 Johanne Dubail
Johanne Dubail Valérie Cormier-Daire
Valérie Cormier-Daire- 1Université de Paris, INSERM UMR 1163, Institut Imagine, Paris, France
- 2Service de Génétique Clinique, Centre de Référence Pour Les Maladies Osseuses Constitutionnelles, AP-HP, Hôpital Necker-Enfants Malades, Paris, France
Chondrodysplasias with multiple dislocations form a group of severe disorders characterized by joint laxity and multiple dislocations, severe short stature of pre- and post-natal onset, hand anomalies, and/or vertebral anomalies. The majority of chondrodysplasias with multiple dislocations have been associated with mutations in genes encoding glycosyltransferases, sulfotransferases, and transporters implicated in the synthesis or sulfation of glycosaminoglycans, long and unbranched polysaccharides composed of repeated disaccharide bond to protein core of proteoglycan. Glycosaminoglycan biosynthesis is a tightly regulated process that occurs mainly in the Golgi and that requires the coordinated action of numerous enzymes and transporters as well as an adequate Golgi environment. Any disturbances of this chain of reactions will lead to the incapacity of a cell to construct correct glycanic chains. This review focuses on genetic and glycobiological studies of chondrodysplasias with multiple dislocations associated with glycosaminoglycan biosynthesis defects and related animal models. Strong comprehension of the molecular mechanisms leading to those disorders, mostly through extensive phenotypic analyses of in vitro and/or in vivo models, is essential for the development of novel biomarkers for clinical screenings and innovative therapeutics for these diseases.
Introduction
In 2019, the Nosology Committee of the International Skeletal Dysplasia Society published a new edition of the Nosology and Classification of Genetic Skeletal Disorders (Mortier et al., 2019). This 2019 version covers 461 different diseases divided into 42 groups according to their clinical, radiographic, and/or molecular phenotypes.
The application of massively parallel sequencing technology has led to the discovery in the last few years of a great number of genetic defects responsible for skeletal disorders. To date, the molecular bases have been identified for 425/461 (92%) of these disorders. In total, pathogenic variants affecting 437 different genes encoding enzymes, extracellular matrix (ECM) proteins, membrane transporters, cilia proteins, signal transduction proteins, and transcription factors have been found.
In this review, we will be focusing on the skeletal dysplasias caused by defects in the glycosaminoglycan (GAG) biosynthesis and, more specifically, on the group of chondrodysplasias with multiple dislocations (CMD), listed in groups 20, 4, and 25 in the International Classification on Genetic Skeletal Disorders (Mortier et al., 2019). They form a group of severe disorders characterized by joint laxity and multiple dislocations affecting large joints (such as hip, knee, and shoulder), severe short stature of pre- and post-natal onset, hand anomalies, and/or vertebral anomalies. Common radiographic features include advanced carpal and tarsal bone age and exaggerated trochanters giving a monkey wrench appearance of the femoral neck (Figure 1). Additional skeletal features, for instance, epiphyseal or metaphyseal changes, specific facial dysmorphisms, and cleft palate, are often part of the clinical presentation. A variable combination of other clinical features such as loose or old-appearing skin, congenital heart defects, teeth anomalies, intellectual disabilities, and obesity can also be observed in those patients. Up to now, more than 25 syndromes with autosomal recessive inheritance patterns have been described (Table 1). CMD has mostly been linked to pathogenic variants in genes implicated in the biosynthesis of proteoglycan (PG). PGs are large macromolecules that are widely expressed in multicellular organisms, present in the ECM or at the cell surface. They consist of a core protein and one or more covalently linked polysaccharides, called GAGs. GAGs are large linear polysaccharides composed of repeated disaccharide units consisting of amino sugar, either N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc), and uronic acid, either glucuronic acid (GlcUA) or iduronic acid (IdoUA), except keratan sulfate (KS) in which disaccharide units consist of GlcNAc and galactose. Hyaluronan (HA) is a non-sulfated glycosaminoglycan and is not attached to any core protein. It is synthesized by a specific synthesis pathway taking place at the cell membrane. Sulfated GAGs are classified into four groups based on the composition of their disaccharide units: chondroitin sulfate (CS), dermatan sulfate (DS), KS, and heparan sulfate. In addition, these GAGs undergo further modifications, such as sulfation at various positions of the chain and epimerization of uronic acid (Lindahl et al., 2017).
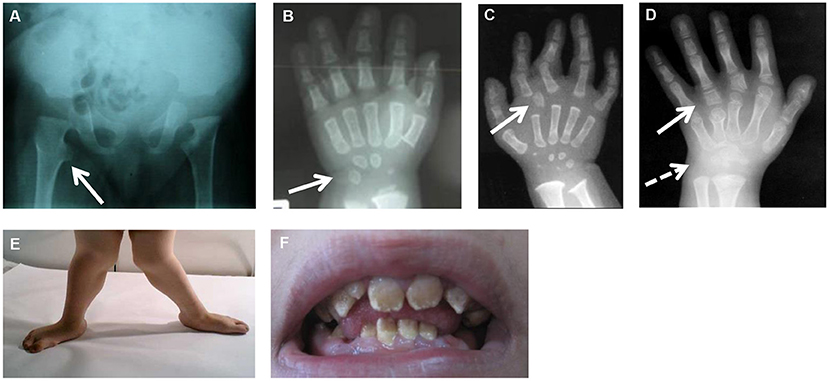
Figure 1. Common and specific clinical features in CMD. (A) Hip X-rays showing a monkey wrench appearance of femur (see arrow). (B) Hand X-rays of a patient with XYLT1 mutations at 8 months of age showing advanced carpal ossification (see arrow). (C) Hand X-rays of a patient with CANT1 mutations at 1 year of age showing presence of a delta phalanx (see arrow). (D) Hand X-rays of a patient with IMPAD1 mutations at 5 years of age showing presence of hyperphalangy (see plain arrow) and carpal synostosis (see dashed arrow). (E) Genu valgum due to joint laxity. (F) Amelogenesis imperfecta in a patient with SLC10A7 mutations.
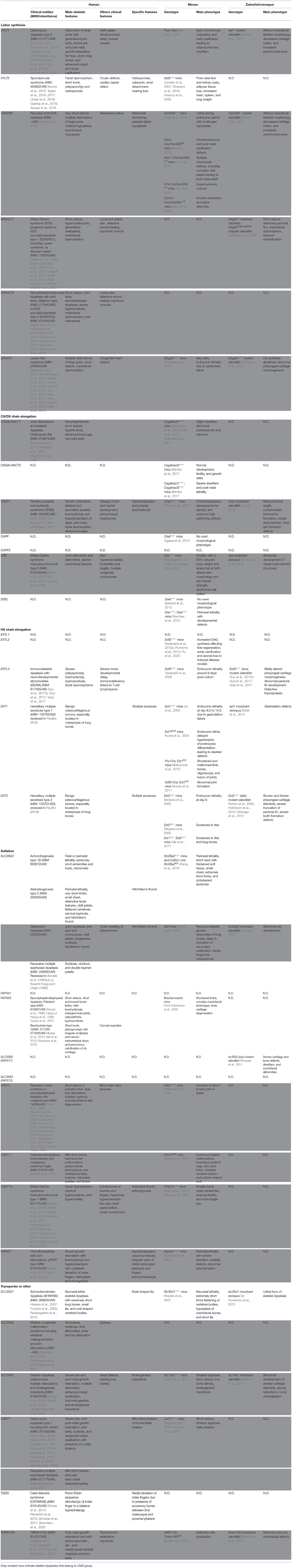
Table 1. Skeletal dysplasias caused by defects in GAG biosynthesis and related animal models.
PGs are highly diverse ECM components. Indeed, they can be composed of different core proteins with one or more GAG chain(s) of various subtypes and are subjected to variable degrees of post-translational modifications, including glycosylation and sulfation. Altogether, this leads PG to have a multitude of biological functions. Indeed, PGs promote ECM assembly by interacting with other ECM components, regulate ECM physical properties, and serve as a reservoir for various growth factors (Schaefer and Schaefer, 2010; Iozzo and Schaefer, 2015). In particular, PGs are highly expressed in cartilage ECM and play a major role in chondrocyte maturation and bone formation through endochondral ossification. PGs are also, through their ability to bind and retain water in the matrix, a critical component of articular cartilage, ensuring adequate mechanical properties and integrity maintain of articular cartilage (Martínez-Moreno et al., 2019).
Chondroitin Sulfate, Dermatan Sulfate, Heparan Sulfate Biosynthesis
The GAG biosynthesis is a complex process implicating the action of multiple enzymes (Figures 2, 3, Table 2) and that, although it is initiated in the endoplasmic reticulum (ER), occurs mainly in the Golgi apparatus cisternae (Prydz, 2015). GAG biosynthesis is initiated in the ER by the attachment of a xylose (Xyl) residue, using uridine diphosphate (UDP)-Xyl as a donor, to specific serine residues of the freshly synthetized PG core protein by β-xylosyltransferases encoded by XYLT1 or XYLT2 (Götting et al., 2007). After Xyl addition and shipment of the xylosylated protein into the Golgi apparatus, a linkage tetrasaccharide is formed by the transfer of two galactose (Gal) residues from UDP-Gal and one GlcUA from UDP-GlcUA via the sequential action of β1,4-galactosyltransferase-I (GalT-I), β1,3-galactosyltransferase-II (GalT-II), and β1,3-glucuronosyltransferase-I (GlcAT-I), encoded by B4GALT7, B3GALT3, and B3GAT3 (Okajima et al., 1999; Pedersen et al., 2000; Bai et al., 2001). Some modifications may occur on the linkage tetrasaccharide, including 2-O-phosphorylation of Xyl residue along with sulfation of the first Gal residue at the C-6 position and of the second Gal residue at the C-4 or C-6 position (Gulberti et al., 2005). The phosphorylation, which can be transient, is catalyzed by a GAG-Xyl kinase encoded by FAM20B (Koike et al., 2009). This phosphorylation together with sulfation may influence the catalytic activity of GalT-I, GalT-II, and GlcAT-I and thus the linkage region assembly and subsequent GAG elongation (Wen et al., 2014).
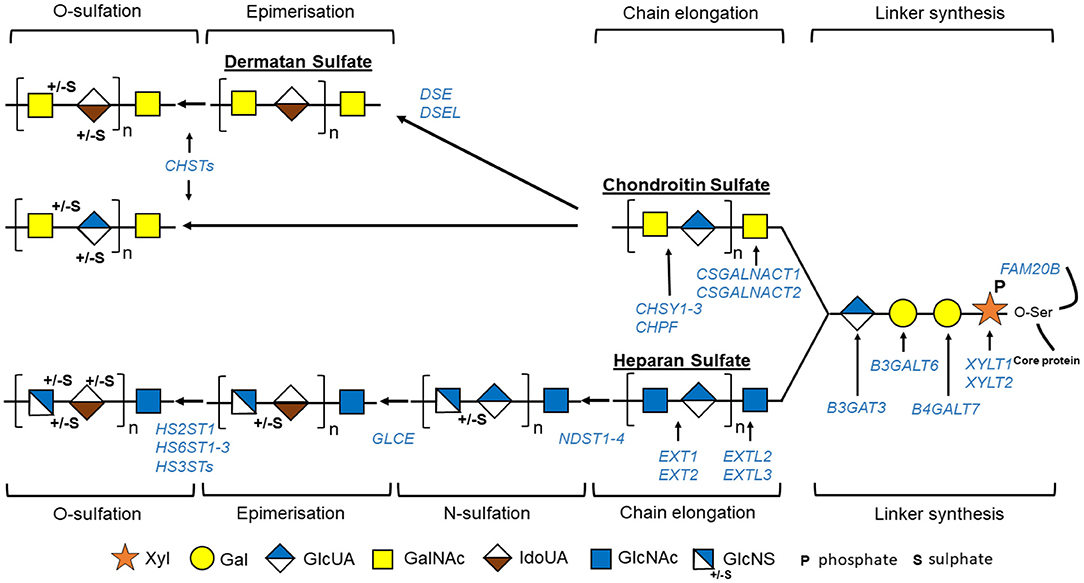
Figure 2. Schematic view of GAG biosynthesis: GAG biosynthesis is initiated by sequential addition to specific serine residues of PG core protein of one xylose (Xyl), two galactoses (Gal), and one glucuronic acid (GlcUA) constituting tetrasaccharide linker region common to tree groups of GAG, i.e., heparan sulfate, chondroitin sulfate, and dermatan sulfate. GAG chains will then be elongated by binding specific repetitive disaccharides [N-acetylgalactosamine (GalNAc) and glucuronic acid (GlcUA) for chondroitin sulfate and N-acetylglucosamine (GlcNAc) and GlcUA for heparan sulfate]. Some residues are modified: epimerization of GlcUA to iduronic acid (IdoUA) that will generate dermatan sulfate from chondroitin sulfate, or N-sulfation/N-deacetylation of GlcNAc to N-sulfoglucosamine (GlcNS) followed by epimerization of GlcUA to IdoUA for heparan sulfate. Finally, GAGs are further modified by O-sulfation. Different enzymes implicated in these processes are indicated in blue. “HS3TSs” include seven HS3ST (HS3ST1, 2, 3A, 3B, 4, 5, and 6), and “CHSTs” include four chondroitin-4-O-sulfotransferases (CHST11-14), two chondroitin-6-O-sulfotransferases (CHST3 and 7), one GalNAc-4-O-sulfate-6-O-sulfotransferase (CHST15), and one uronyl-2-O-sulfotransferase.

Figure 3. Schematic view of enzymes or transporters associated with GAG synthesis: GAG synthesis requires specific substrates, nucleotide sugars used by glycosyltransferases, and PAPS used by sulfotransferases. Nucleotide sugars and PAPS are synthetized in cytoplasm and are transported in Golgi by specific carriers. Then, glycosyltransferase and sulfotransferase activities produce by-products, UDP and PAP, respectively, that have to be degraded to avoid inhibition of catalytic activity of these enzymes via feedback mechanisms. Finally, to assure correct activity of glycosyltransferases, Golgi environment, and in particular its concentration in divalent ions, has to be tightly regulated via import/export of these ions. Enzymes, proteins, or transporters implicated in these processes are indicated in blue.
