 Rakesh Kumar1*†
Rakesh Kumar1*† Vinay Sharma2†
Vinay Sharma2† Srinivas Suresh1
Srinivas Suresh1 Devade Pandurang Ramrao1
Devade Pandurang Ramrao1 Akash Veershetty1
Akash Veershetty1 Sharan Kumar1
Sharan Kumar1 Kagolla Priscilla1BhagyaShree Hangargi1Rahul Narasanna1
Kagolla Priscilla1BhagyaShree Hangargi1Rahul Narasanna1 Manish Kumar Pandey2Gajanana Ramachandra Naik1Sherinmol Thomas3
Manish Kumar Pandey2Gajanana Ramachandra Naik1Sherinmol Thomas3 Anirudh Kumar4*
Anirudh Kumar4*- 1Department of Life Science, Central University of Karnataka, Kalaburagi, India
- 2International Crops Research Institute for the Semi-Arid Tropics, Hyderabad, India
- 3Department of Biosciences & Bioengineering, Indian Institute of Technology Bombay, Mumbai, India
- 4Department of Botany, Indira Gandhi National Tribal University, Amarkantak, India
In the current era, one of biggest challenges is to shorten the breeding cycle for rapid generation of a new crop variety having high yield capacity, disease resistance, high nutrient content, etc. Advances in the “-omics” technology have revolutionized the discovery of genes and bio-molecules with remarkable precision, resulting in significant development of plant-focused metabolic databases and resources. Metabolomics has been widely used in several model plants and crop species to examine metabolic drift and changes in metabolic composition during various developmental stages and in response to stimuli. Over the last few decades, these efforts have resulted in a significantly improved understanding of the metabolic pathways of plants through identification of several unknown intermediates. This has assisted in developing several new metabolically engineered important crops with desirable agronomic traits, and has facilitated the de novo domestication of new crops for sustainable agriculture and food security. In this review, we discuss how “omics” technologies, particularly metabolomics, has enhanced our understanding of important traits and allowed speedy domestication of novel crop plants.
Introduction
The process of crop domestication began approximately 12,000 years ago, and was an important milestone during human civilization and led the foundation of modern agriculture. In the 21st century, most of the cultivated crops are domesticated from their wild ancestral progenitors (Meyer et al., 2012). During the domestication process plants were selected based on visible traits guided by needs of the time which led to the selection of only few desired alleles and dilution of the genetic variation present within the crop (Figure 1). For example, in cereals like wheat and rice, traits such as increase in the number of seeds per plant, uniform seed maturation, and non-shattering of seeds were preferred over the size of kernels during early domestication (Si et al., 2016). However, the selection of such traits varies greatly from plant to plant or between crops. For instance, in fleshy fruits or berries such as tomato, eggplant and apple, the size of the fruits and berries were preferred over overall yield (Zhu et al., 2018). Likewise, in tuber producing plants such as potato the tuber size is one of the preferred traits (Fernie and Yan, 2019). Surprisingly, cultivated plant species represent only 250 fully domesticated species among 2500 species, which have undergone the process of domestication, and represent 160 plant families (Smýkal et al., 2018). This proportion is even starker considering the total plant diversity available for the cultivation or those, which are already being cultivated in different places (semi-cultivated species). For example, around 400,000 semi-cultivated plant species are currently known which can be utilized for designing future crops (Smýkal et al., 2018; Fernie and Yan, 2019).
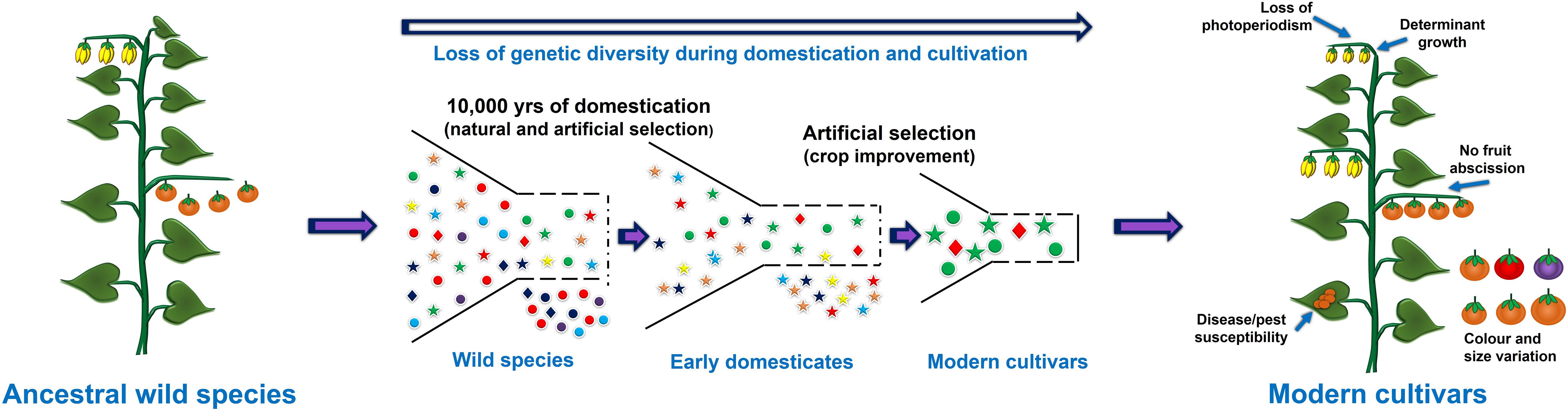
Figure 1. Representation of domestication process and the loss of useful genetic variation due to selective breeding and selection of few alleles.
The process of domestication of a species is a very slow and steady process. In fact, the modern cultivars available were generated following a long series of events: (a) Neolithic Revolution, (b) Columbian Exchange, (c) Industrial Revolution, (d) Green Revolution, and (e) Genomic Revolutions (Smýkal et al., 2018). Presently, to feed an ever-growing global population in the face of climate change is challenge for agriculture especially due to reduction of the arable lands due consistent conversion of lands into semi-arid areas and nutrient deficient land along with increase in soil pH or salinity. Therefore, a more rapid method of developing elite climate smart cultivars with desired traits is required. This could be achieved through integrated OMICS approaches, which include genomics, transcriptomics, proteomics, metabolomics and phenomics integrated with bioinformatics analyses (Kumar et al., 2017, 2018; Sharma et al., 2021). Plant OMICs based research have played very important role in deciphering metabolic pathways and their molecular regulators, which govern key traits and several plant developmental processes (Kumar et al., 2017; Razzaq et al., 2019). In the past decade there has been a significant progress in the field of both sequencing (van Dijk et al., 2018; Kumar et al., 2020; Schmidt et al., 2020) and analytical methods for the detection of molecules (Ren et al., 2018; Gilmore et al., 2019; Macklin et al., 2020), which has not only improved detection throughput but also the accuracy and the sensitivity (Kumar et al., 2017; Chiang et al., 2018; Qi et al., 2019).
In the past, for the selection of traits breeding programs involved phenotypic selection of plants (which are guided by metabolic pathways) (Kiszonas and Morris, 2018). For instance, during the Green Revolution (from 1960 to 1980), development of semi-dwarf high yielding varieties of rice and wheat involved phenotypic selections of improved lines which actually involved selection of gibberellic acid pathway genes including the GA20 oxidase and DELLA protein encoding genes (Silverstone and Sun, 2000). In fact, most of the traits, which were targeted for the Green Revolution, are controlled by one or more metabolic pathways. Therefore, precise editing of these metabolic pathways can help in the development of varieties in a very short time (Rodríguez-Leal et al., 2017; Zhang Y. et al., 2018; Fernie and Yan, 2019). Previously, most of the reviews on plant omics have focused on the instrumentation involved and results obtained by different researchers (Kumar et al., 2017; Mangul et al., 2019; Misra et al., 2019; Tang and Aristilde, 2020). In this review, we represent how this omics knowledge can be utilized for development of improved cultivars by targeting metabolic pathways and also emphasize the use of this information for de novo domestication of wild ancestral species for sustainable food security.
Role of Omics Data in Understanding Plant Traits
Genomics plays an important role in the identification of quantitative trait loci (QTLs) and genes controlling important traits, particularly in domesticated crops (Fernie and Yan, 2019). Moving forward, great insights have been gleaned from genome sequencing and re-sequencing programs examining wild ancestral species of domesticated crops (Unamba et al., 2015). In plant genomics, Next Generation Sequencing (NGS) has played a very important role and provided opportunities in the field of functional genomics due to the availability of reference genomes for several model and crop plant species. These resources combined with high quality re-sequencing offers huge potential for discovery of causal genes and mechanisms associated with the key agronomic traits (Thudi et al., 2016; Chen et al., 2019; Varshney et al., 2019). Re-sequencing also enriched the availability of SNPs data and can be utilized for genomics-based studies such as GWAS (genome wide association study) and QTL-seq (Kumar et al., 2020), both of which are useful tools for trait mapping (Rivas et al., 2011; Zhu et al., 2011; Zhang et al., 2021). With the advent of these technologies combined with advances in metabolomics such as integration of GWAS with metabolomics efficient means for dissecting underlying molecular mechanisms involved in the growth and development are available (Table 1; Fang and Luo, 2019).
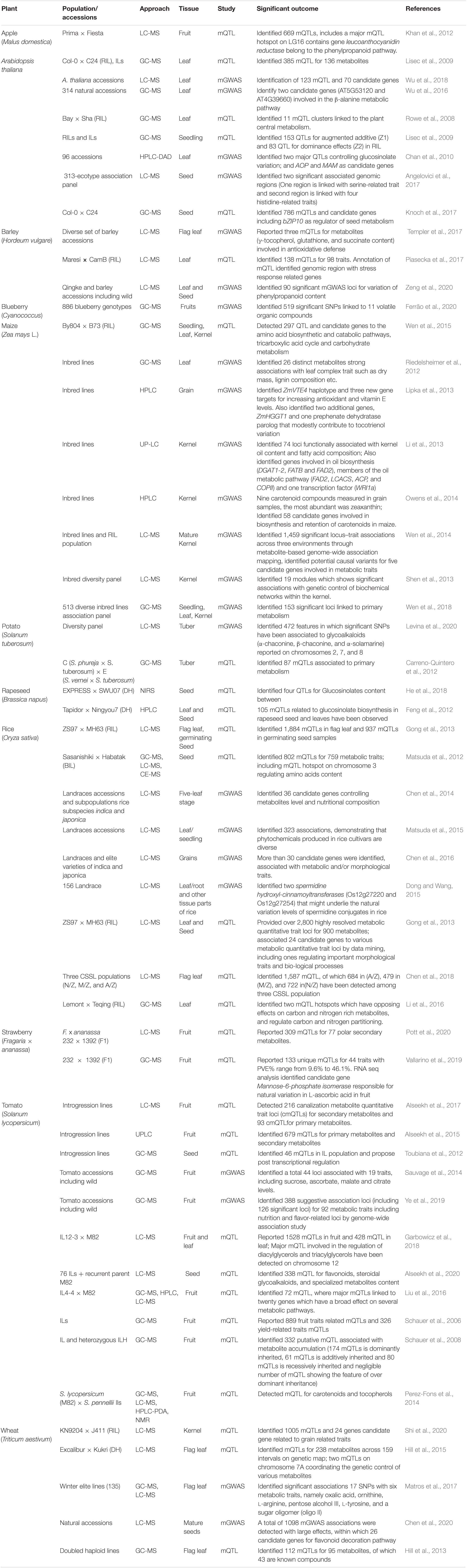
Table 1. List of selected studies involved mQTL and mGWAS approach.
Sequencing and QTL-seq Based Trait Discovery
Presently, QTL-seq is one of the most successful approach developed for the gene discovery and trait dissection (Kumar et al., 2020; Pandey et al., 2020). This approach offers preliminary idea for positional cloning for linked genetic factors or genes, and it has excellent success in marker-assisted selection for crop improvement programs (Xu F. et al., 2015). With the advancements in NGS technologies new approaches like quantitative trait locus sequencing (QTL-seq) has been utilized for exploring rapid QTL or gene identification (Takagi et al., 2013). In QTL-seq approach, the extreme highest and lowest genotypes are selected from the mapping population for target traits, followed by mixing an equal amount of DNA from each bulk to build up two extreme bulk (High bulk and low bulk). Then, each bulk is sequenced and assembled and gene annotation is performed. This approach combined with Bulked segregant analysis, accompanied by whole genome re-sequencing technologies, is more effective and capable than the previous QTL detection methods (Takagi et al., 2013). Utilizing QTL-seq approach several QTLs and genes for different rice phenotypes (Takagi et al., 2013; Daware et al., 2016; Ogiso-Tanaka et al., 2017; Yang et al., 2017; Kadambari et al., 2018; Qin et al., 2018; Bommisetty et al., 2020; Nubankoh et al., 2020), soybean (Song et al., 2017; Zhang X. et al., 2018), chickpea (Singh et al., 2016; Deokar et al., 2019), tomato (Illa-Berenguer et al., 2015), groundnut (Kumar et al., 2020; Luo et al., 2019; Zhao et al., 2020), have been effectively identified. This approach has also been deployed across in several crops due to its inherent ability to understand both qualitative and quantitative traits (Table 2). For instance, Kumar et al. (2020) identified the role of two genes RING-H2 finger protein and zeaxanthin epoxidase in fresh seed dormancy in groundnut; both genes are known to control abscisic acid (ABA) accumulation. Very recently, Ramos et al. (2020) identified three QTLs (genomic regions) viz QtlPC-C04, QtlPC-C11 and QtlPC-C14 (linked to genes R1R2R3) associated with resistance to Phytophthora capsici Leonian which causes crown rot in squash (Cucurbita moschata). The most significant benefit of whole genome sequencing is that it allows the identification of causative mutations in target chromosomal regions. Additionally, this method identifies molecular markers which are located inside the harboring chromosomal region that can also be used to narrow down the genomic region which will help in mining the trait linked genes.
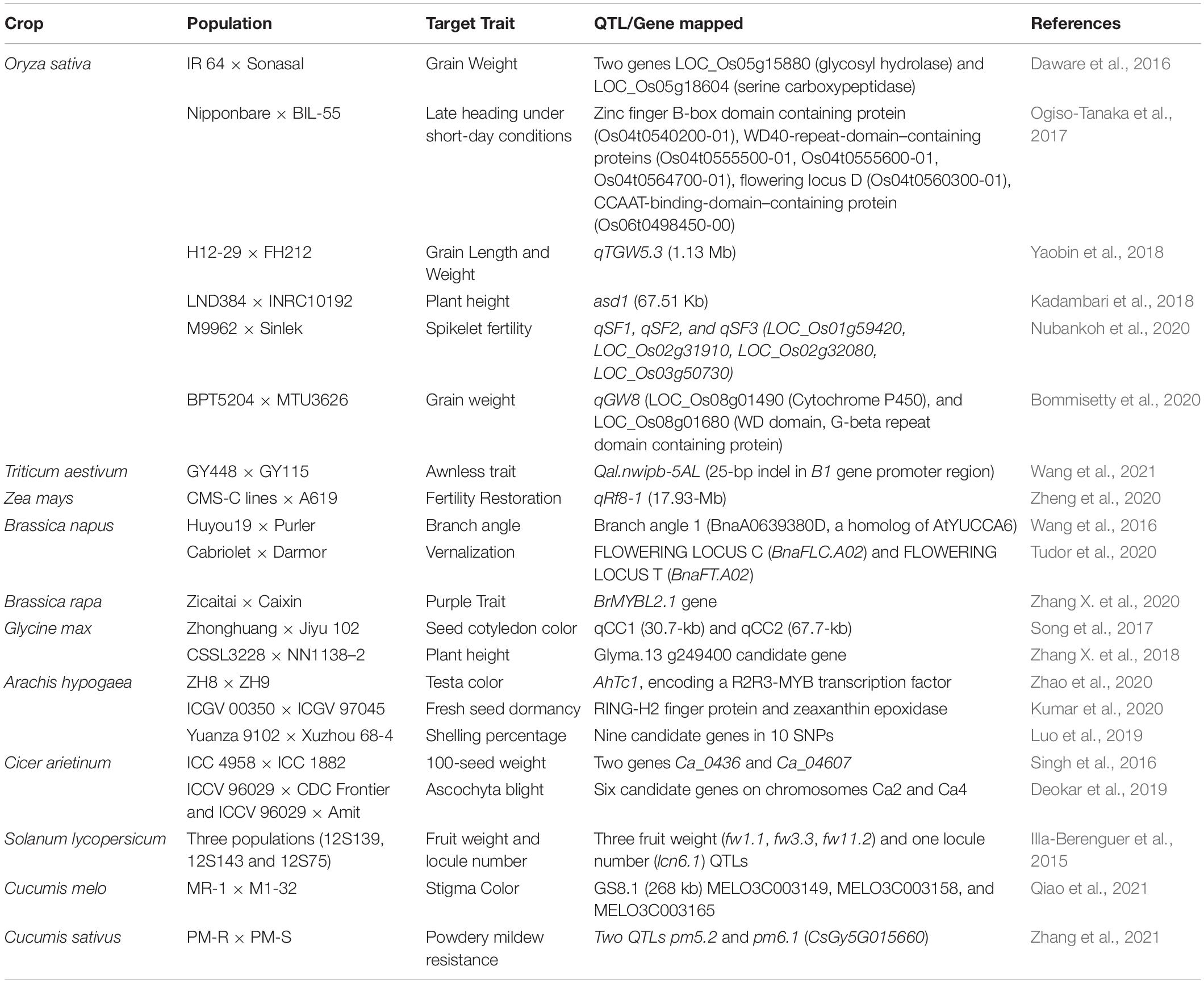
Table 2. List of important QTL-seq studies in crop plants.
RNA-seq Based Trait Discovery
Advances in RNA sequencing (RNA-seq) technologies and approaches have made significant impact toward trait discovery, and have enabled plant developmental studies characterizing expression patterns of all the functional genes as well as regulatory RNAs (Nayak et al., 2019). RNA-seq is a more robust approach for precise measurement of transcripts and has been widely used for transcript profiling in several plant species (Cloonan et al., 2008; Wang et al., 2009). This data is vital for improving genome annotations, and offers precise gene position information for functional characterization and genome editing. The RNA-seq approach has been deployed for molecular characterization of several important agronomic traits such as seed size (Wan et al., 2017), seed coat color (Wan et al., 2018), seed coat cracking (Wan et al., 2016), seed and bud dormancy (Qi et al., 2015; Zhu et al., 2015; Khalil-Ur-Rehman et al., 2017), fatty acid biosynthesis and oil quality (Nayak et al., 2019), nutritional quality traits (Reddy and Ulaganathan, 2015), etc., which can offer precise gene information for developing designer crops for future. Also, RNA-seq have been used to understand the molecular mechanisms associated with salt tolerance in rice (Zhou et al., 2016; Lei et al., 2020); Chinese rye grass (Sun et al., 2013), and maize (Liang and Schnable, 2016). In wheat, RNA-seq study reported the drought responsive molecular pathways along with key candidate genes and molecular markers in the root tissue (Iquebal et al., 2019). RNA-seq has also been shown to be highly useful in combination with other -omics methods for gene discovery and pathway investigations.
Proteomics Enabled Genetic Trait Understanding
Knowledge of proteomics is being used to map the translated genes and loci controlling the expression of respective genes. It helps in identification of proteins responsible for bringing intricate phenotypic variations. High throughput proteomics has gone beyond the identification of individual proteins, to quantitative profiling, post translational modification studies, signaling, protein–protein interaction and many more areas. Photosynthesis plays major role in biomass production and yield. Change in protein profile studies was performed in chlorophyll deficient Brassica napus leaves which established the relationship between chlorophyll biosynthesis and photosynthesis (Chu et al., 2015). Xylem sap proteomics has revealed several insights related to cell wall formation (Zhang M. et al., 2014), leaf senescence (Wang et al., 2012) biotic and abiotic stress response (Alvarez et al., 2008; González et al., 2012), cell to cell communication (Agrawal et al., 2010), and root–shoot communication (Krishnan et al., 2011). The enhanced level of redox proteins, stress and defense related proteins, calcium ion regulation proteins, signaling G-protein and RNA metabolism related proteins were induced in phloem sap study. Recently, proteomics study revealed that low light stress obstructs carbon fixation and OsGAPB overexpression augment tolerance to low light stress conceivably by increasing CO2 assimilation and chlorophyll accumulation in rice (Liu et al., 2020). Interestingly, simultaneous upregulation of both biotic and abiotic stress responsive protein has been observed during bacterial blight infection in rice, which indicate the activation of common pathway (Kumar et al., 2015). Whereas in case of rice-M. oryzae interaction PBZ1, OsPR-10, SalT, Glu1, Glu2, and TLP proteins were up-regulated (Kim et al., 2004). iTRAQ proteomics study of Oryza officinalis provided evidences that proteins related to biosynthesis of secondary metabolites and carbon metabolism were mostly enriched after planthopper infestation (Zhang et al., 2019c). Several proteomics and transcriptomics study conducted on seed dormancy study revealed the important role of antioxidant mechanism, protein thiol and redox regulation along with hormonal signaling in rice, wheat and barley (Hu et al., 2015). Mass spectrometry (MS) based proteomics study demonstrated the cultivar specific induction of proteins in salt stress condition such as glutathione-based detoxification of ROS was highly induced in tolerant variety whereas proteins involved in iron uptakes were more expressed in salt sensitive variety in barley (Witzel et al., 2009). Similarly, the role of OsCYP2 in moderating the antioxidant enzymes was established in transgenic rice overexpressing cyclophilin during salt stress (Ruan et al., 2011). Seed proteomics of various developmental stages during different stresses have revealed the process involved in seed dormancy, seed germination, and seed development (Finnie et al., 2011). Proteomics related to environmental changes and abiotic stress focused on water supply responsive proteins in wheat against drought, high temperature, low temperature, frost, salt and heavy metals have been carried out (Yang et al., 2011; Han et al., 2013; Kosová et al., 2013; Alvarez et al., 2014; Capriotti et al., 2014; Kang et al., 2015). These studies offered novel insights and better understanding of crop physiology and metabolism during various kinds of stresses.
