 Lingling Hou
Lingling Hou Cuifang Hu2
Cuifang Hu2 Min Liang
Min Liang- 1Department of Medical Laboratory, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China
- 2Attardi Institute of Mitochondrial Biomedicine, Wenzhou Medical University, Wenzhou, China
In this study, we assessed three Chinese families with inherited cholecystolithiasis and conducted the clinical, genetic, and molecular characterization of these subjects. Eight of eighteen matrilineal relatives had a clinical phenotype in these three families. Sequence analysis of complete mitochondrial genomes in these probands identified the homoplasmic tRNAPhe 625 G > A mutation and distinct sets of mtDNA polymorphisms belonging to haplogroups H2, F4b, and M10a. The 625G > A mutation disturbed the classic G-C base-pairings at a highly conserved position 49 in the T-stem of mitochondrial tRNAs. Molecular dynamics simulation showed that the structure of tRNAphe with 625 G > A mutation was noticeably remodeled while compared with the isoform of the wild type. The occurrence of tRNAPhe 625 G > A mutation in these various genetically unrelated subjects strongly indicates that this mutation is involved in the pathogenesis of cholecystolithiasis. This is the first evidence that tRNA mutations are associated with cholecystolithiasis, and it provided more insights into the genetic mechanism of cholecystolithiasis.
Introduction
Cholecystolithiasis and its associated complications (e.g., cholecystitis and pancreatitis) have become an increasingly significant public health problem around the globe. ∼20%–30% of adults in developed countries have been diagnosed with cholecystolithiasis (Rebholz et al., 2018). The etiology of cholecystolithiasis is complicated, and age, sex, pregnancy, alcohol, coffee, and other factors are usually involved (Morimoto et al., 2021). In particular, cholecystolithiasis has a strong genetic predisposition, and ethnic differences indicate that genetic factors play an important role in the occurrence of cholecystolithiasis (Gilat et al., 1983; Lammert et al., 2005; Wittenburg et al., 2007; Buch et al., 2010). So far, several nuclear genes have been identified in cholecystolithiasis patients (Buch et al., 2010). However, the association between mitochondrial DNA (mtDNA) and the onset of cholecystolithiasis still remains to be unraveled. The mitochondrion provides energy for various physiological activities of eukaryotic cells and plays an important role in the metabolism of glucose and lipids (Mahmood et al., 2016; García-Berumen et al., 2019). So, mutations in mtDNA can cause many metabolic diseases, including mitochondrial myopathy, mitochondrial encephalomyopathy, hypertension, type 2 diabetes, and dyslipidemia (Goto, 2000; Rahman, 2012; Xue et al., 2016; Rovira-Llopis et al., 2017; Rumora et al., 2018; De Barcelos et al., 2019; Pinti et al., 2019; Radelfahr and Klopstock, 2019; Zhao et al., 2019; Prasun, 2020).To further elucidate the molecular basis of cholecystolithiasis in the Chinese population, a systematic and extended mutational screening of mtDNA has been initiated in a large clinical population of the First Affiliated Hospital of the Wenzhou Medical College (Hou et al., 2018). In the current study, we performed the clinical, genetic, and molecular characterizations of three Chinese families with inherited cholecystolithiasis. Molecular analysis identified the homoplasmic 625G > A mutation, which localized at a highly conserved position 49 in the T-stem in mitochondrial tRNAPhe. To elucidate the role of mitochondrial haplotype in the phenotypic manifestation of the 625G > A mutation, we conducted high-throughput sequencing and Sanger sequencing spanning the entire mitochondrial genome and subsequent DNA sequence analysis of three probands in those Chinese families. Furthermore, we computed the structural analysis and simulations of molecular dynamics of the mitochondrial tRNA gene.
Materials and Methods
Patients
Three Chinese cholecystolithiasis families, as shown in Figure1, were ascertained through the First Affiliated Hospital of Wenzhou Medical University. Informed consent, blood samples, and clinical evaluations were obtained from all participating family members, under protocols approved by the Wenzhou Medical University ethics committee. A comprehensive history, blood biochemistry, imaging examination, and long-term follow-up for these participating subjects were conducted at length to identify both personal or family medical histories of individual cholecystolithiasis disease and other clinical abnormalities.
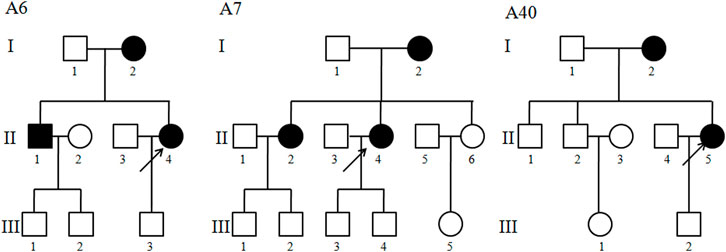
FIGURE 1. Three Han Chinese pedigrees with cholecystolithiasis. Cholecystolithiasis individuals are indicated by filled symbols. An arrow denotes proband.
Mutational Analysis of the Mitochondrial Genome
DNA was isolated from the whole blood of participants by using the DNA extraction reagent (TaKaRa Universal Genomic DNA Extraction Kit Ver3.0) The cholecystolithiasis group DNA samples and the 300 Chinese control samples with concentrations over 87.5 ng/μl were sent to Suichuan Biological Co., Ltd. for VariantProTM Amplicon experiments after evaluation and quality check. This study used the Illumina HisSeq2500 as a high-throughput sequencing method for sequencing. The system uses two patented technologies: OmegaPrimerTM and RelayPCRTM to conduct the entire process from target sequence capture to sequencing library preparation in one step, which has good library uniformity, repeatability, sensitivity, and specificity. To further verify the mutations of the high-throughput sequencing, the entire mitochondrial genome of three probands was PCR amplified in 24 overlapping fragments using sets of the light (L) strand and the heavy (H) strand oligonucleotide primers as described previously (Liang et al., 2009; Zhao et al., 2009). Each fragment was purified and subsequently analyzed by direct sequencing in an ABI 3700 automated DNA sequencer using the Big Dye Terminator Cycle (Applied Biosystems, Foster City, CA) sequencing reaction kit. These sequence results were compared with the updated consensus Cambridge sequence (GenBank accession number: NC_012920) (Brandon et al., 2005). These variants were evaluated for the pathogenicity using the following criteria: 1) present in <1% of the controls; 2) conservation indexes (CI) > 75%; 3) potential structural and functional alterations; and 4)MitoTip Scores >50%(Liang et al., 2009; Zhao et al., 2009).
