 Yuan Jin1†
Yuan Jin1† Xiao-Zhou Liu
Xiao-Zhou Liu Le Xie
Le Xie Sen Chen
Sen Chen Yu Sun
Yu Sun- 1Department of Otorhinolaryngology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Tongji Medical College, Institute of Otorhinolaryngology, Huazhong University of Science and Technology, Wuhan, China
Hearing loss is among the most common congenital sensory impairments. Genetic causes account for more than 50% of the cases of congenital hearing loss. The PTPRQ gene, encoding protein tyrosine phosphatase receptor Q, plays an important role in maintaining the stereocilia structure and function of hair cells. Mutations in the PTPRQ gene have been reported to cause hereditary sensorineural hearing loss. By using next-generation sequencing and Sanger sequencing, we identified a novel compound heterozygous mutation (c.997 G > A and c.6603-3 T > G) of the PTPRQ gene in a Chinese consanguineous family. This is the first report linking these two mutations to recessive hereditary sensorineural hearing loss. These findings contribute to the understanding of the relationship between genotype and hearing phenotype of PTPRQ-related hearing loss, which may be helpful to clinical management and genetic counseling.
Introduction
Hearing loss is the most common sensorineural disorder affecting approximately 6.1% of the world population (Qian et al., 2020; Zhang et al., 2021a; Fu et al., 2021; Lv et al., 2021). It is estimated that more than half of the hearing loss cases are attributable to genetic factors (He et al., 2017; Wang et al., 2022), while the other half of hearing loss cases could be caused by ototoxic drugs, such as aminoglycosides and anti-tumor drugs, aging, excessive noise exposure, and infections (Li et al., 2018a; Li et al., 2018b; Liu et al., 2019a; Cheng et al., 2019; Han et al., 2020; He et al., 2020; Zhong et al., 2020; Zhou et al., 2020; Liu et al., 2021a; Zhang et al., 2021b; Guo et al., 2021; He et al., 2021; Bu et al., 2022; Fu et al., 2022; Jiang et al., 2022). The functions of these hearing loss genes play an essential role in the development and function of hair cells and synaptic transmission of spiral ganglion neurons (Wang et al., 2017; Zhu et al., 2018; Cheng et al., 2021). Thus, hearing loss is often induced by the loss of sensory hair cells and spiral ganglion neurons (Liu et al., 2019b; Guo et al., 2019; He et al., 2019; Qi et al., 2019; Chen et al., 2021; Hu et al., 2021; Wei et al., 2021; Guo et al., 2022; Hu et al., 2022; Jiang et al., 2022) in the inner ear cochlea. Up to 30/8/2021, at least 124 genes have been identified associated with non-syndromic hearing loss genes (https://hereditaryhearingloss.org/).
The PTPRQ gene, located in the DFNB84 region of chromosome 12q21.31, is comprised of 58 exons (Schraders et al., 2010). The transcript levels of PTPRQ are the highest in fetal kidneys, followed by fetal lungs and fetal cochlea (Schraders et al., 2010). In the cochlea, the PTPRQ protein (Protein Tyrosine Phosphatase Receptor Type Q, which encodes 2,299 amino acids) expresses in the basal region of the stereocilia of hair cells (Ozieblo et al., 2019). Particularly, the PTPRQ protein has a higher expression level in the basal turn of the cochlea corresponding to high-frequency hearing (Goodyear et al., 2003). Studies have shown that PTPRQ is indispensable for the formation of hair bundles. In the early postnatal Ptprq −/− mouse model, elongated and fused stereocilia in inner hair cells (IHCs), shortened stereocilia in outer hair cells (OHCs), and loss of hair bundles in both OHCs and IHCs were observed (Goodyear et al., 2003). In the adult Ptprq −/− mice, almost all hair cells had degenerated and even the organ of Corti was missing. In addition, PTPRQ protein forms a complex with myosin VI to tether the membrane of the stereocilia to stereocilia, causing reorganization of the actin cytoskeleton, and plays an important role in the mechanical transduction and adaptation of hair cells (Takenawa and Itoh, 2001; Hirono et al., 2004; Sakaguchi et al., 2008). Studies in families with PTPRQ mutations show that mutants of the PTPRQ gene could cause autosomal recessive or autosomal dominant congenital sensorineural hearing loss, damage all frequency or high frequency, with or without vestibular dysfunction in infancy or early childhood (Li et al., 2018a; Li et al., 2018b; Liu et al., 2019a; Cheng et al., 2019; Zhou et al., 2020; Liu et al., 2021a; Guo et al., 2021; He et al., 2021; Bu et al., 2022; Fu et al., 2022; Jiang et al., 2022). The hearing loss was progressive in some cases. In addition, transcription of PTPRQ was highly expressed in adult lung and heart tissues, and there has been no significant evidence showing dysfunction of organs except that of the cochlea (Schraders et al., 2010).
Until now, cases of PTPRQ-related hearing loss rarely have been reported (summarized in Tables 1, 2). More cases of gene mutation need to be collected to understand the molecular mechanism. Here, we report a novel heterozygous PTPRQ mutation in a Chinese family, which might be helpful to establish a better understanding of the relationship between PTPRQ and the phenotype.
