 Jixia Li1,2*†
Jixia Li1,2*† Maggie L. Kalev-Zylinska1,3*†
Maggie L. Kalev-Zylinska1,3*†- 1Blood and Cancer Biology Laboratory, Department of Molecular Medicine and Pathology, University of Auckland, Auckland, New Zealand
- 2Department of Laboratory Medicine, School of Medicine, Foshan University, Foshan, China
- 3Haematology Laboratory, Department of Pathology and Laboratory Medicine, Auckland City Hospital, Auckland, New Zealand
Myeloid leukemia associated with Down syndrome (ML-DS) has a unique molecular landscape that differs from other subtypes of acute myeloid leukemia. ML-DS is often preceded by a myeloproliferative neoplastic condition called transient abnormal myelopoiesis (TAM) that disrupts megakaryocytic and erythroid differentiation. Over the last two decades, many genetic and epigenetic changes in TAM and ML-DS have been elucidated. These include overexpression of molecules and micro-RNAs located on chromosome 21, GATA1 mutations, and a range of other somatic mutations and chromosomal alterations. In this review, we summarize molecular changes reported in TAM and ML-DS and provide a comprehensive discussion of these findings. Recent advances in the development of CRISPR/Cas9-modified induced pluripotent stem cell-based disease models are also highlighted. However, despite significant progress in this area, we still do not fully understand the pathogenesis of ML-DS, and there are no targeted therapies. Initial diagnosis of ML-DS has a favorable prognosis, but refractory and relapsed disease can be difficult to treat; therapeutic options are limited in Down syndrome children by their stronger sensitivity to the toxic effects of chemotherapy. Because of the rarity of TAM and ML-DS, large-scale multi-center studies would be helpful to advance molecular characterization of these diseases at different stages of development and progression.
Introduction
Myeloid leukemia associated with Down syndrome (ML-DS) is a unique category of acute myeloid leukemia (AML) most often of the megakaryoblastic subtype (i.e., acute megakaryoblastic leukemia (AMKL), formerly known as AML-M7 (Singh et al., 2017). The term ML-DS also includes an antecedent myelodysplastic syndrome (MDS)-like phase. There is no biologic or prognostic difference between MDS (blasts 5–19%) and AML (blasts ≥20%) in Down syndrome (DS) (Lange et al., 1998), therefore this distinction is not being made for ML-DS in the current pathologic classification (Arber et al., 2016).
ML-DS is frequently preceded by transient abnormal myelopoiesis (TAM), a unique myeloproliferative disorder affecting megakaryocytic and erythroid lineages. TAM is a pre-leukemic condition characterized by reduced platelet and increased leukocyte counts, and the presence of blasts in the peripheral blood. TAM diagnosis requires the presence of GATA1 mutations together with increased blasts and/or certain clinical features (in particular hepatosplenomegaly) in a neonate with constitutional trisomy 21, which can be mosaic (Arber et al., 2016). TAM may be indistinguishable from ML-DS but there is a wide spectrum of clinical presentation, ranging from asymptomatic to a stormy course and fatal outcome. Typically, TAM presents in neonates 3–7 days after birth but it may present within 2 months from birth (Singh et al., 2017). Overt TAM (blasts >10%) occurs in approximately 10–15% of DS neonates, but a further 10–15% may have GATA1 mutations detectable only by sensitive methods with no clinical or hematologic manifestations (i.e., silent TAM) (Roberts et al., 2013). Most patients with TAM recover spontaneously within 3 months, but some require cytotoxic therapy. Unfortunately, despite initial TAM resolution, 20–30% of children progress to ML-DS within 4 years (Bombery and Vergilio, 2014).
TAM is extremely rare in neonates without DS but such cases have been well documented (Apollonsky et al., 2008; Tsai et al., 2011; Ono et al., 2015; Yuzawa et al., 2020; Panferova et al., 2021). The molecular pathogenesis and clinical outcomes of TAM in neonates without DS are similar to those with DS (i.e., DS-like). These patients acquire trisomy 21 and GATA1 mutations in the TAM clone (Yuzawa et al., 2020; Panferova et al., 2021). In addition, GATA1 mutations may be germline, as recently reported in familial childhood cases of TAM/AMKL, highlighting a unique functional cooperation between these lesions that may be independent of the order of their acquisition (Hasle et al., 2022). The rates of early death and leukemic progression of TAM in non-DS and DS children are similar, emphasizing the importance of making the diagnosis of DS-like TAM to assist appropriate patient management (Yuzawa et al., 2020). Rare cases of TAM without GATA1 mutations feature in the literature. However, this may be due to technical limitations, in particular prior to the use of sensitive next-generation-sequencing methods (Panferova et al., 2021), small disease clones, or the lack of appropriate diagnostic samples if the condition is not suspected at presentation (Aksu et al., 2020). The expanding use of sensitive sequencing technologies will make the diagnosis of DS-like TAM easier in the future, which should advance our knowledge about this extremely rare condition.
ML-DS often presents with a period of thrombocytopenia reflecting a prodromal MDS-like phase (Lange et al., 1998). ML-DS is characterized by the expansion of megakaryoblasts, frequent bone marrow fibrosis, and the presence of GATA1 mutations in the blasts that drive expression of a truncated (short) GATA1 protein (GATA1s) (Hasle et al., 2008). The median age of patients with ML-DS is 1–1.8 years (Gamis et al., 2003; Bhatnagar et al., 2016). Majority of patients with ML-DS (72%) also carry other cytogenetic changes in addition to trisomy 21 (Forestier et al., 2008; De Souza et al., 2017). The contribution of these changes to disease development and progression is the subject of active research.
ML-DS pathogenesis is understood to follow a multistep clonal evolution process (Figure 1). Trisomy 21 represents a “primary hit”, which alters hematopoiesis during embryonic development; acquisition of somatic GATA1s mutations represents a “secondary hit”, which promotes hematopoietic deregulation and emergence of TAM in DS newborns; additional mutations predominantly affecting chromatin and epigenetic regulators (e.g., the cohesin complex) and signaling mediators (e.g., Janus kinase 2, JAK2) represent a “tertiary hit”, which leads to ML-DS (Labuhn et al., 2019; Garnett et al., 2020; De Castro et al., 2021) (Figure 1). The detailed mechanism of how these events contribute to different stages of disease is still unclear. One of the studies showed that GATA1s mutations lead to TAM when introduced into trisomy 21 long-term hematopoietic stem cells (LT-HSCs), where a subset of chromosome 21 microRNAs (miRNAs) influences predisposition toward pre-leukemia initiation (Wagenblast et al., 2021). However, progression to ML-DS was independent of trisomy 21 in this study, but required synergy between mutations in GATA1 and the cohesin genes, in particular cohesin subunit SA-2 (STAG2) knockout occurring in fetal or early postnatal but not adult HSCs (Wagenblast et al., 2021). Our review was motivated by these and other recent advances in the field that will likely open up new lines of research into ML-DS pathogenesis and targeted treatment development. We provide a comprehensive and up-to-date summary of molecular alterations in ML-DS, with an overriding aim to help guide future mechanistic studies into the pathogenesis of this disease. However, this is a rapidly advancing field, so despite our efforts this review may not be complete.
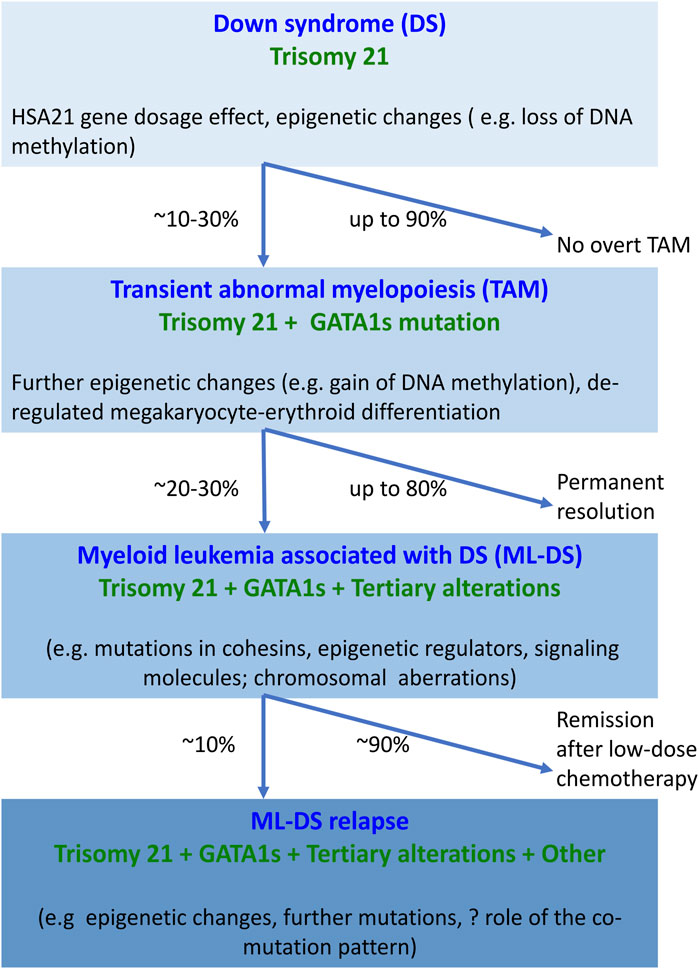
FIGURE 1. Overview of molecular changes reported at different stages of myeloid proliferation associated with Down syndrome. Trisomy 21 alone disturbs hematopoiesis through the increased dosage of HSA21-located genes and alterations in the epigenome, resulting in increased megakaryopoiesis. The combination of trisomy 21 and GATA1s causes expansion of megakaryocytic progenitors. Progression of TAM to ML-DS requires the interaction of GATA1s with additional somatic mutations and chromosomal structural abnormalities. Little is known about the molecular landscape of refractory or relapsed ML-DS. Abbreviations: DS, Down syndrome; GATA1s, GATA1 short; HSA21, human chromosome 21; ML-DS, myeloid leukemia associated with Down syndrome; TAM, transient abnormal myelopoiesis.
Trisomy 21
Trisomy 21 is associated with defects in hematopoiesis and the immune system. Trisomy 21 fetuses have dysregulated development of megakaryocytic, erythroid and B-cell lineages (Laurent et al., 2020; De Castro et al., 2021). The mechanism through which an extra copy of chromosome 21 perturbs hematopoiesis and then how it cooperates with subsequent mutations to lead to TAM and ML-DS are still uncertain. Findings from a humanized model of pre-malignant and malignant stages of ML-DS demonstrated that trisomy 21 was necessary for pre-leukemia initiation but dispensable for leukemia progression (Wagenblast et al., 2021). The predominant current view is that DS-associated myeloproliferations result from deregulation of genes on human chromosome 21 (HSA21) estimated to contain 234 protein-coding genes (Antonarakis, 2017). These include genes critical for myeloid differentiation, such as ETS-related gene (ERG), ETS proto-oncogene 2 (ETS2), runt-related transcription factor 1 (RUNX1), dual specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A), regulator of calcineurin 1 (RCAN1), chromatin assembly factor 1 subunit B (CHAF1B), high mobility group nucleosome binding domain 1 (HMGN1), SON DNA and RNA binding protein (SON), and a subset of miRNAs (Table 1) (Laurent et al., 2020; Vukadin et al., 2021; Wagenblast et al., 2021). The encoded molecules belong to several functional classes, such as transcription factors, signaling effectors, epigenetic regulators, and miRNAs.