Haplogroup Analysis
The Asian mitochondrial haplogroup of these three probands was determined using online software (www.mitotool.org/genomeRSRS.html) or based on the nomenclature of mitochondrial haplogroups previously reported (Tanaka et al., 2004; Kong et al., 2006).
Structural Analysis
The published secondary structures for the tRNAs were used to define the stem and loop structure (Florentz et al., 2003; Ruiz-Pesini and Wallace, 2006; Suzuki and Nagao, 2011). The tRNA sequences and secondary structures of corresponding species were downloaded from the relevant database (http://trna.bioinf.uni-leipzig.de/DataOutput/Search); then, the FASTA format of the sequence was submitted to web servers for RNA secondary structure prediction (http://rna.urmc.rochester.edu/RNAstructureWeb).
Phylogenetic Analysis
A total of 17 vertebrate mitochondrial DNA sequences from the NCBI database were used in the interspecific analysis. The conservation index (CI) was calculated by comparing the human nucleotide variants with other 16 vertebrates. The CI was then defined as the percentage of species from the list of 17 different vertebrates that have the wild-type nucleotide at that position (Liang et al., 2014).
Result
Clinical Presentation
The proband (II-4) with a gallstone in family A6 was found 7 years ago, she suffered persistent pain in the right upper quadrant for 1 month, 1 month later, her back suffered radiation pain, with the help of ultrasound, she was diagnosed as cholecystolithiasis with chronic cholecystitis. As shown in Figure 1 and Table 1, 3 of 4 matrilineal relatives had a history of cholecystolithiasis, and the penetrances of cholecystolithiasis in this family were 75%. In family A7, the proband (II-4) was 42 years old, gallbladder stones were found during a routine physical examination 4 years ago, and now she seeks medical care at the First Affiliated Hospital of Wenzhou Medical University because of the unbearable pain caused by gallstones. In fact, 3 of 9 matrilineal relatives had a history of cholecystolithiasis, and the penetrances in this family were 33.3%. The proband (II-5) of family A40 was 43 years old, suffered paroxysmal cramps in the right upper abdomen for 5 days, and after B type ultrasonic examination and combined with clinical manifestations, and then she was diagnosed with cholecystolithiasis. Interestingly, her mother also had a history of cholecystolithiasis.
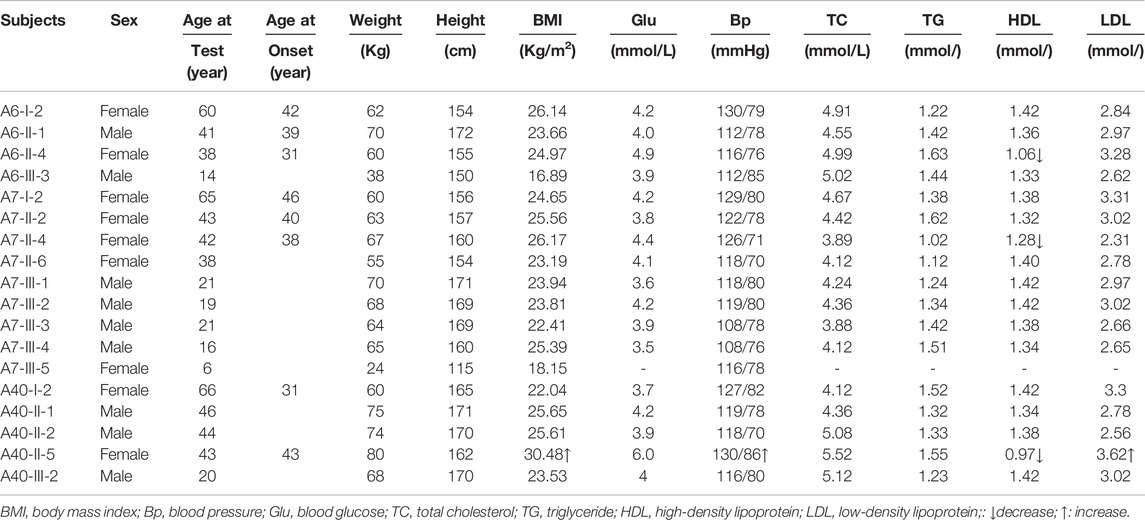
TABLE 1. Summary of the clinical data for eighteen maternal relatives in three Han Chinese cholecystolithiasis pedigrees.
As shown in Table 1, 8 (1 male and 7 female) of 18 matrilineal relatives had a clinical phenotype in these three families, with an average age of onset was 38.7 years. The proband of the A40 family (II-5) weighed 80 kg and had a BMI of 30.48, which indicated that it may be an overweight patient. Blood pressure monitoring showed that the proband of the A6 and A7 family (II-4, II-4) had normal blood pressure, but the proband of the A40 family (II-5) had systolic pressure slightly higher than normal, which was at the critical level. Blood biochemical analysis showed that the blood glucose levels, total cholesterol, and triglyceride concentrations of the three probands were within the normal range, that is the normal values were 3.89–6.11 mmol/L, 2.8–5.17 mmol/L, and 0.56–1.7 mmol/L. The High-Density Lipoprotein (HDL) levels decreased obviously in all three probands, especially A40-II-5, which measured only 0.97 mmol/L, significantly lower than the normal level of 1.29–1.55 mmol/L. Moreover, the Low-Density Lipoprotein (LDH) was measured at 3.62 mmol/L in A40-II-5, higher than the normal 2.07–3.37 mmol/L, while the other two probands have a normal level. The other five patients in these three families have normal blood glucose levels, total cholesterol, and triglyceride concentrations. Furthermore, there is no evidence that any member of those families had any other known cause to account for cholecystolithiasis. Comprehensive family medical histories of these individuals showed no other clinical abnormalities, including diabetes, muscular diseases, hearing impairment, and neurological disorders.
