 Maria Gnoli1†
Maria Gnoli1† Evelise Brizola1†
Evelise Brizola1† Morena Tremosini1
Morena Tremosini1 Elena Pedrini1
Elena Pedrini1 Margherita Maioli1
Margherita Maioli1 Massimiliano Mosca2Alessandra Bassotti3,4
Massimiliano Mosca2Alessandra Bassotti3,4 Paola Castronovo4,5
Paola Castronovo4,5 Cecilia Giunta6Luca Sangiorgi1*
Cecilia Giunta6Luca Sangiorgi1*- 1Department of Rare Skeletal Disorders, IRCCS Istituto Ortopedico Rizzoli, Bologna, Italy
- 2Orthopedic and Traumatologic Clinic, IRCCS Istituto Ortopedico Rizzoli, Bologna, Italy
- 3Regional Center of Ehlers-Danlos Syndrome, Fondazione IRCCS Ca' Granda–Ospedale Maggiore Policlinico, Milan, Italy
- 4Occupational Health Unit, Fondazione IRCCS Ca' Granda–Ospedale Maggiore Policlinico, Milan, Italy
- 5Medical Genetics Laboratory, Fondazione IRCCS Ca' Granda–Ospedale Maggiore Policlinico, Milan, Italy
- 6Connective Tissue Unit, Division of Metabolism and Children's Research Centre, University Children's Hospital, Zurich, Switzerland
Collagen type I mutations are related to wide phenotypic expressions frequently causing an overlap of clinical manifestations, in particular between Osteogenesis Imperfecta (OI) and Ehlers-Danlos syndrome (EDS). Both disorders present inter- and intra-familial clinical variability and several clinical signs are present in both diseases. Recently, after the observation that some individuals first ascertained by a suspicion of EDS resulted then carriers of pathogenic variants of genes known to primarily cause OI, some authors proposed the term “COL1-related overlap disorder” to describe these cases. In this paper, we report clinical, molecular, and biochemical information about an individual with a diagnosis of EDS with severe joint hypermobility who carries a pathogenic heterozygous variant in COL1A2 gene, and a benign variant in COL1A1 gene. The pathogenic variant, commonly ascribed to OI, as well as the benign variant, has been inherited from the individual's mother, who presented only mild signs of OI and the diagnosis of OI was confirmed only after molecular testing. In addition, we reviewed the literature of similar cases of overlapping syndromes caused by COL1 gene mutations. The reported case and the literature review suggest that the COL1-related overlap disorders (OI, EDS and overlapping syndromes) represent a continuum of clinical phenotypes related to collagen type I mutations. The spectrum of COL1-related clinical manifestations, the pathophysiology and the underlying molecular mechanisms support the adoption of the updated proposed term “COL1-related overlap disorder” to describe the overlapping syndromes.
Introduction
COL1A1 and COL1A2 pathogenic variants have been related to several connective tissue disorders (Table 1), including Osteogenesis Imperfecta (OI) and Ehlers-Danlos syndrome (EDS)1,2. Both are heterogeneous groups of disorders sharing several clinical features, as joint hypermobility, osteoarticular abnormalities, and chronic pain.
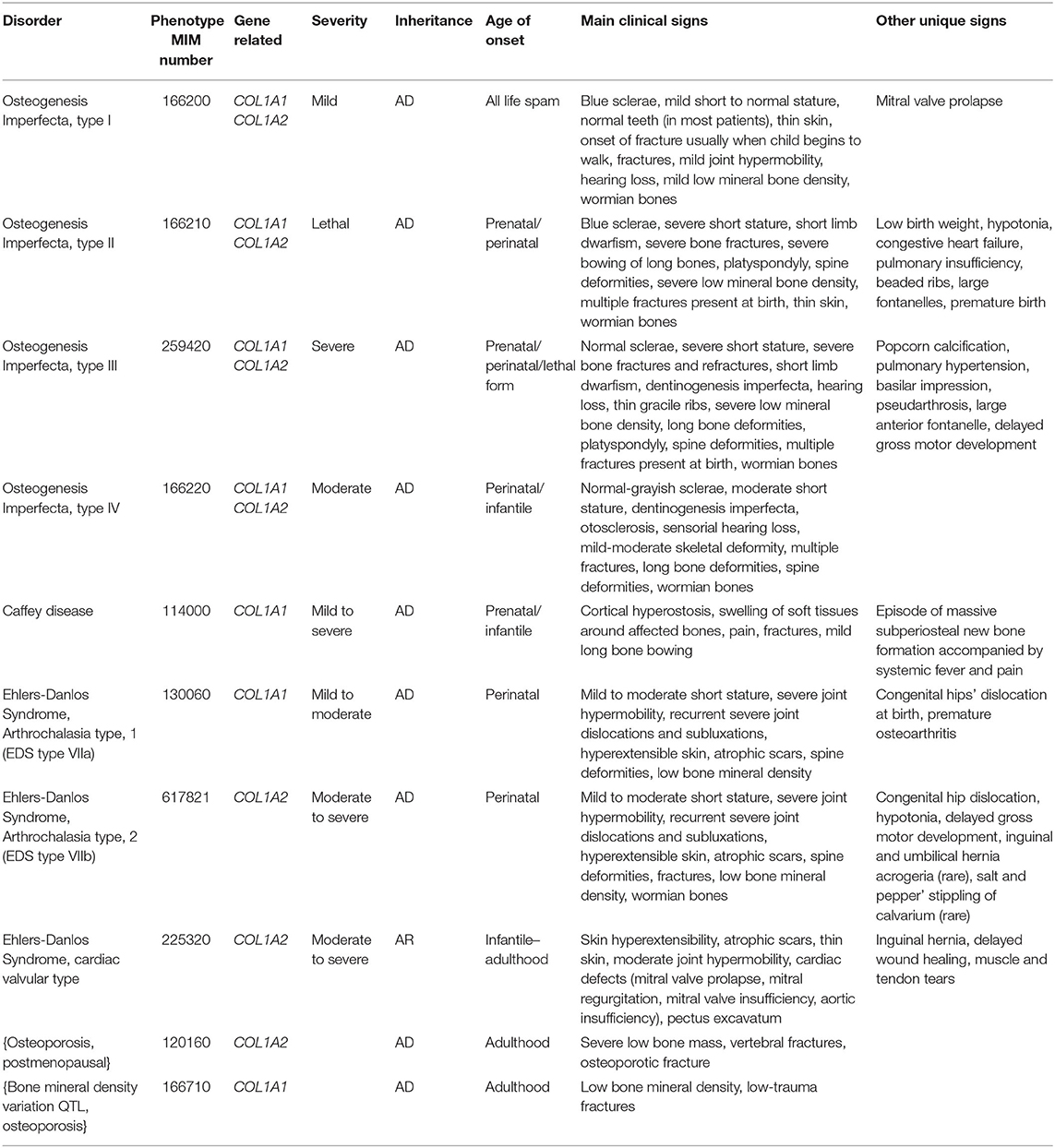
Table 1. Main diseases related to COL1A1 and COL1A2 genes.
OI (MIM numbers#166200, #166210, #259420, #166220) is a rare connective tissue disorder, characterized mainly by bone fragility and by other signs of connective tissue involvement such as joint hypermobility and heart defects (Forlino and Marini, 2016; Maioli et al., 2019). The COL1A1 and COL1A2 genes encode for the α chains of collagen type 1; more than 90% of OI individuals have disease causing variants in these genes (Van Dijk and Sillence, 2014). The most common COL1A1 or COL1A2 sequence abnormalities detected in OI are point mutations that affect a glycine residue within the helical domain (Rauch and Glorieux, 2004; Marini et al., 2007).