Metabolomics Based Trait Understanding
Holistic metabolomics based to study trails in plants were started late, particularly this approach was started through the introduction of untargeted metabolome detection (Alonso et al., 2015). Several studies have been reported where a particular metabolic pathways have been mapped (Sharma et al., 2021). For instance, the substantial changes in the various phytohormones was investigated in poplar leaf (Novák et al., 2008), rice various aerial organs (Kojima et al., 2009), rosemary leaves et al. (Müller and Munné-Bosch, 2011), Arabidopsis developing seeds (Kanno et al., 2010), strawberry fruits (Gu et al., 2019), potato tuber (Peivastegan et al., 2019), wheat developing seeds (Matsuura et al., 2019), watermelon fruit (Kojima et al., 2021), etc. The targeted approach has been also applied to explore the carotenoid pathway (Fernandez-Orozco et al., 2013; Kim et al., 2016; Mibei et al., 2017; Yoo et al., 2017; Price et al., 2018; Di Lena et al., 2019), flavonoid pathways (Karimi et al., 2011; Dong X. et al., 2014; Torres et al., 2019), amino acids (Torres et al., 2019; Praveen et al., 2020), and fatty acids (Talebi et al., 2013; Vidigal et al., 2018). Such profiling studies has helped in improving several important traits in plants by targeting specific pathways. Almost 10 years back Liu et al. (2011) targeted fatty acids biosynthesis pathways for enhancing biofuel production. Very recently and fatty acid desaturase 2 was targeted in several crops such as canola (Okuzaki et al., 2018), peanut (Yuan et al., 2019), rice (Abe et al., 2018), false flax (Morineau et al., 2017), and Soybean (Wu et al., 2020), for enhanced production of oleic acid (C18:1), respectively.
Several un-targeted metabolomics approach has been utilized to target multiple class of metabolites (amines, sugars, organic acids, etc.) from a sample extracted from various tissues of the model and crop plants such as Arabidopsis, apple, groundnut, kiwi fruit, alpine bird’s-foot-trefoil, strawberry, grapes, mango, maize, medicago, orange, pear, sunflower, soybean, tomato, rice, white lupin, etc. (Sharma et al., 2021). Now, the targeted and un-targeted metabolomics approach have been coupled with genomics data for carrying out metabolomics-based quantitative trait locus (mQTL) and metabolic genome-wide association studies (mGWAS) studies (Wen et al., 2015; Chen et al., 2016); which simultaneously identifies the genomic region, causal genes and key metabolites and associated metabolic pathways that govern particular trait in plants. Recently, Li K. et al. (2019) identified 65 primary metabolites viz 22 amino acids, 21 organic acids, 12 sugars, four amines and six miscellaneous metabolites in the leaf of teosinte (an ancestor of maize) and identifies advantageous genes present in the wild relative associated with grain yield and shape trait in maize. In tomato, for one of the important trait accumulation of secondary metabolite in fruit was analyzed, and reported several subset of mQTLs- including mQTLs associated with acyl-sugar, hydroxycinnamates, naringenin chalcone, and a range of glycoalkaloids (Alseekh et al., 2015). Likewise, there are several studies which identified key genomic regions, candidate genes and mQTLs related to important traits through mQTL and mGWAS based studies including some domesticated traits, this was extensively reviewed by Sharma et al. (2021).
Previously, a combined transcriptome, proteome and metabolomics approach was used to investigate the ripening process with a final aim of extending tomato fruit shelf life (Osorio et al., 2011). This study showed a strong relationship between metabolites and their associated transcripts controlling ripening such as sugars, organic acids, and cell wall metabolism pathways. Similar studies have been done for banana which led to identification of genes including ERF1B, fructose-1,6-bisphosphatase and polygalacturonase as key regulators of pulp ripening (Li T. et al., 2019). Recently, a combined transcriptome and metabolome study was deployed to study the molecular aspects of resistance and the interaction between Trichoderma harzianum strain T22 with tomato during defense responses against aphids (Coppola et al., 2019). This study demonstrated the importance of plant transcription factor families such as ZIP, MYB, NAC, AP2-ERF, and WRKY in biotic stress resistance. These examples show the potential of the -omics studies, working in tandem to characterize complex molecular interactions. These data have been used for the development of several gene expression/proteome/metabolome atlases to facilitate omics-driven crop improvement (Table 3).
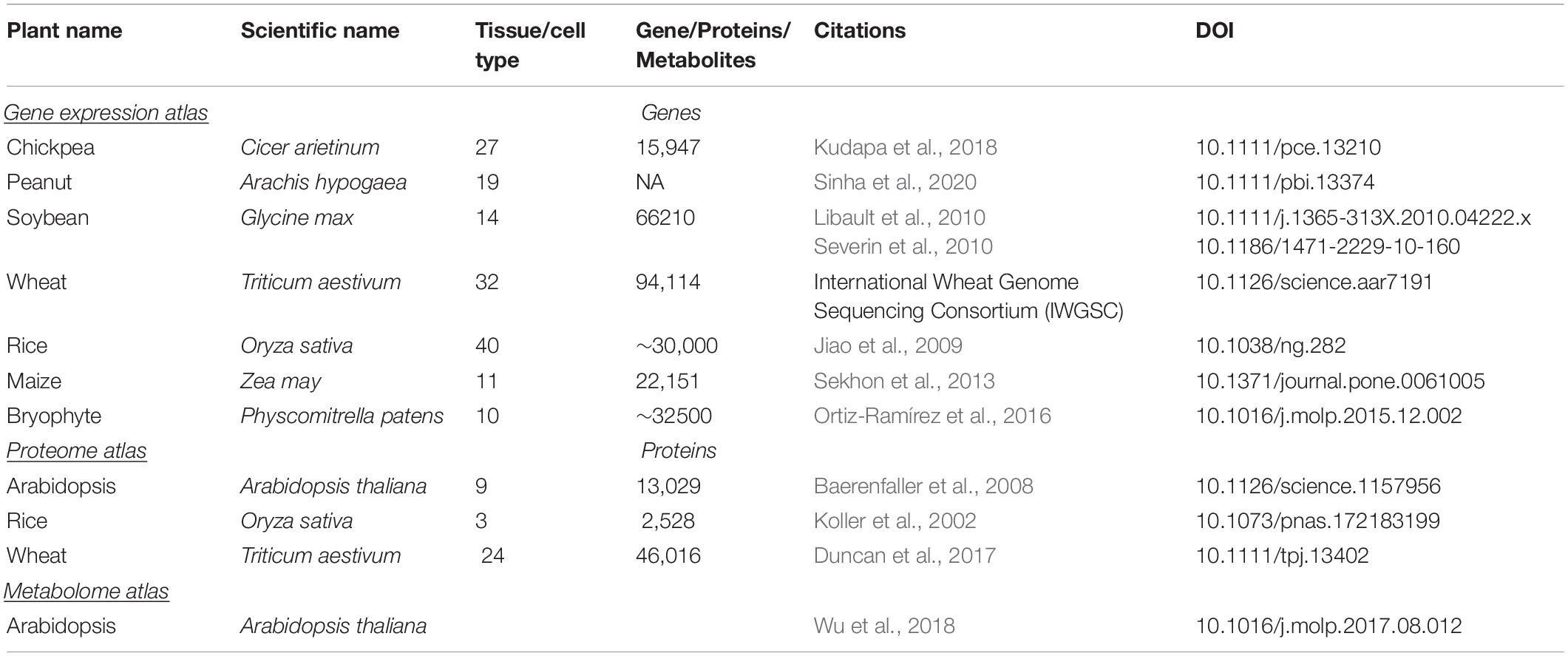
Table 3. List of gene-expression, proteome and metabolome atlas developed in plant.
Molecular Regulations of Domestication Related Traits: Selected Examples
Over the past two decades the molecular regulation and the associated metabolic pathways of several agronomic traits has been revealed because of intensive research and the deployment of omics tools (Table 4). For the major domesticated traits their associated genes pathways have been linked with metabolic networks; however, more focused research is required to understand their specific role in particular metabolic pathways. Here, we review progress in omics-based investigations of several important domestications related traits.
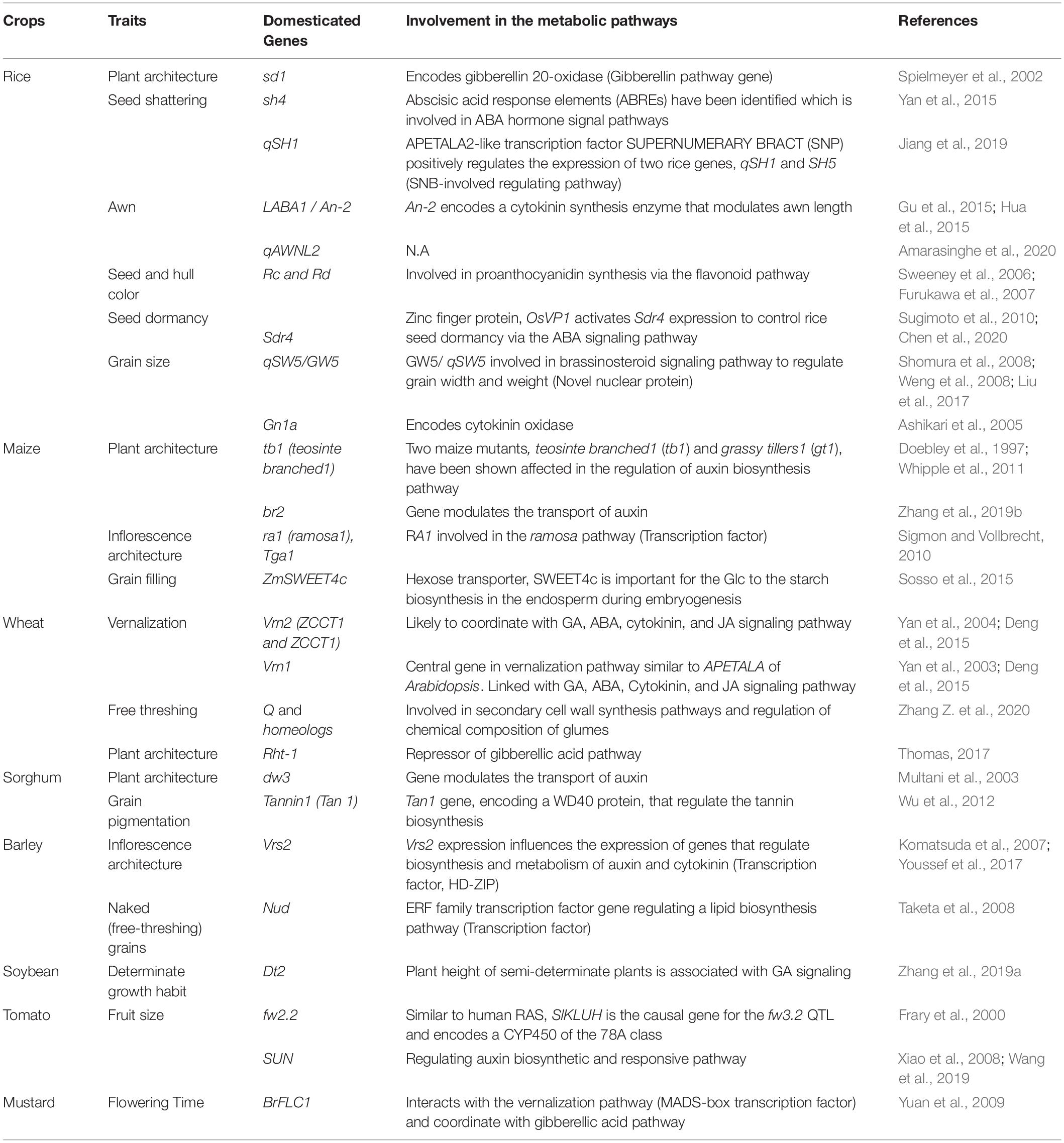
Table 4. List of genes domesticated in the past and associated metabolic pathways.
Transcriptional Control for Loss of Seed Shattering Trait in Cereal
From an evolutionary viewpoint, natural selection allows wild plant species to have specific functions to disperse seeds and fruits. Although from the agronomic viewpoint, natural seed dispersal is an undesirable trait in crops as it leads to significant seed loss in harvest. Consequently, natural seed dispersal was strongly chosen against by ancient humans to ensure productive cultivation during the domestication period (Purugganan and Fuller, 2009; Lenser and Theißen, 2013). The non-shattering traits were considered as the landmark of domestication in seed crops, as it makes the domesticated species mostly rely on human activity for propagation and enables the fixation of other domestication traits (Purugganan and Fuller, 2009). Seed crops have established their reduction of seed shattering ability independently and it is a convergent morphological adaptation to artificial selection (Purugganan and Fuller, 2009; Olsen and Wendel, 2013).
In cereal, seed shattering or fruit dehiscence is enacted through an abscission layer in the lemma-pedicel joint. Various transcription factors (TFs) coding genes were found in rice (Oryza sativa), which are involved in decreasing seed shattering. Shattering4 (Sh4) encodes the TF with Myb3 homology and is important for the formation of a functional abscission layer in the pedicle (Li et al., 2006). A single change of amino acid in DNA binding domain of Sh4 is intimately linked to the reduced seed shattering in domesticated rice. Also, the expression of the domesticated allele has been substantially reduced compared to the wild allele (Li et al., 2006). Thus, the combination of coding and regulatory alteration of Sh4 seems to affect the formation of the abscission layer, and consequently tries to weaken the shattering phenotype (Li et al., 2006). qSH1 is a major QTL on chromosome 1 involved in seed shattering in rice. The main gene, qSH1, codes a homeobox transcription factor-like BEL1 which is homologous to AtRPL (Konishi et al., 2006). A single nucleotide polymorphism (SNP) in the 5′-regulatory region effectively nullifies qSH1 expression in the preliminary abscission layer in the early development stage and contributes to non-shattering traits of rice (Konishi et al., 2006). Interestingly, the regulatory SNP in the homologs of RPL promoter are also amenable for distinct structures of seed dispersal based on natural selection of Brassica species with diminished replum development (Arnaud et al., 2011). These studies show a notable convergent mechanism where the same regulatory SNP could describe developmental variations in seed dispersal structures, which are important for both domestication and natural selection in distant species (Arnaud et al., 2011; Gasser and Simon, 2011). SH5 is another homeobox type BEL1 gene with a high qSH1 homology. SH5 has been expressed in the abscission layer (Yoon et al., 2014). Knockout of SH5 inhibits abscission layer formation and prevents seed shattering. Over-expression of SH5 leads to higher seed shattering, a consequence of decreased pedicel lignin levels (Yoon et al., 2014). The regulatory pathway of abscission layer formation has recently been expanded to include Shattering abortion 1 (SHAT1), an AP2 transcription factor encoding gene (Zhou et al., 2012). SHAT1 is needed for seed shattering by specifying abscission layer. Sh4 positively regulates the SHAT 1 expression in the abscission layer. qSH1 expression is lost in abscission layer in both the shat1 and sh4 mutant background, indicating qSH1 acts downstream of the shat1 and sh4 in the abscission layer establishment (Zhou et al., 2012). Intriguingly, qSH 1 is also needed in the abscission layer for expression of SH1 and Sh4. Thus the qSH 1 possibly takes part in a positive feedback loop of SH1 and Sh4 by establishing the SHAT1 and Sh4 expression in the abscission layer (Zhou et al., 2012). While SH5 and SHAT1 play a role in differentiating the abscission layer, the question remains whether both are artificially selected domestication genes. Like rice, decrease of seed shattering in domesticated sorghum is a result of loss of abscission in the joint that connects the seed hull with the pedicel. In sorghum, seed shattering is regulated by a single gene, Shattering1 (Sh1), which encodes a transcription factor YABBY. The non-shattering trait can be accounted for by any one of the three different loss-of-function mutations selected independently during sorghum domestication process (Lin et al., 2012). The notable mutations in Sh1 orthologs in rice and maize may be related to the shattering decrease in these crops (Lin et al., 2012). Whether Sh1 has been rewired into an SH5-directed seed shattering network in rice remains to be investigated in the future. In a wild relative of sorghum (Sorghum propinquum), seed shattering is conferred by the SpWRKY gene. It is believed that SpWRKY controls cell wall biosynthesis genes negatively in the abscission layer. Even so, SpWRKY was not crafted by artificial selection to contribute to the non-shattering characteristic for domesticated sorghum (Tang et al., 2013). These above studies together have raised a fascinating potential that the convergent domestication of non-shattering crops may have achieved the same underlying genetic goals by parallel selection (Lenser and Theißen, 2013).
In domesticated wheat (Triticum aestivum) free-threshing trait (loss of spike shattering tendency) is conferred by important Q gene (Simons et al., 2006). Q-gene encodes the AP2-family transcription factor. The domesticated Q allele is abundantly transcribed than the wild q allele. Besides, both alleles differ in single amino acid, which significantly improves the homo-dimerization ability of the cultivated allele (Simons et al., 2006). Similar to Sh4, the development of the free-threshing character in cultivated wheat might also have been due to the combination of the coding and regulatory changes in the cultivated gene. The difference of expression between Q and q seems more significant as it can clarify the free threshing character in cultivated wheat (Simons et al., 2006; Zhang et al., 2011). Even though mutation which gives rise to Q has a significant effect on the process of wheat domestication, as it helps farmers to harvest the grain more effectively, the exact cellular cause contributing to free-threshing character is still unclear. Similar research has been progressed in non-cereals crop such as overexpression AtFUL to make the pods shattering resistance in Brassica juncea (Østergaard et al., 2006).
Cross-Talk Between Phytohormones and Related Genes Regulating Seed Shattering and Dehiscence Zones (DZ)
Hormonal homeostasis and interactions have been found recently as direct downstream effects of the core genetic network. As an example indehiscent (IND) expression is involved in the formation of local auxin minimum at the margin of the valve by regulating the auxin efflux in the separation layer cells (Sorefan et al., 2009). Further findings reveal that another b-HLH class SPATULA (SPT) transcription factor, required for carpel fusion early in the female reproductive organ development, may interact physically with IND (Girin et al., 2011). Auxins and cytokinins play an antagonistic role in plant growth and development (Bishopp et al., 2011). This scenario also indicates that the cytokinin signaling pathway is active at the valve margins and such a signaling pathway is interrupted in the shp1/2 and ind mutant. Consequently, local application of cytokinins in the fruit development can help to restore valve margin formation and further enhance dehiscence in shp1/2 and ind mutants, suggesting that cytokinins play a crucial role in valve margin differentiation (Marsch-Martínez et al., 2012). Recent studies reveal gibberellins (GAs) are also involved in the establishment of separation layer cell identity, in addition to auxins and cytokinins (Arnaud et al., 2010). As per the “relief of restraint” model, GA-mediated degradation of DELLA protein is important for GA signaling and also necessary to trigger expression of downstream genes (Harberd, 2003; Sun and Gubler, 2004). GA3ox1, which facilitates the final step in bioactive GAs synthesis, is shown as the direct target of IND. ALC interacts physically with DELLA repressors and local GAs production destabilizes the DELLA protein and relieves ALC to play its role in SL cell specification (Arnaud et al., 2010). In summary, these findings show that many phytohormones participate in the DZ specification and indicate that precise balance between biosynthesis and response is important. Notwithstanding the studies where the function of hormones in the development of DZ have been elucidated, very few studies about how such hormonal signals are coordinated in DZ have been carried out. One of the key challenges is to unravel the complete context of the molecular mechanisms and interactions of plant hormones underlying DZ-specification.
There are many ways for minimizing crop losses due to crop shattering ranging from conventional parental selection with minimum shattering to the screening of mutants and gene editing methods. By advancing the next-generation sequencing and the marker traits associations, many genes involved in pod dehiscence were found, and a series of mutations underlying shattering resistance in several crops and their wild relatives have been identified (Fuller and Allaby, 2009; Dong and Wang, 2015). Attempts have been made to improve shattering resistance in Brassica, which include interfering in the dehiscence process by manipulating the molecular and hormonal control pathways (Fuller and Allaby, 2009; Altpeter et al., 2016) and developing transgenic lines with pod-shattering resistance (Liljegren et al., 2000, 2004). In future, studies should focus, alongside gene-editing methods, on fine-tuning of the degree of shatter-resistance with RNA interference or the use of mutated forms of genes related to shattering in various crops.