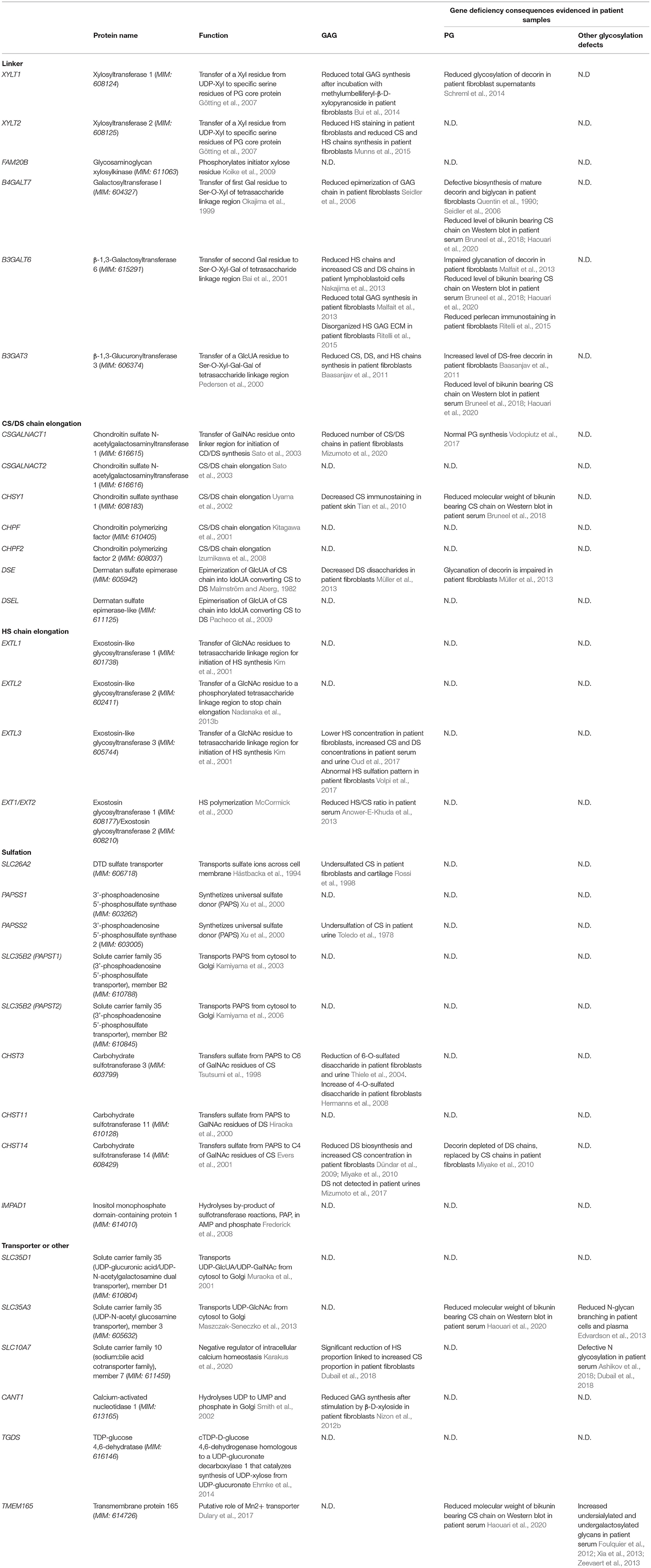
Table 2. GAG biosynthesis enzymes implicated in defects observed in patient samples.
Once the linkage region is completed, two types of reactions occur and determine the type of GAG being synthetized: addition to the linkage tetrasaccharide of either a β4-linked GalNAc, which will initiate CS/DS assembly, or an α4-linked GlcNAc, which will initiate HS assembly (Lindahl et al., 2017).
In CS, the process starts with the transfer of a GalNAc from UDP-GalNAc to the last GlcUA residue of the linkage region by specific β1,4-N-acetlygalactosaminyltransferases, CSGalNAcT1 and CSGalNAcT2 (encoded by CSGALNACT1 and CSGALNACT2, respectively) (Uyama et al., 2002; Sato et al., 2011). CS chains are then polymerized by the action of one or more enzymes having both β3 glucuronosyltransferase and β4 N-acetylgalactosaminyltransferase activities, the chondroitin synthases (CHSY1-3) (Kitagawa et al., 2001). During this step, the chondroitin polymerizing factor, lacking independent activity, will interact with the chondroitin synthases and enhance the CS elongation (Izumikawa et al., 2008). CSs are then subjected to modifications such as epimerization and sulfation throughout the GAG synthesis process or just before PG secretion (Lindahl et al., 2017). CS sulfation in an elaborate process involved multiple sulfotransferases, three chondroitin-4-O-sulfotransferases (CHST11-13) for C4 sulfation of GalNac residues, two chondroitin-6-O-sulfation (CHST3 and 7) for C6 sulfation of GalNac residues, one GalNac-4-O-sulfate-6-O-sulfotransferase (CHST15) for sulfation of disulfated GalNAc, and one uronosyl-2-O-sulfotransferase for C2 sulfation of GlcUA (Mizumoto et al., 2013).
DSs are generated from CS by C5 epimerization of GlcUA to IdoA by two DS epimerases (DSE1–2) and sulfation in distinct positions by dermatan-4-O-sulfotransferase (CHST14) and uronosyl-2-O-sulfotransferase (Malmström and Aberg, 1982).
In HS, the assembly is initiated by the addition of a GlcNAc residue by an α1,4-N-acetylglucosaminyltransferase-I (GlcNAcT-I) encoded by EXTL3 (Kim et al., 2001). HS further elongation is carried out by HS polymerase complex formed by two enzymes with N-acetylglucosaminyltransferase and glucuronyltransferase activities and encoded by EXT1 and EXT2 (McCormick et al., 2000). HS then undergoes extensive modification reaction creating clusters of sulfated domains interspersed with an unsulfated region (Esko and Selleck, 2002). Those modifications are initiated by N-deacetylase/N-sulfotransferases (NDST1–4), which induce the N-sulfation of 40–50% of GlcNAc to N-sulfoglucosamine (GlcNS) followed by conversion of adjacent GlcA to idorunate (IdoA) by a glucuronyl epimerase. O-sulfotransferases can then modify these GlcNS/IdoA rich domains. HS2ST1 catalyzes the 2-O-sulfation of IdoA residues. IdoA(2S)-GlcNS can then be further modified by the addition of 6-O-sulfate and less frequently by addition of 3-0-sulfate groups to the GlcNS residues, by the action of 6-O-sulfotransferases (HS6ST1-3) and seven 3-O-sulfotransferases (HS3ST), respectively.
The sulfation of GAG is a crucial process during PG synthesis and is required for GAG physiological functions. In the Golgi apparatus, sulfotransferases use the 3′-phosphoadenosine 5′-phosphosulfate (PAPS) as a universal sulfate donor to transfer sulfate to specific residues of GAG chains (Paganini et al., 2020). PAPS is synthetized in the cytosol from adenosine triphosphate (ATP) and inorganic sulfate. The latter is transported from the extracellular environment into the cells through a sulfate/chloride antiporter named SLC26A2 (Hästbacka et al., 1994). PAPS synthesis takes place in two sequential steps by the action of a bifunctional enzyme, the PAPS synthase (Xu et al., 2000). The ATP sulfurylase activity firstly catalyzes the production of adenosine 5′-phosphosulfate (APS) from sulfate and ATP; subsequently, APS kinase activity produces PAPS from APS and ATP. Once PAPS is synthetized in the cytosol, it is translocated into the Golgi by two specific PAPS transporters (PAPS transporters 1 and 2) (Kamiyama et al., 2003, 2006). As a consequence of sulfotransferase activity, PAP is released and can inhibit those sulfotransferases via negative feedback. To prevent this, PAP is rapidly degraded into adenosine monophosphate and phosphate by a Golgi resident adenosine 3′, 5′-biphosphate 3′-phosphatase (gPAPP), encoded by IMPAD1 (also known as BPNT2) (Frederick et al., 2008).
Nucleotide sugars such as PAPS are synthetized in the cytoplasm and have to be transported into the Golgi by specific carriers, such as SLC35D1 or SLC35A3, that by an antiport mechanism will export nucleoside monophosphates in the cytosol (Muraoka et al., 2001; Maszczak-Seneczko et al., 2013). This nucleotide sugar import seems to be a rate-limiting step as increased UDP-N-acetylhexosamine availability leads to enhancement of the incorporation into glycoconjugates (Pels Rijcken et al., 1995).
GAG elongation and modification reactions probably colocalized within the Golgi cisternae, most likely by the formation of supramolecular complexes that coordinate these reactions. A correct conformation of Golgi cisternae and organization of their enzymatic content, as well as an adequate Golgi environment, i.e., a properly established pH gradient and concentration of ions such as Ca2+, are also required for correct GAG formation (Prydz, 2015). Any disturbances of this chain of reactions will lead to the incapacity of a cell to construct correct glycanic chains.
Chondrodysplasia With Multiple Dislocation and Associated Animal Models
So far, up to 27 distinct human genetic disorders have been associated with pathogenic variants in 23 genes encoding proteins implicated in GAG biosynthesis (Table 1, Figures 2, 3). With few exceptions, such as EXT1/EXT2 or SLC35D1, the vast majority of the pathological variants identified in these genes are responsible for skeletal dysplasia associating short stature and joint laxity and/or large joint dislocations, characteristic of the CMD group (Table 1, gray rows). Clinical features of inborn errors of GAG biosynthesis, as well as the phenotype of existing related deficient animal models, are described in Table 1. The functional consequences of these inborn errors on GAG or PG synthesis, evidenced in patient samples, are listed in Table 2.
In the following section, we will focus on genes implicated in CMD.
Defective Linker Region Biosynthesis
Disorders due to mutations in XYLT1, B4GALT7, B3GALT6, and B3GAT3, encoding enzymes involved in the synthesis of the common linker region, are now frequently referred to as “linkeropathies.” FAM20B, which encodes a xylose kinase, in which its activity affects the synthesis of the common linker region, will also be described in this section.
XYLT1
Desbuquois dysplasia type 2 (DD 2), called Baratella–Scott syndrome, is caused by homozygous mutations in XYLT1 (Bui et al., 2014). DD 2 is characterized by severe pre- and post-natal growth retardation, dislocation of large joints with generalized joint laxity, short, long bones with a monkey wrench appearance to the proximal femur, advanced ossification of carpal and tarsal bones, and facial dysmorphisms, including flat face with prominent eyes. It is also associated with alterations in the fat distribution, variable degree of intellectual disabilities, and cleft palate. Reduced total GAG synthesis and decorin glycosylation were detected in fibroblasts of affected individuals compared with healthy controls (Bui et al., 2014; Schreml et al., 2014). Before the identification of pathogenic variants in humans, a mutant mouse and a mutant zebrafish were described, both exhibiting skeletal alterations. In pug mice, identified by ENU mutagenesis screen, XYLT1 deficiency due to homozygous missense mutation in Xylt1 is responsible for disproportionate dwarfism due to an early chondrocyte maturation and early ossification (Mis et al., 2014). A mutagenesis screen in zebrafish, isolating mutant fish harboring decreased cartilage matrix and increased perichondral bone, leads to the generation of xylt1 mutants (Eames et al., 2011). These mutant zebrafish failed to produce wild-type levels of CS and exhibited altered craniofacial skeletal morphology.
B4GALT7
Biallelic variants in B4GALT7 cause Ehlers–Danlos syndrome (EDS) progeroid type 1, now called EDS spondylodysplatic type 1 characterized by hypermobile joints, an aged appearance with loose yet elastic skin, poor wound healing, hypotonic muscles, craniofacial dysmorphism, short stature, developmental delays, and generalized osteopenia (Okajima et al., 1999). Homozygous mutations in B4GALT7 are also responsible for a variant of Larsen syndrome frequent on the La Reunion Island, called Larsen of Reunion Island syndrome, which has clinical manifestations including characteristic facial features, multiple dislocations, short stature, and hyperlaxity (Cartault et al., 2015). In patient fibroblasts, B4GALT7 mutations result in abnormal biosynthesis of mature decorin and biglycan with reduced GAG chain epimerization (Quentin et al., 1990; Seidler et al., 2006). Recently, a reduced level of bikunin bearing CS chain was detected by Western blot in patient serum compared with healthy controls (Bruneel et al., 2018; Haouari et al., 2020). Both knockdown (morphant) and mosaic knockdown b4galt7 zebrafish models presented short stature, deformed pectoral fins, craniofacial dysmorphism, and reduced mineralization (Delbaere et al., 2020).
B3GALT6
Mutations in B3GALT6 cause EDS progeroid type 2, also called EDS spondylodysplastic type 2 and spondyloepimetaphyseal dysplasia with joint laxity, Beighton type (Vorster et al., 2015). The main clinical features for this autosomal recessive syndrome include an aged appearance with loose but elastic skin and defective wound healing, hypermobile joints, developmental delay, short stature, craniofacial disproportion, kyphoscoliosis, epimetaphyseal dysplasia, generalized osteopenia, and hypotonic muscles. Fibroblasts from affected individuals exhibited altered GAG synthesis with impaired glycanation of decorin and marked reduction of HS synthesis (Malfait et al., 2013; Nakajima et al., 2013; Ritelli et al., 2015). As for patients with B4GALT7 mutations, a reduced level of bikunin bearing CS chain was detected by Western blot in patient serum compared to healthy controls (Bruneel et al., 2018; Haouari et al., 2020).
B3GAT3
Recessive variants in B3GAT3 cause Larsen-like syndrome characterized by short stature, multiple joint dislocations, scoliosis, osteopenia, and cranial dysmorphisms such as a flattened midface, hypertelorism, depressed nasal bridge, and prominent forehead (Van Damme et al., 2018). Congenital heart defects, including mitral valve prolapse, ventricular defect, and bicuspid aortic valve, can be observed in those patients. Patient fibroblasts exhibited reduced CS, DS, and HS and an increased level of DS-free decorin as compared with healthy controls (Baasanjav et al., 2011). Moreover, a reduced level of bikunin bearing CS chain was detected in patient serum compared with healthy controls (Bruneel et al., 2018; Haouari et al., 2020). B3gat3-deficient mice synthesized a smaller CS and HS chain in their blastocysts than that of heterozygous mice and exhibited an embryonic lethality before the eight-cell stage due to the failure of cytokinesis (Izumikawa et al., 2010). On the other hand, b3gat3 mutant zebrafish presented with abolished CS synthesis and abnormal pharyngeal cartilage morphogenesis (Holmborn et al., 2012).
FAM20B
Recently, compound heterozygous mutations in FAM20B have been described in patients with a lethal form of neonatal short-limb dysplasia characterized by very short stature and multiple dislocations of the large joints, thoracic hypoplasia, respiratory failure, and midface hypoplasia (Kuroda et al., 2019). Fam20b-deficient mice exhibited embryonic lethality at embryonic day 13.5 with multiorgan hypoplasia (Vogel et al., 2012). Furthermore, inactivation of Fam20b in several murine-specific tissues or cell types leading to skeletal defects demonstrated a role of Fam20b in bone development (Tian et al., 2015; Ma et al., 2016, 20; Liu et al., 2018; Saiyin et al., 2019). Similar to xylt1 zebrafish mutant, fam20b zebrafish mutant exhibited altered craniofacial skeletal morphology, decreased cartilage matrix, and increased perichondral bone (Eames et al., 2011).
