 Min-Jee Kim
Min-Jee Kim Mi-Sun Yum1*
Mi-Sun Yum1* Beom Hee Lee
Beom Hee Lee- 1Department of Pediatrics, Asan Medical Center Children’s Hospital, Ulsan University College of Medicine, Seoul, South Korea
- 23billion Inc., Seoul, South Korea
- 3Department of Genetics, Asan Medical Center, Ulsan University College of Medicine, Seoul, South Korea
The complex and evolving nature of clinical phenotypes have made genetically diagnosing pediatric patients with movement disorders difficult. Here, we describe this diverse complexity in the clinical and genetic features of a pediatric cohort examined by whole-exome sequencing (WES) and demonstrate the clinical benefit of WES as a diagnostic tool in a pediatric cohort. We evaluated 75 patients with diverse single or combined movement phenomenologies using WES. WES identified 42 variants in 37 genes (56.0%). The detection rate was highest in patients with dystonia (11/13, 84.6%), followed by ataxia (21/38, 55.3%), myoclonus (3/6, 50.0%), unspecified dyskinesia (1/4, 25.0%), tremor (1/1, 100%), respectively. Most genetically diagnosed patients (90.5%) were affected by other neurologic or systemic manifestations; congenital hypotonia (66.7%), and epilepsy (42.9%) were the most common phenotypes. The genetic diagnosis changed the clinical management for five patients (6.7%), including treatments targeting molecular abnormalities, and other systemic surveillance such as cancer screening. Early application of WES yields a high diagnostic rate in pediatric movement disorders, which can overcome the limitations of the traditional phenotype-driven strategies due to the diverse phenotypic and genetic complexity. Additionally, this early genetic diagnosis expands the patient’s clinical spectrum and provides an opportunity for tailored treatment.
1 Introduction
Movement disorders are a heterogeneous group of neurologic disorders characterized by abnormalities in tone, posture, and initiation or control of voluntary movements, as well as unwanted involuntary movement (Sanger et al., 2010; Pearson and Pons, 2019). Pediatric movement disorders frequently present as complex forms in that different movement phenotypes overlap simultaneously or sequentially in the same patients, and typically coincide with other neurodevelopmental symptoms of diverse neurogenetic disorders (Cordeiro et al., 2018; Graziola et al., 2019). Initial manifestations and clinical course can be variable even in common movement disorders with mutations in the same genes. Therefore, reaching an accurate genetic diagnosis and patient-tailored treatment for pediatric disorders sometimes takes a long journey.
Since the advent of massively parallel, genomic-sequencing techniques in the early 2000s, the increased availability, reduced cost, and improved efficiency of next generation sequencing (NGS) have enhanced gene discovery for neurogenetic disorders and facilitated clinicians’ ability make accurate diagnoses (Rexach et al., 2019; Kim et al., 2020). In particular, either whole exome sequencing (WES) or whole genome sequencing (WGS) can reveal more than 5,000 phenotypically and genetically diverse conditions with a single test. Movement disorders are phenomenological, and each one has heterogeneous genetic causes; NGS is definitely more useful for genetic diagnosis than compared with a single gene test (Neveling et al., 2013). Recent genetic diagnosis of pediatric movement disorders by WES has helped clinicians understand their underlying pathophysiology, expand the spectrum of the genotype–phenotype correlation, and provide personalized treatment (Montaut et al., 2018; Graziola et al., 2019; Rexach et al., 2019; Kim et al., 2020; Papandreou et al., 2020). Moreover, movement disorders itself can be used as the key features for genetic diagnosis of pediatric neurological disorders using NGS.
We investigated the diagnostic yield and clinical characteristics of movement disorders to demonstrate the clinical benefit of WES as a diagnostic tool in a pediatric cohort. Furthermore, we tried to delineated these genetic disorders according to their movement phenotype, and systemic or neurological manifestations to attribute the importance of movement phenomenology in the diagnosis of these complex neurogenetic disorders.
2 Materials and Methods
2.1 Patient Population
We evaluated neurodevelopmental symptoms of a recently built neurodevelopmental cohort for WES of 382 children from the Pediatric Neurology Clinic and Medical Genetics Center at Asan Medical Center Children’s Hospital, Seoul, South Korea, from 2017 to 2020. The neurodevelopmental symptoms in the initial cohort were defined as symptoms that cause neurological referral, such as motor dysfunction, abnormal movement, epilepsy, behavioral problems, macro/microcephaly, or psychomotor delay including failure to achievement of milestone. Pediatric neurologists identified 75 patients with movement disorders as their major symptom in this cohort and those patients were included in this study. We also investigated the clinical features of movement disorders, associated symptoms, previously performed other genetic analyses, neuroimaging tests, and electroencephalographic findings.
2.2 Genetic Analysis
We isolated genomic DNA from whole blood or saliva. We then captured the majority of exons in all human genes (approximately 22,000) using a SureSelect Kit (Version C2; Agilent Technologies, Inc, Santa Clara, CA, United States) and sequenced them using a NovaSeq platform (Illumina, San Diego, CA, United States). We then aligned raw genome sequences to a reference sequence (NCBI genome assembly GRCh37; accessed in February 2009). Variants were annotated, filtered and prioritized using software developed in-house, EVIDENCE (3bilion Inc, Seoul, South Korea). EVIDENCE incorporates daily automatically updated databases, variant classification schema based on the ACMG guidelines, and a symptom similarity scoring system as previously described (Kim et al., 2020; Seo et al., 2020; Seo et al., 2022). All identified variants in this study were confirmed by Sanger sequencing and family study. All subjects gave their informed consent for inclusion before they participated in the study. The Institutional Review Board for Human Research at Asan Medical Center approved this study (IRB number 2017-0988).
