 Aiping Deng1,2†
Aiping Deng1,2† Jinpeng Li1,2†Zebin Yao1,2Gyamfua Afriyie1,2Ziyang Chen1,2Yusong Guo1,2Jie Luo1,2*
Jinpeng Li1,2†Zebin Yao1,2Gyamfua Afriyie1,2Ziyang Chen1,2Yusong Guo1,2Jie Luo1,2* Zhongduo Wang1,2*
Zhongduo Wang1,2*- 1College of Fisheries, Guangdong Ocean University, Zhanjiang, China
- 2Guangdong Provincial Key Laboratory of Aquaculture in South China Sea for Aquatic Economic Animal of Guangdong Higher Education Institutes, Fisheries College, Guangdong Ocean University, Zhanjiang, China
Coelomactra antiquata is an important aquatic economic shellfish with high medicinal value. However, because C. antiquata has no reference genome, a lot of molecular biology research cannot be carried out, so the analysis of its transcripts is an important step to study the regulatory genes of various substances in C. antiquata. In the present study, we conducted the first full-length transcriptome analysis of C. antiquata by using PacBio single-molecule real-time (SMRT) sequencing technology. The results identified a total of 39,209 unigenes with an average length of 2,732 bp, 23,338 CDSs, 251 AS events, 9,881 lncRNAs, 20,106 SSRs, and 2,316 TFs. Subsequently, 59.22% (23,220) of the unigenes were successfully annotated, of which 23,164, 18,711, 15,840, 13,534, and 13,474 unigenes could be annotated using NR, Swiss-prot, KOG, GO, and KEGG databases, respectively. This study lays the foundation for the follow-up research of molecular biology and provides a reference for studying the more medicinal value of C. antiquata.
Introduction
The Coelomactra antiquata is a bivalve marine creature that lives in the bottom sand (Kong and Li, 2009). As a wide temperature shellfish, it is predominantly distributed in the western Pacific Ocean, the Indian Peninsula, Japan, and the coast of China. In China, C. antiquata is distributed from Liaoning province in the north and Guangxi Zhuang autonomous region in the south (Kong et al., 2007). The meat of the C. antiquata is tender, delicious, and nutritious, making it a remarkable species with high economic value (Liu et al., 2006). However, due to excessive fishing in recent decades, the natural population of C. antiquata has gradually decreased (He et al., 2021). Fortunately, the artificial breeding of C. antiquata has gradually matured in recent years of continuous attempts (Liu et al., 2012; Chen., 2018a; Chen., 2018b).
In addition to research on mitochondrial genomes (Meng et al., 2012, 2013; Shen et al., 2016), the content of previous research mainly focused on its morphological research (Kong et al., 2007), population genetic comparison (Kong and Li, 2007), and organizational composition research of C. antiquata (Wu et al., 2019). As well, there was some research on the possible role of this bivalve in disease treatment. For example, treating diabetic mice with different doses of C. antiquata extract can reduce the blood glucose concentration of diabetic mice and increase the antioxidant activity of serum (Wen et al., 2015). Also, using a dose of 30 mg/kg of C. antiquata polysaccharides on human carcinoma of esophagus cells transplanted in nude mice, its inhibitory rate of 28.85% was recorded (Yang et al., 2015).
According to the above description, there have been many reports on the genetic, morphological, and disease treatment effects of C. antiquata, but there are few studies on the transcriptome level. Transcriptome sequencing (RNA-seq) is a technology that uses high-throughput sequencing technology to sequence and analyze all or part of mRNA, small RNA, and no-codingRNA in cells or tissues. RNA-seq can identify genes involved in a variety of biological processes and obtain relevant transcripts in biological processes (De Klerk et al., 2014; Chen et al., 2020). With the continuous development of nucleic acid sequencing technology and the advent of third-generation sequencing technology, the full-length transcriptome can be obtained more simply and accurately (Schadt et al., 2010). Compared with the first-generation and second-generation sequencing technologies, the third-generation sequencing technology can directly obtain the full-length transcript sequence without assembling, which can truly reflect the transcriptome information of the sequenced species (Li et al., 2008; Bleidorn, 2016; Jia et al., 2020). This study used PacBio’s single-molecule real-time (SMRT) sequencing technology to generate comprehensive full-length transcriptome of C. antiquata. We then systematically carried out structural analysis and functional annotation of those full-length transcriptomes to obtain a large amount of usable sequence information. From this sequence information, we can see that many transcripts of C. antiquata have signal transduction, synthesis, and metabolism functions, indicating that there may be many biologically active substances in C. antiquata that participate in the life processes. This study will provide data for follow-up study of certain functional genes, molecular biology research, and exploration of possible biomedical functions of C. antiquata.
Materials and Methods
Sample Collection for Iso-Seq
One C. antiquata (Shell length: 87 mm, shell width: 65 mm, shell height: 40.5 g) sampled from Leizhou in Guangdong Province. Tissues including blood, mantle, adductor muscle, lip, foot, gill, inlet pipe, outlet pipe, kidney, intestine, liver, and gonad were rapidly collected, immediately frozen in liquid nitrogen, and then stored at −80°C for preservation until RNA extraction.