EDS identifies a group of disorders that affect the connective tissues, including skin, bones, blood vessels, and many other organs and tissues. The main clinical features are joint hypermobility, skin hyperextensibility, and tissue fragility (Malfait et al., 2017). The updated classification of EDS linked collagen type I pathogenic variants include: arthrochalasia EDS–aEDS (associated to COL1A1 and COL1A2 mutations); cardiac-valvular–cvEDS (related to biallelic pathogenic COL1A2 variants); vascular EDS–vEDS (rarely associated to specific COL1A1 arginine to cysteine substitutions); and classic EDS–cEDS (rarely related to COL1A1) (Malfait et al., 2017).
Thus, mutations in the COL1A1 gene have been found to cause several forms of EDS, moreover, it has been demonstrated that variants in particular regions are related to an OI/EDS overlapping phenotype (Cabral et al., 2007; Malfait et al., 2013). Moreover, mosaic mutation for a lethal OI variant has been described to cause a mild phenotype, with OI/EDS overlapping clinical manifestations (Symoens et al., 2017). COL1A1 gene mutations have been rarely found in classical EDS (cEDS), whereas COL1A2 pathogenic variants have never been associated to cEDS (Malfait et al., 2007).
Recently, some authors proposed the term “COL1-related overlap disorder” to describe individuals with clinical suspect of EDS and pathogenic variants in genes known to primarily cause OI (Morlino et al., 2020). The authors described 21 affected individuals with multiple soft connective tissue features, five with heterogeneous variant in COL1A1 and eight in COL1A2. These findings suggested a wider spectrum of clinical manifestations of collagen type I pathogenic variants including intermediate or mixed phenotype between EDS and OI.
Herein we report a family with a benign variant in COL1A1 and a severe OI causative mutation in COL1A2 that is related to an EDS phenotype in the proband and to mild OI clinical signs in the proband's mother. Furthermore, we present a review of the literature for similar cases of overlapping syndromes caused by COL1 gene pathogenic variants.
Case Description
Clinical Findings
The 30-year-old female proband came to our attention for genetic counseling related to a previous clinical diagnosis of EDS received at 23-year-old. At the age of 19 years she began to undergo clinical evaluations for general pain and chronic asthenia: a hereditary connective tissue disorder was suspected.
She showed generalized joint hypermobility scoring 9/9 in the Beighton score (Figures 1A–C) with recurrent joint dislocations, in particular of shoulders, wrist, and knees. She referred to be prone to bruising and to present a tendency toward prolonged bleeding. Moreover, she was referred with postural hypotension but an autonomic dysfunction was excluded. She had no blue sclera, dental abnormalities or visual defects. An audiological examination revealed a mild hearing loss at high frequencies. Due to recurrent headache, at the age of 26, she underwent an MRI scan of the brain that did not identify any pathological findings.
