 Weizhong Lu1,2,3
Weizhong Lu1,2,3 Jiawei Shen
Jiawei Shen- 1School of Electronic and Information Engineering, Suzhou University of Science and Technology, Suzhou, China
- 2Suzhou Key Laboratory of Virtual Reality Intelligent Interaction and Application Technology, Suzhou University of Science and Technology, Suzhou, China
- 3Provincial Key Laboratory for Computer Information Processing Technology, Soochow University, Suzhou, China
- 4Suzhou Industrial Park Institute of Services Outsourcing, Suzhou, China
Membrane proteins are an essential part of the body’s ability to maintain normal life activities. Further research into membrane proteins, which are present in all aspects of life science research, will help to advance the development of cells and drugs. The current methods for predicting proteins are usually based on machine learning, but further improvements in prediction effectiveness and accuracy are needed. In this paper, we propose a dynamic deep network architecture based on lifelong learning in order to use computers to classify membrane proteins more effectively. The model extends the application area of lifelong learning and provides new ideas for multiple classification problems in bioinformatics. To demonstrate the performance of our model, we conducted experiments on top of two datasets and compared them with other classification methods. The results show that our model achieves high accuracy (95.3 and 93.5%) on benchmark datasets and is more effective compared to other methods.
1 Introduction
The biological cell’s daily activities are associated with membranes, without which it would not be possible to form a living structure. The essential proteins that make up membranes are the lipids and proteins that are the main components of membranes. In the present biological research, there are eight types of membrane proteins: 1) single-span 1; 2) single-span 2; 3) single-span 3; 4) single-span 4; 5) multi-span; 6) lipid-anchor; 7) GPI-anchor and (8) peripheral (Cedano et al., 1997).
Bioinformatics is present in all aspects of the biological sciences, and how to use computers to classify proteins efficiently and accurately has been a hot research problem in the direction of bioinformatics and computer science. Although traditional physicochemical as well as biological experiments are desirable in terms of predictive accuracy, these methods are too cumbersome and require a great deal of human and material resources. To save time and financial costs, and to better understand the structure and function of membrane proteins, a number of calculations have been developed to efficiently discriminate between protein types (Feng and Zhang, 2000; Cai et al., 2004; Zou et al., 2013; Wei et al., 2017; Zhou et al., 2021; Zou et al., 2020; Qian et al., 2021; Ding et al., 2021; Ding et al., 2021; Zou et al., 2021). The extant methods are in large part improvements on Chou’s algorithm (Chou and Elrod, 1999). Song et al. (Lu et al., 2020) used Chou’s 5-step method to extract evolutionary information to input to a support vector machine for protein prediction. Cao and Lu (Lu et al., 2021) avoided loss of information due to truncation by introducing a fag vector and used a variable length dynamic two-way gated cyclic unit model to predict protein. Yang (Wu et al., 2019) designed a reward function to model the protein input under full-state reinforcement learning. Wu and Huang (Wu et al., 2019) et al. used random forests to build their own model and used binary reordering to make their predictions more efficient. To avoid the limitations of overfitting, Lu and Tang (Lu et al., 2019) et al. used an energy filter to make the sequence length follow the model adaptively.
In most methods of machine learning, predictions are made using fixed models for different kinds of proteins, which generally suffer from two problems. Firstly, they do not allow for incremental learning, and secondly, they do not consider task-to-task connections at the task level. Lifelong learning approaches aim to bridge these two issues. Lifelong machine learning methods were first proposed by Thrun and Mitchell (Thrun and Mitchell, 1995), who viewed each task as a binary problem to classify all tasks. A number of memory-based and neural network-based approaches to lifelong learning were then proposed and refined by Silver (Silver, 1996; Silver and Mercer, 2002; Silver et al., 2015) et al. Ruvolo and Eaton (Ruvolo and Eaton, 2013) proposed the Efficient Lifelong Learning Algorithm (ELLA), which greatly enhanced the algorithm proposed by Kumar (Kumar and Daume, 2012) et al. for multi-task learning (MTL). Ruvolo and Eaton (Ruvolo and Eaton, 2013) viewed lifelong learning as a real-time task selection process, Chen (Chen et al., 2015) proposed a lifelong learning algorithm based on plain Bayesian classification, and Shu (Shu et al., 2017) et al. investigated the direction of lifelong learning by improving the conditional random field model. Mazumder (Chen and Liu, 2014) investigated human-machine conversational machines and enabled chatbots to learn new knowledge in the process of chatting with humans. Chen, Liu and Wang (Wang et al., 2016) proposed a number of lifelong topic modeling methods to mine topics from historical tasks and apply them to new topic discovery. Shu et al. proposed a relaxed labeling approach to solve lifelong unsupervised learning tasks. Chen and Liu (Chen and Liu, 2019) provide more detailed information on the direction of lifelong machine learning in this book.
After a lot of research and careful selection, we ended up using a DSN model based on sequence information and lifetime learning of membrane proteins themselves. First, we processed the membrane protein sequence dataset based on BLAST (Altschul et al., 1997) to obtain the scoring matrix (PSSM) (Dehzangi et al., 2017; Sharma et al., 2018; Yosvany et al., 2018; Chandra et al., 2019). Then, we extracted valid features from the PSSM by the averaging block method (Avblock) (Shen et al., 2019), the discrete wavelet transforms method (DWT) (Shen et al., 2017; Wang et al., 2017), the discrete cosine transforms method (DCT) (Ahmed et al., 1974), the histogram of oriented gradients method (HOG) (Qian et al., 2021)and the Pse-PSSM method. The features extracted by these five methods are then stitched end-to-end and fed into our model for prediction. Finally, the performance is evaluated by random validation tests and independent tests. Through the results we can see that our model achieves good prediction results in the case of predicting membrane protein types. Figure 1 shows a sketch of the main research in this paper.
