 Luis J. Chueca1,2*
Luis J. Chueca1,2* Judith Kochmann1,3
Judith Kochmann1,3 Tilman Schell1
Tilman Schell1 Carola Greve1
Carola Greve1 Axel Janke1,3,4
Axel Janke1,3,4 Markus Pfenninger1,3,5
Markus Pfenninger1,3,5 Sven Klimpel1,3,4
Sven Klimpel1,3,4- 1LOEWE-Centre for Translational Biodiversity Genomics (LOEWE-TBG), Senckenberg Nature Research Society, Frankfurt am Main, Germany
- 2Department of Zoology and Animal Cell Biology, University of the Basque Country (UPV-EHU), Vitoria-Gasteiz, Spain
- 3Senckenberg Biodiversity and Climate Research Centre (SBiK-F), Frankfurt am Main, Germany
- 4Institute for Ecology, Evolution and Diversity, Goethe University, Frankfurt am Main, Germany
- 5Institute of Organismic and Molecular Evolution, Faculty of Biology, Johannes Gutenberg University, Mainz, Germany
Introduction
The raccoon dog, Nyctereutes procyonoides (NCBI Taxonomy ID: 34880, Figure 1a) belongs to the family Canidae, with foxes (genus Vulpes) being their closest relatives (Lindblad-Toh et al., 2005; Sun et al., 2019). Its original distribution in East Asia ranges from south-eastern Siberia to northern Vietnam and the Japanese islands. In the early 20th century, the raccoon dog was introduced into Western Russia for fur breeding and hunting purposes, which led to its widespread establishment in many European countries, Figure 1b. Together with the raccoon (Procyon lotor), it is now listed in Europe as an invasive species of Union concern (Regulation (EU) No. 1143/2014) and member states are required to control pathways of introductions and manage established populations.
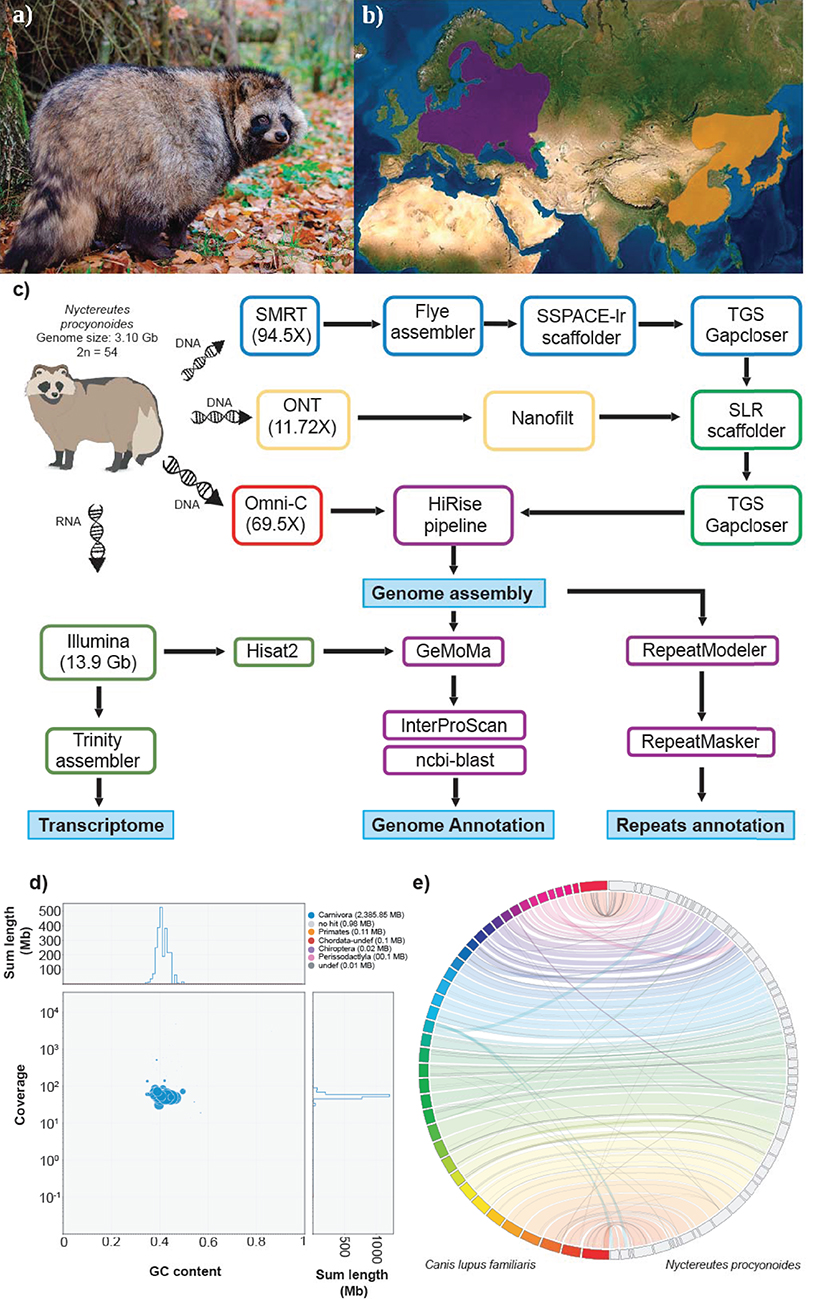
Figure 1. (a) Picture of an adult specimen of raccoon dog (Nyctereutes procyonoides), copyright © Dorian D. Dörge. (b) Native (orange) and introduced (purple) distributions ranges of raccoon dog (source IUCN red list). (c) Workflow of genome assembly and annotation followed in this study. (d) Blob plot showing read depth of coverage, GC content and size of each scaffold. Size of the blobs correspond to size of the scaffold and color corresponds to taxonomic assignment of BLAST (blue = Carnivora). (e) Whole genome synteny, obtained with Jupiterplot, between the Canis lupus familiaris chromosome-level assembly (on the left) and the raccoon dog genome assembly obtained in this study (on the right). The lines indicate aligned regions between the two assemblies.