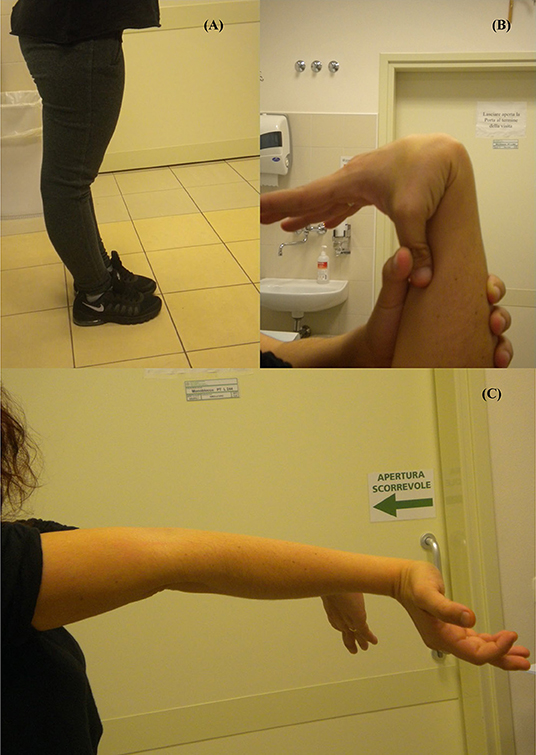
Figure 1. Joint hypermobility: (A) Bilateral active hyperextension of the knee; (B) Passive apposition of thumb to forearm; (C) Active hyperextension of elbow.
Neck vessels ultrasound showed a right carotid artery kinking. She underwent cardiac ultrasound that identified a mild valve insufficiency. At 24-year-old, after surgical removal by laparoscopy of multiple ovarian cysts, previously diagnosed with ultrasounds, she developed an enlarged hypertrophic scar in the site of entry of the laparoscope.
Height was 160 cm (>25°p), she did not present any sign of recent or old bone fractures. At 28-years of age she underwent a dual-energy X-ray absorptiometry (DXA) that revealed an early onset of osteopenia (lumbar and femur neck Z scores −2.1).
Calcium, phosphorus, parathyroid hormone, and alkaline phosphatase levels were in the normal range, while 25-(OH)-vitamin D level was low (value: 15,2 ng/mL; deficiency: <20 ng/mL).
She was born with eutocic delivery, the weight at birth was 4,100 g, length 51 cm. Psychomotor development was regular, no hypotonia or motor delay was diagnosed. She referred having joint hypermobility since childhood, as well as flat feet (clinical report is not available).
After the diagnosis and molecular analysis that identified both investigated COL1A1 and COL1A2 variants in the proband, her mother underwent specific evaluations. The proband's mother, aged 55-year-old, had not been referred for any clinical problems; she had a stature below the mean of Italian general population [155 cm (10°p)] (Cacciari et al., 2006), and a DXA scan showed a mild osteopenia (lumbar Z score −1.3/T score −2.3 and femur neck Z score −0.7/T score −1.4). She did not present any sign of general joint hypermobility, blue sclerae, dentinogenesis imperfecta or recurrent fractures. In addition, no cardiac or hearing abnormalities were detected in the specific performed exams.
Regarding family history, it was found that a paternal cousin of the proband underwent clinical investigation that excluded OI or EDS diagnosis. No other family members presented clinical manifestations suggesting OI or EDS or other connective tissue disorders.
Molecular Findings
At first a diagnosis of EDS was suspected and molecular analysis of a gene panel for collagenopathies was performed. The targeted DNA custom panel, with COL1A1, COL1A2, COL5A1, COL5A2, COL3A1, ADAMTS2, BGALT7, CHST14, PLOD1, PLOD3, and TNXB genes, was designed with the HaloPlex online design tool (SureDesign, Agilent Technologies) and sequenced on MiSeq platform (Illumina, Inc., San Diego, CA, USA) using a Next Generation Sequencing approach.
Due to the complex structure of the TNXB locus, EDS molecular diagnostic workflow should include NGS for the non-homologous TNXB sequence, long PCR/Sanger sequencing for the TNXA/TNXB homology region and TNXB deletion/duplication analysis (Demirdas et al., 2017; Micale et al., 2019). The last two molecular diagnostic steps have not been performed because TNXB-related classical-like EDS (TNXB-clEDS), the ultrarare type of EDS due to biallelic null variants in TNXB, has been excluded by the proband's physical examination that revealed the presence of atrophic scarring.
The analysis failed to find an alteration in COL5A1 and COL5A2 genes, but two heterozygous variants in collagen type I genes were identified: a c.133C>G (p.Leu45Val) in exon 2 of COL1A1 gene and a c.2701G>A (p.Gly901Ser) in exon 42 of COL1A2 gene.
They were confirmed by Sanger sequencing using BigDye Terminator version 1.1 cycle sequencing kit and the 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequence traces were aligned with the GenBank reference sequences of the COL1A1 cDNA (NM_000088.3) and the COL1A2 cDNA (NM_000089.3).