Key Genes Targeted for Dwarfing of Cereal to Enhance the Productivity
The plant architecture is genetically controlled by a set of genes which subsequent affect yield and productivity of crop plant species. Often, mutation or knockdown of a single gene could also lead to significant change in the overall plant growth and development, subsequently plant architecture (Spielmeyer et al., 2002). In 1960s, the agricultural transformation that increased the production of rice and wheat was via the introduction of cultivars with a genetic predisposition to a short stature due to restricted elongation of stem (Silverstone and Sun, 2000). This phenotype enabled a significant partitioning of photosynthate produced from photosynthesis to sink organs like grains (Sun and Frelich, 2011).
Currently introduction of dwarfing genes is the most important aspect deployed in modern cereal breeding. The stems of tall wheat and rice crops are not strong enough to sustain heavy grains of the high yielding cultivars, which result in significant yield losses. In addition, the proportion of assimilates partitioned in grain increases yields. Genes associated with the semi-dwarf growth of the wheat and rice cultivars have been studied. In wheat, Reduced height (Rht) gene has been identified which is shown to interfere with GA signaling transduction pathway (Peng et al., 1999). Subsequently, three research groups investigated semi dwarf1 (SD1) gene from rice and found that the same hormone impair the biosynthesis (Monna et al., 2002; Sasaki et al., 2002; Spielmeyer et al., 2002). Thus, gibberellin hormone appears to be central to plant stature control.
Wheat Rht Gene and Gibberellin Signaling
The Green Revolution’s wheat dwarfing genes originated in Japan (Gale et al., 1985). The Norin 10 dwarfing genes are now available worldwide in 70% of current commercial wheat cultivars. Norin10 contains two dwarfing genes that are semi-dominant homologous alleles on Chromosomes B and D. These alleles are labeled as Rht-B1b (formerly Rht1) and Rht-D1b (Rht2) to reflect their chromosome position (Boerner et al., 1996). The Rht alleles cause a reduced response to the plant hormone GA class (Gale et al., 1985). These plant hormones are diterpenoid carboxylic acids, that are involved in several processes of development in higher plants, including stem elongation (Hooley, 1994). The Rht gene is an ortholog of Arabidopsis GA-Insensitive (GAI) and maize dwarf 8 genes, for which mutations result in GA-insensitive dwarfs (Peng et al., 1999). Rht-1a/d8/GAI (wild type protein) is a subgroup of the GRAS family of proteins that are thought to act as transcriptional regulators (Pysh et al., 1999). Peng et al. (1999) reported base substitutions in the Rht-B1b and Rht-D1b alleles that insert stop codons within the DELLA region. They mentioned that translational re-initiation at one of several methionines which follow the stop codon could lead to the formation of truncated Rht protein without the DELLA domain, which functions as a constituent (GA insensitive) growth repressor. The D8 (Peng et al., 1999) and GAI mutations (Peng et al., 1997) also lead to partial or complete deletion from one or both of the conserved domains. The Rht-1a/d8/GAI proteins thus function as negative GA signaling regulators and suppress GA function, provided N-terminal domains are present (Harberd et al., 1998; Dill et al., 2001). To support this concept, ectopic expression of GAI (Peng et al., 1999) in rice caused dwarfism and loss of function mutations in Rht-like genes in some cases produces an over-growth phenotype (Ikeda et al., 2001; Chandler et al., 2002). Besides d8, Rht-1a orthologs were reported in rice (known as OsGAI or SLR1) (Ogawa et al., 2000; Ikeda et al., 2001) and barley (SLN1) (Chandler et al., 2002). While cereals have a single case of Rht-1a/d8/GAI type proteins, Arabidopsis contains a gene family encoding RGA proteins and three RGA-like proteins (RGL1, -2, -3) in addition to GAI. The Arabidopsis homologues seem to overlap in their function in various GA-regulated developmental processes (Olszewski et al., 2002). It is unknown how a single protein in cereals crops is functionally equivalent to five proteins in Arabidopsis; such variation may indicate major functional redundancy in Arabidopsis or fundamental differences in GA signaling pathways between Arabidopsis and Gramineae members. Recently, some progress was made in understanding the function of Rht-like proteins and their GA repression. RGA (Dill et al., 2001), SLR1 (Itoh et al., 2002), and SLN1 (Gubler et al., 2002) are found in the nucleus and thus rapidly degraded with GA presence, the DELLA domain needed for this process. Rht’s upstream signal transduction pathway is still unknown, but GA-induced degradation is believed to involve ubiquitin-mediated proteolysis (Chandler et al., 2002).
Rice sd1 Gene and Gibberellin Biosynthesis
Unlike Rht, the sd1 mutation of rice is recessive and normal height can be restored in mutants using GA application showing that they have been impaired in GA production (Ashikari et al., 2002). Three research groups independently isolated the sd1 gene and showed it encodes GA 20-oxidase (GA20ox), an enzyme involved in biosynthesis of GA (Monna et al., 2002; Sasaki et al., 2002; Spielmeyer et al., 2002). Two of these research groups have used positional cloning to detect a GA20ox open reading frame close to the sd1 locus on the long chromosome arm (Monna et al., 2002; Spielmeyer et al., 2002). They also reported mutations in corresponding genes from semi-dwarf varieties. The third group, which had inferred the gene’s identity by the effect of GA content mutations, used PCR to amplify DNA fragments, corresponding to two GA20ox genes, one of which mapped to the sd1 loci (Sasaki et al., 2002; Ashikari et al., 2002). Semi-dwarf rice cultivars with Dee-geo-woo-gen sd1 allele contain a 383-bp deletion in the GA20ox gene (known as OsGA20ox2), which incorporates stop codon that is likely to result in a highly truncated, inactive enzyme. Gibberellin 20-oxidases are 2-oxoglutarate-dependent dioxygenases catalyzing carbon-20 depletion in the penultimate stage in biosynthesis of GA (Hedden and Phillips, 2000). These oxidases are encoded by small gene families, members of which have partial functional redundancy due to overlapping (but different) expression profiles or because of movement of the intermediates synthesized by enzymes between tissues. Therefore, loss-of-function GA20ox mutants are relatively less GA-deficient and are semi-dwarfs, unlike significant GA-deficient plants, which are extremely dwarfed and sometimes sterile. Two GA20ox genes were defined in rice: OsGA20ox1 (Toyomasu et al., 1997) and OsGA20ox2. Remarkably, selection for semi-dwarfism in rice has consistently yielded mutations in OsGA20ox2 instead of OsGA20ox1 or another GA-biosynthesis gene (for example, GA 3-oxidase is also encoded by a multi-gene family). Mutations in other genes might have a severe developmental impact or have negative impact on yield, and thus have been not selected in breeding programs. Genetic and functional analyses of SLR1/RHT and SD1 genes in rice and wheat have enormously improved the understanding of GA biosynthesis and signals, resulting in a strong methodology for manipulating the plant height of major crops. Both cases illustrate the central role played by GAs in controlling developmental processes. Therefore, GA signaling pathways (biosynthesis and signal transduction) are key aspects for manipulation in pursuit of further crop yield improvements. The yields of existing cereal crops seem to be approaching their limit, and new interventions are required if population is not to outstrip the food supply. Targeted genetic engineering/modification using newly emerged genomics, genome-editing technologies may be part of the next Green Revolution.
Achieving Submergence Tolerance
The incidences of uncertain rain and flood have been increased due to continued climate change. Today, more than 30 percent of the rice-planting land is vulnerable to flooding resulting in crop loss. In 1960s, the development of semi-dwarf variety was one of greatest achievement which significantly addressed the issue of global hunger threat caused due to human population explosion. The suppression of GAs production in the stem reportedly made high yielding semi-dwarf rice varieties susceptible to one of the most important abiotic stress “water logging.” These developed semi-dwarf rice varieties lacked submergence tolerance. The lower nodes of these varieties unable to produce enough gibberellins to trigger elongation of the internode.
Genomics Based Discovery of Genomic Regions Associated With Submergence Tolerance
Submergence stress causes several adverse impacts on a plant such as low light intensity, hypoxia, nutrient effusion, physical injury, susceptibility to pathogen and pests attacks (Angaji et al., 2010). Several QTL mapping studies reported number of QTLs controlling submergence tolerance (Xu and Mackill, 1996; Nandi et al., 1997; Toojinda et al., 2003). A major QTL (Sub1) for submergence tolerance has been identified on chromosome 9 with LOD 36 and 69% of phenotypic variance explained (PVE) (Xu and Mackill, 1996). Sequencing of Sub1 genomic region identified three genes which encodes a ERFs (Sub1A, Sub1B, and Sub1C) in which Sub1A has been reported as a key component of submergence tolerance (Xu et al., 2006). Further cloning and characterization of Sub1 QTL helping in the detection of responsible genes and also help to discover tightly linked gene-based markers for molecular breeding program (Siangliw et al., 2003; Toojinda et al., 2005; Neeraja et al., 2007). Furthermore, in other studies major QTLs namely qAG9-2 on L.G. 9 and qAG7-1 on L.G. 7 were reported (Angaji et al., 2010; Septiningsih et al., 2013). Later on, qAG9-2 QTL has been fine mapped and found a candidate gene OsTPP7 which encodes a trehalose-6-phosphate phosphatase which is responsible to regulate anaerobic generation (Kretzschmar et al., 2015). Both Sub1 and qAG9-2 major QTLs are widely used in rice breeding programs to improve submergence tolerance at germination and vegetative stages. Utilizing genomics resources several breeding efforts are also made in developing submergence tolerance varieties to sustain rice production. Various landraces and traditional genotypes namely, Kurkaruppan, FR13A, Thavalu, Goda Heenati, etc., were reported to be a suitable source of alleles which is associated with submergence tolerance (Miro and Ismail, 2013).
Precise Characterization of Genes Governing Submergence Tolerance
In recent years significant progressed have been made toward understanding the physiological, biochemical and genetic basis of submergence tolerance, to identify the causal gene(s) that are crucial for submergence tolerance (Oladosu et al., 2020). Recently, Kuroha et al. (2018) identified the gene SD1 (SEMIDWARF) responsible for submergence-induced elongation of internode by producing gibberellins mainly GA4. Another study identified genes SNORKEL 1 (SK1) and SK2 which encodes for ERFs, appeared to trigger submergence tolerance via ethylene signaling (Hattori et al., 2009). Both gene products further facilitate the internode elongation through GAs. Previous study identified a submergence tolerance gene SUB1A (an Ethylene-response-factor-like gene) on chromosome 9 which encodes ERFs (Xu et al., 2006; Fukao et al., 2006). During flash floods, SUB1A inhibits plant elongation at the seedling stage. Flash floods usually last for a few weeks. Cultivars carrying SUB1A tolerance gene show stunted growth and can survive in submerged conditions for a few weeks. Both SNORKEL 1 and SNORKEL 2 (SK1/2) genes and SUB1A encode ERFs which are associated with GAs, but they act in opposite ways in controlling plant development in response to submergence. Further more research is required to uncover the various pathways associated with SK1; SK2 and SUB1A. Furthermore, recently two genes have been identified ACCELERATOR OF INTERNODE ELONGATION 1 (ACE1) and DECELERATOR OF INTERNODE ELONGATION 1 (DEC1) which are responsible to control stem elongation (Nagai et al., 2020). ACE1 gene encoding an unknown function protein which is associated with internodes elongation via GAs, whereas, DEC1 gene encoding a zinc –finger TF, which suppresses internodes elongation. Both the genes influence gibberellin-activated cell division in stem nodes. The expression of ACE1 gene during submergence conditions in rice triggers elongation of internodes within a cell-division zone of the plant. This results in an increased number of elongated internodes and increased plant height. Further gene ACE1C9285 is controlled by SUB1C, a gibberellin-activated TF which is upregulated in response to submergence (Fukao and Bailey-Serres, 2008). SUB1C expression level seemingly low in cultivars that contain the SUB1A-1 regulator gene, a homolog to SUB1C. In short rice cultivars expressing gene SUB1A-1, GAs responsiveness altered, subsequently use carbon pool for leaves elongation, and restrict overall plant development and enter to transient quiescent stage during flooding, an adaptation to overcome deep floods (Fukao et al., 2006; Xu et al., 2006). In semi-dwarf cultivars, internodes elongation only takes place in the upper internodes during growth stage. Nagai et al. (2020) reported a gene ACE1-LIKE1, which triggers upper internodes growth in deep-water. Presently, these omics study based information on the genetic basis of submergence tolerance is the base of rapid improvement of plant architecture to design a high yielding crop tolerant submergence.
Translation of Omics Driven Data for Re-Domestication and De Novo Domestication: Utilization of Genome/Gene Editing Tool
Gene-editing technologies have become choice of a researcher to domesticate neglected crops and wild relatives in a short period (Fernie and Yan, 2019). Traditionally, plant domestication and the development of productive cultivars required decades of breeding, which is also the key reason why so many breeding programs over the last 100 years focused on further improvement of a relatively small number of crops. Recent identification of several major domestication genes and scientific breakthroughs in integrating various genomic changes in plants concurrently with CRISPR/Cas9 editing has allowed re-domestication of existing crop plants and de-novo domestication wild species to be domesticated within a single generation (Figure 2) (Schindele et al., 2020). De-novo domestication has contributed to agro-biodiversity and diet quality, with possible future environmental and nutritional benefits (Singh et al., 2019). In the history of crop domestication amid higher yield selection and breeding, international germplasm exchange; multiple local resistance and resilience genes of wild species have been lost or have never been completely incorporated into breeding lines (Fernie and Yan, 2019). In other words, wild relatives of domesticated plants have significantly higher variable gene pool than that of domesticated ones (Hickey et al., 2019). As we start to uncover more about the framework of crop genomes and the loci of quality traits, there are chances of incorporating valuable characters into existing crop species and ways to quickly re-domesticate new crops. This step can be effectively achieved using breakthrough CRISPR-Cas9 gene-editing technologies, in particular, to introduce beneficial alleles without linkage drag (Li et al., 2018), to produce novel quantitative variations (Rodríguez-Leal et al., 2017), direct deletion of deleterious alleles (Johnsson et al., 2019), and/or higher recombination rates (Mieulet et al., 2018). Recently, gene editing has been shown to enhance plant architecture, flower development, and fruit size in Physalis pruinosa (Lemmon et al., 2018). Gene editing is a promising method to generate diversity and to compensate for the genetic hitchhiking effects in germplasm. For reference, associated selection of traits such as fruit weight and disease resistance altered the tomato metabolome, providing an opportunity for precise breeding to alter nutritional and flavor traits (Zhu et al., 2018). These hitchhiking effects and others, such as those found in rice and maize, represent promising goals for genetic modification to fettle linkage drag (Palaisa et al., 2004). For instance, African rice landrace Kabre possess superior resistance to pests and tolerance to drought; however, during domestication the plant architecture compromised affecting their overall yield potential. To address this Lacchini et al. (2020) targeted multiples genes which control plant architecture (HTD1) and control seed size and/or yield (GS3, GW2, and GN1A) by generating knockouts through multiplex CRISPR/Cas9. In knockouts, mutation in HTD1 gene caused reduced plant high to diminish lodging and improved tillering, whereas mutations in GS3, GW2, and GN1A resulted increased panicle and length along with improved seed girth. Earlier, Hu et al. (2019) demonstrated generation of semi-dwarf rice lines by targeting gene SD1 and Photosensitivity5 (SE5) in elite landraces Kasalath. In this post genomics, the technique CRISPR/Cas has received overwhelming response and till dates several knockouts of rice elite varieties are available with improved traits by targeting specific genes which were characterized due to viability of several omics approached era. Some of the examples for the targeted traits and gene targets in rice are LAZY1 for tiller-spreading, Gn1a, GS3, and DEP1 for improved grain number, size and dense erect panicles, SBEIIb for High amylose content, OsERF922 for enhanced blast resistance, OsSEC3A for resistance against blast causing pathogen Magnaporthe oryzae, OsSWEET13 for bacterial blight resistance, ALS and EPSPS for herbicide resistance, OsPDS, OsMPK2, OsMPK5, OsBADH2, OsAOX1a, OsAOX1b, OsAOX1c, and OsBEL for tolerance against various abiotic stress, OsHAK-1 for low cesium accumulation, and OsPRX2 for potassium deficiency tolerance (Shan et al., 2013; Xie and Yang, 2013; Shan et al., 2014; Xu et al., 2014; Zhang H. et al., 2014; Zhou et al., 2014; Woo et al., 2015; Meng et al., 2017; Nieves-Cordones et al., 2017; Mao et al., 2018; Ma et al., 2018). Likewise, in wheat EDR1, TaMLOA1, TaMLOB1, and TaMLOD1 targeted for resistance to powdery mildew, and GW2 and TaGW2 targeted for increased grain size, weight and protein content (Shan et al., 2014; Wang et al., 2014; Gil-Humanes et al., 2017; Kim et al., 2018; Wang et al., 2018). In orphan crops cassava and flax herbicide resistance has been introduced by targeting a gene EPSPS (Sauer et al., 2016; Hummel et al., 2018); whereas ALS was targeted in soybean (Cai et al., 2015). Similarly, many traits have been introduced or improved by targeting various genes in some economically important crops plants such as maize, tomato, potato, grapes, orange, cucumber, tea, etc. (Adhikari and Poudel, 2020; Bhatta and Malla, 2020).
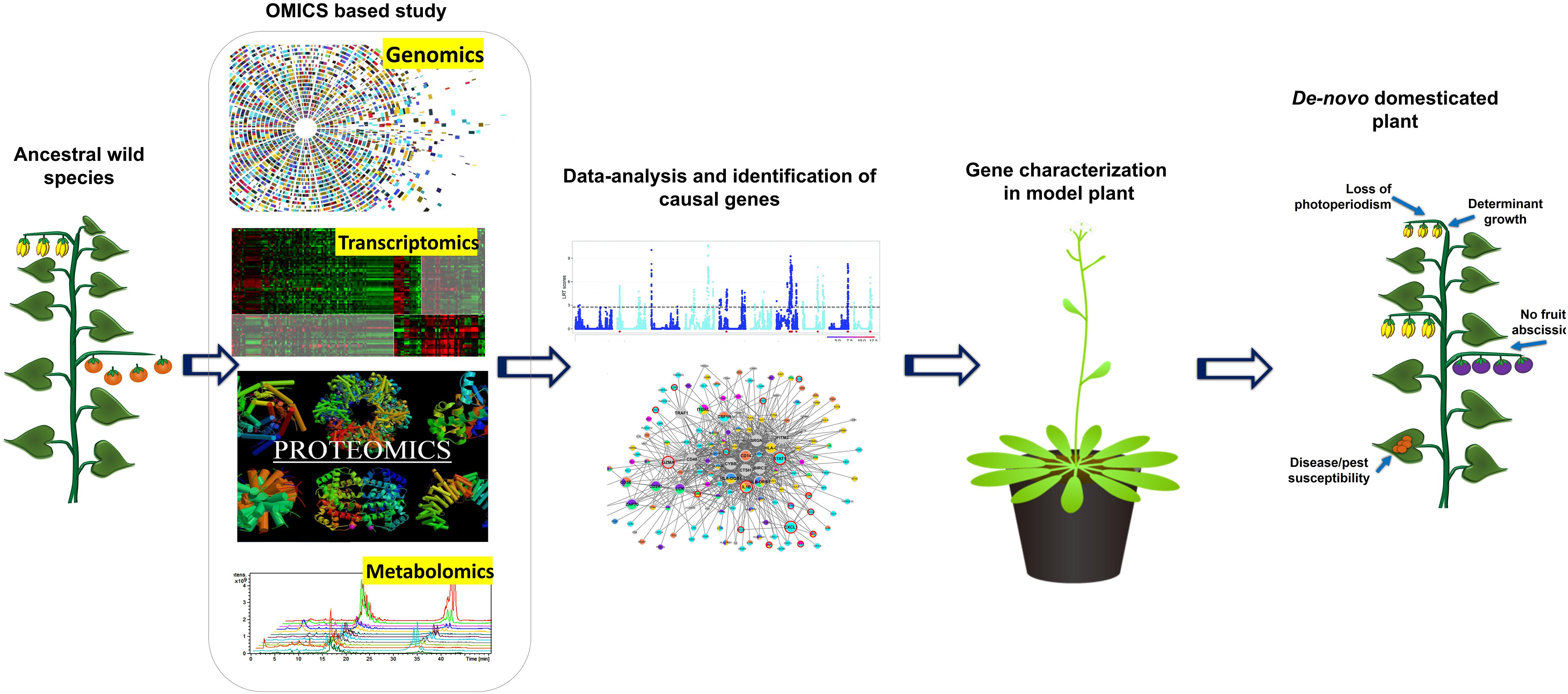
Figure 2. Schematic diagram representing the role of OMICS based research in gene characterization and development of designer crops using de novo domesticated crops approach.
