Abstract
Inherited retinal diseases (IRDs) are a heterogenous group of orphan eye diseases that typically result from monogenic mutations and are considered attractive targets for gene-based therapeutics. Following the approval of an IRD gene replacement therapy for Leber’s congenital amaurosis due to RPE65 mutations, there has been an intensive international research effort to identify the optimal gene therapy approaches for a range of IRDs and many are now undergoing clinical trials. In this review we explore therapeutic challenges posed by IRDs and review current and future approaches that may be applicable to different subsets of IRD mutations. Emphasis is placed on five distinct approaches to gene-based therapy that have potential to treat the full spectrum of IRDs: 1) gene replacement using adeno-associated virus (AAV) and nonviral delivery vectors, 2) genome editing via the CRISPR/Cas9 system, 3) RNA editing by endogenous and exogenous ADAR, 4) mRNA targeting with antisense oligonucleotides for gene knockdown and splicing modification, and 5) optogenetic approaches that aim to replace the function of native retinal photoreceptors by engineering other retinal cell types to become capable of phototransduction.
Introduction
Improvements to global living standards and increased life expectancies over the past century have led to a transition in medical research to noninfectious diseases (COVID19 notwithstanding) and disabilities that impair quality of life. Visual impairment represents a special disability that has profound impact on a person’s ability to interact with the world and has been identified as an area of focus by the World Health Organization (Pizzarello et al., 2004; Bourne, 2020). Recent epidemiological analyses indicate that while major causes of blindness such as cataract, undercorrected refractive error, age-related macular degeneration, glaucoma, and diabetic retinopathy are starting to decline in prevalence, these conditions predominantly affect older age groups (GBD 2019 Blindness and Vision Impairment Collaborators and Vision Loss Expert Group of the Global Burden of Disease Study, 2021). In contrast, blindness among working age adults, which was previously due mainly to diabetic retinopathy in developed nations, is now predominantly due to a very large and heterogenous group of blinding conditions collectively termed inherited retinal diseases (IRDs) (Liew et al., 2014; Heath Jeffery et al., 2021). IRDs have, until very recently, remained essentially untreatable.
IRDs are a heterogeneous group of orphan eye diseases with a prevalence estimated at between 0.06 and 0.2% (Galvin et al., 2020; Gong et al., 2021), with a global IRD caseload in the range of 5–10 million individuals (Hanany et al., 2020). Onset is highly variable, ranging from the first year of life all the way to late adulthood, and presenting symptoms can vary from nyctalopia and photosensitivity to profound vision loss with early-onset nystagmus. The societal burden of IRDs has been quantified in recent studies that investigated the socioeconomic impact of IRDs in the US and western Europe. Despite their scarcity, the societal cost of IRDs in the United Kingdom and United States were estimated at more than USD 700 million and USD 30 billion, respectively (Galvin et al., 2020; Gong et al., 2021). Much of this cost is presumably due to the profound impact that IRDs have on the economic productivity of working-age individuals, where improvements in community eye screening has seen IRDs overtake diabetic retinopathy as the leading cause of blindness among working age adults in some developed nations (Hanany et al., 2020; Heath Jeffery et al., 2021). Among Asian populations the spectrum of IRDs is similarly diverse, although IRD genotypes differ considerably from that in Western populations (Sen et al., 2008; Oishi et al., 2014; Arai et al., 2015; Wang et al., 2018; Yohe et al., 2020; Ma et al., 2021).
The phenotypic heterogeneity of IRDs is brought about by the diverse array of genotypes responsible for IRDs. Presently there are at least 270 discrete genetic loci responsible for IRDs in humans (RetNet www.sph.uth.edu/retnet), each of which may harbor many disease-causing variants with distinct clinical phenotypes, resulting in an almost overwhelming number of IRDs that can be encountered by an ophthalmologist. Despite this, the majority of IRDs can be classified as one of four broad subtypes: 1) rod-cone degenerations; 2) cone-rod degenerations; 3) chorioretinal degenerations; and 4) degenerations involving the macula, with the latter subtype often overlapping with the former subtypes. The clinical presentation of each subtype is usually consistent with the retinal cell types affected. Rod-cone degenerations, the most common of the IRDs, present with nyctalopia and peripheral visual field loss earlier in disease due to preferential degeneration of light-sensitive rods, followed by loss of central visual acuity and color vision later in the disease as cones become affected. Conversely, patients with cone-rod disease will initially present with reduced color vision, photophobia, or reduced visual acuity. Chorioretinal degenerations have variable presentation but typically present with nyctalopia and peripheral field loss with progression to central vision loss later in life. Degenerations involving the macula usually result in symptoms of metamorphopsia, reduced visual acuity, and progressive central and paracentral scotomata, while the peripheral vision is typically spared.
In addition to their ocular manifestations, many IRDs are associated with systemic disease, with the most well-known being Usher syndrome, an important cause of childhood sensorineural deafness (Saihan et al., 2009; Fuster-García et al., 2021). These cases are typically identified by internists and subsequently brought to attention of the ophthalmologist via routine referral for screening, but the ocular manifestations are occasionally the presenting complaint (Ehrenberg et al., 2019; Tatour and Ben-Yosef, 2020).
Progress in our understanding of IRDs at the genetic, biochemical, and cellular level was historically limited by their previously incurable nature. While many researchers spent decades assembling IRD cohorts and publishing phenotypic and genotypic findings for newly characterized IRDs, this was largely an academic pursuit. However, with the advent of high-throughput gene sequencing there has been a dramatic increase the rate of IRD gene discovery (Gao et al., 2019; Koyanagi et al., 2019; Holtan et al., 2020; Pontikos et al., 2020; Weisschuh et al., 2020; Qian et al., 2021). More recently, the pace of IRD research has further increased following the first FDA approval of gene therapy for an IRD (Russell et al., 2017).
In the current work, we review recent developments in our understanding of IRDs and their clinical outcomes before providing an outline of recent approaches taken to treat this important group in blinding retinal diseases.
Pathophysiology of Inherited Retinal Diseases
In rod-cone dystrophies (RCD), or retinitis pigmentosa (RP), patients present with nyctalopia (night blindness), usually within the first or second decade of life. Presentation may be delayed depending on the environment, with patients living in well-lit urban settings often presenting later (Hartong et al., 2006). The second hallmark of RP is gradual, and often insidious, progressive loss of the peripheral visual field. This is seldom noticed by patients until it begins to encroach on the central vision. Field loss in RP patients has been reported to occur at a rate of approximately 5–10% per year (Berson et al., 1985), or 50% over 5 years (Massof et al., 1984). Importantly, in RP, like most IRDs, vision loss is generally symmetrical between two eyes. Central visual acuity can be affected early in the course of disease due to macular edema, which is to some extent treatable (Huckfeldt and Comander, 2017; Bakthavatchalam et al., 2018), or later in the disease as a result of cone involvement with loss of the photoreceptor-containing ellipsoid zone (EZ; Figure 1 and Figure 2). The presence of lyonization among female carriers of X-linked RP can cause asymmetrical disease expression between eyes, making diagnosis more challenging (Wuthisiri et al., 2013; Fahim et al., 2020).
FIGURE 1
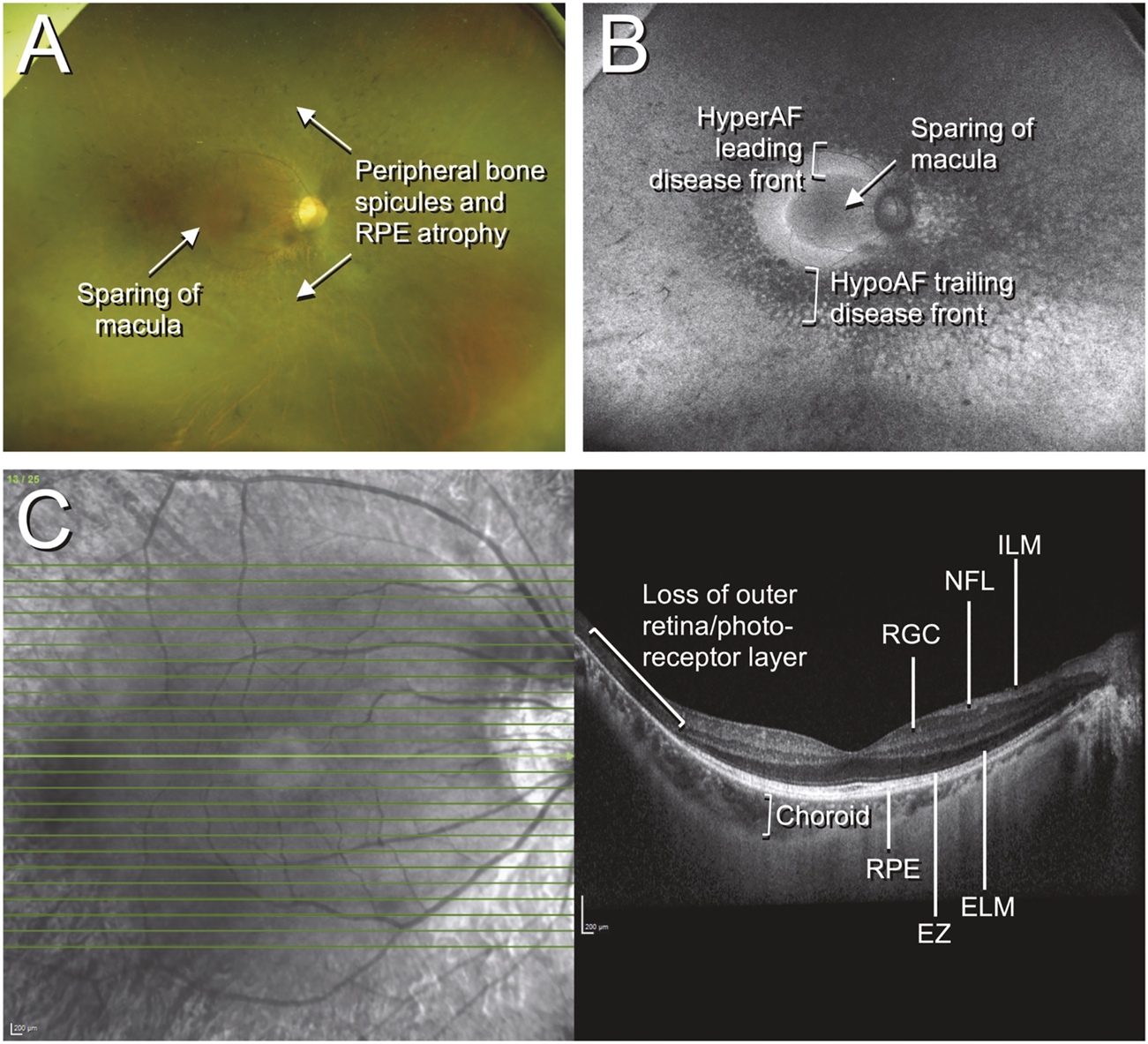
Color fundus photographs, autofluorescence, and optical coherence tomography (OCT) of retinitis pigmentosa. The patient was a 42-year-old Chinese male who presented with an incidental finding of peripheral field loss during an inpatient admission for migraine headache. A long-standing history of nyctalopia was elicited but there was no evidence of neurosensory hearing loss. Whole exome sequencing (WES) uncovered the presence of biallelic pathogenic EYS mutations. (A) Fundus imaging shows peripheral bone spicule-like pigmentary retinopathy with outer retinal atrophy and sparing of the macula; (B) fundus autofluorescence highlights the areas of disease, with the hypoautofluorescent (hypoAF) trailing disease front appearing as dark areas and the hyperautofluorescent (hyperAF) leading disease front appearing as brighter areas; (C) horizontal cross-sectional OCT at the level of the fovea shows intact retinal layers at the fovea with loss of the outer retina and photoreceptor layers in the periphery. Selected retinal layers relevant to gene-based therapy are shown: ILM, internal limiting membrane; NFL, nerve fiber layer; RGC, retinal ganglion cell layer; RPE, retinal pigment epithelium; EZ, ellipsoid zone (photoreceptor inner/outer segments); ELM, external limiting membrane.
FIGURE 2
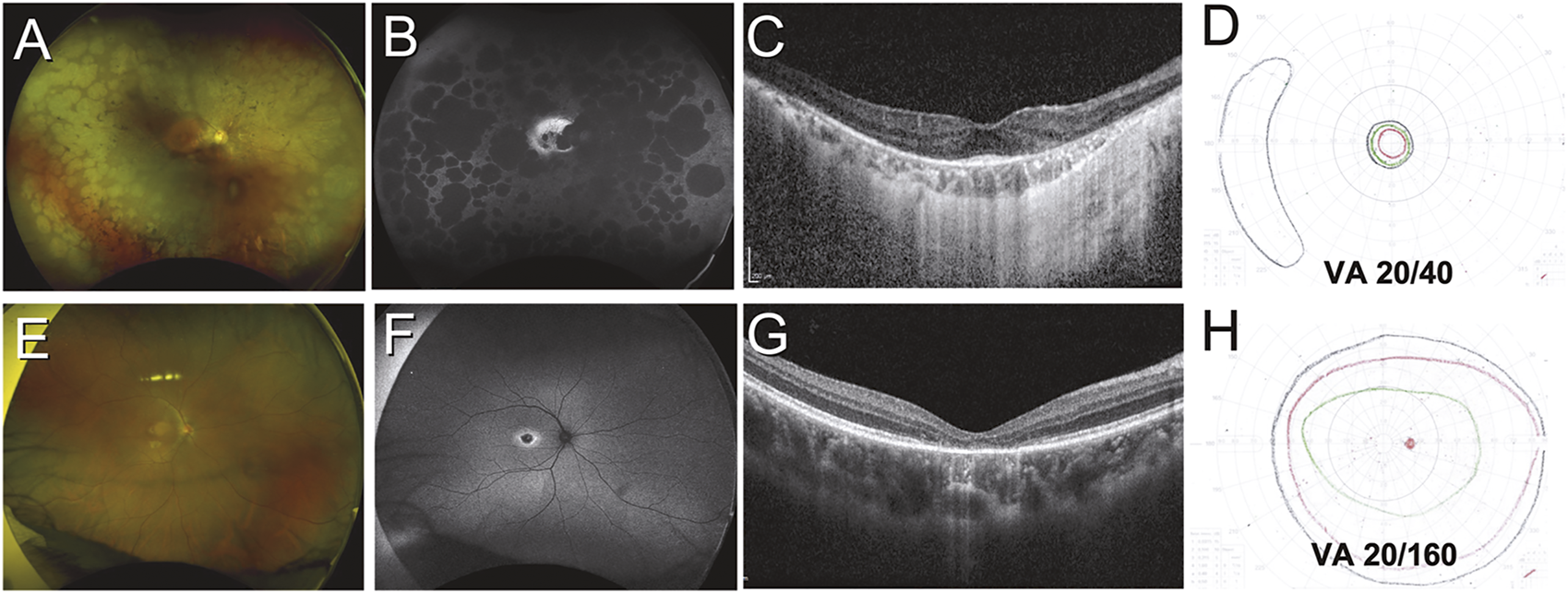
Variation in structural and functional changes due to inherited retinal disease. Case 1 (A–D) was a 51-year-old Indonesian male with retinitis pigmentosa secondary to PRPH2 mutation. Retinal findings include diffuse peripheral RPE atrophy seen on color and autofluorescent imaging (A,B) with sparing of the central macula (B) and a small focus of foveal outer retina intact on OCT imaging (C). Visual acuity was only mildly impaired, but the patient had tunnel vision as demonstrated by Goldmann kinetic perimetry (D). Case 2 (E–H) was a 21-year-old Chinese male with cone dystrophy secondary to PROM1 mutation. Retinal findings are limited to subtle pigmentary changes at the macula (E) which are highlighted on autofluorescent imaging (F), while OCT reveals loss of the outer retina at the foveal region (G). Visual fields are largely preserved but the patient had severely impaired visual acuity (H).