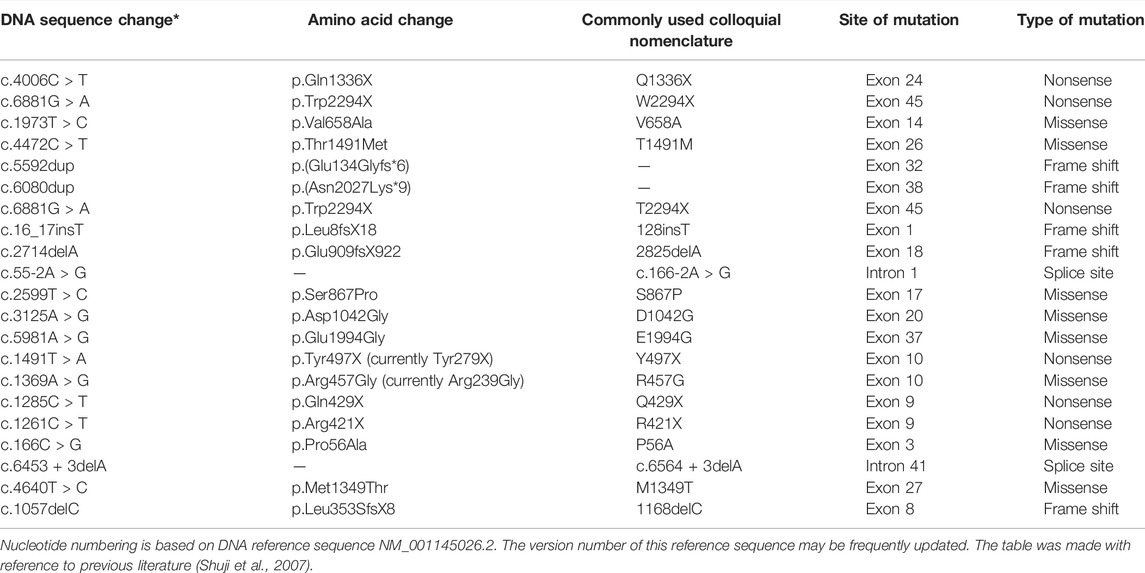
TABLE 1. Standard and Colloquial nomenclature for PTPRQ mutations and variants.
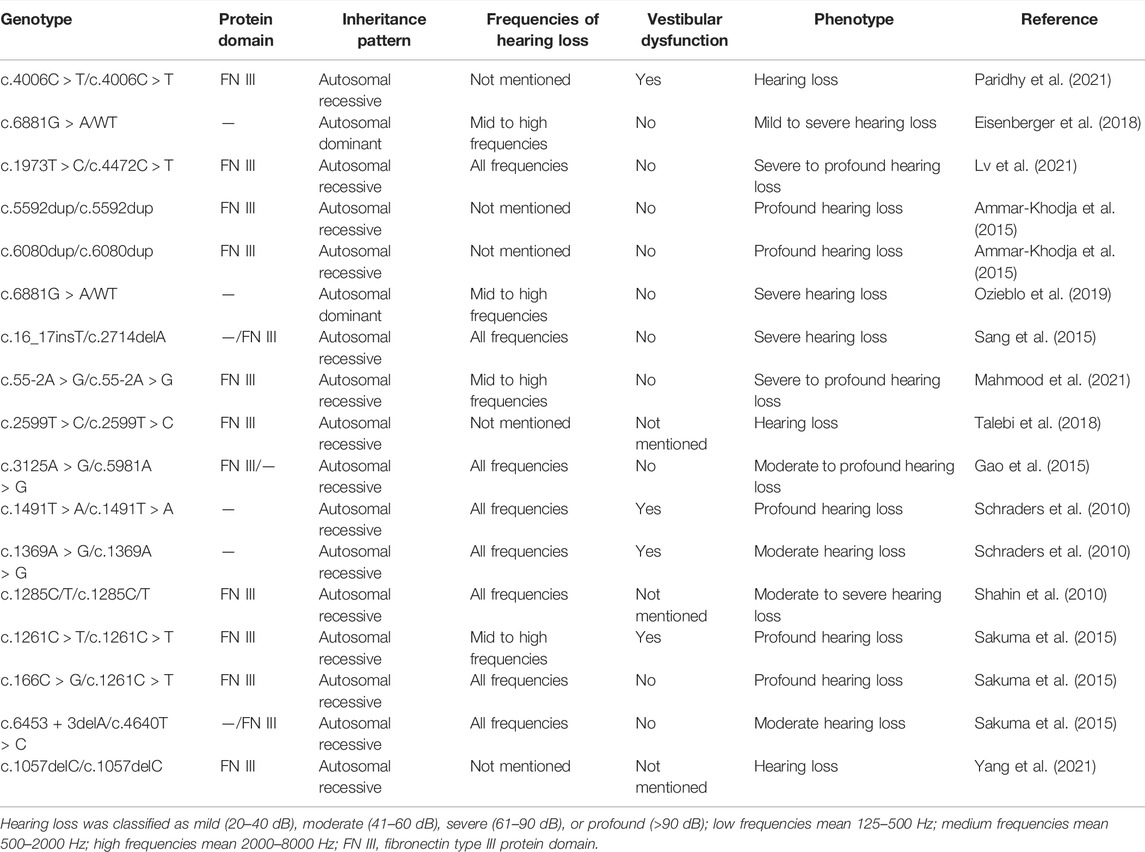
TABLE 2. PTPRQ mutations with hearing phenotypes in families.
Materials and Methods
Family Description
The family members are Han Chinese. Proband II-1 is a 29-year-old female. Proband II-2 is a 23-year-old male. Both probands had failed to pass the hearing screening and were diagnosed with sensorineural hearing loss. Neither parent of two probands exhibited similar hearing loss or vestibular dysfunction (Figure 1).
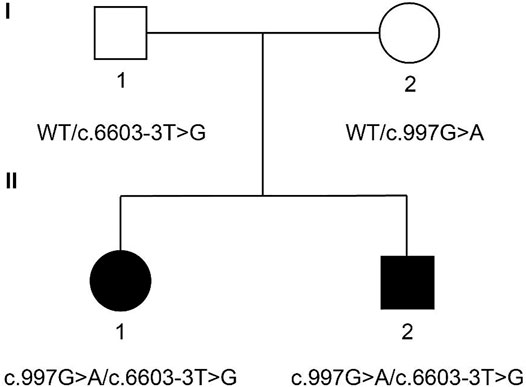
FIGURE 1. Family pedigree of two probands. Probands II-1 and II-2 carry compound heterozygous mutation c.997 G > A (chr12:80862555) and c.6603-3 T > G (chr12:81066945) of PTPRQ. The mother of the probands carries heterozygous mutation c.997 G > A. The father of the probands carries heterozygous mutation c.6603-3 T > G. Probands are marked in black. WT, wild type.
Clinical Examination
Both probands underwent audiological examination such as the otoscopic examination, auditory immittance, and auditory steady-state–evoked responses (ASSR). Computed tomography (CT) and magnetic resonance imaging (MRI) of the temporal bone showed no abnormal malformations. Parents reported no history of miscarriage or stillbirth. The physical examination, otoscopy, and medical history were performed at the outpatient clinic of Wuhan Union Medical College Hospital.