The raccoon dog is a host and vector for a variety of pathogens, including rabies and canine distemper virus. Whether, it is involved in the transmission of coronaviruses to humans is inconclusive (Guan, 2003; Chan and Chan, 2013), but experimental studies have demonstrated that raccoon dogs are susceptible to SARS-CoV-2 infection and its transmission to contact animals (Freuling et al., 2020). However, a recent study using predictions by sequence alignment suggests that the mammalian ACE2 receptor of N. procyonoides binds less effectively to the S-protein of SARS-CoV and SARS-CoV-2 than those of other species like cows and rodents (Luan et al., 2020a,b).
Several subpopulations have been recognized in their current range of distribution in Europe and East Asia based on mtDNA (Kim et al., 2013; Paulauskas et al., 2016), microsatellite (Drygala et al., 2016; Hong et al., 2018), and SNP markers (Nørgaard et al., 2017). Interestingly, continental populations from Asia and Europe seem to have a higher number of chromosomes (2n = 54) than those from Japanese islands (2n = 38) (Wada and Imai, 1991; Wada et al., 1991; Nie et al., 2003). Moreover, the raccoon dog is also known to be one of the few Carnivora species which presents B chromosomes (Bs) in its karyotype (Duke Becker et al., 2011; Makunin et al., 2018). Several mitochondrial genome sequences of wild and bred raccoon dogs are known (Sun et al., 2019), however, a complete nuclear genome is not still available. Apart from its potential role as disease vector, N. procyonoides is of interest because it is the only extant species in the genus Nyctereutes and the only canid known to hibernate.
Here, a first draft genome of a raccoon dog sampled in Germany is presented, which will provide a basis for deeper understanding of its phylogenetic relationships, the evolution and function of B chromosomes in mammals, give insights in the evolution of hibernation, provide markers for future studies on invasive population structures in Europe and serve as a resource for studying gene-disease associations.
Materials and Methods
Sample Collection, Library Construction, Sequencing
One adult female individual of raccoon dog, Nyctereutes procyonoides (Figure 1a), was bagged in February 2020 in Germany (52°06′51.2″N 12°03′03.6″E) according to hunting regulations. Blood samples as well as various types of tissue were immediately stored on dry ice or in RNAlater and kept at −80°C until further processing (Figure 1c).
A SMRTbell library was constructed following the instructions of the SMRTbell Express Prep kit v2.0 with Low DNA Input Protocol (Pacific Biosciences, Menlo Park, CA). Blood (5 ml) was used for high molecular weight DNA extraction using Genomic-tip 100/G (QIAGEN) according to the manufacturers' instructions. One SMRT cell sequencing run was performed in CLR mode on the Sequel System II with the Sequel II Sequencing Kit 2.0. For chromosome-level genome information, genomic DNA was isolated from ear tissue (62 mg) following the OMNI-C Proximity Ligation Assay (Version 1.1) with some modifications. The library was sequenced on the NovaSeq 6000 platform using a 150 paired-end sequencing strategy at Novogene (UK). The fragment size distribution and concentration of each of the final libraries was assessed using the TapeStation (Agilent Technologies) and the Qubit Fluorometer and Qubit dsDNA HS reagents Assay kit (Thermo Fisher Scientific, Waltham, MA), respectively. For more information on the different protocols see Supplementary Information.
To obtain Oxford Nanopore Technologies (ONT) long reads, we ran three flow cells on a MinION portable sequencer (FLO-MIN106). Total genomic DNA was used for library preparation with the Ligation Sequencing kit (SQK-LSK109) from ONT using the manufacturer's protocols. Base calling of the reads from the three MinION flow cells was performed with guppy v4.0.11 (https://nanoporetech.com/nanopore-sequencing-data-analysis), under default settings. Afterwards, ONT reads quality was checked with Nanoplot v1.28.1 (https://github.com/wdecoster/NanoPlot) and reads shorter than 1,000 bases and mean quality below eight were discarded by running Nanofilt v2.6.0 (https://github.com/wdecoster/nanofilt).