RNA Extraction
Total RNA was separately extracted from these tissue samples (Jia et al., 2020; Zheng et al., 2020). The purity, concentration, and absorption peak of the extracted RNA were measured using a NanoDrop 2,000 spectrophotometer (Thermo Fisher Scientific Inc., United States). Agarose gel electrophoresis was mainly used to detect the genomic contamination, purity of samples, and the Agilent 2,100 was used to determine the RIN value accurately detecting the integrity of RNA. When the test results met the requirements, RNA samples from 12 tissue were mixed together for the following library preparation.
Library Preparation and SMRT Sequencing
The Clonetech SMARTerTM PCR cDNA Synthesis Kit was used to reverse transcribe the pooled total RNA into cDNA. Afterwards, polymerase chain reaction (PCR) was employed to amplify the cDNA and using primers with Oligo dT. The amplified cDNA was purified with PB magnetic beads. After purification, all full-length cDNAs were end-repaired and connected with SMRT dumbbell adaptors. Exonuclease digestion was implemented to remove sequences that failed to ligate to the adapters. The resulting sequences were purified again. Finally, a SMRTbell library was constructed. Prior to sequencing, the accurate quantification of the libraries was assessed by Qubit 3.0 and the size of the libraries were detected by Agilent 2,100. Then the full-length transcriptome was sequenced with PacBio sequencer.
Sequencing Data Processing
The raw sequencing data were processed using the SMRTlink (Hon et al., 2020) software with the parameters: --min_passes 3; --min_length 50; --max_length 15,000. The high-quality sequencing reads produced by a single molecule in the sequencing process are called polymerase read, and the polymerase reads remove the sequencing adapters to form subreads. A circular consensus sequence (CCS) was obtained from the subreads. The CCS sequence was checked to see whether it contained 5′primer, 3′primer, and polyA. Their positional relationships were assessed and later divided the CCS sequence into three categories: the full-length sequence (FL), the full-length non-concatemer sequence (FLNC), and the full-length non-chimeric sequence with polyA. ICE of SMRTlink software was used to cluster FLNC sequences and obtain a set of cluster consensus sequences. Further the sequences were polished by Arrow algorithm (Cao et al., 2020) and obtained the FLNC polished high quality consensus Sequences. Finally, CD-HIT (Li & Godzik, 2006) software (parameters: -c 0.99; -G 0; -aL 0.90; -AL 100; -aS 0.99; -AS 30) was used to perform clustering and de-redundancy. The unigenes from high quality full-length transcripts were used for subsequent analysis.
Function Annotation
To obtain basic annotations information, non-redundant transcripts were annotated against six different databases, namely, Non-supervised Orthologous Groups (NR), EuKaryotic Orthologous Groups (KOG), Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-Prot, and Pfam databases. DIAMOND (Buchfink et al., 2014) software (parameters: --more-sensitive; -k 10; -e 1e-5) was used for NR, Swiss-Prot, KOG, GO, KEGG databases analyses, and the Hmmer package (Nguyen et al., 2016) with default parameters utilized for Pfam database analyses.
Structure Analysis
CDS, LncRNA, and TFs Prediction
TransDecoder software (Haas et al., 2013) (parameters: -G universal; -S; -m 100) was used to predict the coding sequences (CDS) of transcripts. Transcripts longer than 200 nucleotides (nts) were used for the long noncoding RNA (lncRNA) prediction. Four methods, Coding Potential Calculator 2 (CPC2) (Kang et al., 2017) (default parameters), Coding-Non-Coding Index (CNCI) (Sun et al., 2013) (parameters: -m pl), Coding Potential Assessment Tool (CPAT) (Wang et al., 2013) (default parameters), and PLEK (Li et al., 2014) (parameters: -minlength 200), were integrated to identify lncRNA in the transcripts and depict the intersection of the results predicted by the four methods. For the TF (transcription factor) analysis, we used DIAMOND (Buchfink et al., 2014) software to align the sequences to the AnimalTFDB (animal transcription factor database) for TFs prediction.
Simple Sequence Repeat Analysis
With the default parameters of MISA 1.0 (Beier et al., 2017), all SSRs present within the transcriptome sequences were identified and count the regional distribution of some SSRs. In the process of identification, the minimum value of repeat number varied with different repeat units per unit sizes and their minimum number of repetitions were: 1–10, 2–6, 3–5, 4–5, 5-5, and 6–5. For instance, 1–10 indicates that a single nucleotide must be repeated at least 10 times to be detected. The SSR were divided into seven types: Mono-, Di-, Tri-, Tetra-, Penta-, Hexa-, and compound SSR.
Alternative Splice Prediction
In this study, IsoSeq_AS_de_novo software (Palareti et al., 2016) with default parameters was used to perform Alternative splice analysis of the non-redundant sequences, and this software used a method that does not require reference sequences to detect AS isoforms.
Result
SMRT Sequencing Data Analysis
By using the PacBio Sequel II sequencing platform, we obtained 956,679 of polymerase read (about 112.26 Gb). In a total of 87,338,730 subreads, an average length of the subread was 1,173 bp and an N50 length of 2,347 bp. After self-correction among subreads, 660,201 CCS reads were gained in which a mean of the CCS read length was 2,416 bp. The amount of CCSs for each transcript ranges from 2 to 24,103, with an average of 9, and the average accuracy of the obtained CCS data was 0.99951. By detecting the sequences, 5,057,806 CCSs were identified as full-length reads and 495,198 were identified as FLNC reads with an average length was 2,423 bp and an N50 length of 3,057 bp. Then, 41,056 polished transcripts were obtained. Finally, the redundant reads were removed by CD-HIT and 39,209 unigenes with a mean length of 2,732 bp were obtained (Table 1). The length distribution of unigenes is as shown in Figure 1. The majority of unigenes were between 1,000 and 4,000 bp (28,643, 73.05%), and the longest unigenes was about 8,000.