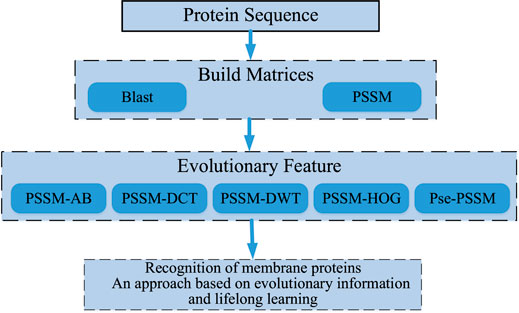
FIGURE 1. The summary of the main research.
2 Materials and Methods
In this experiment, the sequence of discriminating membrane protein types can be broadly divided into three steps: creating the required model, performing training and testing of the model, and making predictions and conducting analysis of the results. Firstly, the features are extracted from the processed dataset. Next, the features are integrated into a lifelong learning model for prediction. Finally, the sequence information of the membrane proteins is transformed into algorithmic information, which is then analyzed and predicted using the model. Figure 2 shows the research infrastructure for this approach.
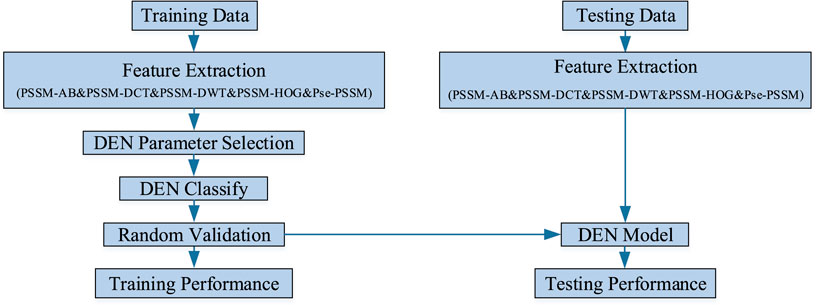
FIGURE 2. The research infrastructure for this approach.
2.1 Analysis of Data Sets
In order to test the performance of our lifelong learning model, we experimentally selected two membrane protein datasets for testing, Data 1 and Data 2. The specifics of the two datasets are shown in Table 1. Data 1 and Data 2 include eight membrane protein types. Data 1 is from the work of Chou (Chou and Shen, 2007) et al. The training and test sets were randomly obtained from Swiss-Prot (Boeckmann et al., 2003) by percentage assignment, which ensured that the quantities of these two sequences are consistent. Data 2 is from the work of Chen (Chen and Li, 2013) et al. where they used the CD-hit (Li and Godzik, 2006) method to remove redundant sequences from dataset 1 so that no two-by-two sequences would have less than 40% identity.
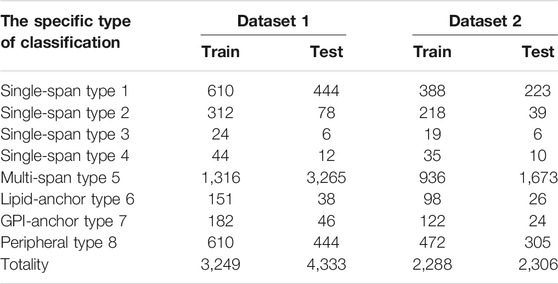
TABLE 1. The sample sizes for the two different data sets used in this experiment.
2.2 Extracting the Message of Evolutionary Conservatism
The PSSM used in this experiment is the “Position-Specific Scoring Matrix.” This scoring matrix stores the sequence information of membrane proteins. We use the PSSM matrix (Shen et al., 2017; Ding et al., 2017; Shen et al., 2019) for membrane protein prediction because it reflects the evolutionary information of membrane proteins very well. For any membrane protein sequence, such as Q, the PSSM can be derived by PSI-BLAST (Altschul et al., 1997), after several iterations. First it forms the PSSM based on the first search result, then it performs the next step which is the second search based on the first search result, then it continues with the second search result for another time and repeats this process until the target is searched for the best result. As the performance of the experimental results is best after three iterations, we generally adopt three iterations as the setting. The value of its E is 0.001. Assume that the sequence Q =
In addition, the expression below shows the representation of
where
2.2.1 Pse-Pssm
Pse-PSSM (pseudo-PSSM) is a feature extraction method often used in membrane protein prediction (Chou and Shen, 2007). PSSM matrices are often used in the characterization of membrane proteins. This feature extraction method, which aims to preserve PSSM biological information through pseudo amino acids, is expressed as follows:
The
where
2.2.2 Average Blocks
The AB method was first proposed by Huang et al. Its full name is the averaging block methodology (AvBlock) (Jong Cheol Jeong et al., 2011). When feature extraction is performed for PSSM, the extracted feature values are diverse because the size of individual features is different and the abundance of amino acids also varies in the individual membrane proteins. To solve this type of problem, we can average the features for the local features of the PSSMs. Inside each module after averaging, 5% of the membrane protein sequences are covered, and this method is the AB feature extraction method. When performing the AB method feature extraction on the PSSM, it is not necessary to consider the sequence length of the membrane proteins. When we split the PSSM matrix by rows, it becomes a block of size L/20 each, with 20 blocks. After this operation, every 20 features form a block. the AB formulation is as follows:
Where N/20 is the size of j blocks and
2.2.3 Discrete Wavelet Transform
We refer to a discrete wavelet transform feature extraction method as DWT, which uses the concepts of frequency and position (Nanni et al., 2012). It is because we can consider the membrane protein sequence as a picture, then matrix the sequence and extract the coefficient information from the matrix by DWT. This method was first suggested by Nanni et al.