A mix of different tissues (liver, heart, gonads, brain, kidney, muscle) was ground into small pieces using steel balls and a Retsch Mill. A total of 120 mg of the tissue was shipped on dry ice to Novogene (UK) for Illumina paired-end 150 RNA-seq of a 250–300 bp insert cDNA library.
Genome Size Estimation
Genome size was estimated following a flow cytometry protocol with propidium iodide-stained nuclei described in Hare and Johnston (2012). Ear tissue of one frozen (−80°C) adult sample of N. procyonoides and neural tissue of the internal reference standard Acheta domesticus (female, 1C = 2 Gb) was mixed and chopped with a razor blade in a petri dish containing 2 ml of ice-cold Galbraith buffer. The suspension was filtered through a 42-μm nylon mesh and stained with the intercalating fluorochrome propidium iodide (PI, Thermo Fisher Scientific) and treated with RNase II A (Sigma-Aldrich), each with a final concentration of 25 μg/ml. The mean red PI fluorescence signal of stained nuclei was quantified using a Beckman-Coulter CytoFLEX flow cytometer with a solid-state laser emitting at 488 nm. Fluorescence intensities of 5,000 nuclei per sample were recorded. We used the software CytExpert 2.3 for histogram analyses. The total quantity of DNA in the sample was calculated as the ratio of the mean red fluorescence signal of the 2C peak of the stained nuclei of the raccoon dog sample divided by the mean fluorescence signal of the 2C peak of the reference standard times the 1C amount of DNA in the standard reference. Six replicates were measured on 6 different days to minimize possible random instrumental errors. Furthermore, we estimated the genome size by coverage from mapping reads used for genome assembly back to the assembly itself using backmap 0.3 (https://github.com/schellt/backmap; Schell et al., 2017). In brief, the method divides the number of mapped nucleotides by the mode of the coverage distribution. By doing so, the length of collapsed regions with many fold increased coverage is taken into account.
Genome Assembly Workflow
SMRT reads longer than 7 kb were assembled under two different approaches (wtdbg v2.5; Ruan and Li, 2020 and Flye v2.7.1; Kolmogorov et al., 2019). The resulting assemblies were compared in terms of contiguity using Quast v5.0.2 (Gurevich et al., 2013), and evaluated for completeness by BUSCO v3.0.2 (Simão et al., 2015) (under short mode) against the laurasiatheria_odb9 data set (Supplementary Table 1). The assembled genome obtained with Flye presented the highest contiguity and completeness of both approaches and was therefore selected for downstream analyses.
Scaffolding and Gap Closing
To further improve the assembly, we applied two rounds of scaffolding and gap closing to the selected genome assembly. The genome was first scaffolded with the SMRT reads by SSPACE-longread v1.1 (Boetzer and Pirovano, 2014) and then with ONT reads by SLR (Luo et al., 2019). TGS gapcloser v1.0.1 (Xu et al., 2019) was run after each scaffolding step. Subsequently, Omni-C reads were employed to further scaffold the draft genome following the HiRise pipeline (Putnam et al., 2016) operated by the Dovetail GenomicsTM team. The assembly was screened for contamination using BlobTools v1.1.1 (Kumar et al., 2013; Laetsch and Blaxter, 2017) by evaluating coverage, GC content and sequence similarity against the NCBI nt database of each sequence (Figure 1d).
Transcriptome Assembly
Quality of raw Illumina sequences was checked with FastQC (Andrews, 2010). Low quality bases and adapter sequences were subsequently trimmed by Trimmomatic v0.39 (Bolger et al., 2014) and the transcriptome was assembled using Trinity v2.9.1 (Haas et al., 2013). The transcriptome assembly was evaluated for completeness by BUSCO v3.0.2 against the laurasiatheria_odb9 data set (C: 81.8% [S: 36.0%, D: 45.8%], F:8.0%, M:10.2%). Moreover, the clean RNA-seq reads from different tissues were aligned against the reference genome by HISAT2 (Kim et al., 2015).
Repeat Annotation
RepeatModeler v2.0 (Smit and Hubley, 2008) was run to construct a de novo repetitive library from the assembly. The specific repetitive library was merged with the canid RepBase (Jurka et al., 2005; http://www.girinst.org/repbase/ 18/10/2020), which was further annotated and masked using RepeatMasker v4.1.0 (http://www.repeatmasker.org/).