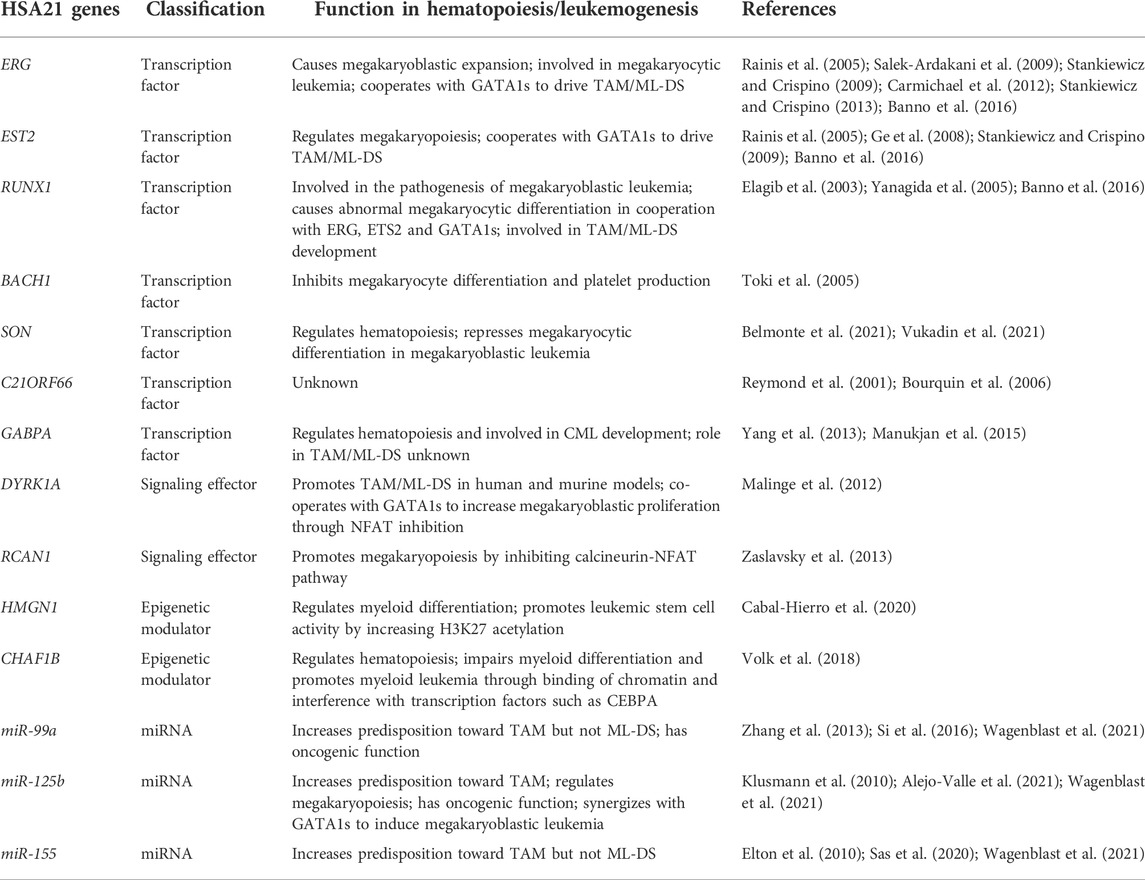
TABLE 1. List of HSA21 genes involved in myeloid proliferation associated with Down syndrome.
Transcription factors
The roles of ERG, ETS2 and RUNX1 in hematopoiesis and leukemogenesis have been thoroughly studied. ERG, an ETS transcription factor, is a megakaryocytic oncogene; its overexpression facilitates megakaryocytic expansion and promotes lymphoid and erythro-megakaryocytic leukemia in vitro and in vivo (Rainis et al., 2005; Salek-Ardakani et al., 2009; Stankiewicz and Crispino, 2009; Carmichael et al., 2012). Increased expression of ERG alone contributes to rapid onset of leukemia in mice (Salek-Ardakani et al., 2009). ERG strongly cooperates with the GATA1s mutated protein to immortalize hematopoietic and megakaryocytic progenitors ex vivo (Salek-Ardakani et al., 2009; Stankiewicz and Crispino, 2009). ERG and protein kinase B (PKB) also crosstalk, which alters GATA1 function (Stankiewicz and Crispino, 2013). Similar to ERG, ETS2 is an ETS transcription factor and a megakaryocytic oncogene (Ge et al., 2008). ETS2 promotes megakaryopoiesis and collaborates with GATA1s to immortalize hematopoietic progenitor cells (HPCs) (Rainis et al., 2005; Stankiewicz and Crispino, 2009). RUNX1 is a crucial transcription factor involved in the regulation of megakaryopoiesis, and its expression and cooperation with GATA1s facilitates megakaryocytic differentiation (Elagib et al., 2003). In 2005, overexpression of RUNX1 was reported in bone marrow of ML-DS children (Langebrake et al., 2006). A subsequent report from 2006 showed RUNX1 expression was lower than anticipated in ML-DS, while it was higher in megakaryoblasts from children with non-DS-AMKL (Bourquin et al., 2006). It appears that SON, another HSA21 gene, inhibits RUNX1 expression (Vukadin et al., 2021), which may neutralize trisomy 21-related overdosage of RUNX1 effects. Evidence from animal studies indicates that RUNX1 overexpression in mice shortens the latency of leukemia development displaying enhanced frequency of megakaryoblastic leukemia, which supports that RUNX1 overexpression is leukemogenic in ML-DS (Yanagida et al., 2005). Data from disease models using human induced pluripotent stem cells (iPSCs) and genome-editing technologies showed that an extra copy of RUNX1 is essential for accelerating early hematopoiesis in the context of trisomy 21, leading to HPC expansion and increased myeloid differentiation (Banno et al., 2016). RUNX1 expression level in trisomy 21 (GATA1 wild type) iPSCs is increased by ∼1.8-fold compared with that in disomy 21 (GATA1 wild type) iPSCs, which is slightly higher than the expected change in gene dosage (Banno et al., 2016). Abnormal megakaryocyte differentiation in TAM is accelerated by trisomy 21. Trisomy 21 up-regulates GATA1s expression leading to aberrant megakaryopoiesis, and the overdosage of RUNX1, ETS2, and ERG accelerates production of aberrantly differentiated cells (Banno et al., 2016). These observations highlight the importance of synergy between trisomy 21 and GATA1s in driving myeloid proliferation in DS children.
Other transcription factor encoding genes located on HSA21 are also highly expressed in ML-DS, including BTB domain and CNC homolog 1 (BACH1) (1.98-fold), SON (1.84-fold), chromosome 21 open reading frame 66 (C21ORF66) (1.64-fold) and GA-binding protein alpha chain (GABPA) (1.53-fold) (Bourquin et al., 2006). BACH1 acts as a transcriptional repressor of normal megakaryopoiesis and is likely a target of GATA1 and SON (Bourquin et al., 2006). Overexpression of BACH1 causes maturation arrest of megakaryocytes resulting in marked peripheral thrombocytopenia (Toki et al., 2005). SON is a gene with homology to the proto-oncogene MYC family, and an RNA splicing factor regulating transcription of leukemia-associated genes. SON is indispensable for proper blood cell formation, as SON knockdown results in lower amounts of all myeloid cells and T cells (Belmonte et al., 2021). Megakaryocytic differentiation in AMKL is impaired by SON inhibiting expression of RUNX1 and other megakaryocytic genes (Vukadin et al., 2021). SON also negatively regulates the expression of the AP-1 complex subunits JUN, JUNB and FOSB, which suggests that overexpression of SON could be pathogenic in ML-DS (Vukadin et al., 2021). C21ORF66 is known as the GC-rich sequence DNA-binding factor candidate (Reymond et al., 2001), but its function is unknown. Further work is needed to elucidate the role of C21ORF66 in hematopoiesis and leukemogenesis. GABPA has a known role in hematopoiesis (Yang et al., 2013). Deletion of GABPA leads to cell cycle arrest in hematopoietic stem cells (HSCs) and profound loss of HPCs (Yang et al., 2013). GABPA is necessary for chronic myeloid leukemia (CML) development through its regulation of protein kinase D2 (PRKD2) (Yang et al., 2013). GABPA expression positively correlates with the BCR::ABL1/ABL1 ratio in cells from patients with CML, and influences imatinib sensitivity in leukemic cell lines (TKI-sensitive K-562 and TKI-resistant NALM-1) (Manukjan et al., 2015). However, the function of GABPA in the setting of trisomy 21 and GATA1 mutations is not clear.
Signaling effectors
DYRK1A belongs to the CMGC kinase group named after the initials of its subgroup members, including cyclin-dependent kinases, mitogen-activated protein kinases (MAPK), glycogen synthase kinases and CDK-like kinases. DYRK1A participates in various cellular functions through the phosphorylation of several substrates such as nuclear factor of activated T cells (NFAT) (Lindberg and Meijer, 2021). DYRK1A is a potent megakaryoblastic tumor-promoting gene, contributing to leukemogenesis in a mouse model containing 33 gene orthologs of HSA21, a GATA1s mutation, and a MPL mutation (Malinge et al., 2012). DYRK1A overexpression induces a marked megakaryoblastic proliferation through the suppression of NFAT in this model (Malinge et al., 2012). RCAN1, also known as Down syndrome critical region gene 1 (DSCR1), is an endogenous calcineurin inhibitor. Overexpression of RCAN1 represses calcineurin-NFAT pathway, which leads to the expansion of megakaryocytes and their progenitors, and a high number of platelets (Zaslavsky et al., 2013). Both DYRK1A and RCAN1 can down-regulate calcineurin-NFAT pathway, but little is known about how these signaling molecules collaborate with other HSA21 genes and GATA1 mutations to initiate megakaryocytic neoplasia.
Epigenetic modulators
HMGN1 is the chromatin accessibility regulator and a target of recurrent DNA copy gains in leukemia (Cabal-Hierro et al., 2020). HMGN1 overexpression blocks myeloid differentiation, increases clonal progenitor expansion, enhances HSC activity and leukemic stem cell (LSC) activity in the presence of RUNX1::RUNX1T1 fusion oncoprotein (Cabal-Hierro et al., 2020). In addition, HMGN1 up-regulation elevates H3K27 acetylation, and in turn histone acetyltransferase CBP/p300 inhibition reverses the HMGN1-induced differentiation arrest. Another epigenetic modulator coded by a gene on HSA21 is CHAF1B, representing the p60 subunit of the chromatin assembly factor complex (Volk et al., 2018). CHAF1B is essential for normal hematopoiesis, whereas its overexpression promotes leukemia by binding chromatin at discrete sites and interfering with the occupancy of CCAAT enhancer binding protein alpha (CEBPA) (Volk et al., 2018). CHAF1B expression is higher in patient cells from ML-DS than those of non-DS-AMKL (Malinge et al., 2012). Reducing CHAF1B activity is sufficient to suppress leukemogenesis in mice without impairing normal hematopoiesis, suggesting CHAF1B is a potential therapeutic target (Volk et al., 2018). Overall, HMGN1 and CHAF1B block myeloid differentiation and promote leukemia growth in other contexts but their roles in the initiation of TAM and progression to ML-DS are not known.
