 Pan Gong
Pan Gong Xianru Jiao
Xianru Jiao Dan Yu2
Dan Yu2 Zhixian Yang
Zhixian Yang- 1Department of Pediatrics, Peking University First Hospital, Beijing, China
- 2Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, China
Objective: KCNT2 gene mutations had been described to cause developmental and epileptic encephalopathies (DEEs). In this study, we presented the detailed clinical features and genetic analysis of two unrelated patients carrying two de novo variants in KCNT2 and reviewed eight different cases available in publications.
Methods: Likely pathogenic variants were identified by whole exome sequencing; clinical data of the patients were retrospectively collected and analyzed.
Results: Our two unrelated patients were diagnosed with Ohtahara syndrome followed by infantile spasms (IS) and possibly the epilepsy of infancy with migrating focal seizures (EIMFS), respectively. They both manifested dysmorphic features with hirsute arms, thick hair, prominent eyebrows, long and thick eyelashes, a broad nasal tip, and short and smooth philtrum. In the eight patients reported previously, two was diagnosed with IS carrying a ‘change-of-function' mutation and a gain-of-function mutation, respectively, two with EIMFS-like carrying a gain-of-function mutation and a loss-of-function mutation, respectively, one with EIMFS carrying a loss-of-function mutation, three with DEE without functional analysis. Among them, two patients with gain-of-function mutations both exhibited dysmorphic features and presented epilepsy phenotype, which was similar to our patients.
Conclusion: Overall, the most common phenotypes associated with KCNT2 mutation were IS and EIMFS. Epilepsy phenotype associated with gain- and loss-of-function mutations could overlap. Additional KCNT2 cases will help to make genotype-phenotype correlations clearer.
Introduction
Developmental and epileptic encephalopathies (DEEs) comprise a heterogeneous group of sever neurological disorders with onset in infancy and childhood, which is characterized by refractory seizures, frequent epileptic activity, and developmental regression or further slowing (Scheffer et al., 2017). Increasing number of genes identified as the cause of DEEs and channelopathies represent an important and broad class (Kumar et al., 2016). A systematic review of neurological disorders and potassium channelopathies revealed pathogenic variants in 19 potassium channel genes, including KCNMA1, KCNN3, KCNT1, KCNT2, KCNB1, KCNJ6, KCNJ10, KCNJ11, KCNA2, KCNA4, KCND3, KCNH1, KCNQ2, KCNAB1, KCNQ3, KCNQ5, KCNC1, KCNC3, and KCTD3 (Kessi et al., 2020). There is a large phenotypic and genetic heterogeneity and the majority of genetic defects are still unknown. Recently, pathogenic variants in KCNT2 gene that encodes the KNa1.2 subunit (Slick or Slo2.1) have been identified in eight cases (Gururaj et al., 2017; Ambrosino et al., 2018; Alagoz et al., 2020; Inuzuka et al., 2020; Mao et al., 2020). The KCNT2-associated DEEs comprises West syndrome, Lennox-Gastaut syndrome (LGS) as well as epilepsy of infancy with migrating focal seizures (EIMFS). In vitro functional analysis suggested that both gain and loss of function variants in KCNT2 may lead to DEEs (McLachlan et al., 2019). Here, we reported two de novo KCNT2 variants in two unrelated patients diagnosed with DEE charactering by profound developmental delay and intractable infantile-onset seizure disorders.
Case Presentation
In total, two unrelated boys were enrolled in our study. Clinical features of affected individuals with KCNT2 variants were summarized in Table 1.