Gene Prediction and Functional Annotation
After the repeat sequences were masked, genes were predicted using the homology-based gene prediction tool GeMoMa v1.7.1 (Keilwagen et al., 2016, 2018) and 11 mammalian species as reference organisms. The selected species were Canis lupus familiaris (GCF_000002285.3; Lindblad-Toh et al., 2005), Vulpes vulpes (GCF_003160815.1; Kukekova et al., 2018), Mustela erminea (GCF_009829155.1), Zalophus californianus (GCF_009762305.2), Ailuropoda melanoleuca (GCF_002007445.1; Li et al., 2010), Ursus maritimus (GCF_000687225.1; Liu et al., 2014), Felis catus (GCF_000181335.3; Pontius et al., 2007), Sus scrofa (GCF_000003025.6; Fang et al., 2012), Bos taurus (GCF_002263795.1; Zimin et al., 2009), Mus musculus (GCF_000001635.26; Church et al., 2009), and Homo sapiens (GCF_000001405.39; Craig Venter et al., 2001). First, from the mapped RNA-seq reads, introns were extracted and filtered by the GeMoMa modules ERE and DenoiseIntrons. Then, we independently ran the module GeMoMa pipeline for each reference species using MMseqs2 (Steinegger and Söding, 2017) as alignment tools and including the mapped RNA-seq data. Finally, the 11 gene annotations were combined into a final annotation by using the GeMoMa modules GAF and AnnotationFinalizer.
Predicted genes were annotated by BLAST search against the Swiss-Prot database with an e-value cutoff of 10−6. InterProScan v5.39.77 (Quevillon et al., 2005) was used to predict motifs and domains, as well as Gene ontology (GO) terms.
Code Availability
The execution of this work involved using many software tools, for which settings and parameters are described below. Software tools indicated within brackets are dependencies employed during the execution of the main indicated tools. All the tools employed in this work are listed in Supplementary Table 3.
Genome Assembly
(1) Flye v2.7.1: parameters: –genome-size 3.198g –asm-coverage 40; (2) sspace-longread v1.1 [bedtools v2.28.0]: all parameters were set as default; (3) TGS-gapcloser v1.0.1 [minimap2 v2.17 racon v1.4.3]: parameters: –tgstype pb; (4) SLR [bwa v0.7.17 samtools v1.10]: parameters: 4.1: bwa index, 4.2 bwa mem, 4.3 samtools view -Sb, 4.4 bwa mem -k11 -W20 -r10 -A1 -B1 -O1 -E1 -L0 -a -Y, 4.5 samtools view -Sb, 4.6 SLR all parameters were set as default; (5) TGS-gapcloser v1.0.1 [minimap2 v2.17 racon v1.4.3]: parameters: –tgstype ont; (6) HiRise pipeline: all parameters were set by Dovetail Genomics team; (7) BUSCO v3.0.2 [python v3.7.4 augustus v3.3.2]: parameters: -l /laurasiatheria_odb9/ -m geno; (8) Blobtools v1.1.1 [samtools v1.10 ncbi-blast v2.10.0]: parameters: 8.1 samtools index, 8.2 blobtools map2cov, 8.3 blastn -task megablast -outfmt '6 qseqid staxids bitscore std' -max_target_seqs 1 -max_hsps 1 -evalue 1e-25, 8.4 blobtools create, view and plot all parameters were set as default; (9) Jupiterplot v1.0 [minimap2 v2.17 samtools v1.10 circos v0.69-9]: parameters: ng=99 t=64 m=2860953 g=1.
Genome Annotation
(1) RepeatModeler v2.0: parameters: -pa 16 -LTRStruct; (2) RepeatMasker v4.1.0: parameters: -s -pa 18 -no_is -xsmall; (3) hisat v2.1.0: parameters: -k 3 –pen-noncansplice 12 -S; (4) GeMoMa v.1.7.1 [java v1.8.0_221]: 4.1 java -Xmx30G -jar GeMoMa-1.7.1.jar CLI ERE c=TRUE; 4.2 java -Xmx30G -jar GeMoMa-1.7.1.jar CLI DenoiseIntrons coverage_unstranded; 4.3 java -Xmx30G -jar GeMoMa-1.7.1.jar CLI GeMoMaPipeline tblastn=false r=EXTRACTED introns coverage_unstranded DenoiseIntrons.m=100000 GeMoMa.m=100000 GeMoMa.Score=ReAlign AnnotationFinalizer.r=NO o=true; 4.4 java -Xmx30G -jar GeMoMa-1.7.1.jar CLI GAF; 4.5 java -Xmx30G -jar GeMoMa-1.7.1.jar CLI AnnotationFinalizer u=YES i c=UNSTRANDED coverage_unstranded; 4.6 java -Xmx30G -jar GeMoMa-1.7.1.jar CLI Extractor p=true c=true; (5) BUSCO v3.0.2 [python v3.7.4 augustus v3.3.2]: parameters: -l /laurasiatheria_odb9/ -m prot; (6) Interproscan v5.39.77: parameters: -f tsv -iprlookup -pa -goterms -exclappl SignalP_GRAM_NEGATIVE,SignalP_GRAM_POSITIVE -dp; (7) ncbi-blast v.2.10.0: parameters: 7.1 makeblastdb -in uniprot_sprot_2020_04.fasta -parse_seqids -dbtype prot, 7.2 blastp -evalue 1e-6 -max_hsps 1 -max_target_seqs 1 -outfmt 6.