In contrast to RCDs, patients with cone-rod dystrophies (CRDs) present with essentially the opposite sequence of symptoms. Visual acuity, impaired color vision, and photosensitivity occur first, followed later by visual field loss and nyctalopia (Figure 2). Predictably, CRDs result in more severe visual impairment earlier on in the course of the disease, although the end-point for both RCDs and CRDs is essentially the same (Hamel, 2007).
Both RCDs and CRDs appear to result in primary defects in rod and cone photoreceptors and animal models have been used to demonstrate that multiple points along the pathways of photoreceptor anabolism and catabolism can give rise to the phenotypes (Roosing et al., 2014; Sahel and Léveillard, 2018; Verbakel et al., 2018).
Inherited chorioretinal degenerations are a group of diseases characterized by early degeneration of the retinal pigment epithelium (RPE) that progresses to involve the choriocapillaris, Haller and Sattler layers of the choroid, and the photoreceptors in the later stages. The presentation of this subgroup of IRDs varies depending on the disease and may involve loss of central vision such as in the case of central areolar choroidal dystrophy (Boon et al., 2009) or progressive loss of peripheral vision in gyrate atrophy and choroideremia (Dimopoulos et al., 2017; Balfoort et al., 2021).
IRDs involving the macula, including Stargardt disease, Best disease, and pattern dystrophies among others, are more clinically heterogeneous than the preceding three subtypes and their diagnosis is usually more challenging because of their overlap with a multitude of acquired diseases. All share the clinical features of disease that is largely confined to the macula or posterior pole with symptoms affecting central vision, though the reasons for their predilection for the macula remains unclear. Current research suggests that differences in the structure of the choroid, Bruch’s membrane, RPE, photoreceptors, light exposure, and localized gene expression patterns may each play a part (Johnson et al., 2017; Tsang and Sharma, 2018).
Measuring Outcomes in Inherited Retinal Diseases Therapy Trials
Before discussing specific gene-based therapeutic approaches for IRDs, it is instructive to review the approaches now being taken in clinical trials to assess the structural and functional outcomes of IRD treatments. Outcomes for common retinal diseases such as neovascular age-related macular degeneration (AMD) and diabetic macular edema (DME) have been relatively straightforward to assess, as the disease progresses on a time scale of months and treatment effects, such as that due to intravitreal anti-vascular endothelial growth factor (anti-VEGF), similarly take place over months or even weeks (Wells et al., 2016; Mitchell et al., 2018). Moreover, the dramatic functional and structural improvements seen after anti-VEGF therapy has meant that changes in visual acuity (measured in LogMAR letters) and direct imaging of the macula by optical coherence tomography (OCT) are typically sufficient endpoints for clinical trials of new drugs (Rofagha et al., 2013; Wong et al., 2016). Inherited retinal diseases, on the other hand, are far more insidious and cause vision loss over years and decades, with vision loss often being confined to the peripheral vision or loss of color vision, night vision, or contrast sensitivity in the early stages. Similarly, the structural changes cause by IRDs can initially be very subtle at disease onset, with easily quantifiable changes taking years to develop. These differences have made the assessment of clinical outcomes for IRD therapies particularly challenging, especially given the heavy financial investment and time constraints of for-profit pharmaceutical ventures. To address these limitations, clinical trials for IRDs often require multiple specialized functional and structural assessments to demonstrate treatment efficacy and these are briefly discussed here.
Functional Assessment in Inherited Retinal Disease Treatment Trials
The best-corrected visual acuity (BCVA) achieved following manifest refraction, and the extent of a test subject’s visual field assessed with static or kinetic perimetry (Figure 2) remain clinical gold standards for functional outcome in IRD treatment trials. The selective loss of rod and/or cone function in many IRDs has prompted the development of specialized perimetry that can preferentially assess each cell type. In standard perimetry a white background is used with presentation to the subject of stimuli of increasing size and luminance to map the visual field. This approach predominantly tests long- and mid-wavelength-sensitive cones (LWC/MWS cones), while chromatic perimetry with monochromatic light has been used to highlight field deficits in cone diseases including achromatopsia and blue cone monochromacy (McGuigan et al., 2016; Bennett et al., 2017). Standard perimetry has been employed in several trials of IRD therapeutics (Schwartz et al., 2015; Maguire et al., 2019), with loss of central or peripheral field in treated eyes being compared with untreated eyes as a primary or secondary outcome. Although disease progression is slow, there is a demonstrable loss of field over a 1–2 years period in common forms of RP including USH2A disease (Xu et al., 2020).
In the majority of IRDs, including RCDs and CRDs, visual field assessment by perimetry is often not feasible as patients progress to perimetric blindness, tunnel vision, or have inadequate fixation for reliable assessment. This is especially true of the early-onset RCDs like LCA. To overcome this problem, clinical trials for IRDs have employed the full-field stimulus test (FST), which can evaluate visual function in patients with severe vision loss (Roman et al., 2021). FST measures the sensitivity of the entire visual field by providing an estimation of the lowest level of luminance that elicits a visual sensation by the subject. The FST presents stimuli of varying luminance according to a prespecified algorithm and the patient presses a button when a visual sensation is perceived. The FST instrument can additionally present varying colors to preferentially test different photoreceptor subsets, or be undertaken following dark adaptation to distinguish between cone and rod deficits (Roman et al., 2021; Tuohy and Megaw, 2021).
The transient pupillary light reflex (TPLR) is another objective measure of retinal function that remains intact even in severe and advanced IRDs (Cideciyan et al., 2021b). This response is typically measured with a high frame rate infrared video camera and specialized recording software under controlled lighting conditions with adjustable light stimuli (Kawasaki et al., 2012; Rukmini et al., 2019). Patients with advanced IRDs that affect both rods and cones display a TPLR reduction of more than 5 log units, offering excellent dynamic range (Rukmini et al., 2019; Cideciyan et al., 2021b) and showed treatment-dependent improvement in phase 1 trials of RPE65 gene replacement therapy in LCA patients (Maguire et al., 2009).
Beyond specialized physiological testing, the need to demonstrate practical benefits for patients undergoing gene-based IRD therapies has driven the development of validated navigational assessment tools. The multi-luminance mobility test (MLMT), originally developed for use in an RPE65 gene therapy trial (see below), requires patients to navigate a short obstacle course that simulates daily walking environments (Chung et al., 2018). MLMT scores are based on accuracy and speed, with a pass defined as completion of the course at a specified background luminance with less than four errors and within 3 mins (Russell et al., 2017; Chung et al., 2018).
Structural Assessment in Inherited Retinal Diseases Treatment Trials
Contemporary structural assessment of the retina in IRDs is centred around the use of two core technologies: optical coherence tomography (OCT), and fundus autofluorescence (FAF). OCT is a non-invasive imaging modality that uses low coherence interferometry to quantify the intensity of light reflected from different structures within the retina. Current high resolution OCT, including spectral domain (SD-OCT) and swept-source (SS-OCT) instruments enable near-histology level resolution of retinal layers and enables both qualitative and quantitative assessment of outer retinal atrophy and photoreceptor loss in IRDs (Georgiou et al., 2020) (Figure 1).
Progressive outer retinal atrophy is a hallmark of the majority of IRDs and this can be readily visualized using FAF imaging. In conditions of high metabolic stress resulting from photoreceptor degeneration, there is an accumulation of fluorophores within the RPE, the predominant species being lipofuscin. FAF imaging enables visualization of these fluorophores as a bright hyperautofluorescent signal (Figure 1). Eventually the RPE and photoreceptors undergo apoptosis, and this signal is lost (Spaide, 2003; Sparrow and Boulton, 2005; Ach et al., 2015), leading to hypoautofluorescence (Figure 1). IRDs often display typical patterns on FAF imaging that are helpful in diagnosis, but the progression of the hyperautofluorescent signal (leading disease front), and the hypoautofluorescent signal (trailing disease front) can also be used to assess the rate of disease progression (Cicinelli et al., 2020; Daich Varela et al., 2021).
Adaptive optics (AO) is a novel imaging modality that measures wavefront aberrations from the retina to produce images of retinal cell layers with extremely high spatial resolution, sufficient to visualize individual photoreceptor cells (Akyol et al., 2021). This technique is non-invasive and has been used to characterize unique morphological changes that occur in various IRDs (Georgiou et al., 2018; Gill et al., 2019). It is expected that AO will become important for patient selection and disease monitoring in future clinical trials of IRD therapeutics.
Approaches to Inherited Retinal Diseases Treatment
Over the past two to three decades numerous research groups worldwide have assembled and meticulously characterized the phenotypes and genotypes of large cohorts of IRD patients, with the largest reported cohorts numbering in the thousands (Gao et al., 2019; Pontikos et al., 2020; Sharon et al., 2020; Weisschuh et al., 2020; Qian et al., 2021). This has been accompanied by the development of cell and animal models of IRDs that have greatly improved our understanding of the molecular events that lead to disease expression for numerous gene variants. This, combined with the uniquely favorable features of the human eye, including ease of access, immune-privileged status, and the robust suite of noninvasive functional and structural investigations currently available, has resulted in IRDs being perceived as one of a limited number of ideal targets for the nascent field of gene-based therapy (Trapani and Auricchio, 2019; Hu et al., 2021). Despite this, the treatment of IRDs with targeted gene therapies presents numerous challenges to both the clinician and basic scientist. In the sections below we will review these challenges and provide examples of treatment approaches that aim to address each of them and lead to effective therapeutic interventions that will provide clinically meaningful benefits to IRD patients.
Spectrum of Genes in Inherited Retinal Diseases
To date approximately 270 unique genes have been identified to be responsible for IRDs, with loci spanning all autosomes and both sex chromosomes. Despite this diversity and the widespread availability of high-throughput DNA sequencing, current cohort studies of IRD patients typically achieve only 50–70% hit rates in genotyping analysis (Van Schil et al., 2018), indicating that a substantial number of unidentified variants are responsible for the global IRD burden. Among known IRD genotypes, only a small number of genes are individually responsible for more than 1% of IRD cases in most published cohorts (Table 1). Among these, variants within ABCA4 (9.3–20.8%), EYS (0.6–23.5%), USH2A (0.6–9.1%), PRPH2 (0.4–4.6%), RHO (0.5–3.4%), and RPGR (1.2–5.7%) account for up to 50% of all successfully genotyped IRD cases across multiple ethnic cohorts (Table 1). Less common but responsible for one of the more severe IRD phenotypes seen in clinical practice is RPE65, whose mutation can cause an early onset RCD termed Leber congenital amaurosis (LCA). Inheritance of IRD genes can follow both recessive and dominant patterns (Daiger et al., 2014) and the size of the responsible genes varies enormously. This variability has important implications for gene-based therapies for IRDs.
TABLE 1
| Contribution of selected gene mutations to IRD burden in study cohort | ||||||||
|---|---|---|---|---|---|---|---|---|
| Affected genes | Retinal phenotype | Germany (n = 1785) | Israel (n = 2,420) | Japan (n = 349) | Taiwan (n = 312) | United Kingdom (n = 3,195) | United States (n = 1,000) | Range |
| Weisschuh et al. (2020) | Sharon et al. (2020) | Arai et al. (2015) | Chen et al. (2021) | Pontikos et al. (2020) | Stone et al. (2017) | |||
| Study diagnostic rate (%) | 70.8 | 56.0 | 45.6 | 57.1 | N/A | 76.0 | ||
| ABCA4 | Stargardt disease, CRD | 10.5 | 11.5 | 0.9 | 9.3 | 20.8 | 17.3 | 0.9–20.8 |
| BEST1 | Best disease | 1.3 | 1.0 | 0 | 1.3 | 3.9 | 2.5 | 0–3.9 |
| CEP290 | LCA, RP | 0.06 | 0.2 | 0 | 2.2 | 0.8 | 1.8 | 0–1.8 |
| CHM | Choroideremia | 2.3 | 0.7 | 0 | 0.6 | 2.7 | 1.4 | 0–2.7 |
| CRB1 | LCA, RP | 1.4 | 1.3 | 1.4 | 1.3 | 2.1 | 2.0 | 1.3–2.1 |
| CYP4V2 | Bietti disease, RCD | 0.1 | 0.2 | 2.0 | 3.8 | 0.6 | 0.0 | 0–3.8 |
| EYS | RP | 1.8 | 2.6 | 23.5 | 7.4 | 1.2 | 0.6 | 0.3–23.5 |
| GUCY2D | LCA, RP, CRD | 0.4 | 1.4 | 0.6 | 1.0 | 1.2 | 0.4 | 0.4–1.4 |
| PROM1 | Macular dystrophy, CRD, RP | 1.2 | 0.1 | 1.2 | 1.6 | 1.2 | 0.6 | 0.1–1.6 |
| PRPF31 | RP | 2.9 | 0.4 | 0 | 2.2 | 1.8 | 1.5 | 0–2.9 |
| PRPH2 | Pattern dystrophy, RP | 2.7 | 0.7 | 4.6 | 1.0 | 4.6 | 3.2 | 0.7–4.6 |
| RDH12 | LCA, RP | 0.4 | 1.3 | 0 | 1.0 | 1.1 | 0.6 | 0–1.3 |
| RHO | RP, stationary night blindness | 3.1 | 0.5 | 2.0 | 1.0 | 3.3 | 3.4 | 0.5–3.4 |
| RLBP1 | Retinitis punctata albescens | 0.2 | 0.2 | 0.3 | 1.9 | 0.2 | 0.1 | 0.1–1.9 |
| RP1 | RP | 1.0 | 0.3 | 0.3 | 1.3 | 3.3 | 1.0 | 0.3–3.3 |
| RPE65 | LCA, RP | 0.3 | 0.8 | 0 | 1.6 | 1.2 | 0.3 | 0–1.6 |
| RPGR | RP, CRD, cone dystrophy | 5.7 | 1.6 | 1.2 | 2.6 | 5.1 | 4.8 | 1.6–5.7 |
| RS1 | X-linked retinoschisis | 1.0 | 0.4 | 0 | 1.0 | 3.3 | 1.3 | 0–3.3 |
| USH2A | RP | 8.5 | 5.5 | 0.6 | 7.0 | 9.1 | 7.6 | 0.6–9.1 |
Contributions of selected genes to the IRD burden among different regional cohorts.