miRNAs
miRNAs, endogenous non-coding RNAs (∼23 nucleotides in length), target mRNA of protein-coding genes to regulate expression, through which they control a range of cellular processes, such as cell proliferation, apoptosis, hematopoiesis and tumorigenesis (Brás et al., 2018). A number of HSA21 miRNA genes are up-regulated in DS, including miR-155, miR-802, miR-125b-2, let-7c and miR-99a. Deregulated expression of miRNAs may contribute to a range of phenotypes in patients with DS, not only leukemia but also brain pathology, congenital heart defects, as well as low incidence of solid tumors in DS individuals (Brás et al., 2018). The miR-99a∼125b cluster, encoding let-7c, miR-99a and miR-125b, is highly expressed in TAM, ML-DS, and non-DS AMKL (Emmrich et al., 2014). The role of some HSA21 miRNAs in TAM/ML-DS pathogenesis has been partially revealed in recent years. (Alejo-Valle et al., 2021). GATA1 mutations and miR-99a∼125b cluster interact to induce the block in megakaryocytic differentiation that leads to the expansion of megakaryocytic progenitors and AMKL in a mouse model (Alejo-Valle et al., 2021). Another study highlighted the role of three HSA21 miRNAs (miR-99a, miR-125b-2, and miR-155) in the development of TAM, but not ML-DS (Wagenblast et al., 2021). Co-expression of miR-99a, miR-125b-2, and miR-155 in normal fetal liver LT-HSCs recapitulates features of a trisomy 21-like hematopoietic state, while deletion of these miRNAs reduces the blast population in the presence of GATA1s. Nevertheless, in the mouse model of ML-DS with and without deletion of HSA21 miRNAs blast numbers are similar (Wagenblast et al., 2021). Other studies suggest that miR-99a plays an oncogenic role through increasing proliferation and colony forming ability, and decreasing apoptosis of hematopoietic progenitors (Zhang et al., 2013; Si et al., 2016). miR-125b-2 is a positive modulator of megakaryopoiesis and an oncogenic miRNA in ML-DS. miR-125b-2 up-regulation promotes proliferation and self-renewal of megakaryocytic and megakaryocytic/erythroid progenitors, while its down-regulation inhibits growth of ML-DS cells (Klusmann et al., 2010). Moreover, miR-125b-mediated repression of the megakaryocytic transcription factor AT-rich interactive domain-containing protein 3A (ARID3A) is a critical event in ML-DS pathogenesis (Alejo-Valle et al., 2021). In the context of miR-125b overexpression and GATA1s mutations, ARID3A is the main target of miR-125b. Down-regulation of ARID3A blocks megakaryocytic differentiation and subsequently AMKL, while restoring ARID3A expression reverses megakaryocytic differentiation arrest in AMKL patient-derived xenografts. This suggests that restoration of ARID3A could be a promising strategy to inhibit megakaryoblastic leukemia growth. miR-155, a known regulator of the immune system, is also a crucial player in TAM through targeting tumor necrosis factor (TNF) superfamily receptors; miR-155 expression increases 2-fold and 3-fold in DS fetal and adult cells, respectively (Elton et al., 2010; Sas et al., 2020). How miR-155-modulated TNF receptor expression promotes TAM/ML-DS remains unknown.
Other effects of trisomy 21
Beyond the direct impact of HSA21 genes on myeloid proliferation, trisomy 21 also alters non-HSA21 gene expression through modulating genome organization (Letourneau et al., 2014; Liu et al., 2015; Ahlfors et al., 2019). Genome-wide studies showed that trisomy 21 has profound effects on DNA methylation in fetal and neonatal hematopoietic cells (Muskens et al., 2021). How these epigenetic changes influence TAM and ML-DS is not yet known. However, it has been shown that prior to the acquisition of GATA1 mutations, trisomy 21 causes loss of DNA methylation at genes linked with the regulation of the cardiovascular, neurological, and endocrine organs. ML-DS has a unique epigenetic pattern characterized by gains of DNA methylation at genes correlated with hematopoiesis, cell proliferation, cell death, and cell cycle, which is distinct from other subtypes of pediatric AML, including non-DS-AMKL (Malinge et al., 2013). Significantly, TAM and ML-DS share the identical landscape of epigenetic changes (Malinge et al., 2013). Hence, it is possible that altered DNA methylation contributes towards development of TAM and ML-DS.
GATA1 mutations
Mutations in GATA1 causing expression of its short isoform (GATA1s) are detected in nearly every case of TAM and ML-DS, implying mutated GATA1 deregulation plays a central role in TAM and ML-DS development (Wechsler et al., 2002; Panferova et al., 2021). The lack of detected GATA1 mutations in ML-DS may be due to technical and sample limitations similar to those listed earlier for TAM. In addition, AMKL is associated with bone marrow fibrosis, which often impacts the quality and quantity of diagnostic bone marrow aspirate samples, in particular blast numbers. Because blasts are the cells that carry GATA1s in ML-DS, their paucity may limit detection. Similar to DS-like TAM, ML-DS-like leukaemia may arise in children without DS where GATA1s and trisomy 21 are somatically acquired in leukemic blasts (Ono et al., 2015; De Rooij et al., 2017; Panferova et al., 2021), or GATA1s mutations may be germline (Hasle et al., 2022). ML-DS-like leukemia is very rare but it shares multiple pathologic and clinical features with ML-DS, including good prognosis (De Rooij et al., 2017), emphasizing the importance of recognizing ML-DS-like leukemia in non-DS children.
GATA1 is encoded by the gene located on chromosome X and acts as a master transcription factor essential for the development of erythroid and megakaryocytic lineages (Pevny et al., 1991). More than 100 types of GATA1 mutations have been reported in DS. These mutations are predominantly insertions, deletions, or duplications occurring in exon 2 or surrounding sequences. GATA1 mutations create an early stop codon that results in an exclusive expression of a short isoform of GATA1 protein (referred to as GATA1s) that lacks the N-terminal activation domain. Rarely, mutations in exon 3 generate GATA1 proteins with internal deletions. GATA1s can bind DNA but fails to initiate transcription, leading to deregulation of many downstream target genes (Wechsler et al., 2002). The cellular stage in which the functional and molecular consequences of GATA1s begin in the embryo has been narrowed down to the erythro-megakaryocytic subpopulation of progenitors with the following immunophenotype: CD34+CD43+CD235-CD11b-CD71+CD41+ (Nishinaka-Arai et al., 2021). The identification of this cellular stage should assist further studies into the pathogenesis of both TAM and ML-DS.
GATA1s promotes megakaryocytic progenitor expansion and disrupts megakaryocytic and erythroid differentiation (Shimizu et al., 2009; Chlon et al., 2015; Banno et al., 2016; Juban et al., 2021). This appears to involve synergistic interactions with other leukemogenic molecules; for example, GATA1s increases expression of miRNA-486-5p, an erythroid oncogenic miRNA (Shaham et al., 2015). In the presence of trisomy 21, GATA1s mutations are sufficient to drive TAM, and these mutations become undetectable when TAM resolves (Shimizu et al., 2009). Evidence from a range of cellular and animal disease models confirmed that TAM is initiated by increased gene dosage from chromosome 21 acting in cooperation with GATA1s. GATA1s mutation alone disrupts differentiation of megakaryocytes and promotes expansion of myeloid and megakaryocytic progenitors, while production of aberrant megakaryoblasts is strengthened on the background of trisomy 21 (Banno et al., 2016; Juban et al., 2021; Matsuo et al., 2021). TAM requires the synergy between trisomy 21 and GATA1s but leukemic transformation may be independent of trisomy 21 (Wagenblast et al., 2021; Arkoun et al., 2022). In contrast, synergy between GATA1s and subsequent “tertiary” molecular alterations is critical for progression of TAM to ML-DS. Evidence from sequential longitudinal studies highlights that pre-leukemic and leukemic clones are truly related, due to the fact that identical GATA1 mutations are found in paired TAM and ML-DS samples (Hitzler et al., 2003; Saida et al., 2013). Although most TAM clones disappear by the age of 3 months, some heterogeneous clones persist during remission, and these carry different leukemia-initiating potential (Saida et al., 2013). ML-DS can be derived from a minor clone with a distinct GATA1s in TAM, but novel clones can also arise and become dominant (Xu et al., 2006; Saida et al., 2013; Labuhn et al., 2019).
So far, there is no solid proof of whether the type of GATA1 mutations, the level of GATA1s expression, or the size of dominant GATA1s-bearing clones can predict progression from TAM to ML-DS (Alford et al., 2011; Grimm et al., 2021). Kanezaki et al. pointed out that the type of GATA1 mutations influences expression of the GATA1s protein, and these expression levels are inversely linked with the risk of progression to ML-DS (Kanezaki et al., 2010). Nonetheless, in the clinical setting, persistence of GATA1s mutations is the most important risk factor associated with progression to ML-DS, even in cases with high GATA1s protein levels (Massey et al., 2006; Pine et al., 2007). The features used in the clinic to predict TAM progression to ML-DS include detection of minimal residual disease by flow cytometry (blasts >0.1%), persistence of patient-specific GATA1s mutation beyond 12 weeks from the initial diagnosis, and the appearance of thrombocytopenia (platelet count less than 100×109/L) (Klusmann et al., 2008; Flasinski et al., 2018).
Tertiary alterations
It has become well accepted that evolution from TAM to ML-DS relies on the acquisition of tertiary somatic mutations and additional chromosomal structural aberrations in GATA1s-mutated cells. Tertiary mutations seen in ML-DS most commonly affect genes encoding the cohesin complex, JAK family kinases, and epigenetic regulators; other mutations occur in genes recurrently mutated in other types of AML, including fms-like tyrosine kinase 3 (FLT3) and TP53 (Table 2) (Yoshida et al., 2013; Labuhn et al., 2019; Panferova et al., 2021). Patients with TAM usually harbor fewer tertiary mutations than those with ML-DS, at the average of 0.4 and 1.6 variants per sample respectively (Labuhn et al., 2019). Most TAM cases carry only GATA1s, while additional somatic mutations are rare. Even if present in TAM, “third hit” mutations appear to be non-functional and un-linked from pre-leukemia or leukemia phenotype (Labuhn et al., 2019). By way of illustration, no autonomous or cytokine-induced signaling was found for JAK1, JAK2, JAK3 or MPL variants by dual-luciferase assays with a signal transducer and activator of transcription 5 (STAT5) reporter at the TAM stage (Labuhn et al., 2019). During leukemic transformation, two to five additional mutations are found in a murine model of ML-DS. The most frequently altered genes encode signaling pathways (34%), members of the cohesin complex or its associated components (28.5%), and epigenetic regulators (22%) (Labuhn et al., 2019). The authors suggest that ML-DS progression is influenced by the cooperation between activated signaling pathways and deregulated epigenetic processes in the context of trisomy 21 and GATA1s. For instance, a remarkable co-occurrence of variants in genes encoding tyrosine kinases (e.g., JAK2-3) and RAS proteins with variants in epigenetic regulators (e.g., enhancer of zeste 2, EZH2) or cohesin genes has been shown in ML-DS mouse models and ML-DS patients (Labuhn et al., 2019). No tertiary mutations were detected in approximately 15–25% of ML-DS patients in relatively large studies reported in the last few years (Labuhn et al., 2019; Panferova et al., 2021). However, it is possible that such mutations will be detected in the future using updated sequencing methodologies. Karyotypic changes other than trisomy 21 may also contribute to ML-DS because such alterations are rarely found at the TAM stage.

TABLE 2. Recurrent somatic mutations reported in myeloid leukemia associated with Down syndrome.
Mutations in the cohesin complex and related components
Cohesin is a multi-subunit complex composed of three main structural proteins (structural maintenance of chromosomes protein 1A (SMC1A), structural maintenance of chromosomes protein 3 (SMC3), and double-strand-break repair protein Rad21 homolog (RAD21)), which bind to either cohesin subunit SA-1 (STAG1) or cohesin subunit SA-2 (STAG2) proteins. Cohesin complex is a ring-shaped structure that surrounds chromosomal DNA and controls its functions, including sister chromatid cohesion, chromatin remodeling, transcriptional regulation, and DNA damage repair (Jann and Tothova, 2021). Nipped-B-like protein (NIPBL) is involved in cohesin loading to chromatin, translocating cohesin along chromatin fibers, and regulating cohesin after loading (Garcia et al., 2021). Cohesin core subunits and its modulators (including STAG2, RAD21, SMC1A, SMC3 and NIPBL) are recurrently mutated in myeloid malignancies. Cohesin mutations are highly prevalent in ML-DS, where they occur in nearly half of patients (Yoshida et al., 2013). STAG2 and RAD21 have the higher mutation frequency than SMC1, SMC3 and NIPBL, with approximately 9.1–18.4% and 11.3–22.4% in ML-DS cases respectively (Nikolaev et al., 2013; Yoshida et al., 2013; Labuhn et al., 2019; Panferova et al., 2021). Each of these mutations results in loss-of-function of the molecules, and in cooperation with GATA1s and trisomy 21 each can drive leukemic transformation.
