 Su Jin Kim1
Su Jin Kim1 Sae-Mi Lee2,3Jong-Moon Choi2
Sae-Mi Lee2,3Jong-Moon Choi2 Ja-Hyun Jang4Hyun Gi Kim5
Ja-Hyun Jang4Hyun Gi Kim5 Jung-Taek Kim6Jae Ho Cho6
Jung-Taek Kim6Jae Ho Cho6 Young Bae Sohn7*
Young Bae Sohn7*- 1Department of Pediatrics, Inha University Hospital, Inha University College of Medicine, Incheon, South Korea
- 2GC Genome, GC Labs, Yongin-si, South Korea
- 3Department of Laboratory Medicine, Kangwon National University School of Medicine, Chuncheon-si, South Korea
- 4Department of Laboratory Medicine and Genetics, Samsung Medical Center, Seoul, South Korea
- 5Department of Radiology, Eunpyeong St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, South Korea
- 6Department of Orthopedic Surgery, Ajou University Hospital, Ajou University School of Medicine, Suwon-si, South Korea
- 7Department of Medical Genetics, Ajou University Hospital, Ajou University School of Medicine, Suwon-si, South Korea
Skeletal dysplasia (SD), a heterogeneous disease group with rare incidence and various clinical manifestations, is associated with multiple causative genes. For clinicians, accurate diagnosis of SD is clinically and genetically difficult. The development of next-generation sequencing (NGS) has substantially aided in the genetic diagnosis of SD. In this study, we conducted a targeted NGS of 437 genes – included in the nosology of SD published in 2019 – in 31 patients with a suspected SD. The clinical and genetic diagnoses were confirmed in 16 out of the 31 patients, and the diagnostic yield was 51.9%. In these patients, 18 pathogenic variants were found in 13 genes (COL2A1, MYH3, COMP, MATN3, CTSK, EBP, CLCN7, COL1A2, EXT1, TGFBR1, SMAD3, FIG4, and ARID1B), of which, four were novel variants. The diagnosis rate was very high in patients with a suspected familial SD and with radiological evidence indicating clinical SD (11 out of 15, 73.3%). In patients with skeletal involvement and other clinical manifestations including dysmorphism or multiple congenital anomalies, and various degrees of developmental delay/intellectual disability, the diagnosis rate was low (5 out of 16, 31.2%) but rare syndromic SD could be diagnosed. In conclusion, NGS-based gene panel sequencing can be helpful in diagnosing SD which has clinical and genetic heterogeneity. To increase the diagnostic yield of suspected SD patients, it is important to categorize patients based on the clinical features, family history, and radiographic evidence.
Introduction
Skeletal dysplasia (SD) is a group of heterogeneous genetic disorders associated with bone and cartilage development. It is characterized by orthopedic symptoms such as short stature, limb or spine deformities, and precocious osteoarthritis, and is sometimes accompanied by pulmonary, genitourinary, visual, auditory, and neurodevelopmental manifestations (Bae et al., 2016; Maddirevula et al., 2018; Uttarilli et al., 2019). In 2019, the International Skeletal Dysplasia Society published an updated version of the nosology of SD comprising 461 genetic skeletal disorders and 437 different causative genes classified into 42 groups based on their clinical, radiographic, and/or molecular phenotypes (Mortier et al., 2019). The clinical diagnosis of SD can be difficult because of its relatively rare incidence; for instance, the incidence rate of osteogenesis imperfecta (OI), which is the most common disease entity of SD, is 1 in 15,000 live births worldwide (Fratzl-Zelman et al., 2015). The varying and non-specific clinical symptoms among SD, especially radiological findings requiring interpretation by experts, constitute another factor hindering clinical diagnosis (Zankl et al., 2007; Calder, 2020). Molecular diagnosis is also complex because variants of one gene can cause different phenotypes, whereas similar clinical manifestations can result from variants of different genes. For example, molecular defects in FGFR3 (MIM#134934), which encodes fibroblast growth factor receptor 3, can lead to various disease spectra of SD. It can present with lethal thanatophoric dysplasia type 1, achondroplasia, craniosynostosis, or other FGFR3-related disorders such as lacrimo-auriculo-dento-digital (LADD) syndrome. On the contrary, OI type 3 (progressively deforming type) can result from mutations in multiple genes including IFITM5, SERPINF1, BMP1, in addition to representative genes such as COL1A1 and COL1A2 (Mortier et al., 2019). For these reasons, determining the gene tests to be performed on patients with a suspected SD is challenging to clinicians. In recent years, next-generation sequencing (NGS) technology, including whole exome sequencing (WES) and whole genome sequencing (WGS), has been a helpful tool for clinicians to diagnose SD (Lazarus et al., 2014; Zhang et al., 2015; Bae et al., 2016). In this study, we performed a targeted NGS in 31 patients with a suspected SD during a 3-year period at a single tertiary center in South Korea and analyzed their clinical and molecular genetic data.
Patients and Methods
Patients and Clinical Evaluation
This study was approved by the Institutional Review Board (AJIRB-BMR-GEN-20-519) of Ajou University Hospital, and all participants and guardians of the pediatric patients provided an informed consent. From March 2017 to February 2020, patients were enrolled in this study when they met one or more of the following criteria: (1) suspected familial SD, (2) radiological abnormalities suggesting SD with or without a family history, and (3) skeletal manifestations with developmental delay (DD)/intellectual disability (ID), or multiple congenital anomalies (MCA), with or without a family history. The exclusion criteria were as follows: known chromosomal aberrations or patients with a confirmed diagnosis by Sanger sequencing due to their characteristic clinical features such as achondroplasia. The patients and available family members were examined by clinical geneticists for detailed clinical phenotypes including past medical histories through a review of medical records, pedigree analyses, physical examination, and laboratory tests. Radiological skeletal surveys were performed by an expert radiologist.
Targeted NGS Panel and Genetic Analysis
Genomic DNA was extracted from peripheral blood. A library was prepared using the xGEN® Inherited Panel (Integrated DNA Technologies, Coralville, IA, United States), which comprised 5,000 genes and 180 SNP sites. Among the 5,000 genes, we analyzed 437 genes listed in the nosology of skeletal dysplasia published in 2019 (Mortier et al., 2019). Massively parallel sequencing was performed using the Illumina NextSeq 500® sequencing platform (Illumina Inc., San Diego, CA, United States). The mean coverage of target regions was 143x, with 99% of bases covered by at least 10 reads. Sequence reads were aligned to hg19 using the Burrow-Wheeler Aligner (version 0.7.12, MEM algorithm). Duplicate reads were removed using Picard-tools version 1.96. Local realignment, base quality score recalibration, and variant calling were performed using the Genome Analysis Toolkit (GATK version 3.5). Variants were annotated using the Variant Effect Predictor and dbNSFP. The identified variants were confirmed by Sanger sequencing. Copy number variants were called from the panel data using XHMM which is based on a depth–based method (Fromer et al., 2012). Familial segregation tests were performed by Sanger sequencing for the indicated cases. Variants with minor allele frequencies over 1% in the 1,000 genomes browser1, NHLBI ESP Exome Variant Server2, genome Aggregation Database (gnomAD)3, and Korean Reference Genome Database (KRGDB)4 were excluded. For the in silico analysis of missense variants, the Sorting Intolerant From Tolerant (SIFT)5, PolyPhen-26, and Mutation Taster7 algorithms were used to predict the variants to damage protein function. The identified variants were classified according to the guidelines for the interpretation of sequence variants by the American College of Medical Genetics and Genomics and Association for Molecular Pathology (Richards et al., 2015). All sequences generated for this project have been submitted to the NCBI SRA (BioProject ID PRJNA718975).