In addition to this, we refer to the projection of the signal
where in the above equation, the scale variable is denoted by a, the translational variable by b, and
where N is the length of the discrete signal and the low-pass and high-pass filters are g and h, respectively.
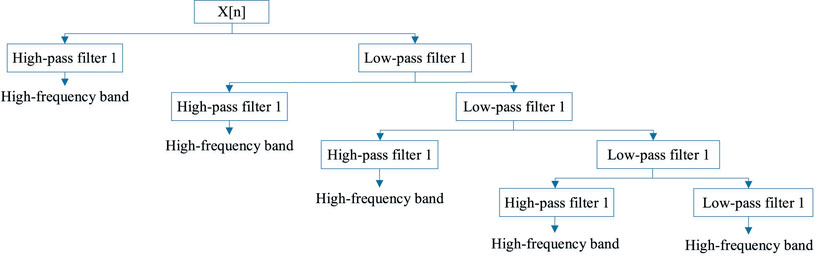
FIGURE 3. The 4-stage DWT structure.
2.2.4 Discrete Cosine Transform
The DCT (Ahmed et al., 1974), known as the Discrete Cosine Transform, converts a signal to its fundamental frequency by means of a linearly separable transform. This method has been widely used in the field of image compression. In this experiment, we compressed the PSSM matrix of membrane proteins using a 2-dimensional DCT (2D-DCT). 2D-DCT is defined as follows:
The mission of the DCT is to convert a uniformly distributed information density into an uneven distribution. Once its length and signal have been converted, the most important part of the information is collected in the low frequency section of the PSSM, that is in the middle and top-left corner.
2.2.5 Histogram of Oriented Gradient
Histogram of Oriented Gradients (HOG), which is a method for describing features, is mainly used in computer vision. In this experiment, to handle a PSSM matrix using the HOG method, it is first necessary to look at it as a particular image matrix. In the first step, the horizontal gradient values and vertical gradient values of the PSSM are used to derive the direction and size of the gradient matrix. In the second step, the gradient matrix is divided into 25 sub-matrices by direction and size. In the third step, conversion of the results generated in the second step is carried out according to the requirement to generate 10 histogram channels per sub-matrix.
2.3 Lifelong Learning
Lifelong learning (Thrun and Mitchell, 1995), like machine learning, can be divided into the directions of lifelong supervised learning, lifelong unsupervised learning and lifelong reinforcement learning. The lifelong machine learning part of the study focuses on whether the model can be extended when new categories of categories are added to the model. When the current model has been classified into n categories, if a new class of data is added, the model can somehow be adaptively expanded to classify n + 1 category. Multi-task learning models and lifelong machine learning are easily translatable to each other if we have all the original data. Whenever a new category is added to the original category and needs to be classified, only one new category needs to be added and then all the training data can be trained again to expand the new category. One obvious disadvantage of this strategy is that it wastes a lot of computational time to compute each new class, and if too many new classes are added, it may lead to changes in the model architecture for multi-task learning. This model therefore uses a dynamically scalable network to better perform incremental learning of the added tasks. Assume that the current model has successfully classified Class 1, Class 2, ... , Class n. When the new data class Class-new is added, the model does not need to train all the data from scratch, but only needs to expand the overall model by adding n new binary classification models. The simple flow of the lifelong learning model is shown in Figure 4.
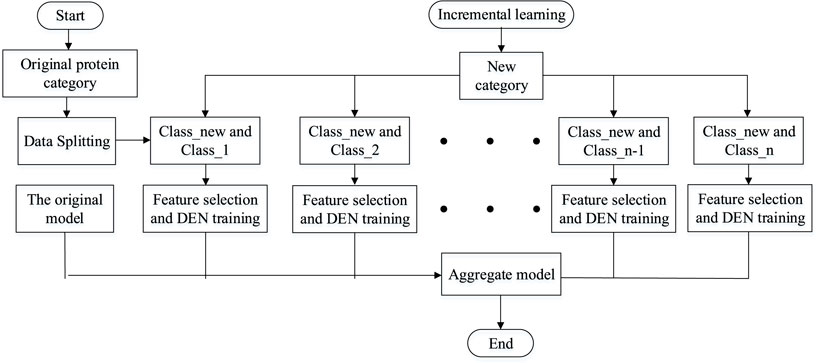
FIGURE 4. The simple flow of the lifelong learning model.