Recent genetic modifications of human iPSC lines derived from DS tissue greatly assisted examination of the cooperativity between GATA1s and cohesin mutations in ML-DS. The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system was used to introduce GATA1s and STAG2 mutations into iPSCs in a sequential manner (Barwe et al., 2020; Barwe et al., 2022). GATA1s and STAG2 knockout cooperatively increased the megakaryocytic population and induced the ML-DS immunophenotype (Barwe et al., 2022). In another study, trisomic 21 iPSC line (Chou et al., 2012) was edited to introduce GATA1s followed by heterozygous inactivation of SMC3 (SMC3+/−) and then, introduction of a gain-of-function MPL mutation (MPLW515K) (Arkoun et al., 2022). It was found that GATA1s impaired megakaryocyte differentiation and that SMC3+/− enhanced this effect independent of trisomy 21. MPLW515K further increased the megakaryocyte output in this model, including through the induction of growth factor independence. Low expression of NFE2 was critical for the induction of megakaryocyte dysplasia by GATA1s (Arkoun et al., 2022). These novel iPSC-based models are likely to rapidly advance our understanding of ML-DS pathogenesis, and assist therapy development.
We include a brief description of the relevant cohesin elements to highlight their roles in haematopoiesis. STAG2, located on the chromosome X, is the most frequently mutated cohesin gene in human cancer (Viny et al., 2019). STAG2 deletion in hematopoietic stem and progenitor cells results in abnormal hematopoietic function, increased self-renewal, and impaired differentiation (Viny et al., 2019). STAG2 loss-of-function decreases cell growth and proliferation, increases cell invasion and metastasis, enhances chemo-resistance, regulates the expression of many immune-related genes, and interplays with RUNX1 deficiency to perturb chromatin looping (Nie et al., 2019; Ochi et al., 2020). Likewise, RAD21 loss-of-function confers enhanced hematopoietic self-renewal and impaired cell differentiation (Fisher et al., 2017; Bisaillon et al., 2020). RAD21 is a regulator of gene expression and epigenetic modulation. For example, RAD21 regulates expression of RUNX1 and methylation of Homeobox a7/Homeobox a9 (Fisher et al., 2017; Bisaillon et al., 2020). NIPBL also regulates RUNX1 expression, thus its loss-of-function impairs RUNX1 expression and consequently, hematopoiesis (Mazzola et al., 2020). NIPBL interplaying with nucleophosmin 1 (NPM1) regulates myeloid differentiation through the WNT (Wingless/Integrated) pathway; the disruption of these interactions has been implicated in leukemogenesis (Mazzola et al., 2019a). SMC3 is the cohesin ATPase subunit, with its dosage controlling embryogenesis and hematopoiesis (Wang et al., 2019; Rivas et al., 2021). Homozygous deletion of SMC3 in mice results in embryonic lethality and the hematopoietic failure (Wang et al., 2019). In comparison, heterozygous SMC3 deletion leads to developmental defects (e.g., abnormal craniofacial morphology), germinal center hyperplasia with increased B-cell proliferation and increased risk of B-cell lymphoma development (Rivas et al., 2021). SMC1A-R586W mutation is known to interfere with cohesin localization and cohesin-mediated DNA loop interaction in AML cells. This mutation confers wide changes to gene expression and genome organization when engineered into murine embryonic stem cells (Carico et al., 2021). Finally, CCCTC-binding factor (CTCF) is a tumor suppressor and involved in many cellular processes, with approximately 11.3–20.4% of ML-DS cases harboring CTCF gene mutations (Yoshida et al., 2013; Labuhn et al., 2019; Panferova et al., 2021). CTCF interacts with the cohesin complex to control genome architecture and gene expression (Zuin et al., 2014; Wang et al., 2020). CTCF and cohesins are known to assist formation of DNA loops, however depletion of either the cohesin complex or CTCF has differential effects on chromatin organization and gene expression in human HEK293T cells (Zuin et al., 2014). Deletion of cohesin caused a general loss of local chromatin interactions but the topological domains remained intact. In contrast, depletion of CTCF both reduced and increased interdomain interactions and distinct groups of genes became dysregulated. Apart from its interplay with cohesins, CTCF is a highly conserved transcription factor implicated in transcriptional activation and repression, insulation, formation of chromatin barrier, gene imprinting, X-chromosome inactivation, and RNA splicing (Bell et al., 1999; Bell and Felsenfeld, 2000; Xu et al., 2007; Shukla et al., 2011; Wang et al., 2020). CTCF is also involved in maintaining genomic methylation patterns through the control of poly (ADP-ribose) polymerase 1 (PARP1) and the activity of DNA methyltransferase 1 (DNMT1) (Zampieri et al., 2012). CTCF haploinsufficiency correlates with altered patterns of DNA methylation and predisposes to cancer in mice (Kemp et al., 2014). CTCF is a critical factor in the control of hematopoiesis and leukemogenesis (Torrano et al., 2005; Kim et al., 2017; Mujahed et al., 2020). In adult mice, conditional CTCF deletion causes an acute loss of HSCs, severe bone marrow failure and increased mortality, highlighting CTCF requirement for the maintenance of the HSC pool (Kim et al., 2017). Abnormal CTCF expression reduces growth and enhances differentiation of the erythroid lineage by down-regulating MYC (Torrano et al., 2005). In AML, CTCF binding was shown to be elevated, compared with normal bone marrow, with increased CTCF binding in promoter regions linked with DNA hypomethylation and increased target gene expression (Mujahed et al., 2020). However, the combination of CTCF loss-of-function with GATA1s and trisomy 21 is unable to drive leukemic transformation, indicating additional events are required (Wagenblast et al., 2021). Collectively, the cohesin complex and CTCF are involved in ML-DS pathogenesis, but the exact roles of these molecules need further elucidation.
Mutations in signaling pathways
Most mutations affecting signaling pathways occur in genes encoding JAK regulators, MPL and KIT (CD117) collectively reported in 48% of ML-DS cases (Labuhn et al., 2019). JAK2 and JAK3 are more frequently mutated (9.9 and 13.5%) than JAK1, MPL and KIT (4.3, 7.1 and 1.4%) (Labuhn et al., 2019). JAK1-3 variants are identified in both ML-DS and TAM samples, however, gain-of-function mutations are only detected in ML-DS, highlighting that aberrant activation of JAK-STAT signaling is important for transition to leukemia (Labuhn et al., 2019). The JAK family of tyrosine kinases (JAK1-3 and tyrosine kinase 2, TYK2) are pivotal mediators of growth factor and cytokine signaling, including downstream of thrombopoietin (TPO) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (De Castro et al., 2021; Moser et al., 2021). JAK1, JAK2 and TYK2, are ubiquitously expressed, whilst JAK3 is predominantly expressed in lymphoid and myeloid cells. JAK3 mutations are more common than of other members of JAK family in ML-DS (Labuhn et al., 2019). Under physiological conditions, JAK-STAT signaling is tightly controlled and involved in a wide range of fundamental biological processes, including cell proliferation, differentiation, apoptosis, inflammation, and blood production (Park et al., 2016; De Castro et al., 2021; Moser et al., 2021). Normal megakaryopoiesis requires TPO-mediated STAT5 activation. Unphosphorylated STAT5 represses megakaryocytic transcriptional program and inhibits megakaryocytic differentiation by competing with ERG for CTCF binding, which can be reversed by TPO-mediated activation of STAT5 (Park et al., 2016). MPL, a receptor of TPO, is also frequently mutated in ML-DS, which contributes to leukemia development. In the presence of trisomy 21 and GATA1s, MPL W515L causes rapid and lethal leukemia in mice (Malinge et al., 2012). Recently, colony stimulating factor 2 receptor subunit beta (CSF2RB) A455D variant was reported in almost 5% of ML-DS children. This variant is mutually exclusive with mutated JAK1-3, MPL or RAS genes, and causes ligand-independent STAT5 activation promoting cytokine-independent cell growth (Labuhn et al., 2019). Upon introduction of the CSF2RB A455D mutant into hematopoietic stem and progenitor cells (HSPCs), megakaryocytic and erythroid proliferation is enhanced, and terminal megakaryocytic maturation is blocked (Labuhn et al., 2019). These alterations are alleviated by the JAK1/2 inhibitor ruxolitinib, emphasizing that aberrant JAK-STAT signaling participates in the CSF2RB A455D-driven leukemogenesis (Labuhn et al., 2019). In addition, CSF2RB binding to FLT3-ITD is found in other AML cell lines and patient cells where CSF2RB deletion decreases STAT5 phosphorylation, inhibits leukemic cell proliferation, and sensitizes cells to FLT3 inhibition (Charlet et al., 2021). These findings demonstrate that CSF2RB is critical for FLT3-ITD-dependent oncogenic signaling and transformation, but its role in ML-DS requires further study.
Mutations in RAS (Rat sarcoma virus) gene family members, such as KRAS, NF1, NRAS, and PTPN11, are found in 14% of ML-DS samples (Labuhn et al., 2019). NRAS and KRAS variants are the most common accounting for 4.5–8.2% and 4.3–9.1% of ML-DS cases respectively (Yoshida et al., 2013; Labuhn et al., 2019; Panferova et al., 2021). Ras belongs to the small GTPase family that binds to guanosine triphosphate (GTP) and hydrolyses it to guanosine diphosphate (GDP), with three distinct isoforms NRas, KRas, and HRas (Padmakumar et al., 2021). Ras is located on the inner surface of the plasma membrane, and acts as a binary molecular switch. Ras can transmit extracellular signals to the nucleus, and cycles between the inactive GDP-bound state and the active GTP-bound state (Zafar et al., 2021). Mutations fix RAS-GTPase proteins in an active GTP-bound state, resulting in constitutive activation of MAPK and PI3K (phosphoinositide 3-kinases) signaling. Consequently, uncontrolled cell proliferation and survival occur in mutated cells. In mouse models, clonal NRAS/KRAS activation increases cell growth, proliferation, and colony formation through a lysine methyltransferase 2A (KMT2A)- polo like kinase 1 (PLK1) axis (Carr et al., 2021). Mutations in RAS have a role in TAM progression to ML-DS, but it is not fully understood how these mutations cooperate with trisomy 21, GATA1s and other mutations in cohesins or epigenetic modulators.