Among more than 300 inherited retinal disease entities caused by variations within more than 270 genes, approximately 70% are inherited in an autosomal recessive manner and 25% are autosomal dominant, with the remainder being X-linked or mitochondrial diseases (RetNet www.sph.uth.edu/retnet). Treatment of autosomal recessive disease is intuitive in the sense that replacement of the defective gene with a functional copy should ameliorate disease, and this approach has already been applied with some success in the case of, for example, RPE65 mutations responsible for LCA (Russell et al., 2017). However, this approach is of limited value in autosomal dominant diseases where gain-of-function mutations are responsible for the disease phenotype. One of the most important causes of autosomal dominant RP is that caused by gain-of-function mutations in the rhodopsin gene, RHO, which is responsible for approximately 15% of all IRDs (Tsai et al., 2018). More than 100 such mutations in RHO are known and the unique challenges posed by these types of IRDs has prompted development of some novel therapeutic strategies which are discussed below.
Disease-causing variants of ABCA4 (ATP binding cassette subfamily A member 4) are responsible for Stargardt disease and RP (Cremers et al., 2020), and in most characterized cohorts it is the single most prevalent gene responsible for IRDs. The ABCA4 gene is just over 135 kbp in size, inclusive of noncoding regions, while the mature mRNA transcript is 7.3 kb (see Table 2). Variants of EYS (eyes shut homolog) and USH2A (usherin) are among the most frequently encountered genetic variants responsible for RP (Gao et al., 2019; Pontikos et al., 2020; Toualbi et al., 2020; Weisschuh et al., 2020; Yang et al., 2020; Qian et al., 2021). The EYS gene is 1.99 Mbp with a coding sequence length of 9.4 kbp, while the USH2A gene is 807 kbp with a coding sequence length of 15.6 kbp. On the smaller end of the gene spectrum, the CYP4V2 (cytochrome P450 family 4 subfamily V member 2) gene responsible for Bietti crystalline retinal dystrophy (Astuti et al., 2015) and the RPE65 (retinoid isomerohydrolase RPE65) gene responsible for LCA are both approximately 28 kbp with coding sequences of 1.6 kbp (Table 2). This variation in gene size is an important driver of the approaches taken to develop gene-based therapies and we have used this to provide a framework for our discussion below.
TABLE 2
| Gene | IRD phenotype | Chromosome location | Gene length (bp) | Coding sequence (bp) | Encoded protein | Protein function | Genbank accession |
|---|---|---|---|---|---|---|---|
| ABCA4 | Stargardt disease | 1 | 135,313 | 6,819 | ATP binding cassette subfamily A member 4 | Photoreceptor transport of all-trans-retinal aldehyde | NG_009073 |
| BEST | Best disease | 11 | 21,580 | 1,755 | Bestrophin 1 | Epithelial chloride ion channel | NG_009033 |
| CEP290 | LCA, RP | 12 | 100,204 | 7,437 | Centrosomal protein 290 | Cilium formation | NG_008417 |
| CHM | Choroideremia | X | 193,383 | 1,959 | CHM Rab escort protein | Rab GTPase | NG_009874 |
| CYP4V2 | Bietti crystalline dystrophy | 4 | 28,939 | 1,575 | Cytochrome P450 family 4 subfamily V member 2 | Fatty acid precursor metabolism | NG_007,965 |
| EYS | RP | 6 | 1,994,246 | 9,432 | Eyes shut homolog | Photoreceptor-specific, secreted matrix protein | NG_023443 |
| RHO | RP, stationary night blindness | 3 | 13,706 | 1,044 | Rhodopsin | Rod-specific phototransducer | NG_009115 |
| RPE65 | LCA, RP | 1 | 28,136 | 1,599 | Retinoid isomerohydrolase RPE65 | Isomerization step of 11-cis retinal synthesis | NG_008472 |
| USH2A | RP, Usher syndrome II | 1 | 807,558 | 15,606 | Usherin | Photoreceptor and auditory hair cell maintenance | NG_009497 |
Features of selected loci responsible for IRDs.
Gene Replacement Strategies
In its simplest implementation, gene replacement for IRDs aims to restore or maintain visual function by introducing a functional copy of a protein coding sequence into a target retinal cell population that is partially or completely deficient of the protein in question. In most cases this refers to biallelic autosomal recessive mutations which account for the majority of IRDs (Sahel et al., 2014; Carss et al., 2017). As a target tissue for gene replacement, the human retina has several advantages over other anatomical sites. Accessing the retina is relatively straightforward for the retinal specialist, and can be accomplished via intravitreal, subretinal, or suprachoroidal routes with outpatient-based surgical or procedural approaches (Figure 3). Additionally, blood-retinal barriers render the retina immune privileged, reducing the risk of immune reactions against gene delivery vectors. That said, given that the majority of IRDs cause pathology at the level of the photoreceptors or RPE (Figure 1), the overlying retinal cell layers including the inner and external limiting membranes form a barrier to the entry of vector particles larger than about 30 nm in size (Teo et al., 2018; Tavakoli et al., 2020), which limits the use of large vectors for intravitreal administration.
FIGURE 3
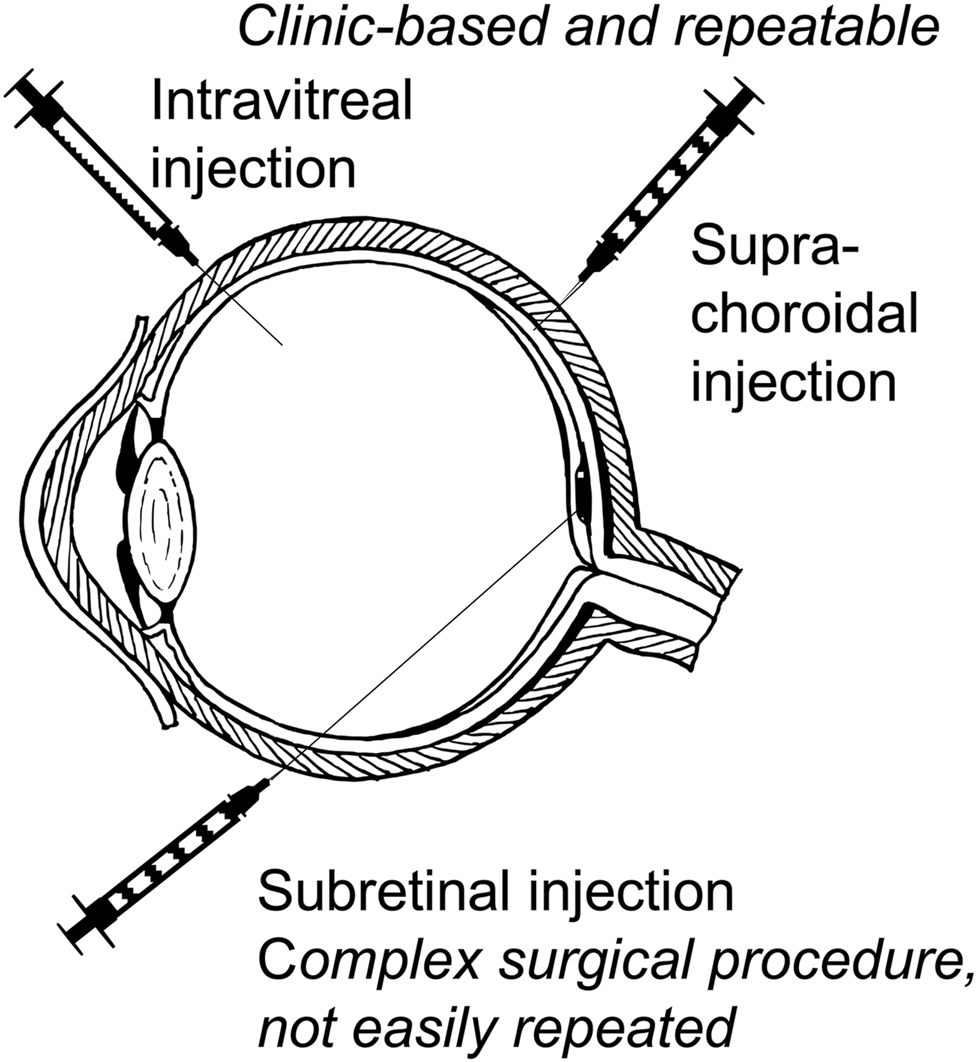
Approaches to retinal delivery of gene-based therapies for inherited retinal diseases. Intravitreal injection and the more recently developed suprachoroidal injection may be given as an outpatient clinic-based procedure with multiple repeat injections possible. Subretinal injection is a formal surgical procedure requiring pars plana vitrectomy and gas tamponade and is not easily repeated.
Current approaches to IRD gene replacement can be divided into viral and nonviral vectors. Several viruses have been investigated as potential vehicles for retinal gene replacement, including adenovirus, lentivirus, herpesvirus, and adeno-associated virus (AAV). The greater safety profile, lower inflammatory response, low incidence of host genome integration, and high efficacy of AAV has made it the vector of choice for retinal gene therapy (Hauswirth, 2014). AAV is a nonenveloped ssDNA parvovirus with a 25 nm particle size that under normal circumstances depends on co-infection by adenovirus or herpesvirus for replication (Balakrishnan and Jayandharan, 2014). The extraordinary number of AAV capsid variants has enabled the development of AAV vectors with a wide variety of host cell specificities, or tropisms. The most common recessive IRDs that can potentially benefit from gene replacement involve mutations in genes expressed mainly in photoreceptors and RPE (Figure 1). Less common IRDs such as congenital stationary night blindness (CSNB) may alternatively involve inner retinal layers (Zeitz et al., 2015).
Addressing the need to target one or several cell types within the retina has been an essential component of gene-based therapeutic development for IRDs. Work from the early 2000’s demonstrated that AAV serotypes displayed highly variable tropism for different human tissues (Wu et al., 2006; Srivastava, 2016; Li and Samulski, 2020). More recent research on AAVs for retinal targeting has highlighted the differences between AAV serotypes for transduction of the numerous cell populations within the human retina. By far the most extensively studied serotype of AAV is type 2 (AAV2), although recent work has demonstrated that the AAV2 viral capsid may not necessarily be the optimal subtype for transduction of all retinal cell types. Using a human retinal explant model, Wiley et al. (2018) demonstrated that AAV serotypes 1 (AAV1) and 4 (AAV4) appeared to have the highest intrinsic affinity for the outer nuclear layer (Table 3), which is where photoreceptor nuclei reside and is thus the preferred target for many IRD therapies. The same group subsequently demonstrated that gene delivery by AAV serotypes also varied based on the route of delivery, with intravitreal delivery resulting in low transduction efficacy with most serotypes and subretinal and suprachoroidal injection yielding high transduction efficacy only with AAV1 when using a mouse model for transgene delivery (Han et al., 2020). Further improvements to the native affinities of the AAV capsid for retinal cell types have been made by directed evolution approaches. The use of both in vitro and in vivo directed evolution has been used to dramatically improve the transduction of nonhuman primate photoreceptors following intravitreal delivery of the evolved AAV vectors (Dalkara et al., 2013; Byrne et al., 2020). Despite these improved vectors, however, only a relatively small percentage of all retinal photoreceptors are typically transduced even when high titers (>1012 virions) of AAV are injected (Dalkara et al., 2013).
TABLE 3
| AAV capsid serotype | Relative transduction efficiency in human retinal explants | ||
|---|---|---|---|
| ONL | INL | GCL and NFL | |
| AAV1 | High | Low | High |
| AAV2 | Intermediate | Intermediate | High |
| AAV4 | High | High | High |
| AAV5 | Intermediate/high | Low | Low |
| AAV6 | Intermediate/high | High | High |
| AAV8 | Low | Intermediate | High |
| AAV9 | Intermediate | Intermediate | High |
Variability in retinal cell transduction by different adeno-associated virus (AAV) capsid serotypes. Relative transduction efficiencies are shown for AAV capsid serotypes each carrying an AAV2 vector with a green fluorescent protein (GFP) gene under the control of a human cytomegalovirus (hCMV) promoter. These were used to transduce cadaveric human retinal explants. Data derived from Wiley et al. (2018).
The first FDA approved IRD gene therapy utilized AAV2 (Russell et al., 2017) to replace defective RPE65 in patients with LCA. In this therapy the 1.6 kb coding sequence of RPE65 was placed under the control of a modified avian β-actin promoter with a cytomegalovirus enhancer sequence (Maguire et al., 2008; Maguire et al., 2009). This cassette was inserted between AAV inverted terminal repeats (ITRs) and the vector propagated in HEK293 cell cultures. In the phase 3 trial of this vector, termed voretigene neparvovec (branded as Luxterna, Spark Therapeutics), LCA patients with biallelic RPE65 mutations aged 3 and above and with visual acuity of 20/60 or worse or visual fields less than 20°, or both, underwent pars plana vitrectomy and subretinal injection of 0.3 ml (1.5 × 1011 vector genomes) of the vector (Russell et al., 2017). This approach had previously been demonstrated to yield efficient photoreceptor transduction in canines and nonhuman primates (Jacobson et al., 2006a; Jacobson et al., 2006b).