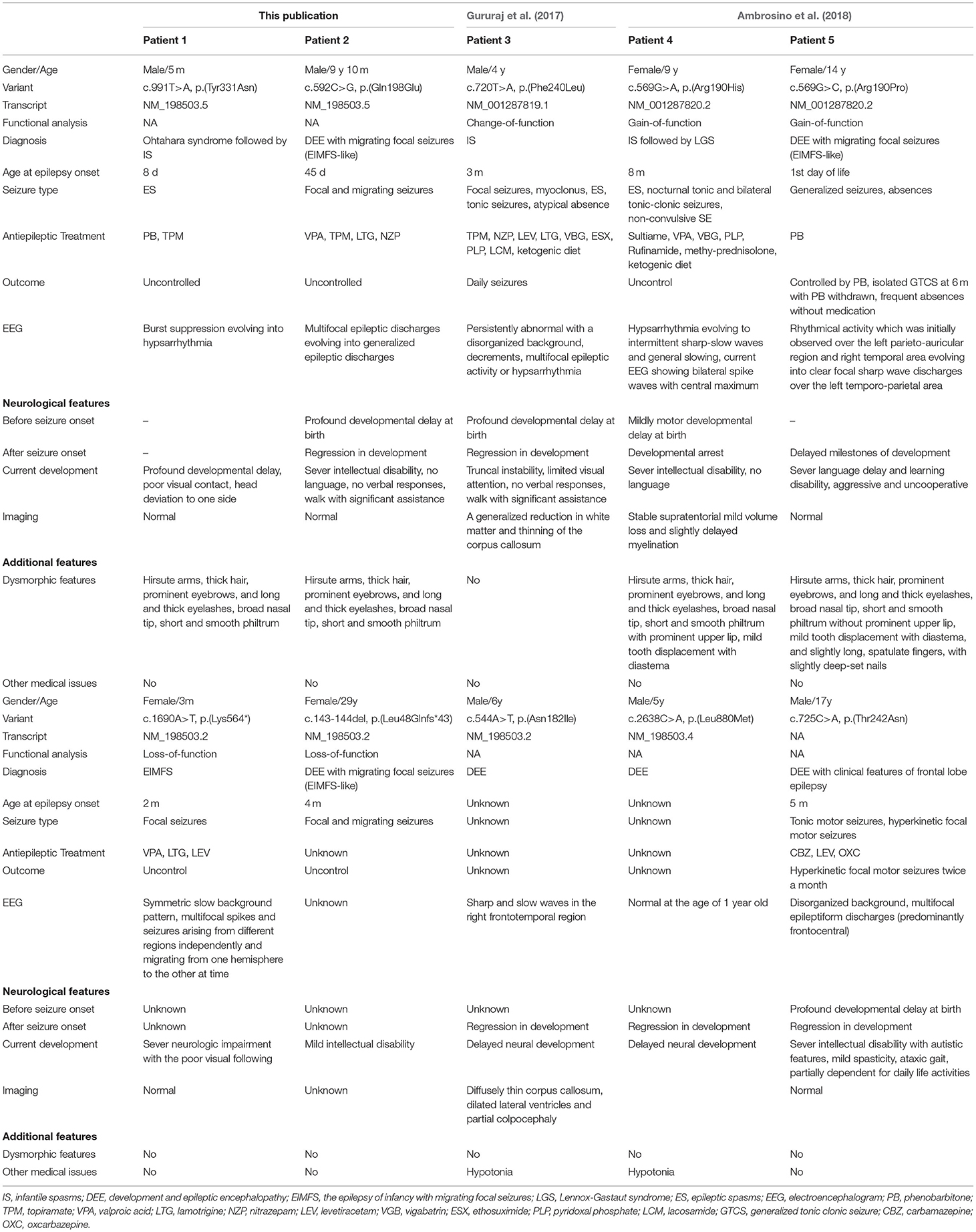
Table 1. Patients with KCNT2 variants in this publication and previous literatures.
Patient #1 was a 5 month-old boy. He was born at 38 weeks of gestation with a birth weight of 2,950 g. He was the first child of healthy non-consanguineous parents. There was no familial history of any neuropsychiatric disease including epilepsy or febrile seizures. He was hypotonic and had a profound delayed development. From 8 days of age, the boy presented with daily clusters of epileptic spasms. The EEG showed burst suppression. At the current age of 5 months, the boy was unable to hold his head with poor visual contact. His body measures were 66 cm and 6.7 kg. The seizures were still uncontrolled on the combination of phenobarbitone and topiramate at the last follow-up. During the course, the semiology evolved from Ohtahara syndrome to West syndrome with severe developmental delay and frequent epileptic spasms. EEG displayed with hypsarrhythmia. Brain magnetic resonance imaging (MRI) at 18 days was normal. The patients manifested dysmorphic features with hirsute arms, thick hair, prominent eyebrows, and long and thick eyelashes and had a broad nasal tip, short and smooth philtrum (Figure 1).
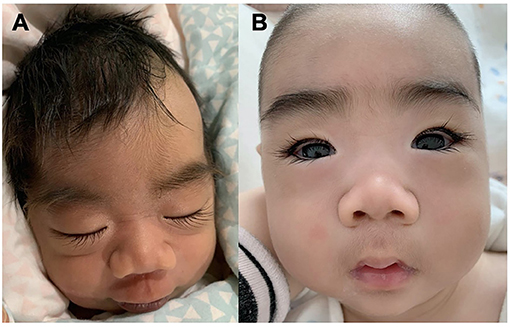
Figure 1. Facial Gestalt of the Patient #1 with de novo KCNT2 variant (A) at birth and (B) at the age of 3 months.