Mutation Detection and Analysis
The method has been described in detail in our previous articles (Chen et al., 2020; Liu et al., 2021b). Briefly, the two probands and their parents each contributed 3–5 ml of venous peripheral blood after the participants had given their informed consent. Genomic DNA was isolated from the blood samples using the QIAamp DNA Blood Midi Kit (Qiagen Inc., Hilden, Germany). Fragmentation of the genomic DNA was performed by using a Covaris LE220 ultrasonicator (Covaris Inc., Woburn, Massachusetts, United States) to generate a paired-end library. The library was enriched after hybridization, elution, and post-capture amplification. The amplified DNA library was sequenced on the BGISEQ-500 platform. Sequencing data were compared with the human genome reference (GRCh37/hg19) to detect target regions, single-nucleotide variants (SNVs), and INDEL calling. Identified SNVs and indels were compared with the information available in multiple databases, such as the National Center for Biotechnology Information GenBank database (https://www.ncbi.nlm.nih.gov/nuccore/), the Database of Single Nucleotide Polymorphisms (dbSNP) (http://www.ncbi.nlm.nih.gov/projects/SNP/), and the 1000 Genomes Database (https://www.internationalgenome.org). According to the sequencing results of two probands, Sanger sequencing was performed to confirm whether their parents had the same mutations. Using online tools such as HSF (http://www.umd.be/HSF3/HSF.shtml), FF (http://www.fruitfly.org/seq_tools/splice.html), SpliceAI (https://spliceailookup.broadinstitute.org/), an assessment was made to determine whether mutations occurring in the introns affected hnRNA splicing.
Results
Clinical Data
Both patients failed the newborn hearing screening and were diagnosed with congenital sensorineural hearing loss. The ASSR of Proband II-1 showed the thresholds of the left ear were 30, 35, 55, 65, 65, and 70 dBnHL at 0.25, 0.5, 1, 2, 4, and 8 kHz, while the thresholds of the right ear were 30, 45, 70, 65, 70, and 70 dBnHL at 0.25, 0.5, 1, 2, 4, and 8 kHz (Figures 2A,B). The ASSR of Proband II-2 showed that the thresholds of the left ear were 55, 70, 65, 70, and 60 dBnHL at 0.25, 0.5, 1, 2, and 4 kHz, while the thresholds of the right ear were 45, 60, 60, 65, and 60 dBnHL at 0.25, 0.5, 1, 2, and 4 kHz (Figures 2C,D). The temporal bone CT scan suggested that the shape and size of the bilateral cochleae were not obviously abnormal. Neither proband reported ever suffering from balance manifestation, tinnitus, or vertigo. In addition, the parents of the probands stated that both probands had no symptoms of falling down or frequent standing instability during their childhood. Physical examinations of the two probands revealed no signs of systemic illness. After wearing a hearing aid, the ASSR of Proband II-1 showed the thresholds of the left ear were 30, 35, 40, 40, and 40 dBnHL at 0.25, 0.5, 1, 2, and 4 kHz, while the thresholds of the right ear were 30, 45, 40, 45, and 40 dBnHL at 0.25, 0.5, 1, 2, and 4 kHz (Figures 2A,B); the ASSR of Proband II-2 showed the thresholds of the left ear were 40, 35, 35, 30, and 30 dBnHL at 0.25, 0.5, 1, 2, and 4 kHz, while the thresholds of the right ear were 35, 30, 35, 30, and 40 dBnHL at 0.25, 0.5, 1, 2, and 4 kHz (Figures 2C,D). Their parents had no history of hearing impairment, nor did their medical history include other organ disorders.
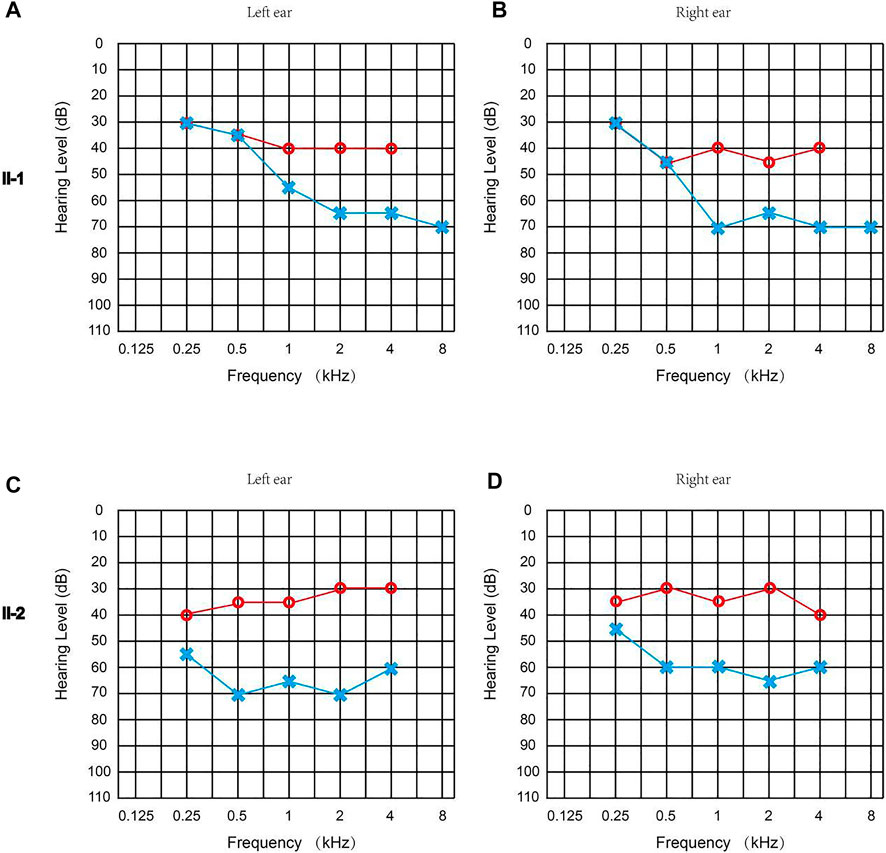
FIGURE 2. Clinical audiology examination of the probands. (A) ASSR of Proband II-1 (left ear): 30, 35, 55, 65, 65, and 70 dBnHL at 0.25, 0.5, 1, 2, 4, and 8 kHz. (B) ASSR of Proband II-1 (right ear): 30, 45, 70, 65, 70, and 70 dBnHL at 0.25, 0.5, 1, 2, 4, and 8 kHz. (C) ASSR of Proband II-2 (left ear): 55, 70, 65, 70, and 60 dBnHL at 0.25, 0.5, 1, 2, and 4 kHz. (D) ASSR of the Proband II-2 (right ear): 45, 60, 60, 65, and 60 dBnHL at 0.25, 0.5, 1, 2, and 4 kHz. The hearing threshold of two probands with a hearing aid is marked in red, and the hearing threshold of two probands without a hearing aid is marked in blue.
