, mCG, and MeCP2 during development is explored further in a review by Lavery and Zoghbi (2019). These advances also underscore the concept that much of MeCP2 function is mediated through co-factor association.
MeCP2 May Function in DNA Repair Processes
The first evidence to suggest that MeCP2 could function in DNA repair processes was first reported in 2005. Valinluck and colleagues found that MeCP2 binds with high affinity to DNA containing halogenated pyrimidines, a modification that can result from inflammation (Valinluck et al., 2005). More recently it was found that neural stem cells from Bird Mecp2–/+ deficient female mice are prone to early senescence and have elevated rates of cell death when exposed to DNA damaging H2O2, UV light and doxorubicin (Alessio et al., 2018). However, the most convincing evidence for the role of MeCP2 in DNA repair comes from an unbiased N-ethyl-N-nitrosourea (ENU) suppressor screen in Bird Mecp2–/y null male mice (Enikanolaiye et al., 2020). Out of 2,498 males born to ENU treated wild-type males mated with Mecp2–/+ females, 96 males had ameliorated neurologic symptoms from which 32 genes which suppressed the Mecp2 null phenotype were identified. Of these 32 genes, Tet1, Birc6, and Spin1 function in the DNA damage response while Rbbp8, Rad50, Fan1, Brca1, and Brca2 encode factors involved in DNA double strand break repair (Enikanolaiye et al., 2020).
Potential Treatments Under Development for Rett Syndrome
The remarkable reversal of Rett like phenotypes in Bird Mecp2–/y null male mice provides a basis for clinical therapies that seek to restore expression of MECP2 (Guy et al., 2007). To this aim, the potential of adeno associated viral (AAV) vectors to deliver MeCP2 to deficient cells in a whole animal model was shown independently in 2013 by two groups. One group led by Stuart Cobb showed that AAV9/MECP2 delivered intravenously in pre-symptomatic Bird Mecp2–/y null male mice extended survival modestly (Gadalla et al., 2013). Gail Mandel led a second study that showed that intravenous AAV9/MeCP2e1 vector reduced motor defects in Bird Mecp2–/+ deficient female mice, which thus had greater relevance to Rett syndrome as 95% of Rett patients are female (Garg et al., 2013). In 2017, after these early proofs of principle, advances in AAV9/MECP2 gene therapy were reported by the combined efforts of Stuart Cobb and Steven J. Gray. The authors in two manuscripts reported improvement in cell transduction with a reduced titer of a second generation AAV9/hMeCP2 virus, a reduction in liver toxicity and prolonged survival in Bird Mecp2–/y null mice by delivery through the cerebral spinal fluid (CSF) or directly into brain (Gadalla et al., 2017; Sinnett et al., 2017). These studies, while they represent significant improvements in MECP2 gene therapy tools, should be performed in Bird Mecp2–/+ deficient heterozygous female deletion mice to be more relevant to Rett. Furthermore, the risks of treating human children with AAV vectors cannot be fully mitigated.
Alternatively, the use of site directed RNA editing is being developed for potential Rett syndrome therapy. RNA editing strategies leverage an Adenosine Deaminase Acting on RNA (ADAR2) RNA editing factors with a guide RNA to repair Mecp2/MECP2 point mutations in RNA. The first report on Mecp2 RNA editing was published by the Gail Mandel lab in 2017 where an exogenous, modified ADAR2 and guide was used to successfully edit 72% G to A mutations in RNA from MecpR106Q mouse neurons (Sinnamon et al., 2017). In 2020 Sinnamon and colleagues successfully edited up to 50% of mRNA in neurons in developing brains of Mecp2 mutant mice using a similar approach (Sinnamon et al., 2020). The potential of RNA editing as a Rett therapy is limited by the fact that although endogenous RNA editing proteins such as ADAR1, exist naturally in the brain, current approaches express a modified RNA editing protein delivered by an AAV virus and the fact that these ADAR2 like proteins primarily edit only G to A mutations (Sinnamon et al., 2017, 2020).
Despite the excitement for MECP2 gene replacement and RNA editing strategies, other therapeutic methods are in development. One strategy explored by the Zoghbi lab in Rett model mice is deep brain stimulation. Electrode based stimulation of symptomatic Mecp2–/+ female mice normalized learning and memory defects (Hao et al., 2015; Pohodich et al., 2018). Another promising area for potential Rett therapies, is the repurposing of existing FDA approved compounds. For example, in 2020, it was found that the diabetes drug, metformin, reduces mitochondrial defects and damage from oxidative stress in Mecp2 308–/+ truncation female mice (Zuliani et al., 2020). Finally, a recently developed compound for Alzheimer’s disease reduced motor defects, learning deficits and breathing abnormalities in Mecp2–/+ female mice with minimal side effects (Kaufmann et al., 2019). Until effective and safe MECP2 gene replacement and/or RNA editing strategies are available, existing or novel chemical compounds could be used to reduce the most severe symptoms in Rett patients.
Discussion
Currently, there is debate as to whether patients with overlapping phenotypes have Rett syndrome or some other disease. For example, patients with mutations in CDKL5 were considered to have a severe variant of Rett, however, this was later categorized as a separate disease (Fehr et al., 2013). Patients with FOXG1 mutations are currently classified with Rett. However, as these rare FOXG1 mutation patients present with disease from birth and have other symptoms that do not overlap with Rett, a proposal to classify these patients as a separate syndrome was recently submitted (Cutri-French et al., 2020). Therefore, with few exceptions, any discussion about Rett syndrome phenotypes is about how defective MECP2/Mecp2 expression presents in patients and animal models and manifests as a neurodevelopmental disease.
