 Wei Zhang1
Wei Zhang1 Jiangfeng Mao
Jiangfeng Mao Min Nie
Min Nie- 1 NHC Key Laboratory of Endocrinology (Peking Union Medical College Hospital), Department of Endocrinology, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
- 2 State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
Purpose: Patients with syndromic 46, XY disorders/differences of sex development (DSD) are characterized by gonadal and phenotypic genders inconsistent with their chromosomal sexes as well as abnormalities of multiple extragonadal organs. They are caused by mutations in specific genes, which are expressed in the affected organs and regulate their development, and over fourteen genes have been identified. In this study, we aimed to determine the underlying cause of a patient with syndromic 46, XY DSD and review the clinical presentations and genetic findings of all reported similar cases.
Methods: Whole-exome sequencing (WES) was performed to find a molecular cause of the patient. In silico tools were used to analyze the pathogenicity of the variants. Reports of cases with similar clinical features and involved genes were summarized by searching through PubMed/MEDLINE using keywords “PPP2R3C” or “G5PR” and “46,XY disorders of sex development”.
Results: Compound heterozygous variants (p.F229del/p.G417E) in PPP2R3C were identified in the 24-year-old female by WES and verified by Sanger sequencing. The patient presents complete testicular dysgenesis, low birth weight, facial deformity, cubitus valgus, and decreasing number of CD19+ B lymphocytes and CD4+ T lymphocytes. A total of thirteen 46, XY DSD cases with four homozygous PPP2R3C mutations (p.Leu103Pro, p.Leu193Ser, p.Phe350Ser, and p.Ser216_Tyr218dup) have been reported previously, and their clinical manifestations are roughly similar to those of our patient.
Conclusion: Novel compound heterozygous variants in PPP2R3C cause specific syndromic 46, XY gonadal dysgenesis, which broadened the pathogenic variants spectrum of PPP2R3C. The typical phenotype of PPP2R3C mutation is complete 46, XY gonadal dysgenesis with multiple extragonadal anomalies, including facial deformities, skeletal system abnormalities, muscle abnormalities, impaired nervous system, impaired hearing and vision, heart and kidney anomalies, and gastrointestinal dysfunction.
Introduction
46, XY disorders/differences of sex development (DSD) is a condition in which phenotypic and/or gonadal sex do not match with 46, XY chromosomal sex. The disease may present a dysgenetic gonadal, testicular, ovotesticular, or ovarian tissue and variable degrees of virilization of the external genitalia, along with structures derived from the Wolffian or Müllerian duct. According to the different pathogenesis, 46, XY DSD is mainly divided into gonadal development abnormalities, insufficiency of androgen synthesis, insensitivity to androgens, or Müllerian duct persistent syndrome (Bashamboo and McElreavey, 2015). The etiology of 46, XY DSD is mutations of genes involved in androgen synthesis, androgen action, or Müllerian duct regression. Currently, more than 35 genes have been identified as genetic causes of 46, XY DSD (Baxter et al., 2015; Yu et al., 2021).
46, XY DSD can be classified as either syndromic or non-syndromic 46, XY DSD based on the presence or absence of extragonadal abnormalities (Hiorta and Gillessen-Kaesbachb, 2009). The incidence of syndromic 46, XY DSD is very low and usually caused by mutations in specific genes that play essential roles in both sex development and extragonadal organ formation. More than 14 genes at present are known to be linked with syndromic 46, XY DSD, such as WT1, NR5A1, SOX9, FGFR2, HOXA13, DHCR7, and XH2, most of which are genes encoding transcript factors (Bagheri-Fam et al., 2015; Tamura et al., 2017).