The wild ancestral species of crop plants such as Solanum pimpinellifolium for tomato; Solanum demissum and S. stoloniferum of potato; Fragaria vesca of strawberry; Teosinte and Tripsacum of maize; Triticum dicoccoides, and T. turgidum L. ssp. Durum of wheat; Oryza rufipogon and O. longistaminata of rice; Manihot glaziovii and M. neosana and Glycine soja of soybean have been used for introgression key agronomic important traits into cultivars though breeding program (Zsögön et al., 2017). Moreover, most of the domesticated related traits and associated genes well characterized and has been linked with the metabolic pathway(s), and/or hormone biosynthesis and signaling (Table 4); therefore, integrated omics approach which also involved metabolomics study has provided insights into the molecular basis of trait domestication. One can target these domesticated genes in wild ancestral plants for their speedy domestication. Now through CRISPR-Cas9 method these wild relative can be directly used for re-domestication or de-novo domestication (Figure 3 and Tables 5, 6). One of the important case study of de novo domestication in tomato has been done by Zsögön et al. (2018) by targeting important domestication related genes through CRISPR-Cas9 in tomato wild ancestral species S. pimpinellifolium. Zsögön et al. (2018) targeted SELFPRUNING (SP, control general plant growth habit), OVATE (O, regulate fruit shape); FASCIATED (FAS), FRUIT WEIGHT 2.2 and CLAVATA3 (CLV3) (control fruit size and locule numbers), MULTIFLORA (MULT, regulate fruit number), and LYCOPENE BETA CYCLASE (CycB). The engineered S. pimpinellifolium lines and achieved remarkable change in the plant overall phenotype with important traits essential for the commercial purpose such as increased lycopene content, enhanced fruit shape and determinant growth of plant; moreover, this was achieved in just single generation. Another study involved editing of multiples genes SP, SP5G (control day-length insensitivity), CLV3, WUSCHEL (WUS) and GDP-L-galactose phosphorylase 1 (GGP1, control biosynthesis of ascorbic acid) in S. pimpinellifolium (Li et al., 2018). This study clearly showed how selective editing of domesticated related genes can completely alter the plant architecture and improves the nutritional quality of fruits and makes convert wild relative into domesticated crop with retained biotic and abiotic stress tolerance properties (Li et al., 2018). Very recently, in the wild strawberry (Fragaria vesca) few attempts has been made to demonstrate the procedure of the re-domestication or de novo domestication (Zhou et al., 2018; Feng et al., 2019). These attempts involved editing of genes tryptophan aminotransferase of Arabidopsis 1 (TAA1, converts tryptophan to indole-3-pyruvic acid), Auxin response factor 8 (ARF8, repressor of auxin signaling) and YUCCA10 (YUC10, family of flavin-containing monooxygenases convert IPyA to IAA), key auxin biosynthetic and signaling pathways genes. Rice has five allotetraploids (BBCC, CCDD, HHJJ, HHKK, and KKLL) wild species which are also valuable genetic resources for improving of elite rice varieties. Among them the CCDD (species from South America genome) possess much stronger biotic and abiotic resistance and larger biomass compared to the cultivated diploid rice. Recently Yu et al. (2021) demonstrated de novo domestication of wild allotetraploid rice PPR1 (O. alta; CCDD type genome) by improving six agronomically important traits viz nutrition use efficiency, abiotic stress tolerance, grain yield and quality, heading date, biotic stress resistance and sterility by genome editing targeting multiple genes including OaSD1-CC, OaSD1-DD, OaAn-1-CC, and OaAn-1-DD by CRISPR/Cas9 method. This suggests that CRISPR/Cas is a promising approach tool for the domestication of crops (Crews and Cattani, 2018), and is highly important for characters of defined selective sweeps in related species. These achievements were possible due to precise prediction of causal genes and metabolic pathways achieved by interpretation of data generated through genomics, transcriptomics, metabolomics, etc.

Figure 3. A schematic representation of a draft model for the selection of target genes for CRISPR/Cas9 mediated domestication of wild ancestral species of monocot.
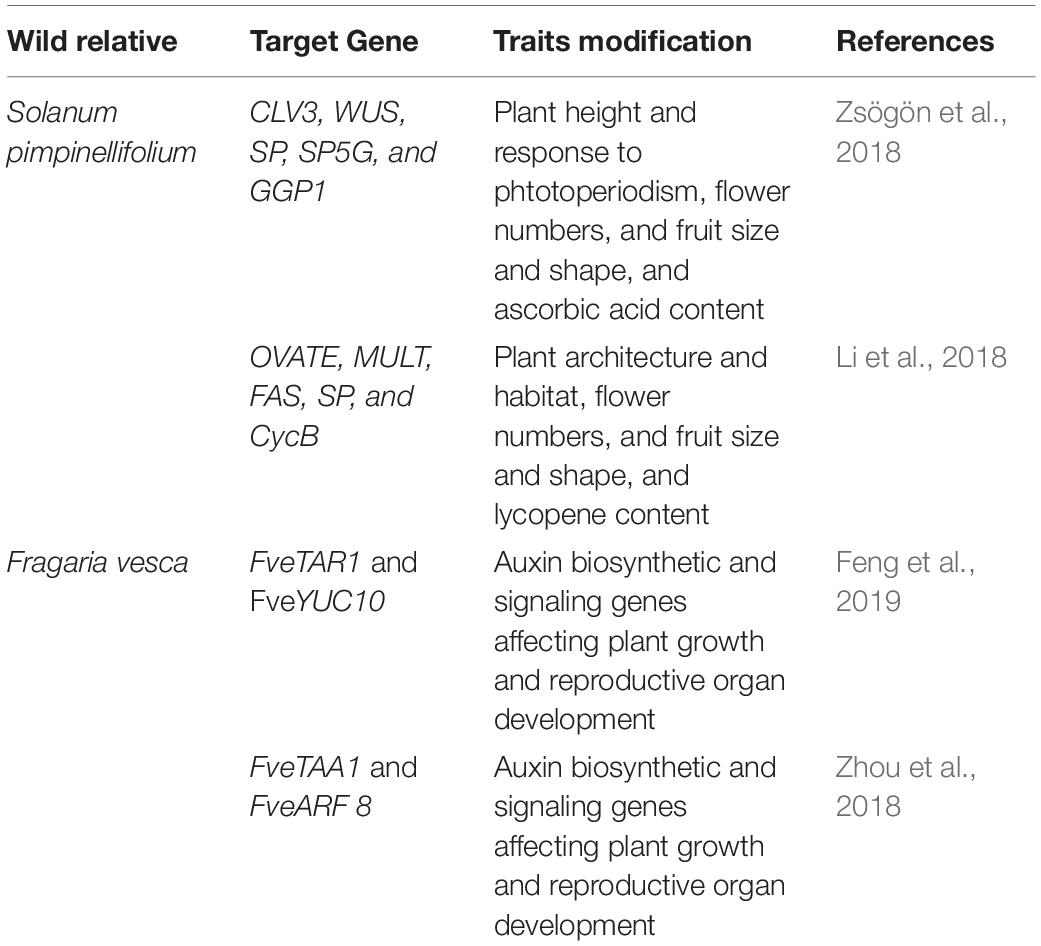
Table 5. List of genes targeted in wild ancestral species of tomato and strawberry to demonstrate de novo domestication.
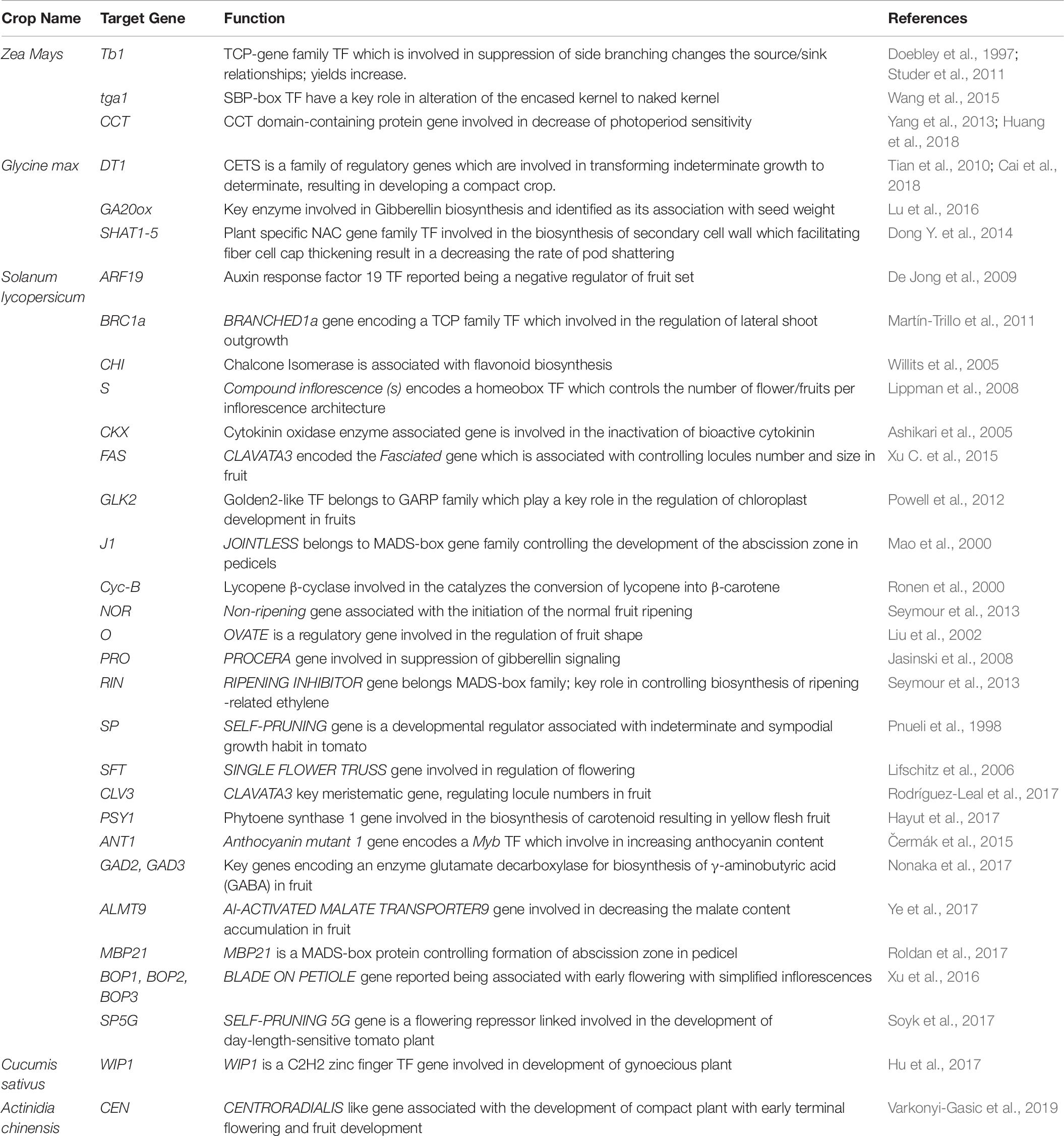
Table 6. A model representing state of art for selecting the genes which can be edited to domesticate crop wild ancestral species through CRISPR/Cas9 approach.
Conclusion
Omics have helped plant biologists to dissect important developmental clues and gene characterization. Presently, multidimensional omics approach where the biological sample can be analyzed for transcriptomics, proteomics and metabolomics in parallel, etc; offers plant biologists a complete understanding of plant metabolism by revisiting the metabolic pathways or identification of newer pathways. In the past 20 years, plant biologists have gathered significant amount of data relevant to genomes, transcriptome, proteome, and metabolome. Recent attempts are on development of gene-expression and proteome atlas. Altogether, this would strengthen the knowledge of the metabolic pathways, which have played crucial role during domestication of crop as well as trait improvement. Now, this knowledge has been translated to develop designer crops with desired traits by editing metabolic pathways of wild ancestral species (rich resource of genetic variations) called as de novo-crop domestication. Domestication of wild or semi domesticated crop (tolerant to stress responses) would be feasible by multi step process were few important traits need to be improved first using genome editing; later the homologous lines can be selected for next level of trait modification. Such approach would be able to deliver a commercial line in 5 to 10 years. The CRISPR/Cas technique need to be explored in full extent by targeting several traits such as bio-fortification of nutrition’s; because the current growing population also demand nutritional security. To achieve this, analysis of resequencing data available for the several crops is important; including GWAS which can identify high quality SNPs and haplotypes associated with target trait. Therefore, we expected in next 20 years’ omics technology driven de-novo crop domestication will play very important role in the field of plant biotechnology.
Author Contributions
RK received the invitation and conceived the plan for the manuscript. RK and VS wrote the manuscript. AK, SS, DR, SK, KP, BH, AV, RK, MP, ST, and GN improved the section and developed the table and figures. ST helped in developing the revised version. All the authors have read the manuscript before submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abe, K., Araki, E., Suzuki, Y., Toki, S., and Saika, H. (2018). Production of high oleic/low linoleic rice by genome editing. Plant Physiol. Biochem. 131, 58–62. doi: 10.1016/j.plaphy.2018.04.033
Adhikari, P., and Poudel, M. (2020). CRISPR-Cas9 in agriculture: Approaches, applications, future perspectives, and associated challenges. Malays. J. Halal Res. 3, 6–16. doi: 10.2478/mjhr-2020-0002
Agrawal, G. K., Jwa, N. S., Lebrun, M. H., Job, D., and Rakwal, R. (2010). Plant secretome: unlocking secrets of the secreted proteins. Proteomics 10, 799–827.
Alonso, A., Marsal, S., and Julià, A. (2015). Analytical methods in untargeted metabolomics: state of the art in 2015. Front. Bioeng. Biotechnol. 3:23. doi: 10.3389/fbioe.2015.00023
Alseekh, S., Ofner, I., Liu, Z., Osorio, S., Vallarino, J., Last, R. L., et al. (2020). Quantitative trait loci analysis of seed-specialized metabolites reveals seed-specific flavonols and differential regulation of glycoalkaloid content in tomato. Plant J. 103, 2007–2024. doi: 10.1111/tpj.14879
Alseekh, S., Tohge, T., Wendenberg, R., Scossa, F., Omranian, N., Li, J., et al. (2015). Identification and mode of inheritance of quantitative trait loci for secondary metabolite abundance in tomato. Plant Cell 27, 485–512. doi: 10.1105/tpc.114.132266
Alseekh, S., Tong, H., Scossa, F., Brotman, Y., Vigroux, F., Tohge, T., et al. (2017). Canalization of tomato fruit metabolism. Plant Cell 29, 2753–2765. doi: 10.1105/tpc.17.00367
Altpeter, F., Springer, N. M., Bartley, L. E., Blechl, A. E., Brutnell, T. P., Citovsky, V., et al. (2016). Advancing crop transformation in the era of genome editing. Plant Cell 28, 1510–1520.
Alvarez, S., Marsh, E. L., Schroeder, S. G., and Schachtman, D. P. (2008). Metabolomic and proteomic changes in the xylem sap of maize under drought. Plant Cell Environ. 31, 325–340. doi: 10.1111/j.1365-3040.2007.01770.x
Alvarez, S., Roy Choudhury, S., and Pandey, S. (2014). Comparative quantitative proteomics analysis of the ABA response of roots of drought-sensitive and drought-tolerant wheat varieties identifies proteomic signatures of drought adaptability. J. Proteome Res. 13, 1688–1701. doi: 10.1021/pr401165b
Amarasinghe, Y. P. J., Kuwata, R., Nishimura, A., Phan, P. D. T., Ishikawa, R., and Ishii, T. (2020). Evaluation of domestication loci associated with awnlessness in cultivated rice. Oryza sativa. Rice 13:26
Angaji, S. A., Septiningsih, E. M., Mackill, D. J., and Ismail, A. M. (2010). QTLs associated with tolerance of flooding during germination in rice (Oryza sativa L.). Euphytica 172, 159–168. doi: 10.1007/s10681-009-0014-5
Angelovici, R., Batushansky, A., Deason, N., Gonzalez-Jorge, S., Gore, M. A., Fait, A., et al. (2017). Network-guided GWAS improves identification of genes affecting free amino acids. Plant Physiol. 173, 872–886. doi: 10.1104/pp.16.01287
Arnaud, N., Girin, T., Sorefan, K., Fuentes, S., Wood, T. A., Lawrenson, T., et al. (2010). Gibberellins control fruit patterning in Arabidopsis thaliana. Genes Dev. 24, 2127–2132. doi: 10.1101/gad.593410
Arnaud, N., Lawrenson, T., Østergaard, L., and Sablowski, R. (2011). The same regulatory point mutation changed seed-dispersal structures in evolution and domestication. Curr. Biol. 21, 1215–1219. doi: 10.1016/j.cub.2011.06.008
Ashikari, M., Sakakibara, H., Lin, S., Yamamoto, T., Takashi, T., Nishimura, A., et al. (2005). Cytokinin oxidase regulates rice grain production. Science 309, 741–745. doi: 10.1126/science.1113373
Ashikari, M., Sasaki, A., Ueguchi-Tanaka, M., Itoh, H., Nishimura, A., Datta, S., et al. (2002). Loss-of-function of a rice gibberellin biosynthetic gene, GA20 oxidase (GA20ox-2), led to the rice ‘green revolution’. Breed. Sci. 52, 143–150. doi: 10.1270/jsbbs.52.143
Baerenfaller, K., Grossmann, J., Grobei, M. A., Hull, R., Hirsch-Hoffmann, M., Yalovsky, S., et al. (2008). Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320, 938–941. doi: 10.1126/science.1157956
Bhatta, B. P., and Malla, S. (2020). Improving horticultural crops via CRISPR/Cas9: current successes and prospects. Plants 9:1360. doi: 10.3390/plants9101360
Bishopp, A., Benková, E., and Helariutta, Y. (2011). Sending mixed messages: auxin-cytokinin crosstalk in roots. Curr. Opin. Plant Biol. 14, 10–16. doi: 10.1016/j.pbi.2010.08.014
Boerner, A., Plaschke, J., Korzun, V., and Worland, A. J. (1996). The relationships between the dwarfing genes of wheat and rye. Euphytica 89, 69–75. doi: 10.1007/bf00015721
Bommisetty, R., Chakravartty, N., Bodanapu, R., Naik, J. B., Panda, S. K., Lekkala, S. P., et al. (2020). Discovery of genomic regions and candidate genes for grain weight employing next generation sequencing based QTL-seq approach in rice (Oryza sativa L.). Mol. Biol. Rep. 47, 8615–8627. doi: 10.1007/s11033-020-05904-7
Cai, Y., Chen, L., Liu, X., Guo, C., Sun, S., Wu, C., et al. (2018). CRISPR/Cas9-mediated targeted mutagenesis of GmFT2a delays flowering time in soya bean. Plant Biotech. J. 16, 176–185. doi: 10.1111/pbi.12758
Cai, Y., Chen, L., Liu, X., Sun, S., Wu, C., Jiang, B., et al. (2015). CRISPR/Cas9-mediated genome editing in soybean hairy roots. PLoS One 10:e0136064. doi: 10.1371/journal.pone.0136064
Capriotti, A. L., Borrelli, G. M., Colapicchioni, V., Papa, R., Piovesana, S., Samperi, R., et al. (2014). Proteomic study of a tolerant genotype of durum wheat under salt-stress conditions. Anal. Bioanal. Chem. 406, 1423–1435. doi: 10.1007/s00216-013-7549-y
Carreno-Quintero, N., Acharjee, A., Maliepaard, C., Bachem, C. W., Mumm, R., Bouwmeester, H., et al. (2012). Untargeted metabolic quantitative trait loci analyses reveal a relationship between primary metabolism and potato tuber quality. Plant Physiol. 158, 1306–1318. doi: 10.1104/pp.111.188441
Čermák, T., Baltes, N. J., Čegan, R., Zhang, Y., and Voytas, D. F. (2015). High-frequency, precise modification of the tomato genome. Genome Biol. 16:232.
Chan, E. K., Rowe, H. C., Hansen, B. G., and Kliebenstein, D. J. (2010). The complex genetic architecture of the metabolome. PLoS Genet. 6:e1001198. doi: 10.1371/journal.pgen.1001198
Chandler, P. M., Marion-Poll, A., Ellis, M., and Gubler, F. (2002). Mutants at the slender1 locus of barley cv Himalaya. molecular and physiological characterization. Plant Physiol. 129, 181–190. doi: 10.1104/pp.010917
Chen, J., Wang, J., Chen, W., Sun, W., Peng, M., Yuan, Z., et al. (2018). Metabolome analysis of multi-connected biparental chromosome segment substitution line populations. Plant Physiol. 178, 612–625. doi: 10.1104/pp.18.00490
Chen, W., Gao, Y., Xie, W., Gong, L., Lu, K., Wang, W., et al. (2014). Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 46:714. doi: 10.1038/ng.3007
Chen, W., Wang, W., Lyu, Y., Wu, Y., Huang, P., Hu, S., et al. (2020). OsVP1 activates Sdr4 expression to control rice seed dormancy via the ABA signaling pathway. Crop J. 9, 68–78. doi: 10.1016/j.cj.2020.06.005
Chen, W., Wang, W., Peng, M., Gong, L., Gao, Y., Wan, J., et al. (2016). Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat. Commun. 7:12767.