2.4 Dynamically Scalable Networks
Dynamically scalable networks are incremental training of deep neural networks for lifelong learning, for which there will be an unknown amount and unknown distribution of data to be trained fed into our model in turn. To expand on this, there are now T a sequence of task learning models, t = 1, .... , t, .... , T is unbounded T in which the tasks at time point t carry training data
where L is task specific loss function,
To counter these problems that arise in the course of lifelong learning, we allow the knowledge generated in previous tasks to be used to the maximum extent possible. At the same time, it is allowed to dynamically extend its capabilities when mechanically accumulated knowledge does not explain well for emerging tasks. Figure 5 and Algorithm 1 illustrate our progressive learning process.
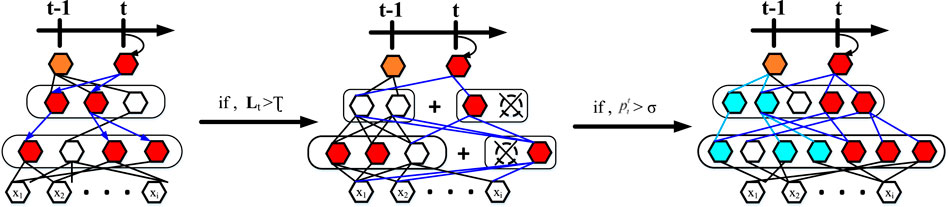
FIGURE 5. Incremental learning for dynamically scalable networks.
3 Experiment Results
In this subsection we will have an analysis of the capabilities of the respective modelling and methodological approaches. Furthermore, the modelling we used in that context was compared with other available methods on separate datasets.
3.1 Assessment Measurements
To evaluate our lifelong learning model better, we chose several parameters: sample all-prediction accuracy (ACC), single sample specificity (SP), single sample sensitivity (SN), and Mathews correlation coefficient. These metrics are widely used in the analysis of biological sequence information:
For the above equation, true trueness (TP) refers to the number of true samples correctly predicted; false positivity (FP) refers to the number of true samples incorrectly predicted; true negativity (TN) refers to the number of negative samples correctly predicted; and false negativity (FN) refers to the number of negative samples erroneously predicted (Chou et al., 2011a; Chou et al., 2011b; Chou, 2013).
3.2 Situational Analysis of Two Data Sets
The lengths of the datasets used in our experiments are shown in Figure 6. Most of the membrane proteins in dataset 1 and dataset 2 have a similar length distribution because of their specific type. To better demonstrate the superiority of lifelong learning for membrane protein classification, we calculated the amino acid frequencies for all protein types in the experiment, as shown in Figure 7.
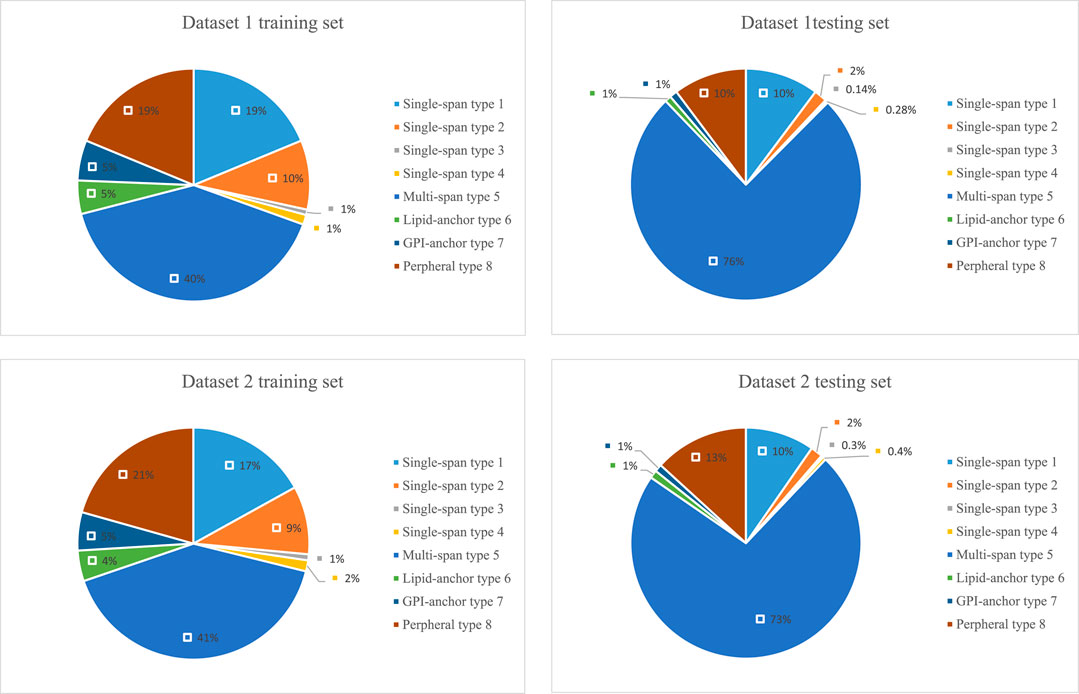
FIGURE 6. Distribution of the lengths of the training and test sets in the two datasets.