3 Results
3.1 Clinical Characteristics of Patients
Among the 382 patients of pediatric cohort who performed WES tests for their diagnosis, 75 patients had movement disorders. Prominent movement phenomenology was ataxia in 38 patients (50.6%), dystonia in 13 patients (17.3%), stereotypy in 12 patients (16.0%), myoclonus in 6 patients (8.0%), unspecified dyskinesia in 4 patients (5.3%), or tremor and chorea in 1 patient (1.4%) (Figure 1). Among these, 32 (42.6%) patients had combined movement phenotypes.
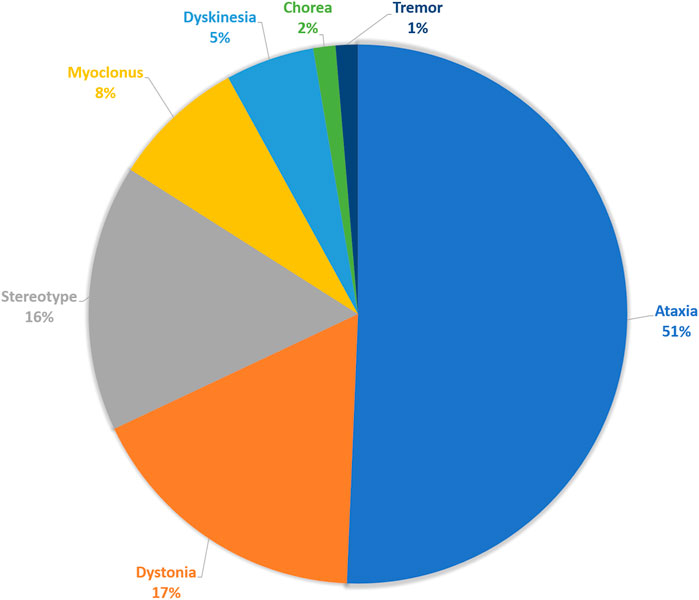
FIGURE 1. Movement phenomenology. The composition of main movement phenomena of the enrolled patients.
Patient demographics, clinical features, and performed tests are described in Table 1. The mean age of symptom onset was 1.8 years, and 66.7% of patients present the movement symptoms before 2 years. Commonly associated neurologic symptoms were congenital hypotonia (42.7%) and epilepsy (34.7%). Of the total patients, 39 (52.0%) showed other systemic symptoms; facial dysmorphism such as prominent or low-set ears, hypertelorism, epicanthal folds, and a flat nasal bridge were most frequently observed in 30.7% of patients, followed by an ocular anomaly (24.0%), and growth delay (14.7%). Before WES, 62 patients (82.7%) underwent genetic testing, including karyotyping, chromosomal microarray (CMA), and single-gene tests, which revealed no specific diagnosis. Additional neuroimaging and EEG study were performed in 90.7% (n = 68/75) and 62.7% (n = 47/75) of patients, respectively. Partial or complete agenesis of the corpus callosum was the most common abnormality observed in neuroimaging (n = 32/43, 74.4%), followed by cerebellar hypoplasia or atrophy (n = 10/43, 23.3%), and malformation of cortical dysplasia (4/43, 9.3%). Epileptiform discharges were observed in 72.3% (n = 34/47) of patients on the EEG.
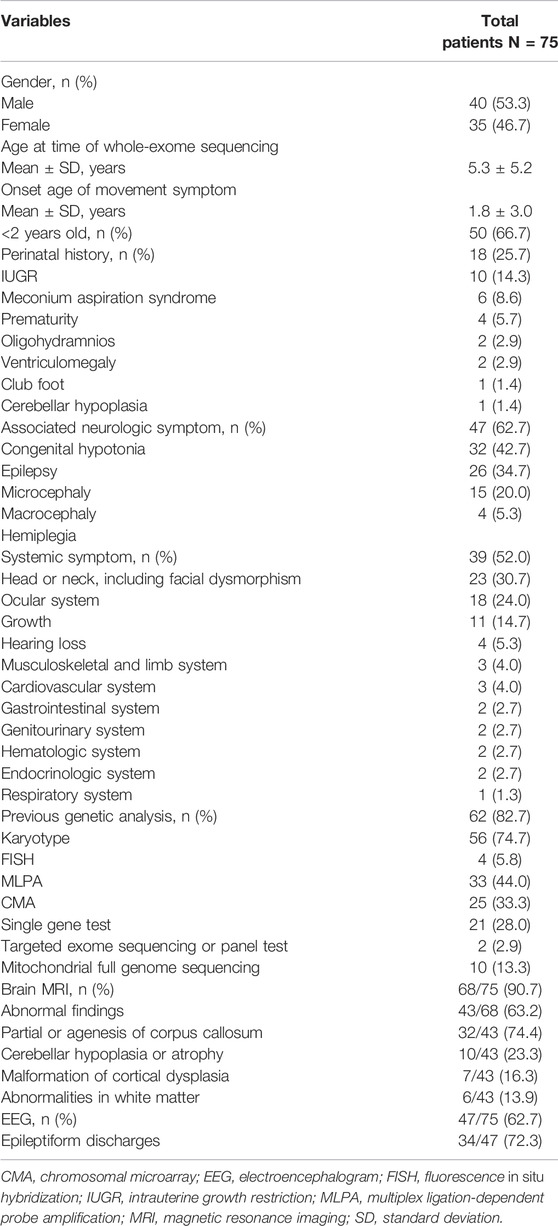
TABLE 1. Clinical characteristics of patients with movement disorders.