Chen, X., Lu, Q., Liu, H., Zhang, J., Hong, Y., Lan, H., et al. (2019). Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Mol. Plant 12, 920–934. doi: 10.1016/j.molp.2019.03.005
Chiang, S., Zhang, W., and Ouyang, Z. (2018). Paper spray ionization mass spectrometry: recent advances and clinical applications. Expert Rev. Proteomics 15, 781–789. doi: 10.1080/14789450.2018.1525295
Chu, P., Yan, G. X., Yang, Q., Zhai, L. N., Zhang, C., Zhang, F. Q., et al. (2015). iTRAQ-based quantitative proteomics analysis of Brassica napus leaves reveals pathways associated with chlorophyll deficiency. J. Proteomics 113, 110–126.
Cloonan, N., Forrest, A. R., Kolle, G., Gardiner, B. B., Faulkner, G. J., Brown, M. K., et al. (2008). Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 5, 613–619. doi: 10.1038/nmeth.1223
Coppola, M., Diretto, G., Digilio, M. C., Woo, S. L., Giuliano, G., Molisso, D., et al. (2019). Transcriptome and metabolome reprogramming in tomato plants by Trichoderma harzianum strain T22 primes and enhances defence responses against aphids. Front. Physiol 10:745. doi: 10.3389/fphys.2019.00745
Crews, T. E., and Cattani, D. J. (2018). Strategies, advances, and challenges in breeding perennial grain crops. Sustainability 10:2192. doi: 10.3390/su10072192
Daware, A., Das, S., Srivastava, R., Badoni, S., Singh, A. K., Agarwal, P., et al. (2016). An efficient strategy combining SSR markers-and advanced QTL-seq-driven QTL mapping unravels candidate genes regulating grain weight in rice. Front. Plant Sci. 7:1535. doi: 10.3389/fpls.2016.01535
De Jong, M., Wolters-Arts, M., Feron, R., Mariani, C., and Vriezen, W. H. (2009). The Solanum lycopersicum auxin response factor 7 (SlARF7) regulates auxin signaling during tomato fruit set and development. Plant J. 57, 160–170. doi: 10.1111/j.1365-313x.2008.03671.x
Deng, W., Casao, M. C., Wang, P., Sato, K., Hayes, P. M., Finnegan, E. J., et al. (2015). Direct links between the vernalization response and other key traits of cereal crops. Nat. Commun. 6:5882
Deokar, A., Sagi, M., Daba, K., and Tar’an, B. (2019). QTL sequencing strategy to map genomic regions associated with resistance to ascochyta blight in chickpea. Plant Biotechnol. J. 17, 275–288. doi: 10.1111/pbi.12964
Di Lena, G., Casini, I., Lucarini, M., and Lombardi-Boccia, G. (2019). Carotenoid profiling of five microalgae species from large-scale production. Food Res. Int. 120, 810–818. doi: 10.1016/j.foodres.2018.11.043
Dill, A., Jung, H. S., and Sun, T. P. (2001). The DELLA motif is essential for gibberellin-induced degradation of RGA. Proc. Natl. Acad. Sci. U.S.A. 98, 14162–14167. doi: 10.1073/pnas.251534098
Doebley, J., Stec, A., and Hubbard, L. (1997). The evolution of apical dominance in maize. Nature 386, 485–488. doi: 10.1038/386485a0
Dong, X., Chen, W., Wang, W., Zhang, H., Liu, X., and Luo, J. (2014). Comprehensive profiling and natural variation of flavonoids in rice. J. Integr. Plant Biol. 56, 876–886. doi: 10.1111/jipb.12204
Dong, Y., and Wang, Y. Z. (2015). Seed shattering: from models to crops. Front. Plant Sci. 6:476. doi: 10.3389/fpls.2015.00476
Dong, Y., Yang, X., Liu, J., Wang, B. H., Liu, B. L., and Wang, Y. Z. (2014). Pod shattering resistance associated with domestication is mediated by a NAC gene in soybean. Nat. Commun. 5:3352.
Duncan, O., Trösch, J., Fenske, R., Taylor, N. L., and Millar, A. H. (2017). Resource: mapping the Triticum aestivum proteome. Plant J. 89, 601–616.
Fang, C., and Luo, J. (2019). Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J. 97, 91–100. doi: 10.1111/tpj.14097
Feng, J., Dai, C., Luo, H., Han, Y., Liu, Z., and Kang, C. (2019). Reporter gene expression reveals precise auxin synthesis sites during fruit and root development in wild strawberry. J. Exp. Bot. 70, 563–574. doi: 10.1093/jxb/ery384
Feng, J., Long, Y., Shi, L., Shi, J., Barker, G., and Meng, J. (2012). Characterization of metabolite quantitative trait loci and metabolic networks that control glucosinolate concentration in the seeds and leaves of Brassica napus. New Phytol. 193, 96–108. doi: 10.1111/j.1469-8137.2011.03890.x
Fernandez-Orozco, R., Gallardo-Guerrero, L., and Hornero-Méndez, D. (2013). Carotenoid profiling in tubers of different potato (Solanum sp) cultivars: accumulation of carotenoids mediated by xanthophyll esterification. Food Chem. 141, 2864–2872. doi: 10.1016/j.foodchem.2013.05.016
Fernie, A. R., and Yan, J. (2019). De novo domestication: an alternative route toward new crops for the future. Mol. Plant 12, 615–631. doi: 10.1016/j.molp.2019.03.016
Ferrão, L. F. V., Johnson, T. S., Benevenuto, J., Edger, P. P., Colquhoun, T. A., and Munoz, P. R. (2020). Genome-wide association of volatiles reveals candidate loci for blueberry flavor. New Phytol. 226, 1725–1737. doi: 10.1111/nph.16459
Finnie, C., Sultan, A., and Grasser, K. D. (2011). From protein catalogues towards targeted proteomics approaches in cereal grains. Phytochemistry 72, 1145–1153. doi: 10.1016/j.phytochem.2010.11.014
Frary, A., Nesbitt, T. C., Grandillo, S., Knaap, E., Cong, B., Liu, J., et al. (2000). fw2. 2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289, 85–88. doi: 10.1126/science.289.5476.85
Fukao, T., and Bailey-Serres, J. (2008). Submergence tolerance conferred by Sub1A is mediated by SLR1 and SLRL1 restriction of gibberellin responses in rice. Proc. Natl. Acad. Sci. U.S.A. 105, 16814–16819. doi: 10.1073/pnas.0807821105
Fukao, T., Xu, K., Ronald, P. C., and Bailey-Serres, J. (2006). A variable cluster of ethylene response factor–like genes regulates metabolic and developmental acclimation responses to submergence in rice. Plant Cell 18, 2021–2034. doi: 10.1105/tpc.106.043000
Fuller, D. Q., and Allaby, R. (2009). Seed dispersal and crop domestication: Shattering, germination and seasonality in evolution under cultivation. Annu. Plant Rev. 38, 238–295. doi: 10.1002/9781444314557.ch7
Furukawa, T., Maekawa, M., Oki, T., Suda, I., Iida, S., Shimada, H., et al. (2007). The Rc and Rd genes are involved in proanthocyanidin synthesis in rice pericarp. Plant J. 49, 91–102. doi: 10.1111/j.1365-313x.2006.02958.x
Gale, M. D., Youssefian, S., and Russell, G. E. (1985). Dwarfing genes in wheat. Progr. Plant Breed. 1, 1–35. doi: 10.1016/b978-0-407-00780-2.50005-9
Garbowicz, K., Liu, Z., Alseekh, S., Tieman, D., Taylor, M., Kuhalskaya, A., et al. (2018). Quantitative trait loci analysis identifies a prominent gene involved in the production of fatty acid-derived flavor volatiles in tomato. Mol. Plant 11, 1147–1165. doi: 10.1016/j.molp.2018.06.003
Gasser, C. S., and Simon, M. K. (2011). Seed dispersal: same gene, different organs. Curr. Biol. 21, R546–R548.
Gil-Humanes, J., Wang, Y., Liang, Z., Shan, Q., Ozuna, C. V., Sánchez-León, S., et al. (2017). High-efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. Plant J. 89, 1251–1262. doi: 10.1111/tpj.13446
Gilmore, I. S., Heiles, S., and Pieterse, C. L. (2019). Metabolic imaging at the single-cell scale: recent advances in mass spectrometry imaging. Annu. Rev. Anal. Chem. 12, 201–224. doi: 10.1146/annurev-anchem-061318-115516
Girin, T., Paicu, T., Stephenson, P., Fuentes, S., Körner, E., O’Brien, M., et al. (2011). INDEHISCENT and SPATULA interact to specify carpel and valve margin tissue and thus promote seed dispersal in Arabidopsis. Plant Cell 23, 3641–3653. doi: 10.1105/tpc.111.090944
Gong, L., Chen, W., Gao, Y., Liu, X., Zhang, H., Xu, C., et al. (2013). Genetic analysis of the metabolome exemplified using a rice population. Proc. Natl. Acad. Sci. U.S.A. 110, 20320–20325. doi: 10.1073/pnas.1319681110
González, J. F., Degrassi, G., Devescovi, G., De Vleesschauwer, D., Höfte, M., Myers, M. P., et al. (2012). A proteomic study of Xanthomonas oryzae pv oryzae in rice xylem sap. J. Proteomics 75, 5911–5919. doi: 10.1016/j.jprot.2012.07.019
Gu, B., Zhou, T., Luo, J., Liu, H., Wang, Y., Shangguan, Y., et al. (2015). An-2 encodes a cytokinin synthesis enzyme that regulates awn length and grain production in rice. Mol. Plant 8, 1635–1650. doi: 10.1016/j.molp.2015.08.001
Gu, T., Jia, S., Huang, X., Wang, L., Fu, W., Huo, G., et al. (2019). Transcriptome and hormone analyses provide insights into hormonal regulation in strawberry ripening. Planta 250, 145–162. doi: 10.1007/s00425-019-03155-w
Gubler, F., Chandler, P. M., White, R. G., Llewellyn, D. J., and Jacobsen, J. V. (2002). Gibberellin signaling in barley aleurone cells. control of SLN1 and GAMYB expression. Plant Physiol. 129, 191–200. doi: 10.1104/pp.010918
Han, Q., Kang, G., and Guo, T. (2013). Proteomic analysis of spring freeze-stress responsive proteins in leaves of bread wheat (Triticum aestivum L.). Plant Physiol. Biochem. 63, 236–244. doi: 10.1016/j.plaphy.2012.12.002
Harberd, N. P. (2003). Relieving DELLA restraint. Science 299, 1853–1854. doi: 10.1126/science.1083217
Harberd, N. P., King, K. E., Carol, P., Cowling, R. J., Peng, J., and Richards, D. E. (1998). Gibberellin: inhibitor of an inhibitor of…? Bioessays 20, 1001–1008. doi: 10.1002/(sici)1521-1878(199812)20:12<1001::aid-bies6>3.0.co;2-o
Hattori, Y., Nagai, K., Furukawa, S., Song, X. J., Kawano, R., and Sakakibara, H. (2009). The ethylene response factors SNORKEL1 and SNORKEL2 allow rice to adapt to deep water. Nature 460, 1026–1030. doi: 10.1038/nature08258
Hayut, S. F., Bessudo, C. M., and Levy, A. A. (2017). Targeted recombination between homologous chromosomes for precise breeding in tomato. Nat. Commun. 8:15605.
He, Y., Fu, Y., Hu, D., Wei, D., and Qian, W. (2018). QTL mapping of seed glucosinolate content responsible for environment in Brassica napus. Front. Plant Sci. 9:891. doi: 10.3389/fpls.2018.00891
Hedden, P., and Phillips, A. L. (2000). Gibberellin metabolism: new insights revealed by the genes. Trends Plant Sci. 5, 523–530. doi: 10.1016/s1360-1385(00)01790-8
Hickey, L. T., Hafeez, A. N., Robinson, H., Jackson, S. A., Leal-Bertioli, S. C., Tester, M., et al. (2019). Breeding crops to feed 10 billion. Nat. Biotechnol. 37, 744–754. doi: 10.1038/s41587-019-0152-9
Hill, C. B., Taylor, J. D., Edwards, J., Mather, D., Bacic, A., Langridge, P., et al. (2013). Whole-genome mapping of agronomic and metabolic traits to identify novel quantitative trait loci in bread wheat grown in a water-limited environment. Plant Physiol. 162, 1266–1281. doi: 10.1104/pp.113.217851
Hill, C. B., Taylor, J. D., Edwards, J., Mather, D., Langridge, P., Bacic, A., et al. (2015). Detection of QTL for metabolic and agronomic traits in wheat with adjustments for variation at genetic loci that affect plant phenology. Plant Sci. 233, 143–154. doi: 10.1016/j.plantsci.2015.01.008
Hooley, R. (1994). Gibberellins: perception, transduction and responses. Plant Mol. Biol. 26, 1529–1555. doi: 10.1007/bf00016489
Hu, B., Li, D., Liu, X., Qi, J., Gao, D., Zhao, S., et al. (2017). Engineering non-transgenic gynoecious cucumber using an improved transformation protocol and optimized CRISPR/Cas9 system. Mol. Plant 10, 1575–1578. doi: 10.1016/j.molp.2017.09.005
Hu, J., Rampitsch, C., and Bykova, N. V. (2015). Advances in plant proteomics toward improvement of crop productivity and stress resistance. Front. Plant Sci. 6:209. doi: 10.3389/fpls.2015.00209
Hu, X., Cui, Y., Dong, G., Feng, A., Wang, D., Zhao, C., et al. (2019). Using CRISPR-Cas9 to generate semi-dwarf rice lines in elite landraces. Sci. Rep. 9:19096.
Hua, L., Wang, D. R., Tan, L., Fu, Y., Liu, F., Xiao, L., et al. (2015). LABA1, a domestication gene associated with long, barbed awns in wild rice. Plant Cell 27, 1875–1888. doi: 10.1105/tpc.15.00260
Huang, C., Sun, H., Xu, D., Chen, Q., Liang, Y., Wang, X., et al. (2018). ZmCCT9 enhances maize adaptation to higher latitudes. Proc. Natl. Acad. Sci. U.S.A. 115, E334–E341.
Hummel, A. W., Chauhan, R. D., Cermak, T., Mutka, A. M., Vijayaraghavan, A., Boyher, A., et al. (2018). Allele exchange at the EPSPS locus confers glyphosate tolerance in cassava. Plant Biotechnol. J. 16, 1275–1282. doi: 10.1111/pbi.12868
Ikeda, A., Ueguchi-Tanaka, M., Sonoda, Y., Kitano, H., Koshioka, M., Futsuhara, Y., et al. (2001). slender rice, a constitutive gibberellin response mutant, is caused by a null mutation of the SLR1 gene, an ortholog of the height-regulating gene GAI/RGA/RHT/D8. Plant Cell 13, 999–1010. doi: 10.2307/3871359
Illa-Berenguer, E., Van Houten, J., Huang, Z., and van der Knaap, E. (2015). Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 128, 1329–1342. doi: 10.1007/s00122-015-2509-x
Iquebal, M. A., Sharma, P., Jasrotia, R. S., Jaiswal, S., Kaur, A., Saroha, M., et al. (2019). RNAseq analysis reveals drought-responsive molecular pathways with candidate genes and putative molecular markers in root tissue of wheat. Sci. Rep. 9:13917.
Itoh, H., Ueguchi-Tanaka, M., Sato, Y., Ashikari, M., and Matsuoka, M. (2002). The gibberellin signaling pathway is regulated by the appearance and disappearance of slender rice1 in nuclei. Plant Cell 14, 57–70. doi: 10.1105/tpc.010319
Jasinski, S., Tattersall, A., Piazza, P., Hay, A., Martinez-Garcia, J. F., Schmitz, G., et al. (2008). Procera encodes a DELLA protein that mediates control of dissected leaf form in tomato. Plant J. 56, 603–612. doi: 10.1111/j.1365-313x.2008.03628.x
Jiang, L., Ma, X., Zhao, S., Tang, Y., Liu, F., Gu, P., et al. (2019). The APETALA2-like transcription factor SUPERNUMERARY BRACT controls rice seed shattering and seed size. Plant Cell 31, 17–36. doi: 10.1105/tpc.18.00304
Jiao, Y., Tausta, S. L., Gandotra, N., Sun, N., Liu, T., Clay, N. K., et al. (2009). A transcriptome atlas of rice cell types uncovers cellular, functional and developmental hierarchies. Nat. Genet. 41, 258–263. doi: 10.1038/ng.282
Johnsson, M., Gaynor, R. C., Jenko, J., Gorjanc, G., de Koning, D. J., and Hickey, J. M. (2019). Removal of alleles by genome editing (RAGE) against deleterious load. Genet. Sel. 51, 1–18.
Kadambari, G., Vemireddy, L. R., Srividhya, A., Nagireddy, R., Jena, S. S., Gandikota, M., et al. (2018). QTL-Seq-based genetic analysis identifies a major genomic region governing dwarfness in rice (Oryza sativa L.). Plant Cell Rep. 37, 677–687. doi: 10.1007/s00299-018-2260-2
Kang, G., Li, G., Wang, L., Wei, L., Yang, Y., Wang, P., et al. (2015). Hg-responsive proteins identified in wheat seedlings using iTRAQ analysis and the role of ABA in Hg stress. J. Proteome Res. 14, 249–267. doi: 10.1021/pr5006873
Kanno, Y., Jikumaru, Y., Hanada, A., Nambara, E., Abrams, S. R., Kamiya, Y., et al. (2010). Comprehensive hormone profiling in developing Arabidopsis seeds: examination of the site of ABA biosynthesis, ABA transport and hormone interactions. Plant Cell Physiol. 51, 1988–2001. doi: 10.1093/pcp/pcq158
Karimi, E., Jaafar, H. Z., and Ahmad, S. (2011). Phenolics and flavonoids profiling and antioxidant activity of three varieties of Malaysian indigenous medicinal herb Labisia pumila Benth. J. Med. Plant Res. 5, 1200–1206.
Khalil-Ur-Rehman, M., Sun, L., Li, C. X., Faheem, M., Wang, W., and Tao, J. M. (2017). Comparative RNA-seq based transcriptomic analysis of bud dormancy in grape. BMC Plant Biol. 17:18. doi: 10.1186/s12870-016-0960-8
Khan, S. A., Chibon, P. Y., de Vos, R. C., Schipper, B. A., Walraven, E., and Beekwilder, J. (2012). Genetic analysis of metabolites in apple fruits indicates an mQTL hotspot for phenolic compounds on linkage group 16. J. Exp. Bot. 63, 2895–2908. doi: 10.1093/jxb/err464
Kim, D., Alptekin, B., and Budak, H. (2018). CRISPR/Cas9 genome editing in wheat. Funct. Integr. 18, 31–41. doi: 10.1007/s10142-017-0572-x
Kim, J. S., An, C. G., Park, J. S., Lim, Y. P., and Kim, S. (2016). Carotenoid profiling from 27 types of paprika (Capsicum annuum L.) with different colors, shapes, and cultivation methods. Food Chem. 201, 64–71. doi: 10.1016/j.foodchem.2016.01.041
Kim, S. T., Kim, S. G., Hwang, D. H., Kang, S. Y., Kim, H. J., Lee, B. H., et al. (2004). Proteomic analysis of pathogen-responsive proteins from rice leaves induced by rice blast fungus, Magnaporthe grisea. Proteomics 4, 3569–3578. doi: 10.1002/pmic.200400999
Kiszonas, A. M., and Morris, C. F. (2018). Wheat breeding for quality: a historical review. Cereal Chem. 95, 17–34.