Defective Glycosaminoglycan Chain Elongation or Epimerization
CSGALNACT1, CHSY1, and DSE Encode for enzymes implicated in CS/DS chain elongation and epimerization of CS to DS. No joint laxity or joint dislocations were described in disorders linked to pathogenic variants encoding for enzymes implicated in HS chains elongation. They will thus not be discussed in this section.
CSGALNACT1
Pathogenic variants in CSGALNACT1 have been identified recently in patients with a skeletal dysplasia characterized by a mild micromelic and non-proportioned stature, joint laxity, and advanced bone age (Vodopiutz et al., 2017; Mizumoto et al., 2020). Altered levels of CS, DS, and HS moieties were observed in patient fibroblasts compared with healthy controls (Vodopiutz et al., 2017; Mizumoto et al., 2020). CSGalNAcT1-deficient mice were described several years before and presented with slight dwarfism and abnormalities in perineural nets and behavior (Watanabe et al., 2010; Sato et al., 2011; Yoshioka et al., 2017).
CHSY1
Temtamy preaxial brachydactyly syndrome is caused by homozygous mutations in CHSY1 (Li et al., 2010). It is characterized by bilateral and symmetric preaxial brachydactyly and hyperphalangism of digits, growth retardation, facial dysmorphism, deafness, and delayed motor and mental development. Patient skin biopsy exhibited decreased CS-specific immunostaining compared with controls, and reduced molecular weight bikunin bearing CS chain was detected by Western blot in patient serum compared with healthy controls (Tian et al., 2010; Bruneel et al., 2018). Chsy1-deficient mice developed a phenotype mimicking the human pathology, presenting with a chondrodysplasia and decreased bone density with severe digit patterning defects (Wilson et al., 2012). In the same way, chsy morphant zebrafish presented with reduced body length, compromised pectoral fin formation, cranial dysmorphism, and inner ear formation defects (Li et al., 2010).
DSE
Homozygous mutations in DSE cause EDS musculocontractural type 2 characterized by joint hypermobility (finger, elbow, and knee), distinctive facial features, multiple congenital contracture contractures (thumbs and feet), and myopathy (Müller et al., 2013). Additional features might include cardiac, valvular, respiratory, gastrointestinal, and ophthalmic complications. Decreased DS level and reduced glycanation of decorin have been evidenced in patient fibroblasts compared with controls (Müller et al., 2013). Dse-deficient mice were smaller, with a kinky tail at birth, altered skin morphology and skin tensile strength, and abdominal wall defects (Maccarana et al., 2009; Gustafsson et al., 2014). On the other hand, dse morphant xenopus showed abnormal development of neural crest-derived structure (Gouignard et al., 2016).
Defective Glycosaminoglycan Sulfation
SLC26A2, CHST3, CHST11, CHST14, and IMPAD1 Encode for transporter and enzymes implicated in sulfation CS/DS chains. Except from a very recent manuscript demonstrating that mutations in HS2ST1 are responsible for a syndrome characterized by developmental delay with corpus callum, skeletal, and renal abnormalities (Schneeberger et al., 2020), pathogenic variants in enzymes implicated in HS sulfation have not been associated with skeletal dysplasia and will not be described here.
SLC26A2
SLC26A2-linked chondrodysplasias form a heterogeneous group of four different skeletal diseases caused by mutations in SLC26A2 (Bonafé et al., 1993a,b,c; Superti-Furga and Unger, 1993). They include decreasing order of severity, from lethal to mild, achondrogenesis type 1B, atelosteogenesis type 2, diastrophic dysplasia (DTD), and recessive multiple epiphyseal dysplasia. Among them, DTD is characterized by short stature, joint deformities and joint contractures, club foot, progressive kyphoscoliosis of the spine, hitchhiker thumb, characteristic ear deformities, and, occasionally, cleft palate. Undersulfated CSs were detected in patient fibroblasts and cartilage biopsies (Rossi et al., 1998). The skeletal phenotype of dtd mice, an animal model of human DTD, included reduced skeletal growth with long bone deformities and osteoporosis. Moreover, growth plate cartilage showed reduced toluidine blue staining, chondrocytes of irregular size, and delayed secondary ossification center formation (Forlino et al., 2005). Slc26a2 morphant zebrafish exhibited, on the other hand, an abnormal otic development (Liu et al., 2015).
CHST3 (CST6)
Loss-of-function mutations in CHST3 cause spondyloepiphyseal dysplasia with congenital joint dislocations also called recessive Larsen syndrome (Thiele et al., 2004). It is characterized by short stature of prenatal onset, large joint dislocations at birth (knees and/or hips), elbow joint dysplasia with subluxation and limited extension, clubfoot, and progressive kyphosis appearing during late childhood. Sulfation defects were detected in patient fibroblasts with a reduction of 6-O-sulfated disaccharides and increased 4-O-sulfated disaccharides compared with controls (Hermanns et al., 2008). Furthermore, a reduction in 6-O-sulfated disaccharides in patient urine was reported (Thiele et al., 2004). Cst6-deficient mice did not develop skeletal dysplasia but exhibited a decreased number of naïve T lymphocytes in the spleen (Uchimura et al., 2002).
CHST11 (C4ST1)
Mutations in CHST11 have been recently identified in patients with osteochondrodysplasia, brachydactyly, and overlapping malformed digits (Shabbir et al., 2018). Individuals with CHST11 mutations have bilateral symmetric skeletal defects affecting primarily the limbs with shortening of the lower leg bones leading to mild short stature, associated with hand and foot malformations, predominantly brachydactyly and overlapping digits. Scoliosis, dislocated patellae and fibulae, and pectus excavatum can also be observed. C4st1 mutant mice, previously generated by gene trap mutagenesis, exhibited a severe chondrodysplasia linked to abnormalities in the long bone growth plate (Klüppel et al., 2005). C4st1 mutant embryos developed several skeletal malformations, including a small rib cage, very short limbs, a twisted vertebral column, and a dome-shaped skull.
CHST14
Mutations in CHST14 cause EDS musculocontractural type 1 that has a similar clinical phenotype to EDS muscolocontractural type 2 due to mutations in CHST3 (Malfait et al., 2010). It is characterized by typical facial appearance, thumb and finger congenital contractures, clubfeet, joint hypermobility, severe kyphoscoliosis, muscular hypotonia, ocular involvement, and characteristic cutaneous features including skin hyperextensibility, thin skin, easy bruisability, atrophic scar, and increased palmar winkling. Decreased dermatan sulfate and increased chondroitin sulfate chain synthesis was measured in patient fibroblasts compared with control, and although DS could be detected in urine from healthy controls, it was not the case in urine from patients with CHST14 mutations (Dündar et al., 2009; Miyake et al., 2010; Mizumoto et al., 2017). Chst14-deficient mice had a smaller body mass, a kinked tail, reduced fertility, and a more fragile skin than wild-type mice (Akyüz et al., 2013; Hirose et al., 2019).
IMPAD1
Mutations in IMPAD1 cause chondrodysplasia with joint dislocations, gPAPP type (Vissers et al., 2011; Nizon et al., 2012a). It is characterized by chondrodysplasia with severe growth retardation with brachydactyly, joint dislocation, and cleft palate with micrognathia. Radiographs of hands and feet revealed abnormal extremities with the presence of many accessory bones, abnormally shaped phalanges, and carpal synostosis. Impad1-deficient mice developed a perinatal lethal phenotype with severe dwarfism, skeletal defects, and abnormal joint formation (Sohaskey et al., 2008).
Defective Activity of Transporters and Other Golgi Proteins
In this section, we focus on genes encoding transporter or protein expressed in the Golgi for which pathogenic variants have been identified in patients with CMD and associated with defects in GAG biosynthesis, even if their exact functions on GAG biosynthesis have not been elucidated.
SLC35A3
Pathogenic variants in SLC35A3 have been identified in patients with epilepsy, mental retardation, and multiple skeletal defects, including shortened long bones, vertebral anomalies, large joint dislocation, and arthrogryposis (Edmondson et al., 2017; Marini et al., 2017). The skeletal features are similar to those observed in the complex vertebral malformation phenotype observed in cattle and due to homozygous missense mutation in bovine Slc35a3 (Thomsen et al., 2006). SLC35A3 encodes a carrier that transports UDP-GlcNAc from the cytosol to the Golgi, where it serves as a substrate for glycosyltransferases (Maszczak-Seneczko et al., 2013). Reduced N-glycan branching has been evidenced in patient cells and plasma compared with control (Edvardson et al., 2013). Moreover, due to its localization and the transporter substrate, it was assumed that SLC35A3 might affect GAG metabolism, and, recently, an abnormal migration profile of bikunin bearing CS chain was observed on Western blot on patient serum (Haouari et al., 2020).
SLC10A7
Biallelic mutations in SLC10A7 have been identified in skeletal dysplasia with amelogenesis imperfecta characterized by a pre- and post-natal short stature, large joint dislocations, luxation of knees with genua valga, hypomineralized amelogenesis imperfecta, decreased bone density, “monkey wrench” appearance of the proximal femora, small epiphyses, advanced carpal ossification abnormal vertebrae, hyperlordosis or kyphoscoliosis, and dysmorphic facial features including Pierre–Robin sequence, micrognathia, and flat face (Ashikov et al., 2018; Dubail et al., 2018). Additional features included heart defects, hearing loss, and obesity. SLC10A7 codes for 10-transmembrane-domain transporter of unknown substrate specificity located at the Golgi and plasma membrane. Although its function remains unknown, it has been demonstrated that it negatively affects intracellular calcium homeostasis and increased calcium intake that have been measured in patient fibroblasts compared with controls (Dubail et al., 2018; Karakus et al., 2020). This inadequate intracellular calcium influx most likely disturbs the Golgi ionic environment and in fine glycosyltransferase activities. Significant reduction of HS proportion linked to increased CS proportion was observed in patient fibroblasts compared with control (Dubail et al., 2018). Interestingly, defective N-glycosylation was also detected in patient serum (Ashikov et al., 2018; Dubail et al., 2018). Slc10a7-deficient mice recapitulated human phenotype with short stature, growth plate disorganization, low bone density, and tooth enamel defects (Dubail et al., 2018). Moreover, alterations of HS/CS content similar to those measured in patient fibroblasts were detected in Slc10a7-deficient mouse cartilage. On the other hand, slc10a7 morphant zebrafish presented with abnormal development of several cartilage elements and a strong reduction in bone mineralization (Ashikov et al., 2018).
CANT1
Mutations in CANT1 cause Desbuquois dysplasia type 1 characterized by severe pre-natal and post-natal growth retardation with short extremities, joint laxity, and progressive scoliosis (Huber et al., 2009). The main radiologic features include short, long bones with metaphyseal flaring, a “monkey wrench” appearance of the femur neck, and advanced carpal and tarsal ossification with the presence of an extra ossification center, a delta phalanx, between the proximal phalanx of the index and bifid distal thumb phalanx. A milder variant of Desbuquois dysplasia type 1, referred to as “Kim variant,” with hands appearing almost normal externally, but that on radiographic analyses are characterized by elongated phalanges, short metacarpals, and remarkably advanced carpal bone age, has also been linked to pathogenic variants in CANT1 (Furuichi et al., 2011). CANT1 codes for calcium-activated nucleotidase, an ER and Golgi nucleotidase that hydrolyses UDP to UMP and phosphate (Smith et al., 2002). Although it has been suggested that CANT1 deficiency would lead to inhibition of glycosyltransferase activities and reduced transport in the Golgi of UDP-sugar via negative feedback resulting from increased Golgi UDP level and that CANT1 is implicated, through inositol 1,4,5-triphosphate receptor activation, in vesicular trafficking in Golgi cisternae by calcium release, both potentially affecting GAG synthesis, its exact function remains unknown (Huber et al., 2009; Nizon et al., 2012b). Demonstrating the implication of CANT1 in GAG biosynthesis regulation, reduced GAG synthesis after stimulation by β-D-xyloside has been measured in patient fibroblasts compared with controls (Nizon et al., 2012b). Cant1-deficient mice recapitulated the human phenotype of patients with Desbuquois dysplasia type 1, with short stature, thoracic kyphosis, and delta phalanx (Paganini et al., 2019). Cant1 deficiency led to altered GAG synthesis, with reduced chain length and increased sulfation and delayed secretion of PG in the ECM.
TMEM165
Congenital disorder of glycosylation (CDG), type IIk or TMEM165-CDG, is caused by biallelic mutations in TMEM165 (Foulquier et al., 2012). The most severe phenotypes observed in patients with TMEM165-CDG present growth retardation resistant to human growth hormone, associated with a psychomotor disability, microcephaly, facial hypoplasia, hypotonia, seizures, and hepatosplenomegaly with increased serum transaminases. Skeletal features of these patients include severe dwarfism, osteoporosis, epi-, meta-, and diaphyseal dysplasia, and joint laxity. TMEM165 encodes the transmembrane protein 165, TMEM165, located in the Golgi membrane and, in a lower proportion, at the plasma membrane, and in late endosomes/lysosomes, and which exact function remains unknown. However, several studies are in favor of a role of TMEM165 in Mn2+ homeostasis in the Golgi, suggesting that TMEM165 could act as a putative Mn2+ transporter. This function on Mn2+ homeostasis is essential for appropriate protein N-glycosylation occurring in the Golgi (Dulary et al., 2017). TMEM165 deficiency has mostly been associated with N-, O-glycosylation defects, with increased undersialylated and undergalactosylated glycans, as well as in high mannose type N-Glycan detected in patient serum compared with healthy control (Foulquier et al., 2012; Xia et al., 2013; Zeevaert et al., 2013). However, recently, an abnormal migration profile of bikunin bearing CS chain was observed on a Western blot on patient serum (Haouari et al., 2020). Moreover, tmem165 deficiency in zebrafish led to alterations in CS PG expression detected by immunohistochemical staining of cartilage (Bammens et al., 2015). Altogether, those results imply that TMEM165 deficiency can also lead to GAG synthesis defects. Analyses of these tmem165 morphant zebrafish demonstrated skeletal abnormalities, more particularly in craniofacial structures with altered bone growth and development. Complete Tmem165 deficiency in mice has not yet been described, but conditional deletion of Tmem165 in the mammary gland led to abnormal milk production due to defective lactose biosynthesis (Snyder et al., 2019).
Glycosaminoglycan Synthesis-Related Disorders Form a Subgroup of Congenital Disorders of Glycosylation
CDG are an expanding group of rare, multisystem, underdiagnosed heterogeneous diseases caused by deficient or improper synthesis or attachment of glycans to proteins and lipids. More than 130 inherited disorders have been identified so far, most of them following an autosomal recessive inheritance pattern (Ondruskova et al., 2021).