Mutation Identification Data
The genomic DNA sequences of the probands were compared with the human genome reference sequence (GRCh37/hg19). Both probands carried compound heterozygous mutations of PTPRQ: c.997 G > A and c.6603-3 T > G. The mutation c.997 G > A occurred in EX7/CDS7 in the PTPRQ gene, causing the substitution of no. 997 nucleotide from guanine to adenine (Figure 3A). The mutation c.6603-3 T > G is a splice mutation in Intron 42, causing the substitution of no. 6603-3 nucleotide from thymine to guanine (Figure 3B). The proband’s father and mother were heterozygous carriers of the c.997 G > A and c.6603-3 T > G mutations, respectively (Figure 1). The mutation c.6603-3 T > G of PTPRQ was inherited from the father and reported a minor allele frequency of 0.000078 in the gnomAD database. The allele frequency of this mutation in the East Asian population is 0.002118. The mutation c.997 G > A of PTPRQ was inherited from the mother, and no information on this mutation was found in the gnomAD database.
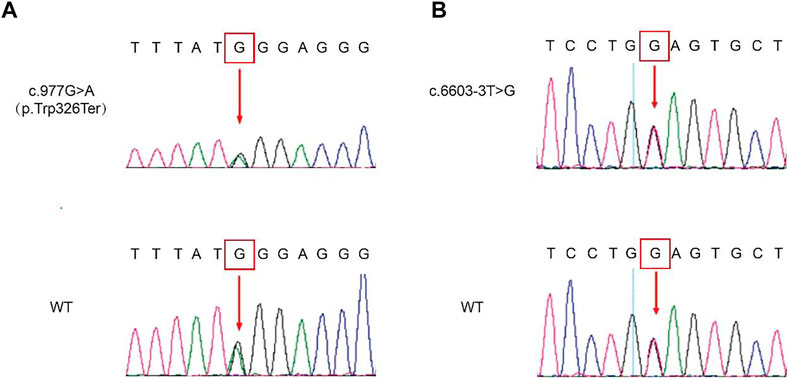
FIGURE 3. Genetic sequencing results of the probands and their parents. Mutated sequences of the identified c.997 G > A (A) and c.6603-3 T > G (B) variant. The red arrow indicates the site of the base deletion or substitution.
Functional Analysis of the Mutant Protein
The PTPRQ protein is composed of three types of domains, namely, 18 fibronectin III repeats domain (FN III domain), transmembrane domain, and tyrosine–protein phosphatase domain (PTPase domain). The mutation c.997 G > A causes the original TGG at nucleotide nos. 976–978 to become TAG, which corresponds to the termination codon (Figure 4A). The mutation c.997 G > A occurs in the second fibronectin III domain. The mutation c.997 G > A resulted in a truncated protein of only 325 proteins. Thus, the following 1,974 amino acids after this site cannot be synthesized (Figures 4A,B). The mutation c.6603-3 T > G causes the original thymine at nucleotide no. 6603-3 in Intron 42 to become guanine, which is near the splice site between Intron 42 and Exon 43 (Figure 4C). This mutation type often results in abnormal splicing of hnRNA. The amino acid encoded by Exon 43 is involved in the formation of the PTPase domain. We assessed whether mutation c.6603-3 T > G affects PTPRQ hnRNA splicing using the online bioinformatics database. All three database tools, HSF(http://www.umd.be/HSF3/HSF.shtml), FF(http://www.fruitfly.org/seq_tools/splice.html), and SpliceAI (https://spliceailookup.broadinstitute.org/), suggest that mutations c.6603-3 T > G will change the original acceptor site and hnRNA splicing. However, patients did not intend to participate in the minigene splicing assay. Therefore, we did not verify the effect of mutation c.6603-3 T > G in in vitro experiments.
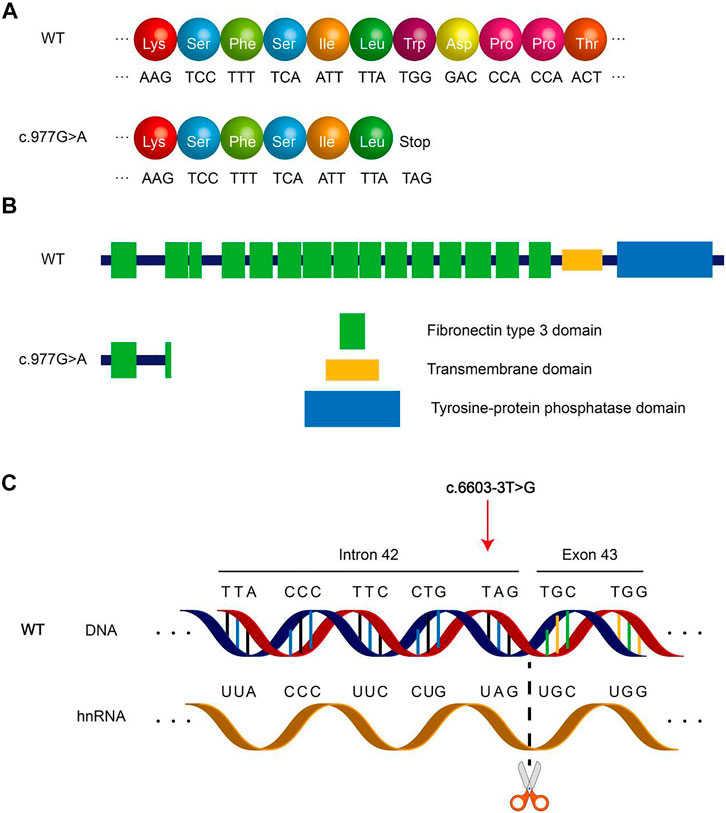
FIGURE 4. Schematic diagram of PTPRQ mutation. (A) Amino acid coding diagram of mutation c.997 G > A. (B) Schematic diagram of PTPRQ peptide chain truncation caused by mutation c.997 G > A. (C) Schematic diagram of mutation c.6603-3 T > G altering hnRNA splicing.