Results
Demographics and Clinical Manifestations of Patients
In total, 31 Korean patients (11 females and 20 males) from 31 non-consanguineous families, were included in this study. The patients were divided into six categories according to their clinical manifestations and family history (Table 1). The detailed demographics and clinical manifestations of the enrolled patients are summarized in Table 2. The median age of the patients was 10.0 years (range, 1 month to 46 years) and the median height standard deviation score (SDS) was −0.95 (range, −4.6 to 2.7). The most common clinical symptoms were familial or non-familial short stature (7/31, 22.5%) and abnormalities of the spine such as scoliosis and kyphosis (7/31, 22.5%) (Table 2).
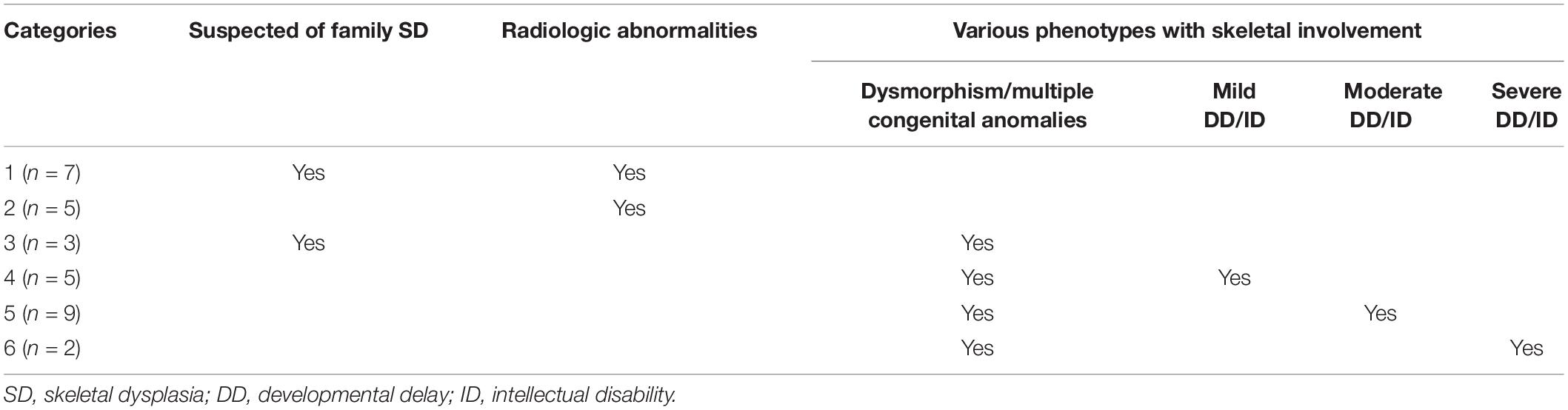
Table 1. Categories of the patients.
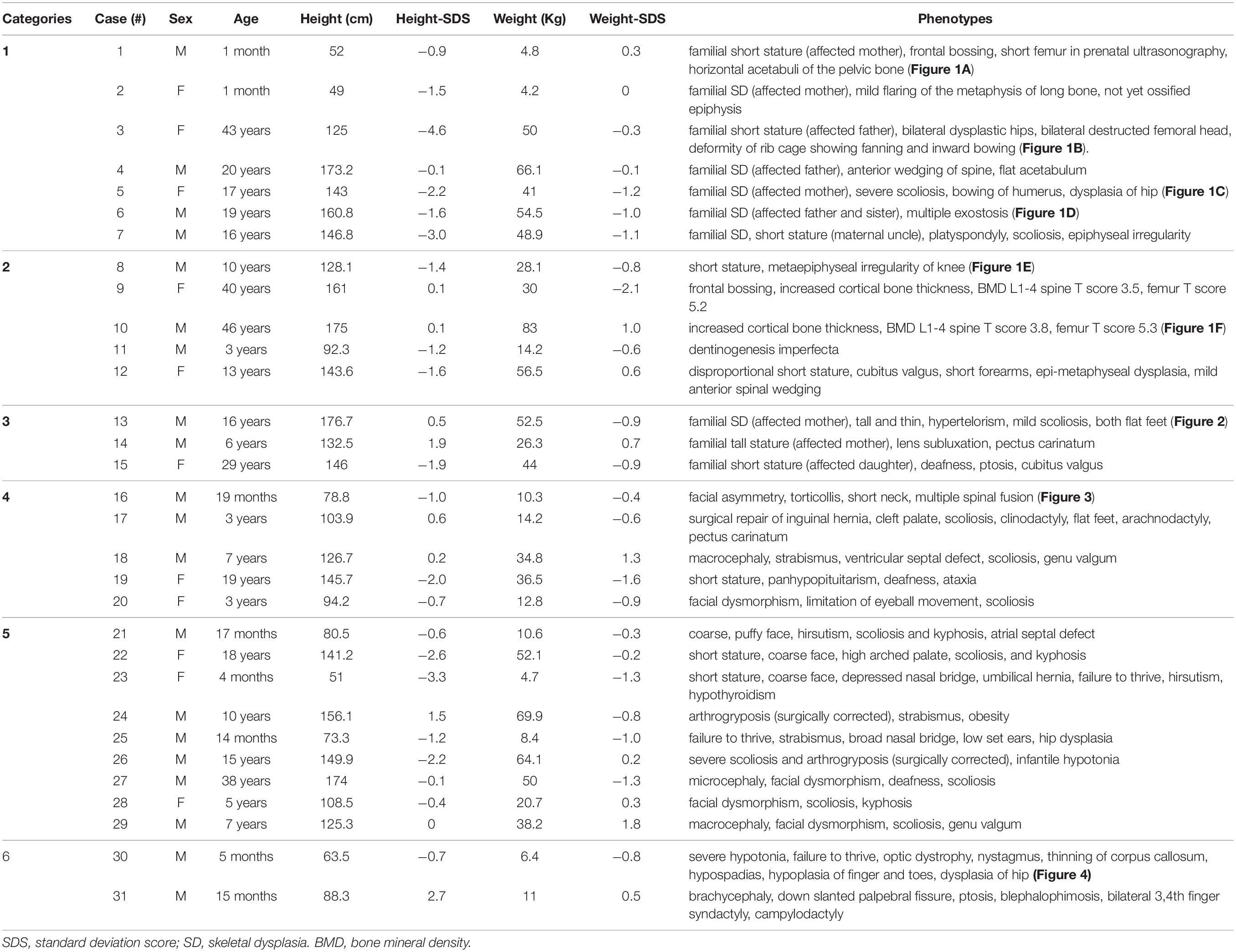
Table 2. Demographics and clinical manifestations of the patients.