Patient #2 was a 9 year-old boy. He was born at full term of gestation with a birth weight of 3,300 g. He was the first child of healthy non-consanguineous parents. There was no familial history of any neuropsychiatric disease including epilepsy or febrile seizures. He had a profound delayed developmental milestone with raising his head at the age of 1 years and starting to walk at the age of 5 years. According to his parents and medical records, the boy presented with daily seizures of awaking from sleep with the head swinging from side to side and the limbs jerking at 45 days of age. Seizures were mainly focal and migrating. EEG showed multifocal epileptic discharges with left temporal predominance. At the current age of 9 years, the boy had severe intellectual disability, was able to walk with assistance, but had no language and no verbal response. His body measures were 118 cm and 20 kg. He still had frequent seizures under the combination of valproate, topiramate, and clonazepam. The current EEG displayed generalized discharges. Brain MRI at 4 years was normal.
The patients manifested dysmorphic features with hirsute arms, thick hair, prominent eyebrows, and long and thick eyelashes and had a broad nasal tip, short and smooth philtrum (Figure 2).
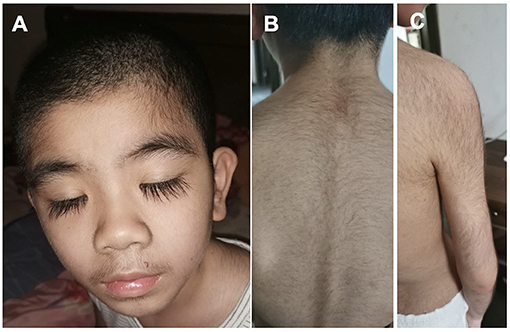
Figure 2. Dysmorphic features of the Patient #2 with de novo KCNT2 variant at the age of 10 years. (A) Facial Gestalt of thick hair, prominent eyebrows, and long and thick eyelashes. (B) Thick hair on the back. (C) Thick hair on the right arm.
Genetic Analysis
Trio-based whole-exome sequencing revealed de novo missense variants of [Chr1:196,395,112A>T, c.991T>A, p.(Tyr331Asn)] and [Chr1:196,465,339G>C, c.592C>G, p.(Gln198Glu)] in KCNT2 gene (NM_198503.5, GRCh37/hg19), respectively, in two patients. Both variants were confirmed by conventional Sanger sequencing. The variants were novel and absent in control populational databases, including 1000 genomes, ExAC, and gnomAD. The pathogenicity of two novel variants was further analyzed using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/). Y331N and Q198E substitutions were shown to be possibly damaging (score 0.619 and 0.598, respectively). MutationTaster program (http://www.mutationtaster.org/) showed that both variants were predicted to cause change in protein function and were disease causing. Y331N was localized in the C-terminal part of KNa1.2 and Q198E was localized in S4-S5 of the protein (Figure 3). Moreover, the phenotype of the patients was consistent with that of KCNT2-associated disease. Therefore, both variants were classified as likely pathogenic based on the American College of Medical Genetics (ACMG) guidelines, both mutations were classified as pathogenic with the PS2, PM2, and PP3 criteria (Richards et al., 2015).
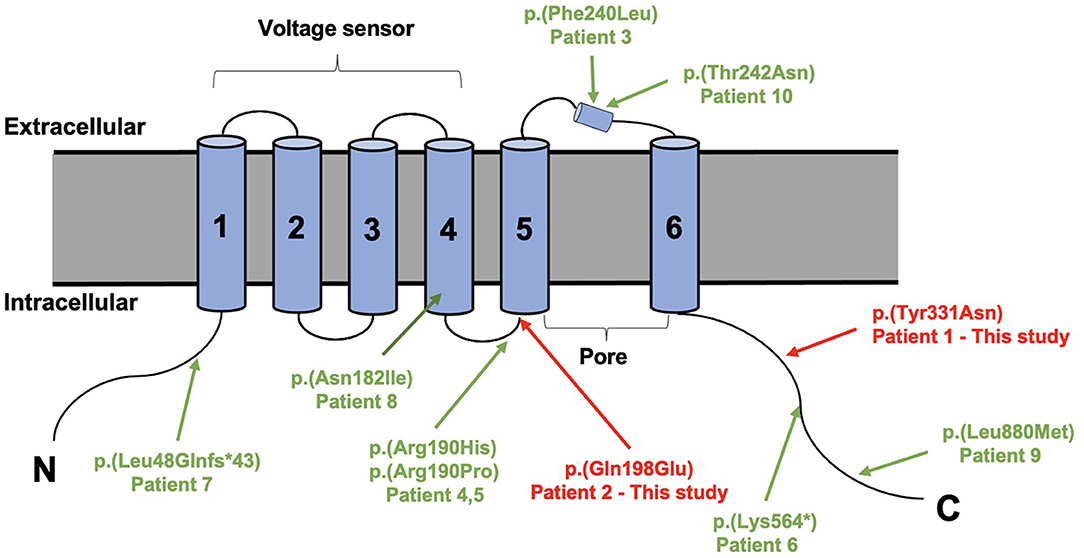
Figure 3. Structure of the sodium-activated potassium (KNa1.2) channel (9606 [NCBI]) with all variants published variants in KCNT2.