TABLE 1. Description of full-length sequencing in C. antiquata.
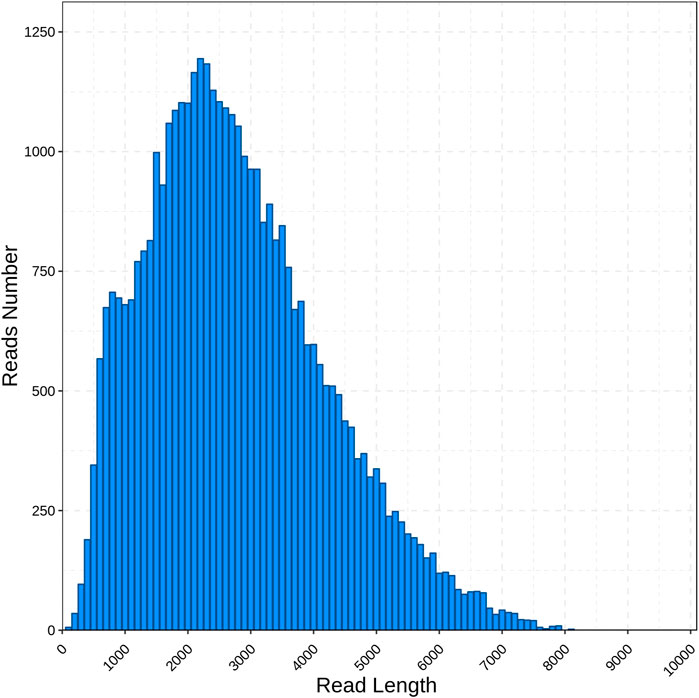
FIGURE 1. Length distribution of unigenes obtained from C. antiquata SMRT library.
Functional Annotation
In all, 39,208 transcripts were annotated in the five databases, of which 23,264 (59.33%), 13,534 (34.52%), 13,474 (34.36%), 15,840 (40.40%), and 18,711 (47.72%) transcripts were, respectively, matched to the NR, GO, KEGG, KOG, and Swiss-Prot databases (Figure 2A). A total of 8,359 (22.84%) transcripts were annotated in all the databases (Figure 2B). Aligning each transcript with the homologous sequence of the NR library, it was determined which species the sequence with the best comparison result belongs to, and count the number of homologous sequences aligned with each species. According to statistics, the species with the most homology was Mizuhopecten yessoensis (6,118 transcripts), followed by Crassostrea gigas (4,276), Crassostrea virginica (3,471), and Lottia gigantea (1,281) (Figure 2C).
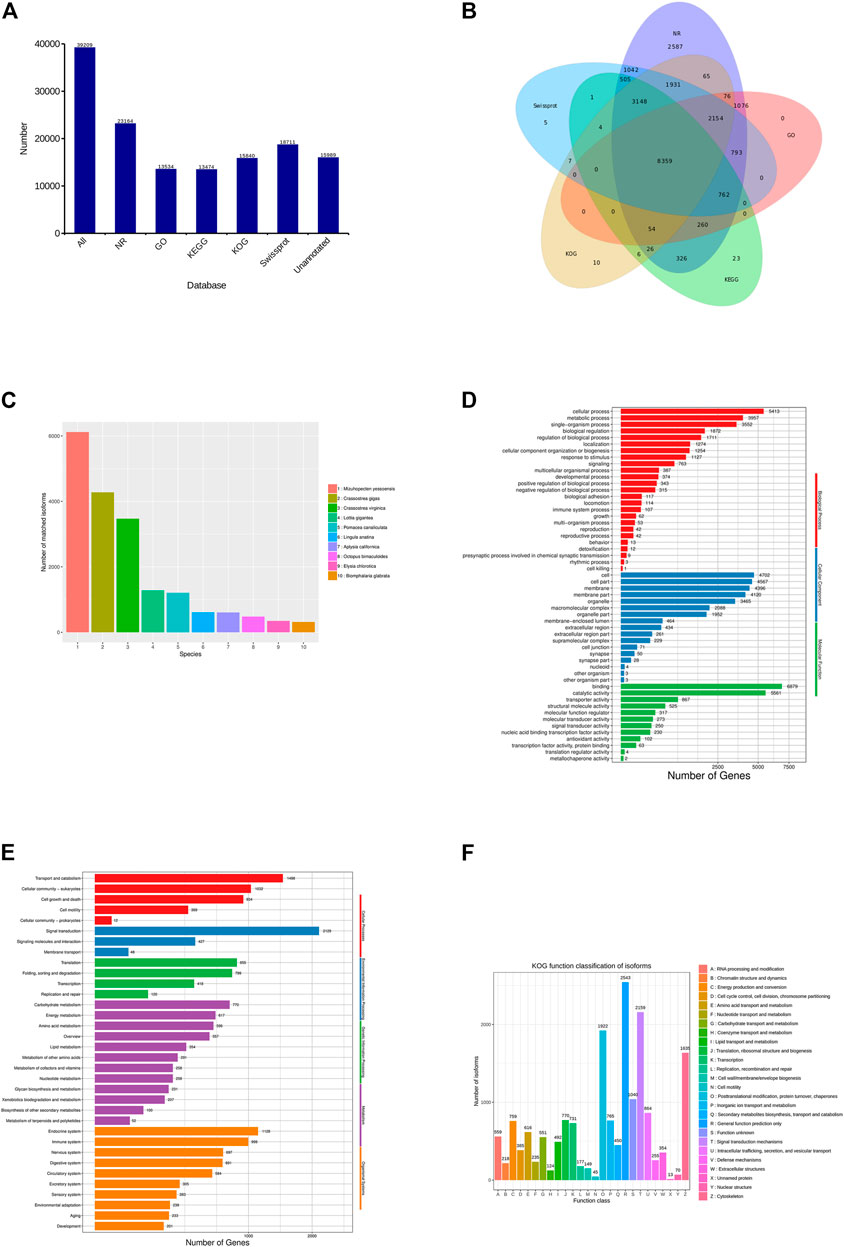
FIGURE 2. Functional annotation in C. antiquata. (A) Statistics of the transcripts annotated in different databases. (B) Venn diagram of annotations in NR, GO, KEGG, KOG, and SwissProt databases. (C) Distribution of the top 10 species with matched transcripts in the NR database. (D) Distribution of GO terms for all annotated transcripts in biological process, cellular component, and molecular function. (E) KEGG pathways enriched by transcripts. (F) COG categories of the transcripts.