Discussion
Both probands failed the newborn hearing screening and were diagnosed with congenital sensorineural hearing loss. The proband’s parents were consanguineous. However, they did not provide more detailed information on family members. By using an approach of next-generation sequencing and the Sanger sequencing method, we identified c.997 G > A and c.6603-3 T > G of PTPRQ in the family, as the probable cause of sensorineural hearing loss.
Mutation c.997 G > A of PTPRQ causes termination of protein synthesis. The mutant protein contains only one complete fibronectin III domain, the other domains are completely deleted (Figure 4B). Such proteins basically lose their function. Usually, the 3′ end splicing site of an intron ends with an AG. Mutations occurring in introns and close to this region may result in changes in the transcription sequence, affecting the nucleotide sequence of the final transcript. The mutation c.6603-3 T > G of PTPRQ occurs in Intron 42, a region near the boundary of Intron 42 and Exon 43, which may lead to changes in hnRNA splicing. Our assessment of this mutation using database tools revealed that the mutation resulted in the disappearance of the original splice site between Intron 42 and Exon 43, possibly resulting in the skipping of Exon 43 during transcription. More seriously, the mutation c.6603-3 T > G of PTPRQ may also lead to all subsequent changes in the amino acid sequence and protein domains. Exon 43 correlates with the synthesis of PTPase domains of PTPRQ proteins. According to the assessment (by HSF, FF, and SpliceAI), mutation c.6603-3 T > G of PTPRQ may cause altered hnRNA splicing, which can lead to changes in the composition and function of the PTPase domains. Neither mutant protein in two probands lacks the PTPase domains that are critical for the normal functioning of the PTPRQ protein. The PTPase domains have phosphatidylinositol phosphatase activity, so the PTPRQ protein can regulate the local phosphoinositides' concentration in a specific area. Furthermore, phosphoinositides play a role in cell growth, polarity, and movement by regulating the reorganization of actin filaments (Sakaguchi et al., 2008). In the inner ear, PTPRQ maintains the stability of the stereocilia bundle on hair cells (Goodyear et al., 2012). Deletion of the PTPase domain may lead to the degradation of stereocilia bundles. The final mutated protein results in impaired mechanotransduction function of hair cells, resulting in hearing loss.
A total of 21 PTPRQ mutations have been reported before this article (Table 1). The nucleotide and amino acid changes for these mutations are summarized in Table 1. Two mutations mentioned in this article, namely, mutations c.997 G > A of PTPRQ and c.6603-3 T > G of PTPRQ, have not been reported before. According to reports, some patients with PTPRQ mutations suffer from mild- to high-frequency hearing loss, while others suffer hearing loss in all frequencies (Han et al., 2020; He et al., 2020; Zhang et al., 2021a; Zhang et al., 2021b; Lv et al., 2021). The relationship between the genotype and hearing phenotype in deaf patients carrying PTPRQ mutations is summarized in Table 2. In the present study, Proband II-1 suffered from mild- to high-frequency hearing loss, while Proband II-2 with the same mutations suffered hearing loss in all frequencies. Although the hearing loss may progress steadily, the low-frequency hearing threshold of Proband II-2 is much higher than Proband II-1. In addition, some patients with PTPRQ mutations have been reported to sustain vestibular dysfunction such as tinnitus or vertigo (Cheng et al., 2019; Zhang et al., 2021b; He et al., 2021; Fu et al., 2022), while others and the probands in this study did not show the same symptoms. We emphasize again that there may be no obvious correlation between genotype and phenotype of PTPRQ.
No obvious disease of other organs was found in participants until now. However, this risk remains due to the limited number of reported cases of PTPRQ mutations. We recommend cochlear, kidney, lung, and thyroid function tests for patients with PTPRQ mutations. In addition, we noticed that the c.6603-3 T > G mutation of PTPRQ was only found in the East Asian population in the GnomAD database. In the future, we will continue to pay attention to PTPRQ-related reports carried out in the East Asian population.
Conclusion
We identified a novel compound heterozygous mutation of PTPRQ (c.997 G > A and c.6603-3 T > G) in a Chinese family with non-syndromic sensorineural hearing loss. Our study expanded the spectrum of PTPRQ mutations. These findings contribute to the understanding of the relationship between genotype and hearing phenotype of PTPRQ-related hearing loss, which may be helpful to clinical management and genetic counseling.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the responsible committee on human experimentation (Tongji Medical College, Huazhong University of Science and Technology). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
YS designed the project and provided clinical examinations for the participants. JY, LX, WX, and SC collected and analyzed the data. JY and X-ZL wrote the article. All authors read and approved the final manuscript.
Funding
This work was supported by a grant from the National Nature Science Foundation of China (82071058).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RC declared a past co-authorship with the authors SC and YS to the handling editor.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the family for their participation in this study.