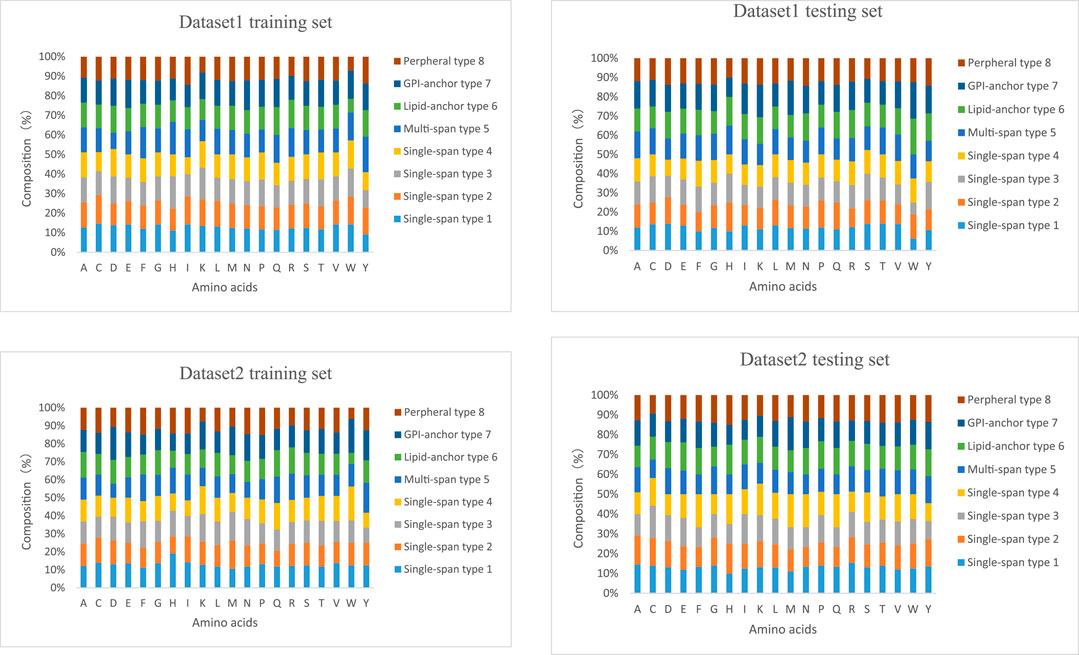
FIGURE 7. Component composition of the training and test sets of the two datasets.
3.3 The Forecasting Results for Dataset 1
The PSSM sequence matrix contains important genetic information required for protein prediction. Many elements of biological evolution, such as the stability of the three-dimensional structure and the aggregation of proteins, can have an impact on the storage and alteration of sequences. These elements demonstrate that PSSM captures important information about ligand binding. Thus, proving the validity of the PSSM characterization method.
We will compare the model methods we used in this use with other existing methods in terms of prediction accuracy on dataset 1. The methods involved in the comparison are MemType-2L (Chou and Shen, 2007) and predMPT (Chen and Li, 2013). Details can be found in Table 2, where it is clear that our model approach has an overall ACC of 95.3%. 3.7% higher than MemType-2L’s 91.6%, 2.7% higher than predMPT’s 92.6%, and 2.4% higher than Average weights’ 92.7%. In the independent test set, our method was superior for membrane protein type 2 (88.4%), type 4 (83.3%), type 5 (96.3%) and type 7 (100%).
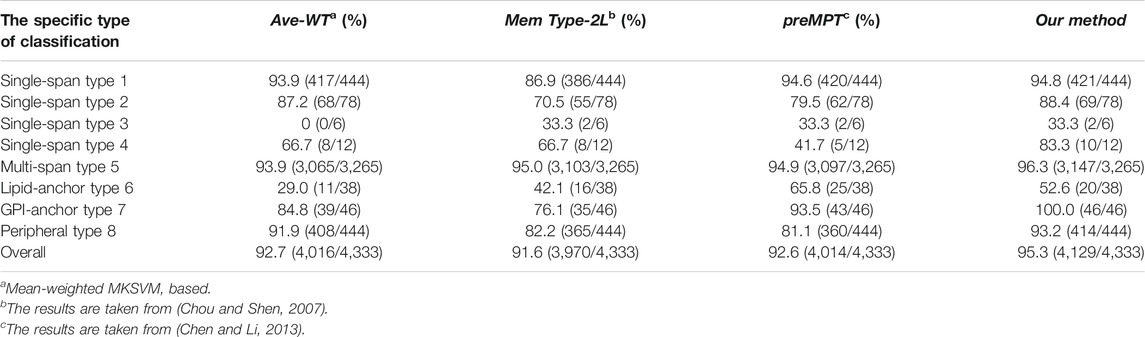
TABLE 2. The performance exhibited by different models on dataset 1.
3.4 The Forecasting Results for Dataset 2
As a solution to the possible problem of untimely updates in Data 1, Chen and Li (Chen and Li, 2013) used the Swissprot annotation method to update Data Set 1, resulting in a new dataset of membrane proteins (Data Set 2). The results of the comparison using Dataset 2 are presented in Table 3. The overall average accuracy of our models was 3.2% higher than the predMPT method (90.3%). Even though they added features such as 3D structure to the predMPT prediction session, our model performance was clearly higher than it. We outperformed it by 2.6% in terms of prediction accuracy for type 1 (94.1 vs. 91.5%). In contrast, analog 5 outperformed it by 1.3% (94.1 vs. 92.8%).

ALGORITHM 1. Incremental Learning for Dynamically Scalable Networks.