The primary endpoint for the phase 3 trial of voretigene neparvovec was patient performance on the abovementioned MLMT. At 1 year after treatment, the MLMT change score was 1.8 compared to 0.2 in the control group (p = 0.0013), with 65% of test subjects and no control subjects able to pass the MLMT at the lowest luminance of 1 lux (Russell et al., 2017). More recently, the three -year outcomes became available and showed a mean MLMT score of 2.4 among the test subjects, with 71% able to pass the MLMT at the lowest luminance, with visual acuity remaining essentially unchanged (Maguire et al., 2021). The most notable complication among the test subjects was a retinal detachment that occurred in a single patient (1 of 21, 4.8%) at around year four, which was probably related to the original surgical intervention and is a known complication of routine vitrectomy. Additional complications, including endophthalmitis, development of macular holes, raised intraocular pressure, macular edema, and macular atrophy can occur following vitrectomy and subretinal injection (Nuzbrokh et al., 2020), and larger cohort studies of treated IRD patients will be required before their incidence can be properly assessed.
Despite the numerous advantages of AAV for retinal gene delivery, its small size places limits on its cargo capacity. The wild-type AAV genome is 4.8 kb in size and contains short 5ʹ and 3ʹ ITRs that flank coding sequences for replication (Rep) and capsid (Cap) proteins (Puppo et al., 2014). Modified cell lines that express Rep and Cap enable for this entire central region to be replaced by an insert size of up to 4.4 kb. For gene replacement therapy this insert needs to include both the protein coding sequence as well as well as upstream and downstream regulatory elements to ensure efficient and appropriate expression of the IRD transgene. Given the limited cargo capacity of AAV and the prevalence of IRDs caused by large gene variants such as USH2A (15.6 kb) and EYS (9.4 kb), researchers have sought to engineer AAV-based vectors that can deliver larger genes. Dual AAV systems enable a near-doubling of deliverable transgene size by dividing the transgene into halves and appending overlapping sequences to the termini to enable homologous recombination (Trapani, 2018). Alternatively, a splicing acceptor and splice donor can be added to the termini to enable intermolecular concatamerization and splicing of the two halves into a single contiguous cDNA construct (Carvalho et al., 2017). In vivo efficacy of a dual AAV vector system has previously been demonstrated in a mouse model of Stargardt disease (McClements et al., 2019). Two fragments of the 6.8 kb ABCA4 coding sequence, 3.7 and 3.3 kb in size and with 207 bases of overlapping sequence were used to develop two vectors, with the upstream fragment being driven by a rhodopsin kinase promoter and the downstream fragment terminated by a hepatitis virus post-transcriptional regulatory element (McClements et al., 2019). Subretinal co-delivery of these constructs yielded robust full-length ABCA4 expression in photoreceptor outer segments and successfully reduced accumulation of toxic bis-retinoids that accumulate in the ABCA4-deficient retina. Human clinical trials involving the use of dual AAV systems are in the planning stage but have yet to be initiated (Piotter et al., 2021).
Nonviral approaches to IRD gene replacement have been explored with the aim of overcoming the gene size limitation of AAV in addition to improving the safety profile and production costs associated with viral vector-based approaches. Synthetic vectors, or nanoparticles, are nonviral vectors comprising a cationic lipid assembly that can encapsulate a transgene of interest up to 20 kb in size and deliver it through nuclear pores to enable gene expression. A variety of nanoparticle materials have been developed and contemporary nanoparticles can transfect both RPE and photoreceptors, albeit with less proven durability than that seen after AAV2 transduction with IRD-related genes (Conley and Naash, 2010; Adijanto and Naash, 2015; Trigueros et al., 2019; Sun et al., 2020).
Genome Editing Strategies
Despite being the predominant treatment modality in current IRD clinical trials (Prado et al., 2020), conventional gene augmentation therapy is limited to the treatment of loss of function genotypes and haploinsufficiency, with no direct modification of the host genome. Moreover, contemporary gene augmentation vectors with demonstrated efficacy for in vivo transduction of retinal photoreceptors are limited in their maximum cargo size (Puppo et al., 2014). This renders gene augmentation unsuitable for many important IRD genotypes, such as RP due to USH2A and EYS variants. Gene editing approaches aim to address these limitations by correcting disease-causing mutations at the level of the host genome.
Human genome editing was originally made possible due to pioneering work in the early 2010’s that demonstrated the use of a clustered regularly interspaced short palindromic repeat (CRISPR) and Cas9 nuclease (CRISPR/Cas9) system derived from Streptococcus pyogenes to introduce site-specific nucleotide alterations in the target genome (Ran et al., 2013; Jiang and Doudna, 2017). In its simplest implementation, the CRISPR/Cas9 system works via a four-step process whereby: 1) the Cas9 protein forms a complex with a sequence-specific guide RNA within the cell; 2) the Cas9-RNA complex anneals to the complementary gDNA sequence; 3) the Cas9-RNA complex creates a double-stranded DNA break in the gDNA; and 4) a modification is made to the gDNA via endogenous DNA repair mechanisms—either homology-directed repair (HDR) or non-homologous end-joining (NHEJ) (Ran et al., 2013). The introduction of exogenous guide DNA enables the selective correction of deleterious mutations via HDR, while NHEJ creates errors at the target site and can selectively inactivate genes (Zhang, 2021).
At present the most important limitation of the CRISPR/Cas9 system is the introduction of unintended mutations at genomic locations containing sequence homology to the target site, also known as off-target editing (Zhang et al., 2015). Several approaches have been taken to overcome this problem for IRD gene editing, including the use of retinal cell-specific promoters to drive the expression of Cas9, directed mutagenesis of the Cas9 nuclease to improve its on-target/off-target editing and reduce its biological half-life, and the use of additional guide RNAs complexed to Cas9 to improve gene targeting (Burnight et al., 2018; Peddle et al., 2020). A second limitation of CRISPR/Cas9 gene editing for IRDs is the efficient delivery of the Cas9 expression cassette and guide RNA into the target retinal cell type. Numerous approaches have been described, including AAV-based vectors that utilize small Cas9 orthologs from other bacterial species (Gasiunas et al., 2020) and dual AAV systems that deliver the different elements of the Cas9, guide RNA, and donor DNA cassettes (Hung et al., 2018).
After several years of intensive basic and pre-clinical research on CRISPR/Cas9-mediated gDNA editing in IRD cell and animal models of disease (Maeder et al., 2019), this approach has now reached clinical trials. LCA type 10 (LCA10), a severe and early-onset IRD caused by bi-allelic loss-of-function mutations in the CEP290 gene (coding sequence size of 7.9 kb). This 7.9 kb coding sequence of CEP290 exceeds AAV insert capacity and gDNA editing, among other approaches, was seen as a viable therapeutic option. The most prevalent CEP290 mutation causing LCA10 is IVS26, which introduces a premature stop codon via alterations to RNA splicing (Burnight et al., 2017). In the phase 1/2 EDIT-101 trial an AAV5 vector is used to deliver SaCas9 (from Staphylococcus aureus) and CEP290-specific guide RNAs to photoreceptor cells by subretinal injection. Wild-type CEP290 mRNA is produced via intronic inversion or deletion mediated by the editing construct. In the case of LCA10, a minimum gDNA editing efficiency of 10% was determined in earlier studies to be required for meaningful vision restoration, and this baseline efficiency was exceeded in mouse and nonhuman primate models (Maeder et al., 2019). Final results from the EDIT-101 trial for LCA10 are expected in 2024, although initial clinical data from the phase 1/2 BRILLIANCE trial (Editas Medicine, Cambridge, Mass.) showed a positive safety profile at 15 months after treatment and limited evidence of clinical efficacy.
Genome editing is also being applied to the treatment of autosomal dominant IRDs, where a point mutation can result in production of a gain-of-function protein that impairs cell functions (Farrar et al., 2012; Athanasiou et al., 2018). Autosomal dominant RP caused by such mutations in RHO has received significant attention due to their relative prevalence in the IRD patient population. An emerging approach for the treatment of these RHO variants involves simultaneous ablation of the mutant RHO allele and replacement with wild-type RHO (Meng et al., 2020). Work by Tsai et al. (2018) demonstrated the use of a pair of AAV2/8 vectors to deliver either 1) guide RNAs that targeted the genomic DNA region up- and downstream of RHO start codon, in addition to a wild-type RHO expression cassette, or 2) a Cas9 expression cassette. The transduced version of the RHO gene resistant to CRISPR/Cas9 ablation complemented the mutant copy, while the endogenous mutated RHO was selectively targeted for ablation using cell and mouse models. This elegant approach ensures that ablation of the dominant mutant RHO gene will only occur in the presence of transduced wild-type RHO. Importantly, this approach is largely agnostic to the RHO mutation responsible for the phenotype. Related approaches that target specific RHO variants using CRISPR/Cas9 approaches were also described recently (Patrizi et al., 2021).
RNA Editing to Treat Inherited Retinal Diseases
Genomic DNA editing has the potential to address the root cause of essentially all IRDs but concerns regarding off target editing and its potentially deleterious impacts on the eye (Smith et al., 2017) has prompted exploration of alternative methods to correct disease-causing variants. RNA editing is a normal biological process that occurs in human cells, including the retina, and can be harnessed to create sequence-specific nucleotide edits of mRNA. In its natural role, RNA editing is performed by adenosine deaminases (adenosine deaminase acting on RNA, or ADAR) and cytidine deaminases (cytidine deaminase acting on RNA, or CDAR) that can catalyze adenosine-to-inosine (A-I) and cytosine-to-uridine (C-U) deamination, which is functionally equivalent to A-to-G and C-to-T editing, respectively (Nishikura, 2010; Gallo et al., 2017). Because these edits can be targeted to a specific mRNA and do not affect genomic DNA, their impact is transient, as would be any potential off-target effects.
Humans possess two ADAR enzymes, ADAR1 and ADAR2, with the former being expressed throughout the retina and the latter being expressed mainly in retinal ganglion cells (Fry et al., 2020). Both enzymes are capable of editing mRNA and the process occurs in the nucleus concurrently with RNA splicing (Eisenberg, 2021).
In its most basic form, the sequence-specific A-to-I editing activity of ADAR requires two components: 1) a guide RNA (specificity domain), analogous to that used in the CRISPR-Cas9 system, which can anneal to the target mRNA, and 2) an ADAR recruiting domain that adopts a dsRNA hairpin structure and promotes ADAR recruitment to the targeted mRNA complex (Eisenberg, 2021). This approach enables the use of endogenous ADAR from host retinal cells, thus avoiding the need to introduce exogenous ADAR expression cassettes like that required for Cas9 (although this approach has been explored for ADAR). Instead, RNA editing can be accomplished with the use of custom antisense oligonucleotides (ASOs) that incorporate the specificity and ADAR recruiting domains, potentially simplifying the development of IRD genotype-specific treatments (Merkle et al., 2019). This ASO-only approach to RNA editing was successfully demonstrated in a variety of human cell lines by Merkle and others (Merkle et al., 2019) and achieved sequence-specific mRNA editing efficacy of 30–70% with endogenously expressed ADAR. Additionally, this approach incorporated the use of chemical modifications to the ASO (2′-O-methyl and phosphorothioate linkages) to improve their biological stability. Such an approach, even with the use of chemically stabilized ASOs would, however, require repeat dosing with the ASO over a patient’s lifetime to maintain efficacy.
The application of RNA editing to IRD treatment is presently limited by editing efficiency and potentially by off-target effects, and currently available tools appear to be primarily suited to the treatment of IRDs caused by a subset of recessive mutations within large genes that are not amenable to AAV-mediated gene augmentation. A recent survey by Fry and coworkers (Fry et al., 2020) found substantial heterogeneity in the proportion of IRD mutations amenable to RNA editing, with 9% of known pathogenic mutations in CEP290 and 32% in ABCA4 being correctable by RNA editing. At present it is unknown what level of RNA editing efficiency will be required to achieve biologically or clinically meaningful improvements in the wide variety of IRDs to which the technique is applicable, although the past several years have seen dramatic improvements in the range of RNA editing tools available for the creation of mutation-specific therapeutics and several industry-led trials of RNA editing for IRD treatment are expected to commence in the coming years.
Antisense Oligonucleotides and RNA Interference for Gene Modulation in Inherited Retinal Disease
Antisense oligonucleotides (ASOs) are small single-stranded RNA or DNA sequences, typically in the range of 15–30 bases, that can be designed to anneal to specific mRNAs and effect gene silencing, inhibit protein translation, or alter mRNA splicing (Crooke et al., 2017; Bennett, 2019). This makes ASOs applicable to dominantly inherited gain-of-function alleles and recessive alleles with splicing defects. Upon annealing to their mRNA target, ASOs can direct RNase H-mediated mRNA cleavage or induce changes in RNA splicing that result in exon-skipping with shortening of the protein product, or nonsense-mediated decay of the mRNA (Maruyama and Yokota, 2020).
This class of IRD therapeutics is particularly attractive because they can be delivered intravitreally and penetrate the outer retinal layers, have an excellent safety profile, and can be produced at scale far more economically than other gene-based therapeutics (Chi et al., 2017; Xue and MacLaren, 2020). Moreover, initial problems with ASO stability in vivo have now been overcome with the development of nuclease-resistant phosphorodiamidate morpholino oligonucleotides (PMOs) that also have dramatically improved cell penetration compared to unmodified ASOs (Smith and Zain, 2019).
Clinical application of ASOs for IRD treatment has recently been demonstrated in several phase I/II clinical trials. An intronic mutation of CEP290 (c.2991 + 1655A > G, or p. Cys998X) is a common cause of LCA10 and results in creation of a cryptic splice donor site, leading to a new exon and in-frame stop codon between the native exons 26 and 27 (Ramsbottom et al., 2018). A 17-mer modified ssRNA, termed Sepofarsen (ProQR Therapeutics, Netherlands), anneals to the mutated CEP290 mRNA splicing site and is effective in restoring normal CEP290 function in cell and animal models (Barny et al., 2018; Ramsbottom et al., 2018). More recently, a phase I/II trial demonstrated rapid and sustained improvements in visual acuity, visual fields, electrophysiological parameters, and retinal structure following a single intravitreal injection of 160 μg or 320 μg of sepofarsen in CEP290 p. Cys998X patients (Cideciyan et al., 2021a). A phase II/III trial of sepofarsen is currently ongoing.