Discussion
As a recently discovered gene associated with epilepsy disorders, a total of eight patients bearing five KCNT2 variants had been reported (Gururaj et al., 2017; Ambrosino et al., 2018; Alagoz et al., 2020; Inuzuka et al., 2020; Mao et al., 2020). The KCNT2-associated DEEs comprises West syndrome as well as epilepsy of infancy with migrating focal seizures (EIMFS). In 2017, Gururaj et al. firstly identified a KCNT2 mutation in an early onset epileptic encephalopathy (EOEE) patient with intractable epileptic spasms by exome sequencing and confirmed the mutation with “change-of-function” effect (Gururaj et al., 2017). At the last follow-up age of 4 years, the patient presented with prolonged tonic seizures, myoclonic jerks and atypical absences with a disorganized background, decrements, multifocal epileptogenic activity or hypsarrhythmia on EEG (Gururaj et al., 2017). We assumed that the semiology of this patient was likely to evolved from infantile spasms (IS) to Lennox-Gastaut syndrome (LGS). In 2018, Ambrosino et al. described two female patients with IS followed by LGS or with DEE with migrating focal seizures. In vitro analysis further suggested that the variants found in patients had gain-of-function effects (Ambrosino et al., 2018). Seizure discharges in the patient with DEE with migrating focal seizures had been observed to migrate from the left to the right hemisphere, which likely corresponded to EIMFS (Ambrosino et al., 2018). Most recently, Mao et al. identified two novel de novo non-sense and frameshift mutations of the KCNT2 gene in two patients diagnosed as EIMFS and early onset epileptic encephalopathy (EOEE) with migrating focal seizures, respectively (Mao et al., 2020). Due to the fact that the latter case was an aged case, diagnosis was not be firmly ascertained and the author described the patient as EIMFS-like EOEE (Mao et al., 2020). They investigated the functional consequence of the two mutations, which showed that both mutations reduced whole-cell potassium current (Mao et al., 2020). Subsequently, there were two reports describing three patients with EOEE caused by mutations in KCNT2 without functional analysis (Alagoz et al., 2020; Inuzuka et al., 2020). Here, our patients showed DEE in common with previous studies. Patient #1 was diagnosed as Ohtahara syndrome subsequently followed by IS with EEG showing burst suppression evolving to hypsarrhythmia. Patient #2 was diagnosed as DEE with migrating focal seizures manifesting profound developmental delay at birth and intractable seizure with developmental regression. Due to incomplete medical records, there was no definitive diagnosis but EIMFS-like EOEE of Patient #2. In general, both gain and loss of function mutations in the KCNT2 gene could lead to DEE, and the most common phenotypes were IS and EIMFS. The epilepsy phenotype associated with gain- and loss-of-function mutations could overlap.
EIMFS is a rare EOEE characterized by polymorphous focal seizures and cognitive, sensory and motor impairment, with arrest of psychomotor development in the first 6 months of life (Coppola et al., 1995, 2006). EIMFS have a genetic origin in which the de novo gain-of-function KCNT1 variants are the most common cause (Barcia et al., 2012; McTague et al., 2013; Rizzo et al., 2016; Villa and Combi, 2016). KCNT1 and KCNT2 respectively encode the KNa1.1 (Slack) and KNa1.2 (Slick) subunits. Both genes belongs to the SLO2 family of the sodium-dependent voltage-gated potassium channel KNa (Bhattacharjee et al., 2003). Functional analysis of mutant channels associated with EIMFS mostly revealed gain-of-function effects (Barcia et al., 2012; Rizzo et al., 2016; Villa and Combi, 2016). Similarly, Ambrosino et al. identified a gain-of-function mutation in the KCNT2 gene in a patient with EIMFS-like DEE. It suggested that pathogenic variants in KCNT1 and KCNT2 therefore might contribute to a similar and overlapping spectrum of DEEs (Ambrosino et al., 2018; Kessi et al., 2020). Meantime, Mao et al. also reported EIMFS in patients carrying loss-of-function mutations in the KCNT2 gene (Mao et al., 2020), which indicated that EIMFS might be caused not only by an increase but also by a decrease in the function of KNa.