Glycosylation is a very important post-translational modification, as it is estimated that at least 2% of the human genome codes for proteins involved in this vital biochemical pathway (Ng and Freeze, 2018). Due to the involvement of glycosylation in various cellular processes, CDG are characterized by multiorgan dysfunction and variable clinical phenotype. Therefore, CDG result in a broad spectrum of pathologies, including skin laxity, skeletal dysplasia, congenital heart defects, neurodevelopmental disorders, and endocrine abnormalities. The most common clinical manifestations include developmental delay, failure to thrive, microcephaly, coagulopathy, and abnormal brain magnetic resonance imaging, including cerebral and/or cerebellar atrophy, cell migration abnormalities, and immune dysfunction (Francisco et al., 2019).
CDG are classified into two groups, CDG I and CDG II. CDG I are defects in the glycan assembly and in the attachment of glycans to proteins in the ER. CDG II are defects in the processing of the already assembled glycans in the Golgi apparatus resulting in truncated and abnormal glycan structures. As it is now considered that glycosylation processes occurring in the ER are required for good folding and stability of the glycoprotein, whereas glycan remodeling in the Golgi will finely regulate the functionality of the protein, CDG I and CDG II will differently affect glycoprotein functions (Freeze, 2007).
Most protein glycosylation disorders are due to defects in the N-glycosylation pathways, but they can also be due to defective O-linked glycans (O-Mannose, O-Glucose, O-Fucose, O-GlcNac, and O-GalNac), GAG, glycosylphosphatidylinositol, and glycolipids (Freeze et al., 2014). As at the Golgi level, the different glycosylation processes coexist, alterations of the Golgi environment/organization, and inadequate supply in common subtrates, will lead to combinations of two or more affected glycosylation pathways. This is well-illustrated by CMD due to mutations in genes coding for transporters or Golgi proteins, i.e., SLC35A3, SLC10A7, and TMEM165, for which alterations in both N-glycosylation and GAG biosynthesis were detected (Foulquier et al., 2012; Edvardson et al., 2013; Bruneel et al., 2018; Dubail et al., 2018).
Diagnosis
The group of CMD includes several complex syndromes with overlapping features and other clinical signs that are often non-specific, such as intellectual disability or cardiac defects. Even if few features are typical for some disorders (Table 1), characteristic hand anomalies in CANT1, IMPAD1, CHSY1, and TGDS-related conditions or amelogenesis imperfecta in dysplasia due to mutations in SLC10A7, for example, may help clinicians to orientate the clinical diagnosis. For the vast majority of CMD, phenotype–genotype correlations are still incomplete. Molecular or enzymatic assays to detect specific defects are thus crucial for final diagnostic. However, simple and affordable laboratory tests for use in screening are still missing, and, currently, clinicians must rely on research laboratories to perform the clinical molecular and biochemical tests to confirm the diagnosis. Many research teams have developed and used various techniques to evaluate quantitatively and qualitatively GAG synthesis. With these techniques, they were able to demonstrate in patient biological samples, for some PGs, a reduction of expression, concentration, or molecular weight or an alteration in the concentration of specific disaccharides, sulfated or unsulfated, constituting the GAG (Table 2). In most cases, analyses were performed on lysates or culture media from fibroblasts, cultured or not with radiolabeled substrates, after separation by high-performance liquid chromatography or gel electrophoresis (sodium dodecyl sulfate–polyacrylamide gel electrophoresis). Alternatively, altered migration of some low molecular weight PG was detected by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and immunoblotting or abnormal GAG expression was revealed by immunohistochemistry or immunofluorescence techniques using primary antibodies against specific GAG residues. Being more easily obtained than fibroblasts, blood and urine samples are more convenient for biochemical testing to perform diagnosis in clinics. Only a few studies have studied the GAG in blood or urine from patients using high-performance liquid chromatography. Recently, abnormal electrophoretic migration profiles of bikunin, a serum PG with a single CS chain, were detected after immunoblotting in serum from patients with specific CMD and, more specifically, from patients with “linkeropathies,” indicating that bikunin is a potential biomarker, easily detectable, for these pathologies (Bruneel et al., 2018; Haouari et al., 2020). Moreover, for patients with mutations in SLC35A3, SLC10A7, or TMEM165, alterations in both N-glycosylation and GAG biosynthesis were detected (Foulquier et al., 2012; Edvardson et al., 2013; Ashikov et al., 2018; Dubail et al., 2018). This demonstrates that it could be useful to look for glycosylation defects in patients with CMD to help refine the diagnosis.
Therapeutic Approaches
As for most of the skeletal and connective tissue disorders, treatments for CMD patients are still restricted to physiotherapy, orthopedic surgery, symptomatic treatment, or monitoring for potential complications to slow down or modify disease progression (Briggs et al., 2015; Marzin and Cormier-Daire, 2020). The development of new innovative therapies requires a strong comprehension of the molecular mechanisms leading to those disorders, mostly through extensive phenotypic analyses of in vitro and/or in vivo models. This is well exemplified by the use of N-acetylcysteine as a new therapeutic approach for patients with DTD. DTD is caused by pathogenic variants in a gene coding for SLC26A, a cell membrane sulfate-chloride antiporter, resulting in defective sulfate uptake leading to low cytosolic sulfate and subsequently PG undersulfation (Rossi et al., 1998). N-Ac, acting as an intracellular sulfate source for macromolecule sulfation, was thus tested, and preclinical studies in DTD mouse model showed promising results (Monti et al., 2015). N-Ac is currently being tested in DTD. Another example is given by TMEM165. Although the function of TMEM165 is still not completely understood, many observations suggest that TMEM165 has a role in Mn2+ homeostasis in the Golgi. As Mn2+ is a cofactor of glycosyltransferases, impaired Golgi Mn2+ homeostasis in the TMEM165-deficient patient is most likely responsible for the glycosylation defects. Strengthening this hypothesis, experiments performed in vitro have demonstrated that Mn2+ supplementation suppressed the glycosylation defects, and Mn2+ supplementation was proposed as potential new therapy for TMEM165-CDG patients (Dulary et al., 2017).
Concluding Remarks
The recent evolution of genomic technologies has allowed a big step forward in the comprehension of pathophysiological mechanisms leading to rare genetic disorders, including skeletal disorders and the group formed by CMD. Studies of this latter group of pathology have proved a central role of GAG biosynthesis in the pathogenesis of these rare disorders. GAG biosynthesis has characteristic biochemical properties and affects many processes such as embryonic development and connective tissue formation and functions. It is also a complex and tightly regulated process that is still not yet completely clarified. Studies performed on patient samples, cell cultures, and animal models for human CMD have provided new insights on the GAG synthesis and on the physiologic functions of GAG in cartilage, bone, and connective tissues. However, further comprehensive approaches to the molecular pathogenesis involving GAG chains, in association or not with other glycosylation defects, are required to facilitate the development of new biomarkers for clinical screenings and innovative therapeutics for these diseases.
Author Contributions
JD and VC-D have contributed to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Agence National de la Recherche funding (SKELGAG−18-CE14-0040-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AR declared a past co-authorship with one of the authors VC-D to the handling editor.
Abbreviations
C4ST, chondroitin-4-O-sulfotransferase; C6ST, chondroitin-6-O-sulfotransferase; CANT1, calcium-activated nucleotidase 1; CDG, congenital disorders of glycosylation; CHSY, chondroitin synthase; CMD, chondrodysplasia with multiple dislocations; CS, chondroitin sulfate; CSGalNAcT, chondroitin N-actelygalactosaminyltranferase; D4ST, dermatan-4-O-sulfotransferase; DD, Desbuquois dysplasia; DES, dermatan sulfate epimerase; DS, dermatan sulfate; DTD, diastrophic dysplasia; DTDST, diastrophic dysplasia sulfate transporter; ECM, extracellular matrix; EDS, Ehlers-Danlos syndrome; ER, endoplasmic reticulum; EXT, exostosin; GAG, glycosaminoglycan; Gal, galactose; GalNAcT-I, β1, 4-N-acetylgalactosaminyltransferase-I; GalT-II, β1, 3-galactosyltransferase-II; GalT-I, β1, 4-galactosyltransferase-I; GalNAc, N-acetylgalactosamine; GlcNAc, N-acetylglucosamine; GlcUA, glucuronic acid; gPAPP, Golgi resident phosphoadenosine phosphate phosphatase; IdoUA, iduronic acid; IMPAD1, inositol monophosphate domain-containing protein 1; KS, keratan sulfate; PAP, phosphoadenosine phosphate; PAPS, 3′-phosphoadenosine 5′-phosphosulfate; PG, proteoglycan; UDP, uridine diphosphate; Xyl, xylose; XYLT, xylosyltransferase.
References
Ahmad, M., Faiyaz Ul Haque, M., Ahmad, W., Abbas, H., Haque, S., Krakow, D., et al. (1998). Distinct, autosomal recessive form of spondyloepimetaphyseal dysplasia segregating in an inbred Pakistani kindred. Am. J. Med. Genet. 78, 468–473. doi: 10.1002/(SICI)1096-8628(19980806)78:5<468::AID-AJMG13>3.0.CO;2-D
Akyüz, N., Rost, S., Mehanna, A., Bian, S., Loers, G., Oezen, I., et al. (2013). Dermatan 4-O-sulfotransferase1 ablation accelerates peripheral nerve regeneration. Exp. Neurol. 247, 517–530. doi: 10.1016/j.expneurol.2013.01.025
Alazami, A. M., Al-Qattan, S. M., Faqeih, E., Alhashem, A., Alshammari, M., Alzahrani, F., et al. (2016). Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 135, 525–540. doi: 10.1007/s00439-016-1660-z
Albuz, B., Çetin, G. O., Özhan, B., Sarikepe, B., Anlaş, Ö., Öztürk, M., et al. (2020). A novel nonsense mutation in CHST3 in a Turkish patient with spondyloepiphyseal dysplasia, Omani type. Clin. Dysmorphol. 29, 61–64. doi: 10.1097/MCD.0000000000000295
Al-Jezawi, N. K., Ali, B. R., and Al-Gazali, L. (2017). Endoplasmic reticulum retention of xylosyltransferase 1 (XYLT1) mutants underlying desbuquois dysplasia type II. Am. J. Med. Genet. A 173, 1773–1781. doi: 10.1002/ajmg.a.38244
Anower-E-Khuda, M. F., Matsumoto, K., Habuchi, H., Morita, H., Yokochi, T., Shimizu, K., et al. (2013). Glycosaminoglycans in the blood of hereditary multiple exostoses patients: Half reduction of heparan sulfate to chondroitin sulfate ratio and the possible diagnostic application. Glycobiology 23, 865–876. doi: 10.1093/glycob/cwt024
Ashikov, A., Abu Bakar, N., Wen, X.-Y., Niemeijer, M., Rodrigues Pinto Osorio, G., Brand-Arzamendi, K., et al. (2018). Integrating glycomics and genomics uncovers SLC10A7 as essential factor for bone mineralization by regulating post-Golgi protein transport and glycosylation. Hum. Mol. Genet. 27, 3029–3045. doi: 10.1093/hmg/ddy213
Baasanjav, S., Al-Gazali, L., Hashiguchi, T., Mizumoto, S., Fischer, B., Horn, D., et al. (2011). Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 89, 15–27. doi: 10.1016/j.ajhg.2011.05.021
Bai, X., Zhou, D., Brown, J. R., Crawford, B. E., Hennet, T., and Esko, J. D. (2001). Biosynthesis of the linkage region of glycosaminoglycans: cloning and activity of galactosyltransferase II, the sixth member of the beta 1,3-galactosyltransferase family (beta 3GalT6). J. Biol. Chem. 276, 48189–48195. doi: 10.1074/jbc.M107339200
Balasubramanian, K., Li, B., Krakow, D., Nevarez, L., Ho, P. J., Ainsworth, J. A., et al. (2017). MED resulting from recessively inherited mutations in the gene encoding calcium-activated nucleotidase CANT1. Am. J. Med. Genet. A 173, 2415–2421. doi: 10.1002/ajmg.a.38349
Bammens, R., Mehta, N., Race, V., Foulquier, F., Jaeken, J., Tiemeyer, M., et al. (2015). Abnormal cartilage development and altered N-glycosylation in Tmem165-deficient zebrafish mirrors the phenotypes associated with TMEM165-CDG. Glycobiology 25, 669–682. doi: 10.1093/glycob/cwv009
Bartolini, B., Thelin, M. A., Rauch, U., Feinstein, R., Oldberg, A., Malmström, A., et al. (2012). Mouse development is not obviously affected by the absence of dermatan sulfate epimerase 2 in spite of a modified brain dermatan sulfate composition. Glycobiology 22, 1007–1016. doi: 10.1093/glycob/cws065
Bloor, S., Giri, D., Didi, M., and Senniappan, S. (2017). Novel splicing mutation in B3GAT3 associated with short stature, GH deficiency, hypoglycaemia, developmental delay, and multiple congenital anomalies. Case Rep. Genet. 2017:3941483. doi: 10.1530/endoabs.51.P021
Bonafé, L., Mittaz-Crettol, L., Ballhausen, D., and Superti-Furga, A. (1993a). “Achondrogenesis type 1B,” in GeneReviews®, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Bonafé, L., Mittaz-Crettol, L., Ballhausen, D., and Superti-Furga, A. (1993b). “Diastrophic dysplasia,” in GeneReviews®, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Bonafé, L., Mittaz-Crettol, L., Ballhausen, D., and Superti-Furga, A. (1993c). “Multiple epiphyseal dysplasia, recessive,” in GeneReviews®, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, et al. (Seattle, WA: University of Washington.