Pathological variants of the USH2A gene, which encodes the usherin protein, are the most common cause of autosomal recessive syndromic and nonsyndromic RP worldwide and among the most common causes of congenital deafness (Toualbi et al., 2020). The c.2299delG and c.2276G > T mutations within exon 13 of USH2A are responsible for up to a third of RP cases in some populations (Pendse et al., 2019). These variants create a premature stop codon and lead to nonfunctional usherin, although removal of the exon 13 equivalent region from mouse USH2A did not appear to interfere with its biological function in a mouse model (Pendse et al., 2019). Moreover, the introduction of a morpholino ASO (QR-421a) that targets exon 13 and promotes exon skipping of this region was able to restore retinal function in zebrafish and mouse disease models (Dulla et al., 2021). The phase I/II STELLAR trial of QR-421a (ProQR Therapeutics, Netherlands) for USH2A exon 13-related RP showed retention of 1–2 lines of visual acuity in eyes treated with a single intravitreal injection of QR-421a, compared to the untreated fellow eyes.
These encouraging early clinical trial findings and the potential convenience with which new targeted ASOs can be developed and manufactured suggests they may play a role in treating not only the more prevalent IRDs like USH2A-related RP, but also rarer IRDs that might not otherwise be considered as viable therapeutic targets due to profit considerations by pharmaceutical companies.
Autosomal dominant IRDs are widely considered a key target for the use of gene knockdown approaches (Farrar et al., 2012; Meng et al., 2020). One promising gene knockdown approach for IRDs is RNA interference (RNAi), an important naturally occurring mechanism of gene suppression in eukaryotes. RNAi is driven by the production of small (average of 22-mer) RNAs, termed microRNAs (miRNAs), that are typically transcribed from the 5ʹ ends of mRNA and can selectively trigger mRNA decay in a sequence-specific fashion (O'Brien et al., 2018). MicroRNA precursor derivatives termed mirtrons are spliced from the 5ʹ end of mRNAs and can similarly knock down target mRNA with high specificity. (Okamura et al., 2007; Ruby et al., 2007). Conveniently, mirtrons can be incorporated into polycistronic expression cassettes that contain both the mirtron and an engineered copy of the target gene that is resistant to mirtron-mediated RNAi, making them particularly useful for autosomal dominant diseases. This approach has been demonstrated in a mouse model of RHO mediated RP, where a single AAV vector containing a mirtron that targeted both the wild-type and mutant copies of RHO for RNAi-mediated decay, and at the same time supplied a RNAi-resistant engineered copy of functional RHO to the transfected cell (Orlans et al., 2021). This mutation-agnostic approach may theoretically be applied to many dominantly inherited IRDs.
Optogenetics as a General-Purpose Therapy for Late Stage Inherited Retinal Disease
Each of the gene based IRD therapies discussed above are limited in their application to patients with a specifically affected gene or gene variant. Given that IRDs are orphan diseases, it is expected that there will only be a relatively small number of individuals in any given population for whom such custom gene therapies are applicable. An important challenge in IRD therapy is thus the development of general-purpose therapies that can be used for a wide variety of IRDs irrespective of the patient’s particular genotype. One such general-purpose therapy approach which is still within the scope of gene-based therapy is optogenetics, which aims to restore vision in late-stage IRDs by targeting genes encoding photosensitive proteins to selected retinal cell types, converting them into a replacement photoreceptor (Duebel et al., 2015; McClements et al., 2020).
Optogenetic techniques were originally developed as research tools to explore the function of mammalian neurons in animal models (Adamantidis et al., 2007; Deisseroth, 2011; Zhang et al., 2011). In this technique, microbially-derived opsin proteins are used to trigger neuron firing in response to specific wavelengths of light (Williams and Deisseroth, 2013). Opsin genes can be inserted into a gene expression cassette and delivered via a viral vector into neurons in vivo, after which the transduced neurons are rendered photosensitive. In many IRDs, including RCDs and CRDs, there is loss of photoreceptors in the outer retina but relative preservation of the inner retinal layers, including the retinal ganglion cell (RGC) layer that coalesces to form the optic nerve head. The greater exposure of this retinal layer to the vitreous cavity, compared to the much deeper photoreceptor layer, makes it an attractive target for virus-mediated gene transduction. Proof-of-principle for optogenetic therapy was provided by Bi and others (Bi et al., 2006), who demonstrated successful phototransduction by bacterial channelrhodopsin (ChR2) in transduced mouse RGCs with signal propagation to the visual cortex.
More recently, optogenetics has been successfully translated into a general purpose IRD therapy. In groundbreaking work by Sahel and others (Sahel et al., 2021), a patient with late-stage RP and visual acuity of perception of light underwent intravitreal injection with an AAV containing an optogenetic expression cassette (AAV2.7m8-CAG-ChrimsonR-tdTomato). Following treatment, the patient underwent training with light-stimulating goggles that converted an external video feed into a pixel map projected onto a 10° circular area of the central retina using a diode light source specific for the optogenetic construct (ChrimsonR, peak wavelength of 595 nm). While the untreated eye remained at baseline vision, the treated eye gained the ability to perceive, locate, and count various objects whilst using the goggles (Sahel et al., 2021). The phase 1/2a PIONEER study will report on the findings of this approach in a small patient cohort, and the primary outcomes are expected to be available in the coming months. Promising early results from this trial have more recently enabled fast track status of this treatment (GS030, from GenSight Biologics, Paris) by the US FDA (FDA, October 2021).
Nonselective optogenetic approaches using RGC transduction are likely sufficient to restore light sensitivity and gross visual function in IRD patients but do not enable image processing by retinal interneurons (Duebel et al., 2015). This places a limit on the quality of vision possible using the approach and for now the method is likely applicable mainly to individuals with severe vision loss. More selective approaches that involve targeted transduction or gene expression in retinal bipolar cells may enable more physiological activation of RGCs and potentially better restoration of visual quality (Gilhooley et al., 2021). An overview of optogenetic and other approaches to gene-based therapy for IRD treatment is shown in Figure 4.
FIGURE 4
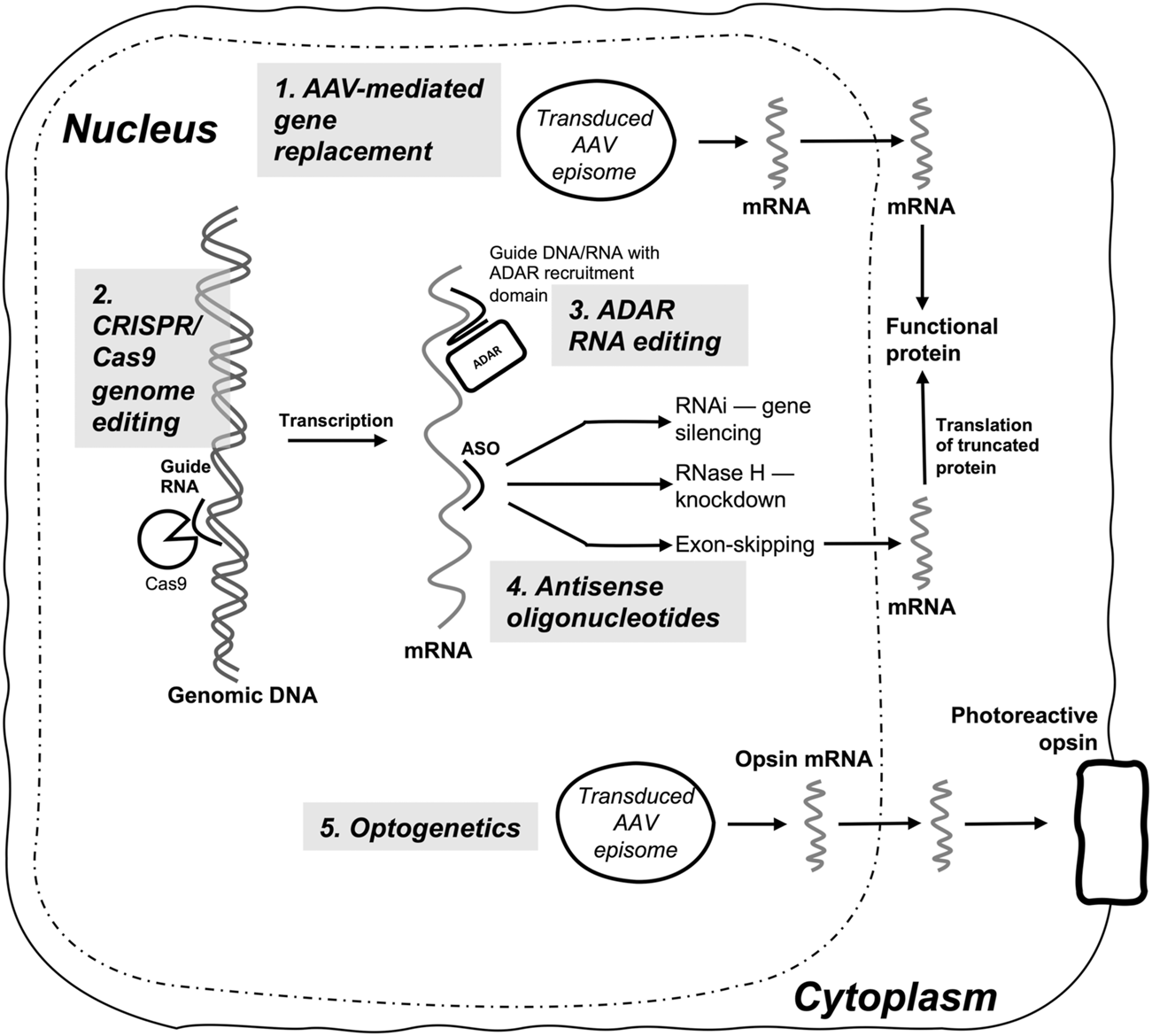
Summary diagram of gene-based therapies currently in clinical use or clinical trials for inherited retinal diseases. (1) AAV-mediated gene replacement therapy is currently the predominant modality, and delivers replacement transgenes (e.g., RPE65), or transgene fragments (dual AAV systems) to produce a functional protein in biallelic autosomal recessive IRDs; (2) CRISPR/Cas9 genome editing requires delivery of CRISPR/Cas9 constructs, most commonly with AAV vector systems, and enables site-directed editing or mutagenesis of IRD target genes (e.g., CEP290); (3) ADAR-mediated RNA editing is used to perform sequence-specific RNA nucleotide edits by utilizing guide DNA or RNA and an ADAR recruitment domain, with either endogenous ADAR or transduced and overexpressed exogenous ADAR enzyme; (4) Antisense oligonucleotides (ASOs) induce sequence specific gene silencing via RNAi, RNase H-mediated mRNA knockdown, or targeted exon skipping (e.g., USH2A exon 13); (5) Optogenetics delivers an engineered phototransducing opsin to a specific retinal cell type (e.g., ganglion cells) to render the cell photosensitive and capable of replacing the light-responsive function of degenerating photoreceptor cells in IRDs.
Conclusion
From the conception of human gene-based therapeutics in the middle of the last century there has been an increasing expectation by many in the medical and research community that, given a critical mass of knowledge and technology, it will eventually be possible to effectively cure genetic diseases (Miller, 1992; Wirth et al., 2013; Dunbar et al., 2018). IRDs are also seen as one of the lowest hanging fruits in this area, given their well-defined and typically monogenic basis and the sophistication of current molecular and surgical techniques. Additionally, the orphan status of IRDs typically facilitates faster regulatory approval and can improve early patient access to treatment (Haffner, 2005). Despite this, recent clinical trials have made it obvious that gene-based therapies still have a host of challenges that must be overcome if IRDs are to become a curable or at least manageable disease. Questions regarding the long-term safety and durability of AAV-based subretinal vector delivery remain, with long term follow up of patients treated with voretigene neparvovec-showing possible safety signals and a suggestion of visual acuity decline after 4 years (Maguire et al., 2021). While the current IRD clinical trial landscape remains dominated by AAV-based approaches requiring subretinal injection, an increasing number of conveniently administered intravitreal therapeutics such as ASOs are being trialed with encouraging early results.
The recent successful trial of genotype-agnostic optogenetic therapy for advanced RP (Sahel et al., 2021) prompts re-evaluation of the value of genotype-specific therapies. Given the large genotypic diversity of IRDs and the high development costs of current therapeutics, it is unlikely that custom gene-based therapies will be developed for all IRD mutations. While optogenetics may afford some level of visual improvement for subjects with advanced IRDs, its current implementation affords only limited visual improvement due to the complexity of retinal neuronal circuitry. General purpose cell-based therapeutics could potentially be applied to any degenerative condition of the outer retina, including IRDs, but have yet reach their potential in clinical trials for IRD patients (Ben M'Barek et al., 2019; Maeda et al., 2019). That said, we envisage general-purpose cell-based therapies and combination therapies involving cell-based and gene-based therapies to become much more dominant in the longer term.
Meanwhile, the recent proliferation of clinical trials for a host of gene based IRD treatments is providing invaluable data that will likely enable a small number of highly efficacious treatment approaches to be applied to the majority of IRDs. Despite many initial setbacks that have arisen in early trials it is becoming increasingly clear that the coming years will be pivotal learning experiences that will pave the way not only for IRD gene therapy, but for the more common and more genetically complex eye diseases.
Statements
Author contributions
BF, TTE, JM, and KT conceived the review. BF drafted the manuscript and its revisions. All authors provided input on the topic coverage and revisions to the manuscript. AB, ST, SY, AT, SL, CMGC and CC provided expert guidance and proofed the manuscript and its revisions.