Molecular Diagnosis of Patients
Next-generation sequencing-based targeted gene analysis and further familial segregation studies identified 13 causative genes in 16 out of a total of 31 patients (51.6% diagnostic yield). The genetic variants of these 16 patients with confirmed clinical and genetic diagnoses are summarized in Table 3. Eighteen pathogenic or likely pathogenic variants in thirteen genes were identified, of which, four were novel (Table 4). The identified genes and final diagnoses of the patients were heterogeneous as follows; two type II collagenopathy including spondylometaphyseal dysplasia congenita and spondyloepiphyseal dysplasia (COL2A1, collagen type II, alpha-1), spondylocarpotarsal synostosis syndrome (MYH3, myosin heavy chain 3), two multiple epiphyseal dysplasia (COMP, cartilage oligomeric matrix protein and MATN3, matrilin-3), pseudoachondroplasia (COMP, cartilage oligomeric matrix protein), pycnodysostosis (CTSK, lysosomal cysteine protease, cathepsin K), chondrodysplasia punctata (EBP, emopamil-binding protein), osteopetrosis (CLCN7, chloride channel 7), osteogenesis imperfecta (COL1A2, collagen type I, alpha-2), multiple cartilaginous exostoses (EXT1, exostosin glycosyltransferase 1), Loeys-Dietz syndrome type 1 (TGFBR1, transforming growth factor- β receptor, type 1), Loeys-Dietz syndrome type 3 (SMAD3, SMAD family member 3), Yunis-Varon dysplasia (FIG4, phosphoinositide 5-phosphatase), and two cases of Coffin-Siris syndrome (ARID1B, at-rich interaction domain-containing protein 1B). One gene (EBP) was inherited in an X-linked dominant manner, two genes (CTSK, FIG4) were inherited in an autosomal recessive manner, and the remaining 10 genetic variants were inherited in an autosomal dominant manner.
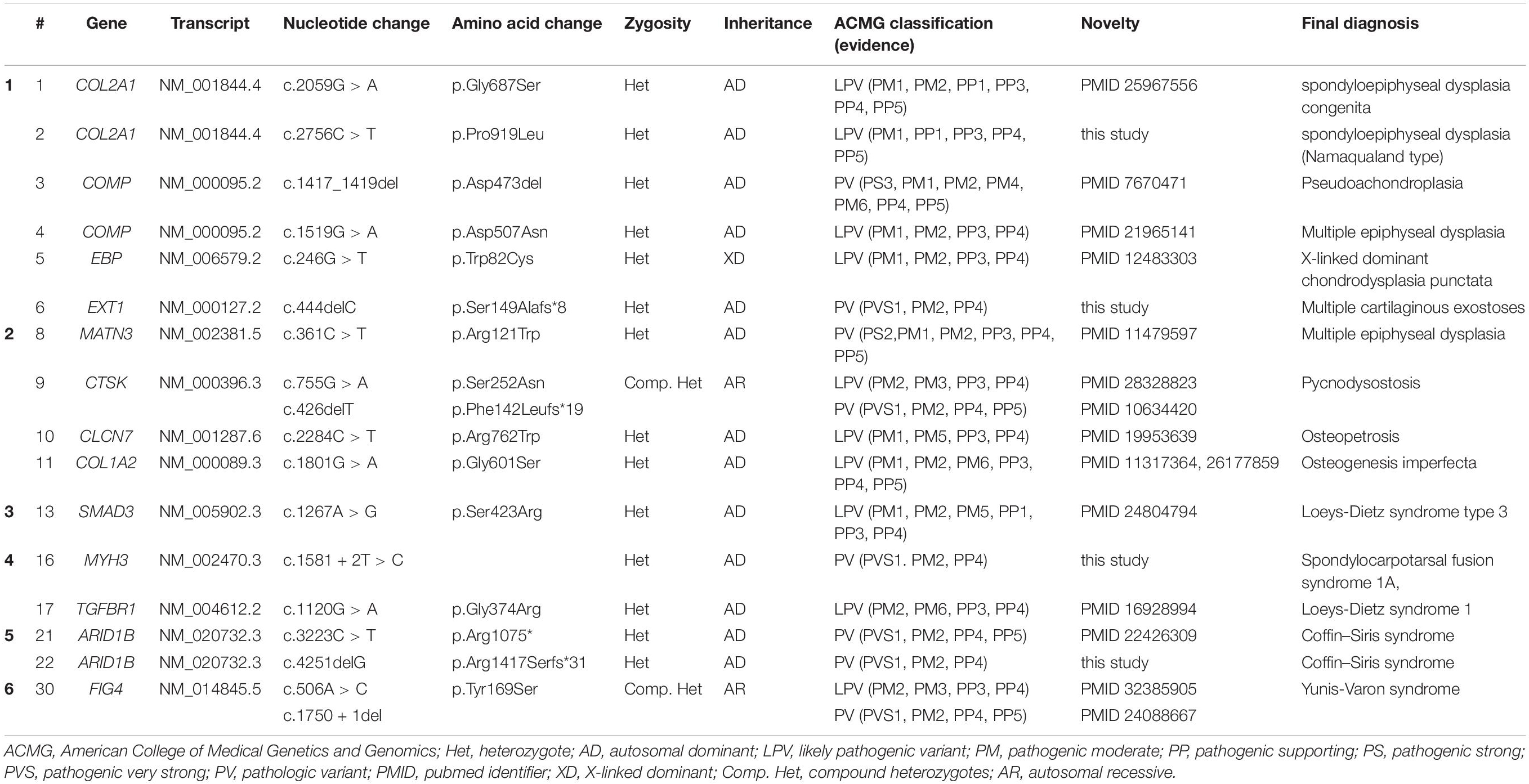
Table 3. Genetic variants of 16 patients with a confirmed diagnosis’.

Table 4. Novel variants of four patients with a confirmed diagnosis.
Representative Cases With a Confirmed Molecular Genetic Diagnosis
The patients in categories 1 and 2 who had radiological abnormalities suggesting SD had a higher diagnostic rate (85.7% and 100%, respectively) than those in the other categories. Figures 1A–F shows the representative radiological findings of patients in categories 1 and 2 with confirmative molecular diagnoses. The frequently observed abnormal findings in the skeletal surveys included shortening and metaphyseal or epiphyseal dysplasia of long bones, dysplastic hips, and dysplasia of the vertebrae. Abnormal bone densitometry (BMD) suggested osteoporotic or osteosclerotic genetic metabolic bone diseases. In category 2, case 9 who was diagnosed with pycnodysostosis and reported previously by us, had an atypical mild presentation with normal height and recurrent fractures (Song et al., 2017). Case 10 had an increased cortical bone thickness in the skeletal survey, and the BMD T scores of the spine and femur were 3.5 and 5.2, respectively. Case 11 presented with a severe dentinogenesis imperfecta only. After the identification of the likely pathogenic variants in COL1A2, BMD surveillance revealed that the BMD of the total body less the head was 0.439 g/cm2 (a normal reference for BMD under the age of 5 was not available). Considering his young age, the definition of the OI type should take into account the clinical symptoms that appear as he grows. Regular bisphosphonate infusion could be started after the molecular diagnosis of OI before he suffered a serious bone fracture.