Genome Size Estimation
(1) backmap.pl v0.3 [minimap2 v2.17, samtools v1.10, qualimap v2.2.1, bedtools 2.28.0, Rscript v3.6.3, multiqc 1.9]: parameters: -pb -v.
Data Validation
Genome Size Validation
The calculated DNA content through flow cytometry experiments was 3.10 Gb, similar to previous flow cytometry studies (3.19 Gb; Wurster-Hill et al., 1988). The genome size estimation by read coverage resulted in 3.23 Gb. Although our draft genome assembly was smaller than the values obtained by flow cytometry and coverage, the assembly length obtained of 2.39 Gb was in the range of other Carnivora genomes (Table 1, Supplementary Table 2) and showed good completeness with 92.9% completely recovered BUSCOs. The difference regarding assembly vs. estimated genome size could be explained by the complex chromosome structure of the raccoon dog which presents large chromatin proximal regions and a fluctuating number of B chromosomes (Duke Becker et al., 2011; Makunin et al., 2018). Both uncommon structures in carnivores are mostly compound by repetitive elements that were most likely not properly resolved and collapsed.
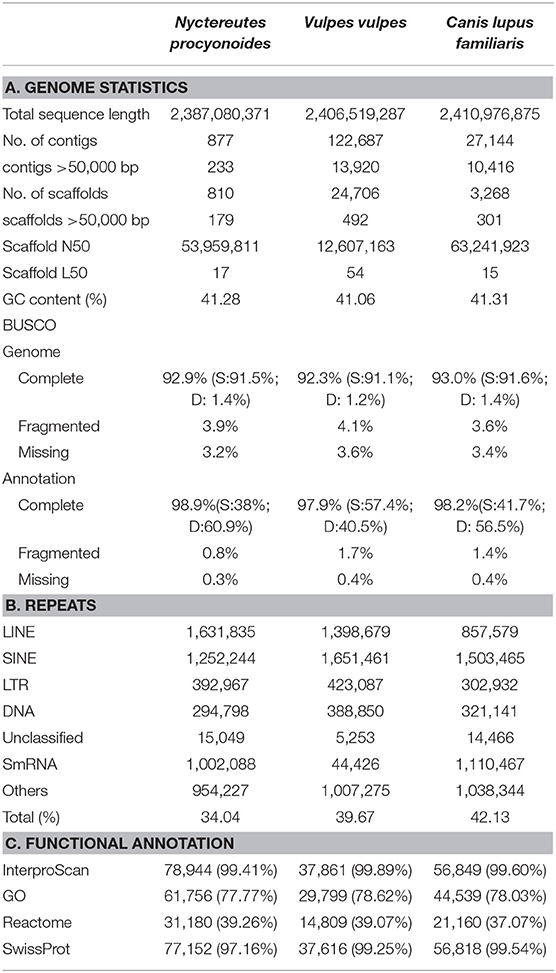
Table 1. A. Genome assembly and annotation statistics for raccoon dog (Nyctereutes procyonoides) and comparison with related species. B. Repeat statistics: De novo and homology based repeat annotations as reported by RepeatMasker and RepeatModeler; Families of repeats included here are long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), long tandem repeats (LTR), DNA repeats (DNA), unclassified (unknown) repeat families, small RNA repeats (SmRNA), and others (consisting of small, but classified repeat groups). The total is the total percentage of base pairs made up of repeats in each genome assembly, respectively. C. Number and percentage of functional annotated predicted protein-coding genes.
Comparison With Other Carnivora Genomes
A total of ~293 Gb raw data, representing 94.5X coverage, was generated using PacBio Sequel II and employed for genome assembly with Flye. After scaffolding with long reads and Omni-C data, we produced a draft genome assembly of 2.39 Gb with a scaffold N50 of 54 Mb (Table 1, section A). The final assembly of the raccoon dog draft genome contained 810 scaffolds (plus mitochondrion), where the largest scaffold was 121,018,622 bp in length which corresponded to the X-sex chromosome. We predicted 27,177 genes in the N. procyonoides genome by using a homology-based gene prediction. Among the identified proteins, 61,756 (77.8%) were annotated to have at least one GO term. Finally, 78,944 proteins (99.4%) were assigned to at least one of the databases from InterProScan (Table 1, section C). BUSCO and functional annotation results indicated high quality (Table 1, Supplementary Table 2). We also compared synteny between raccoon dog and dog genome assemblies by running Jupiterplot v1.0 (https://github.com/JustinChu/JupiterPlot). The Jupiterplot displays the largest 58 raccoon dog scaffolds, which covered more than 99% of the dog genome (Figure 1e). The colored bands represent synteny between both genome assemblies. The plot shows high synteny between both genomes with several genomic rearrangements and break points, some of them previously identified (Duke Becker et al., 2011). All these results makes the N. procyonoides genome the best genome recovered so far for the Vulpini tribe.