At this time it is still unclear how MECP2 expression defects in non-neural tissues contribute to Rett phenotypes. The answer may be complex as illustrated by a report from the Landsberger lab showing that male Bird Mecp2 deletion mice have abnormal muscle tissue while deletion of the Bird Mecp2 allele only in muscle resulted in normal muscle tissue (Conti et al., 2015). The conclusion from this study is that non-cell-autonomous signaling to the muscle by defective neurons in the Bird Mecp2 deletion mice contributes to the muscle defect (Conti et al., 2015). So, there is the possibility that although MECP2 transcripts are abundant in all tissues and cell types, extracellular neuronal signaling could be responsible for some, if not all of the defects.
The recent identification of MeCP2 binding beyond methylated CpG sites has greatly expanded the range of potential functions of the protein. These advances have been driven by the discovery of additional nucleotide modifications in mammalian brain. Yet many questions remain. For example, does MeCP2 binding to 5-hmc (Kriaucionis and Heintz, 2009) in cerebellum have a significant neurologic function (Mellén et al., 2012)? Do MeCP2 isoforms have different 5-hmc binding activity? Some data suggests that MeCP2 protects 5mc from TET1 conversion to 5 hmc (Szulwach et al., 2011) but more study is needed to resolve this important question. The 2014 discovery of DNMT3A dependent CH methylation in mouse brain by the Song lab (Guo et al., 2014) and the description of MeCP2 binding to methyl CpA sites in brain in vivo (Gabel et al., 2015) has been suggested to be key to the pathogenesis of Rett (Lavery et al., 2020). Here again additional research is needed to clearly establish the effect of MeCP2 binding outside of methyl CpG sites.
Another evolving area of research is how MeCP2 functions as a transcriptional repressor and activator. The model of MeCP2 as a transcriptional repressor of single copy genes was developed over twenty years ago (Nan et al., 1997; Jones et al., 1998). Since that time it was found that MeCP2 binds to partially CpG methylated promoters of active genes in neurons (Yasui et al., 2007) and can act as a transcriptional activator by recruitment of CREB in neurons (Chahrour et al., 2008). The latest evidence suggests that MeCP2 modulates gene transcription depending on co-factor interaction. Future gene expression analyses should focus on the differential activity of the MeCP2-e1 and MeCP2-e2 isoforms.
The role of glial cell defects in Rett syndrome pathology is still being described. However, the very fact that glial cells express MECP2/Mecp2 and are defective in Rett patients and Mecp2 null and deficient mice is relevant to neuronal disease phenotypes. As Rett females and Mecp2–/+ deficient heterozygous female mice are mosaic for normal and mutant cells, non-cell autonomous effects are clearly contributing to neuronal dysfunction (Johnson et al., 2017). Analysis of neurons from mosaic Bird Mecp2–/+ deficient female mouse brain and human Rett brain suggest that MeCP2 preferentially represses methylated long genes, Sugino et al. (2014), in a cell autonomous manner in neurons, although non-cell autonomous effects on short genes cannot be excluded (Renthal et al., 2018). As recent study concluded that MeCP2 bias toward long gene regulation may be an artifact of PCR based transcript detection (Raman et al., 2018), single cell analyses using alternatives to current RNA-seq methods in brain cell types will be necessary to resolve the role of non-cell autonomous effects in brains mosaic for wild-type and Mecp2 mutant neurons and glia.
Although it is widely assumed that Rett patients are phenotypically normal at birth this conclusion is based on limited behavioral observation. As there is little incentive and ethical limitations preclude studying apparently normal human infants, a full developmental time course of the disease is needed in Rett model Mecp2–/+ heterozygous mice that more accurately model Rett MECP2 mutations such Mecp2-e1 mutant mice (Yasui et al., 2014). Surprisingly, apart from a study that revealed defects in Bird Mecp2–/y null male embryonic cells, little analysis has been done of early developmental time points (Bedogni et al., 2016). Clearly, a developmental time analyses of both Mecp2–/+ females and Mecp2–/y males at embryonic, pre-disease, early disease, and late disease is needed to identify pathological mechanisms.
There are aspects of MeCP2 function that are oddly neglected. For example, surprisingly, few studies have directly examined the ability of MeCP2 to bind to RNA. The studies that do show a strong in vitro interaction of MeCP2 with mRNA and siRNA via the RG domain between the MBD and TRD (Jeffery and Nakielny, 2004) and in vivo interaction with miRNAs (Khan et al., 2017). A logical progression of these studies would be to examine RNA binding ability of common Rett mutant MeCP2 proteins such as T158M in vitro and in vivo. The Rett field is lacking studies using more unbiased high-through methods such as targets of RNA-binding proteins identified by editing (TRIBE) (McMahon et al., 2016) which fuses MeCP2 to ADAR, an enzyme that modifies RNA where MeCP2 binds. An additional advantage of this approach is that it can be used for in vivo studies.
Finally, unexpected discoveries continue to revitalize the study of MeCP2. Recent cell biology studies reveal that compartmentalization of the nucleus by liquid-liquid phase separation forms critical regulatory compartments such as the nucleolus and heterochromatin (reviewed in Alberti et al., 2019). Now it appears that MeCP2 plays a key role in the formation of nuclear heterochromatin domains according to new in vitro studies (Fan et al., 2020; Wang et al., 2020) and that the MeCP2 intrinsically disorder region plays a key role in this process (Li et al., 2020). These results build on the extensive study of how MeCP2 organizes chromatin in neuronal cells and may provide key insights into Rett syndrome defects.
The most interesting advances in MeCP2 research concern Rett therapies, as restoration of MeCP2 in mouse brain led to disease reversal in a mouse model (Guy et al., 2007). Current MECP2 gene therapy for Rett patients has great potential and some risks. The latest gene therapy treatment of Bird Mecp2–/+ heterozygous mice with AAV9/MECP2 was able to normalize breathing but high viral doses produced severe liver toxicity and death in some animals (Matagne et al., 2020). While RNA editing strategies for repairing mutant MECP2 have great potential as a therapy, delivery of editing proteins to the brain is also as problematic as delivering functional MeCP2. Therefore, considerable time and effort will be needed to refine MECP2 gene replacement and RNA editing based therapies. Until then, alternative treatments are needed to treat Rett.