3.2 Diagnostic Yield and Identified Genetic Variants
We identified 42 variants in 37 genes from the 75 patients and diagnostic yield of patients with movement disorders was 56.0% (42/75; Figure 2 and Table 2). There were no patients with specific family history. Eleven variants (26.2%) were pathogenic, 18 (42.8%) were likely pathogenic, and 13 (31.0%) were variants of uncertain significance (VUS). Patients with VUS were diagnosed with a neurodevelopmental disease according to the results of their parent’s tests, inheritance, and matched clinical phenotype. There were no statistical differences in diagnostic yield between patients who had performed previous genetic tests and those who did not (n = 7/13, 53.8% vs n = 37/62, 59.7%, p = 0.464). Variants were detected in 21 of 38 patients with ataxia (55.3%), 11 of 13 patients with dystonia (84.6%), 5 of 12 patients with stereotypy (41.6%), 3 of 6 patients with myoclonus (50.0%), 1 of 4 patients with unspecified dyskinesia (25.0%), and 1 of 1 patient with tremor (100%), respectively. In regards to prominent movement symptoms, the patients with dystonia had a significantly higher rate of genetic diagnosis compared to patients with other movement disorders (n = 11/13, 84.6% vs n = 2/13, 15.4%, p = 0.034). Among clinical manifestations other than movement symptoms, epilepsy (18/26, 69.2% vs 8/26, 30.8%, p = 0.017), and congenital hypotonia (28/32, 87.5% vs 4/32, 12.5%, p < 0.001) was more frequent in patients with a genetic diagnosis. The presence of systemic symptoms was not significantly associated with the genetic diagnosis rate.
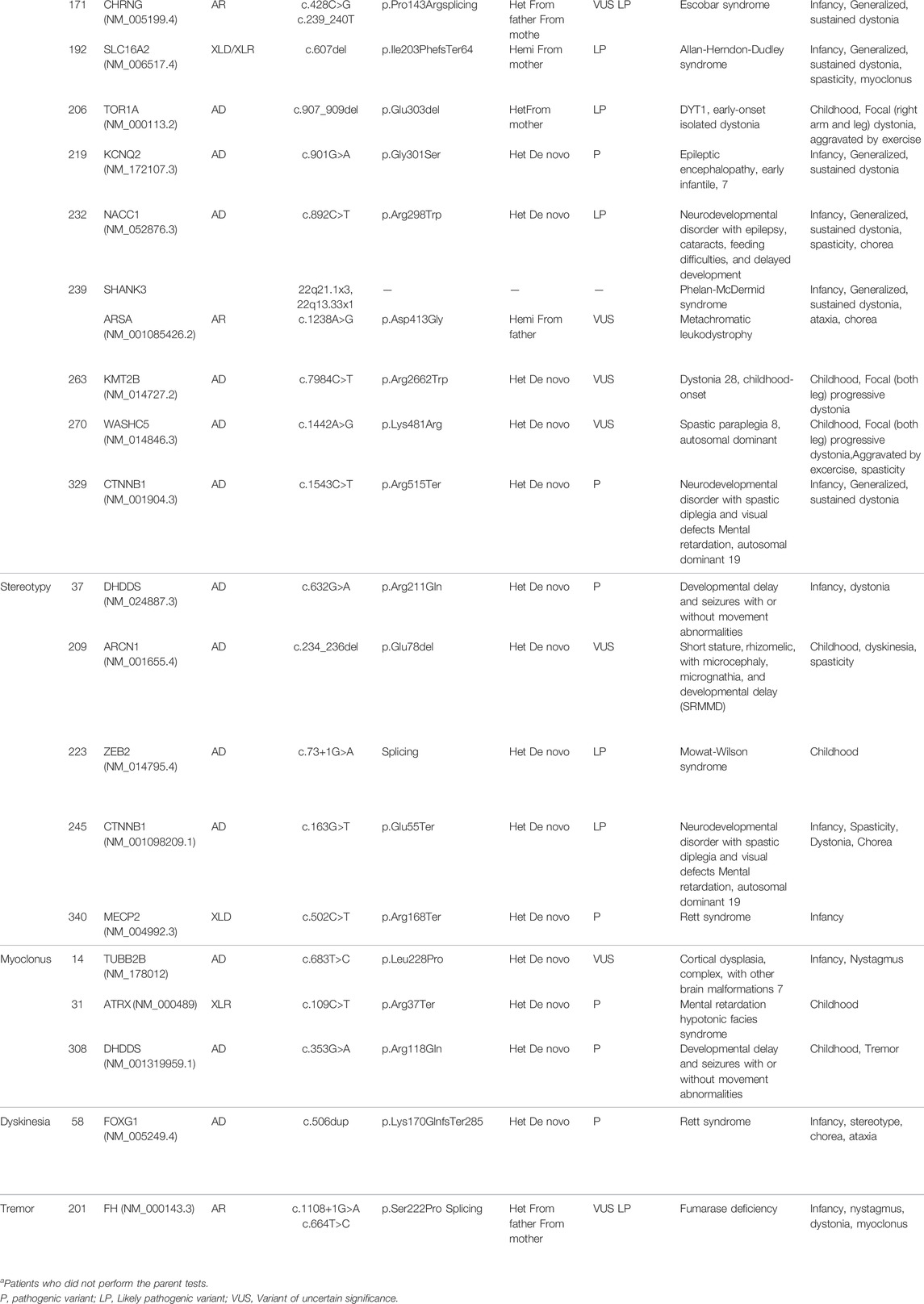
TABLE 2. Identified variants in our cohort classified on the basis of prominent movement disorders.