Mitochondrial DNA Analysis
To elucidate the molecular basis of cholecystolithiasis, we have performed a mutational analysis of the mitochondrial genome in these families. First, PCR amplification of the mitochondrial tRNA gene revealed three families carrying the m.625G > A mutation, which was not present in normal controls. As shown in Figure 2, the homoplasmic m.625G > A mutation affects the G49 residue in the T-stem of the mitochondrial tRNAPhe gene and leads to a disruption of the secondary structure of the tRNA after the loss of the T-stem. This modification could also affect the tertiary structure of the tRNAPhe and had probably a consequence on the aminoacylation. Guanine at this position is a conserved base in sequenced threonine tRNA from bacteria to human mitochondria (Ruiz-Pesini and Wallace, 2006). As shown in Figure 3, among 17 organisms, the Conservative index (CI) of this gene at position 49 in tRNAPhe is 100%. Indeed, MitoTIP Sub-Scoring for variant 625G > A is 81.30%, and it is more likely pathogenic (https://www.mitomap.org/mitomaster/index_snvs.cgi).
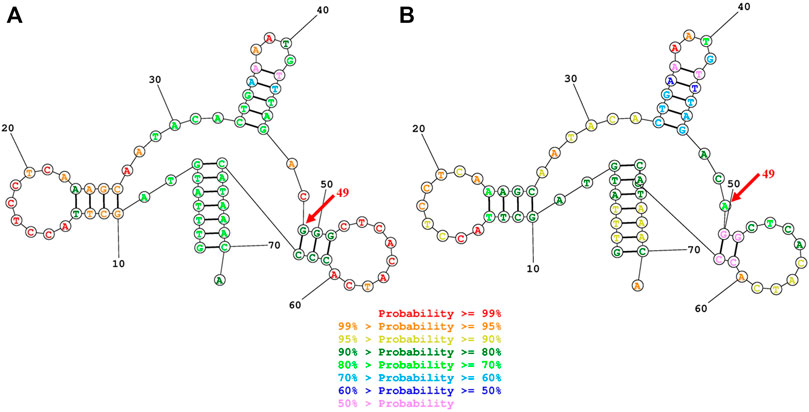
FIGURE 2. Secondary structure prediction of tRNAPhe by the Web Servers for RNA Secondary Structure Prediction (http://rna.urmc.rochester.edu/RNAstructureWeb). (A) G49 residue formed a G-C base pairings in the T-stem of the mitochondrial tRNAPhegene (B)The 625G > A mutation damaged the G-C Base pairing at the 49 residues and leads to a disruption of the secondary structure of the tRNA after the loss of the T-stem.
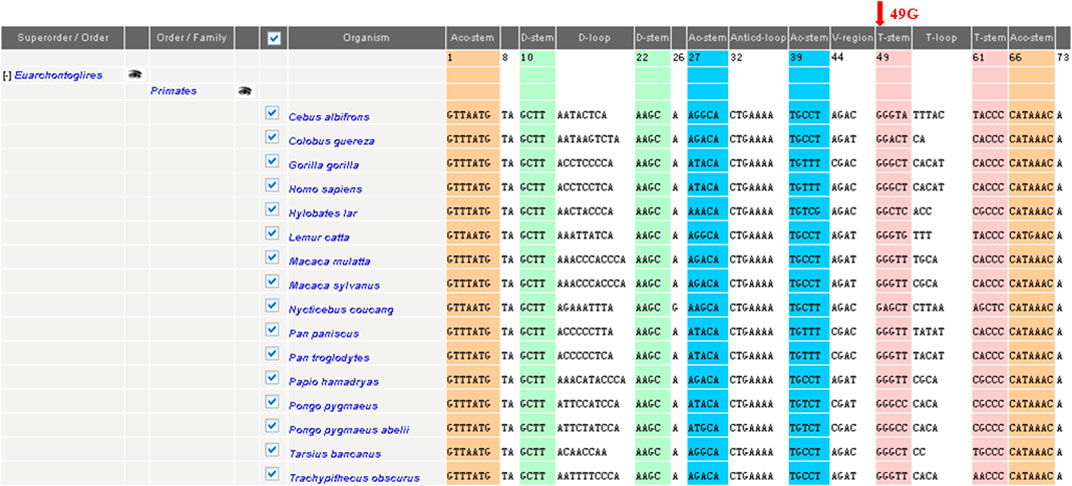
FIGURE 3. Sequence alignment of the mitochondrial tRNAPhe gene in different species. Arrow indicates the conservation of the G nucleotide at position 49 throughout species. The conservation index (CI) at position 49was 100% in 17 vertebrates, by comparing the human nucleotide variants with other 16 vertebrates.
We then conducted high-throughput sequencing and Sanger sequencing spanning the entire mitochondrial genome and subsequent DNA sequence analysis of three probands in those Chinese families. In addition to the identical m.625G > A mutation, as shown in Table 2, these subjects exhibited distinct sets of mtDNA polymorphisms, including eighteen known variants in the D-loop, five known variants, and one unknown variant in the 12S rRNA gene, thirty-four known silent variants in the protein-encoding genes, and fourteen known missense mutations in the protein-encoding genes. These missense mutations are the 5263C > T (A265V) in the ND2 gene, the 8603T > C (F26S), 8701A > G (T59A), 8821T > G (S99A), and 8860A > G (T112A) in ATPase6 gene, the 9966G > A (V254T) in the COIII gene, the 10398A > G (T114A) and 10400C > T (T114A)in the ND3 gene, the 13135G > A (A267T) and 13928G > C (S531T) in the ND5 gene, the 14766C > T (T7I),15071T > C(Y109H), 15218A > G (T158A), and 15326A > G (T194A) in the Cytb gene. These variants in RNAs and polypeptides were further evaluated by phylogenetic analysis of these variants and sequences from other 16 organisms (Liang et al., 2014). However, all these variants showed no evolutionary conservation. Based on the nomenclature of mitochondrial haplogroups (Tanaka et al., 2004; Kong et al., 2006), we used the mtDNA sequence variations among 3 Chinese probands to establish the haplogroup affiliation of each mtDNA. Here, mtDNAs of pedigree A6, A7, and A40 belong to the Eastern Asian halpogroups H2, F4b, and M10a, respectively. To further exclude the influence of nuclear genes, we conducted a whole exon sequencing of the three probands, but we did not find the nuclear susceptibly genes (dates were not shown).