Sex-determining region Y (SRY)-box 9 (SOX9) protein, a very important transcript factor, regulates the onset, development, and maintenance of the testis formation, and it is also essential for the early chondrogenesis in humans. Mutations or deletions of SOX9 can cause a syndromic 46, XY DSD, presenting as 46, XY sex reversal and campomelic dysplasia. The SOX9-phosphoprotein can be translocated into the nucleus of Sertoli cells and maintains differentiation of Sertoli cells (Malki et al., 2005). Human PPP2R3C (Protein Phosphatase 2 Regulatory Subunit B Gamma gene, OMIM: 615902) is located on 14q13.2, which has 13 exons encoding a 453-amino acid protein. Recently, it was found that the PPP2R3C gene regulates the balance of phosphorylation and dephosphorylation of Sox9 protein by forming a protein phosphatase complex. The mutation of the PPP2R3C gene is identified as a novel genetic etiology for syndromic 46, XY DSDs (Guran et al., 2019). Homozygous variants in the PPP2R3C gene can cause a syndromic 46, XY DSD, which manifests specific facial features, myopathy, musculoskeletal abnormalities, and neural motor delay.
In this study, we described the clinical, laboratory characteristics, and novel compound heterozygous PPP2R3C variants in a Chinese patient diagnosed with 46, XY gonadal dysgenesis. The clinical presentations and genetic findings of similar cases reported previously were also reviewed to improve our understanding of the phenotypes and genotypes in patients with PPP2R3C mutations.
Methods
Whole-Exome Sequencing
Patient’s genomic DNA was extracted from 200 μl peripheral blood leukocytes using the Qiagen DNA Blood kit (Qiagen, Dusseldorf, Germany). First, 50 ng DNA was broken into a size of about 200bp by fragmentation enzymes. The DNA fragments were then end-repaired, and the 3’end was added with one A base. Second, the DNA fragments were ligated with barcoded sequencing adaptors, and fragments of about 320bp were collected by XP beads. After PCR amplification, the DNA fragments were hybridized and captured by NanoWES according to the manufacturer’s protocol. The hybrid products were eluted and collected and then subjected to PCR amplification and purification. Next, the libraries were quantified by qPCR, and size distribution was determined using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, United States). Finally, the Novaseq6000 platform (Illumina, San Diego, United States), with 150 bp pair-end sequencing mode, was used for sequencing the genomic DNA of the patient. Raw image files were processed using CASAVA v1.82 for base calling and generating raw data. The sequencing reads were aligned to the human reference genome (hg19/GRCh37) using the Burrows–Wheeler Aligner tool. GATK software was employed for variant calling. Variant annotation and interpretation were conducted by ANNOVAR.
TA Clone and Sanger Sequencing
Exon 7 and exon 13 of the PPP2R3C gene were amplified by PCR, and then the PCR products of exon 7 and exon 13 were purified and underwent Sanger sequencing. Primers were designed using Primer3 software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). Primer sequences were as follows: PPP2R3C-E7-F: 5′-ACTACATATTGGAACTTATCCCTACG-3′, PPP2R3C-E7-R: 5′-GGCAG TGAC AGGAAAATAAATGATA-3′; PPP2R3C-E13-F: 5′-ATTTTATTCCTCGTACTTCAG TGGA-3′, PPP2R3C-E13-R: 5′-GGTCCTATT GCTTCTAAACTATT GC -3′.
TA clone of exon 7 was performed for a small deletion existing in one allele of the patient. The Taq polymerase-amplified PCR products of exon 7 were inserted into the pMD19T vector, and then the constructs were transformed into DH5α-competent cells and grew in the X-Gal and IPTG plates. The white clones were selected and amplified, and their DNA was extracted and sequenced.
Sequencing of PCR products and TA clone was performed using a Taq big dye terminator sequencing kit and an ABI-3730 automated sequencer (Applied Biosystems, Foster City, CA, United States). DNAs from both parents were sequenced to ascertain the inheritance. We read the sequencing results by 4Peaks (Nucleobytes, Netherlands) and then blasted each in NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi) taking the following sequence as references: cDNA: NM_017917.4; protein: NP_060387.2.
Three-Dimensional Visualization and Prediction
Structural models of wild PPP2R3C protein and mutant protein were built by inputting protein sequences in the SWISS-MODEL server (https://swissmodel.expasy.org). Then, we downloaded the PDB file and uploaded it into UCSF ChimeraX (version: 1.2.5), a molecular visualization program. By comparing the changes in the contacts and hydrogen bonds with other amino acids in a spatial model, we can predict the potential pathogenic effects the specific variant brings to the protein.