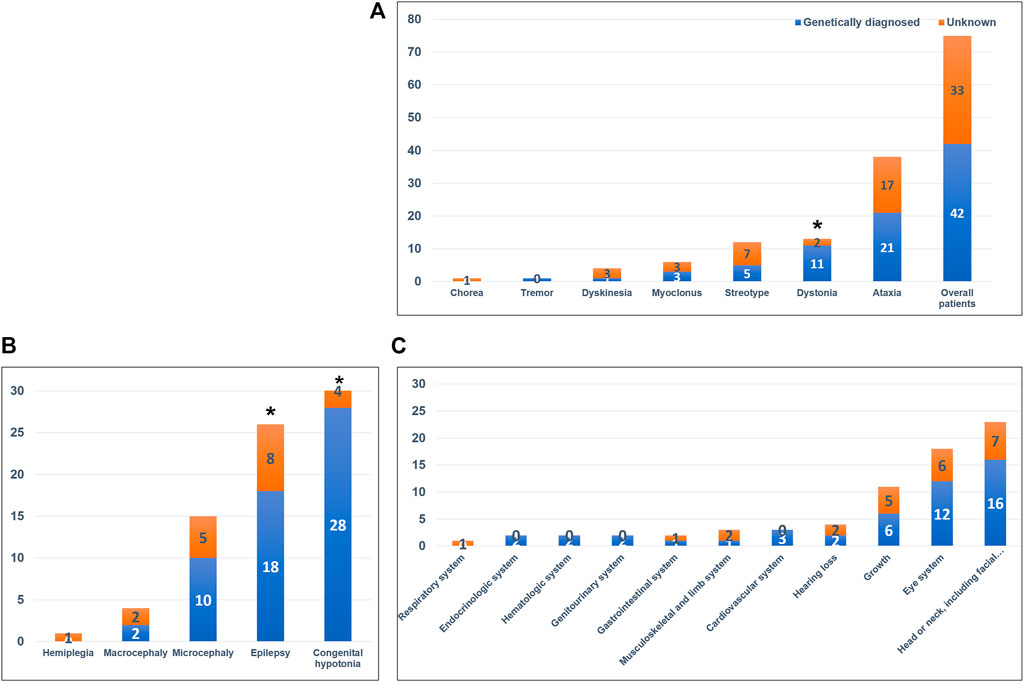
FIGURE 2. Diagnostic yield of whole-exome sequencing and associated neurologic and systemic symptoms. (A) Diagnostic yield in the overall patient cohort and in the different movement disorders. Patients with dystonia had a significantly higher rate of genetic diagnosis compared with patients with other movement disorders (p = 0.034). (B) Associated neurologic symptoms in the overall patient cohort. Patients with congenital hypotonia (p < 0.001) and epilepsy (p = 0.017) had a significantly higher rate of genetic diagnosis. (C) Associated systemic symptoms in the overall patient cohort. *p-value < 0.05.
The identified variants according to the prominent movement disorders are described in Table 2. The variant in most of the patients (n = 40/42, 95.2%) was confirmed by parental studies. Two patients in whom parental tests were not done are diagnosed by previously reported pathogenic variants according to the American College of Medical Genetics guideline and phenotype (Li et al., 2017). Thirty-one patients had de novo variants. Two patients had inherited the genes from their affected parents with autosomal dominant disorders. Biparental inheritance was confirmed in seven patients with autosomal recessive disorders and asymptomatic maternal carrier status in two patients with X-linked disorders.
Variants in the GNB1, CACNA1A, TCF4, DHDDS, and CTNNB1 genes were detected twice, and the other genes were identified only once each.
3.3 Genetic Diagnosis According to Their Movement Phenotypes
3.3.1 Ataxia
In 17 patients with persistent ataxia, the mutation in GNB1(2), KMT2A, UBE3A, GABRG2, SURF1, TCF4, ITPR1, FRRS1L, SLC2A1, MAST1, MED13L, DYNC1H1, DEPDC5, RTEL1, ATM, or EBF3 genes were found to be causative. Congenital hypotonia was the initial presenting sign in most cases, excluding one patient with the SURF1 mutation. Non-progressive ataxia developed between age 6 and 12 months. The patient with the SURF1 mutation showed normal development until age 2 years, when ataxia abruptly developed after a febrile illness.
Episodic ataxia was noted in three patients. Two patients with the CACNA1A mutations experienced recurrent cerebral infarction and episodic ataxia after febrile status epileptics, beginning in infancy or early childhood. Exercise aggravated the episodic ataxia in the patient with KCNA1 mutation since age 3 years. Of note, one patient with an FTL mutation developed progressive ataxia from age 13 years and was ultimately diagnosed with neurodegeneration with brain iron accumulation 3 (OMIM 606159).
3.3.2 Dystonia
In eight patients with generalized dystonia from infancy, the mutation of PIGA, TCF4, CHRNG, SLC16A2, KCNQ2, NACC1, CTNNB1, or ARSA gene were found to be causative. The TOR1A, KMT2B, or WASHC5 mutations were identified as causative gene in three patients with childhood-onset focal dystonia.
3.3.3 Stereotypy
In five patients with stereotypic movement as the only prominent phenotype, DHDDS, ARCN1, ZEB2, CTNNB1, or MECP2 mutations were identified, while other movement phenotypes accompanied three patients. Patients with SRMMD (short stature, rhizomelic, with microcephaly, micrognathia, and developmental delay; OMIM 617164), or Mowat–Wilson syndrome (OMIM 235730) initially developed congenital hypotonia, followed by stereotypy, observed since ages 6 and 2 years, respectively.
3.3.4 Myoclonus
In three patients with myoclonus as primary phenotype, mutation of TUBB2B (OMIM 610031), ATRX, or DHDDS (developmental delay and seizures with or without movement abnormalities; OMIM 617836) gene were found to be causative.
3.3.5 Others
One patient with unspecified dyskinesia had FOXG1 mutation, and a patient with tremor had FH mutation. All patients had a history of early-onset congenital hypotonia.
3.4 Separated or Combined Movement Phenomenology in Patients With Neurogenetic Disease
Seventeen patients (n = 17/42, 40.5%) with neurogenetic disease had complex movement phenomenology (Figure 3). Combined movement phenotypes were noted in seven patients (n = 7/19; 36.8%) with ataxia and nystagmus, three patients (n = 3/11; 27.2%) with dystonia, three patients (n = 3/5; 40.0%) with stereotypy, and two patients (n = 2/3; 66.6%) with myoclonus. Two patients with unspecified dyskinesia or tremor had variants in the FOXG1 or FH genes, combined with other movement phenotypes. Interestingly, among two patients with TCF4 mutation, one showed dystonia only and other represented ataxia and myoclonus. The two patients with DHDDS mutation showed stereotypy with dystonia and myoclonus with tremor, respectively.