Boschann, F., Stuurman, K. E., de Bruin, C., van Slegtenhorst, M., van Duyvenvoorde, H. A., Kant, S. G., et al. (2020). TGDS pathogenic variants cause Catel-Manzke syndrome without hyperphalangy. Am. J. Med. Genet. A 182, 431–436. doi: 10.1002/ajmg.a.61419
Bownass, L., Abbs, S., Armstrong, R., Baujat, G., Behzadi, G., Berentsen, R. D., et al. (2019). PAPSS2-related brachyolmia: clinical and radiological phenotype in 18 new cases. Am. J. Med. Genet. A 179, 1884–1894. doi: 10.1002/ajmg.a.61282
Briggs, M. D., Bell, P. A., Wright, M. J., and Pirog, K. A. (2015). New therapeutic targets in rare genetic skeletal diseases. Expert Opin. Orphan Drugs 3, 1137–1154. doi: 10.1517/21678707.2015.1083853
Bruneel, A., Dubail, J., Roseau, C., Prada, P., Haouari, W., Huber, C., et al. (2018). Serum bikunin is a biomarker of linkeropathies. Clin. Chim. Acta Int. J. Clin. Chem. 485, 178–180. doi: 10.1016/j.cca.2018.06.044
Budde, B. S., Mizumoto, S., Kogawa, R., Becker, C., Altmüller, J., Thiele, H., et al. (2015). Skeletal dysplasia in a consanguineous clan from the island of Nias/Indonesia is caused by a novel mutation in B3GAT3. Hum. Genet. 134, 691–704. doi: 10.1007/s00439-015-1549-2
Bui, C., Huber, C., Tuysuz, B., Alanay, Y., Bole-Feysot, C., Leroy, J. G., et al. (2014). XYLT1 mutations in desbuquois dysplasia type 2. Am. J. Hum. Genet. 94, 405–414. doi: 10.1016/j.ajhg.2014.01.020
Cartault, F., Munier, P., Jacquemont, M.-L., Vellayoudom, J., Doray, B., Payet, C., et al. (2015). Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes larsen of reunion Island syndrome. Eur. J. Hum. Genet. 23, 49–53. doi: 10.1038/ejhg.2014.60
Colman, M., Van Damme, T., Steichen-Gersdorf, E., Laccone, F., Nampoothiri, S., Syx, D., et al. (2019). The clinical and mutational spectrum of B3GAT3 linkeropathy: two case reports and literature review. Orphanet J. Rare Dis. 14, 138. doi: 10.1186/s13023-019-1110-9
Condac, E., Silasi-Mansat, R., Kosanke, S., Schoeb, T., Towner, R., Lupu, F., et al. (2007). Polycystic disease caused by deficiency in xylosyltransferase 2, an initiating enzyme of glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 9416–9421. doi: 10.1073/pnas.0700908104
De Domenico, E., Owens, N. D. L., Grant, I. M., Gomes-Faria, R., and Gilchrist, M. J. (2015). Molecular asymmetry in the 8-cell stage Xenopus tropicalis embryo described by single blastomere transcript sequencing. Dev. Biol. 408, 252–268. doi: 10.1016/j.ydbio.2015.06.010
Delbaere, S., Van Damme, T., Syx, D., Symoens, S., Coucke, P., Willaert, A., et al. (2020). Hypomorphic zebrafish models mimic the musculoskeletal phenotype of β4GalT7-deficient ehlers-danlos syndrome. Matrix Biol. J. Int. Soc. Matrix Biol. 89, 59–75. doi: 10.1016/j.matbio.2019.12.002
Dubail, J., Huber, C., Chantepie, S., Sonntag, S., Tüysüz, B., Mihci, E., et al. (2018). SLC10A7 mutations cause a skeletal dysplasia with amelogenesis imperfecta mediated by GAG biosynthesis defects. Nat. Commun. 9:3087. doi: 10.1038/s41467-018-05191-8
Dulary, E., Potelle, S., Legrand, D., and Foulquier, F. (2017). TMEM165 deficiencies in congenital disorders of glycosylation type II (CDG-II): Clues and evidences for roles of the protein in golgi functions and ion homeostasis. Tissue Cell 49, 150–156. doi: 10.1016/j.tice.2016.06.006
Dündar, M., Müller, T., Zhang, Q., Pan, J., Steinmann, B., Vodopiutz, J., et al. (2009). Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 85, 873–882. doi: 10.1016/j.ajhg.2009.11.010
Duz, M. B., and Topak, A. (2020). Recurrent c.776T>C mutation in CHST3 with four other novel mutations and a literature review. Clin. Dysmorphol. 29, 167–172. doi: 10.1097/MCD.0000000000000329
Eames, B. F., Yan, Y.-L., Swartz, M. E., Levic, D. S., Knapik, E. W., Postlethwait, J. H., et al. (2011). Mutations in fam20b and xylt1 reveal that cartilage matrix controls timing of endochondral ossification by inhibiting chondrocyte maturation. PLoS Genet. 7:e1002246. doi: 10.1371/journal.pgen.1002246
Edmondson, A. C., Bedoukian, E. C., Deardorff, M. A., McDonald-McGinn, D. M., Li, X., He, M., et al. (2017). A human case of SLC35A3-related skeletal dysplasia. Am. J. Med. Genet. A 173, 2758–2762. doi: 10.1002/ajmg.a.38374
Edvardson, S., Ashikov, A., Jalas, C., Sturiale, L., Shaag, A., Fedick, A., et al. (2013). Mutations in SLC35A3 cause autism spectrum disorder, epilepsy and arthrogryposis. J. Med. Genet. 50, 733–739. doi: 10.1136/jmedgenet-2013-101753
Ehmke, N., Caliebe, A., Koenig, R., Kant, S. G., Stark, Z., Cormier-Daire, V., et al. (2014). Homozygous and compound-heterozygous mutations in TGDS cause catel-manzke syndrome. Am. J. Hum. Genet. 95, 763–770. doi: 10.1016/j.ajhg.2014.11.004
Esko, J. D., and Selleck, S. B. (2002). Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71, 435–471. doi: 10.1146/annurev.biochem.71.110601.135458
Evers, M. R., Xia, G., Kang, H. G., Schachner, M., and Baenziger, J. U. (2001). Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 276, 36344–36353. doi: 10.1074/jbc.M105848200
Faden, M., Al-Zahrani, F., Arafah, D., and Alkuraya, F. S. (2010). Mutation of CANT1 causes desbuquois dysplasia. Am. J. Med. Genet. A 152A, 1157–1160. doi: 10.1002/ajmg.a.33404
Faiyaz ul Haque, M., King, L. M., Krakow, D., Cantor, R. M., Rusiniak, M. E., Swank, R. T., et al. (1998). Mutations in orthologous genes in human spondyloepimetaphyseal dysplasia and the brachymorphic mouse. Nat. Genet. 20, 157–162. doi: 10.1038/2458
Faiyaz-Ul-Haque, M., Zaidi, S. H. E., Al-Ali, M., Al-Mureikhi, M. S., Kennedy, S., Al-Thani, G., et al. (2004). A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and ehlers-danlos syndrome resembling the progeroid type. Am. J. Med. Genet. A 128A, 39–45. doi: 10.1002/ajmg.a.30005
Ferencz, B., Condac, E., Poudel, N., Munteanu, M. C., Sivasami, P., Choudhury, B., et al. (2020). Xylosyltransferase 2 deficiency and organ homeostasis. Glycoconj. J. 37, 755–765. doi: 10.1007/s10719-020-09945-9
Ford-Hutchinson, A. F., Ali, Z., Seerattan, R. A., Cooper, D. M. L., Hallgrímsson, B., Salo, P. T., et al. (2005). Degenerative knee joint disease in mice lacking 3'-phosphoadenosine 5'-phosphosulfate synthetase 2 (Papss2) activity: a putative model of human PAPSS2 deficiency-associated arthrosis. Osteoarthritis Cartilage 13, 418–425. doi: 10.1016/j.joca.2004.12.011
Forlino, A., Piazza, R., Tiveron, C., Della Torre, S., Tatangelo, L., Bonaf,è, L., et al. (2005). A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Hum. Mol. Genet. 14, 859–871. doi: 10.1093/hmg/ddi079
Foulquier, F., Amyere, M., Jaeken, J., Zeevaert, R., Schollen, E., Race, V., et al. (2012). TMEM165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 91, 15–26. doi: 10.1016/j.ajhg.2012.05.002
Francisco, R., Marques-da-Silva, D., Brasil, S., Pascoal, C., Dos Reis Ferreira, V., Morava, E., et al. (2019). The challenge of CDG diagnosis. Mol. Genet. Metab. 126, 1–5. doi: 10.1016/j.ymgme.2018.11.003
Frederick, J. P., Tafari, A. T., Wu, S.-M., Megosh, L. C., Chiou, S.-T., Irving, R. P., et al. (2008). A role for a lithium-inhibited Golgi nucleotidase in skeletal development and sulfation. Proc. Natl. Acad. Sci. U.S.A. 105, 11605–11612. doi: 10.1073/pnas.0801182105
Freeze, H. H. (2007). Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr. Mol. Med. 7, 389–396. doi: 10.2174/156652407780831548
Freeze, H. H., Chong, J. X., Bamshad, M. J., and Ng, B. G. (2014). Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am. J. Hum. Genet. 94, 161–175. doi: 10.1016/j.ajhg.2013.10.024
Furuichi, T., Dai, J., Cho, T.-J., Sakazume, S., Ikema, M., Matsui, Y., et al. (2011). CANT1 mutation is also responsible for desbuquois dysplasia, type 2 and kim variant. J. Med. Genet. 48, 32–37. doi: 10.1136/jmg.2010.080226
Furuichi, T., Kayserili, H., Hiraoka, S., Nishimura, G., Ohashi, H., Alanay, Y., et al. (2009). Identification of loss-of-function mutations of SLC35D1 in patients with schneckenbecken dysplasia, but not with other severe spondylodysplastic dysplasias group diseases. J. Med. Genet. 46, 562–568. doi: 10.1136/jmg.2008.065201
Götting, C., Kuhn, J., and Kleesiek, K. (2007). Human xylosyltransferases in health and disease. Cell. Mol. Life Sci. 64, 1498–1517. doi: 10.1007/s00018-007-7069-z
Gouignard, N., Maccarana, M., Strate, I., von Stedingk, K., Malmström, A., and Pera, E. M. (2016). Musculocontractural ehlers-danlos syndrome and neurocristopathies: dermatan sulfate is required for xenopus neural crest cells to migrate and adhere to fibronectin. Dis. Model. Mech. 9, 607–620. doi: 10.1242/dmm.024661
Gulberti, S., Lattard, V., Fondeur, M., Jacquinet, J.-C., Mulliert, G., Netter, P., et al. (2005). Phosphorylation and sulfation of oligosaccharide substrates critically influence the activity of human beta1,4-galactosyltransferase 7 (GalT-I) and beta1,3-glucuronosyltransferase I (GlcAT-I) involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 280, 1417–1425. doi: 10.1074/jbc.M411552200
Guleray, N., Simsek Kiper, P. O., Utine, G. E., Boduroglu, K., and Alikasifoglu, M. (2019). Intrafamilial variability of XYLT2-related spondyloocular syndrome. Eur. J. Med. Genet. 62:103585. doi: 10.1016/j.ejmg.2018.11.019
Guo, L., Elcioglu, N. H., Iida, A., Demirkol, Y. K., Aras, S., Matsumoto, N., et al. (2017a). Novel and recurrent XYLT1 mutations in two turkish families with desbuquois dysplasia, type 2. J. Hum. Genet. 62, 447–451. doi: 10.1038/jhg.2016.143
Guo, L., Elcioglu, N. H., Mizumoto, S., Wang, Z., Noyan, B., Albayrak, H. M., et al. (2017b). Identification of biallelic EXTL3 mutations in a novel type of spondylo-epi-metaphyseal dysplasia. J. Hum. Genet. 62, 797–801. doi: 10.1038/jhg.2017.38
Guo, M. H., Stoler, J., Lui, J., Nilsson, O., Bianchi, D. W., Hirschhorn, J. N., et al. (2013). Redefining the progeroid form of ehlers-danlos syndrome: report of the fourth patient with B4GALT7 deficiency and review of the literature. Am. J. Med. Genet. A 161A, 2519–2527. doi: 10.1002/ajmg.a.36128
Gustafsson, R., Stachtea, X., Maccarana, M., Grottling, E., Eklund, E., Malmström, A., et al. (2014). Dermatan sulfate epimerase 1 deficient mice as a model for human abdominal wall defects. Birt. Defects Res. A Clin. Mol. Teratol. 100, 712–720. doi: 10.1002/bdra.23300
Haouari, W., Dubail, J., Lounis-Ouaras, S., Prada, P., Bennani, R., Roseau, C., et al. (2020). Serum bikunin isoforms in congenital disorders of glycosylation and linkeropathies. J. Inherit. Metab. Dis. 43, 1349–1359. doi: 10.1002/jimd.12291
Hästbacka, J., de la Chapelle, A., Mahtani, M. M., Clines, G., Reeve-Daly, M. P., Daly, M., et al. (1994). The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell 78, 1073–1087. doi: 10.1016/0092-8674(94)90281-X
Hermanns, P., Unger, S., Rossi, A., Perez-Aytes, A., Cortina, H., Bonafé, L., et al. (2008). Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive larsen syndrome and humero-spinal dysostosis. Am. J. Hum. Genet. 82, 1368–1374. doi: 10.1016/j.ajhg.2008.05.006
Hiraoka, N., Nakagawa, H., Ong, E., Akama, T. O., Fukuda, M. N., and Fukuda, M. (2000). Molecular cloning and expression of two distinct human chondroitin 4-O-sulfotransferases that belong to the HNK-1 sulfotransferase gene family. J. Biol. Chem. 275, 20188–20196. doi: 10.1074/jbc.M002443200
Hiraoka, S., Furuichi, T., Nishimura, G., Shibata, S., Yanagishita, M., Rimoin, D. L., et al. (2007). Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat. Med. 13, 1363–1367. doi: 10.1038/nm1655
Hirose, T., Mizumoto, S., Hashimoto, A., Takahashi, Y., Yoshizawa, T., Nitahara-Kasahara, Y., et al. (2020). Systematic investigation of the skin in Chst14-/- mice: a model for skin fragility in musculocontractural ehlers-danlos syndrome caused by CHST14 variants (mcEDS-CHST14). Glycobiology 31, 137–150. doi: 10.1093/glycob/cwaa058
Hirose, T., Takahashi, N., Tangkawattana, P., Minaguchi, J., Mizumoto, S., Yamada, S., et al. (2019). Structural alteration of glycosaminoglycan side chains and spatial disorganization of collagen networks in the skin of patients with mcEDS-CHST14. Biochim. Biophys. Acta Gen. Subj. 1863, 623–631. doi: 10.1016/j.bbagen.2018.12.006
Holmborn, K., Habicher, J., Kasza, Z., Eriksson, A. S., Filipek-Gorniok, B., Gopal, S., et al. (2012). On the roles and regulation of chondroitin sulfate and heparan sulfate in zebrafish pharyngeal cartilage morphogenesis. J. Biol. Chem. 287, 33905–33916. doi: 10.1074/jbc.M112.401646
Huber, C., Oulès, B., Bertoli, M., Chami, M., Fradin, M., Alanay, Y., et al. (2009). Identification of CANT1 mutations in desbuquois dysplasia. Am. J. Hum. Genet. 85, 706–710. doi: 10.1016/j.ajhg.2009.10.001
Iida, A., Simsek-Kiper, P. Ö., Mizumoto, S., Hoshino, T., Elcioglu, N., Horemuzova, E., et al. (2013). Clinical and radiographic features of the autosomal recessive form of brachyolmia caused by PAPSS2 mutations. Hum. Mutat. 34, 1381–1386. doi: 10.1002/humu.22377