Clinical Evaluation
Skeletal deformity and external genital development were assessed by physical examination. Uterine and gonadal development was assessed by laparoscopy. Bone age was assessed by X-rays of hands. In addition, we evaluated the muscular and lymphocytic systems using electromyography and lymphocyte counts. Hormone test results included levels of serum follicle-stimulating hormone (FSH), luteinizing hormone (LH), and testosterone (T), which were measured by chemiluminescent immunoassays (Bayer Diagnostics Corporation, United States).
Literature Review
We searched for reports about PPP2R3C variations in PubMed and MEDLINE, and the keywords for searching included “PPP2R3C” or “G5PR” in combination with “46,XY disorders of sex development”. Available data on clinical evaluations and genetic findings were extracted and summarized.
Results
Compound Heterozygous Variants in PPP2R3C
By WES, we identified two variants (c.684_686delTTC/p.F229del in exon 7 and c.1250G > A/p. G417E in exon 13) of PPP2R3C and confirmed by Sanger sequencing (Figure 1) in the patient, which came from his father and mother, respectively. Potentially pathogenic variants in other genes associated with syndromic 46, XY DSD had not been found. The two wild-type amino acids involved in both variants are highly conserved among ortholog proteins (Figure 2). The deletion of Phe229 will cause an incomplete alpha helix structure and change the condition of four repeated phenylalanines. The Gly417Glu does not affect a significant protein domain, but the number of hydrogen bonds and contacts formed within residues is changed (Figure 2).
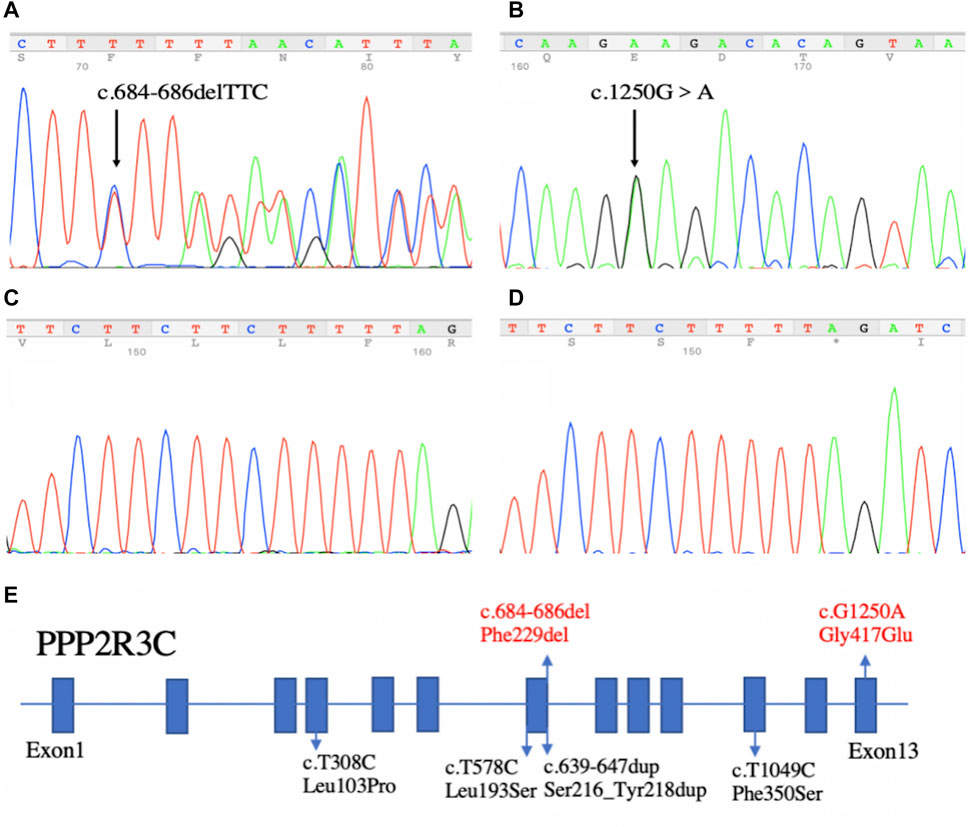
FIGURE 1. Sequencing results of our patient and the summary of PPP2R3C mutations found in patients with 46, XY disorders/differences of sex development. (A) In-frame deletion (c.684_686delTTC) in exon 7 and (B) missense mutation (c.1250G > A) in exon 13. (C,D) Sequencing results of TA-clone. Left panel (C): wild-type allele; Right panel (D): other allele with the in-frame deletion (c.684_686delTTC). (E) Summary of PPP2R3C mutations found in patients with 46, XY disorders of sex development. Blue boxes indicate the exons of PPP2R3C. Four reported variants are depicted in black, and the novel compound heterozygotic variants are depicted in red.