One potential therapy for Rett is deep brain stimulation (DSB) which has been approved by the FDA for epilepsy. Nonetheless, DSB also has its risks as electrodes are implanted into the brain. Existing drugs approved by the FDA for other diseases are being re-directed to treat Rett symptoms. Clinical trials of IGF-1 and IGF-1 analogs that have been approved for short stature are ongoing for Rett and offer some hope of reducing symptoms (Khwaja et al., 2014). Another compound, ANAVEX 2-73 which targets the sigma-1 receptor that affects learning, memory, and neuronal development and is in phase II clinical trials for Alzheimer’s disease is also scheduled for Phase I trials in Rett girls (Kaufmann et al., 2019). In some cases, effective treatment for a specific disease have been employed without fully understanding the mechanisms of the disease process. While this may be the case with Rett syndrome, that should not preclude the continuing research on the evolving molecular function of MeCP2 in neurons and other cell types.
Author Contributions
DY and OS conceived of, planned and wrote the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the NIH NIAA Grant 1RO1AA027075 Neuroimmune interactions in Rett syndrome to Janine M. LaSalle.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, V. H., McBryant, S. J., Wade, P. A., Woodcock, C. L., and Hansen, J. C. (2007). Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J. Biol. Chem. 282, 15057–15064. doi: 10.1074/jbc.M700855200
Alberti, S., Gladfelter, A., and Mittag, T. (2019). Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176, 419–434. doi: 10.1016/j.cell.2018.12.035
Alessio, N., Riccitiello, F., Squillaro, T., Capasso, S., Del Gaudio, S., Di Bernardo, G., et al. (2018). Neural stem cells from a mouse model of Rett syndrome are prone to senescence, show reduced capacity to cope with genotoxic stress, and are impaired in the differentiation process. Exper. Mol. Med. 50:1. doi: 10.1038/s12276-017-0005-x
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Baker, S. A., Chen, L., Wilkins, A. D., Yu, P., Lichtarge, O., and Zoghbi, H. Y. (2013). An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell 152, 984–996. doi: 10.1016/j.cell.2013.01.038
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
Baubec, T., Ivánek, R., Lienert, F., and Schübeler, D. (2013). Methylation-dependent and -independent genomic targeting principles of the mbd protein family. Cell 153, 480–492. doi: 10.1016/j.cell.2013.03.011
Bedogni, F., Gigli, C. C., Pozzi, D., Rossi, R. L., Scaramuzza, L., Rossetti, G., et al. (2016). Defects during Mecp2 null embryonic cortex development precede the onset of overt neurological symptoms. Cereb. Cortex 26, 2517–2529. doi: 10.1093/cercor/bhv078
Bissonnette, J. M., Schaevitz, L. R., Knopp, S. J., and Zhou, Z. (2014). Respiratory phenotypes are distinctly affected in mice with common Rett syndrome mutations MECP2 T158A and R168X. Neuroscience 267, 166–176. doi: 10.1016/j.neuroscience.2014.02.043
Borghi, E., Borgo, F., Severgnini, M., Savini, M. N., Casiraghi, M. C., and Vignoli, A. (2017). Rett syndrome: a focus on gut microbiota. Intern. J. Mol. Sci. 18:344. doi: 10.3390/ijms18020344
Braunschweig, D., Simcox, T., Samaco, R. C., and LaSalle, J. M. (2004). X-chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2-/+ mouse brain. Hum. Mol. Genet. 13, 1275–1286. doi: 10.1093/hmg/ddh142
Brero, A., Easwaran, H. P., Nowak, D., Grunewald, I., Cremer, T., Leonhardt, H., et al. (2005). Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 169, 733–743. doi: 10.1083/jcb.200502062
Bu, Q., Wang, A., Hamzah, H., Waldman, A., Jiang, K., Dong, Q., et al. (2017). CREB signaling is involved in rett syndrome pathogenesis. J. Neurosci. 37, 3671–3685. doi: 10.1523/JNEUROSCI.3735-16.2017
Buchmuller, B. C., Kosel, B., and Summerer, D. (2020). Complete profiling of Methyl-CpG-binding domains for combinations of cytosine modifications at CpG dinucleotides reveals differential read-out in normal and Rett-associated states. Sci. Rep. 10:4053. doi: 10.1038/s41598-020-61030-1
Buchovecky, C. M., Turley, S. D., Brown, H. M., Kyle, S. M., McDonald, J. G., Liu, B., et al. (2013). A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 45, 1013–1020. doi: 10.1038/ng.2714
Chahrour, M., Sung, Y. J., Shaw, C., Zhou, X., Wong, S. T. C., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Chhatbar, K., Cholewa-Waclaw, J., Shah, R., Bird, A., and Sanguinetti, G. (2020). Quantitative analysis questions the role of MeCP2 as a global regulator of alternative splicing. PLoS Genet. 16:1009087. doi: 10.1371/journal.pgen.1009087
Clemens, A. W., Wu, D. Y., Moore, J. R., Christian, D. L., Zhao, G., and Gabel, H. W. (2020). MeCP2 represses enhancers through chromosome topology-associated DNA methylation. Mol. Cell 77, 279–293.e8. doi: 10.1016/j.molcel.2019.10.033
Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, A., Armstrong, A. L., Noebels, J. L., et al. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689. doi: 10.1093/hmg/ddh282