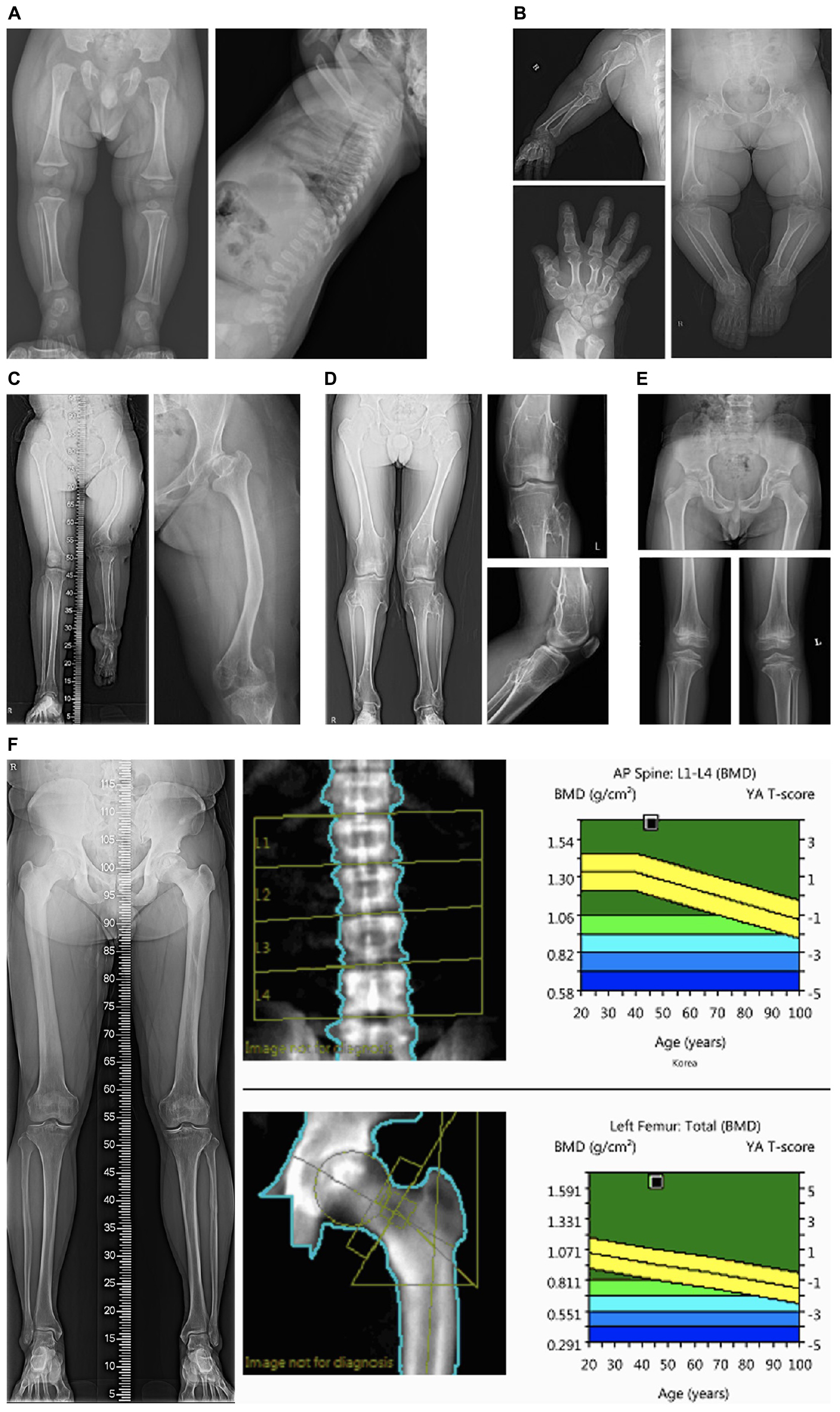
Figure 1. Representative skeletal survey images of patients in categories 1 and 2. (A) Radiographic images of case #1 with type II collagenopathy (spondyloepiphyseal dysplasia congenita) showing horizontal acetabuli of the pelvic bone and a pear-shaped lumbar vertebral body at 8 months of age; (B) Case #3 with pseudoachondroplasia showing dysplastic and relatively short upper and lower extremity bones. Bilateral femur heads are flattened and both acetabuli show degenerative changes; (C) case #5 with X-linked dominant chondrodysplasia punctata showing shortening and bowing of the left femur, dysplastic hips; (D) case #6 with multiple cartilaginous exostoses showing multiple exostoses in the bilateral distal femur and proximal tibia; (E) case #8 with multiple epiphyseal dysplasia showing small and dysplastic both proximal femoral epiphyses and acetabular dysplasia. Both knees show epiphyseal and metaphyseal irregularity; (F) Case #10 with osteopetrosis showing increased cortical bone thickness. The bone densitometry T-scores of the spine and femur were 3.5 and 5.2, respectively.
Case 13 in category 3 had clinical manifestations similar to those of his mother including facial dysmorphism (hypertelorism, dolichocephaly), tall height, and mild scoliosis. His mother had a severe scoliosis and osteoarthritis since her 40’s (Figures 2A–C). Although his skeletal symptoms were not evident, this patient underwent a targeted NGS because of a suspected familial SD and was diagnosed with Loeys-Dietz syndrome type 3 (aneurysms-osteoarthritis syndrome) caused by a pathologic variant in SMAD3, which was inherited from his mother. After diagnosis, he underwent CT angiography for the evaluation of an aneurysm, which revealed a severely dilated root of the ascending aorta (Figure 2D). Bentall’s operation for the aortic aneurysm and severe aortic regurgitation was performed, which enabled the patient to avoid sudden critical vascular events.