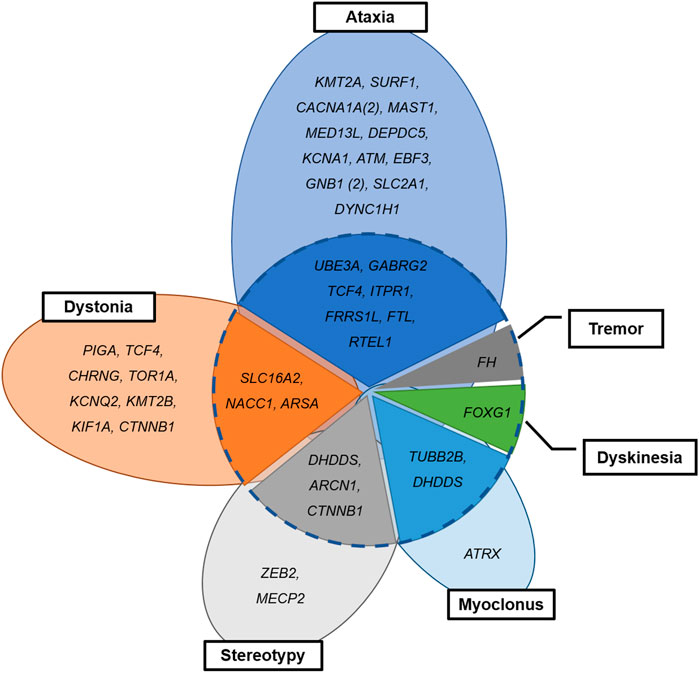
FIGURE 3. Combined or separated movement phenomenology in patients with neurogenetic disease. Dotted circle: combined (inside) and separate phenotypes (outside).
3.5 Genetic Relationship Between Movement Disorders and Other Systemic Manifestations
Thirty-eight patients who diagnosed with neurogenetic disease had other neurologic or systemic manifestation, except three patients with variants in KCNA1, KMT2B, and TOR1A genes (Figure 4); The patient with the KCNA1 mutation had episodic ataxia alone, and those with the KMT2B or TOR1A mutations showed dystonia only.
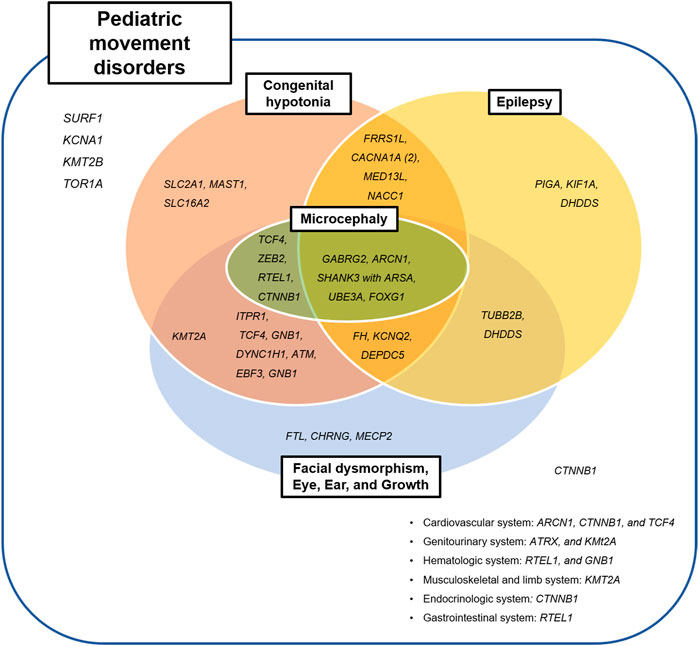
FIGURE 4. Clinical categories of identified pathogenic variants.
The most commonly associated systemic manifestation was dysmorphic face, presented in 18 patients (n = 18/42, 42.9%). Three patients with the CHRNG, MECP2, or ATRX mutations had dysmorphic face only, without other neurologic manifestations. Commonly associated neurologic symptoms included congenital hypotonia (n = 28/42; 66.7%) and epilepsy (n = 18/42; 42.9%). Congenital hypotonia and epilepsy were not observed in two patients with SURF1 or FTL mutations, who had presented with ataxia and childhood-onset cerebellar abnormalities.
3.5.1 History of Congenital Hypotonia
Congenital hypotonia was accompanied in two-third of patients (n = 28/42; 66.7%); 16/19 patients (84.2%) with ataxia (GNB1, KMT2A, UBE3A, GABRG2, CACNA1A (2), TCF4, ITPR1, FRRS1L, SLC2A1, MAST1, MED13L, DEPDC5, RTEL1, ATM, or EBF3), 6/11 patients (54.5%) with dystonia (TCF4, SLC16A2, KCNQ2, NACC1, CTNNB1, or ARSA), 2/5 patients (40.0%) with stereotypy (ARCN1 or ZEB2), 2/2 patients (100%) with spasticity (GNB1 or DYNC1H1), 1/1 patient (100%) with unspecified dyskinesia (FOXG1), and 1/1 patient (100%) with tremor (FH).
3.5.2 Concomitant Epilepsy
Epilepsy was present in almost half of patients (n = 18/42; 42.9%); 7/17 patients (41.2%) with ataxia (CACNA1A (2), UBE3A, GABRG2, FRRS1L, MED13L, or DEPDC5), 5/11 patients (45.5%) with dystonia (PIGA, KCNQ2, NACC1,WASHC5, or ARSA), 2/5 patients (40.0%) with stereotypy (DHDDS or ARCN1), 2/3 patients (50.0%) with myoclonus (TUBB2B or DHDDS), 1/1 patient (100.0%) with unspecified dyskinesia (FOXG1), and 1/1 patient (100.0%) with tremor (FH). The 10/18 patients with epilepsy and genetic diagnosis (55.5%) were diagnosed as having epileptic encephalopathy spectrum including early infantile epileptic encephalopathy, epileptic spasms, and Lennox-Gastaut syndrome. Other eight patients without epileptic encephalopathy also received more than two anti-seizure medications.