Funding
This work was supported by the SingHealth Foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Ach T. Tolstik E. Messinger J. D. Zarubina A. V. Heintzmann R. Curcio C. A. (2015). Lipofuscin Redistribution and Loss Accompanied by Cytoskeletal Stress in Retinal Pigment Epithelium of Eyes with Age-Related Macular Degeneration. Invest. Ophthalmol. Vis. Sci.56, 3242–3252. 10.1167/iovs.14-16274
2
Adamantidis A. R. Zhang F. Aravanis A. M. Deisseroth K. De Lecea L. (2007). Neural Substrates of Awakening Probed with Optogenetic Control of Hypocretin Neurons. Nature450, 420–424. 10.1038/nature06310
3
Adijanto J. Naash M. I. (2015). Nanoparticle-Based Technologies for Retinal Gene Therapy. Eur. J. Pharm. Biopharm.95, 353–367. 10.1016/j.ejpb.2014.12.028
4
Akyol E. Hagag A. M. Sivaprasad S. Lotery A. J. (2021). Adaptive Optics: Principles and Applications in Ophthalmology. Eye35, 244–264. 10.1038/s41433-020-01286-z
5
Arai Y. Maeda A. Hirami Y. Ishigami C. Kosugi S. Mandai M. et al (2015). Retinitis Pigmentosa with EYS Mutations Is the Most Prevalent Inherited Retinal Dystrophy in Japanese Populations. J. Ophthalmol.2015, 819760. 10.1155/2015/819760
6
Astuti G. D. N. Sun V. Bauwens M. Zobor D. Leroy B. P. Omar A. et al (2015). Novel Insights into the Molecular Pathogenesis of CYP 4V2 ‐associated Bietti's Retinal Dystrophy. Mol. Genet. Genomic Med.3, 14–29. 10.1002/mgg3.109
7
Athanasiou D. Aguila M. Bellingham J. Li W. Mcculley C. Reeves P. J. et al (2018). The Molecular and Cellular Basis of Rhodopsin Retinitis Pigmentosa Reveals Potential Strategies for Therapy. Prog. Retin. Eye Res.62, 1–23. 10.1016/j.preteyeres.2017.10.002
8
Bakthavatchalam M. Lai F. H. P. Rong S. S. Ng D. S. Brelen M. E. (2018). Treatment of Cystoid Macular Edema Secondary to Retinitis Pigmentosa: a Systematic Review. Surv. Ophthalmol.63, 329–339. 10.1016/j.survophthal.2017.09.009
9
Balakrishnan B. Jayandharan G. (2014). Basic Biology of Adeno-Associated Virus (AAV) Vectors Used in Gene Therapy. Cgt14, 86–100. 10.2174/1566523214666140302193709
10
Balfoort B. M. Buijs M. J. N. Ten Asbroek A. Bergen A. a. B. Boon C. J. F. Ferreira E. A. et al (2021). A Review of Treatment Modalities in Gyrate Atrophy of the Choroid and Retina (GACR). Mol. Genet. Metab.134, 96–116. 10.1016/j.ymgme.2021.07.010
11
Barny I. Perrault I. Michel C. Soussan M. Goudin N. Rio M. et al (2018). Basal Exon Skipping and Nonsense-Associated Altered Splicing Allows Bypassing Complete CEP290 Loss-Of-Function in Individuals with Unusually Mild Retinal Disease. Hum. Mol. Genet.27, 2689–2702. 10.1093/hmg/ddy179
12
Ben M'Barek K. Habeler W. Regent F. Monville C. (2019). Developing Cell-Based Therapies for RPE-Associated Degenerative Eye Diseases. Adv. Exp. Med. Biol.1186, 55–97. 10.1007/978-3-030-28471-8_3
13
Bennett L. D. Klein M. Locke K. G. Kiser K. Birch D. G. (2017). Dark-Adapted Chromatic Perimetry for Measuring Rod Visual Fields in Patients with Retinitis Pigmentosa. Trans. Vis. Sci. Tech.6, 15. 10.1167/tvst.6.4.15
14
Bennett C. F. (2019). Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med.70, 307–321. 10.1146/annurev-med-041217-010829
15
Berson E. L. Sandberg M. A. Rosner B. Birch D. G. Hanson A. H. (1985). Natural Course of Retinitis Pigmentosa over a Three-Year Interval. Am. J. Ophthalmol.99, 240–251. 10.1016/0002-9394(85)90351-4
16
Bi A. Cui J. Ma Y.-P. Olshevskaya E. Pu M. Dizhoor A. M. et al (2006). Ectopic Expression of a Microbial-type Rhodopsin Restores Visual Responses in Mice with Photoreceptor Degeneration. Neuron50, 23–33. 10.1016/j.neuron.2006.02.026
17
Boon C. J. F. Klevering B. J. Cremers F. P. M. Zonneveld-Vrieling M. N. Theelen T. Den Hollander A. I. et al (2009). Central Areolar Choroidal Dystrophy. Ophthalmology116, 771–782.e1. 10.1016/j.ophtha.2008.12.019
18
Bourne R. R. A. (2020). Vision 2020: where Are We?Curr. Opin. Ophthalmol.31, 81–84. 10.1097/icu.0000000000000647
19
Burnight E. R. Gupta M. Wiley L. A. Anfinson K. R. Tran A. Triboulet R. et al (2017). Using CRISPR-Cas9 to Generate Gene-Corrected Autologous iPSCs for the Treatment of Inherited Retinal Degeneration. Mol. Ther.25, 1999–2013. 10.1016/j.ymthe.2017.05.015
20
Burnight E. R. Giacalone J. C. Cooke J. A. Thompson J. R. Bohrer L. R. Chirco K. R. et al (2018). CRISPR-Cas9 Genome Engineering: Treating Inherited Retinal Degeneration. Prog. Retin. Eye Res.65, 28–49. 10.1016/j.preteyeres.2018.03.003
21
Byrne L. C. Day T. P. Visel M. Strazzeri J. A. Fortuny C. Dalkara D. et al (2020). In Vivo-directed Evolution of Adeno-Associated Virus in the Primate Retina. JCI Insight5, e135112. 10.1172/jci.insight.135112
22
Carss K. J. Arno G. Erwood M. Stephens J. Sanchis-Juan A. Hull S. et al (2017). Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet.100, 75–90. 10.1016/j.ajhg.2016.12.003
23
Carvalho L. S. Turunen H. T. Wassmer S. J. Luna-Velez M. V. Xiao R. Bennett J. et al (2017). Evaluating Efficiencies of Dual AAV Approaches for Retinal Targeting. Front. Neurosci.11, 503. 10.3389/fnins.2017.00503
24
Chen T.-C. Huang D.-S. Lin C.-W. Yang C.-H. Yang C.-M. Wang V. Y. et al (2021). Genetic Characteristics and Epidemiology of Inherited Retinal Degeneration in Taiwan. Npj Genom. Med.6, 16. 10.1038/s41525-021-00180-1
25
Chi X. Gatti P. Papoian T. (2017). Safety of Antisense Oligonucleotide and siRNA-Based Therapeutics. Drug Discov. Today22, 823–833. 10.1016/j.drudis.2017.01.013
26
Chung D. C. Mccague S. Yu Z.-F. Thill S. Distefano-Pappas J. Bennett J. et al (2018). Novel Mobility Test to Assess Functional Vision in Patients with Inherited Retinal Dystrophies. Clin. Exp. Ophthalmol.46, 247–259. 10.1111/ceo.13022
27
Cicinelli M. V. Marchese A. Bordato A. Manitto M. P. Bandello F. Battaglia Parodi M. (2020). Reviewing the Role of Ultra-Widefield Imaging in Inherited Retinal Dystrophies. Ophthalmol. Ther.9, 249–263. 10.1007/s40123-020-00241-1
28
Cideciyan A. V. Jacobson S. G. Ho A. C. Garafalo A. V. Roman A. J. Sumaroka A. et al (2021a). Durable Vision Improvement after a Single Treatment with Antisense Oligonucleotide Sepofarsen: a Case Report. Nat. Med.27, 785–789. 10.1038/s41591-021-01297-7
29
Cideciyan A. V. Krishnan A. K. Roman A. J. Sumaroka A. Swider M. Jacobson S. G. (2021b). Measures of Function and Structure to Determine Phenotypic Features, Natural History, and Treatment Outcomes in Inherited Retinal Diseases. Annu. Rev. Vis. Sci.7, 747–772. 10.1146/annurev-vision-032321-091738
30
Conley S. M. Naash M. I. (2010). Nanoparticles for Retinal Gene Therapy. Prog. Retin. Eye Res.29, 376–397. 10.1016/j.preteyeres.2010.04.004
31
Cremers F. P. M. Lee W. Collin R. W. J. Allikmets R. (2020). Clinical Spectrum, Genetic Complexity and Therapeutic Approaches for Retinal Disease Caused by ABCA4 Mutations. Prog. Retin. Eye Res.79, 100861. 10.1016/j.preteyeres.2020.100861
32
Crooke S. T. Wang S. Vickers T. A. Shen W. Liang X.-h. (2017). Cellular Uptake and Trafficking of Antisense Oligonucleotides. Nat. Biotechnol.35, 230–237. 10.1038/nbt.3779
33
Daich Varela M. Esener B. Hashem S. A. Cabral De Guimaraes T. A. Georgiou M. Michaelides M. (2021). Structural Evaluation in Inherited Retinal Diseases. Br. J. Ophthalmol.105, 1623–1631. 10.1136/bjophthalmol-2021-319228
34
Daiger S. P. Bowne S. J. Sullivan L. S. (2014). Genes and Mutations Causing Autosomal Dominant Retinitis Pigmentosa. Cold Spring Harb Perspect. Med.5, a017129. 10.1101/cshperspect.a017129
35
Dalkara D. Byrne L. C. Klimczak R. R. Visel M. Yin L. Merigan W. H. et al (2013). In Vivo-directed Evolution of a New Adeno-Associated Virus for Therapeutic Outer Retinal Gene Delivery from the Vitreous. Sci. Transl Med.5, 189ra76. 10.1126/scitranslmed.3005708
36
Deisseroth K. (2011). Optogenetics. Nat. Methods8, 26–29. 10.1038/nmeth.f.324
37
Dimopoulos I. S. Radziwon A. St. Laurent C. D. Macdonald I. M. (2017). Choroideremia. Curr. Opin. Ophthalmol.28, 410–415. 10.1097/icu.0000000000000392
38
Duebel J. Marazova K. Sahel J.-A. (2015). Optogenetics. Curr. Opin. Ophthalmol.26, 226–232. 10.1097/icu.0000000000000140
39
Dulla K. Slijkerman R. Van Diepen H. C. Albert S. Dona M. Beumer W. et al (2021). Antisense Oligonucleotide-Based Treatment of Retinitis Pigmentosa Caused by USH2A Exon 13 Mutations. Mol. Ther.29, 2441–2455. 10.1016/j.ymthe.2021.04.024
40
Dunbar C. E. High K. A. Joung J. K. Kohn D. B. Ozawa K. Sadelain M. (2018). Gene Therapy Comes of Age. Science359, eaan4672. 10.1126/science.aan4672
41
Ehrenberg M. Weiss S. Orenstein N. Goldenberg-Cohen N. Ben-Yosef T. (2019). The Co-Occurrence of Rare Non-ocular Phenotypes in Patients with Inherited Retinal Degenerations. Mol. Vis.25, 691–702.