The pathogenic nature of the variants was evaluated by a series of in silico tools included in Varsome that displays automated variant classification according to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) (Richards et al., 2015; Kopanos et al., 2019). The first variant is classified as Likely Benign, principally because it has been observed in healthy adults and has a benign computational verdict based on nine benign predictions vs. one pathogenic prediction.
The second one has been reported in the Leiden Open Variation Database (LOVD) and Human Gene Mutation Database (HGMD) as pathogenetic and was related to moderate-severe and lethal OI (two affected individuals, respectively OI type IV and type II) (Nuytinck et al., 1997; Marini et al., 2007; Pyott et al., 2011). Varsome classifies it as a Variant of Unknown Significance (VOUS).
The variants affecting glycine residue in collagen type I are usually damaging and related to the more severe forms of the disease. Both variants in the collagen type I genes were found also in the proband's mother, in whom no evidence of mosaic status was found investigating blood and saliva samples.
To better investigate the effect of the variants the proband underwent a skin biopsy for biochemical screening of collagens.
Biochemical Findings
Collagen was prepared from cultured dermal fibroblasts of the affected individual by metabolic labeling, with [1,2 14C] proline (Perkin Elmer), followed by digestion with pepsin, separation by 5% SDS–PAGE and visualization by fluorography (Lindert et al., 2018). The biochemical analysis of collagen revealed alterations in the production and secretion of type I collagen, which suggest a clinical diagnosis of OI.
Figure 2 shows the routine diagnostic SDS-PAGE gel obtained by loading 50 μl of 14C proline-labeled collagens extracted from cultured fibroblasts of the patient P and three different controls (C1, C2, and C3). No volume adjustment to show equal band intensity between the different samples has been done. On SDS-PAGE (Figure 2, medium fraction) in the patient fibroblasts (P) both α1(I) and α2(I) chains in the medium fraction as well as in the cell layer presented as overmodified bands with a slower migration and a broader appearance compared to controls (C1, C2, and C3). Furthermore, the cell layer from the patient fibroblasts (P) presented an additional broad α'1(I) band that migrates above the overmodified α1(I) band and appears to be intracellularly retained (α'1(I) in Figure 2, cell layer).
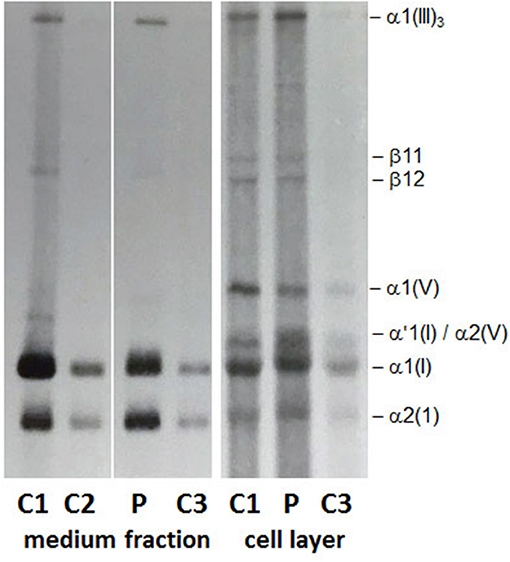
Figure 2. Collagen biochemical analysis: The α1(I) and α2(I) chains in the medium fraction and in the cell layer show a slower migration and a broader appearance on SDS-PAGE. In the cell layer a strong and broad band migrating at the position of a pNα1(I) chain appears to be retained intracellularly.
Although, the presence of an undetected variant in either COL1A1 or a collagen chaperon/posttranslational modifying enzyme, that could explain the abnormal findings detected by SDS-PAGE, cannot be completely excluded, we have a tentative alternative hypothesis to explain these abnormal findings. The overmodified α1(I) and α2(I) chains detected on SDS-PAGE in the medium fraction (Figure 2) correspond to the fully processed and secreted triple-helical chains and likely result from the slower formation of the collagen triple helix as a consequence of the COL1A2 p.Gly901Ser mutation. Whereas, the overmodified and intracellularly retained α'1(I) band (Figure 2, cell layer) may represent those overmodified α1(I) chains harboring the COL1A1 c.133C>G (p.Leu45Val) variant. How this variant in the N-terminal propetide of COL1A1 could interfere with the secretion of the overmodified α1(I) chains should be further investigated and the hypothesis confirmed.
Review of Similar Cases
A literature review of similar cases performed in PubMed lead to 15 papers published between 1997 and 2020. A summary of the findings and the cases is reported in Table 2.