References
Ammar-Khodja, F., Bonnet, C., Dahmani, M., Ouhab, S., Lefèvre, G. M., Ibrahim, H., et al. (2015). Diversity of the Causal Genes in Hearing Impaired Algerian Individuals Identified by Whole Exome Sequencing. Mol. Genet. Genomic Med. 3 (3), 189–196. doi:10.1002/mgg3.131
Bu, C., Xu, L., Han, Y., Wang, M., Wang, X., Liu, W., et al. (2022). c-Myb Protects Cochlear Hair Cells from Cisplatin-Induced Damage via the PI3K/Akt Signaling Pathway. Cell Death Discov. 8 (1), 78. doi:10.1038/s41420-022-00879-9
Chen, S., Jin, Y., Xie, L., Xie, W., Xu, K., Qiu, Y., et al. (2020). A Novel Spontaneous Mutation of the SOX10 Gene Associated with Waardenburg Syndrome Type II. Neural Plast. 2020, 9260807. doi:10.1155/2020/9260807
Chen, Y., Gu, Y., Li, Y., Li, G.-L., Chai, R., Li, W., et al. (2021). Generation of Mature and Functional Hair Cells by Co-expression of Gfi1, Pou4f3, and Atoh1 in the Postnatal Mouse Cochlea. Cell Rep. 35 (3), 109016. doi:10.1016/j.celrep.2021.109016
Cheng, C., Wang, Y., Guo, L., Lu, X., Zhu, W., Muhammad, W., et al. (2019). Age-related Transcriptome Changes in Sox2+ Supporting Cells in the Mouse Cochlea. Stem Cell Res. Ther. 10 (1), 365. doi:10.1186/s13287-019-1437-0
Cheng, C., Hou, Y., Zhang, Z., Wang, Y., Lu, L., Zhang, L., et al. (2021). Disruption of the Autism-Related Gene Pak1 Causes Stereocilia Disorganization, Hair Cell Loss, and Deafness in Mice. J. Genet. Genomics 48 (4), 324–332. doi:10.1016/j.jgg.2021.03.010
Eisenberger, T., Di Donato, N., Decker, C., Delle Vedove, A., Neuhaus, C., Nürnberg, G., et al. (2018). A C-Terminal Nonsense Mutation Links PTPRQ with Autosomal-Dominant Hearing Loss, DFNA73. Genet. Med. 20 (6), 614–621. doi:10.1038/gim.2017.155
Fu, X., An, Y., Wang, H., Li, P., Lin, J., Yuan, J., et al. (2021). Deficiency of Klc2 Induces Low-Frequency Sensorineural Hearing Loss in C57BL/6 J Mice and Human. Mol. Neurobiol. 58 (9), 4376–4391. doi:10.1007/s12035-021-02422-w
Fu, X., Li, P., Zhang, L., Song, Y., An, Y., Zhang, A., et al. (2022). Activation of Rictor/mTORC2 Signaling Acts as a Pivotal Strategy to Protect against Sensorineural Hearing Loss. Proc. Natl. Acad. Sci. U. S. A. 119 (10), e2107357119. doi:10.1073/pnas.2107357119
Gao, X., Su, Y., Chen, Y.-L., Han, M.-Y., Yuan, Y.-Y., Xu, J.-C., et al. (2015). Identification of Two Novel Compound Heterozygous PTPRQ Mutations Associated with Autosomal Recessive Hearing Loss in a Chinese Family. PLoS One 10 (4), e0124757. doi:10.1371/journal.pone.0124757
Goodyear, R. J., Legan, P. K., Wright, M. B., Marcotti, W., Oganesian, A., Coats, S. A., et al. (2003). A Receptor-like Inositol Lipid Phosphatase Is Required for the Maturation of Developing Cochlear Hair Bundles. J. Neurosci. 23 (27), 9208–9219. doi:10.1523/jneurosci.23-27-09208.2003
Goodyear, R. J., Jones, S. M., Sharifi, L., Forge, A., and Richardson, G. P. (2012). Hair Bundle Defects and Loss of Function in the Vestibular End Organs of Mice Lacking the Receptor-like Inositol Lipid Phosphatase PTPRQ. J. Neurosci. 32 (8), 2762–2772. doi:10.1523/jneurosci.3635-11.2012
Guo, R., Ma, X., Liao, M., Liu, Y., Hu, Y., Qian, X., et al. (2019). Development and Application of Cochlear Implant-Based Electric-Acoustic Stimulation of Spiral Ganglion Neurons. ACS Biomater. Sci. Eng. 5 (12), 6735–6741. doi:10.1021/acsbiomaterials.9b01265
Guo, L., Cao, W., Niu, Y., He, S., Chai, R., and Yang, J. (2021). Autophagy Regulates the Survival of Hair Cells and Spiral Ganglion Neurons in Cases of Noise, Ototoxic Drug, and Age-Induced Sensorineural Hearing Loss. Front. Cell. Neurosci. 15, 760422. doi:10.3389/fncel.2021.760422
Guo, R., Xiao, M., Zhao, W., Zhou, S., Hu, Y., Liao, M., et al. (2022). 2D Ti3C2TxMXene Couples Electrical Stimulation to Promote Proliferation and Neural Differentiation of Neural Stem Cells. Acta Biomater. 139, 105–117. doi:10.1016/j.actbio.2020.12.035
Han, S., Xu, Y., Sun, J., Liu, Y., Zhao, Y., Tao, W., et al. (2020). Isolation and Analysis of Extracellular Vesicles in a Morpho Butterfly Wing-Integrated Microvortex Biochip. Biosens. Bioelectron. 154, 112073. doi:10.1016/j.bios.2020.112073
He, Z., Guo, L., Shu, Y., Fang, Q., Zhou, H., Liu, Y., et al. (2017). Autophagy Protects Auditory Hair Cells against Neomycin-Induced Damage. Autophagy 13 (11), 1884–1904. doi:10.1080/15548627.2017.1359449
He, Z., Fang, Q., Li, H., Shao, B., Zhang, Y., Zhang, Y., et al. (2019). The Role of FOXG1 in the Postnatal Development and Survival of Mouse Cochlear Hair Cells. Neuropharmacology 144, 43–57. doi:10.1016/j.neuropharm.2018.10.021
He, Z.-h., Zou, S.-y., Li, M., Liao, F.-l., Wu, X., Sun, H.-y., et al. (2020). The Nuclear Transcription Factor FoxG1 Affects the Sensitivity of Mimetic Aging Hair Cells to Inflammation by Regulating Autophagy Pathways. Redox Biol. 28, 101364. doi:10.1016/j.redox.2019.101364