Moreover, it was noticed that the two patients confirmed to carry gain-of-function mutations both exhibited not only severe developmental delay, but also dysmorphic features with hirsute arms, thick hair, prominent eyebrows, and long and thick eyelashes, broad nasal tip, short and smooth philtrum with prominent upper lip, mild tooth displacement with diastema (Ambrosino et al., 2018). Similarly, both of our patients had prominent dysmorphic features with hirsute arms, thick hair, prominent eyebrows, and long and thick eyelashes. We presumed that the dysmorphic features might be the distinguishing characterizations in patients with gain-of-function mutations in KCNT2. However, we could not confirm the mutations in our patients in the absence of functional analysis. Given the small number of patients reported so far, it is difficult to make genotype-phenotype correlations. Additional KCNT2 cases will help clarify the potential association between gain-of-function and dysmorphic features.
Seizures in all patients were intractable and there was a poor prognosis in development. The oldest patient at the age of 29 years still had seizures and showed mild intellectual disability (Mao et al., 2020). Quinidine as an underlying precision medicine for epilepsy syndromes due to gain of function mutations in KCNT1 aroused particular clinical interest (Bearden et al., 2014). Therefore, Ambrosino et al. confirmed by functional analysis that the patients carried quinidine-responsive gain-of-function mutations in the KCNT2 gene and treated one patient with quinidine add-on therapy and achieved marked clinical improvements, including EEG, vigilance, alertness as well as developmental progression (Ambrosino et al., 2018). It suggested that quinidine could be a potential personalized medicine approach for KCNT2-related DEE.
Conclusion
In this study, we presented the detailed clinical features and genetic analysis of two unrelated patients with KCNT2-related DEE and provided a comprehensive outline of available publications regarding KCNT2 mutations. Our data further expanded the spectrum of KCNT2 mutation. So far, with our case, a total of ten cases carrying KCNT2 mutations have been reported. IS and EIMFS were the most common phenotypes caused by pathogenic mutations in KCNT2. Both gain and loss of function mutations could lead to EIMFS. Additional KCNT2 cases will help to make genotype-phenotype correlations clearer. Quinidine could be a potential personalized medicine approach for KCNT2-related DEE. Clinical follow-up of additional patients will further define the clinical spectrum of KCNT2-related DEE and the long-time efficacy on seizures and development of quinidine as an underlying precision medicine for epilepsy syndromes due to gain-of-function mutations.
Data Availability Statement
The sequencing data are available on https://www.ncbi.nlm.nih.gov/bioproject/PRJNA689060.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Committee of Peking University First Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
ZY conceptualized and designed the study, coordinated the study overall, and revised the manuscript. PG co-designed the study, drafted the initial manuscript, and revised the manuscript. XJ and DY helped to collect and summarize data and revised the manuscript. All authors approved the final revision of the article.
Funding
This work was supported by National Nature Science Foundation of China (81771393), Beijing Municipal Science & Technology Commission (Z171100001017125), Beijing Natural Science Foundation (7202210), Capital's Funds for Health Improvement and Research (2020-2-4077).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We extend our appreciation to the patients and their families who participated in this study. We would like to thank Dr. Xiaodong Wang from Cipher Gene, Ltd. for assisting in manuscript editing.