Knoch, D., Riewe, D., Meyer, R. C., Boudichevskaia, A., Schmidt, R., and Altmann, T. (2017). Genetic dissection of metabolite variation in Arabidopsis seeds: evidence for mQTL hotspots and a master regulatory locus of seed metabolism. J. Exp. Bot. 68, 1655–1667. doi: 10.1093/jxb/erx049
Kojima, K., Andou, D., and Ito, M., (2021). Plant hormone changes in growing small watermelon fruit. Hort. J. UTD-209. doi: 10.2503/hortj.UTD-209
Kojima, M., Kamada-Nobusada, T., Komatsu, H., Takei, K., Kuroha, T., Mizutani, M., et al. (2009). Highly sensitive and high-throughput analysis of plant hormones using MS-probe modification and liquid chromatography–tandem mass spectrometry: an application for hormone profiling in Oryza sativa. Plant Cell Physiol. 50, 1201–1214. doi: 10.1093/pcp/pcp057
Koller, A., Washburn, M. P., Lange, B. M., Andon, N. L., Deciu, C., Haynes, P. A., et al. (2002). Proteomic survey of metabolic pathways in rice. Proc. Natl. Acad. Sci. U.S.A. 99, 11969–11974. doi: 10.1073/pnas.172183199
Komatsuda, T., Pourkheirandish, M., He, C., Azhaguvel, P., Kanamori, H., and Perovic, D. et al. (2007). Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. U.S.A. 104, 1424–1429. doi: 10.1073/pnas.0608580104
Konishi, S., Izawa, T., Lin, S. Y., Ebana, K., Fukuta, Y., Sasaki, T., et al. (2006). An SNP caused loss of seed shattering during rice domestication. Science 312, 1392–1396. doi: 10.1126/science.1126410
Kosová, K., Vítámvás, P., Planchon, S., Renaut, J., Vanková, R., and Prášil, I. T. (2013). Proteome analysis of cold response in spring and winter wheat (Triticum aestivum) crowns reveals similarities in stress adaptation and differences in regulatory processes between the growth habits. J. Proteome Res. 12, 4830–4845. doi: 10.1021/pr400600g
Kretzschmar, T., Pelayo, M. A. F., Trijatmiko, K. R., Gabunada, L. F. M., Alam, R., and Jimenez, R. R. et al. (2015). A trehalose-6-phosphate phosphatase enhances anaerobic germination tolerance in rice. Nat. Plants 1, 1–5.
Krishnan, H. B., Natarajan, S. S., Bennett, J. O., and Sicher, R. C. (2011). Protein and metabolite composition of xylem sap from field-grown soybeans (Glycine max). Planta 233, 921–931. doi: 10.1007/s00425-011-1352-9
Kudapa, H., Garg, V., Chitikineni, A., and Varshney, R. K. (2018). The RNA-seq-based high resolution gene expression atlas of chickpea (Cicer arietinum L.) reveals dynamic spatio-temporal changes associated with growth and development. Plant Cell Environ. 41, 2209–2225.
Kumar, A., Bimolata, W., Kannan, M., Kirti, P. B., Qureshi, I. A., and Ghazi, I. A. (2015). Comparative proteomics reveals differential induction of both biotic and abiotic stress response associated proteins in rice during Xanthomonas oryzae pv. oryzae infection. Funct. Integr. Genomic 15, 425–437. doi: 10.1007/s10142-014-0431-y
Kumar, R., Bohra, A., Pandey, A. K., Pandey, M. K., and Kumar, A. (2017). Metabolomics for plant improvement: status and prospects. Front. Plant Sci. 8:1302. doi: 10.3389/fpls.2017.01302
Kumar, R., Janila, P., Vishwakarma, M. K., Khan, A. W., Manohar, S. S., and Gangurde, S. S. et al. (2020). Whole-genome resequencingbased QTL-seq identified candidate genes and molecular markers for fresh seed dormancy in groundnut. Plant Biotechnol. J. 18, 992–1003. doi: 10.1111/pbi.13266
Kumar, R., Tamboli, V., Sharma, R., and Sreelakshmi, Y. (2018). NAC-NOR mutations in tomato Penjar accessions attenuate multiple metabolic processes and prolong the fruit shelf life. Food Chem. 259, 234–244. doi: 10.1016/j.foodchem.2018.03.135
Kuroha, T., Nagai, K., Gamuyao, R., Wang, D. R., Furuta, T., Nakamori, M., et al. (2018). Ethylene-gibberellin signaling underlies adaptation of rice to periodic flooding. Science 361, 181–186. doi: 10.1126/science.aat1577
Lacchini, E., Kiegle, E., Castellani, M., Adam, H., Jouannic, S., Gregis, V., et al. (2020). CRISPR-mediated accelerated domestication of African rice landraces. PLoS One 15:e0229782. doi: 10.1371/journal.pone.0229782
Lei, L., Zheng, H., Bi, Y., Yang, L., Liu, H., Wang, J., et al. (2020). Identification of a Major QTL and candidate gene analysis of salt tolerance at the bud burst stage in rice (Oryza sativa L.) using QTL-seq and RNA-seq. Rice 13:55.
Lemmon, Z. H., Reem, N. T., Dalrymple, J., Soyk, S., Swartwood, K. E., Rodriguez-Leal, D., et al. (2018). Rapid improvement of domestication traits in an orphan crop by genome editing. Nat. Plants 4, 766–770. doi: 10.1038/s41477-018-0259-x
Lenser, T., and Theißen, G. (2013). Molecular mechanisms involved in convergent crop domestication. Trends Plant Sci. 18, 704–714. doi: 10.1016/j.tplants.2013.08.007
Levina, A. V., Hoekenga, O., Gordin, M., Broeckling, C., and De Jong, W. S. (2020). Genetic analysis of potato tuber metabolite composition: genome-wide association studies applied to a non-targeted metabolome. Crop Sci. 61, 591-603. doi: 10.1002/csc2.20398
Li, B., Zhang, Y., Mohammadi, S. A., Huai, D., Zhou, Y., and Kliebenstein, D. J. (2016). An integrative genetic study of rice metabolism, growth and stochastic variation reveals potential C/N partitioning loci. Sci. Rep. 6:30143.
Li, C., Zhou, A., and Sang, T. (2006). Rice domestication by reducing shattering. Science 311, 1936–1939. doi: 10.1126/science.1123604
Li, H., Peng, Z., Yang, X., Wang, W., Fu, J., Wang, J., et al. (2013). Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50. doi: 10.1038/ng.2484
Li, K., Wen, W., Alseekh, S., Yang, X., Guo, H., and Li, W. (2019). Large-scale metabolite quantitative trait locus analysis provides new insights for high-quality maize improvement. Plant J. 99, 216–230.
Li, T., Yang, X., Yu, Y., Si, X., Zhai, X., Zhang, H., et al. (2018). Domestication of wild tomato is accelerated by genome editing. Nat. Biotechnol. 36, 1160–1163. doi: 10.1038/nbt.4273
Li, T., Yun, Z., Wu, Q., Qu, H., Duan, X., and Jiang, Y. (2019). Combination of transcriptomic, proteomic, and metabolomic analysis reveals the ripening mechanism of banana pulp. Biomolecules 9:523. doi: 10.3390/biom9100523
Liang, Z., and Schnable, J. C. (2016). RNA-seq based analysis of population structure within the maize inbred B73. PLoS One 11:e0157942. doi: 10.1371/journal.pone.0157942
Libault, M., Farmer, A., Joshi, T., Takahashi, K., Langley, R. J., and Franklin, L. D. (2010). An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J. 63, 86–99.
Lifschitz, E., Eviatar, T., Rozman, A., Shalit, A., Goldshmidt, A., and Amsellem, Z. et al. (2006). The tomato FT ortholog triggers systemic signals that regulate growth and flowering and substitute for diverse environmental stimuli. Proc. Natl. Acad. Sci. U.S.A. 103, 6398–6403. doi: 10.1073/pnas.0601620103
Liljegren, S. J., Ditta, G. S., Eshed, Y., Savidge, B., Bowman, J. L., and Yanofsky, M. F. (2000). SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404, 766–770. doi: 10.1038/35008089
Liljegren, S. J., Roeder, A. H., Kempin, S. A., Gremski, K., Østergaard, L., Guimil, S., et al. (2004). Control of fruit patterning in Arabidopsis by INDEHISCENT. Cell J. 116, 843–853. doi: 10.1016/s0092-8674(04)00217-x
Lin, Z., Li, X., Shannon, L. M., Yeh, C. T., Wang, M. L., Bai, G., et al. (2012). Parallel domestication of the shattering1 genes in cereals. Nat. Genet. 44, 720–724. doi: 10.1038/ng.2281
Lipka, A. E., Gore, M. A., Magallanes-Lundback, M., Mesberg, A., Lin, H., Tiede, T., et al. (2013). Genome-wide association study and pathway-level analysis of tocochromanol levels in maize grain. G3 3, 1287–1299. doi: 10.1534/g3.113.006148
Lippman, Z. B., Cohen, O., Alvarez, J. P., Abu-Abied, M., Pekker, I., and Paran, I. et al. (2008). The making of a compound inflorescence in tomato and related nightshades. PLoS Biol. 6:288. doi: 10.1371/journal.pbio.0060288
Lisec, J., Steinfath, M., Meyer, R. C., Selbig, J., Melchinger, A. E., Willmitzer, L., et al. (2009). Identification of heterotic metabolite QTL in Arabidopsis thaliana RIL and IL populations. Plant J. 59, 777–788. doi: 10.1111/j.1365-313x.2009.03910.x
Liu, J., Chen, J., Zheng, X., Wu, F., Lin, Q., Heng, Y., et al. (2017). GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants 3:17043.
Liu, J., Van Eck, J., Cong, B., and Tanksley, S. D. (2002). A new class of regulatory genes underlying the cause of pear-shaped tomato fruit. Proc. Natl. Acad. Sci. U.S.A. 99, 13302–13306. doi: 10.1073/pnas.162485999
Liu, X., Sheng, J., and Curtiss, R. III (2011). Fatty acid production in genetically modified cyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 108, 6899–6904. doi: 10.1073/pnas.1103014108
Liu, Y., Pan, T., Tang, Y., Zhuang, Y., Liu, Z., Li, P., et al. (2020). Proteomic Analysis of rice subjected to low light stress and overexpression of OsGAPB increases the stress tolerance. Rice 13:30.
Liu, Z., Alseekh, S., Brotman, Y., Zheng, Y., Fei, Z., Tieman, D. M., et al. (2016). Identification of a Solanum pennellii chromosome 4 fruit flavor and nutritional quality-associated metabolite QTL. Front. Plant Sci. 7:1671. doi: 10.3389/fpls.2016.01671
Lu, X., Li, Q. T., Xiong, Q., Li, W., Bi, Y. D., Lai, Y. C., et al. (2016). The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 86, 530–544. doi: 10.1111/tpj.13181
Luo, H., Pandey, M. K., Khan, A. W., Guo, J., Wu, B., Cai, Y., et al. (2019). Discovery of genomic regions and candidate genes controlling shelling percentage using QTL-seq approach in cultivated peanut (Arachis hypogaea L.). Plant Biotechnol. J. 17, 1248–1260. doi: 10.1111/pbi.13050
Ma, J., Chen, J., Wang, M., Ren, Y., Wang, S., Lei, C., et al. (2018). Disruption of OsSEC3A increases the content of salicylic acid and induces plant defense responses in rice. J. Exp. Bot. 69, 1051–1064. doi: 10.1093/jxb/erx458
Macklin, A., Khan, S., and Kislinger, T. (2020). Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin. Proteomics 17:17.
Mangul, S., Martin, L. S., Hill, B. L., Lam, A. K. M., Distler, M. G., Zelikovsky, A., et al. (2019). Systematic benchmarking of omics computational tools. Nat. Commun. 10:1393.
Mao, L., Begum, D., Chuang, H. W., Budiman, M. A., Szymkowiak, E. J., Irish, E. E., et al. (2000). JOINTLESS is a MADS-box gene controlling tomato flower abscission zone development. Nature 406, 910–913. doi: 10.1038/35022611
Mao, X., Zheng, Y., Xiao, K., Wei, Y., Zhu, Y., Cai, Q., et al. (2018). OsPRX2 contributes to stomatal closure and improves potassium deficiency tolerance in rice. Biochem. Biophys. Res. Commun. 495, 461–467. doi: 10.1016/j.bbrc.2017.11.045
Marsch-Martínez, N., Ramos-Cruz, D., Irepan Reyes-Olalde, J., Lozano-Sotomayor, P., Zúñiga-Mayo, V. M., and de Folter, S. (2012). The role of cytokinin during Arabidopsis gynoecia and fruit morphogenesis and patterning. Plant J. 72, 222–234. doi: 10.1111/j.1365-313x.2012.05062.x
Martín-Trillo, M., Grandío, E. G., Serra, F., Marcel, F., Rodríguez-Buey, M. L., Schmitz, G., et al. (2011). Role of tomato BRANCHED1-like genes in the control of shoot branching. Plant J. 67, 701–714. doi: 10.1111/j.1365-313x.2011.04629.x
Matros, A., Liu, G., Hartmann, A., Jiang, Y., Zhao, Y., Wang, H., et al. (2017). Genome–metabolite associations revealed low heritability, high genetic complexity, and causal relations for leaf metabolites in winter wheat (Triticum aestivum). J. Exp. Bot. 68, 415–428.
Matsuda, F., Nakabayashi, R., Yang, Z., Okazaki, Y., Yonemaru, J., Ebana, K., et al. (2015). Metabolome-genome-wide association study (mGWAS) dissects genetic architecture for generating natural variation in rice secondary metabolism. Plant J. 81, 13–23. doi: 10.1111/tpj.12681
Matsuda, F., Okazaki, Y., Oikawa, A., Kusano, M., Nakabayashi, R., Kikuchi, J., et al. (2012). Dissection of genotype–phenotype associations in rice grains using metabolome quantitative trait loci analysis. Plant J. 70, 624–636. doi: 10.1111/j.1365-313x.2012.04903.x
Matsuura, T., Mori, I. C., Himi, E., and Hirayama, T. (2019). Plant hormone profiling in developing seeds of common wheat (Triticum aestivum L.). Breed. Sci. 69, 601–610. doi: 10.1270/jsbbs.19030
Meng, Y., Hou, Y., Wang, H., Ji, R., Liu, B., Wen, J., et al. (2017). Targeted mutagenesis by CRISPR/Cas9 system in the model legume medicago truncatula. Plant Cell Rep. 36, 371–374. doi: 10.1007/s00299-016-2069-9
Meyer, R. S., DuVal, A. E., and Jensen, H. R. (2012). Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol. 196, 29–48. doi: 10.1111/j.1469-8137.2012.04253.x
Mibei, E. K., Ambuko, J., Giovannoni, J. J., Onyango, A. N., and Owino, W. O. (2017). Carotenoid profiling of the leaves of selected African eggplant accessions subjected to drought stress. Food Sci. Nutr. 5, 113–122. doi: 10.1002/fsn3.370
Mieulet, D., Aubert, G., Bres, C., Klein, A., Droc, G., Vieille, E., et al. (2018). Unleashing meiotic crossovers in crops. Nat. Plants 4, 1010–1016. doi: 10.1038/s41477-018-0311-x
Miro, B., and Ismail, A. M. (2013). Tolerance of anaerobic conditions caused by flooding during germination and early growth in rice (Oryza sativa L.). Front. Plant Sci. 4:269. doi: 10.3389/fpls.2013.00269
Misra, B. B., Langefeld, C., Olivier, M., and Cox, L. A. (2019). Integrated omics: tools, advances and future approaches. J. Mol. Endocrinol. 62, R21–R45.
Monna, L., Kitazawa, N., Yoshino, R., Suzuki, J., Masuda, H., Maehara, Y., et al. (2002). Positional cloning of rice semidwarfing gene, sd-1: rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 9, 11–17. doi: 10.1093/dnares/9.1.11
Morineau, C., Bellec, Y., Tellier, F., Gissot, L., Kelemen, Z., Nogué, F., et al. (2017). Selective gene dosage by CRISPR-Cas9 genome editing in hexaploid Camelina sativa. Plant Biotechnol. J. 15, 729–739. doi: 10.1111/pbi.12671
Müller, M., and Munné-Bosch, S. (2011). Rapid and sensitive hormonal profiling of complex plant samples by liquid chromatography coupled to electrospray ionization tandem mass spectrometry. Plant Methods 7, 1–11.
Multani, D. S., Briggs, S. P., Chamberlin, M. A., Blakeslee, J. J., Murphy, A. S., and Johal, G. S. (2003). Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 302, 81–84. doi: 10.1126/science.1086072
Nagai, K., Mori, Y., Ishikawa, S., Furuta, T., Gamuyao, R., Niimi, Y., et al. (2020). Antagonistic regulation of the gibberellic acid response during stem growth in rice. Nature 584, 109–114. doi: 10.1038/s41586-020-2501-8
Nandi, S., Subudhi, P. K., Senadhira, D., Manigbas, N. L., Sen-Mandi, S., and Huang, N. (1997). Mapping QTLs for submergence tolerance in rice by AFLP analysis and selective genotyping. Mol. Gen. Genet. 255, 1–8. doi: 10.1007/s004380050468
Nayak, S. N., Hebbal, V., Soni, P., Kumar, R., Pandey, A. K., Wan, L., et al. (2019). Groundnut Kernel Transcriptome. In Comprehensive Foodomics. Amsterdam: Elsevier.
Neeraja, C. N., Maghirang-Rodriguez, R., Pamplona, A., Heuer, S., Collard, B. C., Septiningsih, E. M., et al. (2007). A marker-assisted backcross approach for developing submergence-tolerant rice cultivars. Theor. Appl. Genet. 115, 767–776. doi: 10.1007/s00122-007-0607-0
Nieves-Cordones, M., Mohamed, S., Tanoi, K., Kobayashi, N. I., Takagi, K., Vernet, A., et al. (2017). Production of low-Cs+ rice plants by inactivation of the K+ transporter Os HAK 1 with the CRISPR-Cas system. Plant J. 92, 43–56. doi: 10.1111/tpj.13632
Nonaka, S., Arai, C., Takayama, M., Matsukura, C., and Ezura, H. (2017). Efficient increase of γ-aminobutyric acid (GABA) content in tomato fruits by targeted mutagenesis. Sci. Rep. 7:7057.
Novák, O., Hauserová, E., Amakorová, P., Doležal, K., and Strnad, M. (2008). Cytokinin profiling in plant tissues using ultra-performance liquid chromatography–electrospray tandem mass spectrometry. Phytochemistry 69, 2214–2224. doi: 10.1016/j.phytochem.2008.04.022
Nubankoh, P., Wanchana, S., Saensuk, C., Ruanjaichon, V., Cheabu, S., Vanavichit, A., et al. (2020). QTL-seq reveals genomic regions associated with spikelet fertility in response to a high temperature in rice (Oryza sativa L.). Plant Cell Rep. 39, 149–162. doi: 10.1007/s00299-019-02477-z
Ogawa, M., Kusano, T., Katsumi, M., and Sano, H. (2000). Rice gibberellin-insensitive gene homolog, OsGAI, encodes a nuclear-localized protein capable of gene activation at transcriptional level. Gene 245, 21–29. doi: 10.1016/s0378-1119(00)00018-4
Ogiso-Tanaka, E., Tanaka, T., Tanaka, K., Nonoue, Y., Sasaki, T., Fushimi, E., et al. (2017). Detection of novel QTLs qDTH4. 5 and qDTH6. 3, which confer late heading under short-day conditions, by SSR marker-based and QTL-seq analysis. Breed. Sci. 67, 101–109. doi: 10.1270/jsbbs.16096
Okuzaki, A., Ogawa, T., Koizuka, C., Kaneko, K., Inaba, M., Imamura, J., et al. (2018). CRISPR/Cas9-mediated genome editing of the fatty acid desaturase 2 gene in Brassica napus. Plant Physiol. Biochem. 131, 63–69. doi: 10.1016/j.plaphy.2018.04.025
Oladosu, Y., Rafii, M. Y., Arolu, F., Chukwu, S. C., Muhammad, I., Kareem, I., et al. (2020). Submergence tolerance in rice: review of mechanism, breeding and, future prospects. Sustainability 12:1632. doi: 10.3390/su12041632
Olsen, K. M., and Wendel, J. F. (2013). A bountiful harvest: genomic insights into crop domestication phenotypes. Annu. Rev. Plant Biol. 64, 47–70. doi: 10.1146/annurev-arplant-050312-120048
Olszewski, N., Sun, T. P., and Gubler, F. (2002). Gibberellin signalling: biosynthesis, catabolism, and response pathways. Plant Cell 14(Suppl. 1), S61–S80.