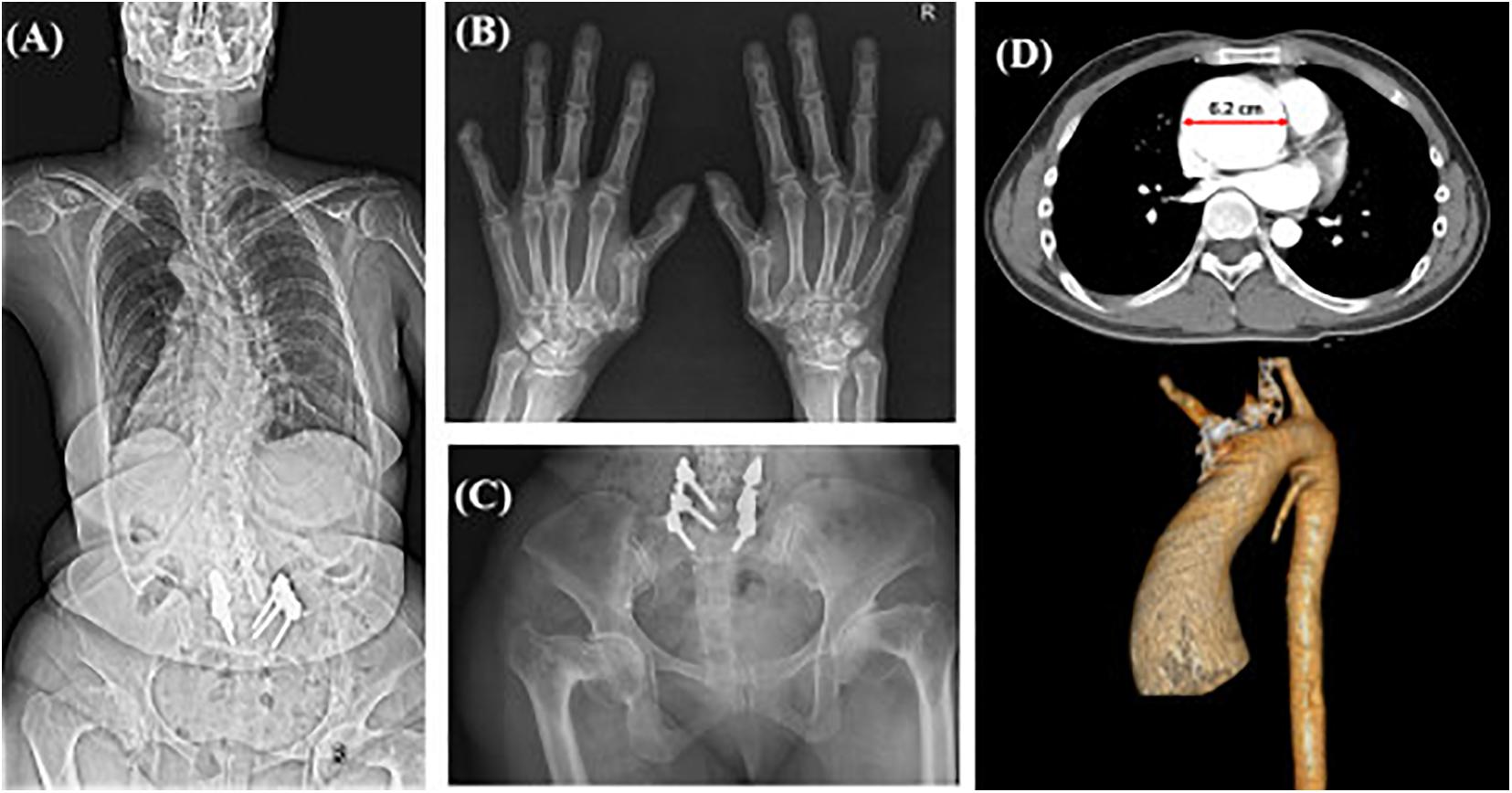
Figure 2. Radiographic images of case #13 and affected mother with aneurysms-osteoarthritis syndrome. X-ray images of the patient’s affected mother showing (A) severe scoliosis despite surgical correction, (B) degenerative osteoarthritis of the hand joints with subluxation of the 1st carpometacarpal joints, (C) bilateral severe coxa vara deformities; (D) CT scan with 3D reconstruction of Case #13 showing aneurysm of the ascending aorta with a dilated root (diameter: 6.2 cm).
Among the 16 patients in categories 4–6, five patients were diagnosed with SD (31.3% diagnostic yield) (Table 3). Case 16 presented a mild developmental delay, an asymmetric face, torticollis, a short neck, and scoliosis. X-ray and CT scans of the spine revealed narrowing of the disk space and multiple fusions of the spinal processes (Figures 3A,B). Two patients (cases 21 and 22) with skeletal manifestations such as scoliosis and kyphosis and moderate developmental delay were diagnosed with ARID1B-related Coffin-Siris syndrome. Genetic counseling and early intervention with special education could be initiated, especially in younger patients (17 months of age) after confirmative diagnosis.
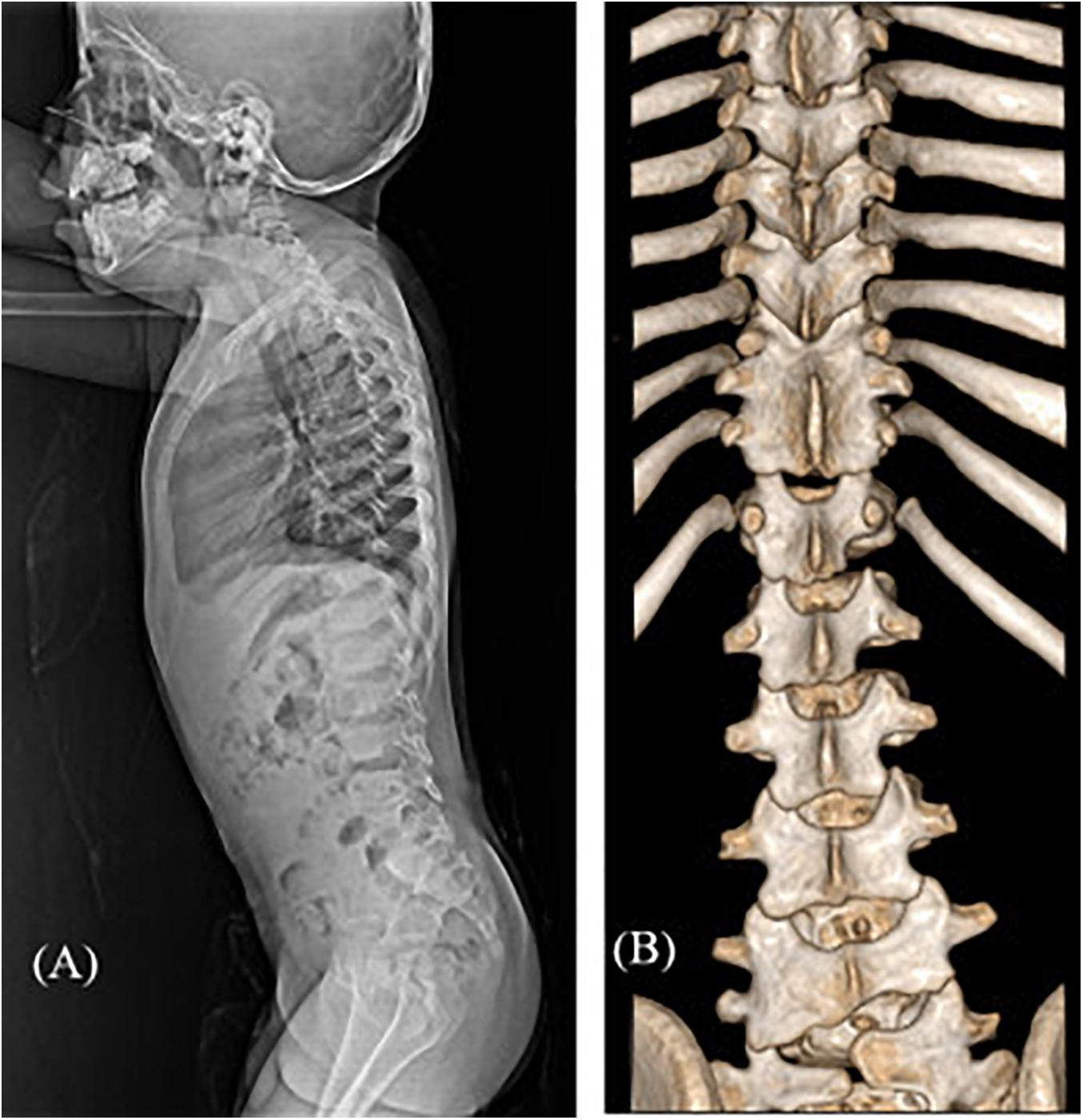
Figure 3. Radiographic images of case #16 with spondylocarpotarsal fusion syndrome 1A. (A) Lateral spine X-ray showing mildly narrowed disk spaces (B) 3D reconstruction CT scan showing fusion of T9-10-11 bilateral facet joints and spinous processes and L4-5 left facet joints.