Mutations in epigenetic regulators
Loss-of-function mutations in epigenetic regulators are emerging as critical contributors to ML-DS progression. Such mutations are reported in approximately 36–45% of ML-DS samples and affect a range of regulators, including additional sex combs-like 1 (ASXL1), BCL6 corepressor (BCOR), DNMT1, DNMT3A, embryonic ectoderm development (EED), E1A binding protein P300 (EP300), EZH2, KAT8 regulatory NSL complex subunit 1 (KANSL1), lysine demethylase 6A (KDM6A), lysine methyltransferase 2C (KMT2C), N-acetyltransferase 6 (NAT6), SUZ12, and tet methylcytosine dioxygenase 2 (TET2) (Nikolaev et al., 2013; Yoshida et al., 2013; Labuhn et al., 2019; Panferova et al., 2021). Mutations in KANSL1, EZH2 and SUZ12 were seen at the highest frequency, in 6.1–12.1%, 2.3–32.7% and 2–6.4% of ML-DS cases respectively (Nikolaev et al., 2013; Yoshida et al., 2013; Labuhn et al., 2019; Panferova et al., 2021). KANSL1 is essential for the activity of the histone acetylation complex, which takes part in the acetylation of histone H4 lysine 16 and eventually leads to transcriptional activation. Loss-of-function mutations in KANSL1 are detected in both ML-DS and non-DS-AMKL (Yoshida et al., 2013; Labuhn et al., 2019). KANSL1 mutations combined with trisomy 21 and GATA1s drive leukemic engraftment in mice (Wagenblast et al., 2021). EZH2 forms polycomb repressive complex 2 (PRC2) together with SUZ12, EED and RB binding protein 4 (RBBP4). PRC2 is mainly responsible for the methylation of lysine 27 in the tail of histone H3 family proteins (H3K27me3), which subsequently silences its target gene expression. Thus, EZH2 is a transcriptional repressor with methyltransferase activity, whereas SUZ12 is essential for the structural integrity of PRC2 and the facilitation of chromatin binding (Chen et al., 2018; Zeisig and So, 2021). EZH2 is unable to perform this enzymatic function alone, and the interplay with EED and SUZ12 enables PRC2 function (Chen et al., 2018; Zeisig and So, 2021). In megakaryopoiesis, EZH2 inhibition accelerates megakaryocytic differentiation and blocks megakaryocytic proliferation (Mazzi et al., 2021). EZH2 and SUZ12 act as tumor suppressors; mutations in either gene lead to loss-of-function of PRC2 core subunits and a deficit of H3K27me3 (Ntziachristos et al., 2012). Murine ML-DS leukemia models and ML-DS patients show loss-of-function mutations in EZH2 and other PRC2 members, supporting the PRC2 role in transition from TAM to ML-DS (Labuhn et al., 2019). Although the importance of mutated epigenetic modifiers in ML-DS has been recognized, their pathologic functions and clinical impact remain unclear.
Chromosomal abnormalities
Beyond trisomy 21, additional cytogenetic changes are observed in the majority of children with ML-DS, but rarely in TAM (Malinge et al., 2008; Labuhn et al., 2019). Therefore, these changes could play a role in the development of ML-DS. Cytogenetic changes reported in ML-DS include gains and losses of whole chromosomes or their arms, or chromosomal rearrangements (Table 3). Common chromosomal gains are trisomies: +2, +8, +11, +13, +14, +19, +22, or tetrasomies: +14, +21. Chromosomal losses include monosomies: −1, −3, −4, −5, −7, −9, −16, and −18. Other aberrations include: add(1q), add(6p), add (6q), add (7q), add(8p), add(11q), add (16q), add (19p); or deletions: del(5p), del(5q), del(6q), del(7p), del(7q), del(11p), del(13q), del(16q), del(17p), del(17q), and del(22q). The most common structural abnormalities are del(7p)/del(7q)/−7, del(16q), trisomy 8, and tetrasomy 21. However, none of these changes offer clear insights into the molecular pathogenesis of ML-DS, and their prognostic impact is also largely unknown (Forestier et al., 2008; De Souza et al., 2017; Labuhn et al., 2019). One recent study points out that +8 can be associated with inferior event-free survival in ML-DS (Uffmann et al., 2017). More work is required to elucidate the pathogenetic role and clinical impact of chromosomal abnormalities in ML-DS.
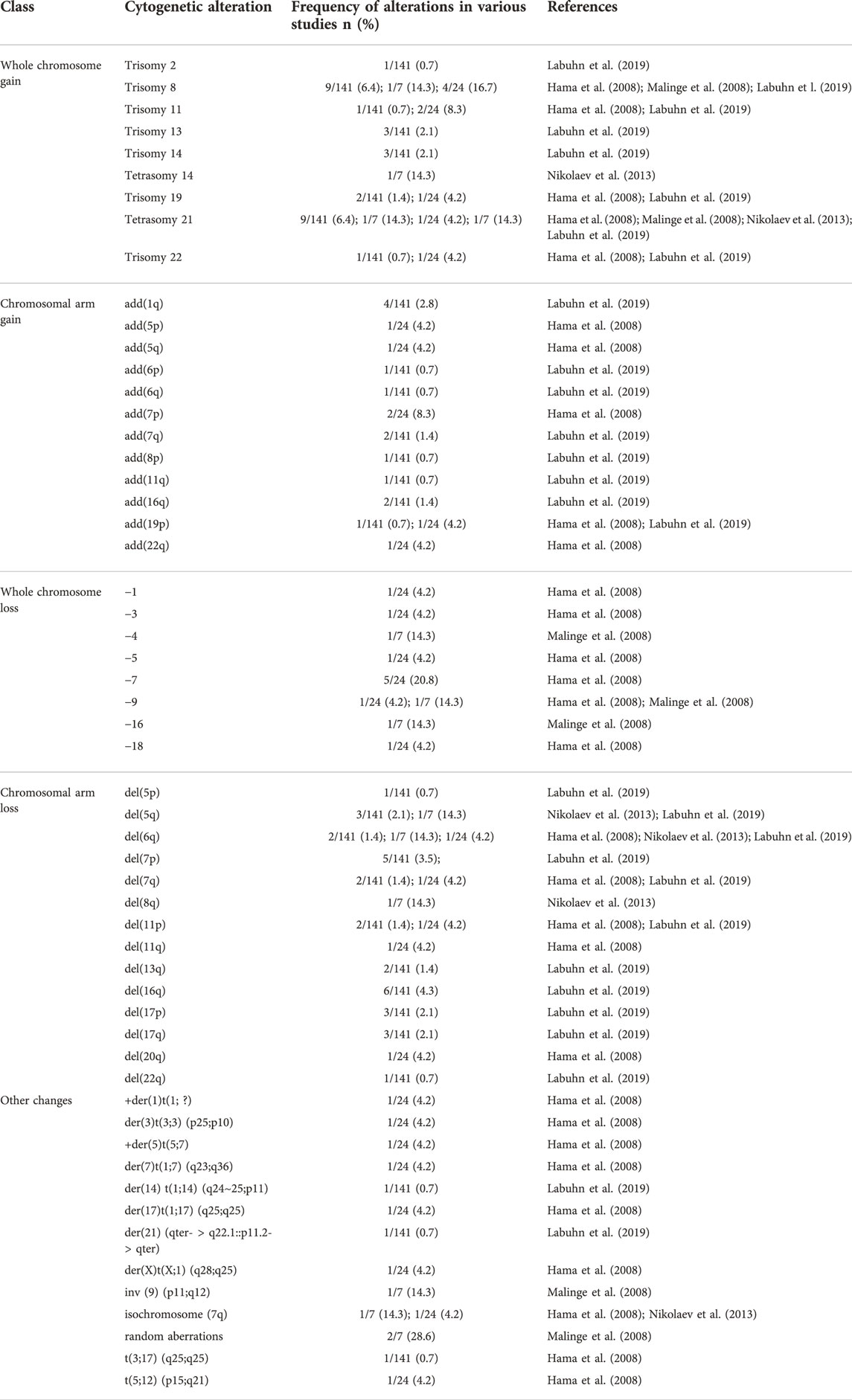
TABLE 3. Chromosomal abnormalities reported in myeloid leukemia associated with Down syndrome.
Co-occurrence patterns of additional somatic mutations other than GATA1s
Transformation of TAM to ML-DS often arises on the background of activating signaling mutations interacting with deregulated epigenetic modifiers. For instance, there is a significant co-occurrence of variants in genes encoding tyrosine kinases and RAS proteins with variants in epigenetic regulators or cohesins both in ML-DS mouse models and patient samples (Labuhn et al., 2019). CTCF and EZH2 mutations alone are insufficient to drive ML-DS in the presence of trisomy 21 and GATA1s, implying other somatic mutations are required (Wagenblast et al., 2021). The frequent co-occurrence of variants in EZH2 and CB1 is identified in a murine model of ML-DS, while NF1 mutations appear mutually exclusive with CB1, EZH2, and CTCF variants (Labuhn et al., 2019). Co-occurrence of additional mutations is important for leukemic progression, but their patterns, functional effects, and clinical significance need further investigation.
Mutational landscape of relapsed myeloid leukemia associated with Down syndrome
ML-DS usually has a low incidence of relapse, seen in approximately 5–6% of patients in developed countries, mostly because the initial disease is very sensitive to chemotherapy (Uffmann et al., 2017). However, when relapse occurs in ML-DS patients, the prognosis is less favorable. Little is known about the molecular underpinnings leading to relapse and current treatment options are less effective in relapsed patients. One study showed that in a cohort of 170 pediatric patients with ML-DS, five of 7 relapsed cases harbored trisomy 8, while the other two carried isochromosome 7 and additional material on chromosome 16 respectively (Uffmann et al., 2017). As for somatic mutations, the sequencing data from one paired sample (diagnostic and relapsed) demonstrated the presence of EZH2 F562S, JAK2 V617F and MTNR1B R316H in the relapsed sample but not at the time of diagnosis, while SMC1A R711Q, MPL W515S, JAK2 F694S and EZH2 H206fs were detected at both time-points (Labuhn et al., 2019). A lot more work will need to be done in this area in the future.
Novel therapeutic targets
As the genomic, epigenomic and transcriptomic changes are uncovered in TAM and ML-DS, new molecular targets for prevention and treatment are being proposed. Mutations in signaling effectors are one of the most frequent events in ML-DS associated with the overactivation of pathways such as JAK-STAT, RAS/MEK/ERK and PI3K/PKB (Labuhn et al., 2019). Inhibition of these pathways may help treat ML-DS. FDA-approved JAK1/2 inhibitors, ruxolitinib and momelotinib (Sureau et al., 2021), could be considered for patients with activating JAK-STAT mutations. Similarly, drugs targeting RAS and PI3K/PKB signaling could be trialed in children with mutations in these pathways (De Castro et al., 2021). CD117/KIT expression is a marker of GATA1s-induced pre-leukemia- and GATA1s/STAG2-knock-out-induced leukemia-initiating cells. The maintenance and expansion of those cells rely on KIT signaling; thus, KIT inhibitors have emerged as potential therapeutic targets (Wagenblast et al., 2021). Further, mutations in cohesin subunits and cohesin regulators are crucial for ML-DS pathogenesis (Labuhn et al., 2019), thus targeting cohesin-mutant cells has been suggested to be a new therapeutic strategy. There are three distinct approaches through which cohesin mutated cells can be targeted: 1) direct modulation of cohesin subunits and its regulators; 2) targeting cohesin-induced deregulated signaling; and 3) targeting altered DNA damage repair mechanisms (Antony et al., 2021). STAG1 inhibition may be a suitable therapy for patients with STAG2 mutations because it is synthetically lethal with STAG2 variants. ML-DS displays frequent gains of DNA methylation, thus epigenetic therapies may be useful. In support, lysine-specific demethylase inhibitor T-3775440 inhibits growth of patient-derived blasts ex vivo (Labuhn et al., 2019). Finally, three HSA21 miRNAs (miR-99a, miR-125b and miR-155) are overexpressed in blast cells from ML-DS, and their blockage inhibits GATA1s-induced pre-leukemia development (Wagenblast et al., 2021). Hence, miRNAs could also become potential therapeutic targets in the future.