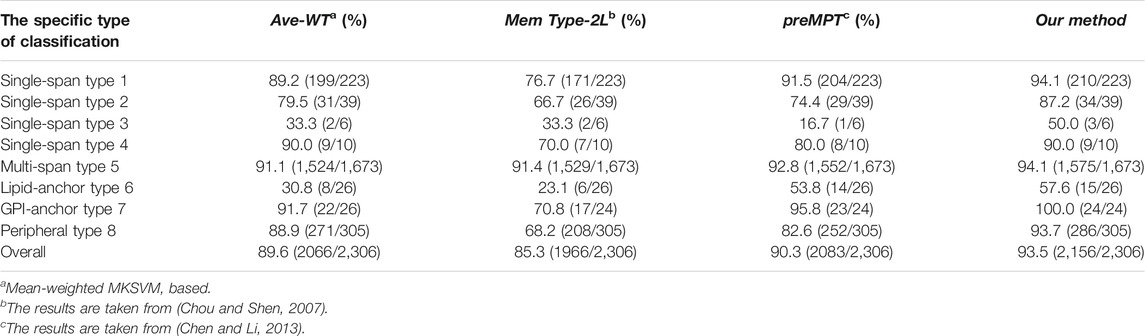
TABLE 3. The performance exhibited by different models on dataset 2.
4 Conclusion and Discussion
In previous work, investigators have often used the PseAAC (Chou, 2001) approach to identify membrane protein types, and this approach has indeed performed well in the field of protein classification. Using Chou’s operation (Chou and Shen, 2007) on feature extraction from PSSM, we were inspired to use the five methods of Pse-pssm, DCT, AvBlock, HOG and DWT to extract features. In order to avoid the low accuracy of a single feature extraction method, we integrated the above five methods together and fed the integrated features into our DSN model method.
Our constructed lifelong learning dynamic network model proved to achieve superior results on different datasets (95.3 and 93.5%). However, the prediction of some small sample affiliations by the methodology has not been as accurate as we had anticipated. In order to improve the performance of this model, we will consider improving our own features and combining some other feature extraction methods, and adjusting the parameters of our model in our future research.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://github.com/sjw-cmd/DATA.git.
Author Contributions
WL proposed the original idea. JS and HW designed the framework and the experiments. WL, JS and YZ performed the experiments and the primary data analysis. WL and JS wrote the manuscript. YQ, QF and XC modified the codes and the manuscript. All authors contributed to the manuscript. All authors read and approved the final manuscript.
Funding
This paper is supported by the National Natural Science Foundation of China (No.61902272, 62073231, 61772357, 62176175, 61876217, 61902271), National Research Project (2020YFC2006602), Provincial Key Laboratory for Computer Information Processing Technology, Soochow University (KJS2166).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge and thank the reviewers for their suggestions that allowed the improvement of our manuscript.
References
Ahmed, N., Natarajan, T., and Rao, K. R. (1974). Discrete Cosine Transform. IEEE Trans. Comput. C-23 (1), 90–93. doi:10.1109/t-c.1974.223784
Altschul, S., Madden, T., Schffer, A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped Blast and Psi-Blast: a New Generation of Protein Database Search Programs. Nucleic Acids Res. 25 (17), 3389–3402. doi:10.1093/nar/25.17.3389
Boeckmann, B., Bairoch, A., Apweiler, R., et al. (2003). The Swiss-Prot Protein Knowledgebase and its Supplement Trembl in 2003. Nucleic Acids Res. 31 (1), 365–370. doi:10.1093/nar/gkg095
Cai, Y.-D., Ricardo, P.-W., Jen, C.-H., and Chou, K.-C. (2004). Application of Svm to Predict Membrane Protein Types. J. Theor. Biol. 226 (4), 373–376. doi:10.1016/j.jtbi.2003.08.015
Cedano, J., Aloy, P., Pérez-Pons, J. A., and Querol, E. (1997). Relation between Amino Acid Composition and Cellular Location of Proteins 1 1Edited by F. E. Cohen. J. Mol. Biol. 266 (3), 594–600. doi:10.1006/jmbi.1996.0804
Chandra, A. A., Sharma, A., Dehzangi, A., and Tsunoda, T. (2019). Evolstruct-phogly: Incorporating Structural Properties and Evolutionary Information from Profile Bigrams for the Phosphoglycerylation Prediction. BMC Genomics 19 (9), 984. doi:10.1186/s12864-018-5383-5
Chen, Y.-K., and Li, K.-B. (2013). Predicting Membrane Protein Types by Incorporating Protein Topology, Domains, Signal Peptides, and Physicochemical Properties into the General Form of Chou's Pseudo Amino Acid Composition. J. Theor. Biol. 318 (1), 1–12. doi:10.1016/j.jtbi.2012.10.033
Chen, Z., and Liu, B. (2019). Lifelong Machine learning[M]. San Rafael, CA: Morgan & Claypool Publishers, 1207.
Chen, Z., and Liu, B. (2014). “Topic Modeling Using Topics from many Domains, Lifelong Learning and Big Data[C],” in Proceedings of the 31th International Conference on Machine Learning (Beijing), 703711.
Chen, Z., Ma, N., and Liu, B. (2015). “Lifelong Learning for Sentiment Classification[C],” in Proceedings of the 53rd Annual Meeting of the Association for Computational Linguistics (Beijing), 750756.