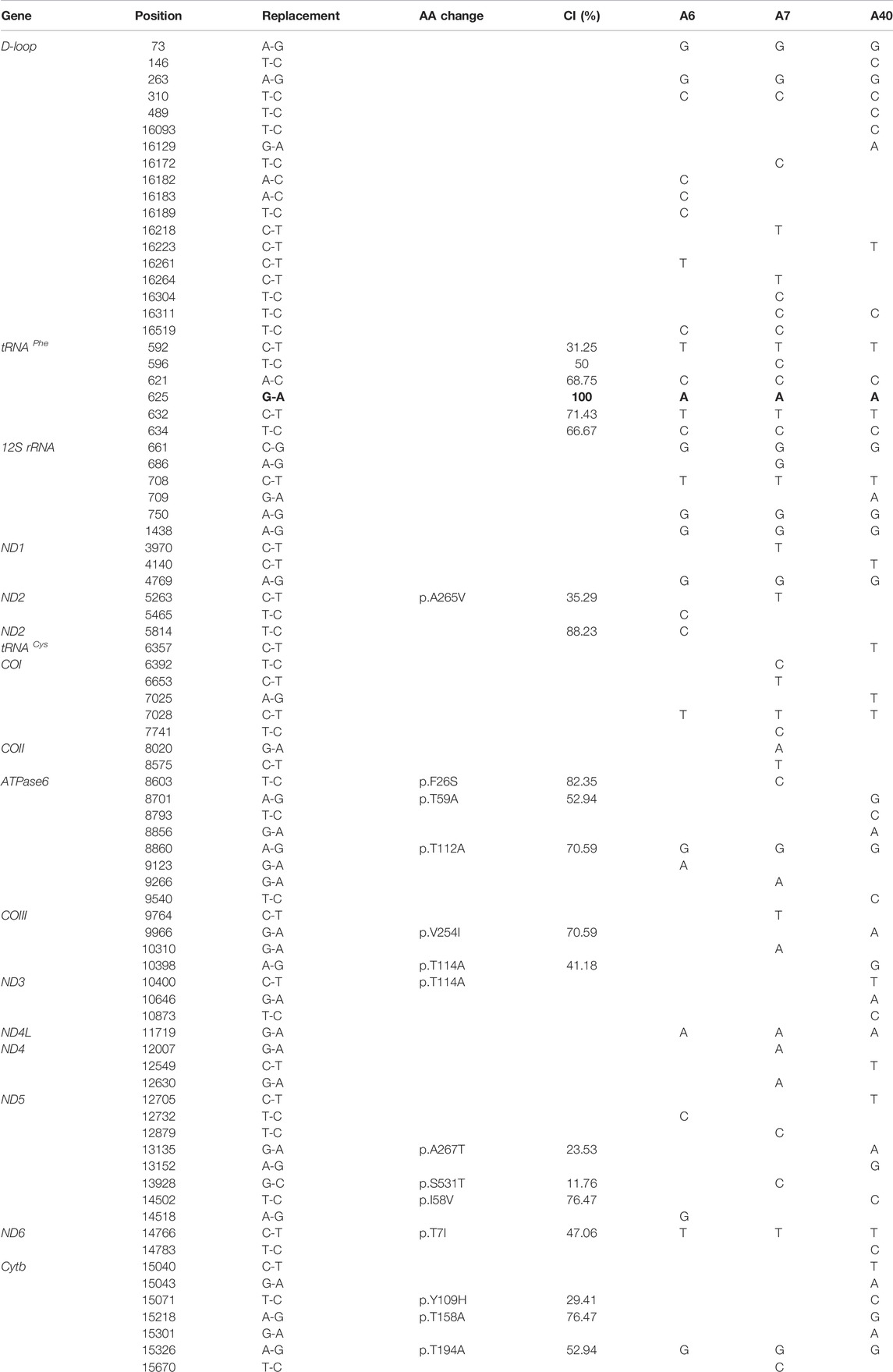
TABLE 2. mtDNA variants in three Chinese pedigrees with cholecystolithiasis.
Disccusion
Cholecystolithiasis is very common in the general population and is an important cause of hospitalization (Marschall and Einarsson, 2007; Schirmer et al., 2005; Di Ciaula et al., 2018, 2019; Gutt et al., 2020). The clinical presentation of cholecystolithiasis varied from the asymptomatic carrier status, to uncomplicated symptomatic disease with pain attacks, and complicated symptomatic disease, including acute cholecystitis, common bile duct stones, pancreatitis, cholangitis, and bowel obstruction(Shabanzadeh, 2018). In the current study, we have conducted the clinical, genetic, and molecular characterization of three Chinese families with cholecystolithiasis. Of these, 8 of 18 matrilineal relatives had a clinical phenotype, the penetrances of cholecystolithiasis were 75%, 33.3%, and 40%, respectively. Furthermore, the age at onset for cholecystolithiasis in those subjects varied from 31 to 43 years old, with an average of 38.7 years, slightly younger than the reported average age of onset(Chen et al., 2014). The majority of cholecystolithiasis patients show abnormal levels of cholesterol and lipoprotein in the blood (Shabanzadeh et al., 2016), but in our study, the cholesterol levels of all three pedigrees probands were normal while their HDL levels decreased significantly, especially inpatient A40.