Connelly, J. C., Cholewa, J., Webb, S., Steccanella, V., Waclaw, B., and Bird, A. (2020). Absence of MeCP2 binding to non-methylated GT sequences in vivo. Nucleic Acids Research 48, 3542–3552. doi: 10.1093/nar/gkaa102
Connolly, D. R., and Zhou, Z. (2019). Genomic insights into MeCP2 function: a role for the maintenance of chromatin architecture. Curr. Opin. Neurobiol. 59, 174–179. doi: 10.1016/j.conb.2019.07.002
Conti, V., Gandaglia, A., Galli, F., Tirone, M., Bellini, E., Campana, L., et al. (2015). MeCP2 affects skeletal muscle growth and morphology through non cell-autonomous mechanisms. PLoS One 10:e0130183. doi: 10.1371/journal.pone.0130183
Cutri-French, C., Armstrong, D., Saby, J., Gorman, C., Lane, J., Fu, C., et al. (2020). Comparison of core features in four developmental encephalopathies in the Rett natural history study. Ann. Neurol. 88, 396–406. doi: 10.1002/ana.25797
Del Gaudio, D., Fang, P., Scaglia, F., Ward, P. A., Craigen, W. J., Glaze, D. G., et al. (2006). Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 8, 784–792. doi: 10.1097/01.gim.0000250502.28516.3c
Derecki, N. C., Cronk, J. C., Lu, Z., Xu, E., Abbott, S. B. G., Guyenet, P. G., et al. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109. doi: 10.1038/nature10907
Djuric, U., Cheung, A. Y. L., Zhang, W., Mok, R. S., Lai, W., Piekna, A., et al. (2015). MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis. 76, 37–45. doi: 10.1016/j.nbd.2015.01.001
Dong, Q., Liu, Q., Li, R., Wang, A., Bu, Q., Wang, K. H., et al. (2018). Mechanism and consequence of abnormal calcium homeostasis in rett syndrome astrocytes. eLife 7:33417. doi: 10.7554/eLife.33417
Enikanolaiye, A., Ruston, J., Zeng, R., Taylor, C., Schrock, M., Buchovecky, C. M., et al. (2020). Suppressor mutations in Mecp2-null mice implicate the DNA damage response in Rett syndrome pathology. Genome Res. 30, 540–552. doi: 10.1101/gr.258400.119
Fan, C., Zhang, H., Fu, L., Li, Y., Du, Y., Qiu, Z., et al. (2020). Rett mutations attenuate phase separation of MeCP2. Cell Discov. 6:38. doi: 10.1038/s41421-020-0172-0
Fehr, S., Wilson, M., Downs, J., Williams, S., Murgia, A., Sartori, S., et al. (2013). The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 21, 266–273. doi: 10.1038/ejhg.2012.156
Free, A., Wakefield, R. I. D., Smith, B. O., Drydenll, D. T. F., Barlow, P. N., and Bird, A. P. (2001). DNA Recognition by the Methyl-CpG binding domain of MeCP2. J. Biol. Chem. 276, 3353–3360. doi: 10.1074/jbc.M007224200
Fyffe, S. L., Neul, J. L., Samaco, R. C., Chao, H. T., Ben-Shachar, S., Moretti, P., et al. (2008). Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 59, 947–958. doi: 10.1016/j.neuron.2008.07.030
Gabel, H. W., Kinde, B., Stroud, H., Gilbert, C. S., Harmin, D. A., Kastan, N. R., et al. (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. doi: 10.1038/nature14319
Gadalla, K. K., Bailey, M. E., Spike, R. C., Ross, P. D., Woodard, K. T., Kalburgi, S. N., et al. (2013). Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol. Therapy 21, 18–30. doi: 10.1038/mt.2012.200
Gadalla, K. K. E., Vudhironarit, T., Hector, R. D., Sinnett, S., Bahey, N. G., Bailey, M. E. S., et al. (2017). Development of a Novel AAV Gene therapy cassette with improved safety features and efficacy in a mouse model of Rett syndrome. Mol. Ther. Methods Clin. Dev. 5, 180–190. doi: 10.1016/j.omtm.2017.04.007
Garg, S. K., Lioy, D. T., Cheval, H., McGann, J. C., Bissonnette, J. M., Murtha, M. J., et al. (2013). Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J. Neurosci. 33, 13612–13620. doi: 10.1523/JNEUROSCI.1854-13.2013
Georgel, P. T., Horowitz-Scherer, R. A., Adkins, N., Woodcock, C. L., Wade, P. A., and Hansen, J. C. (2003). Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 278, 32181–32188. doi: 10.1074/jbc.M305308200
Ghosh, R. P., Nikitina, T., Horowitz-Scherer, R. A., Gierasch, L. M., Uversky, V. N., Hite, K., et al. (2010). Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 49, 4395–4410. doi: 10.1021/bi9019753
Giacometti, E., Luikenhuis, S., Beard, C., and Jaenisch, R. (2007). Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc. Natl. Acad. Sci. U.S.A. 104, 1931–1936. doi: 10.1073/pnas.0610593104
Gianakopoulos, P. J., Zhang, Y., Pencea, N., Orlic-Milacic, M., Mittal, K., Windpassinger, C., et al. (2012). Mutations in MECP2 exon 1 in classical rett patients disrupt MECP2_e1 transcription, but not transcription of MECP2_e2. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 159B, 210–216. doi: 10.1002/ajmg.b.32015
Goffin, D., Allen, M., Zhang, L., Amorim, M., Wang, I. T. J., Reyes, A. R. S., et al. (2012). Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci. 15, 274–283. doi: 10.1038/nn.2997
Guo, J. U., Su, Y., Shin, J. H., Shin, J., Li, H., Xie, B., et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17, 215–222. doi: 10.1038/nn.3607
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Hao, S., Tang, B., Wu, Z., Ure, K., Sun, Y., Tao, H., et al. (2015). Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature 526, 430–434. doi: 10.1038/nature15694