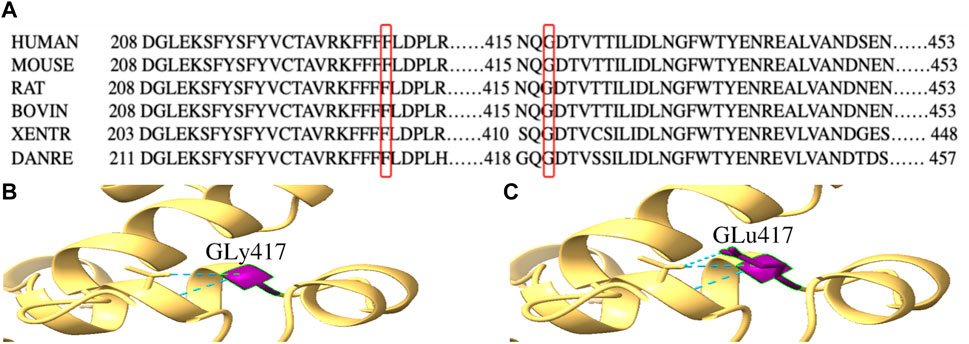
FIGURE 2. Conservation analysis of PPP2R3C 229F and 417G and three-dimensional molecular models of p.G417E PPP2R3C. (A) Partial alignment of PPP2R3C amino acid sequence, showing high conservation of phenylalanine (at position 229) and glycine (at position 417); (B,C) Gly417 and Glu417 residues are depicted in purple. The mutation of p.G417E will change the number of hydrogen bonds (blue).
Clinical Manifestation
A 24-year-old female of Chinese Han descent was diagnosed with 46, XY gonadal dysgenesis at age 18, when she was presented with poor development of secondary sexual characteristics and primary amenorrhea. She was born of a non-consanguineous marriage at term by vaginal delivery without any perinatal complications and was raised female gender. She was born with a birth weight of around 1 kg and normal body length. There was no history of feeding difficulties or developmental retardation in childhood. At ten years of age, she presented retardation of bone age about two years, and the closure of bone epiphysis did not appear till over 20 years old. The patient was 148 cm tall (-3SD) at 18 years old and was diagnosed with short stature. She received growth hormone therapy for three months and gained the height increase of about 2.5 cm in the following two years. The patient did not receive other therapy till presentation.
On physical examination, she had a height of 155 cm, a weight of 48 kg, and a blood pressure of 124/75 mmHg. Tanner staging was B1P2. External genitalia revealed the clitoris of normal and puerile female genitalia. There were two distinct openings in the urethra and vagina. No gonads or lumps were found in the inguinal region. The patient did not have facial hair, acne, male torso, body hairs, and deepening of voice. Systemic examination revealed remarkable facial features including unruly scalp hair, thin eyebrows, thin lips, and long and smooth philtrum. The patient had hypoplastic ala nasi and beaked nose, low-set ears, and facial pigmented nevus which were non-ignorable facial characteristics as well. The systemic examination also revealed abnormalities of the musculoskeletal system including a short fifth phalanx, webbing of the neck, and cubitus valgus (Figures 3A–C).