SARS-CoV-2
Animal cell infection by SARS-CoV-2 is determined by specificity between the receptor-binding domain (RBD) spike protein (S-protein) of SARS-CoV-2 and the membrane proteins ACE2 (peptidase domain of angiostensin I converting enzyme 2) and TMPRSS2 (transmembrane serine protease) (Lam et al., 2020). We identified both proteins in the raccoon dog genome annotation, showing high similarity with dog and fox orthologues. ACE2 protein alignments between dog and raccoon dog showed 99.3% of similarity, with only 6 of 894 different amino acids (Supplementary Figure 1). Moreover, the affinity in the binding process between S-protein from SARS-CoV-2 and ACE2 have been found to be smaller for groups like canids (Canis, Vulpes), chiroptera (Rhinophus, Pteropus) and pangolins (Manis) among others due to the matching of 14 of the 20 key amino acids in human ACE2 protein (Luan et al., 2020a). However, the reported infections of SARS-CoV-2 in domestic dogs and ferrets (Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017; Shi et al., 2020) indicated that the raccoon dog can be considered as a potential host and vector for this virus along its natural distribution range in East Asia and also in its introduced populations within Europe.
Data Availability Statement
All raw data generated for this study (PacBio, MinION, Omni-C and RNA-seq reads) are available at the European Nucleotide Archive database (ENA) under the Project number: PRJEB41734, https://www.ebi.ac.uk/ena, PRJEB41734. The final genome assembly and annotation can be found under the accession number GCA_905146905, https://www.ebi.ac.uk/ena, GCA_905146905.
Ethics Statement
Ethical review and approval was not required for the animal study because the animal was culled in full accordance to German hunting laws (waidgerecht), which means that unnecessary suffering was avoided. Moreover, the individual was not killed for the study. We used one that was killed anyway in accordance to the Convention on Biological Diversity CBD (in § 8h), that stipulates precaution, control and eradication of invasive species as a goal and task of nature conservation under international law. In 2000, the states committed themselves to developing national strategies in Decision V/8(6).
Author Contributions
SK, JK, and MP conceived this study. JK and CG prepared the samples. CG conducted lab work. LC performed bioinformatic analyses and data statistics with support of TS. LC, JK, AJ, TS, and MP discussed and interpreted the data. LC wrote the manuscript and all authors commented and revised the manuscript.
Funding
The present study is a result of the Centre for Translational Biodiversity Genomics (LOEWE-TBG) and was supported through the program LOEWE–Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz of Hesse's Ministry of Higher Education, Research, and the Arts. This study was also supported by the German Federal Environmental Foundation (DBU, Grant number 35524/01) and by Uniscientia Stiftung Vaduz (P 180-2021). LC was supported by a Post-doctoral Fellowship awarded by the Department of Education, Universities and Research of the Basque Government (Ref.: POS_2018_1_0012).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Genome Technology Center (RGTC) at Radboudumc for the use of the Sequencing Core Facility (Nijmegen, The Netherlands), which provided the PacBio SMRT sequencing service on the Sequel II platform. We also thank Damian Baranski for help with the DNA isolation and library preparations, and Norbert Peter and Dorian D. Dörge for providing samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.658256/full#supplementary-material
References
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Babraham Inst. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Boetzer, M., and Pirovano, W. (2014). SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15, 1–9. doi: 10.1186/1471-2105-15-211
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chan, P. K. S., and Chan, M. C. W. (2013). Tracing the SARS-coronavirus. J. Thorac. Dis. 5, 118–121. doi: 10.3978/j.issn.2072-1439.2013.06.19
Church, D. M., Goodstadt, L., Hillier, L. W., Zody, M. C., Goldstein, S., She, X., et al. (2009). Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 7:e1000112. doi: 10.1371/journal.pbio.1000112
Craig Venter, J., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., et al. (2001). The sequence of the human genome. Science 291, 1304–1351. doi: 10.1126/science.1058040