Ortiz-Ramírez, C., Hernandez-Coronado, M., Thamm, A., Catarino, B., Wang, M., and Dolan, L. (2016). A transcriptome atlas of Physcomitrella patens provides insights into the evolution and development of land plants. Mol. Plant 9, 205–220. doi: 10.1016/j.molp.2015.12.002
Osorio, S., Alba, R., Damasceno, C. M., Lopez-Casado, G., Lohse, M., Zanor, M. I., et al. (2011). Systems biology of tomato fruit development: combined transcript, protein, and metabolite analysis of tomato transcription factor (nor, rin) and ethylene receptor (Nr) mutants reveals novel regulatory interactions. Plant Physiol. 157, 405–425. doi: 10.1104/pp.111.175463
Østergaard, L., Kempin, S. A., Bies, D., Klee, H. J., and Yanofsky, M. F. (2006). Pod shatter-resistant Brassica fruit produced by ectopic expression of the FRUITFULL gene. Plant Biotechnol. J. 4, 45–51. doi: 10.1111/j.1467-7652.2005.00156.x
Owens, B. F., Lipka, A. E., Magallanes-Lundback, M., Tiede, T., Diepenbrock, C. H., Kandianis, C. B., et al. (2014). A foundation for provitamin A biofortification of maize: genome-wide association and genomic prediction models of carotenoid levels. Genetics 198, 1699–1716. doi: 10.1534/genetics.114.169979
Palaisa, K., Morgante, M., Tingey, S., and Rafalski, A. (2004). Long-range patterns of diversity and linkage disequilibrium surrounding the maize Y1 gene are indicative of an asymmetric selective sweep. Proc. Natl. Acad. Sci. U.S.A. 101, 9885–9890. doi: 10.1073/pnas.0307839101
Pandey, M. K., Pandey, A. K., Kumar, R., Nwosu, C. V., Guo, B., Wright, G. C., et al. (2020). Translational genomics for achieving higher genetic gains in groundnut. Theore. Appl. Genet. 133, 1679–1702. doi: 10.1007/s00122-020-03592-2
Peivastegan, B., Hadizadeh, I., Nykyri, J., Nielsen, K. L., Somervuo, P., Sipari, N., et al. (2019). Effect of wet storage conditions on potato tuber transcriptome, phytohormones and growth. BMC Plant Biol. 19:262. doi: 10.1186/s12870-019-1875-y
Peng, J., Carol, P., Richards, D. E., King, K. E., Cowling, R. J., Murphy, G. P., et al. (1997). The Arabidopsis GAI gene defines a signalling pathway that negatively regulates gibberellin responses. Genes Dev. 11, 3194–3205. doi: 10.1101/gad.11.23.3194
Peng, J., Richards, D. E., Hartley, N. M., Murphy, G. P., Devos, K. M., Flintham, J. E., et al. (1999). ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 400, 256–261. doi: 10.1038/22307
Perez-Fons, L., Wells, T., Corol, D. I., Ward, J. L., Gerrish, C., et al. (2014). A genome-wide metabolomic resource for tomato fruit from Solanum pennellii. Sci. Rep. 4:3859.
Piasecka, A., Sawikowska, A., Kuczyńska, A., Ogrodowicz, P., Mikołajczak, K., Krystkowiak, K., et al. (2017). Drought-related secondary metabolites of barley (Hordeum vulgare L.) leaves and their metabolomic quantitative trait loci. Plant J. 89, 898–913. doi: 10.1111/tpj.13430
Pnueli, L., Carmel-Goren, L., Hareven, D., Gutfinger, T., Alvarez, J., Ganal, M., et al. (1998). The SELF-PRUNING gene of tomato regulates vegetative to reproductive switching of sympodial meristems and is the ortholog of CEN and TFL1. Development 125, 1979–1989.
Pott, D. M., Vallarino, J. G., Cruz-Rus, E., Willmitzer, L., Sánchez-Sevilla, J. F., Amaya, I., et al. (2020). Genetic analysis of phenylpropanoids and antioxidant capacity in strawberry fruit reveals mQTL hotspots and candidate genes. Sci. Rep. 10:20197.
Powell, A. L., Nguyen, C. V., Hill, T., Cheng, K. L., Figueroa-Balderas, R., Aktas, H., et al. (2012). Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science 336, 1711–1715. doi: 10.1126/science.1222218
Praveen, A., Pandey, A., and Gupta, M. (2020). Protective role of nitric oxide on nitrogen-thiol metabolism and amino acids profiling during arsenic exposure in Oryza sativa L. Ecotoxicology 29, 825–836. doi: 10.1007/s10646-020-02250-z
Price, E. J., Bhattacharjee, R., Lopez-Montes, A., and Fraser, P. D. (2018). Carotenoid profiling of yams: clarity, comparisons and diversity. Food Chem. 259, 130–138. doi: 10.1016/j.foodchem.2018.03.066
Purugganan, M. D., and Fuller, D. Q. (2009). The nature of selection during plant domestication. Nature 457, 843–848. doi: 10.1038/nature07895
Pysh, L. D., Wysocka-Diller, J. W., Camilleri, C., Bouchez, D., and Benfey, P. N. (1999). The GRAS gene family in Arabidopsis: sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J. 18, 111–119. doi: 10.1046/j.1365-313x.1999.00431.x
Qi, C., Jiang, H., Xiong, J., Yuan, B., and Feng, Y. (2019). On-line trapping/capillary hydrophilic-interaction liquid chromatography/mass spectrometry for sensitive determination of RNA modifications from human blood. Chin. Chem. Lett. 30, 553–557. doi: 10.1016/j.cclet.2018.11.029
Qi, J., Sun, P., Liao, D., Sun, T., Zhu, J., and Li, X. (2015). Transcriptomic analysis of American ginseng seeds during the dormancy release process by RNA-Seq. PLoS One 10:e0118558. doi: 10.1371/journal.pone.0118558
Qiao, A., Fang, X., Liu, S., Liu, H., Gao, P., and Luan, F. (2021). QTL-seq identifies major quantitative trait loci of stigma color in melon. Hortic. Plant J. doi: 10.1016/j.hpj.2021.01.004
Qin, Y., Cheng, P., Cheng, Y., Feng, Y., Huang, D., Huang, T., et al. (2018). QTL-Seq identified a major QTL for grain length and weight in rice using near isogenic F2 population. Rice Sci. 25, 121–131. doi: 10.1016/j.rsci.2018.04.001
Ramos, A., Fu, Y., Michael, V., and Meru, G. (2020). QTL-seq for identification of loci associated with resistance to Phytophthora crown rot in squash. Sci. Rep. 10:5326.
Razzaq, A., Sadia, B., Raza, A., Khalid Hameed, M., and Saleem, F. (2019). Metabolomics: a way forward for crop improvement. Metabolites 9:303. doi: 10.3390/metabo9120303
Reddy, M. M., and Ulaganathan, K. (2015). RNA-Seq analysis of urea nutrition responsive transcriptome of Oryza sativa elite indica cultivar RP Bio 226. Genom. Data 6, 112–113. doi: 10.1016/j.gdata.2015.08.025
Ren, J. L., Zhang, A. H., Kong, L., and Wang, X. J. (2018). Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC Adv. 8, 22335–22350. doi: 10.1039/c8ra01574k
Riedelsheimer, C., Lisec, J., Czedik-Eysenberg, A., Sulpice, R., Flis, A., Grieder, C., et al. (2012). Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. U.S.A. 109, 8872–8877. doi: 10.1073/pnas.1120813109
Rivas, M. A., Beaudoin, M., Gardet, A., Stevens, C., Sharma, Y., Zhang, C. K., et al. (2011). Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073. doi: 10.1038/ng.952
Rodríguez-Leal, D., Lemmon, Z. H., Man, J., Bartlett, M. E., and Lippman, Z. B. (2017). Engineering quantitative trait variation for crop improvement by genome editing. Cell 171, 470–480. doi: 10.1016/j.cell.2017.08.030
Roldan, M. V. G., Périlleux, C., Morin, H., Huerga-Fernandez, S., Latrasse, D., Benhamed, M., et al. (2017). Natural and induced loss of function mutations in SlMBP21 MADS-box gene led to jointless-2 phenotype in tomato. Sci. Rep. 7:4402.
Ronen, G., Carmel-Goren, L., Zamir, D., and Hirschberg, J. (2000). An alternative pathway to β-carotene formation in plant chromoplasts discovered by map-based cloning of Beta and old-gold color mutations in tomato. Proc. Natl. Acad. Sci. U.S.A. 97, 11102–11107. doi: 10.1073/pnas.190177497
Rowe, H. C., Hansen, B. G., Halkier, B. A., and Kliebenstein, D. J. (2008). Biochemical networks and epistasis shape the Arabidopsis thaliana metabolome. Plant Cell 20, 1199–1216. doi: 10.1105/tpc.108.058131
Ruan, S. L., Ma, H. S., Wang, S. H., Fu, Y. P., Xin, Y., Liu, W. Z., et al. (2011). Proteomic identification of OsCYP2 a rice cyclophilin that confers salt tolerance in rice (Oryza sativa L.) seedlings when overexpressed. BMC Plant Biol. 11:34. doi: 10.1186/1471-2229-11-34
Sasaki, A., Ashikari, M., Ueguchi-Tanaka, M., Itoh, H., Nishimura, A., Swapan, D., et al. (2002). A mutant gibberellin-synthesis gene in rice. Nature 416, 701–702. doi: 10.1038/416701a
Sauer, N. J., Narváez-Vásquez, J., Mozoruk, J., Miller, R. B., Warburg, Z. J., Woodward, M. J., et al. (2016). Oligonucleotide-mediated genome editing provides precision and function to engineered nucleases and antibiotics in plants. Plant Physiol. 170, 1917–1928. doi: 10.1104/pp.15.01696
Sauvage, C., Segura, V., Bauchet, G., Stevens, R., Do, P. T., Nikoloski, Z., et al. (2014). Genome-wide association in tomato reveals 44 candidate loci for fruit metabolic traits. Plant Physiol. 165, 1120–1132. doi: 10.1104/pp.114.241521
Schauer, N., Semel, Y., Balbo, I., Steinfath, M., Repsilber, D., Selbig, J., et al. (2008). Mode of inheritance of primary metabolic traits in tomato. Plant Cell 20, 509–523. doi: 10.1105/tpc.107.056523
Schauer, N., Semel, Y., Roessner, U., Gur, A., Balbo, I., Carrari, F., et al. (2006). Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 24, 447–454. doi: 10.1038/nbt1192
Schindele, A., Dorn, A., and Puchta, H. (2020). CRISPR/Cas brings plant biology and breeding into the fast lane. Curr. Opin. Biotechnol. 61, 7–14. doi: 10.1016/j.copbio.2019.08.006
Schmidt, J., Blessing, F., Fimpler, L., and Wenzel, F. (2020). Nanopore sequencing in a clinical routine laboratory: challenges and opportunities. Clin. Lab. 66:1097.
Sekhon, R. S., Briskine, R., Hirsch, C. N., Myers, C. L., Springer, N. M., Buell, C. R., et al. (2013). Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS One 8:e61005. doi: 10.1371/journal.pone.0061005
Septiningsih, E. M., Ignacio, J. C. I., Sendon, P. M., Sanchez, D. L., Ismail, A. M., and Mackill, D. J. (2013). QTL mapping and confirmation for tolerance of anaerobic conditions during germination derived from the rice landrace Ma-Zhan Red. Theor. Appl. Genet. 126, 1357–1366. doi: 10.1007/s00122-013-2057-1
Severin, A. J., Woody, J. L., Bolon, Y. T., Joseph, B., Diers, B. W., Farmer, A. D., et al. (2010). RNA-seq atlas of glycine max: a guide to the soybean transcriptome. BMC Plant Biol. 10:160. doi: 10.1186/1471-2229-10-160
Seymour, G. B., Chapman, N. H., Chew, B. L., and Rose, J. K. (2013). Regulation of ripening and opportunities for control in tomato and other fruits. Plant Biotech. J. 11, 269–278. doi: 10.1111/j.1467-7652.2012.00738.x
Shan, Q., Wang, Y., Li, J., and Gao, C. (2014). Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 9, 2395–2410. doi: 10.1038/nprot.2014.157
Shan, Q., Wang, Y., Li, J., Zhang, Y., Chen, K., Liang, Z., et al. (2013). Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 31, 686–688. doi: 10.1038/nbt.2650
Sharma, V., Gupta, P., Priscilla, K., SharanKumar, A., Hangargi, B., Veershetty, A., et al. (2021). metabolomics intervention towards better understanding of plant traits. Cells 10:346. doi: 10.3390/cells10020346
Shen, M., Broeckling, C. D., Chu, E. Y., Ziegler, G., Baxter, I. R., Prenni, J. E., et al. (2013). Leveraging non-targeted metabolite profiling via statistical genomics. PLoS One 8:e57667. doi: 10.1371/journal.pone.0057667
Shi, T., Zhu, A., Jia, J., Hu, X., Chen, J., Liu, W., et al. (2020). Metabolomics analysis and metabolite-agronomic trait associations using kernels of wheat (Triticum aestivum) recombinant inbred lines. Plant J. 103, 279–292. doi: 10.1111/tpj.14727
Shomura, A., Izawa, T., Ebana, K., Ebitani, T., Kanegae, H., Konishi, S., et al. (2008). Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 40, 1023–1028. doi: 10.1038/ng.169
Si, L., Chen, J., Huang, X., Gong, H., Luo, J., Hou, Q., et al. (2016). OsSPL13 controls grain size in cultivated rice. Nat. Genet. 48, 447–456. doi: 10.1038/ng.3518
Siangliw, M., Toojinda, T., Tragoonrung, S., and Vanavichit, A. (2003). Thai jasmine rice carrying QTLch9 (Sub QTL) is submergence tolerant. Ann. Bot. 91, 255–261. doi: 10.1093/aob/mcf123
Sigmon, B., and Vollbrecht, E. (2010). Evidence of selection at the ramosa1 locus during maize domestication. Mol. Ecol. 19, 1296–1311. doi: 10.1111/j.1365-294x.2010.04562.x
Silverstone, A. L., and Sun, T. (2000). Gibberellins and the green revolution. Trends Plant Sci. 5, 1–2. doi: 10.1016/s1360-1385(99)01516-2
Simons, K. J., Fellers, J. P., Trick, H. N., Zhang, Z., Tai, Y. S., Gill, B. S., et al. (2006). Molecular characterization of the major wheat domestication gene Q. Genetics 172, 547–555. doi: 10.1534/genetics.105.044727
Singh, A., Dubey, P. K., Chaurasia, R., Dubey, R. K., Pandey, K. K., Singh, G. S., et al. (2019). Domesticating the undomesticated for global food and nutritional security: four steps. Agron J. 9:491. doi: 10.3390/agronomy9090491
Singh, V. K., Khan, A. W., Jaganathan, D., Thudi, M., Roorkiwal, M., Takagi, H., et al. (2016). QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotechnol. J. 14, 2110–2119. doi: 10.1111/pbi.12567
Sinha, P., Bajaj, P., Pazhamala, L., Nayak, S., Pandey, M. K., Chitikineni, A., et al. (2020). The Arachis hypogaea gene expression atlas (AhGEA) for accelerating translational research in cultivated groundnut. Plant Biotechnol. J.
Smýkal, P., Nelson, M. N., Berger, J. D., and Von Wettberg, E. J. (2018). The impact of genetic changes during crop domestication. Agronomy 8:119. doi: 10.3390/agronomy8070119
Song, J., Li, Z., Liu, Z., Guo, Y., and Qiu, L. J. (2017). Next-generation sequencing from bulked-segregant analysis accelerates the simultaneous identification of two qualitative genes in soybean. Front. Plant Sci. 8:919. doi: 10.3389/fpls.2017.00919
Sorefan, K., Girin, T., Liljegren, S. J., Ljung, K., Robles, P., Galván-Ampudia, C. S., et al. (2009). A regulated auxin minimum is required for seed dispersal in Arabidopsis. Nature 459, 583–586. doi: 10.1038/nature07875
Sosso, D., Luo, D., Li, Q. B., Sasse, J., Yang, J., Gendrot, G., et al. (2015). Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 47, 1489–1493. doi: 10.1038/ng.3422
Soyk, S., Müller, N. A., Park, S. J., Schmalenbach, I., Jiang, K., Hayama, R., et al. (2017). Variation in the flowering gene SELF PRUNING 5G promotes day-neutrality and early yield in tomato. Nat. Genet. 49, 162–168. doi: 10.1038/ng.3733
Spielmeyer, W., Ellis, M. H., and Chandler, P. M. (2002). Semidwarf (sd-1),“green revolution” rice, contains a defective gibberellin 20-oxidase gene. Proc. Natl. Acad. Sci. U.S.A. 99, 9043–9048. doi: 10.1073/pnas.132266399
Studer, A., Zhao, Q., Ross-Ibarra, J., and Doebley, J. (2011). Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 43, 1160–1163. doi: 10.1038/ng.942
Sugimoto, K., Takeuchi, Y., Ebana, K., Miyao, A., Hirochika, H., Hara, N., et al. (2010). Molecular cloning of Sdr4, a regulator involved in seed dormancy and domestication of rice. Proc. Natl. Acad. Sci. U.S.A. 107, 5792–5797. doi: 10.1073/pnas.0911965107
Sun, S., and Frelich, L. E. (2011). Flowering phenology and height growth pattern are associated with maximum plant height, relative growth rate and stem tissue mass density in herbaceous grassland species. J. Ecol. 99, 991–1000. doi: 10.1111/j.1365-2745.2011.01830.x
Sun, T. P., and Gubler, F. (2004). Molecular mechanism of gibberellin signaling in plants. Annu. Rev. Plant Biol. 55, 197–223. doi: 10.1146/annurev.arplant.55.031903.141753
Sun, Y., Wang, F., Wang, N., Dong, Y., Liu, Q., Zhao, L., et al. (2013). Transcriptome exploration in Leymus chinensis under saline-alkaline treatment using 454 pyrosequencing. PLoS One 8:e53632. doi: 10.1371/journal.pone.0053632
Sweeney, M. T., Thomson, M. J., Pfeil, B. E., and McCouch, S. (2006). Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 18, 283–294. doi: 10.1105/tpc.105.038430
Takagi, H., Uemura, A., Yaegashi, H., Tamiru, M., Abe, A., Mitsuoka, C., et al. (2013). MutMap-Gap: whole-genome resequencing of mutant F 2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol. 200, 276–283. doi: 10.1111/nph.12369
Taketa, S., Amano, S., Tsujino, Y., Sato, T., Saisho, D., Kakeda, K., et al. (2008). Barley grain with adhering hulls is controlled by an ERF family transcription factor gene regulating a lipid biosynthesis pathway. Proc. Natl. Acad. Sci. U.S.A. 105, 4062–4067. doi: 10.1073/pnas.0711034105
Talebi, A. F., Mohtashami, S. K., Tabatabaei, M., Tohidfar, M., Bagheri, A., Zeinalabedini, M., et al. (2013). Fatty acids profiling: a selective criterion for screening microalgae strains for biodiesel production. Algal Res. 2, 258–267. doi: 10.1016/j.algal.2013.04.003
Tang, H., Cuevas, H. E., Das, S., Sezen, U. U., Zhou, C., Guo, H., et al. (2013). Seed shattering in a wild Sorghum is conferred by a locus unrelated to domestication. Proc. Natl. Acad. Sci. 110, 15824–15829. doi: 10.1073/pnas.1305213110
Tang, Y. J., and Aristilde, L. (2020). Editorial overview: analytical biotechnology in the era of high-performance omics, synthetic biology, and machine learning. Curr. Opin. Biotechnol. 64, iii–vi. doi: 10.1016/j.copbio.2020.07.009
Templer, S. E., Ammon, A., Pscheidt, D., Ciobotea, O., Schuy, C., McCollum, C., et al. (2017). Metabolite profiling of barley flag leaves under drought and combined heat and drought stress reveals metabolic QTLs for metabolites associated with antioxidant defense. J. Exp. Bot. 68, 1697–1713. doi: 10.1093/jxb/erx038
Thomas, S. G. (2017). Novel Rht-1 dwarfing genes: tools for wheat breeding and dissecting the function of DELLA proteins. J. Exp. Bot. 68, 354–358. doi: 10.1093/jxb/erw509
Thudi, M., Chitikineni, A., Liu, X., He, W., Roorkiwal, M., Yang, W., et al. (2016). Recent breeding programs enhanced genetic diversity in both desi and kabuli varieties of chickpea (Cicer arietinum L.). Sci. Rep. 6:38636.
Tian, Z., Wang, X., Lee, R., Li, Y., Specht, J. E., Nelson, R. L., et al. (2010). Artificial selection for determinate growth habit in soybean. Proc. Natl. Acad. Sci. U.S.A. 107, 8563–8568. doi: 10.1073/pnas.1000088107
Toojinda, T., Siangliw, M., Tragoonrung, S., and Vanavichit, A. (2003). Molecular genetics of submergence tolerance in rice: QTL analysis of key traits. Ann. Bot. 91, 243–253. doi: 10.1093/aob/mcf072
Toojinda, T., Tragoonrung, S., Vanavichit, A., Siangliw, J. L., Pa-In, N., et al. (2005). Molecular breeding for rainfed lowland rice in the Mekong region. Plant Prod. Sci. 8, 330–333. doi: 10.1626/pps.8.330
Torres, N., Hilbert, G., Antolín, M. C., and Goicoechea, N. (2019). Aminoacids and flavonoids profiling in Tempranillo berries can be modulated by the arbuscular mycorrhizal fungi. Plants 8:400. doi: 10.3390/plants8100400
Toubiana, D., Semel, Y., Tohge, T., Beleggia, R., Cattivelli, L., Rosental, L., et al. (2012). Metabolic profiling of a mapping population exposes new insights in the regulation of seed metabolism and seed, fruit, and plant relations. PLoS Genet. 8:e1002612. doi: 10.1371/journal.pgen.1002612
Toyomasu, T., Kawaide, H., Sekimoto, H., Von Numers, C., Phillips, A. L., Hedden, P., et al. (1997). Cloning and characterization of a cDNA encoding gibberellin 20-oxidase from rice (Oryza sativa) seedlings. Physiol. Plant 99, 111–118. doi: 10.1034/j.1399-3054.1997.990116.x
Tudor, E. H., Jones, D. M., He, Z., Bancroft, I., Trick, M., Wells, R., et al. (2020). QTL-seq identifies BnaFT. A02 and BnaFLC. A02 as candidates for variation in vernalization requirement and response in winter oilseed rape (Brassica napus). Plant Biotechnol. J. 18, 2466–2481. doi: 10.1111/pbi.13421
Unamba, C. I., Nag, A., and Sharma, R. K. (2015). Next generation sequencing technologies: the doorway to the unexplored genomics of non-model plants. Front. Plant Sci. 6:1074. doi: 10.3389/fpls.2015.01074
Vallarino, J. G., Pott, D. M., Cruz-Rus, E., Miranda, L., Medina-Minguez, J. J., Valpuesta, V., et al. (2019). Identification of quantitative trait loci and candidate genes for primary metabolite content in strawberry fruit. Hortic. Res. 6:4.