Conclusions and future directions
ML-DS has three major molecular features: trisomy 21, GATA1s mutations, and tertiary alterations (Figure 1). Trisomy 21 drives megakaryocytic expansion through the increased gene dosage effect, but trisomy 21 may not be required for progression to ML-DS. GATA1s mutations are acquired during fetal liver hematopoiesis in susceptible HSCs with high proliferative potential, which leads to abnormal megakaryocytic proliferation and impaired erythroid differentiation. GATA1s effects begin in an immunophenotypically distinct population of fetal erythro-megakaryocytic cells. The development of ML-DS requires acquisition and selection of clones with additional somatic mutations and chromosomal structural abnormalities. Substantial progress has been made over the last 20 years in the molecular characterization of ML-DS, but some important questions remain unanswered. How do trisomy 21, GATA1s, additional somatic mutations and chromosomal alterations cooperate to drive ML-DS? In the context of trisomy 21 and GATA1s, what is the relevant co-occurrence pattern of somatic mutations and cytogenetic changes? What is the biological role and the clinical impact of such changes? Recent application of CRISPR/Cas9 technology in iPSC-based models of ML-DS started to provide some essential answers to these questions.
From the clinical standpoint, new therapies are needed for children with refractory and relapsed disease, in particular as high-dose chemotherapy causes unacceptable toxicity in DS children. To test emerging therapeutic targets, we need to advance pre-clinical disease models of ML-DS. Chromosome 21-encoded proteins and miRNAs are important players in ML-DS. Is it possible to target these molecules alone or do we need to simultaneously target secondary and tertiary genetic changes to control leukemia growth? The work needs to continue to better elucidate disease mechanisms and to develop more effective therapies.
Author contributions
JL drafted the manuscript. MK-Z provided supervision and guidance, helped write and revise the manuscript. Both authors approved the submitted version.
Funding
Anne Norman and Victoria Nicholls Leukaemia and Lymphoma Research Fund.
Acknowledgments
Special thanks to Taryn Green for her careful proof-reading and technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahlfors, H., Anyanwu, N., Pakanavicius, E., Dinischiotu, N., Lana-Elola, E., Watson-Scales, S., et al. (2019). Gene expression dysregulation domains are not a specific feature of Down syndrome. Nat. Commun. 10 (1), 2489. doi:10.1038/s41467-019-10129-9
Aksu, T., Gumruk, F., and Unal, S. (2020). Comment on: Clinical, cytogenetic, and molecular analyses of 17 neonates with transient abnormal myelopoiesis and nonconstitutional trisomy 21. Pediatr. Blood Cancer 67 (11), e28289. doi:10.1002/pbc.28289
Alejo-Valle, O., Weigert, K., Bhayadia, R., Ng, M., Issa, H., Beyer, C., et al. (2021). The megakaryocytic transcription factor ARID3A suppresses leukemia pathogenesis. Blood 139, 651–665. doi:10.1182/blood.2021012231
Alford, K. A., Reinhardt, K., Garnett, C., Norton, A., Böhmer, K., von Neuhoff, C., et al. (2011). Analysis of GATA1 mutations in Down syndrome transient myeloproliferative disorder and myeloid leukemia. Blood 118 (8), 2222–2238. doi:10.1182/blood-2011-03-342774
Antonarakis, S. E. (2017). Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 18 (3), 147–163. doi:10.1038/nrg.2016.154
Antony, J., Chin, C. V., and Horsfield, J. A. (2021). Cohesin mutations in cancer: Emerging therapeutic targets. Int. J. Mol. Sci. 22 (13), 6788. doi:10.3390/ijms22136788
Apollonsky, N., Shende, A., Ouansafi, I., Brody, J., Atlas, M., Aygun, B., et al. (2008). Transient myeloproliferative disorder in neonates with and without down syndrome: A tale of 2 syndromes. J. Pediatr. Hematol. Oncol. 30 (11), 860–864. doi:10.1097/MPH.0b013e31818a953e
Arber, D. A., Orazi, A., Hasserjian, R., Thiele, J., Borowitz, M. J., Le Beau, M. M., et al. (2016). The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20), 2391–2405. doi:10.1182/blood-2016-03-643544
Arkoun, B., Robert, E., Boudia, F., Mazzi, S., Dufour, V., Siret, A., et al. (2022). Stepwise GATA1 and SMC3 mutations alter megakaryocyte differentiation in a Down syndrome leukemia model. J. Clin. Invest. 1, e156290. doi:10.1172/JCI156290
Banno, K., Omori, S., Hirata, K., Nawa, N., Nakagawa, N., Nishimura, K., et al. (2016). Systematic cellular disease models reveal synergistic interaction of trisomy 21 and GATA1 mutations in hematopoietic abnormalities. Cell Rep. 15 (6), 1228–1241. doi:10.1016/j.celrep.2016.04.031
Barwe, S. P., Sebastian, A., Sidhu, I., Kolb, E. A., and Gopalakrishnapillai, A. (2022). Modeling down syndrome myeloid leukemia by sequential introduction of GATA1 and STAG2 mutations in induced pluripotent stem cells with trisomy 21. Cells 11 (4), 628. doi:10.3390/cells11040628
Barwe, S. P., Sidhu, I., Kolb, E. A., and Gopalakrishnapillai, A. (2020). Modeling transient abnormal myelopoiesis using induced pluripotent stem cells and CRISPR/Cas9 technology. Mol. Ther. Methods Clin. Dev. 19, 201–209. doi:10.1016/j.omtm.2020.09.007
Bazinet, A., Heath, J., Chong, A. S., Simo-Cheyou, E. R., Worme, S., Rivera Polo, B., et al. (2021). Common clonal origin of chronic myelomonocytic leukemia and B-cell acute lymphoblastic leukemia in a patient with a germline CHEK2 variant. Cold Spring Harb. Mol. Case Stud. 7 (3), a006090. doi:10.1101/mcs.a006090
Bell, A. C., and Felsenfeld, G. (2000). Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405 (6785), 482–485. doi:10.1038/35013100
Bell, A. C., West, A. G., and Felsenfeld, G. (1999). The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98 (3), 387–396. doi:10.1016/s0092-8674(00)81967-4
Belmonte, R. L., Engbretson, I. L., Kim, J. H., Cajias, I., Ahn, E. E., Stachura, D. L., et al. (2021). Son is necessary for proper vertebrate blood development. PLoS One 16 (2), e0247489. doi:10.1371/journal.pone.0247489
Benetatos, L., Benetatou, A., and Vartholomatos, G. (2020). Enhancers and MYC interplay in hematopoiesis. J. Mol. Med. 98 (4), 471–481. doi:10.1007/s00109-020-01891-1
Bhatnagar, N., Nizery, L., Tunstall, O., Vyas, P., and Roberts, I. (2016). Transient abnormal myelopoiesis and AML in down syndrome: An update. Curr. Hematol. Malig. Rep. 11 (5), 333–341. doi:10.1007/s11899-016-0338-x
Bisaillon, R., Moison, C., Thiollier, C., Krosl, J., Bordeleau, M. E., Lehnertz, B., et al. (2020). Genetic characterization of ABT-199 sensitivity in human AML. Leukemia 34 (1), 63–74. doi:10.1038/s41375-019-0485-x
Bombery, M., and Vergilio, J. A. (2014). Transient abnormal myelopoiesis in neonates: GATA get the diagnosis. Arch. Pathol. Lab. Med. 138 (10), 1302–1306. doi:10.5858/arpa.2014-0304-CC
Bourquin, J. P., Subramanian, A., Langebrake, C., Reinhardt, D., Bernard, O., Ballerini, P., et al. (2006). Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc. Natl. Acad. Sci. U. S. A. 103 (9), 3339–3344. doi:10.1073/pnas.0511150103
Brás, A., Rodrigues, A. S., Gomes, B., and Rueff, J. (2018). Down syndrome and microRNAs. Biomed. Rep. 8 (1), 11–16. doi:10.3892/br.2017.1019
Cabal-Hierro, L., van Galen, P., Prado, M. A., Higby, K. J., Togami, K., Mowery, C. T., et al. (2020). Chromatin accessibility promotes hematopoietic and leukemia stem cell activity. Nat. Commun. 11 (1), 1406. doi:10.1038/s41467-020-15221-z
Carico, Z. M., Stefan, H. C., Justice, M., Yimit, A., and Dowen, J. M. (2021). A cohesin cancer mutation reveals a role for the hinge domain in genome organization and gene expression. PLoS Genet. 17 (3), e1009435. doi:10.1371/journal.pgen.1009435
Carmichael, C. L., Metcalf, D., Henley, K. J., Kruse, E. A., Di Rago, L., Mifsud, S., et al. (2012). Hematopoietic overexpression of the transcription factor Erg induces lymphoid and erythro-megakaryocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 109 (38), 15437–15442. doi:10.1073/pnas.1213454109
Carr, R. M., Vorobyev, D., Lasho, T., Marks, D. L., Tolosa, E. J., Vedder, A., et al. (2021). RAS mutations drive proliferative chronic myelomonocytic leukemia via a KMT2A-PLK1 axis. Nat. Commun. 12 (1), 2901. doi:10.1038/s41467-021-23186-w
Charlet, A., Kappenstein, M., Keye, P., Kläsener, K., Endres, C., Poggio, T., et al. (2021). The IL-3, IL-5, and GM-CSF common receptor beta chain mediates oncogenic activity of FLT3-ITD-positive AML. Leukemia 36, 701–711. doi:10.1038/s41375-021-01462-4
Chattopadhyaya, S., and Ghosal, S. (2022). DNA methylation: A saga of genome maintenance in hematological perspective. Hum. Cell 35 (2), 448–461. doi:10.1007/s13577-022-00674-9
Chen, S., Jiao, L., Shubbar, M., Yang, X., and Liu, X. (2018). Unique structural platforms of Suz12 dictate distinct classes of PRC2 for chromatin binding. Mol. Cell 69 (5), 840–852. e845. doi:10.1016/j.molcel.2018.01.039
Chlon, T. M., McNulty, M., Goldenson, B., Rosinski, A., and Crispino, J. D. (2015). Global transcriptome and chromatin occupancy analysis reveal the short isoform of GATA1 is deficient for erythroid specification and gene expression. Haematologica 100 (5), 575–584. doi:10.3324/haematol.2014.112714
Chou, S. T., Byrska-Bishop, M., Tober, J. M., Yao, Y., Vandorn, D., Opalinska, J. B., et al. (2012). Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc. Natl. Acad. Sci. U. S. A. 109 (43), 17573–17578. doi:10.1073/pnas.1211175109
De Castro, C. P. M., Cadefau, M., and Cuartero, S. (2021). The mutational landscape of myeloid leukaemia in down syndrome. Cancers (Basel) 13 (16), 4144. doi:10.3390/cancers13164144
De Rooij, J. D., Branstetter, C., Ma, J., Li, Y., Walsh, M. P., Cheng, J., et al. (2017). Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet. 49 (3), 451–456. doi:10.1038/ng.3772
De Souza, D. C., de Figueiredo, A. F., Ney Garcia, D. R., da Costa, E. S., Othman, M. A. K., Liehr, T., et al. (2017). A unique set of complex chromosomal abnormalities in an infant with myeloid leukemia associated with Down syndrome. Mol. Cytogenet. 10, 35. doi:10.1186/s13039-017-0335-3