According to GO classification statistics of the transcripts, the annotated results included three broad categories: Biological process (22,917 transcripts), Cellular component (26,837) and Molecular function (15,073). The Cellular process (5,413, 39.99%), Cell (4,702, 34.74%), and Binding (6,879, 50.82%) were the most annotated transcripts in the three categories mentioned above (Figure 2D).
In KEGG pathways, the transcripts were assigned to five main categories: Cellular processes (3,845 transcripts), Environmental information processing (2,604), Genetic information processing (2,192), Metabolism (4,291), and Organismal systems (5,359). Signal transduction (2,129, 15.80%) was the largest group of transcripts, followed by Transport and catabolism (1,498, 11.12%) (Figure 2E).
The KOG classifications of the transcripts obtained clusters of 26 functional categories (Figure 2F). A total of 2,543 (16.05%) transcripts were annotated in General function prediction only, which is the most among functional categories. Next was the Signal transduction mechanisms (2,159, 13.63%).
Structure Analysis
CDS Prediction
The number and length of 5′UTR, 3′UTR, and CDS were identified by transdecoder software. In total, 15,555 transcripts were predicted in the 5′UTR, 20,550 in the 3′UTR, and 23,338 in the CDS. As shown in Figure 3, most of the CDS, 19,643 transcripts (84.16%) lengths, were less than 2,000 nt, 14.13% ranged from 2,000 to 4,000 nt (3,299 transcripts), and only 396 transcripts representing 1.69% were over 4,000 nt.
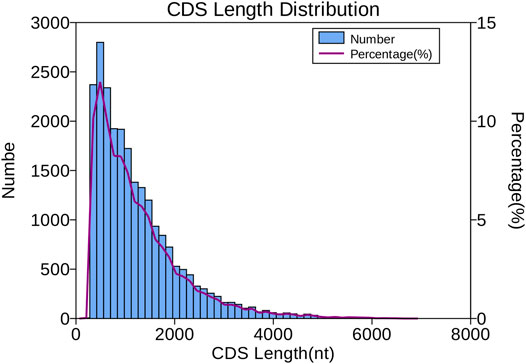
FIGURE 3. Length distribution of CDSs.
Identification of lncRNAs
We identified 14,493, 13,827, 14,027, and 11,074 lncRNAs by CNCI, CPAT, CPC2, and PLEK, respectively. The results of these four methods were integrated and 9,881 lncRNA transcripts were predicted totally (Figure 4A). By comparing the length distribution density of lncRNA and original mRNAs, it was found that there were more lncRNAs with lengths between 1,000 and 2,000 nt than mRNAs, and the longest predicted lncRNA does not exceed 8,000 nt (Figure 4B).
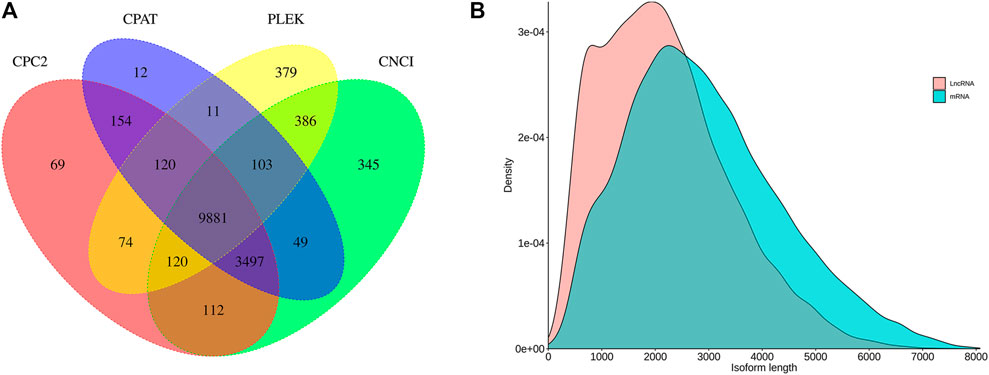
FIGURE 4. Long non-coding RNA (lncRNA) prediction. (A) Venn diagram of lncRNAs predicted by CNCI, CPAT, CPC2, and PLEK methods. (B) Length distribution of identified lncRNAs and mRNA in C. antiquata.