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D., and Thermes, C. (2018). The third revolution in sequencing technology. Trends Genet. 34, 666–681. doi: 10.1016/j.tig.2018.05.008
Varkonyi-Gasic, E., Wang, T., Voogd, C., Jeon, S., Drummond, R. S., Gleave, A. P., et al. (2019). Mutagenesis of kiwifruit CENTRORADIALIS-like genes transforms a climbing woody perennial with long juvenility and axillary flowering into a compact plant with rapid terminal flowering. Plant Biotech. J. 17, 869–880. doi: 10.1111/pbi.13021
Varshney, R. K., Thudi, M., Roorkiwal, M., He, W., Upadhyaya, H. D., Yang, W., et al. (2019). Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nat. Genet. 51, 857–864. doi: 10.1038/s41588-019-0401-3
Vidigal, P., Duarte, B., Cavaco, A. R., Caçador, I., Figueiredo, A., Matos, A. R., et al. (2018). Preliminary diversity assessment of an undervalued tropical bean (Lablab purpureus (L.) Sweet) through fatty acid profiling. Plant Physiol. Biochem. 132, 508–514. doi: 10.1016/j.plaphy.2018.10.001
Wan, L., Li, B., Lei, Y., Yan, L., Huai, D., Kang, Y., et al. (2018). Transcriptomic profiling reveals pigment regulation during peanut testa development. Plant Physiol. Biochem. 125, 116–125. doi: 10.1016/j.plaphy.2018.01.029
Wan, L., Li, B., Lei, Y., Yan, L., Ren, X., Chen, Y., et al. (2017). Mutant transcriptome sequencing provides insights into pod development in peanut (Arachis hypogaea L.). Front. Plant Sci. 8:1900. doi: 10.3389/fpls.2017.01900
Wan, L., Li, B., Pandey, M. K., Wu, Y., Lei, Y., Yan, L., et al. (2016). Transcriptome analysis of a new peanut seed coat mutant for the physiological regulatory mechanism involved in seed coat cracking and pigmentation. Front. Plant Sci. 7:1491. doi: 10.3389/fpls.2016.01491
Wang, D., Cao, D., Zong, Y., Li, Y., Wang, J., Li, Z., et al. (2021). Bulked QTL-Seq identified a major QTL for the awnless trait in spring wheat cultivars in Qinghai. China. Biotechnol. Biotechnol. Equip. 35, 124–130. doi: 10.1080/13102818.2020.1857661
Wang, H., Cheng, H., Wang, W., Liu, J., Hao, M., Mei, D., et al. (2016). Identification of BnaYUCCA6 as a candidate gene for branch angle in Brassica napus by QTL-seq. Sci. Rep. 6:38493.
Wang, H., Studer, A. J., Zhao, Q., Meeley, R., and Doebley, J. F. (2015). Evidence that the origin of naked kernels during maize domestication was caused by a single amino acid substitution in tga1. Genetics 200, 965–974. doi: 10.1534/genetics.115.175752
Wang, T., Wei, J. J., Sabatini, D. M., and Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. doi: 10.1126/science.1246981
Wang, W., Pan, Q., He, F., Akhunova, A., Chao, S., Trick, H., et al. (2018). Transgenerational CRISPR-Cas9 activity facilitates multiplex gene editing in allopolyploid wheat. CRISPR J. 1, 65–74. doi: 10.1089/crispr.2017.0010
Wang, Y., Clevenger, J. P., Illa-Berenguer, E., Meulia, T., van der Knaap, E., and Sun, L. (2019). A comparison of sun, ovate, fs8. 1 and auxin application on tomato fruit shape and gene expression. Plant Cell Physiol. 60, 1067–1081. doi: 10.1093/pcp/pcz024
Wang, Y., Li, B., Du, M., Eneji, A. E., Wang, B., Duan, L., et al. (2012). Mechanism of phytohormone involvement in feedback regulation of cotton leaf senescence induced by potassium deficiency. J. Exp. Bot. 63, 5887–5901. doi: 10.1093/jxb/ers238
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wen, W., Jin, M., Li, K., Liu, H., Xiao, Y., Zhao, M., et al. (2018). An integrated multi-layered analysis of the metabolic networks of different tissues uncovers key genetic components of primary metabolism in maize. Plant J. 93, 1116–1128. doi: 10.1111/tpj.13835
Wen, W., Li, D., Li, X., Gao, Y., Li, W., Li, H., et al. (2014). Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 5:3438.
Wen, W., Li, K., Alseekh, S., Omranian, N., Zhao, L., Zhou, Y., et al. (2015). Genetic determinants of the network of primary metabolism and their relationships to plant performance in a maize recombinant inbred line population. Plant Cell 27, 1839–1856. doi: 10.1105/tpc.15.00208
Weng, J., Gu, S., Wan, X., Gao, H., Guo, T., Su, N., et al. (2008). Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 18, 1199–1209. doi: 10.1038/cr.2008.307
Whipple, C. J., Kebrom, T. H., Weber, A. L., Yang, F., Hall, D., Meeley, R., et al. (2011). grassy tillers1 promotes apical dominance in maize and responds to shade signals in the grasses. Proc. Natl. Acad. Sci. U.S.A. 108, E506–E512.
Willits, M. G., Kramer, C. M., Prata, R. T., De Luca, V., Potter, B. G., et al. (2005). Utilization of the genetic resources of wild species to create a non-transgenic high flavonoid tomato. J. Agric. Food Chem. 53, 1231–1236. doi: 10.1021/jf049355i
Witzel, K., Weidner, A., Surabhi, G. K., Börner, A., and Mock, H. P. (2009). Salt stress-induced alterations in the root proteome of barley genotypes with contrasting response towards salinity. J. Exp. Bot. 60, 3545–3557. doi: 10.1093/jxb/erp198
Woo, J. W., Kim, J., Kwon, S. I., Corvalán, C., Cho, S. W., Kim, H., et al. (2015). DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat. Biotechnol. 33, 1162–1164. doi: 10.1038/nbt.3389
Wu, N., Lu, Q., Wang, P., Zhang, Q., Zhang, J., Qu, J., et al. (2020). Construction and analysis of GmFAD2-1A and GmFAD2-2A soybean fatty acid desaturase mutants based on CRISPR/Cas9 technology. Int. J. Mol. Sci. 21:1104. doi: 10.3390/ijms21031104
Wu, S., Alseekh, S., Cuadros-Inostroza, Á, Fusari, C. M., Mutwil, M., Kooke, R., et al. (2016). Combined use of genome-wide association data and correlation networks unravels key regulators of primary metabolism in Arabidopsis thaliana. PLoS Genet. 12:e1006363. doi: 10.1371/journal.pgen.1006363
Wu, S., Tohge, T., Cuadros-Inostroza, Á, Tong, H., Tenenboim, H., Kooke, R., et al. (2018). Mapping the Arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Mol. Plant 11, 118–134. doi: 10.1016/j.molp.2017.08.012
Wu, Y., Li, X., Xiang, W., Zhu, C., Lin, Z., Wu, Y., et al. (2012). Presence of tannins in sorghum grains is conditioned by different natural alleles of Tannin1. Proc. Natl. Acad. Sci. U.S.A. 109, 10281–10286. doi: 10.1073/pnas.1201700109
Xiao, H., Jiang, N., Schaffner, E., Stockinger, E. J., and Van Der Knaap, E. (2008). A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319, 1527–1530. doi: 10.1126/science.1153040
Xie, K., and Yang, Y. (2013). RNA-guided genome editing in plants using a CRISPR–Cas system. Mol. Plant. 6, 1975–1983. doi: 10.1093/mp/sst119
Xu, C., Liberatore, K. L., MacAlister, C. A., Huang, Z., Chu, Y. H., Jiang, K., et al. (2015). A cascade of arabinosyltransferases controls shoot meristem size in tomato. Nat. Genet. 47, 784–792. doi: 10.1038/ng.3309
Xu, C., Park, S. J., Van Eck, J., and Lippman, Z. B. (2016). Control of inflorescence architecture in tomato by BTB/POZ transcriptional regulators. Genes Dev. 30, 2048–2061. doi: 10.1101/gad.288415.116
Xu, F., Sun, X., Chen, Y., Huang, Y., Tong, C., and Bao, J. (2015). Rapid identification of major QTLs associated with rice grain weight and their utilization. PloS One 10:e0122206. doi: 10.1371/journal.pone.0122206
Xu, K., and Mackill, D. J. (1996). A major locus for submergence tolerance mapped on rice chromosome 9. Mol. Breeding 2, 219–224. doi: 10.1007/bf00564199
Xu, K., Xu, X., Fukao, T., Canlas, P., Maghirang-Rodriguez, R., Heuer, S., et al. (2006). Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 442, 705–708. doi: 10.1038/nature04920
Xu, R., Li, H., Qin, R., Wang, L., Li, L., Wei, P., et al. (2014). Gene targeting using the Agrobacterium tumefaciens-mediated CRISPR-Cas system in rice. Rice 7:5.
Yan, H., Ma, L., Wang, Z., Lin, Z., Su, J., and Lu, B. R. (2015). Multiple tissue-specific expression of rice seed-shattering gene SH4 regulated by its promoter pSH4. Rice 8:12.
Yan, L., Loukoianov, A., Blechl, A., Tranquilli, G., Ramakrishna, W., et al. (2004). The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303, 1640–1644. doi: 10.1126/science.1094305
Yan, L., Loukoianov, A., Tranquilli, G., Helguera, M., Fahima, T., and Dubcovsky, J. (2003). Positional cloning of the wheat vernalization gene VRN1. Proc. Natl. Acad. Sci. U.S.A. 100, 6263–6268. doi: 10.1073/pnas.0937399100
Yang, F., Jørgensen, A. D., Li, H., Søndergaard, I., Finnie, C., Svensson, B., et al. (2011). Implications of high-temperature events and water deficits on protein profiles in wheat (Triticum aestivum L. cv. Vinjett) grain. Proteomics 11, 1684–1695. doi: 10.1002/pmic.201000654
Yang, Q., Li, Z., Li, W., Ku, L., Wang, C., Ye, J., et al. (2013). CACTA-like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the post-domestication spread of maize. Proc. Natl. Acad. Sci. U.S.A. 110, 16969–16974. doi: 10.1073/pnas.1310949110
Yang, X., Xia, X., Zhang, Z., Nong, B., Zeng, Y., Xiong, F., et al. (2017). QTL mapping by whole genome re-sequencing and analysis of candidate genes for nitrogen use efficiency in rice. Front. Plant Sci. 8:1634. doi: 10.3389/fpls.2017.01634
Yaobin, Q., Peng, C., Yichen, C., Yue, F., Derun, H., Tingxu, H., et al. (2018). QTL-Seq identified a major QTL for grain length and weight in rice using near isogenic F2 population. Rice Sci. 25, 121–131. doi: 10.1016/j.rsci.2018.04.001
Ye, J., Li, W., Ai, G., Li, C., Liu, G., Chen, W., et al. (2019). Genome-wide association analysis identifies a natural variation in basic helix-loop-helix transcription factor regulating ascorbate biosynthesis via D-mannose/L-galactose pathway in tomato. PLoS Genet. 15:e1008149. doi: 10.1371/journal.pgen.1008149
Ye, J., Wang, X., Hu, T., Zhang, F., Wang, B., Li, C., et al. (2017). An InDel in the promoter of Al-ACTIVATED MALATE TRANSPORTER9 selected during tomato domestication determines fruit malate contents and aluminum tolerance. Plant Cell 29, 2249–2268. doi: 10.1105/tpc.17.00211
Yoo, H. J., Park, W. J., Lee, G. M., Oh, C. S., Yeam, I., Won, D. C., et al. (2017). Inferring the genetic determinants of fruit colors in tomato by carotenoid profiling. Molecules 22:764. doi: 10.3390/molecules22050764
Yoon, J., Cho, L. H., Kim, S. L., Choi, H., Koh, H. J., and An, G. (2014). The BEL 1-type homeobox gene SH 5 induces seed shattering by enhancing abscission-zone development and inhibiting lignin biosynthesis. Plant J. 79, 717–728. doi: 10.1111/tpj.12581
Youssef, H. M., Eggert, K., Koppolu, R., Alqudah, A. M., Poursarebani, N., Fazeli, A., et al. (2017). VRS2 regulates hormone-mediated inflorescence patterning in barley. Nat. Genet. 49, 157–161. doi: 10.1038/ng.3717
Yu, H., Lin, T., Meng, X., Du, H., Zhang, J., Liu, G., et al. (2021). A route to de novo domestication of wild allotetraploid rice. Cell184, 1156–1170.
Yuan, M., Zhu, J., Gong, L., He, L., Lee, C., Han, S., et al. (2019). Mutagenesis of FAD2 genes in peanut with CRISPR/Cas9 based gene editing. BMC Biotechnol. 19:24. doi: 10.1186/s12896-019-0516-8
Yuan, Y. X., Wu, J., Sun, R. F., Zhang, X. W., Xu, D. H., Bonnema, G., et al. (2009). A naturally occurring splicing site mutation in the Brassica rapa FLC1 gene is associated with variation in flowering time. J. Exp. Bot. 60, 1299–1308. doi: 10.1093/jxb/erp010
Zeng, X., Yuan, H., Dong, X., Peng, M., Jing, X., Xu, Q., et al. (2020). Genome-wide dissection of co-selected UV-B responsive pathways in the UV-B adaptation of qingke. Mol. Plant 13, 112–127. doi: 10.1016/j.molp.2019.10.009
Zhang, C., Badri Anarjan, M., Win, K. T., Begum, S., and Lee, S. (2021). QTL-seq analysis of powdery mildew resistance inmildew resistance in a Korean cucumber inbred line. Theor. Appl. Genet. 134, 435–451. doi: 10.1007/s00122-020-03705-x
Zhang, D., Wang, X., Li, S., Wang, C., Gosney, M. J., Mickelbart, M. V., et al. (2019a). A post-domestication mutation, Dt2, triggers systemic modification of divergent and convergent pathways modulating multiple agronomic traits in soybean. Mol. Plant 12, 1366–1382. doi: 10.1016/j.molp.2019.05.010
Zhang, H., Zhang, J., Wei, P., Zhang, B., Gou, F., Feng, Z., et al. (2014). The CRISPR/C as9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol. J. 12, 797–807. doi: 10.1111/pbi.12200
Zhang, M., Lv, D., Ge, P., Bian, Y., Chen, G., Zhu, G., et al. (2014). Phosphoproteome analysis reveals new drought response and defense mechanisms of seedling leaves in bread wheat (Triticum aestivum L.). J. Proteomics 109, 290–308. doi: 10.1016/j.jprot.2014.07.010
Zhang, X., Hou, X., Liu, Y., Zheng, L., Yi, Q., Zhang, H., et al. (2019b). Maize brachytic2 (br2) suppresses the elongation of lower internodes for excessive auxin accumulation in the intercalary meristem region. BMC Plant Biol. 19:589. doi: 10.1186/s12870-019-2200-5
Zhang, X., Wang, W., Guo, N., Zhang, Y., Bu, Y., Zhao, J., et al. (2018). Combining QTL-seq and linkage mapping to fine map a wild soybean allele characteristic of greater plant height. BMC Genom. 19:226. doi: 10.1186/s12864-018-4582-4
Zhang, X., Yin, F., Xiao, S., Jiang, C., Yu, T., Chen, L., et al. (2019c). Proteomic analysis of the rice (Oryza officinalis) provides clues on molecular tagging of proteins for brown planthopper resistance. BMC Plant Biol. 19:30. doi: 10.1186/s12870-018-1622-9
Zhang, X., Zhang, K., Wu, J., Guo, N., Liang, J., Wang, X., et al. (2020). QTL-Seq and sequence assembly rapidly mapped the gene BrMYBL2. 1 for the purple trait in Brassica rapa. Sci. Rep. 10:2328.
Zhang, Y., Massel, K., Godwin, I. D., and Gao, C. (2018). Applications and potential of genome editing in crop improvement. Genome Biol. 19:210.
Zhang, Z., Belcram, H., Gornicki, P., Charles, M., Just, J., Huneau, C., et al. (2011). Duplication and partitioning in evolution and function of homoeologous Q loci governing domestication characters in polyploid wheat. Proc. Natl. Acad. Sci. U.S.A. 108, 18737–18742. doi: 10.1073/pnas.1110552108
Zhang, Z., Li, A., Song, G., Geng, S., Gill, B. S., Faris, J. D., et al. (2020). Comprehensive analysis of Q gene near-isogenic lines reveals key molecular pathways for wheat domestication and improvement. Plant J. 102, 299–310. doi: 10.1111/tpj.14624
Zhao, Y., Ma, J., Li, M., Deng, L., Li, G., Xia, H., et al. (2020). Whole-genome resequencing-based QTL-seq identified AhTc1 gene encoding a R2R3-MYB transcription factor controlling peanut purple testa colour. Plant Biotechnol. J. 18, 96–105. doi: 10.1111/pbi.13175
Zheng, M., Yang, T., Liu, X., Lü, G., Zhang, P., Jiang, B., et al. (2020). qRf8-1, a Novel QTL for the fertility restoration of maize CMS-C identified by QTL-seq. G3 10, 2457–2464. doi: 10.1534/g3.120.401192
Zhou, H., Liu, B., Weeks, D. P., Spalding, M. H., and Yang, B. (2014). Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 42, 10903–10914. doi: 10.1093/nar/gku806
Zhou, J., Wang, G., and Liu, Z. (2018). Efficient genome editing of wild strawberry genes, vector development and validation. Plant Biotech. J. 16, 1868–1877. doi: 10.1111/pbi.12922
Zhou, Y., Lu, D., Li, C., Luo, J., Zhu, B. F., Zhu, J., et al. (2012). Genetic control of seed shattering in rice by the APETALA2 transcription factor shattering abortion1. Plant Cell 24, 1034–1048. doi: 10.1105/tpc.111.094383
Zhou, Y., Yang, P., Cui, F., Zhang, F., Luo, X., and Xie, J. (2016). Transcriptome analysis of salt stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). PLoS One 11:e0146242. doi: 10.1371/journal.pone.0146242
Zhu, C., Li, X., and Yu, J. (2011). Integrating rare-variant testing, function prediction, and gene network in composite resequencing-based genome-wide association studies (CR-GWAS). G3 1, 233–243. doi: 10.1534/g3.111.000364
Zhu, G., Wang, S., Huang, Z., Zhang, S., Liao, Q., Zhang, C., et al. (2018). Rewiring of the fruit metabolome in tomato breeding. Cell 172, 249–261. doi: 10.1016/j.cell.2017.12.019
Zhu, Y., Li, Y., Xin, D., Chen, W., Shao, X., Wang, Y., et al. (2015). RNA-Seq-based transcriptome analysis of dormant flower buds of Chinese cherry (Prunus pseudocerasus). Gene 555, 362–376. doi: 10.1016/j.gene.2014.11.032
Zsögön, A., Čermák, T., Naves, E. R., Notini, M. M., Edel, K. H., Weinl, S., et al. (2018). De novo domestication of wild tomato using genome editing. Nat. Biotechnol. 36, 1211–1216. doi: 10.1038/nbt.4272
Keywords: omics, metabolomics, de novo domestication, crop improvement, domesticated-genes
Citation: Kumar R, Sharma V, Suresh S, Ramrao DP, Veershetty A, Kumar S, Priscilla K, Hangargi B, Narasanna R, Pandey MK, Naik GR, Thomas S and Kumar A (2021) Understanding Omics Driven Plant Improvement and de novo Crop Domestication: Some Examples. Front. Genet. 12:637141. doi: 10.3389/fgene.2021.637141
Received: 02 December 2020; Accepted: 02 March 2021;
Published: 06 April 2021.
Edited by:
Fatemeh Maghuly, University of Natural Resources and Life Sciences, Vienna, AustriaReviewed by:
Uday Chand Jha, Indian Institute of Pulses Research (ICAR), IndiaAtsushi Fukushima, RIKEN, Japan
Copyright © 2021 Kumar, Sharma, Suresh, Ramrao, Veershetty, Kumar, Priscilla, Hangargi, Narasanna, Pandey, Naik, Thomas and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rakesh Kumar, cmFrZXNoa3VtYXJAY3VrLmFjLmlu; Anirudh Kumar, YW5pcnVkaC5rdW1hckBpZ250dS5hYy5pbg==
†These authors have contributed equally to this work