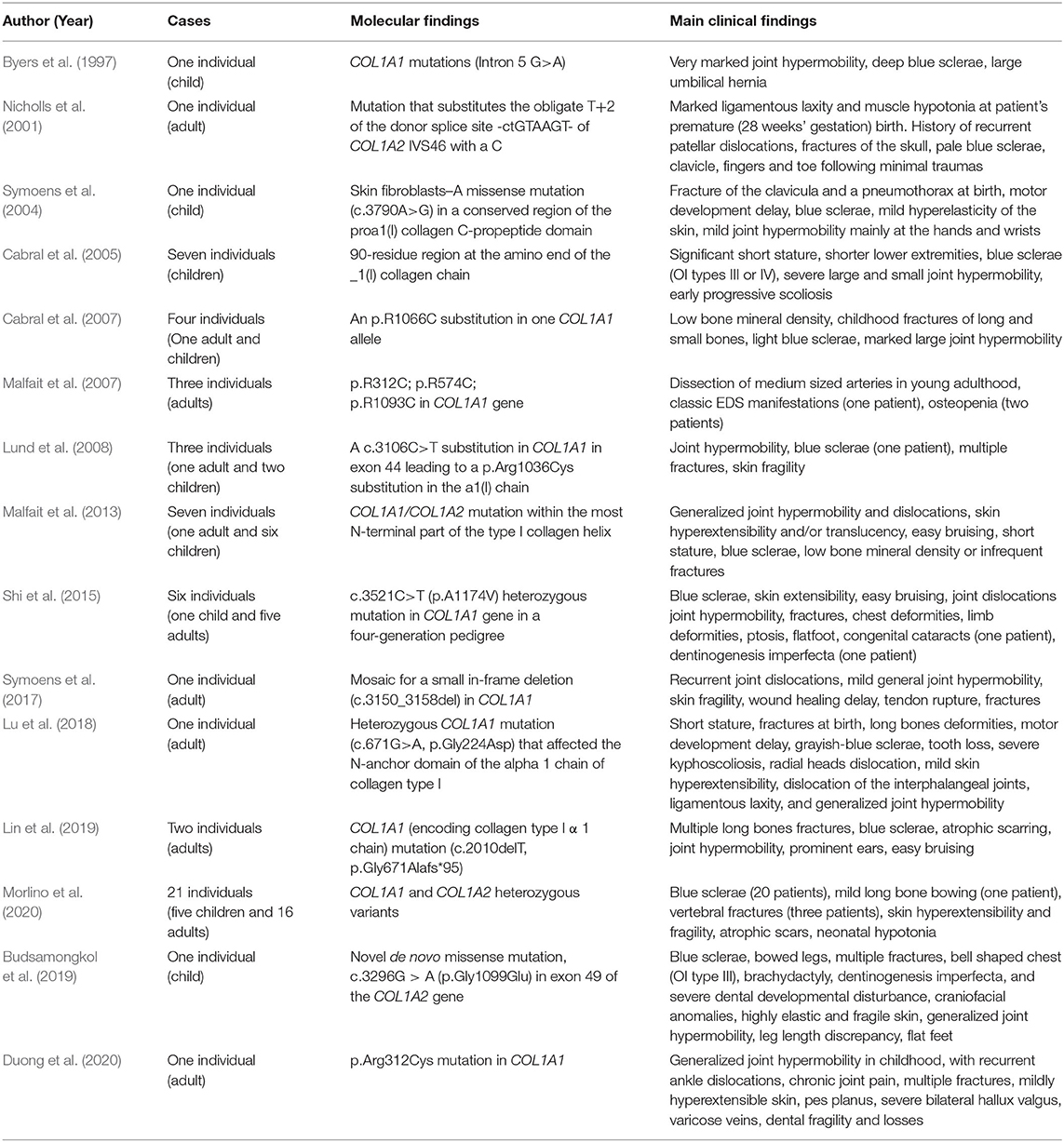
Table 2. Summary of OI/EDS overlap cases reported in the literature.
Discussion
Collagen genes mutations are related to several disorders and to a wide range of clinical and radiological features. Over 1,600 distinct pathogenic variants have been reported in the two genes COL1A1 and COL1A2 that encode the two types of polypeptide chain of the type I procollagen proα1(I) and proα2(I) heterotrimer3.
Clinical expression variability is a common trait of OI and EDS. Both disorders are collagenopathies and the affected individuals may show many similar features, however the two diseases generally present distinct characteristics. In this article we presented clinical, molecular and biochemical information of an individual with clinical diagnosis of EDS, but a COL1A2 pathogenic variant usually related to a severe/moderate form of OI and, in addition, the affected individual's mother who had the same COL1A2 mutation, but only very mild clinical signs of OI.
Since the first case suggesting an overlapping of EDS/OI phenotype was published in 1997 (Byers et al., 1997), other cases have been reported in the literature (Table 2). The use of the term “COL1-related overlap disorder” covers the EDS/OI overlapping phenotypes, other than the common clinical types of EDS or OI already included in the respective classifications (Malfait et al., 2017; Morlino et al., 2020). The mixed phenotype may vary from mild to moderate/severe and major clinical features may include blue sclerae, generalized joint hypermobility, flatfeet with valgus deformity and significantly soft and/or hyperextensible skin. Minor criteria may include hearing loss, short stature, atrophic scars, fractures, joint dislocations, dolichostenomelia, and muscle/ligaments/tendons rupture. Dentinogenesis imperfecta, progressive/severe heart valve disease, congenital fractures, and long bone deformities are considered as exclusion criteria for COL1-related overlap disorder (Morlino et al., 2020).