SSR Analysis
SSR analysis of the transcriptome revealed a total of 20,106 SSRs using MISA 1.0 software. Upon careful scrutiny of the obtained SSRs, the most predominant was the mono-nucleotide repeats (9,443), which accounted for 46.96%, followed by the di-nucleotide repeats (5,932), representing 29.50%, and tri-nucleotide repeats (3,379), accounted for 16.80%. However, tetra-nucleotide, penta-nucleotide, and hexa-nucleotide repeats accounted for a very small number, 4.92, 1.15, and 0.64%, respectively. Besides, the number of repetitions for most SSRs were 5-8 and 9–12 (Figure 5). Since some transcripts cannot predict the CDS, the total number of SSRs that can be counted in different regions was 9,804. Among the 9,804 SSRs, the number in the 3′UTR was the most (8,024), followed by the CDS (1,354), and the 5′UTR (426) was the least (Table 2).
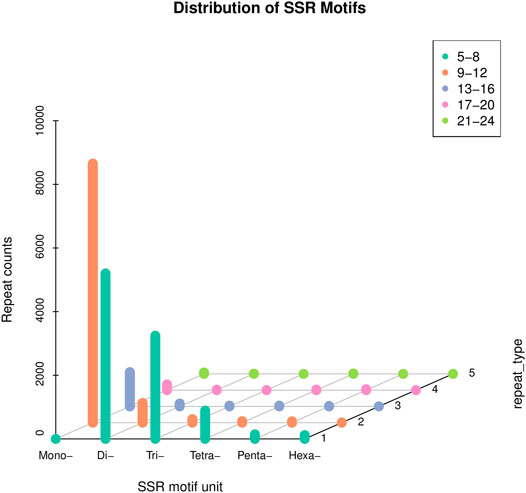
FIGURE 5. Summary of SSR types in the C. antiquata transcriptome.
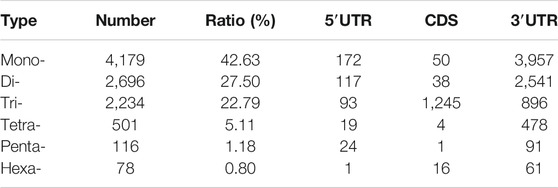
TABLE 2. Regional distribution of some SSRs in the full-length transcriptome of C. antiquata.
Transcription Factor Prediction
TF is a key factor in regulating gene expression in animals. In this study, 2,316 TFs from 59 TF families were identified by DIAMOND software. List the top 20 TF families in Figure 6, the BHLH family (369, 15.93%) was the most represented, followed by the zf-C2H2 family (278, 12.00%).
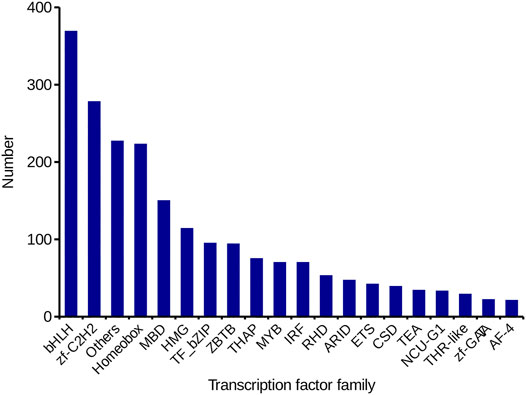
FIGURE 6. Identification of TFs inC. antiquata. The number and family of TFs were predicted by SMRT.
Alternative Splice Prediction
A total of 251 AS events were detected via the IsoSeq_AS_de_novo in all unigenes obtained by SMRT sequencing. Due to the lack of an available C. antiquata reference genome, it is necessary to further characterize the types of AS events in future studies.
Discussion
PacBio RNA-seq has fast sequencing speed, high accuracy, and long readings. Because of the advantages of PacBio RNA-seq, it has been widely used in various species research (Ma et al., 2019; Xue et al., 2019; Luo et al., 2020; Zheng et al., 2020; Chen et al., 2021). Because C. antiquata is an important economic shellfish with high medicinal potential, there are many studies now. The present study provides the first full-length transcriptome resource for C. antiquata using PacBio single-molecule long-read sequencing technology. By processing and analyzing the sequenced data, a total of 39,209 unigenes were finally identified, with an average length of 2,732 bp. In previous studies, some researchers used the Illumina platform to sequence the transcriptome, and obtained the second-generation transcriptome data of the C. antiquata (Yi et al., 2019). The second-generation sequencing identified 214,732 unigenes with a mean length of 616.2 bp. Compared with the results measured by second-generation sequencing, the total number of transcripts obtained by the SMRT sequencing technology in this study is larger and the average length is longer.
In addition, the unigenes obtained were functionally annotated in databases, and 59.22% of the unigenes were successfully annotated. The percentage is not very high, and the possible reason is that there are few studies on molecular biology of this shellfish in the past, the data collected in the database is incomplete, and the genomic information of C. antiquata has not yet been referenced. According to the NR annotation situation, Mizuhopecten yessoensis has the most homologous sequences annotated, which reflects the high affinity between C. antiquata and Mizuhopecten yessoensis. It also provides a valuable data basis for the detailed comparison of gene expression between the two species in the future. Among the function statistics against KOG, KEGG, and GO database, the number of transcripts annotated in KOG was the largest. More transcripts are involved in intracellular signal transduction and play a role in the endocrine system to participate in the metabolism of various substances, which proves that there may be many biologically active substances in C. antiquata that can be excavated and used in biomedicine.