Chou, K.-C., and Elrod, D. W. (1999). Prediction of Membrane Protein Types and Subcellular Locations. Proteins 34 (1), 137–153. doi:10.1002/(sici)1097-0134(19990101)34:1<137:aid-prot11>3.0.co;2-o
Chou, K.-C., and Shen, H.-B. (2007). Memtype-2L: a Web Server for Predicting Membrane Proteins and Their Types by Incorporating Evolution Information through Pse-Pssm. Biochem. Biophysical Res. Commun. 360 (2), 339–345. doi:10.1016/j.bbrc.2007.06.027
Chou, K.-C. (2013). Some Remarks on Predicting Multi-Label Attributes in Molecular Biosystems. Mol. Biosyst. 9 (6), 1092–1100. doi:10.1039/c3mb25555g
Chou, K.-C., Wu, Z.-C., and Xiao, X. (2011). iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins. Plos One 6 (3), e18258. doi:10.1371/journal.pone.0018258
Chou, K. C. (2001). Prediction of Protein Cellular Attributes Using Pseudo-amino Acid Composition. Proteins 43 (6), 246–255. doi:10.1002/prot.1035
Chou, K. C., Wu, Z. C., and Xiao, X. (2011). Iloc-hum: Using the Accumulation-Label Scale to Predict Subcellular Locations of Human Proteins with Both Single and Multiple Sites. Mol. Biosyst. 8 (2), 629–641. doi:10.1039/c1mb05420a
Dehzangi, A., López, Y., Lal, S. P., Taherzadeh, G., Michaelson, J., Sattar, A., et al. (2017). Pssm-suc: Accurately Predicting Succinylation Using Position Specific Scoring Matrix into Bigram for Feature Extraction. J. Theor. Biol. 425, 97–102. doi:10.1016/j.jtbi.2017.05.005
Ding, Y., Tang, J., and Guo, F. (2021). Identification of Drug-Target Interactions via Multi-View Graph Regularized Link Propagation Model. Neurocomputing 461, 618–631. doi:10.1016/j.neucom.2021.05.100
Ding, Y., Tang, J., and Guo, F. (2017). Identification of Protein-Ligand Binding Sites by Sequence Information and Ensemble Classifier. J. Chem. Inf. Model. 57 (12), 3149–3161. doi:10.1021/acs.jcim.7b00307
Ding, Y., Yang, C., Tang, J., and Guo, F. (2021). Identification of Protein-Nucleotide Binding Residues via Graph Regularized K-Local Hyperplane Distance Nearest Neighbor Model. Appl. Intell. (10), 1–15. doi:10.1007/s10489-021-02737-0
Feng, Z.-P., and Zhang, C.-T. (2000). Prediction of Membrane Protein Types Based on the Hydrophobic index of Amino Acids. J. Protein Chem. 19 (4), 269–275. doi:10.1023/a:1007091128394
Jong Cheol Jeong, J., Xiaotong Lin, X., and Xue-Wen Chen, X. (2011). On Position-specific Scoring Matrix for Protein Function Prediction. Ieee/acm Trans. Comput. Biol. Bioinf. 8 (2), 308–315. doi:10.1109/tcbb.2010.93
Kumar, A., and DaumeIII, H. (2012). “Learning Task Grouping and Overlap in Multitask Learning[C],” in Proceedings of the 29th International Conference on Machine Learning (Edinburgh), 112.
Li, W., and Godzik, A.(2006). Cd-hit: a Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 22 (13), 1658–1659. doi:10.1093/bioinformatics/btl158
Lu, W., Song, Z., Ding, Y., Wu, H., Cao, Y., Zhang, Y., et al. (2020). Use Chou's 5-Step Rule to Predict DNA-Binding Proteins with Evolutionary Information. Biomed. Res. Int. 2020, 6984045. doi:10.1155/2020/6984045
Lu, W., Cao, Y., Wu, H., Ding, Y., Song, Z., Zhang, Y., et al. (2021). Research on RNA Secondary Structure Predicting via Bidirectional Recurrent Neural Network. BMC Bioinformatics 22, 431. doi:10.1186/s12859-021-04332-z
Lu, W., Tang, Y., Wu, H., Huang, H., Fu, Q., Qiu, J., et al. (2019). Predicting RNA Secondary Structure via Adaptive Deep Recurrent Neural Networks with Energy-Based Filter. BMC Bioinformatics 20, 684. doi:10.1186/s12859-019-3258-7
Nanni, L., Brahnam, S., and Lumini, A. (2012). Wavelet Images and Chou's Pseudo Amino Acid Composition for Protein Classification. Amino Acids 43 (2), 657–665. doi:10.1007/s00726-011-1114-9
Qian, Y., Meng, H., Lu, W., Liao, Z., Ding, Y., and Wu, H. (2021). Identification of DNA-Binding Proteins via Hypergraph Based Laplacian Support Vector Machine. Cbio 16. doi:10.2174/1574893616666210806091922
Ruvolo, P., and Eaton, E. (2013). “Active Task Selection for Lifelong Machine Learning[C],” in Proceedings of the Twenty-seventh AAAI Conference on Artificial Intelligence (Washington), 862868.
Ruvolo, P., and Eaton, E. (2013). “Ella: An Efficient Lifelong Learning Algorithm[C],” in Proceedings of the 30th International Conference on Machine Learning (Atlanta), 507515.