Inoue, S., Ishii, A., Shirotani, G., Tsutsumi, M., Ohta, E., Nakamura, M., et al. (2014). Case of Desbuquois dysplasia type 1: potentially lethal skeletal dysplasia. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 56, e26–29. doi: 10.1111/ped.12383
Iozzo, R. V., and Schaefer, L. (2015). Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. J. Int. Soc. Matrix Biol. 42, 11–55. doi: 10.1016/j.matbio.2015.02.003
Izumikawa, T., Kanagawa, N., Watamoto, Y., Okada, M., Saeki, M., Sakano, M., et al. (2010). Impairment of embryonic cell division and glycosaminoglycan biosynthesis in glucuronyltransferase-I-deficient mice. J. Biol. Chem. 285, 12190–12196. doi: 10.1074/jbc.M110.100941
Izumikawa, T., Koike, T., Shiozawa, S., Sugahara, K., Tamura, J., and Kitagawa, H. (2008). Identification of chondroitin sulfate glucuronyltransferase as chondroitin synthase-3 involved in chondroitin polymerization: chondroitin polymerization is achieved by multiple enzyme complexes consisting of chondroitin synthase family members. J. Biol. Chem. 283, 11396–11406. doi: 10.1074/jbc.M707549200
Jamsheer, A., Olech, E. M., Kozłowski, K., Niedziela, M., Sowińska-Seidler, A., Obara-Moszyńska, M., et al. (2016). Exome sequencing reveals two novel compound heterozygous XYLT1 mutations in a polish patient with desbuquois dysplasia type 2 and growth hormone deficiency. J. Hum. Genet. 61, 577–583. doi: 10.1038/jhg.2016.30
Job, F., Mizumoto, S., Smith, L., Couser, N., Brazil, A., Saal, H., et al. (2016). Functional validation of novel compound heterozygous variants in B3GAT3 resulting in severe osteopenia and fractures: expanding the disease phenotype. BMC Med. Genet. 17:86. doi: 10.1186/s12881-016-0344-9
Jones, K. L., Schwarze, U., Adam, M. P., Byers, P. H., and Mefford, H. C. (2015). A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am. J. Med. Genet. A 167A, 2691–2696. doi: 10.1002/ajmg.a.37209
Kamiyama, S., Sasaki, N., Goda, E., Ui-Tei, K., Saigo, K., Narimatsu, H., et al. (2006). Molecular cloning and characterization of a novel 3'-phosphoadenosine 5'-phosphosulfate transporter, PAPST2. J. Biol. Chem. 281, 10945–10953. doi: 10.1074/jbc.M508991200
Kamiyama, S., Suda, T., Ueda, R., Suzuki, M., Okubo, R., Kikuchi, N., et al. (2003). Molecular cloning and identification of 3'-phosphoadenosine 5'-phosphosulfate transporter. J. Biol. Chem. 278, 25958–25963. doi: 10.1074/jbc.M302439200
Karakus, E., Wannowius, M., Müller, S. F., Leiting, S., Leidolf, R., Noppes, S., et al. (2020). The orphan solute carrier SLC10A7 is a novel negative regulator of intracellular calcium signaling. Sci. Rep. 10:7248. doi: 10.1038/s41598-020-64006-3
Kausar, M., Chew, E. G. Y., Ullah, H., Anees, M., Khor, C. C., Foo, J. N., et al. (2019). A novel homozygous frameshift variant in XYLT2 causes spondyloocular syndrome in a consanguineous pakistani family. Front. Genet. 10:144. doi: 10.3389/fgene.2019.00144
Kim, B. T., Kitagawa, H., Tamura, J., Saito, T., Kusche-Gullberg, M., Lindahl, U., et al. (2001). Human tumor suppressor EXT gene family members EXTL1 and EXTL3 encode alpha 1,4- N-acetylglucosaminyltransferases that likely are involved in heparan sulfate/heparin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 98, 7176–7181. doi: 10.1073/pnas.131188498
Kitagawa, H., Uyama, T., and Sugahara, K. (2001). Molecular cloning and expression of a human chondroitin synthase. J. Biol. Chem. 276, 38721–38726. doi: 10.1074/jbc.M106871200
Klüppel, M., Wight, T. N., Chan, C., Hinek, A., and Wrana, J. L. (2005). Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Dev. Camb. Engl. 132, 3989–4003. doi: 10.1242/dev.01948
Kodama, K., Takahashi, H., Oiji, N., Nakano, K., Okamura, T., Niimi, K., et al. (2020). CANT1 deficiency in a mouse model of desbuquois dysplasia impairs glycosaminoglycan synthesis and chondrocyte differentiation in growth plate cartilage. FEBS Open Bio. 10, 1096–1103. doi: 10.1002/2211-5463.12859
Koike, T., Izumikawa, T., Tamura, J.-I., and Kitagawa, H. (2009). FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem. J. 421, 157–162. doi: 10.1042/BJ20090474
Koziel, L., Kunath, M., Kelly, O. G., and Vortkamp, A. (2004). Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev. Cell 6, 801–813. doi: 10.1016/j.devcel.2004.05.009
Kuroda, Y., Murakami, H., Enomoto, Y., Tsurusaki, Y., Takahashi, K., Mitsuzuka, K., et al. (2019). A novel gene (FAM20B encoding glycosaminoglycan xylosylkinase) for neonatal short limb dysplasia resembling desbuquois dysplasia. Clin. Genet. 95, 713–717. doi: 10.1111/cge.13530
LaCroix, A. J., Stabley, D., Sahraoui, R., Adam, M. P., Mehaffey, M., Kernan, K., et al. (2019). GGC repeat expansion and exon 1 methylation of XYLT1 Is a common pathogenic variant in baratela-scott syndrome. Am. J. Hum. Genet. 104, 35–44. doi: 10.1016/j.ajhg.2018.11.005
Laugel-Haushalter, V., Bär, S., Schaefer, E., Stoetzel, C., Geoffroy, V., Alembik, Y., et al. (2019). A new SLC10A7 homozygous missense mutation responsible for a milder phenotype of skeletal dysplasia with amelogenesis imperfecta. Front. Genet. 10:504. doi: 10.3389/fgene.2019.00504
Lautrup, C. K., Teik, K. W., Unzaki, A., Mizumoto, S., Syx, D., Sin, H. H., et al. (2020). Delineation of musculocontractural ehlers-danlos syndrome caused by dermatan sulfate epimerase deficiency. Mol. Genet. Genomic Med. 8:e1197. doi: 10.1002/mgg3.1197
Li, Y., Laue, K., Temtamy, S., Aglan, M., Kotan, L. D., Yigit, G., et al. (2010). Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of BMP signaling. Am. J. Hum. Genet. 87, 757–767. doi: 10.1016/j.ajhg.2010.10.003
Lin, X., Wei, G., Shi, Z., Dryer, L., Esko, J. D., Wells, D. E., et al. (2000). Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev. Biol. 224, 299–311. doi: 10.1006/dbio.2000.9798
Lindahl, U., Couchman, J., Kimata, K., and Esko, J. D. (2017). “Proteoglycans and sulfated glycosaminoglycans,” in Essentials of Glycobiology (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), 207–221.
Liu, F., Xia, W., Hu, J., Wang, Y., Yang, F., Sun, S., et al. (2015). Solute carrier family 26 member a2 (slc26a2) regulates otic development and hair cell survival in zebrafish. PLoS ONE 10:e0136832. doi: 10.1145/2818302
Liu, X., Li, N., Zhang, H., Liu, J., Zhou, N., Ran, C., et al. (2018). Inactivation of Fam20b in the neural crest-derived mesenchyme of mouse causes multiple craniofacial defects. Eur. J. Oral Sci. 126, 433–436. doi: 10.1111/eos.12563
Ma, P., Yan, W., Tian, Y., Wang, J., Feng, J. Q., Qin, C., et al. (2016). Inactivation of Fam20B in joint cartilage leads to chondrosarcoma and postnatal ossification defects. Sci. Rep. 6:29814. doi: 10.1038/srep29814
Maccarana, M., Kalamajski, S., Kongsgaard, M., Magnusson, S. P., Oldberg, A., and Malmström, A. (2009). Dermatan sulfate epimerase 1-deficient mice have reduced content and changed distribution of iduronic acids in dermatan sulfate and an altered collagen structure in skin. Mol. Cell. Biol. 29, 5517–5528. doi: 10.1128/MCB.00430-09
Malfait, F., Kariminejad, A., Van Damme, T., Gauche, C., Syx, D., Merhi-Soussi, F., et al. (2013). Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic ehlers-danlos-syndrome-like connective tissue disorder. Am. J. Hum. Genet. 92, 935–945. doi: 10.1016/j.ajhg.2013.04.016
Malfait, F., Syx, D., Vlummens, P., Symoens, S., Nampoothiri, S., Hermanns,-L.ê, T., et al. (2010). Musculocontractural ehlers-danlos syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 31, 1233–1239. doi: 10.1002/humu.21355
Malmström, A., and Aberg, L. (1982). Biosynthesis of dermatan sulphate. Assay and properties of the uronosyl C-5 epimerase. Biochem. J. 201, 489–493. doi: 10.1042/bj2010489
Marini, C., Hardies, K., Pisano, T., May, P., Weckhuysen, S., Cellini, E., et al. (2017). Recessive mutations in SLC35A3 cause early onset epileptic encephalopathy with skeletal defects. Am. J. Med. Genet. A 173, 1119–1123. doi: 10.1002/ajmg.a.38112
Martínez-Moreno, D., Jiménez, G., Gálvez-Martín, P., Rus, G., and Marchal, J. A. (2019). Cartilage biomechanics: a key factor for osteoarthritis regenerative medicine. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1067–1075. doi: 10.1016/j.bbadis.2019.03.011
Marzin, P., and Cormier-Daire, V. (2020). New perspectives on the treatment of skeletal dysplasia. Ther. Adv. Endocrinol. Metab. 11:2042018820904016. doi: 10.1177/2042018820904016
Maszczak-Seneczko, D., Sosicka, P., Olczak, T., Jakimowicz, P., Majkowski, M., and Olczak, M. (2013). UDP-N-acetylglucosamine transporter (SLC35A3) regulates biosynthesis of highly branched N-glycans and keratan sulfate. J. Biol. Chem. 288, 21850–21860. doi: 10.1074/jbc.M113.460543
Matsumoto, Y., Matsumoto, K., Irie, F., Fukushi, J., Stallcup, W. B., and Yamaguchi, Y. (2010). Conditional ablation of the heparan sulfate-synthesizing enzyme Ext1 leads to dysregulation of bone morphogenic protein signaling and severe skeletal defects. J. Biol. Chem. 285, 19227–19234. doi: 10.1074/jbc.M110.105338
McCormick, C., Duncan, G., Goutsos, K. T., and Tufaro, F. (2000). The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. U.S.A. 97, 668–673. doi: 10.1073/pnas.97.2.668
Menzies, L., Cullup, T., Calder, A., Wilson, L., and Faravelli, F. (2019). A novel homozygous variant in CANT1 in a patient with kim-type desbuquois dysplasia. Clin. Dysmorphol. 28, 219–223. doi: 10.1097/MCD.0000000000000291
Mihalic Mosher, T., Zygmunt, D. A., Koboldt, D. C., Kelly, B. J., Johnson, L. R., McKenna, D. S., et al. (2019). Expansion of B4GALT7 linkeropathy phenotype to include perinatal lethal skeletal dysplasia. Eur. J. Hum. Genet. 27, 1569–1577. doi: 10.1038/s41431-019-0464-8
Mis, E. K., Liem, K. F. J., Kong, Y., Schwartz, N. B., Domowicz, M., and Weatherbee, S. D. (2014). Forward genetics defines Xylt1 as a key, conserved regulator of early chondrocyte maturation and skeletal length. Dev. Biol. 385, 67–82. doi: 10.1016/j.ydbio.2013.10.014
Miyake, N., Elcioglu, N. H., Iida, A., Isguven, P., Dai, J., Murakami, N., et al. (2012). PAPSS2 mutations cause autosomal recessive brachyolmia. J. Med. Genet. 49, 533–538. doi: 10.1136/jmedgenet-2012-101039
Miyake, N., Kosho, T., Mizumoto, S., Furuichi, T., Hatamochi, A., Nagashima, Y., et al. (2010). Loss-of-function mutations of CHST14 in a new type of ehlers-danlos syndrome. Hum. Mutat. 31, 966–974. doi: 10.1002/humu.21300
Mizumoto, S., Ikegawa, S., and Sugahara, K. (2013). Human genetic disorders caused by mutations in genes encoding biosynthetic enzymes for sulfated glycosaminoglycans. J. Biol. Chem. 288, 10953–10961. doi: 10.1074/jbc.R112.437038
Mizumoto, S., Janecke, A. R., Sadeghpour, A., Povysil, G., McDonald, M. T., Unger, S., et al. (2020). CSGALNACT1-congenital disorder of glycosylation: a mild skeletal dysplasia with advanced bone age. Hum. Mutat. 41, 655–667. doi: 10.1002/humu.23952
Mizumoto, S., Kosho, T., Hatamochi, A., Honda, T., Yamaguchi, T., Okamoto, N., et al. (2017). Defect in dermatan sulfate in urine of patients with ehlers-danlos syndrome caused by a CHST14/D4ST1 deficiency. Clin. Biochem. 50, 670–677. doi: 10.1016/j.clinbiochem.2017.02.018
Monti, L., Paganini, C., Lecci, S., De Leonardis, F., Hay, E., Cohen-Solal, M., et al. (2015). N-acetylcysteine treatment ameliorates the skeletal phenotype of a mouse model of diastrophic dysplasia. Hum. Mol. Genet. 24, 5570–5580. doi: 10.1093/hmg/ddv289
Mortier, G. R., Cohn, D. H., Cormier-Daire, V., Hall, C., Krakow, D., Mundlos, S., et al. (2019). Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 179, 2393–2419. doi: 10.1002/ajmg.a.61366
Müller, T., Mizumoto, S., Suresh, I., Komatsu, Y., Vodopiutz, J., Dundar, M., et al. (2013). Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural ehlers-danlos syndrome. Hum. Mol. Genet. 22, 3761–3772. doi: 10.1093/hmg/ddt227
Mundy, C., Yasuda, T., Kinumatsu, T., Yamaguchi, Y., Iwamoto, M., Enomoto-Iwamoto, M., et al. (2011). Synovial joint formation requires local Ext1 expression and heparan sulfate production in developing mouse embryo limbs and spine. Dev. Biol. 351, 70–81. doi: 10.1016/j.ydbio.2010.12.022
Munns, C. F., Fahiminiya, S., Poudel, N., Munteanu, M. C., Majewski, J., Sillence, D. O., et al. (2015). Homozygosity for frameshift mutations in XYLT2 result in a spondylo-ocular syndrome with bone fragility, cataracts, and hearing defects. Am. J. Hum. Genet. 96, 971–978. doi: 10.1016/j.ajhg.2015.04.017
Muraoka, M., Kawakita, M., and Ishida, N. (2001). Molecular characterization of human UDP-glucuronic acid/UDP-N-acetylgalactosamine transporter, a novel nucleotide sugar transporter with dual substrate specificity. FEBS Lett. 495, 87–93. doi: 10.1016/S0014-5793(01)02358-4
Muys, J., Blaumeiser, B., Jacquemyn, Y., and Janssens, K. (2017). Prenatal homozygosity mapping detects a novel mutation in CHST3 in a fetus with skeletal dysplasia and joint dislocations. Clin. Case Rep. 5, 440–445. doi: 10.1002/ccr3.800