Cholecystolithiasis is a multifactorial disease, the pathogenesis involves both genetic and environmental factors, of these, genetic factors account on an average for 25–29% of the risk (Nakeeb et al., 2002; Stokes et al., 2011; 2014). The nuclear gene ABCB4 (ATP-binding cassette B4) is closely related to the occurrence of cholecystolithiasis (Jirsa et al., 2014; Elderman et al., 2015). In addition, the variants of ABCG8 and UGT1A1 have been identified to be involved, it may contribute to 21.2% the male population (Buch et al., 2010; Buch et al., 2010). Cholecystolithiasis are currently found in the maternal lineages of these families, suggesting that mtDNA mutations may be the molecular basis of this disease. The sequence analysis of the complete mitochondrial genomes in these pedigrees identified 625G > A mutation in the T-loop of the tRNAphe gene. Indeed, Transfer RNAs (tRNAs), acting as adapters between nucleic acid and protein, plays an important central role in translation, and the secondary and tertiary structure of tRNAs are highly conserved (Shi and Moore, 2000; Machnicka et al., 2014). Mitochondrial tRNA mutations are thought to be associated with many multisystem disorders among the Chinese population. Of these, the two most common pathogenic mt-tRNA variants: m.3243A > G (within MT-TL1, encoding mt-tRNALeu(UUR)) and m.8344A > G (within MT-TK, encoding mt-tRNALys), which account for the vast majority of all mt-tRNA-related diseases (Richteret al., 2021). In addition, mitochondrial tRNA mutations are involved in other isolated organ-specific diseases, such as deafness, tic disorders, Leber’s hereditary optic neuropathy, coronary heart disease, hypertension, etc., (Qin et al., 2014; Xue et al., 2016; Jiang et al., 2020; Zheng et al., 2020; Shuaiet al., 2021) The T-loop in tRNA, as a promoter for both the heavy and light strands of the mtDNA, is critical for tRNA structural stability (Anderson et al., 1981). The phenylalanine at position 49 in tRNA is extremely conserved among different organisms. This site mutation may lead to instability of the T-stem, which in turn leads to the destruction of the tRNA secondary structure. In addition to the 625G > A mutation, the pathogenic mitochondrial mutation site that has been reported is located in T-stem, such as 1662C > T, 4310A > G, 5558A > G, 5562U > A, 5772C > T, 5773C > T, 5783C > T, 5836T > C, 5846G > A, 10049A > T, 12254A > T, 14682G > T,14683T > C, 14696T > C, 15942T > C, 15944delT, 15941T > C, 15965T > C, and 15968A > G (http://www.mtdb.igp.uu.se/).
MitoTIP Sub-Scoring shows that m.625G > A mutation is more likely pathogenic. Indeed, the 625G > A mutation has been reported to be associated with deafness and epilepsy families (Sudo et al., 2011). In our study, the homoplasmic 625G > A mutation present only in the maternal lineage of those pedigrees but not other members of these families strongly indicates that this mutation may be associated with the pathogenesis of cholecystolithiasis. The varying degrees of penetrance of cholecystolithiasis indicated that the m.625G > A mutation, similar to other mitochondrial mutations (Liang et al., 2009; Liang et al., 2014), is itself insufficient to produce the clinical phenotype, the modifier factors including mitochondrial haplotypes are necessary for the phenotypic manifestation (zhao et al., 2009; Liang et al., 2014; Ji et al., 2014).
Mitochondrial haplotypes are defined by the combinations of single nuclear polymorphisms (SNPs) in mtDNA inherited from a common ancestor(Wallace. 2013). The tree of Native American mtDNA showed that almost all identified haplotypes clustered into only A, B, C, and D four distinct basal branches. After Native Americans, the Asian-specific haplogroups E, F, and G were identified in Tibetans, haplogroups H, I, J, and K in North Americans of European descent, haplogroups L, L1, and L2, the most ancient of all continent-specific haplogroups, were found in Sub-Saharan Africans, the Asian-specific haplogroup M was then identified, haplogroups T, U, V, W, and X were identified in Europeans (Antonio et al., 2020). Mitochondrial haplotype plays an important role in the phenotypic expression of diseases. Mitochondrial haplogroup G is associated with nonalcoholic fatty liver disease (Gusdonet al., 2021). The specific mtDNA background B5a1 was significantly associated with Southeast Asian G11778A LHON and appeared to modify the risk of visual loss (Kaewsutthi et al., 2011). Mitochondrial haplogroup J was associated with a higher risk of obesity in the Qatari population(Dashti et al., 2021). Mitochondrial DNA Haplogroup M7 Confers Disability in a Chinese Aging Population(Sun et al., 2020). Here, these mitochondrial genomes of A6, A7, and A40 pedigrees belong to the Eastern Asian haplogroups H2, F4b, and M10a, respectively. In the other studies, two Chinese cholecystolithiasis pedigrees carrying the m.827A > G belonged to the haplogroups B4a and B4c (Sun et al., 2021). This implied that the m.625G > A mutation, similar to other mitochondrial mutations, occurred sporadically and multiplied through the evolution of the mtDNA (Liang et al., 2014).
At present, the pathogenesis of cholecystolithiasis has not been well explained. The most commonly accepted hypothesis is gallbladder dyskinesia, caused by a pathological change in the gallbladder wall (Cafasso and Smith, 2014). However, the mechanism of gallbladder dyskinesia is not well understood. In fact, mitochondria provide a constant source of energy for human physiological activities, mitochondrial mutations can affect the ATP production. The m.625G > A mutation disturbed the tRNA secondary structure, and it may affect protein translation and thus energy production. Therefore, we speculate that the gallbladder sphincter muscle is unable to contract well due to a lack of ATP, resulting in gallbladder stones that cannot be discharged and cleared. On the other hand, mitochondrial dysfunction may also affect lipid metabolism. These may be the important reason for the occurrence of cholecystolithiasis. However, other genetic factors, other than 625G > A mutation, and environmental factors also play an important role in cholecystolithiasis disease.