42
Eisenberg E. (2021). Proteome Diversification by RNA Editing. Methods Mol. Biol.2181, 229–251. 10.1007/978-1-0716-0787-9_14
43
Fahim A. T. Sullivan L. S. Bowne S. J. Jones K. D. Wheaton D. K. H. Khan N. W. et al (2020). X-Chromosome Inactivation Is a Biomarker of Clinical Severity in Female Carriers of RPGR-Associated X-Linked Retinitis Pigmentosa. Ophthalmol. Retina4, 510–520. 10.1016/j.oret.2019.11.010
44
Farrar G. J. Millington-Ward S. Chadderton N. Humphries P. Kenna P. F. (2012). Gene-based Therapies for Dominantly Inherited Retinopathies. Gene Ther.19, 137–144. 10.1038/gt.2011.172
45
Fry L. E. Peddle C. F. Barnard A. R. Mcclements M. E. Maclaren R. E. (2020). RNA Editing as a Therapeutic Approach for Retinal Gene Therapy Requiring Long Coding Sequences. Int. J. Mol. Sci.21, 777. 10.3390/ijms21030777
46
Fuster-García C. García-Bohórquez B. Rodríguez-Muñoz A. Aller E. Jaijo T. Millán J. M. et al (2021). Usher Syndrome: Genetics of a Human Ciliopathy. Int. J. Mol. Sci.22, 6723. 10.3390/ijms22136723
47
Gallo A. Vukic D. Michalík D. O’Connell M. A. Keegan L. P. (2017). ADAR RNA Editing in Human Disease; More to it Than Meets the I. Hum. Genet.136, 1265–1278. 10.1007/s00439-017-1837-0
48
Galvin O. Chi G. Brady L. Hippert C. Del Valle Rubido M. Daly A. et al (2020). The Impact of Inherited Retinal Diseases in the Republic of Ireland (ROI) and the United Kingdom (UK) from a Cost-Of-Illness Perspective. Opth14, 707–719. 10.2147/opth.s241928
49
Gao F.-J. Li J.-K. Chen H. Hu F.-Y. Zhang S.-H. Qi Y.-H. et al (2019). Genetic and Clinical Findings in a Large Cohort of Chinese Patients with Suspected Retinitis Pigmentosa. Ophthalmology126, 1549–1556. 10.1016/j.ophtha.2019.04.038
50
Gasiunas G. Young J. K. Karvelis T. Kazlauskas D. Urbaitis T. Jasnauskaite M. et al (2020). A Catalogue of Biochemically Diverse CRISPR-Cas9 Orthologs. Nat. Commun.11, 5512. 10.1038/s41467-020-19344-1
51
GBD 2019 Blindness and Vision Impairment Collaborators Vision Loss Expert Group of the Global Burden of Disease Study (2021). Causes of Blindness and Vision Impairment in 2020 and Trends over 30 years, and Prevalence of Avoidable Blindness in Relation to VISION 2020: the Right to Sight: an Analysis for the Global Burden of Disease Study. Lancet Glob. Health9, e144–e160. 10.1016/S2214-109X(20)30489-7
52
Georgiou M. Kalitzeos A. Patterson E. J. Dubra A. Carroll J. Michaelides M. (2018). Adaptive Optics Imaging of Inherited Retinal Diseases. Br. J. Ophthalmol.102, 1028–1035. 10.1136/bjophthalmol-2017-311328
53
Georgiou M. Fujinami K. Michaelides M. (2020). Retinal Imaging in Inherited Retinal Diseases. Ann. Eye Sci.5, 25. 10.21037/aes-20-81
54
Gilhooley M. J. Hickey D. G. Lindner M. Palumaa T. Hughes S. Peirson S. N. et al (2021). ON-bipolar Cell Gene Expression during Retinal Degeneration: Implications for Optogenetic Visual Restoration. Exp. Eye Res.207, 108553. 10.1016/j.exer.2021.108553
55
Gill J. S. Moosajee M. Dubis A. M. (2019). Cellular Imaging of Inherited Retinal Diseases Using Adaptive Optics. Eye33, 1683–1698. 10.1038/s41433-019-0474-3
56
Gong J. Cheung S. Fasso-Opie A. Galvin O. Moniz L. S. Earle D. et al (2021). The Impact of Inherited Retinal Diseases in the United States of America (US) and Canada from a Cost-Of-Illness Perspective. Opth15, 2855–2866. 10.2147/opth.s313719
57
Haffner M. E. (2005). The Food and Drug Administration's Office of Orphan Products Development: Incentives, grants, and Special Designations Speed Therapies for Orphan Diseases. Retina25, S89–s90. 10.1097/00006982-200512001-00045
58
Hamel C. P. (2007). Cone Rod Dystrophies. Orphanet J. Rare Dis.2, 7. 10.1186/1750-1172-2-7
59
Han I. C. Cheng J. L. Burnight E. R. Ralston C. L. Fick J. L. Thomsen G. J. et al (2020). Retinal Tropism and Transduction of Adeno-Associated Virus Varies by Serotype and Route of Delivery (Intravitreal, Subretinal, or Suprachoroidal) in Rats. Hum. Gene Ther.31, 1288–1299. 10.1089/hum.2020.043
60
Hanany M. Rivolta C. Sharon D. (2020). Worldwide Carrier Frequency and Genetic Prevalence of Autosomal Recessive Inherited Retinal Diseases. Proc. Natl. Acad. Sci. USA117, 2710–2716. 10.1073/pnas.1913179117
61
Hartong D. T. Berson E. L. Dryja T. P. (2006). Retinitis Pigmentosa. Lancet368, 1795–1809. 10.1016/s0140-6736(06)69740-7
62
Hauswirth W. W. (2014). Retinal Gene Therapy Using Adeno-Associated Viral Vectors: Multiple Applications for a Small Virus. Hum. Gene Ther.25, 671–678. 10.1089/hum.2014.2530
63
Heath Jeffery R. C. Mukhtar S. A. Mcallister I. L. Morgan W. H. Mackey D. A. Chen F. K. (2021). Inherited Retinal Diseases Are the Most Common Cause of Blindness in the Working-Age Population in Australia. Ophthalmic Genet.42, 431–439. 10.1080/13816810.2021.1913610
64
Holtan J. P. Selmer K. K. Heimdal K. R. Bragadóttir R. (2020). Inherited Retinal Disease in Norway - a Characterization of Current Clinical and Genetic Knowledge. Acta Ophthalmol.98, 286–295. 10.1111/aos.14218
65
Hu M. L. Edwards T. L. O’Hare F. Hickey D. G. Wang J.-H. Liu Z. et al (2021). Gene Therapy for Inherited Retinal Diseases: Progress and Possibilities. Clin. Exp. Optom.104, 444–454. 10.1080/08164622.2021.1880863
66
Huckfeldt R. M. Comander J. (2017). Management of Cystoid Macular Edema in Retinitis Pigmentosa. Semin. Ophthalmol.32, 43–51. 10.1080/08820538.2016.1228404
67
Hung S. S. Li F. Wang J.-H. King A. E. Bui B. V. Liu G.-S. et al (2018). Methods for In Vivo CRISPR/Cas Editing of the Adult Murine Retina. Methods Mol. Biol.1715, 113–133. 10.1007/978-1-4939-7522-8_9
68
Jacobson S. G. Acland G. M. Aguirre G. D. Aleman T. S. Schwartz S. B. Cideciyan A. V. et al (2006a). Safety of Recombinant Adeno-Associated Virus Type 2-RPE65 Vector Delivered by Ocular Subretinal Injection. Mol. Ther.13, 1074–1084. 10.1016/j.ymthe.2006.03.005
69
Jacobson S. G. Boye S. L. Aleman T. S. Conlon T. J. Zeiss C. J. Roman A. J. et al (2006b). Safety in Nonhuman Primates of Ocular AAV2-RPE65, a Candidate Treatment for Blindness in Leber Congenital Amaurosis. Hum. Gene Ther.17, 845–858. 10.1089/hum.2006.17.845
70
Jiang F. Doudna J. A. (2017). CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys.46, 505–529. 10.1146/annurev-biophys-062215-010822
71
Johnson A. A. Guziewicz K. E. Lee C. J. Kalathur R. C. Pulido J. S. Marmorstein L. Y. et al (2017). Bestrophin 1 and Retinal Disease. Prog. Retin. Eye Res.58, 45–69. 10.1016/j.preteyeres.2017.01.006
72
Kawasaki A. Crippa S. V. Kardon R. Leon L. Hamel C. (2012). Characterization of Pupil Responses to Blue and Red Light Stimuli in Autosomal Dominant Retinitis Pigmentosa Due toNR2E3Mutation. Invest. Ophthalmol. Vis. Sci.53, 5562–5569. 10.1167/iovs.12-10230
73
Koyanagi Y. Akiyama M. Nishiguchi K. M. Momozawa Y. Kamatani Y. Takata S. et al (2019). Genetic Characteristics of Retinitis Pigmentosa in 1204 Japanese Patients. J. Med. Genet.56, 662–670. 10.1136/jmedgenet-2018-105691
74
Li C. Samulski R. J. (2020). Engineering Adeno-Associated Virus Vectors for Gene Therapy. Nat. Rev. Genet.21, 255–272. 10.1038/s41576-019-0205-4
75
Liew G. Michaelides M. Bunce C. (2014). A Comparison of the Causes of Blindness Certifications in England and Wales in Working Age Adults (16-64 Years), 1999-2000 with 2009-2010. BMJ Open4, e004015. 10.1136/bmjopen-2013-004015
76
Ma D. J. Lee H.-S. Kim K. Choi S. Jang I. Cho S.-H. et al (2021). Whole-Exome Sequencing in 168 Korean Patients with Inherited Retinal Degeneration. BMC Med. Genomics14, 74. 10.1186/s12920-021-00874-6
77
Maeda A. Mandai M. Takahashi M. (2019). Gene and Induced Pluripotent Stem Cell Therapy for Retinal Diseases. Annu. Rev. Genom. Hum. Genet.20, 201–216. 10.1146/annurev-genom-083118-015043
78
Maeder M. L. Stefanidakis M. Wilson C. J. Baral R. Barrera L. A. Bounoutas G. S. et al (2019). Development of a Gene-Editing Approach to Restore Vision Loss in Leber Congenital Amaurosis Type 10. Nat. Med.25, 229–233. 10.1038/s41591-018-0327-9
79
Maguire A. M. Simonelli F. Pierce E. A. Pugh E. N. Jr. Mingozzi F. Bennicelli J. et al (2008). Safety and Efficacy of Gene Transfer for Leber's Congenital Amaurosis. N. Engl. J. Med.358, 2240–2248. 10.1056/nejmoa0802315
80
Maguire A. M. High K. A. Auricchio A. Wright J. F. Pierce E. A. Testa F. et al (2009). Age-dependent Effects of RPE65 Gene Therapy for Leber's Congenital Amaurosis: a Phase 1 Dose-Escalation Trial. Lancet374, 1597–1605. 10.1016/s0140-6736(09)61836-5
81
Maguire A. M. Russell S. Wellman J. A. Chung D. C. Yu Z.-F. Tillman A. et al (2019). Efficacy, Safety, and Durability of Voretigene Neparvovec-Rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy. Ophthalmology126, 1273–1285. 10.1016/j.ophtha.2019.06.017
82
Maguire A. M. Russell S. Chung D. C. Yu Z.-F. Tillman A. Drack A. V. et al (2021). Durability of Voretigene Neparvovec for Biallelic RPE65-Mediated Inherited Retinal Disease: Phase 3 Results at 3 and 4 Years. Ophthalmology128, 1460–1468. 10.1016/j.ophtha.2021.03.031
83
Maruyama R. Yokota T. (2020). Knocking Down Long Noncoding RNAs Using Antisense Oligonucleotide Gapmers. Methods Mol. Biol.2176, 49–56. 10.1007/978-1-0716-0771-8_3
84
Massof R. W. Wu L. Finkelstein D. Perry C. Starr S. J. Johnson M. A. (1984). Properties of Electroretinographic Intensity-Response Functions in Retinitis Pigmentosa. Doc Ophthalmol.57, 279–296. 10.1007/bf00143087
85
McClements M. E. Barnard A. R. Singh M. S. Charbel Issa P. Jiang Z. Radu R. A. et al (2019). An AAV Dual Vector Strategy Ameliorates the Stargardt Phenotype in AdultAbca4−/−Mice. Hum. Gene Ther.30, 590–600. 10.1089/hum.2018.156
86
McClements M. E. Staurenghi F. Maclaren R. E. Cehajic-Kapetanovic J. (2020). Optogenetic Gene Therapy for the Degenerate Retina: Recent Advances. Front. Neurosci.14, 570909. 10.3389/fnins.2020.570909
87
McGuigan D. B. 3rd Roman A. J. Cideciyan A. V. Matsui R. Gruzensky M. L. Sheplock R. et al (2016). Automated Light- and Dark-Adapted Perimetry for Evaluating Retinitis Pigmentosa: Filling a Need to Accommodate Multicenter Clinical Trials. Invest. Ophthalmol. Vis. Sci.57, 3118–3128. 10.1167/iovs.16-19302
88
Meng D. Ragi S. D. Tsang S. H. (2020). Therapy in Rhodopsin-Mediated Autosomal Dominant Retinitis Pigmentosa. Mol. Ther.28, 2139–2149. 10.1016/j.ymthe.2020.08.012
89
Merkle T. Merz S. Reautschnig P. Blaha A. Li Q. Vogel P. et al (2019). Precise RNA Editing by Recruiting Endogenous ADARs with Antisense Oligonucleotides. Nat. Biotechnol.37, 133–138. 10.1038/s41587-019-0013-6
90
Miller A. D. (1992). Human Gene Therapy Comes of Age. Nature357, 455–460. 10.1038/357455a0
91
Mitchell P. Liew G. Gopinath B. Wong T. Y. (2018). Age-Related Macular Degeneration. Lancet392, 1147–1159. 10.1016/s0140-6736(18)31550-2
92
Nishikura K. (2010). Functions and Regulation of RNA Editing by ADAR Deaminases. Annu. Rev. Biochem.79, 321–349. 10.1146/annurev-biochem-060208-105251
93
Nuzbrokh Y. Kassotis A. S. Ragi S. D. Jauregui R. Tsang S. H. (2020). Treatment-Emergent Adverse Events in Gene Therapy Trials for Inherited Retinal Diseases: A Narrative Review. Ophthalmol. Ther.9, 709–724. 10.1007/s40123-020-00287-1
94
O'Brien J. Hayder H. Zayed Y. Peng C. (2018). Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol.9, 402. 10.3389/fendo.2018.00402
95
Oishi M. Oishi A. Gotoh N. Ogino K. Higasa K. Iida K. et al (2014). Comprehensive Molecular Diagnosis of a Large Cohort of Japanese Retinitis Pigmentosa and Usher Syndrome Patients by Next-Generation Sequencing. Invest. Ophthalmol. Vis. Sci.55, 7369–7375. 10.1167/iovs.14-15458
96
Okamura K. Hagen J. W. Duan H. Tyler D. M. Lai E. C. (2007). The Mirtron Pathway Generates microRNA-Class Regulatory RNAs in Drosophila. Cell130, 89–100. 10.1016/j.cell.2007.06.028
97
Orlans H. O. Mcclements M. E. Barnard A. R. Martinez-Fernandez De La Camara C. Maclaren R. E. (2021). Mirtron-Mediated RNA Knockdown/replacement Therapy for the Treatment of Dominant Retinitis Pigmentosa. Nat. Commun.12, 4934. 10.1038/s41467-021-25204-3
98
Patrizi C. Llado M. Benati D. Iodice C. Marrocco E. Guarascio R. et al (2021). Allele-specific Editing Ameliorates Dominant Retinitis Pigmentosa in a Transgenic Mouse Model. Am. J. Hum. Genet.108, 295–308. 10.1016/j.ajhg.2021.01.006
99
Peddle C. F. Fry L. E. Mcclements M. E. Maclaren R. E. (2020). CRISPR Interference-Potential Application in Retinal Disease. Int. J. Mol. Sci.21, 2329. 10.3390/ijms21072329
100
Pendse N. D. Lamas V. Pawlyk B. S. Maeder M. L. Chen Z.-Y. Pierce E. A. et al (2019). In Vivo Assessment of Potential Therapeutic Approaches for USH2A-Associated Diseases. Adv. Exp. Med. Biol.1185, 91–96. 10.1007/978-3-030-27378-1_15