He, Z.-H., Li, M., Fang, Q.-J., Liao, F.-L., Zou, S.-Y., Wu, X., et al. (2021). FOXG1 Promotes Aging Inner Ear Hair Cell Survival through Activation of the Autophagy Pathway. Autophagy 17 (12), 4341–4362. doi:10.1080/15548627.2021.1916194
Hirono, M., Denis, C. S., Richardson, G. P., and Gillespie, P. G. (2004). Hair Cells Require Phosphatidylinositol 4,5-bisphosphate for Mechanical Transduction and Adaptation. Neuron 44 (2), 309–320. doi:10.1016/j.neuron.2004.09.020
Hu, Y., Li, D., Wei, H., Zhou, S., Chen, W., Yan, X., et al. (2021). Neurite Extension and Orientation of Spiral Ganglion Neurons Can Be Directed by Superparamagnetic Iron Oxide Nanoparticles in a Magnetic Field. Ijn 16, 4515–4526. doi:10.2147/ijn.s313673
Hu, Y., Chen, W., Yin, H., Chen, X., Cai, J., Guo, J., et al. (2022). Super-aligned Carbon Nanotubes and GelMA Hydrogel Composite Scaffolds Promote Spiral Ganglion Neuron Growth and Orientation. Mater. Today Nano 18, 100181. doi:10.1016/j.mtnano.2022.100181
Jiang, P., Ma, X., Han, S., Ma, L., Ai, J., Wu, L., et al. (2022). Characterization of the microRNA Transcriptomes and Proteomics of Cochlear Tissue-Derived Small Extracellular Vesicles from Mice of Different Ages after Birth. Cell. Mol. Life Sci. 79 (3), 154. doi:10.1007/s00018-022-04164-x
Li, A., You, D., Li, W., Cui, Y., He, Y., Li, W., et al. (2018). Novel Compounds Protect Auditory Hair Cells against Gentamycin-Induced Apoptosis by Maintaining the Expression Level of H3K4me2. Drug Deliv. 25 (1), 1033–1043. doi:10.1080/10717544.2018.1461277
Li, H., Song, Y., He, Z., Chen, X., Wu, X., Li, X., et al. (2018). Meclofenamic Acid Reduces Reactive Oxygen Species Accumulation and Apoptosis, Inhibits Excessive Autophagy, and Protects Hair Cell-like HEI-OC1 Cells from Cisplatin-Induced Damage. Front. Cell. Neurosci. 12, 139. doi:10.3389/fncel.2018.00139
Liu, W., Xu, X., Fan, Z., Sun, G., Han, Y., Zhang, D., et al. (2019). Wnt Signaling Activates TP53-Induced Glycolysis and Apoptosis Regulator and Protects against Cisplatin-Induced Spiral Ganglion Neuron Damage in the Mouse Cochlea. Antioxidants Redox Signal. 30 (11), 1389–1410. doi:10.1089/ars.2017.7288
Liu, Y., Qi, J., Chen, X., Tang, M., Chu, C., Zhu, W., et al. (2019). Critical Role of Spectrin in Hearing Development and Deafness. Sci. Adv. 5 (4), eaav7803. doi:10.1126/sciadv.aav7803
Liu, W., Xu, L., Wang, X., Zhang, D., Sun, G., Wang, M., et al. (2021). PRDX1 Activates Autophagy via the PTEN-AKT Signaling Pathway to Protect against Cisplatin-Induced Spiral Ganglion Neuron Damage. Autophagy 17 (12), 4159–4181. doi:10.1080/15548627.2021.1905466
Liu, X., Chen, S., Sun, Y., and Kong, W. (2021). [Nonsyndromic Deafness Due to Compound Heterozygous Mutation of the CDH23 Gene]. Lin. Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 35 (3), 22933794607–22933795237. Chinese. doi:10.13201/j.issn.2096-7993.2021.03.008
Lv, J., Fu, X., Li, Y., Hong, G., Li, P., Lin, J., et al. (2021). Deletion of Kcnj16 in Mice Does Not Alter Auditory Function. Front. Cell Dev. Biol. 9, 630361. doi:10.3389/fcell.2021.630361
Mahmood, U., Shazia, A. B., Muhammad, A., Zubair, M. A., and Saima, R. (2021). Identification of Hearing Loss-Associated Variants of PTPRQ, MYO15A, and SERPINB6 in Pakistani Families. Biomed. Res. Int. 2021, 5584788. doi:10.1155/2021/5584788
Ozieblo, D., Anna, S., Marcin, L. L., Birgit, S. B., Grażyna, H., Nataliya, D. D., et al. (2019). First Confirmatory Study on PTPRQ as an Autosomal Dominant Non-syndromic Hearing Loss Gene. J. Transl. Med. 17 (1), 351. doi:10.1186/s12967-019-2099-5
Paridhy, V. S., Jayasankaran, C., Justin, M. J., Tom, R., Zippora, B., and Mathuravalli, K. (2021). PTPRQ, and SLC12A2-Associated Genetic and Phenotypic Heterogeneity Among Hearing Impaired Assortative Mating Families in Southern India. Ann. Hum. Genet. 86 (1), 1–13. doi:10.1111/ahg.12442
Qi, J., Liu, Y., Chu, C., Chen, X., Zhu, W., Shu, Y., et al. (2019). A Cytoskeleton Structure Revealed by Super-resolution Fluorescence Imaging in Inner Ear Hair Cells. Cell Discov. 5, 12. doi:10.1038/s41421-018-0076-4
Qian, F., Wang, X., Yin, Z., Xie, G., Yuan, H., Liu, D., et al. (2020). The Slc4a2b Gene Is Required for Hair Cell Development in Zebrafish. aging 12 (19), 18804–18821. doi:10.18632/aging.103840
Sakaguchi, H., Tokita, J., Naoz, M., Bowen-Pope, D., Gov, N. S., and Kachar, B. (2008). Dynamic Compartmentalization of Protein Tyrosine Phosphatase Receptor Q at the Proximal End of Stereocilia: Implication of Myosin VI-based Transport. Cell Motil. Cytoskelet. 65 (7), 528–538. doi:10.1002/cm.20275