El Hussein, S., DiNardo, C. D., Takahashi, K., Khoury, J. D., Fang, H., Furudate, K., et al. (2022). Acquired WT1 mutations contribute to relapse of NPM1-mutated acute myeloid leukemia following allogeneic hematopoietic stem cell transplant. Bone Marrow Transpl. 57, 370–376. doi:10.1038/s41409-021-01538-w
Elagib, K. E., Racke, F. K., Mogass, M., Khetawat, R., Delehanty, L. L., Goldfarb, A. N., et al. (2003). RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood 101 (11), 4333–4341. doi:10.1182/blood-2002-09-2708
Elton, T. S., Sansom, S. E., and Martin, M. M. (2010). Trisomy-21 gene dosage over-expression of miRNAs results in the haploinsufficiency of specific target proteins. RNA Biol. 7 (5), 540–547. doi:10.4161/rna.7.5.12685
Emmrich, S., Rasche, M., Schöning, J., Reimer, C., Keihani, S., Maroz, A., et al. (2014). miR-99a/100∼125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGFβ and Wnt signaling. Genes Dev. 28 (8), 858–874. doi:10.1101/gad.233791.113
Fasouli, E. S., and Katsantoni, E. (2021). JAK-STAT in early hematopoiesis and leukemia. Front. Cell Dev. Biol. 9, 669363. doi:10.3389/fcell.2021.669363
Fisher, J. B., Peterson, J., Reimer, M., Stelloh, C., Pulakanti, K., Gerbec, Z. J., et al. (2017). The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia 31 (3), 712–719. doi:10.1038/leu.2016.240
Flasinski, M., Scheibke, K., Zimmermann, M., Creutzig, U., Reinhardt, K., Verwer, F., et al. (2018). Low-dose cytarabine to prevent myeloid leukemia in children with down syndrome: TMD prevention 2007 study. Blood Adv. 2 (13), 1532–1540. doi:10.1182/bloodadvances.2018018945
Fontana, D., Mauri, M., Renso, R., Docci, M., Crespiatico, I., Røst, L. M., et al. (2020). ETNK1 mutations induce a mutator phenotype that can be reverted with phosphoethanolamine. Nat. Commun. 11 (1), 5938. doi:10.1038/s41467-020-19721-w
Forestier, E., Izraeli, S., Beverloo, B., Haas, O., Pession, A., Michalová, K., et al. (2008). Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with down syndrome: An iBFM-SG study. Blood 111 (3), 1575–1583. doi:10.1182/blood-2007-09-114231
Fujiwara, T. (2017). GATA transcription factors: Basic principles and related human disorders. Tohoku J. Exp. Med. 242 (2), 83–91. doi:10.1620/tjem.242.83
Gamis, A. S., Woods, W. G., Alonzo, T. A., Buxton, A., Lange, B., Barnard, D. R., et al. (2003). Increased age at diagnosis has a significantly negative effect on outcome in children with down syndrome and acute myeloid leukemia: A report from the Children's cancer group study 2891. J. Clin. Oncol. 21 (18), 3415–3422. doi:10.1200/JCO.2003.08.060
Garcia, P., Fernandez-Hernandez, R., Cuadrado, A., Coca, I., Gomez, A., Maqueda, M., et al. (2021). Disruption of NIPBL/Scc2 in Cornelia de Lange Syndrome provokes cohesin genome-wide redistribution with an impact in the transcriptome. Nat. Commun. 12 (1), 4551. doi:10.1038/s41467-021-24808-z
Garnett, C., Cruz Hernandez, D., and Vyas, P. (2020). GATA1 and cooperating mutations in myeloid leukaemia of Down syndrome. IUBMB Life 72 (1), 119–130. doi:10.1002/iub.2197
Ge, Y., LaFiura, K. M., Dombkowski, A. A., Chen, Q., Payton, S. G., Buck, S. A., et al. (2008). The role of the proto-oncogene ETS2 in acute megakaryocytic leukemia biology and therapy. Leukemia 22 (3), 521–529. doi:10.1038/sj.leu.2405066
George, B., Kantarjian, H., Baran, N., Krocker, J. D., and Rios, A. (2021). TP53 in acute myeloid leukemia: Molecular aspects and patterns of mutation. Int. J. Mol. Sci. 22 (19), 10782. doi:10.3390/ijms221910782
Gilliland, D. G., and Griffin, J. D. (2002). The roles of FLT3 in hematopoiesis and leukemia. Blood 100 (5), 1532–1542. doi:10.1182/blood-2002-02-0492
Gonzales, F., Barthélémy, A., Peyrouze, P., Fenwarth, L., Preudhomme, C., Duployez, N., et al. (2021). Targeting RUNX1 in acute myeloid leukemia: Preclinical innovations and therapeutic implications. Expert Opin. Ther. Targets 25 (4), 299–309. doi:10.1080/14728222.2021.1915991
Grafone, T., Palmisano, M., Nicci, C., and Storti, S. (2012). An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 6 (1), e8. doi:10.4081/oncol.2012.e8
Grimm, J., Heckl, D., and Klusmann, J. H. (2021). Molecular mechanisms of the genetic predisposition to acute megakaryoblastic leukemia in infants with down syndrome. Front. Oncol. 11, 636633. doi:10.3389/fonc.2021.636633
Gu, Z., Liu, Y., Cai, F., Patrick, M., Zmajkovic, J., Cao, H., et al. (2019). Loss of EZH2 reprograms BCAA metabolism to drive leukemic transformation. Cancer Discov. 9 (9), 1228–1247. doi:10.1158/2159-8290.CD-19-0152
Hama, A., Yagasaki, H., Takahashi, Y., Nishio, N., Muramatsu, H., Yoshida, N., et al. (2008). Acute megakaryoblastic leukaemia (AMKL) in children: A comparison of AMKL with and without down syndrome. Br. J. Haematol. 140 (5), 552–561. doi:10.1111/j.1365-2141.2007.06971.x
Hamarsheh, S., Osswald, L., Saller, B. S., Unger, S., De Feo, D., Vinnakota, J. M., et al. (2020). Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun. 11 (1), 1659. doi:10.1038/s41467-020-15497-1
Hasle, H., Abrahamsson, J., Arola, M., Karow, A., O'Marcaigh, A., Reinhardt, D., et al. (2008). Myeloid leukemia in children 4 years or older with Down syndrome often lacks GATA1 mutation and cytogenetics and risk of relapse are more akin to sporadic AML. Leukemia 22 (7), 1428–1430. doi:10.1038/sj.leu.2405060
Hasle, H., Kline, R. M., Kjeldsen, E., Nik-Abdul-Rashid, N. F., Bhojwani, D., Verboon, J. M., et al. (2022). Germline GATA1s-generating mutations predispose to leukemia with acquired trisomy 21 and Down syndrome-like phenotype. Blood 139 (21), 3159–3165. doi:10.1182/blood.2021011463
Haupt, S., Mitchell, C., Corneille, V., Shortt, J., Fox, S., Pandolfi, P. P., et al. (2013). Loss of PML cooperates with mutant p53 to drive more aggressive cancers in a gender-dependent manner. Cell Cycle 12 (11), 1722–1731. doi:10.4161/cc.24805
Hitzler, J. K., Cheung, J., Li, Y., Scherer, S. W., and Zipursky, A. (2003). GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11), 4301–4304. doi:10.1182/blood-2003-01-0013
Hong, Q., Li, Y., Chen, X., Ye, H., Tang, L., Zhou, A., et al. (2016). CDKN2B, SLC19A3 and DLEC1 promoter methylation alterations in the bone marrow of patients with acute myeloid leukemia during chemotherapy. Exp. Ther. Med. 11 (5), 1901–1907. doi:10.3892/etm.2016.3092
Ikeda, K., Ueda, T., Yamasaki, N., Nakata, Y., Sera, Y., Nagamachi, A., et al. (2016). Maintenance of the functional integrity of mouse hematopoiesis by EED and promotion of leukemogenesis by EED haploinsufficiency. Sci. Rep. 6, 29454. doi:10.1038/srep29454
Izzo, F., Lee, S. C., Poran, A., Chaligne, R., Gaiti, F., Gross, B., et al. (2020). DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet. 52 (4), 378–387. doi:10.1038/s41588-020-0595-4
Jann, J. C., and Tothova, Z. (2021). Cohesin mutations in myeloid malignancies. Blood 138, 649–661. doi:10.1182/blood.2019004259
Joshi, S. K., Qian, K., Bisson, W. H., Watanabe-Smith, K., Huang, A., Bottomly, D., et al. (2020). Discovery and characterization of targetable NTRK point mutations in hematologic neoplasms. Blood 135 (24), 2159–2170. doi:10.1182/blood.2019003691
Juban, G., Sakakini, N., Chagraoui, H., Cruz Hernandez, D., Cheng, Q., Soady, K., et al. (2021). Oncogenic Gata1 causes stage-specific megakaryocyte differentiation delay. Haematologica 106 (4), 1106–1119. doi:10.3324/haematol.2019.244541
Kanezaki, R., Toki, T., Terui, K., Xu, G., Wang, R., Shimada, A., et al. (2010). Down syndrome and GATA1 mutations in transient abnormal myeloproliferative disorder: Mutation classes correlate with progression to myeloid leukemia. Blood 116 (22), 4631–4638. doi:10.1182/blood-2010-05-282426
Kawara, H., Akahori, R., Wakasugi, M., Sancar, A., and Matsunaga, T. (2019). DCAF7 is required for maintaining the cellular levels of ERCC1-XPF and nucleotide excision repair. Biochem. Biophys. Res. Commun. 519 (1), 204–210. doi:10.1016/j.bbrc.2019.08.147
Kelly, M. J., So, J., Rogers, A. J., Gregory, G., Li, J., Zethoven, M., et al. (2019). Bcor loss perturbs myeloid differentiation and promotes leukaemogenesis. Nat. Commun. 10 (1), 1347. doi:10.1038/s41467-019-09250-6
Kemp, C. J., Moore, J. M., Moser, R., Bernard, B., Teater, M., Smith, L. E., et al. (2014). CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 7 (4), 1020–1029. doi:10.1016/j.celrep.2014.04.004
Kim, T. G., Kim, S., Jung, S., Kim, M., Yang, B., Lee, M. G., et al. (2017). CCCTC-binding factor is essential to the maintenance and quiescence of hematopoietic stem cells in mice. Exp. Mol. Med. 49 (8), e371. doi:10.1038/emm.2017.124
Kiyoi, H., Yamaji, S., Kojima, S., and Naoe, T. (2007). JAK3 mutations occur in acute megakaryoblastic leukemia both in Down syndrome children and non-Down syndrome adults. Leukemia 21 (3), 574–576. doi:10.1038/sj.leu.2404527
Klusmann, J. H., Creutzig, U., Zimmermann, M., Dworzak, M., Jorch, N., Langebrake, C., et al. (2008). Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood 111 (6), 2991–2998. doi:10.1182/blood-2007-10-118810
Klusmann, J. H., Li, Z., Böhmer, K., Maroz, A., Koch, M. L., Emmrich, S., et al. (2010). miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev. 24 (5), 478–490. doi:10.1101/gad.1856210
Klusmann, J. H., Reinhardt, D., Hasle, H., Kaspers, G. J., Creutzig, U., Hahlen, K., et al. (2007). Janus kinase mutations in the development of acute megakaryoblastic leukemia in children with and without Down's syndrome. Leukemia 21 (7), 1584–1587. doi:10.1038/sj.leu.2404694
Labuhn, M., Perkins, K., Matzk, S., Varghese, L., Garnett, C., Papaemmanuil, E., et al. (2019). Mechanisms of progression of myeloid preleukemia to transformed myeloid leukemia in children with down syndrome. Cancer Cell 36 (2), 123–138. e110. doi:10.1016/j.ccell.2019.06.007
Lange, B. J., Kobrinsky, N., Barnard, D. R., Arthur, D. C., Buckley, J. D., Howells, W. B., et al. (1998). Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children's Cancer Group Studies 2861 and 2891. Blood 91 (2), 608–615.
Langebrake, C., Klusmann, J. H., Wortmann, K., Kolar, M., Puhlmann, U., Reinhardt, D., et al. (2006). Concomitant aberrant overexpression of RUNX1 and NCAM in regenerating bone marrow of myeloid leukemia of Down's syndrome. Haematologica 91 (11), 1473–1480.