Similar to other OI/EDS overlap described cases, the affected individual presented joint hypermobility (typical of both OI and EDS), joint dislocations (typical of EDS), skin fragility (typical of EDS), and osteopenia (typical of OI). The proband presented minor signs of vascular involvement such as right carotid artery kinking and a mild valve insufficiency. Cases with vascular involvement, in particular aortic dissection, have been described in OI (Byra et al., 2008; Balasubramanian et al., 2019). A higher rate of vascular events was estimated in overlap OI/EDS cases, in particular, Arginine substitutions in collagen type I have been described in EDS with propensity to arterial rupture (Malfait et al., 2007). Nevertheless, also cases of cEDS without any vascular complication have been reported (Colombi et al., 2017, Duong et al., 2020). The case here reported is comparable to previous reported ones, when initially there is suspicion of EDS on clinical ground but molecular testing for COL5A1 or COL5A2 (related to classical EDS) variants are negative and, oddly, variants in COL1A1 and COL1A2, primarily ascribed as causative of OI, are confirmed (Shi et al., 2015; Malfait et al., 2017; Morlino et al., 2020).
The genotype for COL1A1 and COL1A2 variants is the same in both proband and proband's mother. The same COL1A2 mutation has been reported in a case of OI type II and in mosaic state in the clinically not affected parent of the proband (Pyott et al., 2011). However, in this family, no evidence of mosaic status (that might justify the clinical heterogeneity and the very mild expression of the disease in the proband's mother) was found when mutations in DNA extracted from blood sample and saliva was assessed.
The investigation performed to clarify the biochemical outcome of the COL1A2 and COL1A1 genetic variants demonstrated alterations in the production and secretion of type I collagen (Figure 2), which could be only partially explained by the known pathogenic effect of the COL1A2-mutation. In fact, both α1(I) and α2(I) in the medium and the cell layer presented as overmodified bands, as expected by glycine substitutions within the triple helical domain of collagen that delay collagen triple helix formation (Raghunath et al., 1994). However, the overmodified and slower migrating α'1(I) band that is detected in the cell layer only may represent fully processed and strongly overmodified α1(I)chains that are retained intracellularly and likely degraded. As a consequence, thereof, a reduced amount of overmodified type I collagen molecules is secreted outside of the cell and participate in the formation of mature collagen fibrils and fibers in the extracellular matrix. Based on the hypothesis, yet to be demonstrated, that the p.Leu45Val variant may interfere with the secretion of the strongly overmodified α1(I) chains, we propose to consider it a disease modifier variant with a beneficial effect on the otherwise severe clinical outcome of the COL1A2 (p.Gly901Ser) variant. This is a speculative interpretation of the unexpected outcome of the biochemical study. However, additional investigations are required to prove this hypothesis, which does not take into account the possibility of the existence of undetected variants in any of the collagen posttranslational modifying enzymes and/or chaperones.
As already highlighted by Morlino et al. (2020), the term OI/EDS overlap may lead to confusion for both non-specialist professionals and affected individuals and, even though it is acceptable on the molecular background, a more specific term such as “COL1-related overlap disorder” could benefit medical management and genetic counseling to for these individuals. These findings corroborate the available literature, suggesting that there is still much more information about the genotype-phenotype correlation in type 1 collagen mutations to be discovered in the future. The case described here reinforces the need of an updated classification for these overlapping EDS/OI phenotypes as “COL1-related overlap disorder” (Morlino et al., 2020).
Conclusion
We reported the case of an individual with a diagnosis of EDS with severe joint hypermobility who carried a pathogenic heterozygous variant in COL1A2 gene, and a benign variant in COL1A1 gene. The pathogenic variant, commonly ascribed to OI as well as the variant with a benign effect on the phenotype, have been inherited from the individual's mother, who showed only very mild signs of OI and the diagnosis of OI could be made only after molecular testing. The literature was reviewed for similar cases of overlapping syndromes caused by COL1 gene mutations. The reported case and the literature review confirmed an extensive clinical expression variability as part of a wider spectrum of clinical phenotypes related to collagen type I pathogenic variants that could justify the adoption of an updated classification as “COL1-related overlap disorder.”
Data Availability Statement
The pathogenic variant in COL1A2 is reported in the Human Gene Mutation Database – HGMD (http://www.hgmd.cf.ac.uk/ac/index.php). The COL1A1 variant is described on ExAC browser (48277279) (http://exac.broadinstitute.org) and on dbSNP (rs546629502) (https://www.ncbi.nlm.nih.gov/snp/).
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MG, EB, EP, MMa, MT, MMo, AB, PC, and CG: conception, analysis, collection, and interpretation of data. MG, EB, MMa, PC, CG, and LS: drafting the manuscript. All the authors reviewed and approved the final version of the manuscript.
Funding
This work has been supported by ERN BOND–European Reference Network for rare BONe Diseases–Project ID No. 739543.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FM declared a past co-authorship with one of the authors CG to the handling editor.
Acknowledgments
We would like to thank the patient and her family for their participation and Timothée Ndarugendamwo for collagen biochemical analysis.