Harikrishnan, K. N., Chow, M. Z., Baker, E. K., Pal, S., Bassal, S., Brasacchio, D., et al. (2005). Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat. Genet. 37, 254–264. doi: 10.1038/ng1516
He, Y. F., Li, B. Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. doi: 10.1126/science.1210944
Herrera, J. A., Ward, C. S., Wehrens, X. H. T., and Neul, J. L. (2016). Methyl-CpG binding-protein 2 function in cholinergic neurons mediates cardiac arrhythmogenesis. Hum. Mol. Genet. 25, 4983–4995. doi: 10.1093/hmg/ddw326
Horike, S. I., Cai, S., Miyano, M., Cheng, J. F., and Kohwi-Shigematsu, T. (2005). Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37, 31–40. doi: 10.1038/ng1491
Itoh, M., Tahimic, C. G. T., Ide, S., Otsuki, A., Sasaoka, T., Noguchi, S., et al. (2012). Methyl CpG-binding protein isoform MeCP2-e2 is dispensable for rett syndrome phenotypes but essential for embryo viability and placenta development. J. Biol. Chem. 287, 13859–13867. doi: 10.1074/jbc.M111.309864
Ito-Ishida, A., Baker, S. A., Sillitoe, R. V., Sun, Y., Zhou, J., Ono, Y., et al. (2020). MeCP2 levels regulate the 3d structure of heterochromatic foci in mouse neurons. J. Neurosci. 40, 8746–8766. doi: 10.1523/JNEUROSCI.1281-19.2020
Ito-Ishida, A., Ure, K., Chen, H., Swann, J. W., and Zoghbi, H. Y. (2015). Loss of MeCP2 in Parvalbumin-and Somatostatin-expressing neurons in mice leads to distinct rett syndrome-like phenotypes. Neuron 88, 651–658. doi: 10.1016/j.neuron.2015.10.029
Jeffery, L., and Nakielny, S. (2004). Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 279, 49479–49487. doi: 10.1074/jbc.M409070200
Johnson, B. S., Zhao, Y. T., Fasolino, M., Lamonica, J. M., Kim, Y. J., Georgakilas, G., et al. (2017). Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat. Med. 23, 1203–1214. doi: 10.1038/nm.4406
Jones, P. L., Veenstra, G. J. C., Wade, P. A., Vermaak, D., Kass, S. U., Landsberger, N., et al. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191. doi: 10.1038/561
Jung, B. P., Jugloff, D. G. M., Zhang, G., Logan, R., Brown, S., and Eubanks, J. H. (2003). The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J. Neurobiol. 55, 86–96. doi: 10.1002/neu.10201
Kahanovitch, U., Patterson, K. C., Hernandez, R., and Olsen, M. L. (2019). Glial dysfunction in meCP2 deficiency models: implications for rett syndrome. Intern. J. Mol. Sci. 20:3813. doi: 10.3390/ijms20153813
Kaufmann, W. E., Sprouse, J., Rebowe, N., Hanania, T., Klamer, D., and Missling, C. U. (2019). ANAVEX® 2-73 (blarcamesine), a Sigma-1 receptor agonist, ameliorates neurologic impairments in a mouse model of Rett syndrome. Pharmacol. Biochem. Behav. 187:172796. doi: 10.1016/j.pbb.2019.172796
Kernohan, K. D., Vernimmen, D., Gloor, G. B., and Bérubé, N. G. (2014). Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping. Nucleic Acids Res. 42, 8356–8368. doi: 10.1093/nar/gku564
Kerr, B., Alvarez-saavedra, M., Sáez, M. A., Saona, A., and Young, J. I. (2008). Defective body-weight regulation, motor control and abnormal social interactions in Mecp2 hypomorphic mice. Hum. Mol. Genet. 17, 1707–1717. doi: 10.1093/hmg/ddn061
Khan, A. W., Ziemann, M., Rafehi, H., Maxwell, S., Ciccotosto, G. D., and El-Osta, A. (2017). MeCP2 interacts with chromosomal microRNAs in brain. Epigenetics 12, 1028–1037. doi: 10.1080/15592294.2017.1391429
Khwaja, O. S., Ho, E., Barnes, K. V., O’Leary, H. M., Pereira, L. M., Finkelstein, Y., et al. (2014). Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, 4596–4601. doi: 10.1073/pnas.1311141111
Kishi, N., and Macklis, J. D. (2010). MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exper. Neurol. 222, 51–58. doi: 10.1016/j.expneurol.2009.12.007
Klose, R. J., Sarraf, S. A., Schmiedeberg, L., McDermott, S. M., Stancheva, I., and Bird, A. P. (2005). DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 19, 667–678. doi: 10.1016/j.molcel.2005.07.021
Kriaucionis, S., and Bird, A. (2004). The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 32, 1818–1823. doi: 10.1093/nar/gkh349
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 324, 929–930. doi: 10.1126/science.1169786
Kyle, S. M., Saha, P. K., Brown, H. M., Chan, L. C., and Justice, M. J. (2016). MeCP2 co-ordinates liver lipid metabolism with the NCoR1/HDAC3 corepressor complex. Hum. Mol. Genet. 25, 3029–3041. doi: 10.1093/hmg/ddw156
Kyle, S. M., Vashi, N., and Justice, M. J. (2018). Rett syndrome: a neurological disorder with metabolic components. Open Biol. 8:170216. doi: 10.1098/rsob.170216
Lavery, L. A., Ure, K., Wan, Y. W., Luo, C., Trostle, A. J., Wang, W., et al. (2020). Losing dnmt3a dependent methylation in inhibitory neurons impairs neural function by a mechanism impacting rett syndrome. eLife 9:52981. doi: 10.7554/eLife.52981
Lavery, L. A., and Zoghbi, H. Y. (2019). The distinct methylation landscape of maturing neurons and its role in Rett syndrome pathogenesis. Curr. Opin. Neurobiol. 59, 180–188. doi: 10.1016/j.conb.2019.08.001