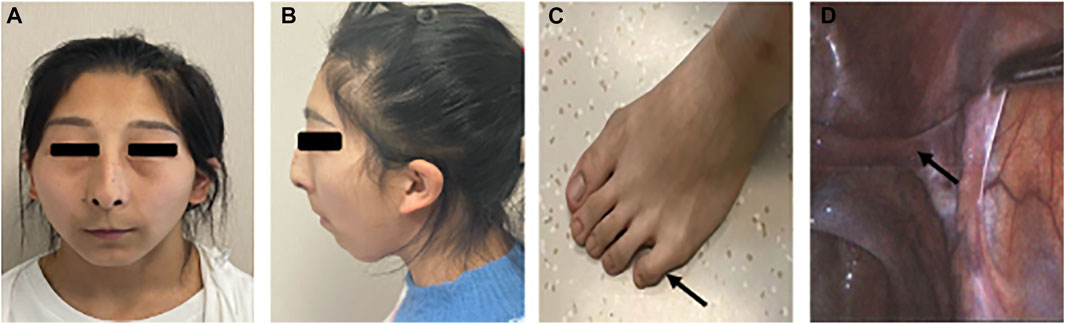
FIGURE 3. Partial clinical presentations of the patient. (A) Facial frontal photo of the patient and (B) facial profile photo of the patient: flat face, thin lips, low ears, mandibular retrusion, dysplasia of the alar nose, (C) short fifth phalanx, and (D) oviduct-like structure in laparoscopy.
On investigation, the hormone profile showed LH 23.22IU/L (normal range 1.2–8.6), FSH 70.88IU/L (normal range 1.2–19.2), T 0.34 ng/ml (normal range 1.75–7.81), E2 11 pg/ml (normal range <39), 17-OHP 1.19 ng/ml (-), FT4 1.19 ng/dl (normal range 0.81–1.89), FT3 2.95 pg/ml (normal range 1.80–4.10), and TSH1.2μIU/ml (normal range 0.38–4.34) (Table.1). Lab evaluation also included a normal white blood cell count, normal hemoglobin concentration, and normal lymphocyte count and proportion. PPP2R3C gene is highly expressed in lymphocytes and plays a role in the survival of CD19+ B cells. Gene knockout (PPP2R3C-/-) mice by conditional targeting in CD19+ B cells showed a deficit in B-cell survival and a reduced number of mature B cells. After we identified the PPP2R3C variants of our patient, we tested her T/B cell subsets. The analysis of T/B cell subsets revealed a decreased number of CD19+ B cells (1.6%, normal range: 8.5%–14.5%) and CD4+ T cells (21.5%, normal range: 30.0–46.0%) (Table.2). The karyotype was 46, XY. Ultrasound and laparoscopy revealed the dysgenesis of the uterus and bilateral oviduct-like structure (Figure 3D).

TABLE 1. Sex hormones of the patient.
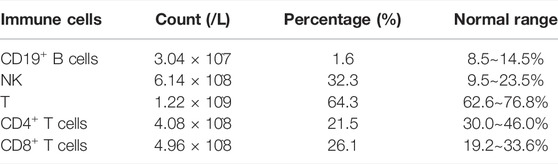
TABLE 2. Lymphocyte subsets of the patient.
Considering the risk of gonadal malignancy, she underwent bilateral gonadectomy at twenty years of age, and streak gonads and bilateral oviducts were found during the operation. Histopathology of gonadectomy material showed an oviduct-like structure bilaterally. After gonadectomy, she was treated with Femoston (estradiol/dydrogesterone: 1/10 mg), with a dose of one tablet daily for almost four years.
Literature Review
Thirteen 46, XY DSD patients had been reported in three articles previously (Guran et al., 2019; Cicek et al., 2021; Altunoglu et al., 2022). All the patients came from the consanguineous marriage and had been identified as homozygous PPP2R3C variants. Eleven out of thirteen 46, XY DSD patients were presented with complete gonadal dysgenesis, complete female vulva with hypoplastic labium major, and hypoplastic uterus. Sex hormone levels were compatible with complete gonadal dysgenesis. One patient presented with partial testicular dysgenesis, and he had ambiguous genitalia, micropenis, and left cryptorchidism. PPP2R3C variants are also reported to be associated with syndromic 46, XX DSD. Three such patients were presented with complete gonadal dysgenesis, recognizable facial features, and multiple extragenital malformations. All patients with PPP2R3C mutations had some similar extragonadal system manifestations, such as facial deformity (16 of 16,100%), retardation of bone age (15 of 16, 93.7%), and delayed development of the nervous system (14 of 16, 87.5%). The typical facial deformity includes abnormal eyebrows, low small-ears, and dysplasia of the alar nose. Among sixteen patients, more than half of the cases had special clinical manifestations, such as impaired vision (10 of 16, 62.5%), low birth weight (9 of 16, 56.2%), and myopathy (8 of 16, 50%). Symptoms including, renal agenesis (6 of 16, 37.5%), gastrointestinal dysfunction (6 of 16, 37.5%), sensorineural hearing loss (4 of 16, 25%), and cardiac defect (3 of 16, 18.7%) also appeared in some patients (Table 3).