Drygala, F., Korablev, N., Ansorge, H., Fickel, J., Isomursu, M., Elmeros, M., et al. (2016). Homogenous population genetic structure of the non-native raccoon dog (Nyctereutes procyonoides) in Europe as a result of rapid population expansion. PLoS ONE 11:e0153098. doi: 10.1371/journal.pone.0153098
Duke Becker, S. E., Thomas, R., Trifonov, V. A., Wayne, R. K., Graphodatsky, A. S., and Breen, M. (2011). Anchoring the dog to its relatives reveals new evolutionary breakpoints across 11 species of the Canidae and provides new clues for the role of B chromosomes. Chromosom. Res. 19, 685–708. doi: 10.1007/s10577-011-9233-4
Elbe, S., and Buckland-Merrett, G. (2017). Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob. Challenges 1, 33–46. doi: 10.1002/gch2.1018
Fang, X., Mou, Y., Huang, Z., Li, Y., Han, L., Zhang, Y., et al. (2012). The sequence and analysis of a Chinese pig genome. Gigascience 1, 1–11. doi: 10.1186/2047-217X-1-16
Freuling, C. M., Breithaupt, A., Müller, T., Sehl, J., Balkema-Buschmann, A., Rissmann, M., et al. (2020). Susceptibility of raccoon dogs for experimental SARS-CoV-2 infection. Emerging Infect. Dis. 26, 2982–2985. doi: 10.3201/eid2612.203733
Guan, Y. (2003). Isolation and characterization of viruses related to the SARS Coronavirus from animals in Southern China. Science 302, 276–278. doi: 10.1126/science.1087139
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hare, E. E., and Johnston, J. S. (2012). Chapter 1 of propidium iodide-stained nuclei. Methods 772, 3–12. doi: 10.1007/978-1-61779-228-1_1
Hong, Y., Kim, K. S., Kimura, J., Kauhala, K., Voloshina, I., Goncharuk, M. S., et al. (2018). Genetic diversity and population structure of East Asian Raccoon Dog (Nyctereutes procyonoides): genetic features in central and marginal populations. Zool. Sci. 35, 249–259. doi: 10.2108/zs170140
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O., and Grau, J. (2018). Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics 19:189. doi: 10.1186/s12859-018-2203-5
Keilwagen, J., Wenk, M., Erickson, J. L., Schattat, M. H., Grau, J., and Hartung, F. (2016). Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44:e89. doi: 10.1093/nar/gkw092
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, S. I., Park, S. K., Lee, H., Oshida, T., Kimura, J., Kim, Y. J., et al. (2013). Phylogeography of Korean raccoon dogs: implications of peripheral isolation of a forest mammal in East Asia. J. Zool. 290, 225–235. doi: 10.1111/jzo.12031
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kukekova, A. V., Johnson, J. L., Xiang, X., Feng, S., Liu, S., Rando, H. M., et al. (2018). Red fox genome assembly identifies genomic regions associated with tame and aggressive behaviours. Nat. Ecol. Evol. 2, 1479–1491. doi: 10.1038/s41559-018-0611-6
Kumar, S., Jones, M., Koutsovoulos, G., Clarke, M., and Blaxter, M. (2013). Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front. Genet. 4, 1–12. doi: 10.3389/fgene.2013.00237
Laetsch, D. R., and Blaxter, M. L. (2017). BlobTools: interrogation of genome assemblies [version 1; peer review : 2 approved with reservations]. F1000Research 6:1287. doi: 10.12688/f1000research.12232.1
Lam, S. D., Bordin, N., Waman, V. P., Scholes, H. M., Ashford, P., Sen, N., et al. (2020). SARS-CoV-2 spike protein predicted to form complexes with host receptor protein orthologues from a broad range of mammals. Sci. Rep. 10, 1–14. doi: 10.1038/s41598-020-71936-5
Li, R., Fan, W., Tian, G., Zhu, H., He, L., Cai, J., et al. (2010). The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317. doi: 10.1038/nature08696
Lindblad-Toh, K., Wade, C. M., Mikkelsen, T. S., Karlsson, E. K., Jaffe, D. B., Kamal, M., et al. (2005). Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819. doi: 10.1038/nature04338
Liu, S., Lorenzen, E. D., Fumagalli, M., Li, B., Harris, K., Xiong, Z., et al. (2014). Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157, 785–794. doi: 10.1016/j.cell.2014.03.054
Luan, J., Jin, X., Lu, Y., and Zhang, L. (2020a). SARS-CoV-2 spike protein favors ACE2 from Bovidae and Cricetidae. J. Med. Virol. 92, 1649–1656. doi: 10.1002/jmv.25817
Luan, J., Lu, Y., Jin, X., and Zhang, L. (2020b). Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 526, 165–169. doi: 10.1016/j.bbrc.2020.03.047
Luo, J., Lyu, M., Chen, R., Zhang, X., Luo, H., and Yan, C. (2019). SLR: a scaffolding algorithm based on long reads and contig classification. BMC Bioinformatics 20:539. doi: 10.1186/s12859-019-3114-9