To our knowledge, case 30 in category 6 was the first case diagnosed with Yunis-Varon dysplasia in South Korea. He presented MCA including hypopigmented skin and hair, facial dysmorphism, ocular symptoms (optic nerve dystrophy, nystagmus), hypoplasia, and dysplastic distal phalanx of the fingers and toes (Figures 4A,B), developmental dysplasia of hip (DDH), hypospadias, and a bifid scrotum. The patient suffered respiratory difficulty with laryngomalacia, severe developmental delay with muscular hypotonia, and failure to thrive. A brain MRI taken at 8 months of age showed structural brain anomalies including diffuse corpus callosal thinning, a prominent subarachnoid space at the frontotemporal convexity, dilation of the 3rd ventricle, and T2 high signal intensity of the bilateral medulla oblongata in the inferior olivary nuclei (Figure 4C). In the targeted NGS, compound heterozygous variants of the FIG4 gene were identified (Table 3) in the patient, and his parents were heterozygous carriers. His developmental milestone did not progress at all; he could not control his head completely or establish eye contact until 15 months of age. He died at of 15 months of age due to respiratory failure.
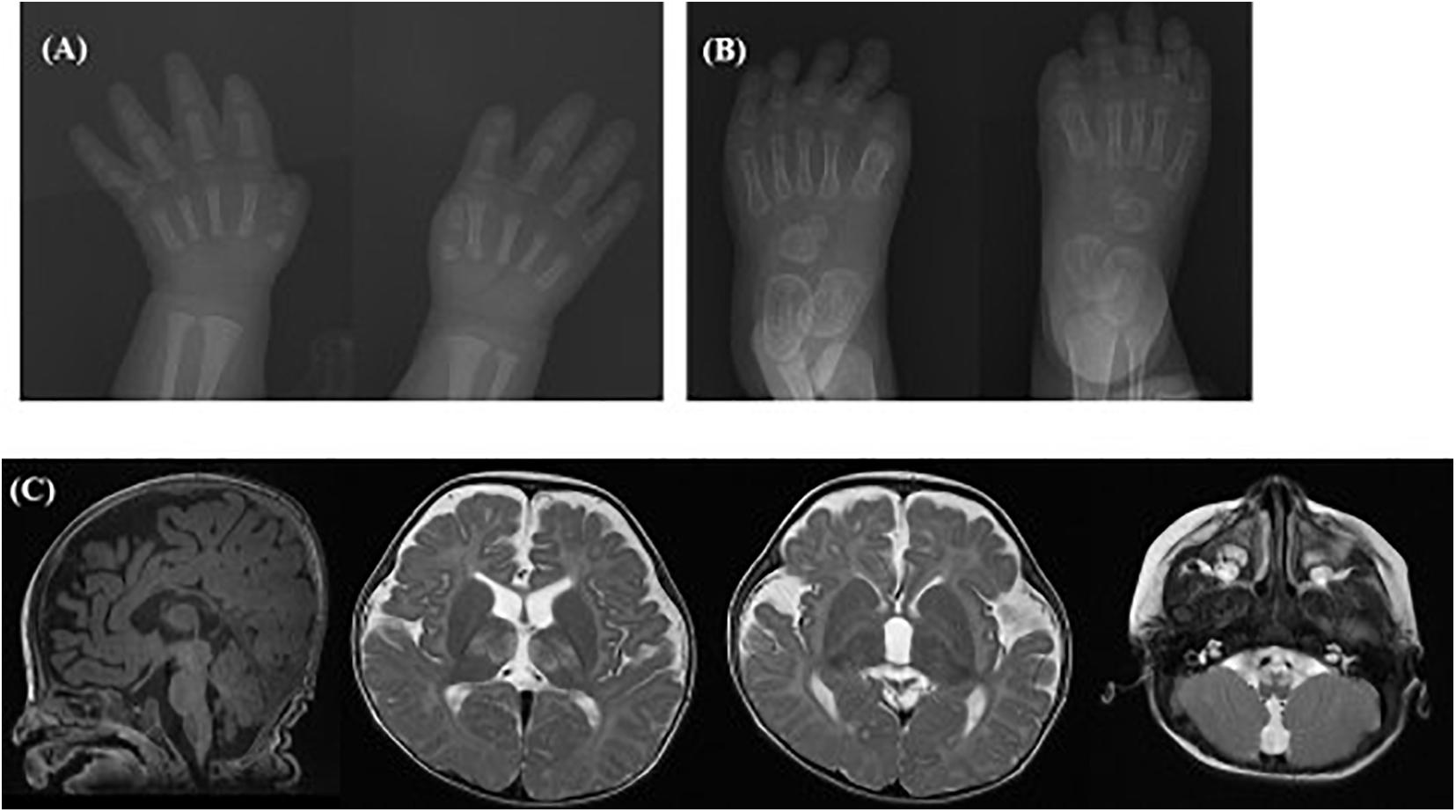
Figure 4. Radiographic images of case #30 with Yunis-Varon dysplasia. X-ray showing hypoplasia and agenesis of the distal phalanx of the (A) fingers and (B) toes. (C) Brain MRI showing diffuse corpus callosal thinning, prominent subarachnoidal space at the frontotemporal convexity, and dilation of the 3rd ventricle, T2 high signal intensity of the bilateral thalami and medulla oblongata of inferior olivary nuclei.
Discussion
This study explored the diagnoses of various spectrums of SD through a targeted NGS in 31 patients with a suspected SD at a single tertiary center in South Korea over a 3-year period. In this study, we could make a definite diagnosis in 16 patients based on the clinical and molecular findings using NGS. In total, 18 pathogenic/likely pathogenic variants of 13 different genes were found in 16 patients, out of which, 10 genes were found in one patient each, and three genes (COL2A1, COMP, and ARID1B) were found in two patients each. These results suggest that SD is a genetically heterogeneous group. Four novel variants were found among the eighteen identified variants, which would be helpful in expanding the genotype–phenotype correlations.
In previous studies that attempted to diagnose patients with a suspected SD using NGS, the diagnostic yield was reported to vary widely depending on the genes included in the NGS panel, and the criteria used for selecting the patients. Zhang et al. (2015) performed a study in 82 patients with a suspected SD using an NGS panel containing 61 genes, which were selected according to the nosology and classification of genetic skeletal disorders in 2010. They found mutations in 13 different genes in 44 out of 82 patients, and reported a diagnostic yield of 54%. Polla et al. (2015) executed an NGS panel consisting of 309 genes in 69 patients with a suspected SD and reported a diagnostic yield of 18.8%. Bae et al. (2016) carried out the targeted exome sequencing (TES) for 255 genes in 185 patients with SD. Pathogenic variants were detected in 74% (71 out of 96) of the patients with an assured clinical diagnosis, and 20.3% (13 out of 64) of the patients with uncertain clinical diagnoses were detected in TES.
In our study, the total diagnostic yield was 51.6%. However, the diagnosis rates differed among the patient categories according to their clinical characteristics. The diagnostic yield was very high (11 out of 12, 91.6%) in patients with radiologic evidence that could clinically suggest SD (categories 1 and 2). Patients with a suspected familial SD (Categories 1 and 3) also showed a high diagnosis rate of 70% (7 out of 10). In contrast, the diagnostic yield for categories 4–6 was 31.2% (5 out of 16). Therefore, clinical phenotyping would still be important for a higher diagnostic yield of the NGS-based analysis. Among the patients in categories 1–3 in this study, pathogenic variants were found in genes such as COL2A1, COMP, and MATN3, which are the causative genes of type II collagenopathy and multiple epiphyseal dysplasia (MED). Although SD is a genetically heterogeneous group, type II collagenopathy or MED has been reported relatively frequently in most studies. In addition, patients with genetic mutations in ACAN, PHEX, TRPV4, FBN1, and FGFR3 have also often been observed (Lazarus et al., 2014; Polla et al., 2015; Bae et al., 2016; Barat-Houari et al., 2016; Sentchordi-Montane et al., 2018; Uttarilli et al., 2019). Therefore, in these patients, a gene panel consisting of representative SD genes can reduce diagnostic costs.