Data Availability Statement
The original contributions presented in the study are publicly available.The datasets generated for this study can be found in the GSA for Human and the accession number is: HRA002404, https://bigd.big.ac.cn/gsa-human/browse/HRA002404.
Ethics Statement
The studies involving human participants were reviewed and approved by the Wenzhou Medical University ethics committee. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
ML designed the experiments and monitored the project progression, data analysis, and interpretation. LH carried out the clinical evaluation and recruited patients. CH performed the data analysis. LJ prepared the initial draft of the manuscript. QW performed the secondary structure analysis. ML made the final version of the manuscript. All authors reviewed the manuscript.
Funding
This work was supported by grant 81600769 from the Natural Science Foundation of China, grant LQ19H120004 from the Natural Science Foundation of Zhejiang Province, and grant qnyc-077 from the First Affiliated Hospital of Wenzhou Medical University to ML and grant Y20190475 from the Science and Technology Bureau of Wenzhou to LH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful to the patients and their family members for their participation, and thanks for the guidance from Dr. Min-Xin Guan.
References
Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H. L., Coulson, A. R., Drouin, J., et al. (1981). Sequence and Organization of the Human Mitochondrial Genome. Nature 290 (5806), 457–465. doi:10.1038/290457a0
Antonio, T., Alessandro, A., Anna, O., and Ornella, S. (2020). “Haplogroups and the History of Human Evolution Through mtDNA,” in The Human Mitochondrial Genome. Editors G. Giuseppe, and M. P. Anna (Academic Press), 111–129. doi:10.1016/B978-0-12-819656-4.00005-X
Brandon, M. C., Lott, M. T., Nguyen, K. C., Spolim, S., Navathe, S. B., Baldi, P., et al. (2004). MITOMAP: a Human Mitochondrial Genome Database--2004 Update. Nucleic Acids Res. 33, D611–D613. Database issue). doi:10.1093/nar/gki079
Buch, S., Schafmayer, C., Völzke, H., Seeger, M., Miquel, J. F., Sookoian, S. C., et al. (2010). Loci from a Genome-wide Analysis of Bilirubin Levels Are Associated with Gallstone Risk and Composition. Gastroenterology 139 (6), 1942–1951. doi:10.1053/j.gastro.2010.09.003
Cafasso, D. E., and Smith, R. R. (2014). Symptomatic Cholelithiasis and Functional Disorders of the Biliary Tract. Surg. Clin. N. Am. 94 (2), 233–256. doi:10.1016/j.suc.2013.12.001
Chen, J.-Y., Hsu, C.-T., Liu, J.-H., and Tung, T.-H. (2014). Clinical Predictors of Incident Gallstone Disease in a Chinese Population in Taipei, Taiwan. BMC Gastroenterol. 14, 83. doi:10.1186/1471-230x-14-83
Dashti, M., Alsaleh, H., Rodriguez-Flores, J. L., Eaaswarkhanth, M., Al-Mulla, F., and Thanaraj, T. A. (2021). Mitochondrial Haplogroup J Associated with Higher Risk of Obesity in the Qatari Population. Sci. Rep. 11 (1), 1091. doi:10.1038/s41598-020-80040-7
De Barcelos, I. P., Emmanuele, V., and Hirano, M. (2019). Advances in Primary Mitochondrial Myopathies. Curr. Opin. Neurol. 32 (5), 715–721. doi:10.1097/wco.0000000000000743
Di Ciaula, A., Wang, D. Q.-H., and Portincasa, P. (2018). An Update on the Pathogenesis of Cholesterol Gallstone Disease. Expert Rev. Gastroenterology Hepatology 34 (2), 71–80. doi:10.1097/mog.0000000000000423
Di Ciaula, A., Wang, D. Q., and Portincasa, P. (2019). Cholesterol Cholelithiasis: Part of a Systemic Metabolic Disease, Prone to Primary Prevention. Expert Rev. Gastroenterol. Hepatol. 13 (2), 157–171. doi:10.1080/17474124.2019.1549988
Elderman, J. H., ter Borg, P. C. J., Dees, J., and Dees, A. (2015). Pregnancy and ABCB4 Gene Mutation: Risk of Recurrent Cholelithiasis. Case Rep. 2015, bcr2014206919. doi:10.1136/bcr-2014-206919
Florentz, C., Sohm, B., Tryoen-Tth, P., Potz, J., and Sissler, M. (2003). Human Mitochondrial tRNAs in Health and Disease. Cell. Mol. Life Sci. (CMLS) 60 (7), 1356–1375. doi:10.1007/s00018-003-2343-1
García-Berumen, C. I., Ortiz-Avila, O., Vargas-Vargas, M. A., Del Rosario-Tamayo, B. A., Guajardo-López, C., Saavedra-Molina, A., et al. (2019). The Severity of Rat Liver Injury by Fructose and High Fat Depends on the Degree of Respiratory Dysfunction and Oxidative Stress Induced in Mitochondria. Lipids Health Dis. 18 (1), 78. doi:10.1186/s12944-019-1024-5
Gilat, T., Feldman, C., Halpern, Z., Dan, M., and Bar-Meir, S. (1983). An Increased Familial Frequency of Gallstones. Gastroenterology 84 (2), 242–246. doi:10.1016/s0016-5085(83)80118-8
Goto, Y.-i. (2000). Mitochondrial Encephalomyopathy. Neuropathology 20 (Suppl. l), 82–84. doi:10.1046/j.1440-1789.2000.00304.x
Gusdon, A. M., Hui, Y., Chen, J., Mathews, C. E., and Qu, S. (2021). Mitochondrial Haplogroup G Is Associated with Nonalcoholic Fatty Liver Disease, while Haplogroup A Mitigates the Effects of PNPLA3. Endocrinol. Diab Metab. 4 (1). e00187. doi:10.1002/edm2.187
Gutt, C., Schläfer, S., and Lammert, F. (2020). The Treatment of Gallstone Disease. Dtsch. Arztebl Int. 117 (9), 148–158. doi:10.3238/arztebl.2020.0148
Hou, L., Jiang, F., and Liang, M. (2018). Analysis of Mitochondria tRNA Gene Mutations in 88 Cases with Cholecystolithiasis. Chin. J. Health Lab. Tec. 28 (17), 1309–1317.