Nadanaka, S., Kagiyama, S., and Kitagawa, H. (2013a). Roles of EXTL2, a member of the EXT family of tumour suppressors, in liver injury and regeneration processes. Biochem. J. 454, 133–145. doi: 10.1042/BJ20130323
Nadanaka, S., Zhou, S., Kagiyama, S., Shoji, N., Sugahara, K., Sugihara, K., et al. (2013b). EXTL2, a member of the EXT family of tumor suppressors, controls glycosaminoglycan biosynthesis in a xylose kinase-dependent manner. J. Biol. Chem. 288, 9321–9333. doi: 10.1074/jbc.M112.416909
Nakajima, M., Mizumoto, S., Miyake, N., Kogawa, R., Iida, A., Ito, H., et al. (2013). Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 92, 927–934. doi: 10.1016/j.ajhg.2013.04.003
Ng, B. G., and Freeze, H. H. (2018). Perspectives on glycosylation and its congenital disorders. Trends Genet. 34, 466–476. doi: 10.1016/j.tig.2018.03.002
Nizon, M., Alanay, Y., Tuysuz, B., Kiper, P. O. S., Geneviève, D., Sillence, D., et al. (2012a). IMPAD1 mutations in two catel-manzke like patients. Am. J. Med. Genet. A 158A, 2183–2187. doi: 10.1002/ajmg.a.35504
Nizon, M., Huber, C., De Leonardis, F., Merrina, R., Forlino, A., Fradin, M., et al. (2012b). Further delineation of CANT1 phenotypic spectrum and demonstration of its role in proteoglycan synthesis. Hum. Mutat. 33, 1261–1266. doi: 10.1002/humu.22104
Norton, W. H. J., Ledin, J., Grandel, H., and Neumann, C. J. (2005). HSPG synthesis by zebrafish Ext2 and Extl3 is required for Fgf10 signalling during limb development. Dev. Camb. Engl. 132, 4963–4973. doi: 10.1242/dev.02084
Ogawa, H., Hatano, S., Sugiura, N., Nagai, N., Sato, T., Shimizu, K., et al. (2012). Chondroitin sulfate synthase-2 is necessary for chain extension of chondroitin sulfate but not critical for skeletal development. PLoS ONE 7:e43806. doi: 10.1371/journal.pone.0043806
Okajima, T., Fukumoto, S., Furukawa, K., and Urano, T. (1999). Molecular basis for the progeroid variant of ehlers-danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 274, 28841–28844. doi: 10.1074/jbc.274.41.28841
Ondruskova, N., Cechova, A., Hansikova, H., Honzik, T., and Jaeken, J. (2021). Congenital disorders of glycosylation: still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 1865:129751. doi: 10.1016/j.bbagen.2020.129751
Oud, M. M., Tuijnenburg, P., Hempel, M., van Vlies, N., Ren, Z., Ferdinandusse, S., et al. (2017). Mutations in EXTL3 cause neuro-immuno-skeletal dysplasia syndrome. Am. J. Hum. Genet. 100, 281–296. doi: 10.1016/j.ajhg.2017.01.013
Pacheco, B., Malmström, A., and Maccarana, M. (2009). Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem. 284, 9788–9795. doi: 10.1074/jbc.M809339200
Pacifici, M. (2018). The pathogenic roles of heparan sulfate deficiency in hereditary multiple exostoses. Matrix Biol. J. Int. Soc. Matrix Biol. 71–72, 28–39. doi: 10.1016/j.matbio.2017.12.011
Paganini, C., Gramegna Tota, C., Superti-Furga, A., and Rossi, A. (2020). Skeletal dysplasias caused by sulfation defects. Int. J. Mol. Sci. 21:2710. doi: 10.3390/ijms21082710
Paganini, C., Monti, L., Costantini, R., Besio, R., Lecci, S., Biggiogera, M., et al. (2019). Calcium activated nucleotidase 1 (CANT1) is critical for glycosaminoglycan biosynthesis in cartilage and endochondral ossification. Matrix Biol. J. Int. Soc. Matrix Biol. 81, 70–90. doi: 10.1016/j.matbio.2018.11.002
Pedersen, L. C., Tsuchida, K., Kitagawa, H., Sugahara, K., Darden, T. A., and Negishi, M. (2000). Heparan/chondroitin sulfate biosynthesis. Structure and mechanism of human glucuronyltransferase I. J. Biol. Chem. 275, 34580–34585. doi: 10.1074/jbc.M007399200
Pels Rijcken, W. R., Overdijk, B., Van den Eijnden, D. H., and Ferwerda, W. (1995). The effect of increasing nucleotide-sugar concentrations on the incorporation of sugars into glycoconjugates in rat hepatocytes. Biochem. J. 305 (Pt. 3), 865–870. doi: 10.1042/bj3050865
Pferdehirt, R., Jain, M., Blazo, M. A., Lee, B., and Burrage, L. C. (2015). Catel-Manzke syndrome: further delineation of the phenotype associated with pathogenic variants in TGDS. Mol. Genet. Metab. Rep. 4, 89–91. doi: 10.1016/j.ymgmr.2015.08.003
Prydz, K. (2015). Determinants of glycosaminoglycan (GAG) structure. Biomolecules 5, 2003–2022. doi: 10.3390/biom5032003
Pu, A., Mishra, M. K., Dong, Y., Ghorbanigazar, S., Stephenson, E. L., Rawji, K. S., et al. (2020). The glycosyltransferase EXTL2 promotes proteoglycan deposition and injurious neuroinflammation following demyelination. J. Neuroinflammation 17:220. doi: 10.1186/s12974-020-01895-1
Purnomo, E., Emoto, N., Nugrahaningsih, D. A. A., Nakayama, K., Yagi, K., Heiden, S., et al. (2013). Glycosaminoglycan overproduction in the aorta increases aortic calcification in murine chronic kidney disease. J. Am. Heart Assoc. 2:e000405. doi: 10.1161/JAHA.113.000405
Quentin, E., Gladen, A., Rodén, L., and Kresse, H. (1990). A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. U.S.A. 87, 1342–1346. doi: 10.1073/pnas.87.4.1342
Rautengarten, C., Quarrell, O. W., Stals, K., Caswell, R. C., De Franco, E., Baple, E., et al. (2019). A hypomorphic allele of SLC35D1 results in schneckenbecken-like dysplasia. Hum. Mol. Genet. 28, 3543–3551. doi: 10.1093/hmg/ddz200
Ritelli, M., Chiarelli, N., Zoppi, N., Dordoni, C., Quinzani, S., Traversa, M., et al. (2015). Insights in the etiopathology of galactosyltransferase II (GalT-II) deficiency from transcriptome-wide expression profiling of skin fibroblasts of two sisters with compound heterozygosity for two novel B3GALT6 mutations. Mol. Genet. Metab. Rep. 2, 1–15. doi: 10.1016/j.ymgmr.2014.11.005
Ritelli, M., Cinquina, V., Giacopuzzi, E., Venturini, M., Chiarelli, N., and Colombi, M. (2019). Further defining the phenotypic spectrum of b3gat3 mutations and literature review on linkeropathy syndromes. Genes 10:631. doi: 10.3390/genes10090631
Ritelli, M., Dordoni, C., Cinquina, V., Venturini, M., Calzavara-Pinton, P., and Colombi, M. (2017). Expanding the clinical and mutational spectrum of B4GALT7-spondylodysplastic ehlers-danlos syndrome. Orphanet J. Rare Dis. 12:153. doi: 10.1186/s13023-017-0704-3
Rossi, A., Kaitila, I., Wilcox, W. R., Rimoin, D. L., Steinmann, B., Cetta, G., et al. (1998). Proteoglycan sulfation in cartilage and cell cultures from patients with sulfate transporter chondrodysplasias: relationship to clinical severity and indications on the role of intracellular sulfate production. Matrix Biol. J. Int. Soc. Matrix Biol. 17, 361–369. doi: 10.1016/S0945-053X(98)90088-9
Saiyin, W., Li, L., Zhang, H., Lu, Y., and Qin, C. (2019). Inactivation of FAM20B causes cell fate changes in annulus fibrosus of mouse intervertebral disc and disc defects via the alterations of TGF-β and MAPK signaling pathways. Biochim. Biophys. Acta Mol. Basis Dis. 1865:165555. doi: 10.1016/j.bbadis.2019.165555
Salter, C. G., Davies, J. H., Moon, R. J., Fairhurst, J., Bunyan, D., and Foulds, N. (2016). Further defining the phenotypic spectrum of B4GALT7 mutations. Am. J. Med. Genet. A 170, 1556–1563. doi: 10.1002/ajmg.a.37604
Sandal, S., Kaur, A., and Panigrahi, I. (2018). Novel mutation in the CHST14 gene causing musculocontractural type of ehlers-danlos syndrome. BMJ Case Rep. 2018:bcr2018226165. doi: 10.1136/bcr-2018-226165
Sato, T., Gotoh, M., Kiyohara, K., Akashima, T., Iwasaki, H., Kameyama, A., et al. (2003). Differential roles of two N-acetylgalactosaminyltransferases, CSGalNAcT-1, and a novel enzyme, CSGalNAcT-2. Initiation and elongation in synthesis of chondroitin sulfate. J. Biol. Chem. 278, 3063–3071. doi: 10.1074/jbc.M208886200
Sato, T., Kudo, T., Ikehara, Y., Ogawa, H., Hirano, T., Kiyohara, K., et al. (2011). Chondroitin sulfate N-acetylgalactosaminyltransferase 1 is necessary for normal endochondral ossification and aggrecan metabolism. J. Biol. Chem. 286, 5803–5812. doi: 10.1074/jbc.M110.159244
Schaefer, L., and Schaefer, R. M. (2010). Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 339, 237–246. doi: 10.1007/s00441-009-0821-y
Schneeberger, P. E., von Elsner, L., Barker, E. L., Meinecke, P., Marquardt, I., Alawi, M., et al. (2020). Bi-allelic pathogenic variants in HS2ST1 cause a syndrome characterized by developmental delay and corpus callosum, skeletal, and renal abnormalities. Am. J. Hum. Genet. 107, 1044–1061. doi: 10.1016/j.ajhg.2020.10.007
Schoner, K., Bald, R., Horn, D., Rehder, H., Kornak, U., and Ehmke, N. (2017). Mutations in TGDS associated with additional malformations of the middle fingers and halluces: atypical catel-manzke syndrome in a fetus. Am. J. Med. Genet. A 173, 1694–1697. doi: 10.1002/ajmg.a.38209
Schreml, J., Durmaz, B., Cogulu, O., Keupp, K., Beleggia, F., Pohl, E., et al. (2014). The missing “link”: an autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Hum. Genet. 133, 29–39. doi: 10.1007/s00439-013-1351-y
Schulte Althoff, S., Grüneberg, M., Reunert, J., Park, J. H., Rust, S., Mühlhausen, C., et al. (2016). TMEM165 deficiency: postnatal changes in glycosylation. JIMD Rep. 26, 21–29. doi: 10.1007/8904_2015_455
Seidler, D. G., Faiyaz-Ul-Haque, M., Hansen, U., Yip, G. W., Zaidi, S. H. E., Teebi, A. S., et al. (2006). Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an ehlers-danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (beta4GalT-7). J. Mol. Med. Berl. Ger. 84, 583–594. doi: 10.1007/s00109-006-0046-4
Shabbir, R. M. K., Nalbant, G., Ahmad, N., Malik, S., and Tolun, A. (2018). Homozygous CHST11 mutation in chondrodysplasia, brachydactyly, overriding digits, clino-symphalangism and synpolydactyly. J. Med. Genet. 55, 489–496. doi: 10.1136/jmedgenet-2017-105003
Sher, G., and Naeem, M. (2014). A novel CHSY1 gene mutation underlies temtamy preaxial brachydactyly syndrome in a pakistani family. Eur. J. Med. Genet. 57, 21–24. doi: 10.1016/j.ejmg.2013.11.001
Shieh, Y.-E., Wells, D. E., and Sater, A. K. (2014). Zygotic expression of Exostosin1 (Ext1) is required for BMP signaling and establishment of dorsal-ventral pattern in xenopus. Int. J. Dev. Biol. 58, 27–34. doi: 10.1387/ijdb.130257as
Shimbo, M., Suzuki, R., Fuseya, S., Sato, T., Kiyohara, K., Hagiwara, K., et al. (2017). Postnatal lethality and chondrodysplasia in mice lacking both chondroitin sulfate N-acetylgalactosaminyltransferase-1 and−2. PLoS ONE 12:e0190333. doi: 10.1371/journal.pone.0190333
Shimizu, K., Okamoto, N., Miyake, N., Taira, K., Sato, Y., Matsuda, K., et al. (2011). Delineation of dermatan 4-O-sulfotransferase 1 deficient ehlers-danlos syndrome: observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. A 155A, 1949–1958. doi: 10.1002/ajmg.a.34115
Silveira, C., Leal, G. F., and Cavalcanti, D. P. (2016). Desbuquois dysplasia type II in a patient with a homozygous mutation in XYLT1 and new unusual findings. Am. J. Med. Genet. A 170, 3043–3047. doi: 10.1002/ajmg.a.37858
Singh, A., Kim, O.-H., Iida, A., Park, W.-Y., Ikegawa, S., and Kapoor, S. (2015). A novel CANT1 mutation in three Indian patients with desbuquois dysplasia kim type. Eur. J. Med. Genet. 58, 105–110. doi: 10.1016/j.ejmg.2014.11.006
Sivasami, P., Poudel, N., Munteanu, M. C., Hudson, J., Lovern, P., Liu, L., et al. (2019). Adipose tissue loss and lipodystrophy in xylosyltransferase II deficient mice. Int. J. Obes. 43, 1783–1794. doi: 10.1038/s41366-019-0324-1
Smith, T. M., Hicks-Berger, C. A., Kim, S., and Kirley, T. L. (2002). Cloning, expression, and characterization of a soluble calcium-activated nucleotidase, a human enzyme belonging to a new family of extracellular nucleotidases. Arch. Biochem. Biophys. 406, 105–115. doi: 10.1016/S0003-9861(02)00420-4
Snyder, N. A., Palmer, M. V., Reinhardt, T. A., and Cunningham, K. W. (2019). Milk biosynthesis requires the golgi cation exchanger TMEM165. J. Biol. Chem. 294, 3181–3191. doi: 10.1074/jbc.RA118.006270
Sohaskey, M. L., Yu, J., Diaz, M. A., Plaas, A. H., and Harland, R. M. (2008). JAWS coordinates chondrogenesis and synovial joint positioning. Dev. Camb. Engl. 135, 2215–2220. doi: 10.1242/dev.019950
Srivastava, P., Pandey, H., Agarwal, D., Mandal, K., and Phadke, S. R. (2017). Spondyloepiphyseal dysplasia omani type: CHST3 mutation spectrum and phenotypes in three Indian families. Am. J. Med. Genet. A 173, 163–168. doi: 10.1002/ajmg.a.37996
Stachtea, X. N., Tykesson, E., van Kuppevelt, T. H., Feinstein, R., Malmström, A., Reijmers, R. M., et al. (2015). Dermatan sulfate-free mice display embryological defects and are neonatal lethal despite normal lymphoid and non-lymphoid organogenesis. PLoS ONE 10:e0140279. doi: 10.1371/journal.pone.0140279
Stickens, D., Zak, B. M., Rougier, N., Esko, J. D., and Werb, Z. (2005). Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Dev. Camb. Engl. 132, 5055–5068. doi: 10.1242/dev.02088
Superti-Furga, A., and Unger, S. (1993). “Atelosteogenesis type 2,” in GeneReviews®, eds. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, et al. (Seattle WA: University of Washington, Seattle).