We also analyzed the structure of the de-redundancy transcripts. A total of 20,106 SSRs and 9,881 lncRNAs were predicted. SSR is widely used in genetic diversity testing, genetic map construction, Gene expression regulation, etc. (Tranbarger et al., 2012; Chen et al., 2014). Compared with the number of SSRs and SSR types obtained by sequencing of the transcriptome of the Tegillarca granosa, the number of SSR present in C. antiquata was less, and its SSR di-nucleotide repeats were the most (H. Chen et al., 2017). These differences may be related to the different tissue specificities of the two shellfishes. Besides, this study predicted the number of TFs and the detailed family classification of C. antiquata, The BHIH family has the largest number. BHLH TFs are the most widespread category in eukaryotes, and they can participate in various processes in cells, such as regulating carbohydrate response genes (Yu et al., 2021), which may indirectly affect the synthesis of various biologically active substances.
In summary, this study successfully constructed a high-quality full-length transcriptome of C. antiquata, and preliminarily analyzed its transcriptome structure and functional characteristics, obtained the relevant annotation function of transcripts in the database, and enriched the genetic information of this species. It has laid a solid foundation for the mining and utilization of later functional genes and other molecular biology research.
Conclusion
We applied PacBio SMRT sequencing platform to obtain a large number of full-length transcriptome data of C. antiquata for the first time. The number and mean length of the unigenes from SMRT sequencing were much better than those from Illumina sequencing. And through structural analysis and functional annotation of the obtained full-length transcripts, gene function and gene structure information can be obtained more comprehensively. The acquisition of the full-length transcripts provides molecular biology data for C. antiquata, which lacks genomic information. As a species with high medicinal value, in the future, the full-length transcriptome data can be combined with the second-generation sequencing results to conduct further research on the medical effects of its internal substances.
Data Availability Statement
The datasets provided in this study can be found in the online repository: NCBI SRA database, with the accession number SRR15211944. The repository and accession number can be found at https://www.ncbi.nlm.nih.gov/sra.
Author Contributions
AD and JL are the main executives of the experimental design and experimental research of this study, completing the experimental data analysis and writing the first draft of the paper; ZY and ZC participated in some experiments; GA and YG checked and corrected the draft; JL and ZW as the project leader, instructed experimental design, data analysis, reviewed the draft of the paper and approved the final draft. All authors have read and agreed to the final text.
Funding
This work was supported by the project of Zhanjiang Agricultural and Rural Bureau under Zhancainong (2019) No. 152; Zhanjiang Municipal Bureau of Agriculture and Rural Affairs, the central governments 2019 fishery product oil price subsidy municipal-level overall funding project, research on artificial breeding and breeding technology of Coelomactra antiquata, Zhan Cainong (2019) No. 152.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a Web Server for Microsatellite Prediction. Bioinformatics (Oxford, England) 33, 2583–2585. doi:10.1093/bioinformatics/btx198
Bleidorn, C. (2016). Third Generation Sequencing: Technology and its Potential Impact on Evolutionary Biodiversity Research. Syst. Biodiversity 14 (1), 1–8. doi:10.1080/14772000.2015.1099575
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 12 (1), 59–60. doi:10.1038/nmeth.3176
Cao, M., Zhang, M., Yang, N., Fu, Q., Su, B., Zhang, X., et al. (2020). Full Length Transcriptome Profiling Reveals Novel Immune-Related Genes in Black Rockfish (Sebastes Schlegelii). Fish Shellfish Immunol. 106 (2020), 1078–1086. doi:10.1016/j.fsi.2020.09.015
Chen, H., Xiao, G., Chai, X., Lin, X., Fang, J., and Teng, S. (2017). Transcriptome Analysis of Sex-Related Genes in the Blood Clam Tegillarca Granosa. PLoS ONE 12 (9), e0184584–21. doi:10.1371/journal.pone.0184584
Chen, J., Yu, Y., Kang, K., and Zhang, D. (2020). SMRT Sequencing of the Full-Length Transcriptome of the white-backed Planthopper Sogatella Furcifera. PeerJ 8 (6), e9320. doi:10.7717/peerj.9320