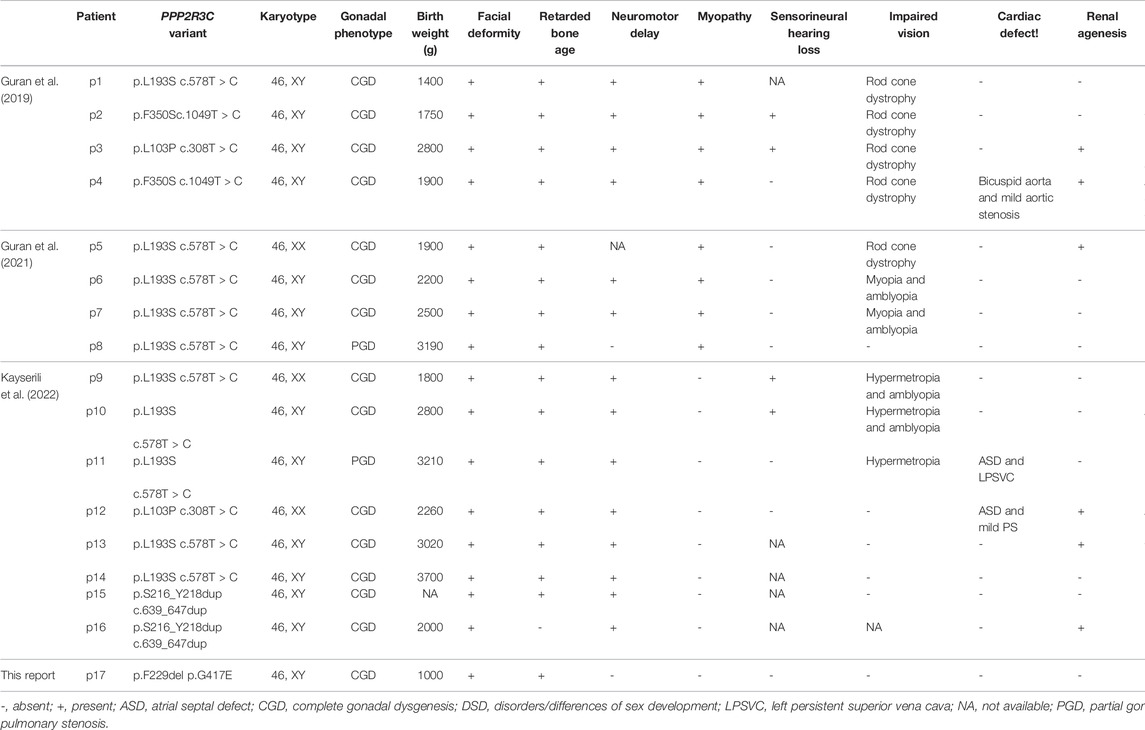
TABLE 3. Clinical and molecular findings of 17 DSD patients with PPP2R3C mutations.
A total of four PPP2R3C variants had been identified in DSD patients. Specifically, 10 patients had homozygous p.Leu193Ser variant, and 2 of the other 6 patients had homozygous p.Leu103Pro, p.Phe350Ser, or p.Ser216_Tyr218dup variants, respectively. The association between the genotype and phenotype cannot be found based on the current data (Table 3).