References
Alagoz, M., Kherad, N., Bozkurt, S., and Yuksel, A. (2020). New mutations in KCNT2 gene causing early infantile epileptic encephalopathy type 57: case study and literature review. Acta Biochim. Pol. 67, 431–434. doi: 10.18388/abp.2020_5364
Ambrosino, P., Soldovieri, M. V., Bast, T., Turnpenny, P. D., Uhrig, S., Biskup, S., et al. (2018). De novo gain-of-function variants in KCNT2 as a novel cause of developmental and epileptic encephalopathy. Ann. Neurol. 83, 1198–1204. doi: 10.1002/ana.25248
Barcia, G., Fleming, M. R., Deligniere, A., Gazula, V. R., Brown, M. R., Langouet, M., et al. (2012). De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 44, 1255–1259. doi: 10.1038/ng.2441
Bearden, D., Strong, A., Ehnot, J., DiGiovine, M., Dlugos, D., and Goldberg, E. M. (2014). Targeted treatment of migrating partial seizures of infancy with quinidine. Ann. Neurol. 76, 457–461. doi: 10.1002/ana.24229
Bhattacharjee, A., Joiner, W. J., Wu, M., Yang, Y., Sigworth, F. J., and Kaczmarek, L. K. (2003). Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23, 11681–11691. doi: 10.1523/JNEUROSCI.23-37-11681.2003
Coppola, G., Plouin, P., Chiron, C., Robain, O., and Dulac, O. (1995). Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia 36, 1017–1024. doi: 10.1111/j.1528-1157.1995.tb00961.x
Coppola, G., Veggiotti, P., Del Giudice, E. M., Bellini, G., Longaretti, F., Taglialatela, M., et al. (2006). Mutational scanning of potassium, sodium and chloride ion channels in malignant migrating partial seizures in infancy. Brain Dev. 28, 76–79. doi: 10.1016/j.braindev.2005.05.002
Gururaj, S., Palmer, E. E., Sheehan, G. D., Kandula, T., Macintosh, R., Ying, K., et al. (2017). A De novo mutation in the sodium-activated potassium channel KCNT2 Alters ion selectivity and causes epileptic encephalopathy. Cell Rep. 21, 926–933. doi: 10.1016/j.celrep.2017.09.088
Inuzuka, L. M., Macedo-Souza, L. I., Della-Ripa, B., Monteiro, F. P., Ramos, L., Kitajima, J. P., et al. (2020). Additional observation of a de novo pathogenic variant in KCNT2 leading to epileptic encephalopathy with clinical features of frontal lobe epilepsy. Brain Dev. 42, 691–695. doi: 10.1016/j.braindev.2020.05.003
Kessi, M., Chen, B., Peng, J., Tang, Y., Olatoutou, E., He, F., et al. (2020). Intellectual disability and potassium channelopathies: a systematic review. Front. Genet. 11:614. doi: 10.3389/fgene.2020.00614
Kumar, P., Kumar, D., Jha, S. K., Jha, N. K., and Ambasta, R. K. (2016). Ion channels in neurological disorders. Adv. Prot. Chem. Struct. Biol. 103, 97–136. doi: 10.1016/bs.apcsb.2015.10.006
Mao, X., Bruneau, N., Gao, Q., Becq, H., Jia, Z., Xi, H., et al. (2020). The epilepsy of infancy with migrating focal seizures: identification of de novo mutations of the KCNT2 gene that exert inhibitory effects on the corresponding heteromeric KNa1.1/KNa1.2 potassium channel. Front. Cell Neurosci. 14:1. doi: 10.3389/fncel.2020.00001
McLachlan, F., Sires, A. M., and Abbott, C. M. (2019). The role of translation elongation factor eEF1 subunits in neurodevelopmental disorders. Hum. Mutat. 40, 131–141. doi: 10.1002/humu.23677
McTague, A., Appleton, R., Avula, S., Cross, J. H., King, M. D., Jacques, T. S., et al. (2013). Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain. 136(Pt. 5), 1578–1591. doi: 10.1093/brain/awt073
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rizzo, F., Ambrosino, P., Guacci, A., Chetta, M., Marchese, G., Rocco, T., et al. (2016). Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol. Cell Neurosci. 72, 54–63. doi: 10.1016/j.mcn.2016.01.004
Scheffer, I. E., Berkovic, S., Capovilla, G., Connolly, M. B., French, J., Guilhoto, L., et al. (2017). ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 58, 512–521. doi: 10.1111/epi.13709
Keywords: epilepsy, KCNT2, genetic, seizures, encephalopathy
Citation: Gong P, Jiao X, Yu D and Yang Z (2021) Case Report: Causative De novo Variants of KCNT2 for Developmental and Epileptic Encephalopathy. Front. Genet. 12:649556. doi: 10.3389/fgene.2021.649556
Received: 27 January 2021; Accepted: 12 April 2021;
Published: 30 June 2021.
Edited by:
Zhichao Liu, National Center for Toxicological Research (FDA), United StatesReviewed by:
Catarina M. Quinzii, Columbia University, United StatesNicole J. Lake, Yale University, United States
Copyright © 2021 Gong, Jiao, Yu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhixian Yang, emhpeGlhbi55YW5nQDE2My5jb20=