Chen, Q. C. (2018b). Coelomactra Antiquata Seedling Cultivation Technology. Aquaculture 39 (03), 30–32. doi:10.3969/j.issn.1004-2091.2018.03.013
Chen, Q. C. (2018a). Cultivation and Spawning Technology of Coelomactra Antiquata Parent Shellfish. Scientific fish farming 08, 50–51. doi:10.14184/j.cnki.issn1004-843x.2018.08.029
Chen, S., Chen, W., Shen, X., Yang, Y., Qi, F., Liu, Y., et al. (2014). Analysis of the Genetic Diversity of Garlic (Allium Sativum L.) by Simple Sequence Repeat and Inter Simple Sequence Repeat Analysis and Agro-Morphological Traits. Biochem. Syst. Ecol. 55, 260–267. doi:10.1016/j.bse.2014.03.021
Chen, Y., Wu, X., Lai, J., Liu, Y., Song, M., Li, F., et al. (2021). Full-length Transcriptome Sequencing and Identification and Immune Response of TRIM Genes in Dabry's sturgeon (Acipenser Dabryanus). Aquaculture 538 (August 2020), 736599. doi:10.1016/j.aquaculture.2021.736599
De Klerk, E., Den Dunnen, J. T., and ‘t Hoen, P. A. C. (2014). RNA Sequencing: From Tag-Based Profiling to Resolving Complete Transcript Structure. Cell. Mol. Life Sci. 71 (18), 3537–3551. doi:10.1007/s00018-014-1637-9
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De Novo transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 8, 1494–1512. doi:10.1038/NPROT.2013.084
He, Z., Su, Y., and Wang, T. (2021). Full-length Transcriptome Analysis of Four Different Tissues of Cephalotaxus Oliveri. International Journal of Molecular Sciences 22 (2), 787. doi:10.3390/ijms22020787
Hon, T., Mars, K., Young, G., Tsai, Y.-C., Karalius, J. W., Landolin, J. M., et al. (2020). Highly Accurate Long-Read HiFi Sequencing Data for Five Complex Genomes. Sci. Data 7, 1. doi:10.1038/s41597-020-00743-4
Jia, X., Tang, L., Mei, X., Liu, H., Luo, H., Deng, Y., et al. (2020). Single-molecule Long-Read Sequencing of the Full-Length Transcriptome of Rhododendron Lapponicum L. Sci. Rep. 10 (1), 1–11. doi:10.1038/s41598-020-63814-x
Kang, Y.-J., Yang, D.-C., Kong, L., Hou, M., Meng, Y.-Q., Wei, L., et al. (2017). CPC2: a Fast and Accurate Coding Potential Calculator Based on Sequence Intrinsic Features. Nucleic Acids Res. 45, W12–W16. doi:10.1093/nar/gkx428
Kong, L., and Li, Q. (2007). Genetic Comparison of Cultured and Wild Populations of the Clam Coelomactra Antiquata (Spengler) in China Using AFLP Markers. Aquaculture 271 (1–4), 152–161. doi:10.1016/j.aquaculture.2007.06.007
Kong, L., and Li, Q. (2009). Genetic Evidence for the Existence of Cryptic Species in an Endangered Clam Coelomactra Antiquata. Mar. Biol. 156 (7), 1507–1515. doi:10.1007/s00227-009-1190-5
Kong, L., Li, Q., and Qiu, Z. (2007). Genetic and Morphological Differentiation in the Clam Coelomactra Antiquata (Bivalvia: Veneroida) along the Coast of China. J. Exp. Mar. Biol. Ecol. 343 (1), 110–117. doi:10.1016/j.jembe.2006.12.003
Li, A., Zhang, J., and Zhou, Z. (2014). PLEK: a Tool for Predicting Long Non-coding RNAs and Messenger RNAs Based on an Improved K-Mer Scheme. BMC bioinformatics 15. doi:10.1186/1471-2105-15-311
Li, J.-J., Li, Q., and Kong, L.-F. (2008). Isolation and Characterization of Microsatellite Loci in the Xishishe Clam Coelomactra Antiquata (Bivalvia: Veneroida). Conserv Genet. 9 (2), 453–455. doi:10.1007/s10592-007-9329-8
Li, W., and Godzik, A. (2006). Cd-hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 22 (13), 1658–1659. doi:10.1093/bioinformatics/btl158
Liu, F., Lu, Y., Hao, Z., Mou, Z., Guo, J., Kong, X., et al. (2012). Artificial Propagation Technology of Coelomactra Antiquata in Rizhao Coast. Hebei Fish. 04, 29–30. doi:10.3969/j.issn.1004-6755.2012.04.011
Liu, H., Zhu, J. X., Sun, H. L., Fang, J. G., Gao, R. C., and Dong, S. L. (2006). The Clam, Xishi Tongue Coelomactra Antiquata (Spengler), a Promising New Candidate for Aquaculture in China. Aquaculture 255 (1–4), 402–409. doi:10.1016/j.aquaculture.2005.12.027
Luo, W., Wu, Q., Wang, T., Xu, Z., Wang, D., Wang, Y., et al. (2020). Full-length Transcriptome Analysis of Misgurnus anguillicaudatus. Mar. Genomics 54 (January), 100785. doi:10.1016/j.margen.2020.100785
Ma, J.-E., Jiang, H.-Y., Li, L.-M., Zhang, X.-J., Li, H.-M., Li, G.-Y., et al. (2019). SMRT Sequencing of the Full-Length Transcriptome of the Sunda Pangolin (Manis Javanica). Gene 692 (January), 208–216. doi:10.1016/j.gene.2019.01.008
Meng, X., Shen, X., Zhao, N., Tian, M., Liang, M., Hao, J., et al. (2013). Mitogenomics Reveals Two Subspecies inCoelomactra antiquata(Mollusca: Bivalvia). Mitochondrial DNA 24 (2), 102–104. doi:10.3109/19401736.2012.726620