Genes previously been reported to cause epilepsy with movement disorders (KCNA1, KMT2B, CACNA1A, FRRS1L, KCNQ2, SLC2A1, UBE3A, GABRG2, GNB1, FOXG1, or MECP2) were also observed in our study; however, epilepsy was not associated with KCNA1, KMT2B, SLC2A1, GNB1, or MECP2 in our cohort. On the other hand, ten additional genes were associated with epilepsy in our study (MED13L, DEPDC5, PIGA, NACC1, ARSA, KIF1A, DHDDS, TUBB2B, ARCN1, or FH), for which there is no previously reported association with epilepsy-dyskinesia disorders (Figure 4).
3.5.3 Clinical Management According to Genetic Diagnosis
Genetic diagnosis changed the clinical care of five patients (n = 5/75, 6.7%). They were treated with targeting the identified abnormalities at the molecular level with a ketogenic diet (with a variant in SLC2A1), acetazolamide (with two variants in CACNA1A), and vitamin E and alpha-lipoic acid (with a variant in ATM). Cancer screening was performed in two patient with the FH and ATM mutation (Chan et al., 2017; Garg and Rabban, 2021). The patient with a variant in SLC2A1 showed significant improvement of their ataxia after ketogenic diet recommendation and was able to walk alone at 5 years of age. The two patients with variants in CACNA1A showed decreased episodic ataxia events after acetazolamide treatment. The patient with the ATM variant reports some improved effect on their ataxia after alpha-lipoic acid administration. There were no abnormal cancer related findings after screening in the two patients with the FH and ATM variants. Annual follow-up was scheduled.
4 Discussion
Movement disorders sometimes can be key features in the differential diagnosis of neurogenetic disorders; however, their phenotypes have received less attention owing to their vague and complex features.
In the current study, we described the clinical and genetic features of the movement phenotypes presenting in a cohort of pediatric patients with neurogenetic disorders. Despite the small size of patient cohort, WES revealed high diagnostic rate. Patients with a genetic diagnosis more often accompanied the movement phenotype of dystonia, congenital hypotonia, and epilepsy. Importantly, our study dissected the complexity between diverse phenotypic and genetic heterogeneity, offering some new clinical and genetic insights. Additionally, we provide the chance to patients-tailored treatment according to the genetic diagnosis in patients with movement phenotype.
We identified 42 variants in 37 different genes, which yielded a diagnostic rate of 56.0%. Six studies have identified genetic disorders using NGS, including targeted or WES in adults (Neveling et al., 2013; van Egmond et al., 2017; Montaut et al., 2018; Reale et al., 2018) and pediatric patients (Cordeiro et al., 2018; Graziola et al., 2019) with movement disorders, and the diagnostic yield varied from 11.3 to 51%. The rate of diagnosis was higher in studies with a pediatric cohort (28–51%) than with adult patients (11–22%). In addition, studies using WES ((Neveling et al., 2013; Cordeiro et al., 2018), 20–51%) yielded a higher diagnostic rate than those using panel gene testing ((Montaut et al., 2018; Reale et al., 2018; Graziola et al., 2019), 11–28%).
Pediatric movement disorders show complex, evolving phenotype patterns, and are frequently observed as part of multiple neurological or systemic manifestations. For these reasons, the genetic diagnosis of pediatric movement disorders is challenging. In our study, multidisciplinary approach to dissect the patient phenotypes, systemic as well as neurological, by a pediatric neurologist and clinical geneticist, and the application of specific NGS technology using an artificial-intelligence-based, automated variant-priority analysis system (Seo et al., 2020) might have helped to achieve a higher diagnostic rate. By broadening the association-linkage between human phenotypes and genes, this automated system might have been able to detect variants that could otherwise have been missed.
Previously studies have noted differences in the distributions of the causing gene between adults and pediatric cohorts. The PARKIN, LRRK2, GBA, PLA2G6, VSP13A, and ATP13A2 genes have been considered the major causative genes in adult populations (van Egmond et al., 2017; Montaut et al., 2018; Reale et al., 2018), whereas the causative genes were diverse in pediatric cohorts; The 14 dyskinesia patients with the PRRT2 or SLC2A1 mutations (Graziola et al., 2019), 6 dystonia patients with the ATP1A3 mutations (Graziola et al., 2019), and 4 ataxia with the STXBP1 mutations (Cordeiro et al., 2018). In our pediatric patients, the genetic spectrum was more diverse and complex, and the main movement phenomenology was heterogeneous even among the same gene-defect carriers (GNB1, TCF4, DHDDS, or CTNNB1 genes).
In our patient cohort, the most common phenomenologies were ataxia and dystonia. Patients with dystonia had a higher genetic risk profile compared with other movement phenotypes (Graziola et al., 2019). In contrast with previous studies (Cordeiro et al., 2018; Graziola et al., 2019), only a small proportion of patients had dyskinesia (5.3%) and no patient was identified carrying the PRRT2 mutation which is a well-known, most-common cause of the paroxysmal dyskinesia (Erro et al., 2014; De Gusmao and Silveira-Moriyama, 2019). We performed a single gene test for PRRT2 for patients with paroxysmal dyskinesia who were not eligible for WES, which may be a reason for these results.
In line with other studies (Cordeiro et al., 2018; Montaut et al., 2018; Graziola et al., 2019), half of our patients with a neurogenetic diagnosis had combined movement phenotypes. Traditionally, genetic dystonia has been divided into three main categories according to the combined movement phenotype: isolated dystonia, dystonia combined with myoclonus or parkinsonism, and complex, in which dystonia is a feature of complex neurological syndromes (Pearson and Pons, 2019). In our study, importantly, these combined phenotypes were noted in patients affected by the neurodegenerative diseases caused by the SLC16A2, ARSA, FOXG1, or FH mutations (Figure 3), which warrants the monitoring of disease progression in the affected patients.