Lewis, J. D., Meehan, R. R., Henzel, W. J., Maurer-Fogy, I., Jeppesen, P., Klein, F., et al. (1992). Purification, sequence, and cellular localization of a novel chromosomal protein that binds to Methylated DNA. Cell 69, 905–914. doi: 10.1016/0092-8674(92)90610-O
Li, C. H., Coffey, E. L., Dall’Agnese, A., Hannett, N. M., Tang, X., Henninger, J. E., et al. (2020). MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature 586, 440–444. doi: 10.1038/s41586-020-2574-4
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett-syndrome. Nature 475, 497–500. doi: 10.1038/nature10214
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. doi: 10.1038/nature08514
Liu, M., Movahed, S., Dangi, S., Pan, H., Kaur, P., Bilinovich, S. M., et al. (2020). DNA looping by two 5-methylcytosine-binding proteins quantified using nanofluidic devices. Epigenet. Chrom. 13:18. doi: 10.1186/s13072-020-00339-7
Lorincz, M. C., Schübeler, D., and Groudine, M. (2001). Methylation-mediated proviral silencing is associated with MeCP2 recruitment and localized Histone H3 deacetylation. Mol. Cell. Biol. 21, 7913–7922. doi: 10.1128/mcb.21.23.7913-7922.2001
Maezawa, I., and Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010
Maezawa, I., Swanberg, S., Harvey, D., LaSalle, J. M., and Jin, L. W. (2009). Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 29, 5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009
Martínez De Paz, A., Khajavi, L., Martin, H., Claveria-Gimeno, R., Tom Dieck, S., Cheema, M. S., et al. (2019). MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenet. Chrom. 12:63. doi: 10.1186/s13072-019-0298-1
Matagne, V., Borloz, E., Ehinger, Y., Saidi, L., Villard, L., and Roux, J.-C. (2020). Severe offtarget effects following intravenous delivery of AAV9-MECP2 in a female mouse model of Rett syndrome. Neurobiol. Dis. 149:105235. doi: 10.1016/j.nbd.2020.105235
Maunakea, A. K., Chepelev, I., Cui, K., and Zhao, K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. doi: 10.1038/cr.2013.110
Maxwell, S. S., Pelka, G. J., Tam, P. P., and El-Osta, A. (2013). Chromatin context and ncRNA highlight targets of MeCP2 in brain. RNA Bio. 10, 1741–1757. doi: 10.4161/rna.26921
McCauley, M. D., Wang, T., Mike, E., Herrera, J., Beavers, D. L., Huang, T. W., et al. (2011). Rett syndrome: pathogenesis of lethal cardiac arrhythmias in Mecp2 mutant mice: implication for therapy in Rett syndrome. Sci. Transl. Med. 3:2982. doi: 10.1126/scitranslmed.3002982
McGraw, C. M., Samaco, R. C., and Zoghbi, H. Y. (2011). Adult neural function requires MeCP2. Science 333:186. doi: 10.1126/science.1206593
McMahon, A. C., Rahman, R., Jin, H., Shen, J. L., Fieldsend, A., Luo, W., et al. (2016). TRIBE: hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell 165, 742–753. doi: 10.1016/j.cell.2016.03.007
Meehan, R., Lewis, J. D., and Bird, A. P. (1992). Characterization of MECP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 20, 5085–5092. doi: 10.1093/nar/20.19.5085
Meguro-Horike, M., Yasui, D. H. D. H., Powell, W., Schroeder, D. I., Oshimura, M., LaSalle, J. M., et al. (2011). Neuron-specific impairment of inter-chromosomal pairing and transcription in a novel model of human 15q-duplication syndrome. Hum. Mol. Genet. 20, 3798–3810. doi: 10.1093/hmg/ddr298
Mellén, M., Ayata, P., Dewell, S., Kriaucionis, S., and Heintz, N. (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430. doi: 10.1016/j.cell.2012.11.022
Millar-Büchner, P., Philp, A. R., Gutierrez, N., Villanueva, S., Kerr, B., and Flores, C. A. (2016). Severe changes in colon epithelium in the Mecp2-null mouse model of Rett syndrome. Mol. Cell. Pediatr. 3:37. doi: 10.1186/s40348-016-0065-3
Mnatzakanian, G. N., Lohi, H., Munteanu, I., Alfred, S. E., Yamada, T., MacLeod, P. J. M., et al. (2004). A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 36, 339–341. doi: 10.1038/ng1327
Muotri, A. R., Marchetto, M. C. N., Coufal, N. G., Oefner, R., Yeo, G., Nakashima, K., et al. (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446. doi: 10.1038/nature09544
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481. doi: 10.1016/S0092-8674(00)81887-5
Nikitina, T., Ghosh, R. P., Horowitz-Scherer, R. A., Hansen, J. C., Grigoryev, S. A., and Woodcock, C. L. (2007). MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 282, 28237–28245. doi: 10.1074/jbc.M704304200
Olson, C. O., Zachariah, R. M., Ezeonwuka, C. D., Liyanage, V. R. B., and Rastegar, M. (2014). Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS One 9:e0090645. doi: 10.1371/journal.pone.0090645
Osenberg, S., Karten, A., Sun, J., Li, J., Charkowick, S., Felice, C. A., et al. (2018). Activity-dependent aberrations in gene expression and alternative splicing in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 115, E5363–E5372. doi: 10.1073/pnas.1722546115
Piccolo, F. M., Liu, Z., Dong, P., Hsu, C. L., Stoyanova, E. I., Rao, A., et al. (2019). MeCP2 nuclear dynamics in live neurons results from low and high affinity chromatin interactions. eLife 8:e051449. doi: 10.7554/eLife.51449
Pohodich, A. E., Yalamanchili, H., Raman, A. T., Wan, Y. W., Gundry, M., Hao, S., et al. (2018). Forniceal deep brain stimulation induces gene expression and splicing changes that promote neurogenesis and plasticity. eLife 7:e034021. doi: 10.7554/eLife.34031