In our study, 31.2% (5 out of 16) of the patients with skeletal involvement and other clinical manifestations including dysmorphism or MCA, and various degrees of DD/ID (categories 4–6) were able to obtain a definite diagnosis. Skeletal symptoms such as craniofacial abnormalities, scoliosis, kyphosis, or joint contractures are often accompanied with various syndromic DD/ID, and this should be considered by clinicians. Therefore, we included patients with DD/ID and various skeletal manifestations including scoliosis, kyphosis, multiple spinal fusion, syndactyly, clinodactyly, arachnodactyly, arthrogryposis, and DDH. Although the diagnosis rate was lower than that of patients with a high clinical suspicion of SD, the diagnosed patients in these categories had a rare syndromic SD such as spondylocarpotarsal fusion syndrome 1A (case 16), Loeys-Dietz syndrome 1 (case 17), Coffin–Siris syndrome (cases 21 and 22), and Yunis-Varon syndrome (case 30). In particular, the Yunis-Varon syndrome is extremely rare. Since its first report in 1980, fewer than 30 cases have been reported (Siddique et al., 2019; Wright et al., 2020). This is the first case reported in South Korea. It is difficult for clinicians to suspect these extremely rare diseases based on the clinical features or radiographic evidence, and other concomitant symptoms might not suggest SD. Therefore, NGS-based gene analysis might be helpful in increasing the diagnostic yield and expanding the knowledge regarding the clinical phenotypes for these rare diseases.
In this study, 43.7% (7 out of 16) of the diagnosed patients had a confirmed genetic diagnosis at under 3 years of age. For SD patients, especially those with other symptoms, definite diagnosis at an early age through genetic analysis could help in predicting undiscovered symptoms and in managing SD patients throughout their life. It is also important to understand the pathophysiology of SD, where symptoms change with the maturation of the skeletal system (Falardeau et al., 2017). Accurate genetic diagnosis facilitates family planning through genetic counseling and helps patients and their families accept the situation. In case 11, before all clinical symptoms of OI appeared, a genetic diagnosis was made, and complications such as fractures were sufficiently explained to the parents, a regular bisphosphonate infusion was started. In case 13, after a definite diagnosis through genetic study, we could find and correct a critical cardiac valve regurgitation and aortic aneurysm before the symptoms developed through surveillance for the involvement of other organs that may be accompanying the specific SD (Loeys-Dietz syndrome type 3). Two cases of Coffin-Siris syndrome diagnosed in this study were also evaluated and managed by a multidisciplinary team approach.
However, causative genetic variants were not identified in 15 patients (48.4%) in our cohort. For these patients, further interpretation of the NGS data, other than the 437 genes related to SD, could identify the causative genes. For instance, a patient with Cantu syndrome (case 23) caused by a heterozygous variant in KCNJ8 (NM_004982.3: c.41T > G, p.Leu14Arg) was diagnosed through a further analysis of the NGS data. She presented facial dysmorphism (coarse face, depressed nasal bridge, hypertrichosis), umbilical hernia, growth retardation, and developmental delay since infancy. Although flaring of the metaphyseal plate was noted in the skeletal survey, initial analysis of the NGS data for SD did not identify any pathogenic variants. After Cantu syndrome was diagnosed by the interpretation of the expanded target genes, “reverse phenotyping” confirmed the diagnosis. Additional genetic investigations, including copy number variation (CNV) analysis, WES or WGS, are needed for other patients whose final diagnosis could not be made by the re-interpretation of the NGS data for the expanded target genes. In general, a considerable number of SD cases are known to be single gene disorders rather than disorders caused by CNVs such as duplications or microdeletions, and CNV has been studied more in DD/ID and in autism-spectrum disorders. However, some SDs have recently been reported because of the total deletion of genes included in the microdeletion of specific chromosomal loci (van Dijk et al., 2010; Jennes et al., 2011; Su et al., 2015; Puvabanditsin et al., 2018). In the third-generation sequencing, using CNV calling algorithms, it is possible to detect several CNVs in the NGS data; however, their sensitivity is generally lower than that of chromosomal microarray (CMA) and multiplex ligation-dependent probe amplification (MLPA). It is thus necessary to confirm with CMA or MLPA when CNV is suspected in NGS or WES. Furthermore, there is a possibility of finding new candidate genes and expanding the phenotypes of SD when WES or WGS is performed in these patients. In fact, with the development of the NGS technology including WES or WGS, more genes responsible for SD were identified, and 226 causative genes were included in the 2010 revision of the nosology and classification of genetic skeletal disorders (Warman et al., 2011), and this number was increased to 364 in 2015 (Bonafe et al., 2015) and 437 in 2019 (Mortier et al., 2019). In the newest version of the nosology of genetic skeletal disorders, compared with the previous version, pathogenic variants of 437 disease-causing genes have been found in 92% (425/461) of all disorders. A study on a large cohort in India has proposed that several genes not included in the nosology of genetic skeletal disorders 2019 are candidate genes for SD based on WES in patients with a suspected SD (Shaheen et al., 2016; Maddirevula et al., 2018; Uttarilli et al., 2019).
In conclusion, we performed a targeted NGS, including all 437 causative genes listed in the nosology of skeletal dysplasia published in 2019 (Mortier et al., 2019), in 31 patients with a suspected SD. We reported four novel variants, atypical clinical symptoms of known diseases, and extremely rare disease entities of SD to help expand the genotype–phenotype correlation of SD. To increase the diagnostic yield of suspected SD patients, it is important to categorize patients according to their clinical characteristics and to use the targeted NGS or WES appropriately. The limitation of this study is that this single-center study was carried out for a relatively short period of time, and the number of patients was not sufficient to demonstrate consistent characteristics or the genotype–phenotype correlations of patients in each category. Further research is thus needed to properly classify patients according to their clinical characteristics, family history, and radiological evidence to develop an applicable NGS panel based on patient classification, or to determine which patients need WES or WGS.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA, BioProject ID PRJNA718975, https://www.ncbi.nlm.nih.gov/sra/PRJNA718975.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of Ajou University Hospital (AJIRB-BMR-GEN-20-519). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
SK analyzed and interpreted the data, and prepared and corrected the manuscript. S-ML, J-MC, and J-HJ reviewed the analysis of genetic information, and involved in the revision of the manuscript. HK, J-TK, and JC enrolled the participants and analyzed their clinical or radiological data. YS designed the study and approved the submitted version of the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2017R1C 1B5016225).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all the patients and their caregivers for their involvement in the present study.