Footnotes
1. ^https://www.omim.org/entry/120150?search=COL1A1&highlight=col1a1
2. ^https://www.omim.org/entry/120160?search=COL1A2&highlight=col1a2
References
Balasubramanian, M., Verschueren, A., Kleevens, S., Luyckx, I., Perik, M., Schirwani, S., et al. (2019). Aortic aneurysm/dissection and osteogenesis imperfecta: four new families and review of the literature. Bone 121, 191–195. doi: 10.1016/j.bone.2019.01.022
Budsamongkol, T., Intarak, N., Theerapanon, T., Yodsanga, S., Porntaveetus, T., and Shotelersuk, V. (2019). A novel mutation in COL1A2 leads to osteogenesis imperfecta/Ehlers-Danlos overlap syndrome with brachydactyly. Genes Dis. 6, 138–146. doi: 10.1016/j.gendis.2019.03.001
Byers, P. H., Duvic, M., Atkinson, M., Robinow, M., Smith, L. T., Krane, S. M., et al. (1997). Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am. J. Med. Genet. 72, 94–105.
Byra, P., Chillag, S., and Petit, S. (2008). Osteogenesis imperfecta and aortic dissection. Am. J. Med. Sci. 336, 70–72. doi: 10.1097/MAJ.0b013e318158e981
Cabral, W. A., Makareeva, E., Colige, A., Letocha, A. D., Ty, J. M., Yeowell, H. N., et al. (2005). Mutations near amino end of alpha1(I) collagen cause combined osteogenesis imperfecta/Ehlers-Danlos syndrome by interference with N-propeptide processing. J. Biol. Chem. 280, 19259–19269. doi: 10.1074/jbc.M414698200
Cabral, W. A., Makareeva, E., Letocha, A. D., Scribanu, N., Fertala, A., Steplewski, A., et al. (2007). Y-position cysteine substitution in type I collagen (alpha1(I) R888C/p.R1066C) is associated with osteogenesis imperfecta/Ehlers-Danlos syndrome phenotype. Hum. Mutat. 28, 396–405. doi: 10.1002/humu.20456
Cacciari, E., Milani, S., Balsamo, A., Spada, E., Bona, G., Cavallo, L., et al. (2006). Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J Endocrinol. Investig. 29, 581–593. doi: 10.1007/BF03344156
Colombi, M., Dordoni, C., Venturini, M., Zanca, A., Calzavara-Pinton, P., and Ritelli, M. (2017). Delineation of Ehlers-Danlos syndrome phenotype due to the c.934C>T, p.(Arg312Cys) mutation in COL1A1: report on a three-generation family without cardiovascular events, and literature review. Am. J. Med. Genet. A 173, 524–530. doi: 10.1002/ajmg.a.38035
Demirdas, S., Dulfer, E., Robert, L., Kempers, M., van Beek, D., Micha, D., et al. (2017). Recognizing the tenascin-X deficient type of Ehlers-Danlos syndrome: a cross-sectional study in 17 patients. Clin. Genet. 91, 411–425. doi: 10.1111/cge.12853
Duong, J., Rideout, A., MacKay, S., Beis, J., Parkash, S., Schwarze, U., et al. (2020). A family with Classical Ehlers-Danlos Syndrome (cEDS), mild bone fragility, and without vascular complications, caused by the p.Arg312Cys mutation in COL1A1. Eur. J. Med. Genet. 63:103730. doi: 10.1016/j.ejmg.2019.103730
Forlino, A., and Marini, J. C. (2016). Osteogenesis imperfecta. Lancet 387, 1657–1671. doi: 10.1016/S0140-6736(15)00728-X
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: the human genomic variant search engine. Bioinformatics 35, 1978–1980. doi: 10.1093/bioinformatics/bty897
Lin, Z., Zeng, J., and Wang, X. (2019). Compound phenotype of osteogenesis imperfecta and Ehlers-Danlos syndrome caused by combined mutations in COL1A1 and COL5A1. Biosci. Rep. 39:BSR20181409. doi: 10.1042/BSR20181409
Lindert, U., Gnoli, M., Maioli, M., Bedeschi, M. F., Sangiorgi, L., Rohrbach, M., et al. (2018). Insight into the pathology of a COL1A1 signal peptide heterozygous mutation leading to severe osteogenesis imperfecta. Calcif. Tissue Int. 102, 373–379. doi: 10.1007/s00223-017-0359-z
Lu, Y., Wang, Y., Rauch, F., Li, H., Zhang, Y., Zhai, N., et al. (2018). Osteogenesis imperfecta type III/Ehlers-Danlos overlap syndrome in a Chinese man. Intractable Rare Dis. Res. 7, 37–41. doi: 10.5582/irdr.2018.01010
Lund, A., Joensen, F., Christensen, E., Dunø, M., Skovby, F., and Schwartz, M. (2008). A novel arginine-to-cysteine substitution in the triple helical region of the alpha1(I) collagen chain in a family with an osteogenesis imperfecta/Ehlers-Danlos phenotype. Clin. Genet. 73, 97–101. doi: 10.1111/j.1399-0004.2007.00926.x
Maioli, M., Gnoli, M., Boarini, M., Tremosini, M., Zambrano, A., Pedrini, E., et al. (2019). Genotype-phenotype correlation study in 364 osteogenesis imperfecta Italian patients. Eur. J. Hum. Genet. 27, 1090–1100. doi: 10.1038/s41431-019-0373-x