Rajavelu, A., Lungu, C., Emperle, M., Dukatz, M., Bröhm, A., Broche, J., et al. (2018). Chromatin-dependent allosteric regulation of DNMT3A activity by MeCP2. Nucleic Acids Res. 46, 9044–9056. doi: 10.1093/nar/gky715
Raman, A. T., Pohodich, A. E., Wan, Y. W., Yalamanchili, H. K., Lowry, W. E., Zoghbi, H. Y., et al. (2018). Apparent bias toward long gene misregulation in MeCP2 syndromes disappears after controlling for baseline variations. Nat. Commun. 9:3225. doi: 10.1038/s41467-018-05627-1
Reichow, B., George-Puskar, A., Lutz, T., Smith, I. C., and Volkmar, F. R. (2015). Brief report: systematic review of Rett syndrome in males. J. Autism Dev. Disord. 45, 3377–3383. doi: 10.1007/s10803-015-2519-1
Renthal, W., Boxer, L. D., Hrvatin, S., Li, E., Silberfeld, A., Nagy, M. A., et al. (2018). Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci. 21, 1670–1679. doi: 10.1038/s41593-018-0270-6
Ross, P. D., Guy, J., Selfridge, J., Kamal, B., Bahey, N., Elizabeth Tanner, K., et al. (2016). Exclusive expression of MeCP2 in the nervous system distinguishes between brain and peripheral Rett syndrome-like phenotypes. Hum. Mole. Genet. 25, 4389–4404. doi: 10.1093/hmg/ddw269
Rube, H. T. T., Lee, W., Hejna, M., Chen, H., Yasui, D. H. D. H., Hess, J. F. J. F., et al. (2016). Sequence features accurately predict genome-wide MeCP2 binding in vivo. Nat. Commun. 7:11025. doi: 10.1038/ncomms11025
Samaco, R. C., Fryer, J. D., Ren, J., Fyffe, S., Chao, H. T., Sun, Y., et al. (2008). A partial loss of function allele of Methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum. Mol. Genet. 17, 1718–1727. doi: 10.1093/hmg/ddn062
Samaco, R. C., Mandel-Brehm, C., Chao, H. T., Ward, C. S., Fyffe-Maricich, S. L., Ren, J., et al. (2009). Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc. Natl. Acad. Sci. U.S.A. 106, 21966–21971. doi: 10.1073/pnas.0912257106
Saunders, C. J., Minassian, B. E., Chow, E. W. C., Zhao, W., and Vincent, J. B. (2009). Novel exon 1 mutations in MECP2 implicate isoform MeCP2-e1 in classical rett syndrome. Am. J. Med. Genet. Part A 149, 1019–1023. doi: 10.1002/ajmg.a.32776
Schaevitz, L. R., Gómez, N. B., Zhen, D. P., and Berger-Sweeney, J. E. (2013). MeCP2 R168X male and female mutant mice exhibit Rett-like behavioral deficits. Genes Brain Behav. 12, 732–740. doi: 10.1111/gbb.12070
Schafer, D. P., Heller, C. T., Gunner, G., Heller, M., Gordon, C., Hammond, T., et al. (2016). Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. eLife 5:e015224. doi: 10.7554/eLife.15224
Schmidt, A., Zhang, H., and Cardoso, M. C. (2020). MeCP2 and chromatin compartmentalization. Cells 9:878. doi: 10.3390/cells9040878
Sekul, E. A., Moak, J. P., Schultz, R. J., Glaze, D. G., Dunn, J. K., and Percy, A. K. (1994). Electrocardiographic findings in Rett syndrome: an explanation for sudden death? J. Pediatr. 125, 80–82. doi: 10.1016/S0022-3476(94)70128-8
Sinnamon, J. R., Kim, S. Y., Corson, G. M., Song, Z., Nakai, H., Adelman, J. P., et al. (2017). Site-directed RNA repair of endogenous Mecp2 RNA in neurons. Proc. Natl. Acad. Sci. U.S.A. 114, E9395–E9402. doi: 10.1073/pnas.1715320114
Sinnamon, J. R., Kim, S. Y., Fisk, J. R., Song, Z., Nakai, H., Jeng, S., et al. (2020). In vivo repair of a protein underlying a neurological disorder by programmable RNA editing. Cell Rep. 32:107878. doi: 10.1016/j.celrep.2020.107878
Sinnett, S. E., Hector, R. D., Gadalla, K. K. E., Heindel, C., Chen, D., Zaric, V., et al. (2017). Improved MECP2 gene therapy extends the survival of MeCP2-null mice without apparent toxicity after intracisternal delivery. Mol. Therapy Methods Clin. Dev. 5, 106–115. doi: 10.1016/j.omtm.2017.04.006
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R. W., James, K. D., Turner, D. J., et al. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37, 457–468. doi: 10.1016/j.molcel.2010.01.030
Strati, F., Cavalieri, D., Albanese, D., De Felice, C., Donati, C., Hayek, J., et al. (2016). Altered gut microbiota in Rett syndrome. Microbiome 4:41. doi: 10.1186/s40168-016-0185-y
Stuss, D. P., Cheema, M., Ng, M. K., Martinez De Paz, A., Williamson, B., Missiaen, K., et al. (2013). Impaired in vivo binding of MeCP2 to chromatin in the absence of its DNA methyl-binding domain. Nucleic Acids Res. 41, 4888–4900. doi: 10.1093/nar/gkt213
Sugino, K., Hempel, C. M., Okaty, B. W., Arnson, H. A., Kato, S., Dani, V. S., et al. (2014). Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J. Neurosci. 34, 12877–12883. doi: 10.1523/JNEUROSCI.2674-14.2014
Symons, F. J., Byiers, B., Tervo, R. C., and Beisang, A. (2013). Parent-reported pain in Rett syndrome. Clin. J. Pain 29, 744–746. doi: 10.1097/AJP.0b013e318274b6bd
Szulwach, K. E., Li, X., Li, Y., Song, C. X., Wu, H., Dai, Q., et al. (2011). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616. doi: 10.1038/nn.2959
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Trappe, R., Laccone, F., Cobilanschi, J., Meins, M., Huppke, P., Hanefeld, F., et al. (2001). MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 68, 1093–1101. doi: 10.1086/320109