Discussion
In the present study, we reported a syndromic 46, XY DSD due to the compound heterozygous variants in the PPP2R3C gene. The variant p.Gly417Glu was not found in gnomAD, ExAC, or 1000 Genomes databases. p.Phe229del was not found in the 1000 Genomes database but showed low frequency in ExAC (0.0000753) and gnomAD (0.000194452) databases. These two variants were predicted to be harmful to protein function by REVEL, an ensemble method for predicting the pathogenicity of variants. So far, four other variants (p.Leu103Pro, p.Leu193Ser, p.Phe350Ser, and p.Ser216_Tyr218dup) in PPP2R3C have been reported in cases with similar clinical characteristics. Three missense mutations (p.Leu103Pro, p.Leu193Ser, and p.Phe350Ser) were predicted to be pathogenic according to in silico tools, conserved among species, and not found in gnomAD, ExAC, or 1000 Genomes databases. Therefore, the pathogenic variants of PPP2R3C cause a syndromic 46, XY complete gonadal dysgenesis in an autosomal recessive inheritance pattern.
46, XY DSD patients with PPP2R3C variants manifested gonadal dysgenesis and complete female external genitalia along with the hypoplastic uterus seen in ultrasound or laparoscopy. Extragonadal multisystem abnormalities were also shared in these patients. They were presented with flat faces, poorly developed nasal alars, thin lips, and low ears. The facial abnormalities might be related to dysplasia of cartilage in ears and nose in the early chondrogenesis. The SOX9 protein played a significant role in early chondrogenesis, and it was suggested that pathogenic variants in PPP2R3C can impair SOX9 signaling by upregulating the catalytic function of PP2A (Guran et al., 2019). In addition to facial deformities, low birth weight, retardation of bone age, and limited elbow extension also are common characteristics in patients with PPP2R3C variants. Previously reported patients did not show any clinical immunodeficiency symptoms, and serum immunoglobulin concentrations and lymphocyte counts were normal. Our patient’s CD19+ B lymphocyte counts showed an obvious decrease, which may be due to the decreased viability and increased apoptosis. Gene knockout (PPP2R3C-/-) mice by conditional targeting in CD19+ B cells showed a deficit in B-cell survival and low level of mature B cells (Xing et al., 2005). It is suggested that PPP2R3C is essential for the maintenance of B cells through the regulation status of the JNK-mediated apoptosis signal (Xing et al., 2008). Significantly, our patient also presented the decreased CD4+ T lymphocytes and increased NK cells, and this condition suggested that PPP2R3C plays a role in the survival of multiple lymphocytes and identified PPP2R3C variants as a potential cause of the disorder of immune cells.
Conclusion
In conclusion, novel compound heterozygous variants in PPP2R3C cause specific syndromic 46, XY gonadal dysgenesis, which broadened the pathogenic variant spectrum of PPP2R3C. The typical phenotype of PPP2R3C mutation is complete gonadal dysgenesis with multiple extragonadal anomalies, including facial deformities, skeletal system abnormalities, muscle abnormalities, impaired nervous system, impaired hearing and vision, heart and kidney anomalies, and gastrointestinal dysfunction. In addition, we found a decreased number of CD19+ B cells and CD4+ T cells in our patient, which is a new phenotype in syndromic 46, XY gonadal dysgenesis.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Human Research of Peking Union Medical College Hospital (No. JS-2111). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the relevant individual for the publication of any potentially identifiable images or data included in this article.
Author Contributions
XW and MN designed the study. JM and XW collected blood samples and clinical data. WZ, BS, ZZ, and XZ performed the experiments. WZ and MN analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81971375), the CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1-003), the Natural Science Foundation of Beijing (Grant Nos. 7212080 and 7202151), and the Nonprofit Central Research Institute Fund of the Chinese Academy of Medical Sciences (Nos. 2018PT32001 and 2019PT320007).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the patient for participating in the study.