Ji, Y., Liang, M., Zhang, J., Zhang, M., Zhu, J., Meng, X., et al. (2014). Mitochondrial Haplotypes May Modulate the Phenotypic Manifestation of the LHON-Associated ND1 G3460A Mutation in Chinese Families. J. Hum. Genet. 59, 134–140. doi:10.1038/jhg.2013.134
Jiang, P., Ling, Y., Zhu, T., Luo, X., Tao, Y., Meng, F., et al. (2020). Mitochondrial tRNA Mutations in Chinese Children with Tic Disorders. Biosci. Rep. 40 (12), 1856. doi:10.1042/bsr20201856
Jirsa, M., Bronský, J., Dvořáková, L., Šperl, J., Šmajstrla, V., Horák, J., et al. (2014). ABCB4mutations Underlie Hormonal Cholestasis but Not Pediatric Idiopathic Gallstones. World. J. Gastrenterol. 20 (19), 5867–5874. doi:10.3748/wjg.v20.i19.5867
Kaewsutthi, S., Phasukkijwatana, N., Joyjinda, Y., Chuenkongkaew, W., Kunhapan, B., Tun, A. W., et al. (2011). Mitochondrial Haplogroup Background May Influence Southeast Asian G11778A Leber Hereditary Optic Neuropathy. Invest.. Ophthalmol. Vis. Sci. 52 (7), 4742–4748. doi:10.1167/iovs.10-5816
Kong, Q.-P., Bandelt, H.-J., Sun, C., Yao, Y.-G., Salas, A., Achilli, A., et al. (2006). Updating the East Asian mtDNA Phylogeny: a Prerequisite for the Identification of Pathogenic Mutations. Hum. Mol. Genet. 15 (13), 2076–2086. doi:10.1093/hmg/ddl130
Lammert, F., and Sauerbruch, T. (2005). Mechanisms of Disease: the Genetic Epidemiology of Gallbladder Stones. Nat. Rev. Gastroenterol. Hepatol. 2 (9), 423–433. doi:10.1038/ncpgasthep0257
Liang, M., Guan, M., Zhao, F., Zhou, X., Yuan, M., Tong, Y., et al. (2009). Leber's Hereditary Optic Neuropathy Is Associated with Mitochondrial ND1 T3394C Mutation. Biochem. Biophysical Res. Commun. 383 (3), 286–292. doi:10.1016/j.bbrc.2009.03.097
Liang, M., Jiang, P., Li, F., Zhang, J., Ji, Y., He, Y., et al. (2014). Frequency and Spectrum of MitochondrialND6Mutations in 1218 Han Chinese Subjects with Leber's Hereditary Optic Neuropathy. Invest.. Ophthalmol. Vis. Sci. 55 (3), 1321–1331. doi:10.1167/iovs.13-13011
Machnicka, M. A., Olchowik, A., Grosjean, H., and Bujnicki, J. M. (2014). Distribution and Frequencies of Post-transcriptional Modifications in tRNAs. RNA Biol. 11 (12), 1619–1629. doi:10.4161/15476286.2014.992273
Mahmood, S., Birkaya, B., Rideout, T. C., and Patel, M. S. (2016). Lack of Mitochondria-Generated Acetyl-CoA by Pyruvate Dehydrogenase Complex Downregulates Gene Expression in the Hepatic De Novo Lipogenic Pathway. Am. J. Physiology-Endocrinology Metabolism 311 (1), E117–E127. doi:10.1152/ajpendo.00064.2016
Marschall, H. U., and Einarsson, C. (2007). Gallstone Disease. J. Intern. Med. 261, 529–542. doi:10.1111/j.1365-2796.2007.01783.x
Morimoto, M., Matsuo, T., and Mori, N. (2021). Management of Porcelain Gallbladder, its Risk Factors, and Complications: A Review. Diagnostics 11 (6), 1073. doi:10.3390/diagnostics11061073
Pinti, M. V., Fink, G. K., Hathaway, Q. A., Durr, A. J., Kunovac, A., and Hollander, J. M. (2019). Mitochondrial Dysfunction in Type 2 Diabetes Mellitus: an Organ-Based Analysis. Am. J. Physiology-Endocrinology Metabolism 316 (2), E268–E285. doi:10.1152/ajpendo.00314.2018
Prasun, P. (2020). Mitochondrial Dysfunction in Metabolic Syndrome. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1866 (10), 165838. doi:10.1016/j.bbadis.2020.165838
Qin, Y., Xue, L., Jiang, P., Xu, M., He, Y., Shi, S., et al. (2014). Mitochondrial tRNA Variants in Chinese Subjects with Coronary Heart Disease. J. Am. Heart. Soc. 3 (1). e000437. doi:10.1161/jaha.113.000437
Radelfahr, F., and Klopstock, T. (2019). Mitochondriale Erkrankungen. Nervenarzt 90 (2), 121–130. doi:10.1007/s00115-018-0666-2
Rahman, S. (2012). Mitochondrial Disease and Epilepsy. Dev. Med. Child. Neurol. 54 (5), 397–406. doi:10.1111/j.1469-8749.2011.04214.x
Rebholz, C., Krawczyk, M., and Lammert, F. (2018). Genetics of Gallstone Disease. Eur. J. Clin. Invest. 48 (7). e12935. doi:10.1111/eci.12935
Richter, U., McFarland, R., Taylor, R. W., and Pickett, S. J. (2021). The Molecular Pathology of Pathogenic Mitochondrial tRNA Variants. FEBS Lett. 595 (8), 1003–1024. doi:10.1002/1873-3468.14049
Rovira-Llopis, S., Bañuls, C., Diaz-Morales, N., Hernandez-Mijares, A., Rocha, M., and Victor, V. M. (2017). Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox Biol. 11, 637–645. doi:10.1016/j.redox.2017.01.013
Ruiz-Pesini, E., and Wallace, D. C. (2006). Evidence for Adaptive Selection Acting on the tRNA and rRNA Genes of Human Mitochondrial DNA. Hum. Mutat. 27 (11), 1072–1081. doi:10.1002/humu.20378