Valinluck, V., Liu, P., Kang, J. I., Burdzy, A., and Sowers, L. C. (2005). 5-Halogenated pyrimidine lesions within a CpG sequence context mimic 5-methylcytosine by enhancing the binding of the methyl-CpG-binding domain of methyl-CpG-binding protein 2 (MeCP2). Nucleic Acids Res. 33, 3057–3064. doi: 10.1093/nar/gki612
Valinluck, V., Tsai, H. H., Rogstad, D. K., Burdzy, A., Bird, A., and Sowers, L. C. (2004). Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 32, 4100–4108. doi: 10.1093/nar/gkh739
Veeraragavan, S., Wan, Y.-W., Connolly, D. R., Hamilton, S. M., Ward, C. S., Soriano, S., et al. (2015). Loss of MeCP2 in the rat models regression, impaired sociability and transcriptional deficits of Rett syndrome. Hum. Mol. Genet. 25:ddw178. doi: 10.1093/hmg/ddw178
Vogel Ciernia, A., Pride, M. C., Durbin-Johnson, B., Noronha, A., Chang, A., Yasui, D. H. D. H., et al. (2017). Early motor phenotype detection in a female mouse model of Rett syndrome is improved by cross-fostering. Hum. Mol. Genet. 26, 1839–1854. doi: 10.1093/hmg/ddx087
Vogel Ciernia, A., Yasui, D. H., Pride, M. C., Durbin-Johnson, B., Noronha, A. B., Chang, A., et al. (2018). MeCP2 isoform e1 mutant mice recapitulate motor and metabolic phenotypes of Rett syndrome. Hum. Mol. Genet. 27, 4077–4093. doi: 10.1093/hmg/ddy301
Wahba, G., Schock, S. C., Claridge, E., Bettolli, M., Grynspan, D., Humphreys, P., et al. (2015). MeCP2 in the enteric nervous system. Neurogastroenterol. Motil. 27, 1156–1161. doi: 10.1111/nmo.12605
Wang, L., Hu, M., Zuo, M. Q., Zhao, J., Wu, D., Huang, L., et al. (2020). Rett syndrome-causing mutations compromise MeCP2-mediated liquid-liquid phase separation of chromatin. Cell Res. 30, 393–407. doi: 10.1038/s41422-020-0288-7
Wasag, P., and Lenartowski, R. (2016). Nuclear matrix - structure, function and pathogenesis. Postepy Hog. Med. Dosw. 70, 1206–1219.
Weitzel, J. M., Buhrmester, H., and Strätling, W. H. (1997). Chicken MAR-binding protein ARBP is homologous to rat methyl-CpG-binding protein MeCP2. Mol. Cell. Biol. 17, 5656–5666. doi: 10.1128/mcb.17.9.5656
Wong, J. J. L., Gao, D., Nguyen, T. V., Kwok, C. T., Van Geldermalsen, M., Middleton, R., et al. (2017). Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat. Commun. 8:134. doi: 10.1038/ncomms15134
Yasui, D. H., Gonzales, M. L., Aflatooni, J. O., Crary, F. K., Hu, D. J., Gavino, B. J., et al. (2014). Mice with an isoform-ablating Mecp2exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum. Mole. Genet. 23, 2447–2458. doi: 10.1093/hmg/ddt640
Yasui, D. H., Peddada, S., Bieda, M. C., Vallero, R. O., Hogart, A., Nagarajan, R. P., et al. (2007). Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. U.S.A. 104, 9416–9421. doi: 10.1073/pnas.0707442104
Yasui, D. H., Scoles, H. A., Horike, S.-I., Meguro-Horike, M., Dunaway, K. W., Schroeder, D. I., et al. (2011). 15q11.2-13.3 chromatin analysis reveals epigenetic regulation of CHRNA7 with deficiencies in Rett and autism brain. Hum. Mol. Genet. 20, 4311–4323. doi: 10.1093/hmg/ddr357
Young, J. I., Hong, E. P., Castle, J. C., Crespo-Barreto, J., Bowman, A. B., Rose, M. F., et al. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U.S.A. 102, 17551–17558. doi: 10.1073/pnas.0507856102
Yu, F., Zingler, N., Schumann, G., and Strätling, W. H. (2001). Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res. 29, 4493–4501. doi: 10.1093/nar/29.21.4493
Zachariah, R. M., Olson, C. O., Ezeonwuka, C., and Rastegar, M. (2012). Novel MeCP2 isoform-specific antibody reveals the endogenous MeCP2E1 expression in murine brain, primary neurons and Astrocytes. PLoS One 7:e0049763. doi: 10.1371/journal.pone.0049763
Zhang, P., Ludwig, A. K., Hastert, F. D., Rausch, C., Lehmkuhl, A., Hellmann, I., et al. (2017). L1 retrotransposition is activated by Ten-eleven-translocation protein 1 and repressed by methyl-CpG binding proteins. Nucleus 8, 548–562.
Zhao, B., Wu, Q., Ye, A. Y., Guo, J., Zheng, X., Yang, X., et al. (2019). Somatic LINE-1 retrotransposition in cortical neurons and non-brain tissues of Rett patients and healthy individuals. PLoS Genet. 15:1008043. doi: 10.1371/journal.pgen.1008043
Zoghbi, H. Y. (2005). MeCP2 dysfunction in humans and mice. J. Child Neurol. 20, 736–740. doi: 10.1177/08830738050200090701
Keywords: Rett syndrome, epigenetic, DNA, chromatin, gene expression, MeCP2
Citation: Sharifi O and Yasui DH (2021) The Molecular Functions of MeCP2 in Rett Syndrome Pathology. Front. Genet. 12:624290. doi: 10.3389/fgene.2021.624290
Received: 31 October 2020; Accepted: 08 February 2021;
Published: 23 April 2021.
Edited by:
Andrei V. Chernov, University of California, San Diego, United StatesReviewed by:
Nicoletta Landsberger, University of Milan, ItalyWei Li, University of Alabama at Birmingham, United States
Alessandra Pecorelli, North Carolina State University, United States
Copyright © 2021 Sharifi and Yasui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dag H. Yasui, ZGh5YXN1aUB1Y2RhdmlzLmVkdQ==