Sharma, R., Raicar, G., Tsunoda, T., Patil, A., and Sharma, A. (2018). Opal: Prediction of Morf Regions in Intrinsically Disordered Protein Sequences. Bioinformatics 34 (11), 1850–1858. doi:10.1093/bioinformatics/bty032
Shen, C., Ding, Y., Tang, J., Song, J., and Guo, F. (2017). Identification of DNA-Protein Binding Sites through Multi-Scale Local Average Blocks on Sequence Information. Molecules 22 (12), 2079. doi:10.3390/molecules22122079
Shen, C., Ding, Y., Tang, J., Xu, X., and Guo, F. (2017). An Ameliorated Prediction of Drug-Target Interactions Based on Multi-Scale Discrete Wavelet Transform and Network Features. Ijms 18 (8), 1781. doi:10.3390/ijms18081781
Shen, Y., Tang, J., and Guo, F. (2019). Identification of Protein Subcellular Localization via Integrating Evolutionary and Physicochemical Information into Chou's General PseAAC. J. Theor. Biol. 462, 230–239. doi:10.1016/j.jtbi.2018.11.012
Shu, L., Xu, H., and Liu, B. (2017). “Lifelong Learning Crf for Supervised Aspect Extraction[C],” in Proceedings of the 55th Annual Meeting of the Association for Computational Linguistics (Vancouver), 148154.
Silver, D. L., Mason, G., and Eljabu, L. (2015). “Consolidation Using Sweep Task Rehearsal: Overcoming the Stability Plasticity Problem[C],” in Canadian Conference on Artificial Intelligence (Halifax), 307322.
Silver, D. L., and Mercer, R. E. (2002). “The Task Rehearsal Method of Lifelong Learning: Overcoming Impoverished Data[C],” in Conference of the Canadian Society for Computational Studies of Intelligence (Calgary), 90101.
Silver, D. L. (1996). The Parallel Transfer of Task Knowledge Using Dynamic Learning Rates Based on a Measure of Relatedness[J]. Connect. Sci. 8 (2), 277294. doi:10.1080/095400996116929
Thrun, S., and Mitchell, T. M. (1995). Lifelong Robot Learning[J]. Robotics Autonomous Syst. 15 (12), 2546. doi:10.1016/0921-8890(95)00004-Y
Wang, S., Chen, Z., and Liu, B. (2016). “Mining Aspect-specific Opinion Using a Holistic Lifelong Topic Model[C],” in Proceedings of the 25th International Conference on World Wide Web (Montreal), 167176.
Wang, Y., Ding, Y., Guo, F., Wei, L., and Tang, J. (2017). Improved Detection of Dna-Binding Proteins via Compression Technology on Pssm Information. Plos One 12 (9), e0185587. doi:10.1371/journal.pone.0185587
Wei, L., Tang, J., and Zou, Q. (2017). Local-dpp: an Improved Dna-Binding Protein Prediction Method by Exploring Local Evolutionary Information. Inf. Sci. 384, 135–144. doi:10.1016/j.ins.2016.06.026
Wu, H., Yang, R., Fu, Q., Chen, J., Lu, W., and Li, H. (2019a). Research on Predicting 2D-HP Protein Folding Using Reinforcement Learning with Full State Space. BMC Bioinformatics 20, 685. doi:10.1186/s12859-019-3259-6
Wu, H., Huang, H., Lu, W., Fu, Q., Ding, Y., Qiu, J., et al. (2019b). Ranking Near-Native Candidate Protein Structures via Random forest Classification. BMC Bioinformatics 20, 683. doi:10.1186/s12859-019-3257-8
Yosvany, L., Alok, S., Abdollah, D., Pranit, L. S., Ghazaleh, T., Abdul, S., et al. (2018). Success: Evolutionary and Structural Properties of Amino Acids Prove Effective for Succinylation Site Prediction. BMC Genom. 19 (Suppl. 1), 923. doi:10.1186/s12864-017-4336-8
Zhou, H., Wang, H., Ding, Y., and Tang, J. (2021). Multivariate Information Fusion for Identifying Antifungal Peptides with Hilbert-Schmidt Independence Criterion. Cbio 16. doi:10.2174/1574893616666210727161003
Zou, Q., Li, X., Jiang, Y., Zhao, Y., and Wang, G. (2013). Binmempredict: a Web Server and Software for Predicting Membrane Protein Types. Cp 10 (1), 2–9. doi:10.2174/1570164611310010002
Zou, Y., Ding, Y., Peng, L., and Zou, Q. (2021). FTWSVM-SR: DNA-Binding Proteins Identification via Fuzzy Twin Support Vector Machines on Self-Representation. Interdiscip. Sci. Comput. Life Sci.. doi:10.1007/s12539-021-00489-6
Keywords: lifelong learning, membrane proteins, dynamically scalable networks, position specific scoring matrix, evolutionary features
Citation: Lu W, Shen J, Zhang Y, Wu H, Qian Y, Chen X and Fu Q (2022) Identifying Membrane Protein Types Based on Lifelong Learning With Dynamically Scalable Networks. Front. Genet. 12:834488. doi: 10.3389/fgene.2021.834488
Received: 13 December 2021; Accepted: 21 December 2021;
Published: 14 March 2022.
Edited by:
Leyi Wei, Shandong University, ChinaReviewed by:
Chunhua Li, Beijing University of Technology, ChinaJieming Ma, Xi’an Jiaotong-Liverpool University, China
Copyright © 2022 Lu, Shen, Zhang, Wu, Qian, Chen and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongjie Wu, aG9uZ2ppZXd1QHVzdHMuZWR1LmNu