References
Altunoglu, U., Börklü, E., Shukla, A., Escande‐Beillard, N., Ledig, S., Azaklı, H., et al. (2022). Expanding the Spectrum of Syndromic PPP2R3C ‐Related XY Gonadal Dysgenesis to XX Gonadal Dysgenesis. Clin. Genet. 101 (2), 221–232. doi:10.1111/cge.14086
Bagheri-Fam, S., Ono, M., Li, L., Zhao, L., Ryan, J., Lai, R., et al. (2015). FGFR2mutation in 46,XY Sex Reversal with Craniosynostosis. Hum. Mol. Genet. 24 (23), 6699–6710. doi:10.1093/hmg/ddv374
Bashamboo, A., and McElreavey, K. (2015). Human Sex-Determination and Disorders of Sex-Development (DSD). Seminars Cell Dev. Biol. 45, 77–83. doi:10.1016/j.semcdb.2015.10.030
Baxter, R. M., Arboleda, V. A., Lee, H., Barseghyan, H., Adam, M. P., Fechner, P. Y., et al. (2015). Exome Sequencing for the Diagnosis of 46,XY Disorders of Sex Development. J. Clin. Endocrinol. Metab. 100 (2), E333–E344. doi:10.1210/jc.2014-2605
Cicek, D., Warr, N., Yesil, G., Kocak Eker, H., Bas, F., Poyrazoglu, S., et al. (2021). Broad-Spectrum XX and XY Gonadal Dysgenesis in Patients with a Homozygous L193S Variant in PPP2R3C. Eur. J. Endocrinol. 186 (1), 65–72. doi:10.1530/EJE-21-0910
Guran, T., Yesil, G., Turan, S., Atay, Z., Bozkurtlar, E., Aghayev, A., et al. (2019). PPP2R3C Gene Variants Cause Syndromic 46,XY Gonadal Dysgenesis and Impaired Spermatogenesis in Humans. Eur. J. Endocrinol. 180 (5), 291–309. doi:10.1530/eje-19-0067
Hiorta, O., and Gillessen-Kaesbachb, G. (2009). Disorders of Sex Development in Developmental Syndromes. Endocr. Dev. 14, 174–180. doi:10.1159/000207486
Malki, S., Nef, S., Notarnicola, C., Thevenet, L., Gasca, S., Méjean, C., et al. (2005). Prostaglandin D2 Induces Nuclear Import of the Sex-Determining Factor SOX9 via its cAMP-PKA Phosphorylation. EMBO J. 24 (10), 1798–1809. doi:10.1038/sj.emboj.7600660
Tamura, M., Isojima, T., Kasama, T., Mafune, R., Shimoda, K., Yasudo, H., et al. (2017). Novel DHCR7 Mutation in a Case of Smith-Lemli-Opitz Syndrome Showing 46,XY Disorder of Sex Development. Hum. Genome Var. 4, 17015. doi:10.1038/hgv.2017.15
Xing, Y., Igarashi, H., Wang, X., and Sakaguchi, N. (2005). Protein Phosphatase Subunit G5PR Is Needed for Inhibition of B Cell Receptor-Induced Apoptosis. J. Exp. Med. 202 (5), 707–719. doi:10.1084/jem.20050637
Xing, Y., Wang, X., Igarashi, H., Kawamoto, H., and Sakaguchi, N. (2008). Protein Phosphatase Subunit G5PR that Regulates the JNK-Mediated Apoptosis Signal Is Essential for the Survival of CD4 and CD8 Double-Positive Thymocytes. Mol. Immunol. 45 (7), 2028–2037. doi:10.1016/j.molimm.2007.10.028
Keywords: disorders/differences of sex development(DSD), syndromic 46 XY DSD, gonadal dysgenesis, facial deformity, PPP2R3C mutations, compound heterozygous variants
Citation: Zhang W, Mao J, Wang X, Sun B, Zhao Z, Zhang X, Nie M and Wu X (2022) Case Report: Novel Compound Heterozygotic Variants in PPP2R3C Gene Causing Syndromic 46, XY Gonadal Dysgenesis and Literature Review. Front. Genet. 13:871328. doi: 10.3389/fgene.2022.871328
Received: 19 March 2022; Accepted: 11 May 2022;
Published: 23 June 2022.
Edited by:
Tulay Guran, Marmara University, TurkeyReviewed by:
Laura Audí, Vall d’Hebron University Hospital, SpainSorahia Domenice, University of São Paulo, Brazil
Copyright © 2022 Zhang, Mao, Wang, Sun, Zhao, Zhang, Nie and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Nie, bm1fcHVtY2hAYWxpeXVuLmNvbQ==; Xueyan Wu, d3NoZXlhbkB2aXAuc2luYS5jb20=