Footnotes
- ^ http://browser.1000genomes.org/
- ^ http://evs.gs.washington.edu/EVS/
- ^ http://gnomad.broadinstitute.org/
- ^ http://coda.nih.go.kr/coda/KRGDB/index.jsp
- ^ http://sift.jcvi.org/
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://www.mutationtaster.org/
References
Bae, J. S., Kim, N. K., Lee, C., Kim, S. C., Lee, H. R., Song, H. R., et al. (2016). Comprehensive genetic exploration of skeletal dysplasia using targeted exome sequencing. Genet. Med. 18, 563–569. doi: 10.1038/gim.2015.129
Barat-Houari, M., Dumont, B., Fabre, A., Them, F. T., Alembik, Y., Alessandri, J. L., et al. (2016). The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur. J. Hum. Genet. 24, 992–1000. doi: 10.1038/ejhg.2015.250
Bonafe, L., Cormier-Daire, V., Hall, C., Lachman, R., Mortier, G., Mundlos, S., et al. (2015). Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. A 167A, 2869–2892. doi: 10.1002/ajmg.a.37365
Calder, A. D. (2020). The changing world of skeletal dysplasia. Lancet Child Adolesc. Health 4, 253–254. doi: 10.1016/s2352-4642(20)30056-0
Falardeau, F., Camurri, M. V., and Campeau, P. M. (2017). Genomic approaches to diagnose rare bone disorders. Bone 102, 5–14. doi: 10.1016/j.bone.2016.07.020
Fratzl-Zelman, N., Misof, B. M., Roschger, P., and Klaushofer, K. (2015). Classification of osteogenesis imperfecta. Wien. Med. Wochenschr. 165, 264–270. doi: 10.1007/s10354-015-0368-3
Fromer, M., Moran, J. L., Chambert, K., Bank, E., Bergen, S. E., Ruderfer, D. M., et al. (2012). Discovery and statistical genotyping of copy-number variation from whole-exome sequencingdepth. Am. J. Hum. Genet. 91, 597–607. doi: 10.1016/j.ajhg.2012.08.005
Jennes, I., de Jong, D., Mees, K., Hogendoorn, P. C., Szuhai, K., and Wuyts, W. (2011). Breakpoint characterization of large deletions in EXT1 or EXT2 in 10 multiple osteochondromas families. BMC Med. Genet. 12:85. doi: 10.1186/1471-2350-12-85
Lazarus, S., Zankl, A., and Duncan, E. L. (2014). Next-generation sequencing: a frameshift in skeletal dysplasia gene discovery. Osteoporos. Int. 25, 407–422. doi: 10.1007/s00198-013-2443-1
Maddirevula, S., Alsahli, S., Alhabeeb, L., Patel, N., Alzahrani, F., Shamseldin, H. E., et al. (2018). Expanding the phenome and variome of skeletal dysplasia. Genet. Med. 20, 1609–1616. doi: 10.1038/gim.2018.50
Mortier, G. R., Cohn, D. H., Cormier-Daire, V., Hall, C., Krakow, D., Mundlos, S., et al. (2019). Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 179, 2393–2419. doi: 10.1002/ajmg.a.61366
Polla, D. L., Cardoso, M. T., Silva, M. C., Cardoso, I. C., Medina, C. T., Araujo, R., et al. (2015). Use of Targeted Exome Sequencing for Molecular Diagnosis of Skeletal Disorders. PLoS One 10:e0138314. doi: 10.1371/journal.pone.0138314
Puvabanditsin, S., February, M., Mayne, J., McConnell, J., and Mehta, R. (2018). Cleidocranial Dysplasia with 6p21.1-p12.3 Microdeletion: a Case Report and Literature Review. Cleft. Palate. Craniofac. J. 55, 891–894. doi: 10.1597/15-306
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Sentchordi-Montane, L., Aza-Carmona, M., Benito-Sanz, S., Barreda-Bonis, A. C., Sanchez-Garre, C., Prieto-Matos, P., et al. (2018). Heterozygous aggrecan variants are associated with short stature and brachydactyly: description of 16 probands and a review of the literature. Clin. Endocrinol. 88, 820–829. doi: 10.1111/cen.13581
Shaheen, R., Patel, N., Shamseldin, H., Alzahrani, F., Al-Yamany, R., ALMoisheer, A., et al. (2016). Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort. Genet. Med. 18, 686–695. doi: 10.1038/gim.2015.147
Siddique, A. W., Ahmed, Z., Haider, A., Khalid, H., and Karim, T. (2019). Yunis-Varon Syndrome. J. Ayub. Med. Coll. Abbottabad. 31, 290–292.
Song, H. K., Sohn, Y. B., Choi, Y. J., Chung, Y. S., and Jang, J. H. (2017). A case report of pycnodysostosis with atypical femur fracture diagnosed by next-generation sequencing of candidate genes. Medicine 96:e6367. doi: 10.1097/MD.0000000000006367
Su, P., Wang, Y., Cooper, D. N., Zhu, W., Huang, D., Ferec, C., et al. (2015). Disclosing the Hidden Structure and Underlying Mutational Mechanism of a Novel Type of Duplication CNV Responsible for Hereditary Multiple Osteochondromas. Hum. Mutat. 36, 758–763. doi: 10.1002/humu.22815
Uttarilli, A., Shah, H., Bhavani, G. S., Upadhyai, P., Shukla, A., and Girisha, K. M. (2019). Phenotyping and genotyping of skeletal dysplasias: evolution of a center and a decade of experience in India. Bone 120, 204–211. doi: 10.1016/j.bone.2018.10.026
van Dijk, F. S., Huizer, M., Kariminejad, A., Marcelis, C. L., Plomp, A. S., Terhal, P. A., et al. (2010). Complete COL1A1 allele deletions in osteogenesis imperfecta. Genet. Med. 12, 736–741. doi: 10.1097/GIM.0b013e3181f01617
Warman, M. L., Cormier-Daire, V., Hall, C., Krakow, D., Lachman, R., LeMerrer, M., et al. (2011). Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet A 155A, 943–968. doi: 10.1002/ajmg.a.33909
Wright, G. C., Brown, R., Grayton, H., Livingston, J. H., Park, S. M., Parker, A. P. J., et al. (2020). Clinical and radiological characterization of novel FIG4-related combined system disease with neuropathy. Clin. Genet. 98, 147–154. doi: 10.1111/cge.13771
Zankl, A., Jackson, G. C., Crettol, L. M., Taylor, J., Elles, R., Mortier, G. R., et al. (2007). Preselection of cases through expert clinical and radiological review significantly increases mutation detection rate in multiple epiphyseal dysplasia. Eur. J. Hum. Genet. 15, 150–154. doi: 10.1038/sj.ejhg.5201744
Keywords: skeletal dysplasia, next-generation sequencing, genetic heterogeneity, skeletal genetics, molecular genetic test
Citation: Kim SJ, Lee S-M, Choi J-M, Jang J-H, Kim HG, Kim J-T, Cho JH and Sohn YB (2021) Genetic Analysis Using a Next Generation Sequencing-Based Gene Panel in Patients With Skeletal Dysplasia: A Single-Center Experience. Front. Genet. 12:670608. doi: 10.3389/fgene.2021.670608
Received: 22 February 2021; Accepted: 15 April 2021;
Published: 26 May 2021.
Edited by:
Przemko Tylzanowski, KU Leuven, BelgiumReviewed by:
Leslie Matalonga, Centre for Genomic Regulation (CRG), SpainOlivia J. Veatch, University of Kansas Medical Center, United States
Copyright © 2021 Kim, Lee, Choi, Jang, Kim, Kim, Cho and Sohn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Young Bae Sohn, eWJzb2huQGFqb3UuYWMua3I=