Two third of our patients coexisted with other neurologic manifestation, and it is of note that congenital hypotonia were highly detected in the genetically diagnosed patients. This could be explained by the fact that the brain underdevelopment contributes to hypotonia development during neonatal and infantile periods and then, the motor symptoms such as movement phenomenologies evolve with age as the brain develops, even when the underlying pathology is non-progressive (Pearson and Pons, 2019).
Additionally, epilepsy was also a commonly associated neurologic manifestation in diagnosed patients. Recently, the researchers are becoming aware of the increasing number of neurogenetic comorbid conditions presenting with both epilepsy and atypical movements (Papandreou et al., 2020). These disorders within the ‘genetic epilepsy-dyskinesia’ spectrums are clinically and genetically heterogeneous from isolated movement disorders to severe infantile epileptic encephalopathy (de Gusmao and Waugh, 2018; De Gusmao and Silveira-Moriyama, 2019; Papandreou et al., 2020). Over 60 genes have been reported as associated with epilepsy-dyskinesia phenotype (de Gusmao and Waugh, 2018; De Gusmao and Silveira-Moriyama, 2019; Papandreou et al., 2020), and the list is expanding. In our study, MED13L, DEPDC5, PIGA, NACC1, ARSA, WASHC5, DHDDS, TUBB2B, or ARCN1 can be added to this category (Figure 4).
Importantly, the genetic diagnosis changed the clinical care for five patients (6.7%) in our study. Four patients received targeted molecular therapy. For the patient with a mutation in the ATM gene mutation, encoding ATM serine/threonine kinase protein which is involved in the DNA repair machinery, antioxidants including vitamin E and alpha-lipoic acid were given to reduce the increased oxidative stress (Maciejczyk et al., 2017; Blignaut et al., 2021). For the patient with SLC2A1 mutation, the ketogenic diet, the first line treatment for GLUT1 deficiency syndrome, were offered to produce ketone bodies as an alternative energy source for brain metabolism and to bypass the GLUT defect (Koch and Weber, 2019). The CACNA1A mutation results in reduced calcium current and decreased inhibitory effects of Purkinje cells. Acetazolamide responsiveness is a hallmark of the disease and about 50–75% of patients show rapid improvement in episode frequency and severity (Kotagal, 2012). We performed tumor surveillance for the patient with a FH (HLRCC; OMIM #150800) and ATM mutations.
The clinical features and course of movement phenotypes in pediatric patients with neurogenetic disorders are diverse, difficulting the clinical classification of pediatric movement disorders. Due to the diverse and complex range of phenotypic and genetic heterogeneities, genome sequencing (WES or WGS) should be considered for patients with movement disorders. Additionally, WES/WGS may be preferred over panel-gene testing for revealing and understanding this complexity. Since most patients in our cohort underwent other genetic testing including karyotyping, CMA, or single gene sequencing prior to WES, the diagnostic yield of WES can be misleading and the exact superiority of WES could not be assessed. However, as shown in our study, the early application of WES with its rapid interpretation by automated system yields the acknowledgeable diagnostic rate and physicians can overcome the limitations of the usual phenotype-driven strategies and avoid the long journey for diagnosis. Furthermore, phenotypic reanalysis after genetic confirmation help to understand the expanding clinical spectrum of a genetic disease and identification of the patients in their early disease state, despite not exhibiting the full spectrum of the disease.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ebi.ac.uk/ena/browser/view/PRJEB51291.
Ethics Statement
The studies involving human participants were reviewed and approved by The Institutional Review Board for Human Research at Asan Medical Center approved this study (IRB number 2017-0988). Written informed consent to participate in this study was provided by the participants’ legal guardian.
Author Contributions
Research project: M-SY and BL; Statistical Analysis: M-JK, M-SY, and BL; Writing of the first draft by M-JK, M-SY, and GS; Review and Critique by M-SY, BL, GS, and T-SK.
Funding
This work was supported by an Institute for Information and Communications Technology Promotion (IITP) grant funded by the Korean government (MSIT) (2018-0-00861, Intelligent SW Technology Development for Medical Data Analysis) and Bigdata analysis.
Conflict of Interest
Author GS is employed by the company 3billion Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all participants and their family members for taking part in this study.