Syx, D., Van Damme, T., Symoens, S., Maiburg, M. C., van de Laar, I., Morton, J., et al. (2015). Genetic heterogeneity and clinical variability in musculocontractural ehlers-danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 36, 535–547. doi: 10.1002/humu.22774
Takahashi, I., Noguchi, N., Nata, K., Yamada, S., Kaneiwa, T., Mizumoto, S., et al. (2009). Important role of heparan sulfate in postnatal islet growth and insulin secretion. Biochem. Biophys. Res. Commun. 383, 113–118. doi: 10.1016/j.bbrc.2009.03.140
Tanteles, G. A., Dixit, A., Dhar, S., and Suri, M. (2013). Two Somali half-siblings with CHST3-related chondrodysplasia illustrating the phenotypic spectrum and intrafamilial variability. Am. J. Med. Genet. A 161A, 2588–2593. doi: 10.1002/ajmg.a.36094
Taylan, F., Costantini, A., Coles, N., Pekkinen, M., Héon, E., Siklar, Z., et al. (2016). Spondyloocular syndrome: novel mutations in XYLT2 gene and expansion of the phenotypic spectrum. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 31, 1577–1585. doi: 10.1002/jbmr.2834
Taylan, F., Yavaş Abali, Z., Jäntti, N., Güneş, N., Darendeliler, F., Baş, F., et al. (2017). Two novel mutations in XYLT2 cause spondyloocular syndrome. Am. J. Med. Genet. A 173, 3195–3200. doi: 10.1002/ajmg.a.38470
Thiele, H., Sakano, M., Kitagawa, H., Sugahara, K., Rajab, A., Höhne, W., et al. (2004). Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. U.S.A. 101, 10155–10160. doi: 10.1073/pnas.0400334101
Thomsen, B., Horn, P., Panitz, F., Bendixen, E., Petersen, A. H., Holm, L.-E., et al. (2006). A missense mutation in the bovine SLC35A3 gene, encoding a UDP-N-acetylglucosamine transporter, causes complex vertebral malformation. Genome Res. 16, 97–105. doi: 10.1101/gr.3690506
Tian, J., Ling, L., Shboul, M., Lee, H., O'Connor, B., Merriman, B., et al. (2010). Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling. Am. J. Hum. Genet. 87, 768–778. doi: 10.1016/j.ajhg.2010.11.005
Tian, Y., Ma, P., Liu, C., Yang, X., Crawford, D. M., Yan, W., et al. (2015). Inactivation of Fam20B in the dental epithelium of mice leads to supernumerary incisors. Eur. J. Oral Sci. 123, 396–402. doi: 10.1111/eos.12222
Toledo, S. P., Mourão, P. A., Lamego, C., Alves, C. A., Dietrich, C. P., Assis, L. M., et al. (1978). Recessively inherited, late onset spondylar dysplasia and peripheral corneal opacity with anomalies in urinary mucopolysaccharides: a possible error of chondroitin-6-sulfate synthesis. Am. J. Med. Genet. 2, 385–395. doi: 10.1002/ajmg.1320020408
Trejo, P., Rauch, F., Glorieux, F. H., Ouellet, J., Benaroch, T., and Campeau, P. M. (2017). Spondyloepimetaphysial dysplasia with joint laxity in three siblings with B3GALT6 mutations. Mol. Syndromol. 8, 303–307. doi: 10.1159/000479672
Tsutsumi, K., Shimakawa, H., Kitagawa, H., and Sugahara, K. (1998). Functional expression and genomic structure of human chondroitin 6-sulfotransferase. FEBS Lett. 441, 235–241. doi: 10.1016/S0014-5793(98)01532-4
Tuysuz, B., Mizumoto, S., Sugahara, K., Celebi, A., Mundlos, S., and Turkmen, S. (2009). Omani-type spondyloepiphyseal dysplasia with cardiac involvement caused by a missense mutation in CHST3. Clin. Genet. 75, 375–383. doi: 10.1111/j.1399-0004.2009.01167.x
Tüysüz, B., Yilmaz, S., Gül, E., Kolb, L., Bilguvar, K., Evliyaoglu, O., et al. (2013). Spondyloepimetaphyseal dysplasia pakistani type: expansion of the phenotype. Am. J. Med. Genet. A 161A, 1300–1308. doi: 10.1002/ajmg.a.35906
Uchimura, K., Kadomatsu, K., Nishimura, H., Muramatsu, H., Nakamura, E., Kurosawa, N., et al. (2002). Functional analysis of the chondroitin 6-sulfotransferase gene in relation to lymphocyte subpopulations, brain development, and oversulfated chondroitin sulfates. J. Biol. Chem. 277, 1443–1450. doi: 10.1074/jbc.M104719200
Uehara, M., Kosho, T., Yamamoto, N., Takahashi, H. E., Shimakura, T., Nakayama, J., et al. (2018). Spinal manifestations in 12 patients with musculocontractural ehlers-danlos syndrome caused by CHST14/D4ST1 deficiency (mcEDS-CHST14). Am. J. Med. Genet. A 176, 2331–2341. doi: 10.1002/ajmg.a.40507
Uehara, M., Oba, H., Hatakenaka, T., Ikegami, S., Kuraishi, S., Takizawa, T., et al. (2020). Posterior spinal fusion for severe spinal deformities in musculocontractural ehlers-danlos syndrome: detailed observation of a novel case and review of 2 reported cases. World Neurosurg. 143, 454–461. doi: 10.1016/j.wneu.2020.08.085
Umair, M., Eckstein, G., Rudolph, G., Strom, T., Graf, E., Hendig, D., et al. (2018). Homozygous XYLT2 variants as a cause of spondyloocular syndrome. Clin. Genet. 93, 913–918. doi: 10.1111/cge.13179
Unger, S., Lausch, E., Rossi, A., Mégarbané, A., Sillence, D., Alcausin, M., et al. (2010). Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features. Am. J. Med. Genet. A 152A, 2543–2549. doi: 10.1002/ajmg.a.33641
Uyama, T., Kitagawa, H., Tamura Ji, J., and Sugahara, K. (2002). Molecular cloning and expression of human chondroitin N-acetylgalactosaminyltransferase: the key enzyme for chain initiation and elongation of chondroitin/dermatan sulfate on the protein linkage region tetrasaccharide shared by heparin/heparan sulfate. J. Biol. Chem. 277, 8841–8846. doi: 10.1074/jbc.M111434200
Van Damme, T., Pang, X., Guillemyn, B., Gulberti, S., Syx, D., De Rycke, R., et al. (2018). Biallelic B3GALT6 mutations cause spondylodysplastic ehlers-danlos syndrome. Hum. Mol. Genet. 27, 3475–3487. doi: 10.1093/hmg/ddy234
van Roij, M. H. H., Mizumoto, S., Yamada, S., Morgan, T., Tan-Sindhunata, M. B., Meijers-Heijboer, H., et al. (2008). Spondyloepiphyseal dysplasia, omani type: further definition of the phenotype. Am. J. Med. Genet. A 146A, 2376–2384. doi: 10.1002/ajmg.a.32482
Vissers, L. E. L. M., Lausch, E., Unger, S., Campos-Xavier, A. B., Gilissen, C., Rossi, A., et al. (2011). Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am. J. Hum. Genet. 88, 608–615. doi: 10.1016/j.ajhg.2011.04.002
Vodopiutz, J., Mizumoto, S., Lausch, E., Rossi, A., Unger, S., Janocha, N., et al. (2017). Chondroitin sulfate N-acetylgalactosaminyltransferase-1 (CSGalNAcT-1) deficiency results in a mild skeletal dysplasia and joint laxity. Hum. Mutat. 38, 34–38. doi: 10.1002/humu.23070
Voermans, N. C., Kempers, M., Lammens, M., van Alfen, N., Janssen, M. C., Bönnemann, C., et al. (2012). Myopathy in a 20-year-old female patient with D4ST-1 deficient ehlers-danlos syndrome due to a homozygous CHST14 mutation. Am. J. Med. Genet. A 158A, 850–855. doi: 10.1002/ajmg.a.35232
Vogel, P., Hansen, G. M., Read, R. W., Vance, R. B., Thiel, M., Liu, J., et al. (2012). Amelogenesis imperfecta and other biomineralization defects in Fam20a and Fam20c null mice. Vet. Pathol. 49, 998–1017. doi: 10.1177/0300985812453177
Volpi, S., Yamazaki, Y., Brauer, P. M., van Rooijen, E., Hayashida, A., Slavotinek, A., et al. (2017). EXTL3 mutations cause skeletal dysplasia, immune deficiency, and developmental delay. J. Exp. Med. 214, 623–637. doi: 10.1084/jem.20161525
von Oettingen, J. E., Tan, W.-H., and Dauber, A. (2014). Skeletal dysplasia, global developmental delay, and multiple congenital anomalies in a 5-year-old boy-report of the second family with B3GAT3 mutation and expansion of the phenotype. Am. J. Med. Genet. A 164A, 1580–1586. doi: 10.1002/ajmg.a.36487
Vorster, A. A., Beighton, P., and Ramesar, R. S. (2015). Spondyloepimetaphyseal dysplasia with joint laxity (Beighton type); mutation analysis in eight affected South African families. Clin. Genet. 87, 492–495. doi: 10.1111/cge.12413
Waryah, A. M., Shahzad, M., Shaikh, H., Sheikh, S. A., Channa, N. A., Hufnagel, R. B., et al. (2016). A novel CHST3 allele associated with spondyloepiphyseal dysplasia and hearing loss in Pakistani kindred. Clin. Genet. 90, 90–95. doi: 10.1111/cge.12694
Watanabe, Y., Takeuchi, K., Higa Onaga, S., Sato, M., Tsujita, M., Abe, M., et al. (2010). Chondroitin sulfate N-acetylgalactosaminyltransferase-1 is required for normal cartilage development. Biochem. J. 432, 47–55. doi: 10.1042/BJ20100847
Wen, J., Xiao, J., Rahdar, M., Choudhury, B. P., Cui, J., Taylor, G. S., et al. (2014). Xylose phosphorylation functions as a molecular switch to regulate proteoglycan biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 111, 15723–15728. doi: 10.1073/pnas.1417993111
Wilson, D. G., Phamluong, K., Lin, W. Y., Barck, K., Carano, R. A. D., Diehl, L., et al. (2012). Chondroitin sulfate synthase 1 (Chsy1) is required for bone development and digit patterning. Dev. Biol. 363, 413–425. doi: 10.1016/j.ydbio.2012.01.005
Wiweger, M. I., Avramut, C. M., de Andrea, C. E., Prins, F. A., Koster, A. J., Ravelli, R. B. G., et al. (2011). Cartilage ultrastructure in proteoglycan-deficient zebrafish mutants brings to light new candidate genes for human skeletal disorders. J. Pathol. 223, 531–542. doi: 10.1002/path.2824
Wiweger, M. I., Zhao, Z., van Merkesteyn, R. J. P., Roehl, H. H., and Hogendoorn, P. C. W. (2012). HSPG-deficient zebrafish uncovers dental aspect of multiple osteochondromas. PLoS ONE 7:e29734. doi: 10.1371/journal.pone.0029734
Xia, B., Zhang, W., Li, X., Jiang, R., Harper, T., Liu, R., et al. (2013). Serum N-glycan and O-glycan analysis by mass spectrometry for diagnosis of congenital disorders of glycosylation. Anal. Biochem. 442, 178–185. doi: 10.1016/j.ab.2013.07.037
Xu, Z. H., Otterness, D. M., Freimuth, R. R., Carlini, E. J., Wood, T. C., Mitchell, S., et al. (2000). Human 3'-phosphoadenosine 5'-phosphosulfate synthetase 1 (PAPSS1) and PAPSS2: gene cloning, characterization and chromosomal localization. Biochem. Biophys. Res. Commun. 268, 437–444. doi: 10.1006/bbrc.2000.2123
Yauy, K., Tran Mau-Them, F., Willems, M., Coubes, C., Blanchet, P., Herlin, C., et al. (2018). B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genet. Med. Off. J. Am. Coll. Med. Genet. 20, 269–274. doi: 10.1038/gim.2017.109
Yoshioka, N., Miyata, S., Tamada, A., Watanabe, Y., Kawasaki, A., Kitagawa, H., et al. (2017). Abnormalities in perineuronal nets and behavior in mice lacking CSGalNAcT1, a key enzyme in chondroitin sulfate synthesis. Mol. Brain 10:47. doi: 10.1186/s13041-017-0328-5
Zak, B. M., Schuksz, M., Koyama, E., Mundy, C., Wells, D. E., Yamaguchi, Y., et al. (2011). Compound heterozygous loss of Ext1 and Ext2 is sufficient for formation of multiple exostoses in mouse ribs and long bones. Bone 48, 979–987. doi: 10.1016/j.bone.2011.02.001
Zeevaert, R., de Zegher, F., Sturiale, L., Garozzo, D., Smet, M., Moens, M., et al. (2013). Bone dysplasia as a key feature in three patients with a novel congenital disorder of glycosylation (CDG) type II due to a deep intronic splice mutation in TMEM165. JIMD Rep. 8, 145–152. doi: 10.1007/8904_2012_172
Keywords: chondrodysplasia, dislocations, glycosaminoglycan, congenital disorder of glycosylation, genotype-phenotype correlation
Citation: Dubail J and Cormier-Daire V (2021) Chondrodysplasias With Multiple Dislocations Caused by Defects in Glycosaminoglycan Synthesis. Front. Genet. 12:642097. doi: 10.3389/fgene.2021.642097
Received: 15 December 2020; Accepted: 04 May 2021;
Published: 16 June 2021.
Edited by:
Vicki Rosen, Harvard University, United StatesCopyright © 2021 Dubail and Cormier-Daire. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johanne Dubail, am9oYW5uZS5kdWJhaWxAaW5zZXJtLmZy