Sakuma, N., Moteki, H., Azaiez, H., Booth, K. T., Takahashi, M., Arai, Y., et al. (2015). Novel PTPRQ Mutations Identified in Three Congenital Hearing Loss Patients with Various Types of Hearing Loss. Ann. Otol. Rhinol. Laryngol. 124 (Suppl. 1), 184S–92S. doi:10.1177/0003489415575041
Sang, Q., Mei, H., Kuermanhan, A., Feng, R., Guo, L., Qu, R., et al. (2015). Identification of a Novel Compound Heterozygous Mutation in PTPRQ in a DFNB84 Family with Prelingual Sensorineural Hearing Impairment. Mol. Genet. Genomics 290 (3), 1135–1139. doi:10.1007/s00438-014-0979-1
Schraders, M., Oostrik, J., Huygen, P. L. M., Strom, T. M., van Wijk, E., Kunst, H. P. M., et al. (2010). Mutations in PTPRQ Are a Cause of Autosomal-Recessive Nonsyndromic Hearing Impairment DFNB84 and Associated with Vestibular Dysfunction. Am. J. Hum. Genet. 86 (4), 604–610. doi:10.1016/j.ajhg.2010.02.015
Shahin, H., Rahil, M., Rayan, A. A., Avraham, K. B., King, M.-C., Kanaan, M., et al. (2010). Nonsense Mutation of the Stereociliar Membrane Protein Gene PTPRQ in Human Hearing Loss DFNB84. J. Med. Genet. 47 (9), 643–645. doi:10.1136/jmg.2009.075697
Shuji, O., Margaret, L. G., Johan, T. D., and Robert, B. W. (2007). Standard Mutation Nomenclature in Molecular Diagnostics: Practical and Educational Challenges. Association for Molecular Patholpogy Training and Education Committtee 11 (5), 494. doi:10.2353/jmoldx.2007.060081
Takenawa, T., and Itoh, T. (2001). Phosphoinositides, Key Molecules for Regulation of Actin Cytoskeletal Organization and Membrane Traffic from the Plasma Membrane. Biochimica Biophysica Acta (BBA) - Mol. Cell Biol. Lipids 1533 (3), 190–206. doi:10.1016/s1388-1981(01)00165-2
Talebi, F., Ghanbari Mardasi, F., Mohammadi Asl, J., Tizno, S., and Najafvand Zadeh, M. (2018). Identification of Novel PTPRQ and MYO1A Mutations in an Iranian Pedigree with Autosomal Recessive Hearing Loss. Cell J. 20 (1), 127–131. doi:10.22074/cellj.2018.4805
Wang, Y., Li, J., Yao, X., Li, W., Du, H., Tang, M., et al. (2017). Loss of CIB2 Causes Profound Hearing Loss and Abolishes Mechanoelectrical Transduction in Mice. Front. Mol. Neurosci. 10, 401. doi:10.3389/fnmol.2017.00401
Wang, M., Dong, Y., Gao, S., Zhong, Z., Cheng, C., Qiang, R., et al. (2022). Hippo/YAP Signaling Pathway Protects against Neomycin-Induced Hair Cell Damage in the Mouse Cochlea. Cell. Mol. Life Sci. 79 (2), 79. doi:10.1007/s00018-021-04029-9
Wei, H., Chen, Z., Hu, Y., Cao, W., Ma, X., Zhang, C., et al. (2021). Topographically Conductive Butterfly Wing Substrates for Directed Spiral Ganglion Neuron Growth. Small 17 (38), e2102062. doi:10.1002/smll.202102062
Yang, G., Yin, Y., Tan, Z., Liu, J., Deng, X., and Yang, Y. (2021). Whole-exome Sequencing Identified a Novel Heterozygous Mutation of SALL1 and a New Homozygous Mutation of PTPRQ in a Chinese Family with Townes-Brocks Syndrome and Hearing Loss. BMC Med. Genomics 14 (1), 24. doi:10.1186/s12920-021-00871-9
Zhang, S., Dong, Y., Qiang, R., Zhang, Y., Zhang, X., Chen, Y., et al. (2021). Characterization of Strip1 Expression in Mouse Cochlear Hair Cells. Front. Genet. 12, 625867. doi:10.3389/fgene.2021.625867
Zhang, Y., Li, Y., Fu, X., Wang, P., Wang, Q., Meng, W., et al. (2021). The Detrimental and Beneficial Functions of Macrophages after Cochlear Injury. Front. Cell Dev. Biol. 9, 631904. doi:10.3389/fcell.2021.631904
Zhong, Z., Fu, X., Li, H., Chen, J., Wang, M., Gao, S., et al. (2020). Citicoline Protects Auditory Hair Cells against Neomycin-Induced Damage. Front. Cell Dev. Biol. 8, 712. doi:10.3389/fcell.2020.00712
Zhou, H., Qian, X., Xu, N., Zhang, S., Zhu, G., Zhang, Y., et al. (2020). Disruption of Atg7-dependent Autophagy Causes Electromotility Disturbances, Outer Hair Cell Loss, and Deafness in Mice. Cell Death Dis. 11 (10), 913. doi:10.1038/s41419-020-03110-8
Keywords: PTPRQ gene, novel compound heterozygous mutation, targeted next-generation sequencing, hearing loss, autosomal recessive inheritance
Citation: Jin Y, Liu X-Z, Xie L, Xie W, Chen S and Sun Y (2022) Targeted Next-Generation Sequencing Identified Novel Compound Heterozygous Variants in the PTPRQ Gene Causing Autosomal Recessive Hearing Loss in a Chinese Family. Front. Genet. 13:884522. doi: 10.3389/fgene.2022.884522
Received: 26 February 2022; Accepted: 26 May 2022;
Published: 08 July 2022.
Edited by:
Naglaa M. Kamal, Cairo University, EgyptReviewed by:
Renjie Chai, Southeast University, ChinaMuhammad Jawad Hassan, National University of Medical Sciences (NUMS), Pakistan
Copyright © 2022 Jin, Liu, Xie, Xie, Chen and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Sun, c3VueXVAaHVzdC5lZHUuY24=
†These authors have contributed equally to this work