Rumora, A. E., Lentz, S. I., Hinder, L. M., Jackson, S. W., Valesano, A., Levinson, G. E., et al. (2018). Dyslipidemia Impairs Mitochondrial Trafficking and Function in Sensory Neurons. FASEB J. 32 (1), 195–207. doi:10.1096/fj.201700206R
Schirmer, B. D., Winters, K. L., and Edlich, R. F. (2005). Cholelithiasis and Cholecystitis. J. Long. Term. Eff. Med. Implants 15 (3), 329–338. doi:10.1615/jlongtermeffmedimplants.v15.i3.90
Shabanzadeh, D. M. (2018). Incidence of Gallstone Disease and Complications. Curr. Opin. Gastroenterol. 34 (2), 81–89. doi:10.1097/mog.0000000000000418
Shabanzadeh, D. M., Sørensen, L. T., and Jørgensen, T. (2016). Determinants for Gallstone Formation - a New Data Cohort Study and a Systematic Review with Meta-Analysis*. Scand. J. Gastroenterology 51 (10), 1239–1248. doi:10.1080/00365521.2016.1182583
Shi, H., and Moore, P. B. (2000). The Crystal Structure of Yeast Phenylalanine tRNA at 1.93 Å Resolution: A Classic Structure Revisited. RNA 6 (8), 1091–1105. doi:10.1017/s1355838200000364
Shuai, J., Shi, J., Liang, Y., Ji, F., Gu, L., and Yuan, Z. (2021). Mutational Analysis of Mitochondrial tRNA Genes in 138 Patients with Leber's Hereditary Optic Neuropathy. Ir. J. Med. Sci. 191, 865–876. doi:10.1007/s11845-021-02656-6
Stokes, C. S., Krawczyk, M., and Lammert, F. (2011). Gallstones: Environment, Lifestyle and Genes. Dig. Dis. 29 (2), 191–201. doi:10.1159/000323885
Sudo, A., Takeichi, N., Hosoki, K., and Saitoh, S. (2011). Successful Cochlear Implantation in a Patient with Mitochondrial Hearing Loss and m.625G>A Transition. J. Laryngol. Otol. 125 (12), 1282–1285. doi:10.1017/s0022215111002453
Sun, D., Niu, Z., Zheng, H.-X., Wu, F., Jiang, L., Han, T.-Q., et al. (2021). A Mitochondrial DNA Variant Elevates the Risk of Gallstone Disease by Altering Mitochondrial Function. Cell. Mol. Gastroenterology Hepatology 11 (4), 1211–1226. doi:10.1016/j.jcmgh.2020.11.015
Sun, D., Yao, S., Wu, F., Deng, W., Ma, Y., Jin, L., et al. (2020). Mitochondrial DNA Haplogroup M7 Confers Disability in a Chinese Aging Population. Front. Genet. 11, 577795. doi:10.3389/fgene.2020.577795
Suzuki, T., Nagao, A., and Suzuki, T. (2011). Human Mitochondrial tRNAs: Biogenesis, Function, Structural Aspects, and Diseases. Annu. Rev. Genet. 45, 299–329. doi:10.1146/annurev-genet-110410-132531
Tanaka, M., Cabrera, V. M., González, A. M., Larruga, J. M., Takeyasu, T., Fuku, N., et al. (2004). Mitochondrial Genome Variation in Eastern Asia and the Peopling of Japan. Genome Res. 14 (10A), 1832–1850. doi:10.1101/gr.2286304
Wallace, D. C. (2013). Bioenergetics in Human Evolution and Disease: Implications for the Origins of Biological Complexity and the Missing Genetic Variation of Common Diseases. Phil. Trans. R. Soc. B 368 (1622), 20120267. doi:10.1098/rstb.2012.0267
Wittenburg, H., and Lammert, F. (2007). Genetic Predisposition to Gallbladder Stones. Semin. Liver Dis. 27 (1), 109–121. doi:10.1055/s-2006-960174
Xue, L., Wang, M., Li, H., Wang, H., Jiang, F., Hou, L., et al. (2016). Mitochondrial tRNA Mutations in 2070 Chinese Han Subjects with Hypertension. Mitochondrion 30, 208–221. doi:10.1016/j.mito.2016.08.008
Zhao, F., Guan, M., Zhou, X., Yuan, M., Liang, M., Liu, Q., et al. (2009). Leber's Hereditary Optic Neuropathy Is Associated with Mitochondrial ND6 T14502C Mutation. Biochem. Biophysical Res. Commun. 389 (3), 466–472. doi:10.1016/j.bbrc.2009.08.168
Zhao, X., Cui, L., Xiao, Y., Mao, Q., Aishanjiang, M., Kong, W., et al. (2019). Hypertension-associated Mitochondrial DNA 4401A>G Mutation Caused the Aberrant Processing of tRNAMet, All 8 tRNAs and ND6 mRNA in the Light-Strand Transcript. Nucleic Acids Res. 47 (19), 10340–10356. doi:10.1093/nar/gkz742
Keywords: cholecystolithiasis, mitochondrial, tRNA, mutation, pedigree
Citation: Hou L, Hu C, Ji L, Wang Q and Liang M (2022) The Mitochondrial tRNAPhe 625G>A Mutation in Three Han Chinese Families With Cholecystolithiasis. Front. Genet. 13:814729. doi: 10.3389/fgene.2022.814729
Received: 18 November 2021; Accepted: 19 April 2022;
Published: 27 May 2022.
Edited by:
Pramod Tiwari, Jiwaji University, IndiaReviewed by:
Mouna Tabebi, Linköping University Hospital, SwedenBalraj Mittal, British Burn Association, United Kingdom
Copyright © 2022 Hou, Hu, Ji, Wang and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Liang, bGlhbmdtaW44NTY4NUAxMjYuY29t