References
Blignaut, M., Harries, S., Lochner, A., and Huisamen, B. (2021). Ataxia Telangiectasia Mutated Protein Kinase: a Potential Master Puppeteer of Oxidative Stress-Induced Metabolic Recycling. Oxid. Med. Cell Longev. 2021, 8850708. doi:10.1155/2021/8850708
Chan, M. M. Y., Barnicoat, A., Mumtaz, F., Aitchison, M., Side, L., Brittain, H., et al. (2017). Cascade Fumarate Hydratase Mutation Screening Allows Early Detection of Kidney Tumour: a Case Report. BMC Med. Genet. 18, 79. doi:10.1186/s12881-017-0436-1
Cordeiro, D., Bullivant, G., Siriwardena, K., Evans, A., Kobayashi, J., Cohn, R. D., et al. (2018). Genetic Landscape of Pediatric Movement Disorders and Management Implications. Neurol. Genet. 4, e265. doi:10.1212/nxg.0000000000000265
De Gusmao, C. M., and Silveira-Moriyama, L. (2019). Paroxysmal Movement Disorders - Practical Update on Diagnosis and Management. Expert Rev. Neurother. 19, 807–822. doi:10.1080/14737175.2019.1648211
de Gusmao, C. M., and Waugh, J. L. (2018). Inherited and Acquired Choreas. Seminars Pediatr. Neurology 25, 42–53. doi:10.1016/j.spen.2018.01.002
Erro, R., Sheerin, U.-M., and Bhatia, K. P. (2014). Paroxysmal Dyskinesias Revisited: a Review of 500 Genetically Proven Cases and a New Classification. Mov. Disord. 29, 1108–1116. doi:10.1002/mds.25933
Garg, K., and Rabban, J. (2021). Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome Associated Uterine Smooth Muscle Tumors: Bridging Morphology and Clinical Screening. Genes Chromosom. Cancer 60, 210–216. doi:10.1002/gcc.22905
Graziola, F., Garone, G., Stregapede, F., Bosco, L., Vigevano, F., Curatolo, P., et al. (2019). Diagnostic Yield of a Targeted Next-Generation Sequencing Gene Panel for Pediatric-Onset Movement Disorders: a 3-year Cohort Study. Front. Genet. 10, 1026. doi:10.3389/fgene.2019.01026
Kim, M.-J., Yum, M.-S., Seo, G. H., Lee, Y., Jang, H. N., Ko, T.-S., et al. (2020). Clinical Application of Whole Exome Sequencing to Identify Rare but Remediable Neurologic Disorders. Jcm 9, 3724. doi:10.3390/jcm9113724
Koch, H., and Weber, Y. G. (2019). The Glucose Transporter Type 1 (Glut1) Syndromes. Epilepsy & Behav. 91, 90–93. doi:10.1016/j.yebeh.2018.06.010
Kotagal, V. (2012). Acetazolamide-responsive Ataxia. Semin. Neurol. 32, 533–537. doi:10.1055/s-0033-1334475
Li, M. M., Datto, M., Duncavage, E. J., Kulkarni, S., Lindeman, N. I., Roy, S., et al. (2017). Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagnostics 19, 4–23. doi:10.1016/j.jmoldx.2016.10.002
Maciejczyk, M., Mikoluc, B., Pietrucha, B., Heropolitanska - Pliszka, E., Pac, M., Motkowski, R., et al. (2017). Oxidative Stress, Mitochondrial Abnormalities and Antioxidant Defense in Ataxia-Telangiectasia, Bloom Syndrome and Nijmegen Breakage Syndrome. Redox Biol. 11, 375–383. doi:10.1016/j.redox.2016.12.030
Montaut, S., Tranchant, C., Drouot, N., Rudolf, G., Guissart, C., Tarabeux, J., et al. (2018). Assessment of a Targeted Gene Panel for Identification of Genes Associated With Movement Disorders. JAMA Neurol. 75, 1234–1245. doi:10.1001/jamaneurol.2018.1478
Neveling, K., Feenstra, I., Gilissen, C., Hoefsloot, L. H., Kamsteeg, E.-J., Mensenkamp, A. R., et al. (2013). A Post-hoc Comparison of the Utility of Sanger Sequencing and Exome Sequencing for the Diagnosis of Heterogeneous Diseases. Hum. Mutat. 34, 1721–1726. doi:10.1002/humu.22450
Papandreou, A., Danti, F. R., Spaull, R., Leuzzi, V., McTague, A., and Kurian, M. A. (2020). The Expanding Spectrum of Movement Disorders in Genetic Epilepsies. Dev. Med. Child. Neurol. 62, 178–191. doi:10.1111/dmcn.14407
Pearson, T. S., and Pons, R. (2019). Movement Disorders in Children. CONTINUUM Lifelong Learn. Neurology 25, 1099–1120. doi:10.1212/con.0000000000000756
Reale, C., Panteghini, C., Carecchio, M., and Garavaglia, B. (2018). The Relevance of Gene Panels in Movement Disorders Diagnosis: a Lab Perspective. Eur. J. Paediatr. Neurology 22, 285–291. doi:10.1016/j.ejpn.2018.01.013
Rexach, J., Lee, H., Martinez-Agosto, J. A., Németh, A. H., and Fogel, B. L. (2019). Clinical Application of Next-Generation Sequencing to the Practice of Neurology. Lancet Neurology 18, 492–503. doi:10.1016/s1474-4422(19)30033-x
Sanger, T. D., Chen, D., Fehlings, D. L., Hallett, M., Lang, A. E., Mink, J. W., et al. (2010). Definition and Classification of Hyperkinetic Movements in Childhood. Mov. Disord. 25, 1538–1549. doi:10.1002/mds.23088
Seo, G. H., Kim, T., Choi, I. H., Park, J. y., Lee, J., Kim, S., et al. (2020). Diagnostic Yield and Clinical Utility of Whole Exome Sequencing Using an Automated Variant Prioritization System, EVIDENCE. Clin. Genet. 98, 562–570. doi:10.1111/cge.13848
Seo, G. H., Lee, H., Lee, J., Han, H., Cho, Y. K., Kim, M., et al. (2022). Diagnostic Performance of Automated, Streamlined, Daily Updated Exome Analysis in Patients with Neurodevelopmental Delay. Mol. Med. 28, 38. doi:10.1186/s10020-022-00464-x
Keywords: movement disorders, whole-exome sequencing, next-generation sequencing, neurogenetic disorders, pediatric neurological disease
Citation: Kim M-J, Yum M-S, Seo GH, Ko T-S and Lee BH (2022) Phenotypic and Genetic Complexity in Pediatric Movement Disorders. Front. Genet. 13:829558. doi: 10.3389/fgene.2022.829558
Received: 06 December 2021; Accepted: 06 May 2022;
Published: 01 June 2022.
Edited by:
Gustavo Glusman, Institute for Systems Biology (ISB), United StatesReviewed by:
Alessio Di Fonzo, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, ItalyPaulo Victor Sgobbi Souza, Federal University of São Paul, Brazil
Claudia Gonzaga-Jauregui, Universidad Nacional Autónoma de México, Mexico
Copyright © 2022 Kim, Yum, Seo, Ko and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mi-Sun Yum, bWlzdW4ueXVtQGdtYWlsLmNvbQ==; Beom Hee Lee, YmhsZWVAYW1jLnNlb3VsLmty