Meng, X., Zhao, N., Shen, X., Hao, J., Liang, M., Zhu, X., et al. (2012). Complete Mitochondrial Genome of Coelomactra Antiquata (Mollusca: Bivalvia): the First Representative from the Family Mactridae with Novel Gene Order and Unusual Tandem Repeats. Comp. Biochem. Physiol. D: Genomics Proteomics 7 (2), 175–179. doi:10.1016/j.cbd.2012.02.001
Nguyen, N.-p., Nute, M., Mirarab, S., and Warnow, T. (2016). HIPPI: Highly Accurate Protein Family Classification with Ensembles of HMMs. BMC Genomics 17 (Suppl. 10), 89–100. doi:10.1186/s12864-016-3097-0
Palareti, G., Legnani, C., Cosmi, B., Antonucci, E., Erba, N., Poli, D., et al. (2016). Comparison between Different D - D Imer Cutoff Values to Assess the Individual Risk of Recurrent Venous Thromboembolism: Analysis of Results Obtained in the DULCIS Study. Int. Jnl. Lab. Hem. 38 (1), 42–49. doi:10.1111/ijlh.12426
Schadt, E. E., Turner, S., and Kasarskis, A. (2010). A Window into Third-Generation Sequencing. Hum. Mol. Genet. 19 (R2), R227–R240. doi:10.1093/hmg/ddq416
Shen, X., Meng, X., Tian, M., Yan, B., Cheng, H., Lu, W., et al. (2016). The First Mitochondrial Genome of Coelomactra Antiquata (Mollusca: Veneroida: Mactridae) from Guangxi (China) and Potential Molecular Markers. Mitochondrial DNA A 27 (5), 3642–3643. doi:10.3109/19401736.2015.1079835
Sun, L., Luo, H., Bu, D., Zhao, G., Yu, K., Zhang, C., et al. (2013). Utilizing Sequence Intrinsic Composition to Classify Protein-Coding and Long Non-coding Transcripts. Nucleic Acids Res. 41, e166. doi:10.1093/nar/gkt646
Tranbarger, T., Kluabmongkol, W., Sangsrakru, D., Morcillo, F., Tregear, J. W., Tragoonrung, S., et al. (2012). SSR Markers in Transcripts of Genes Linked to post-transcriptional and Transcriptional Regulatory Functions during Vegetative and Reproductive Development of Elaeis Guineensis. BMC Plant Biol. 12 (January), 1. doi:10.1186/1471-2229-12-1
Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J.-P., and Li, W. (2013). CPAT: Coding-Potential Assessment Tool Using an Alignment-free Logistic Regression Model. Nucleic Acids Res. 41, e74. doi:10.1093/nar/gkt006
Wen, Y. M., Wan, D. J., Xie, Y. H., Luo, C. L., and Cheng, J. (2015). Influence of Surf Clam Shell on Blood Lipid and Antioxidant Activity of Diabetic Mice. West. Traditional Chin. Med. 28 (10), 19–21. doi:10.3969/j.issn.1004-6852.2015.10.006
Wu, J., Guo, X., Liu, H., and Chen, L. (2019). Isolation and Comparative Study on the Characterization of Guanidine Hydrochloride Soluble Collagen and Pepsin Soluble Collagen from the Body of Surf Clam Shell (Coelomactra Antiquata). Foods 8 (1), 11. doi:10.3390/foods8010011
Xue, T., Zhang, H., Zhang, Y., Wei, S., Chao, Q., Zhu, Y., et al. (2019). Full-length Transcriptome Analysis of Shade-Induced Promotion of Tuber Production in Pinellia Ternata. BMC Plant Biol. 19 (1), 1–13. doi:10.1186/s12870-019-2197-9
Yang, W. Q., Wen, Y. M., Lin, W. D., Xie, Y. H., and Chen, C. M. (2015). Effect of Polysaccharides from Coelomactra Antiquata on Human Carcinoma of Esophagus Cells Transplanted in Nude Mice. Nat. Product. Res. Development 27 (08), 1402–1406. doi:10.16333/j.1001-6880.2015.08.016
Yi, L., Ma, K. Y., Qin, J., Chu, K. H., Shen, X., and Meng, X. (2019). Insights into Cryptic Diversity and Adaptive Evolution of the Clam Coelomactra Antiquata (Spengler, 1802) from Comparative Transcriptomics. Mar. Biodiv 49 (5), 2311–2322. doi:10.1007/s12526-019-00964-w
Yu, J. Q., Gu, K. D., Sun, C. H., Zhang, Q. Y., Wang, J. H., Ma, F. F., et al. (2021). The Apple bHLH Transcription Factor MdbHLH3 Functions in Determining the Fruit Carbohydrates and Malate. Plant Biotechnol. J. 19 (2), 285–299. doi:10.1111/pbi.13461
Keywords: Coelomactra antiquata, full-length transcriptome, SMRT sequencing, RNA-seq, function annotation
Citation: Deng A, Li J, Yao Z, Afriyie G, Chen Z, Guo Y, Luo J and Wang Z (2021) SMRT Sequencing of the Full-Length Transcriptome of the Coelomactra antiquata. Front. Genet. 12:741243. doi: 10.3389/fgene.2021.741243
Received: 14 July 2021; Accepted: 24 September 2021;
Published: 14 October 2021.
Edited by:
Youri I Pavlov, University of Nebraska Medical Center, United StatesReviewed by:
Natalie Saini, Medical University of South Carolina, United StatesLefei Yi, Jiangsu Ocean Universiity, China
Copyright © 2021 Deng, Li, Yao, Afriyie, Chen, Guo, Luo and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Luo, bHVvakBnZG91LmVkdS5jbg==; Zhongduo Wang, d2FuZ3pkQGdkb3UuZWR1LmNu
†These authors have contributed equally to this work