Malfait, F., Francomano, C., Byers, P., Belmont, J., Berglund, B., Black, J., et al. (2017). The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 175, 8–26. doi: 10.1002/ajmg.c.31547
Malfait, F., Symoens, S., De Backer, J., Hermanns,-L.ê, T., Sakalihasan, N., Lapière, C. M., et al. (2007). Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum. Mutat. 28, 387–395. doi: 10.1002/humu.20455
Malfait, F., Symoens, S., Goemans, N., Gyftodimou, Y., Holmberg, E., López-González, V., et al. (2013). Helical mutations in type I collagen that affect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers-Danlos Syndrome overlap syndrome. Orphanet. J. Rare Dis. 8:78. doi: 10.1186/1750-1172-8-78
Marini, J. C., Forlino, A., Cabral, W. A., Barnes, A. M., San Antonio, J. D., Milgrom, S., et al. (2007). Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 28, 209–221. doi: 10.1002/humu.20429
Micale, L., Guarnieri, V., Augello, B., Palumbo, O., Agolini, E., Sofia, V. M., et al. (2019). Novel TNXB variants in two italian patients with classical-like Ehlers-Danlos syndrome. Genes (Basel) 10:967. doi: 10.3390/genes10120967
Morlino, S., Micale, L., Ritelli, M., Rohrbach, M., Zoppi, N., Vandersteen, A., et al. (2020). COL1-related overlap disorder: a novel connective tissue disorder incorporating the osteogenesis imperfecta/Ehlers-Danlos syndrome overlap. Clin. Genet. 97, 396–406. doi: 10.1111/cge.13683
Nicholls, A. C., Valler, D., Wallis, S., and Pope, F. M. (2001). Homozygosity for a splice site mutation of the COL1A2 gene yields a non-functional pro(alpha)2(I) chain and an EDS/OI clinical phenotype. J. Med. Genet. 38, 132–136. doi: 10.1136/jmg.38.2.132
Nuytinck, L., Wettinck, K., Freund, M., Van Maldergem, L., Fabry, G., and De Paepe, A. (1997). Osteogenesis imperfecta phenotypes resulting from serine for glycine substitutions in the alpha2(I) collagen chain. Eur. J. Hum. Genet. 5, 161–167. doi: 10.1159/000484752
Pyott, S. M., Pepin, M. G., Schwarze, U., Yang, K., Smith, G., and Byers, P. H. (2011). Recurrence of perinatal lethal osteogenesis imperfecta in sibships: parsing the risk between parental mosaicism for dominant mutations and autosomal recessive inheritance. Genet. Med. 13, 125–130. doi: 10.1097/GIM.0b013e318202e0f6
Raghunath, M., Bruckner, P., and Steinmann, B. (1994). Delayed triple helix formation of mutant collagen from patients with osteogenesis imperfecta. J. Mol. Biol. 236, 940–949. doi: 10.1006/jmbi.1994.1199
Rauch, F., and Glorieux, F. H. (2004). Osteogenesis imperfecta. Lancet 363, 1377–1385. doi: 10.1016/S0140-6736(04)16051-0
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Shi, X., Lu, Y., Wang, Y., Zhang, Y. A., Teng, Y., Han, W., et al. (2015). Heterozygous mutation of c.3521C>T in COL1A1 may cause mild osteogenesis imperfecta/Ehlers-Danlos syndrome in a Chinese family. Intractable Rare Dis. Res. 4, 49–53. doi: 10.5582/irdr.2014.01039
Symoens, S., Nuytinck, L., Legius, E., Malfait, F., Coucke, P. J., and De Paepe, A. (2004). Met>Val substitution in a highly conserved region of the pro-alpha1(I) collagen C-propeptide domain causes alternative splicing and a mild EDS/OI phenotype. J. Med. Genet. 41:e96. doi: 10.1136/jmg.2003.014589
Symoens, S., Steyaert, W., Demuynck, L., De Paepe, A., Diderich, K. E., Malfait, F., et al. (2017). Tissue-specific mosaicism for a lethal osteogenesis imperfecta COL1A1 mutation causes mild OI/EDS overlap syndrome. Am. J. Med. Genet. A 173, 1047–1050. doi: 10.1002/ajmg.a.38135
Keywords: COL1, collagen, collagen type I, overlap, clinical signs, musculoskeletal diseases, rare diseases
Citation: Gnoli M, Brizola E, Tremosini M, Pedrini E, Maioli M, Mosca M, Bassotti A, Castronovo P, Giunta C and Sangiorgi L (2021) COL1-Related Disorders: Case Report and Review of Overlapping Syndromes. Front. Genet. 12:640558. doi: 10.3389/fgene.2021.640558
Received: 11 December 2020; Accepted: 08 April 2021;
Published: 07 May 2021.
Edited by:
Ivan Duran, University of Malaga, SpainReviewed by:
Marco Castori, Casa Sollievo della Sofferenza (IRCCS), ItalyGerard Pals, Amsterdam University Medical Center, Netherlands
Fransiska Malfait, Ghent University, Belgium
Copyright © 2021 Gnoli, Brizola, Tremosini, Pedrini, Maioli, Mosca, Bassotti, Castronovo, Giunta and Sangiorgi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Sangiorgi, bHVjYS5zYW5naW9yZ2lAaW9yLml0
†These authors share first authorship