Makunin, A. I., Romanenko, S. A., Beklemisheva, V. R., Perelman, P. L., Druzhkova, A. S., Petrova, K. O., et al. (2018). Sequencing of supernumerary chromosomes of red fox and raccoon dog confirms a non-random gene acquisition by B chromosomes. Genes 9, 1–14. doi: 10.3390/genes9080405
Nie, W., Wang, J., Perelman, P., Graphodatsky, A. S., and Yang, F. (2003). Comparative chromosome painting defines the karyotypic relationships among the domestic dog, Chinese raccoon dog and Japanese raccoon dog. Chromosom. Res. 11, 735–740. doi: 10.1023/B:CHRO.0000005760.03266.29
Nørgaard, L. S., Mikkelsen, D. M. G., Elmeros, M., Chriél, M., Madsen, A. B., Nielsen, J. L., et al. (2017). Population genomics of the raccoon dog (Nyctereutes procyonoides) in Denmark: insights into invasion history and population development. Biol. Invasions 19, 1637–1652. doi: 10.1007/s10530-017-1385-5
Paulauskas, A., Griciuviene, L., Radzijevskaja, J., and Gedminas, V. (2016). Genetic characterization of the raccoon dog (Nyctereutes procyonoides), an alien species in the baltic region. Turkish J. Zool. 40, 933–943. doi: 10.3906/zoo-1502-34
Pontius, J. U., Mullikin, J. C., Smith, D. R., Lindblad-Toh, K., Gnerre, S., Clamp, M., et al. (2007). Initial sequence and comparative analysis of the cat genome. Genome Res. 17, 1675–1689. doi: 10.1101/gr.6380007
Putnam, N. H., O'Connell, B. L., Stites, J. C., Rice, B. J., Blanchette, M., Calef, R., et al. (2016). Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350. doi: 10.1101/gr.193474.115
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, 116–120. doi: 10.1093/nar/gki442
Ruan, J., and Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158. doi: 10.1038/s41592-019-0669-3
Schell, T., Feldmeyer, B., Schmidt, H., Greshake, B., Tills, O., Truebano, M., et al. (2017). An annotated draft genome for Radix auricularia (Gastropoda, Mollusca). Genome Biol. Evol. 9, 585–592. doi: 10.1093/gbe/evx032
Shi, J., Wen, Z., Zhong, G., Yang, H., Wang, C., Huang, B., et al. (2020). Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 368, 1016–1020. doi: 10.1126/science.abb7015
Shu, Y., and McCauley, J. (2017). GISAID: global initiative on sharing all influenza data – from vision to reality. Eurosurveillance 22, 2–4. doi: 10.2807/1560-7917.ES.2017.22.13.30494
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smit, A., and Hubley, R. (2008). RepeatModeler Open-1.0. Available online at: http://www.repeatmasker.org (accessed August 10, 2020).
Steinegger, M., and Söding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. doi: 10.1038/nbt.3988
Sun, L. W., Yang, Y., and Li, G. Y. (2019). The complete mitochondrial genome of the raccoon dogs (Canidae: Nyctereutes ussurienusis) and intraspecific comparison of three Asian raccoon dogs. Mitochondrial DNA B Resour. 4, 670–671. doi: 10.1080/23802359.2017.1419081
Wada, M. Y., and Imai, H. T. (1991). On the Robertsonian polymorphism found in the Japanese raccoon dog (Nyctereutes procyonoides viverrinus). Jpn. J. Genet. 66, 1–11. doi: 10.1266/jjg.66.1
Wada, M. Y., Lim, Y., and Wurster-Hill, D. H. (1991). Banded karyotype of a wild-caught male Korean raccoon dog, Nyctereutes procyonoides koreensis. Genome 34, 302–306. doi: 10.1139/g91-049
Wurster-Hill, D. H., Ward, O. G., Davis, B. H., Park, J. P., Moyzis, R. K., and Meyne, J. (1988). Fragile sites, telomeric DNA sequences, B chromosomes, and DNA content in raccoon dogs, Nyctereutes procyonoides, with comparative notes on foxes, coyote, wolf, and raccoon. Cytogenet. Genome Res. 49, 278–281. doi: 10.1159/000132677
Xu, M., Guo, L., Gu, S., Wang, O., Zhang, R., Fan, G., et al. (2019). TGS-GapCloser: fast and accurately passing through the Bermuda in large genome using error-prone third-generation long reads. bioRxiv. doi: 10.1101/831248v1
Keywords: genome assembly and annotation, SARS-CoV-2, Carnivora, raccoon dog (Nyctereutes procyonoides), B chromosome
Citation: Chueca LJ, Kochmann J, Schell T, Greve C, Janke A, Pfenninger M and Klimpel S (2021) De novo Genome Assembly of the Raccoon Dog (Nyctereutes procyonoides). Front. Genet. 12:658256. doi: 10.3389/fgene.2021.658256
Received: 25 January 2021; Accepted: 24 March 2021;
Published: 29 April 2021.
Edited by:
Gabriele Bucci, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Andrea Spitaleri, San Raffaele Hospital (IRCCS), ItalyShilpa Garg, Harvard Medical School, United States
Copyright © 2021 Chueca, Kochmann, Schell, Greve, Janke, Pfenninger and Klimpel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis J. Chueca, bHVpc2phdmllci5jaHVlY2FAZWh1LmV1cw==