101
Piotter E. Mcclements M. E. Maclaren R. E. (2021). Therapy Approaches for Stargardt Disease. Biomolecules11, 1179. 10.3390/biom11081179
102
Pizzarello L. Abiose A. Ffytche T. Duerksen R. Thulasiraj R. Taylor H. et al (2004). VISION 2020: The Right to Sight: A Global Initiative to Eliminate Avoidable Blindness. Arch. Ophthalmol.122, 615–620. 10.1001/archopht.122.4.615
103
Pontikos N. Arno G. Jurkute N. Schiff E. Ba-Abbad R. Malka S. et al (2020). Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology127, 1384–1394. 10.1016/j.ophtha.2020.04.008
104
Prado D. A. Acosta-Acero M. Maldonado R. S. (2020). Gene Therapy beyond Luxturna: a New Horizon of the Treatment for Inherited Retinal Disease. Curr. Opin. Ophthalmol.31, 147–154. 10.1097/icu.0000000000000660
105
Puppo A. Cesi G. Marrocco E. Piccolo P. Jacca S. Shayakhmetov D. M. et al (2014). Retinal Transduction Profiles by High-Capacity Viral Vectors. Gene Ther.21, 855–865. 10.1038/gt.2014.57
106
Qian X. Wang J. Wang M. Igelman A. D. Jones K. D. Li Y. et al (2021). Identification of Deep-Intronic Splice Mutations in a Large Cohort of Patients With Inherited Retinal Diseases. Front. Genet.12, 647400. 10.3389/fgene.2021.647400
107
Ramsbottom S. A. Molinari E. Srivastava S. Silberman F. Henry C. Alkanderi S. et al (2018). Targeted Exon Skipping of a CEP290 Mutation Rescues Joubert Syndrome Phenotypes In Vitro and in a Murine Model. Proc. Natl. Acad. Sci. USA115, 12489–12494. 10.1073/pnas.1809432115
108
Ran F. A. Hsu P. D. Wright J. Agarwala V. Scott D. A. Zhang F. (2013). Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc.8, 2281–2308. 10.1038/nprot.2013.143
109
Rofagha S. Bhisitkul R. B. Boyer D. S. Sadda S. R. Zhang K. (2013). Seven-Year Outcomes in Ranibizumab-Treated Patients in ANCHOR, MARINA, and HORIZON. Ophthalmology120, 2292–2299. 10.1016/j.ophtha.2013.03.046
110
Roman A. J. Cideciyan A. V. Wu V. Garafalo A. V. Jacobson S. G. (2021). Full-field Stimulus Testing: Role in the Clinic and as an Outcome Measure in Clinical Trials of Severe Childhood Retinal Disease. Prog. Retin. Eye Res., 101000. 10.1016/j.preteyeres.2021.101000
111
Roosing S. Thiadens A. A. H. J. Hoyng C. B. Klaver C. C. W. Den Hollander A. I. Cremers F. P. M. (2014). Causes and Consequences of Inherited Cone Disorders. Prog. Retin. Eye Res.42, 1–26. 10.1016/j.preteyeres.2014.05.001
112
Ruby J. G. Jan C. H. Bartel D. P. (2007). Intronic microRNA Precursors that Bypass Drosha Processing. Nature448, 83–86. 10.1038/nature05983
113
Rukmini A. V. Milea D. Gooley J. J. (2019). Chromatic Pupillometry Methods for Assessing Photoreceptor Health in Retinal and Optic Nerve Diseases. Front. Neurol.10, 76. 10.3389/fneur.2019.00076
114
Russell S. Bennett J. Wellman J. A. Chung D. C. Yu Z.-F. Tillman A. et al (2017). Efficacy and Safety of Voretigene Neparvovec (AAV2-hRPE65v2) in Patients with RPE65 -Mediated Inherited Retinal Dystrophy: a Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet390, 849–860. 10.1016/s0140-6736(17)31868-8
115
Sahel J.-A. Léveillard T. (2018). Maintaining Cone Function in Rod-Cone Dystrophies. Adv. Exp. Med. Biol.1074, 499–509. 10.1007/978-3-319-75402-4_62
116
Sahel J.-A. Marazova K. Audo I. (2014). Clinical Characteristics and Current Therapies for Inherited Retinal Degenerations. Cold Spring Harbor Perspect. Med.5, a017111. 10.1101/cshperspect.a017111
117
Sahel J.-A. Boulanger-Scemama E. Pagot C. Arleo A. Galluppi F. Martel J. N. et al (2021). Partial Recovery of Visual Function in a Blind Patient after Optogenetic Therapy. Nat. Med.27, 1223–1229. 10.1038/s41591-021-01351-4
118
Saihan Z. Webster A. R. Luxon L. Bitner-Glindzicz M. (2009). Update on Usher Syndrome. Curr. Opin. Neurol.22, 19–27. 10.1097/wco.0b013e3283218807
119
Schwartz S. D. Regillo C. D. Lam B. L. Eliott D. Rosenfeld P. J. Gregori N. Z. et al (2015). Human Embryonic Stem Cell-Derived Retinal Pigment Epithelium in Patients with Age-Related Macular Degeneration and Stargardt's Macular Dystrophy: Follow-Up of Two Open-Label Phase 1/2 Studies. Lancet385, 509–516. 10.1016/s0140-6736(14)61376-3
120
Sen P. Bhargava A. George R. Ramesh S. V. Hemamalini A. Prema R. et al (2008). Prevalence of Retinitis Pigmentosa in South Indian Population Aged above 40 Years. Ophthalmic Epidemiol.15, 279–281. 10.1080/09286580802105814
121
Sharon D. Ben‐Yosef T. Goldenberg‐Cohen N. Pras E. Gradstein L. Soudry S. et al (2020). A Nationwide Genetic Analysis of Inherited Retinal Diseases in Israel as Assessed by the Israeli Inherited Retinal Disease Consortium (IIRDC). Hum. Mutat.41, 140–149. 10.1002/humu.23903
122
Smith C. I. E. Zain R. (2019). Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol.59, 605–630. 10.1146/annurev-pharmtox-010818-021050
123
Smith A. J. Carter S. P. Kennedy B. N. (2017). Genome Editing: the Breakthrough Technology for Inherited Retinal Disease?Expert Opin. Biol. Ther.17, 1245–1254. 10.1080/14712598.2017.1347629
124
Spaide R. F. (2003). Fundus Autofluorescence and Age-Related Macular Degeneration. Ophthalmology110, 392–399. 10.1016/s0161-6420(02)01756-6
125
Sparrow J. R. Boulton M. (2005). RPE Lipofuscin and its Role in Retinal Pathobiology. Exp. Eye Res.80, 595–606. 10.1016/j.exer.2005.01.007
126
Srivastava A. (2016). In Vivo tissue-tropism of Adeno-Associated Viral Vectors. Curr. Opin. Virol.21, 75–80. 10.1016/j.coviro.2016.08.003
127
Stone E. M. Andorf J. L. Whitmore S. S. Deluca A. P. Giacalone J. C. Streb L. M. et al (2017). Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology124, 1314–1331. 10.1016/j.ophtha.2017.04.008
128
Sun D. Schur R. M. Sears A. E. Gao S.-Q. Vaidya A. Sun W. et al (2020). Non-viral Gene Therapy for Stargardt Disease with ECO/pRHO-ABCA4 Self-Assembled Nanoparticles. Mol. Ther.28, 293–303. 10.1016/j.ymthe.2019.09.010
129
Tatour Y. Ben-Yosef T. (2020). Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics (Basel)10, 779. 10.3390/diagnostics10100779
130
Tavakoli S. Peynshaert K. Lajunen T. Devoldere J. Del Amo E. M. Ruponen M. et al (2020). Ocular Barriers to Retinal Delivery of Intravitreal Liposomes: Impact of Vitreoretinal Interface. J. Control. Release328, 952–961. 10.1016/j.jconrel.2020.10.028
131
Teo K. Y. C. Lee S. Y. Barathi A. V. Tun S. B. B. Tan L. Constable I. J. (2018). Surgical Removal of Internal Limiting Membrane and Layering of AAV Vector on the Retina under Air Enhances Gene Transfection in a Nonhuman Primate. Invest. Ophthalmol. Vis. Sci.59, 3574–3583. 10.1167/iovs.18-24333
132
Toualbi L. Toms M. Moosajee M. (2020). USH2A-Retinopathy: From Genetics to Therapeutics. Exp. Eye Res.201, 108330. 10.1016/j.exer.2020.108330
133
Trapani I. Auricchio A. (2019). Has Retinal Gene Therapy Come of Age? from Bench to Bedside and Back to Bench. Hum. Mol. Genet.28, R108–r118. 10.1093/hmg/ddz130
134
Trapani I. (2018). Dual AAV Vectors for Stargardt Disease. Methods Mol. Biol.1715, 153–175. 10.1007/978-1-4939-7522-8_11
135
Trigueros S. Domènech E. B. Toulis V. Marfany G. (2019). Vitro Gene Delivery in Retinal Pigment Epithelium Cells by Plasmid DNA-Wrapped Gold Nanoparticles. Genes (Basel)10, 289. 10.3390/genes10040289
136
Tsai Y.-T. Wu W.-H. Lee T.-T. Wu W.-P. Xu C. L. Park K. S. et al (2018). Clustered Regularly Interspaced Short Palindromic Repeats-Based Genome Surgery for the Treatment of Autosomal Dominant Retinitis Pigmentosa. Ophthalmology125, 1421–1430. 10.1016/j.ophtha.2018.04.001
137
Tsang S. H. Sharma T. (2018). Stargardt Disease. Adv. Exp. Med. Biol.1085, 139–151. 10.1007/978-3-319-95046-4_27
138
Tuohy G. P. Megaw R. (2021). A Systematic Review and Meta-Analyses of Interventional Clinical Trial Studies for Gene Therapies for the Inherited Retinal Degenerations (IRDs). Biomolecules11, 760. 10.3390/biom11050760
139
Van Schil K. Naessens S. Van De Sompele S. Carron M. Aslanidis A. Van Cauwenbergh C. et al (2018). Mapping the Genomic Landscape of Inherited Retinal Disease Genes Prioritizes Genes Prone to Coding and Noncoding Copy-Number Variations. Genet. Med.20, 202–213. 10.1038/gim.2017.97
140
Verbakel S. K. Van Huet R. A. C. Boon C. J. F. Den Hollander A. I. Collin R. W. J. Klaver C. C. W. et al (2018). Non-syndromic Retinitis Pigmentosa. Prog. Retin. Eye Res.66, 157–186. 10.1016/j.preteyeres.2018.03.005
141
Wang L. Zhang J. Chen N. Wang L. Zhang F. Ma Z. et al (2018). Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes (Basel)9, 360. 10.3390/genes9070360
142
Weisschuh N. Obermaier C. D. Battke F. Bernd A. Kuehlewein L. Nasser F. et al (2020). Genetic Architecture of Inherited Retinal Degeneration in Germany: A Large Cohort Study from a Single Diagnostic center over a 9‐year Period. Hum. Mutat.41, 1514–1527. 10.1002/humu.24064
143
Wells J. A. Glassman A. R. Ayala A. R. Jampol L. M. Bressler N. M. Bressler S. B. et al (2016). Aflibercept, Bevacizumab, or Ranibizumab for Diabetic Macular Edema:Two-Year Results from a Comparative Effectiveness Randomized Clinical Trial. Ophthalmology123, 1351–1359. 10.1016/j.ophtha.2016.02.022
144
Wiley L. A. Burnight E. R. Kaalberg E. E. Jiao C. Riker M. J. Halder J. A. et al (2018). Assessment of Adeno-Associated Virus Serotype Tropism in Human Retinal Explants. Hum. Gene Ther.29, 424–436. 10.1089/hum.2017.179
145
Williams S. C. P. Deisseroth K. (2013). Optogenetics. Proc. Natl. Acad. Sci.110, 16287. 10.1073/pnas.1317033110
146
Wirth T. Parker N. Ylä-Herttuala S. (2013). History of Gene Therapy. Gene525, 162–169. 10.1016/j.gene.2013.03.137
147
Wong T. Y. Cheung C. M. G. Larsen M. Sharma S. Simó R. (2016). Diabetic Retinopathy. Nat. Rev. Dis. Primers2, 16012. 10.1038/nrdp.2016.12
148
Wu Z. Asokan A. Samulski R. J. (2006). Adeno-associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther.14, 316–327. 10.1016/j.ymthe.2006.05.009
149
Wuthisiri W. Lingao M. D. Capasso J. E. Levin A. V. (2013). Lyonization in Ophthalmology. Curr. Opin. Ophthalmol.24, 389–397. 10.1097/icu.0b013e3283641f91
150
Xu M. Zhai Y. Macdonald I. M. (2020). Visual Field Progression in Retinitis Pigmentosa. Invest. Ophthalmol. Vis. Sci.61, 56. 10.1167/iovs.61.6.56
151
Xue K. MacLaren R. E. (2020). Antisense Oligonucleotide Therapeutics in Clinical Trials for the Treatment of Inherited Retinal Diseases. Expert Opin. Investig. Drugs29, 1163–1170. 10.1080/13543784.2020.1804853
152
Yang L. Fujinami K. Fujinami K. Ueno S. Kuniyoshi K. Hayashi T. et al (2020). Genetic Spectrum of EYS-Associated Retinal Disease in a Large Japanese Cohort: Identification of Disease-Associated Variants with Relatively High Allele Frequency. Sci. Rep.10, 5497. 10.1038/s41598-020-62119-3
153
Yohe S. Sivasankar M. Ghosh A. Ghosh A. Holle J. Murugan S. et al (2020). Prevalence of Mutations in Inherited Retinal Diseases: A Comparison between the United States and India. Mol. Genet. Genomic Med.8, e1081. 10.1002/mgg3.1081
154
Zeitz C. Robson A. G. Audo I. (2015). Congenital Stationary Night Blindness: an Analysis and Update of Genotype-Phenotype Correlations and Pathogenic Mechanisms. Prog. Retin. Eye Res.45, 58–110. 10.1016/j.preteyeres.2014.09.001
155
Zhang F. Vierock J. Yizhar O. Fenno L. E. Tsunoda S. Kianianmomeni A. et al (2011). The Microbial Opsin Family of Optogenetic Tools. Cell147, 1446–1457. 10.1016/j.cell.2011.12.004
156
Zhang X.-H. Tee L. Y. Wang X.-G. Huang Q.-S. Yang S.-H. (2015). Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. - Nucleic Acids4, e264. 10.1038/mtna.2015.37
157
Zhang B. (2021). CRISPR/Cas Gene Therapy. J. Cel. Physiol.236, 2459–2481. 10.1002/jcp.30064
Summary
Keywords
inherited retinal diseases (IRDs), gene tharapy, adeno-associated virus, RNA editing, CRISPR, optogenetic, antisense oligonucleotides, retina
Citation
Fenner BJ, Tan T-E, Barathi AV, Tun SBB, Yeo SW, Tsai ASH, Lee SY, Cheung CMG, Chan CM, Mehta JS and Teo KYC (2022) Gene-Based Therapeutics for Inherited Retinal Diseases. Front. Genet. 12:794805. doi: 10.3389/fgene.2021.794805
Received
14 October 2021
Accepted
14 December 2021
Published
07 January 2022
Volume
12 - 2021
Edited by
Jesse Sengillo, University of Miami Health System, United States
Reviewed by
Jiaxing Wang, Emory University, United States
Qingnan Liang, Baylor College of Medicine, United States
Updates
Copyright
© 2022 Fenner, Tan, Barathi, Tun, Yeo, Tsai, Lee, Cheung, Chan, Mehta and Teo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelvin Y. C. Teo, kelvin.teo.y.c@singhealth.com.sg
This article was submitted to Human and Medical Genomics, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.