Laurent, A. P., Kotecha, R. S., and Malinge, S. (2020). Gain of chromosome 21 in hematological malignancies: Lessons from studying leukemia in children with down syndrome. Leukemia 34 (8), 1984–1999. doi:10.1038/s41375-020-0854-5
Letourneau, A., Santoni, F. A., Bonilla, X., Sailani, M. R., Gonzalez, D., Kind, J., et al. (2014). Domains of genome-wide gene expression dysregulation in Down's syndrome. Nature 508 (7496), 345–350. doi:10.1038/nature13200
Lindberg, M. F., and Meijer, L. (2021). Dual-specificity, tyrosine phosphorylation-regulated kinases (DYRKs) and cdc2-like kinases (CLKs) in human disease, an overview. Int. J. Mol. Sci. 22 (11), 6047. doi:10.3390/ijms22116047
Liu, B., Filippi, S., Roy, A., and Roberts, I. (2015). Stem and progenitor cell dysfunction in human trisomies. EMBO Rep. 16 (1), 44–62. doi:10.15252/embr.201439583
Lorenzini, P. A., Chew, R. S. E., Tan, C. W., Yong, J. Y., Zhang, F., Zheng, J., et al. (2019). Human PRPF40B regulates hundreds of alternative splicing targets and represses a hypoxia expression signature. RNA 25 (8), 905–920. doi:10.1261/rna.069534.118
Loscocco, G. G., Guglielmelli, P., and Vannucchi, A. M. (2020). Impact of mutational profile on the management of myeloproliferative neoplasms: A short review of the emerging data. Onco. Targets. Ther. 13, 12367–12382. doi:10.2147/OTT.S287944
Lu, H. Y., Sharma, M., Sharma, A. A., Lacson, A., Szpurko, A., Luider, J., et al. (2021). Mechanistic understanding of the combined immunodeficiency in complete human CARD11 deficiency. J. Allergy Clin. Immunol. 148 (6), 1559–1574. e13.e1513. doi:10.1016/j.jaci.2021.04.006
Magrin, L., Fanale, D., Brando, C., Fiorino, A., Corsini, L. R., Sciacchitano, R., et al. (2021). POLE, POLD1, and NTHL1: The last but not the least hereditary cancer-predisposing genes. Oncogene 40 (40), 5893–5901. doi:10.1038/s41388-021-01984-2
Majewski, I. J., Blewitt, M. E., de Graaf, C. A., McManus, E. J., Bahlo, M., Hilton, A. A., et al. (2008). Polycomb repressive complex 2 (PRC2) restricts hematopoietic stem cell activity. PLoS Biol. 6 (4), e93. doi:10.1371/journal.pbio.0060093
Malinge, S., Bliss-Moreau, M., Kirsammer, G., Diebold, L., Chlon, T., Gurbuxani, S., et al. (2012). Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J. Clin. Invest. 122 (3), 948–962. doi:10.1172/JCI60455
Malinge, S., Chlon, T., Doré, L. C., Ketterling, R. P., Tallman, M. S., Paietta, E., et al. (2013). Development of acute megakaryoblastic leukemia in Down syndrome is associated with sequential epigenetic changes. Blood 122 (14), e33–43. doi:10.1182/blood-2013-05-503011
Malinge, S., Ragu, C., Della-Valle, V., Pisani, D., Constantinescu, S. N., Perez, C., et al. (2008). Activating mutations in human acute megakaryoblastic leukemia. Blood 112 (10), 4220–4226. doi:10.1182/blood-2008-01-136366
Man, N., Mas, G., Karl, D. L., Sun, J., Liu, F., Yang, Q., et al. (2021). p300 suppresses the transition of myelodysplastic syndromes to acute myeloid leukemia. JCI Insight 6 (19), e138478. doi:10.1172/jci.insight.138478
Manukjan, G., Ripperger, T., Santer, L., von Neuhoff, N., Ganser, A., Schambach, A., et al. (2015). Expression of the ETS transcription factor GABPα is positively correlated to the BCR-ABL1/ABL1 ratio in CML patients and affects imatinib sensitivity in vitro. Exp. Hematol. 43 (10), 880–890. doi:10.1016/j.exphem.2015.05.011
Maslah, N., Cassinat, B., Verger, E., Kiladjian, J. J., and Velazquez, L. (2017). The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 31 (8), 1661–1670. doi:10.1038/leu.2017.139
Massey, G. V., Zipursky, A., Chang, M. N., Doyle, J. J., Nasim, S., Taub, J. W., et al. (2006). A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12), 4606–4613. doi:10.1182/blood-2005-06-2448
Matsuo, S., Nishinaka-Arai, Y., Kazuki, Y., Oshimura, M., Nakahata, T., Niwa, A., et al. (2021). Pluripotent stem cell model of early hematopoiesis in Down syndrome reveals quantitative effects of short-form GATA1 protein on lineage specification. PLoS One 16 (3), e0247595. doi:10.1371/journal.pone.0247595
Maurya, S., Yang, W., Tamai, M., Zhang, Q., Erdmann-Gilmore, P., Bystry, A., et al. (2021). Loss of KMT2C reprograms the epigenomic landscape in hPSCs resulting in NODAL overexpression and a failure of hemogenic endothelium specification. Epigenetics 17, 220–238. doi:10.1080/15592294.2021.1954780
Maxson, J. E., and Tyner, J. W. (2017). Genomics of chronic neutrophilic leukemia. Blood 129 (6), 715–722. doi:10.1182/blood-2016-10-695981
Mazzi, S., Dessen, P., Vieira, M., Dufour, V., Cambot, M., El Khoury, M., et al. (2021). Dual role of EZH2 in megakaryocyte differentiation. Blood 138 (17), 1603–1614. doi:10.1182/blood.2019004638
Mazzola, M., Deflorian, G., Pezzotta, A., Ferrari, L., Fazio, G., Bresciani, E., et al. (2019a). Nipbl: A new player in myeloid cell differentiation. Haematologica 104 (7), 1332–1341. doi:10.3324/haematol.2018.200899
Mazzola, M., Deflorian, G., Pezzotta, A., Ferrari, L., Fazio, G., Bresciani, E., et al. (2019b). Nipbl: A new player in myeloid cell differentiation. Haematologica 104 (7), 1332–1341. doi:10.3324/haematol.2018.200899
Mazzola, M., Pezzotta, A., Fazio, G., Rigamonti, A., Bresciani, E., Gaudenzi, G., et al. (2020). Dysregulation of NIPBL leads to impaired RUNX1 expression and haematopoietic defects. J. Cell. Mol. Med. 24 (11), 6272–6282. doi:10.1111/jcmm.15269
Moles, R., Bai, X. T., Chaib-Mezrag, H., and Nicot, C. (2016). WRN-targeted therapy using inhibitors NSC 19630 and NSC 617145 induce apoptosis in HTLV-1-transformed adult T-cell leukemia cells. J. Hematol. Oncol. 9 (1), 121. doi:10.1186/s13045-016-0352-4
Moser, B., Edtmayer, S., Witalisz-Siepracka, A., and Stoiber, D. (2021). The ups and downs of STAT inhibition in acute myeloid leukemia. Biomedicines 9 (8), 1051. doi:10.3390/biomedicines9081051
Muffels, I. J. J., Wiame, E., Fuchs, S. A., Massink, M. P. G., Rehmann, H., Musch, J. L. I., et al. (2021). NAA80 bi-allelic missense variants result in high-frequency hearing loss, muscle weakness and developmental delay. Brain Commun. 3 (4), fcab256. doi:10.1093/braincomms/fcab256
Mujahed, H., Miliara, S., Neddermeyer, A., Bengtzén, S., Nilsson, C., Deneberg, S., et al. (2020). AML displays increased CTCF occupancy associated with aberrant gene expression and transcription factor binding. Blood 136 (3), 339–352. doi:10.1182/blood.2019002326
Muskens, I. S., Li, S., Jackson, T., Elliot, N., Hansen, H. M., Myint, S. S., et al. (2021). The genome-wide impact of trisomy 21 on DNA methylation and its implications for hematopoiesis. Nat. Commun. 12 (1), 821. doi:10.1038/s41467-021-21064-z
Nagase, R., Inoue, D., Pastore, A., Fujino, T., Hou, H. A., Yamasaki, N., et al. (2018). Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 215 (6), 1729–1747. doi:10.1084/jem.20171151
Nakamura-Ishizu, A., and Suda, T. (2020). Multifaceted roles of thrombopoietin in hematopoietic stem cell regulation. Ann. N. Y. Acad. Sci. 1466 (1), 51–58. doi:10.1111/nyas.14169
Nie, Z., Gao, W., Zhang, Y., Hou, Y., Liu, J., Li, Z., et al. (2019). STAG2 loss-of-function mutation induces PD-L1 expression in U2OS cells. Ann. Transl. Med. 7 (7), 127. doi:10.21037/atm.2019.02.23
Nikolaev, S. I., Santoni, F., Vannier, A., Falconnet, E., Giarin, E., Basso, G., et al. (2013). Exome sequencing identifies putative drivers of progression of transient myeloproliferative disorder to AMKL in infants with Down syndrome. Blood 122 (4), 554–561. doi:10.1182/blood-2013-03-491936
Nishinaka-Arai, Y., Niwa, A., Matsuo, S., Kazuki, Y., Yakura, Y., Hiroma, T., et al. (2021). Down syndrome-related transient abnormal myelopoiesis is attributed to a specific erythro-megakaryocytic subpopulation with GATA1 mutation. Haematologica 106 (2), 635–640. doi:10.3324/haematol.2019.242693
Ntziachristos, P., Tsirigos, A., Van Vlierberghe, P., Nedjic, J., Trimarchi, T., Flaherty, M. S., et al. (2012). Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 18 (2), 298–301. doi:10.1038/nm.2651
Ochi, Y., Kon, A., Sakata, T., Nakagawa, M. M., Nakazawa, N., Kakuta, M., et al. (2020). Combined cohesin-RUNX1 deficiency synergistically perturbs chromatin looping and causes myelodysplastic syndromes. Cancer Discov. 10 (6), 836–853. doi:10.1158/2159-8290.CD-19-0982
Ono, R., Hasegawa, D., Hirabayashi, S., Kamiya, T., Yoshida, K., Yonekawa, S., et al. (2015). Acute megakaryoblastic leukemia with acquired trisomy 21 and GATA1 mutations in phenotypically normal children. Eur. J. Pediatr. 174 (4), 525–531. doi:10.1007/s00431-014-2430-3
Padmakumar, D., Chandraprabha, V. R., Gopinath, P., Vimala Devi, A. R. T., Anitha, G. R. J., Sreelatha, M. M., et al. (2021). A concise review on the molecular genetics of acute myeloid leukemia. Leuk. Res. 111, 106727. doi:10.1016/j.leukres.2021.106727
Pandey, R., Saxena, M., and Kapur, R. (2017). Role of SHP2 in hematopoiesis and leukemogenesis. Curr. Opin. Hematol. 24 (4), 307–313. doi:10.1097/MOH.0000000000000345
Panferova, A., Gaskova, M., Nikitin, E., Baryshev, P., Timofeeva, N., Kazakova, A., et al. (2021). GATA1 mutation analysis and molecular landscape characterization in acute myeloid leukemia with trisomy 21 in pediatric patients. Int. J. Lab. Hematol. 43 (4), 713–723. doi:10.1111/ijlh.13451
Park, H. J., Li, J., Hannah, R., Biddie, S., Leal-Cervantes, A. I., Kirschner, K., et al. (2016). Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 35 (6), 580–594. doi:10.15252/embj.201592383
Pevny, L., Simon, M. C., Robertson, E., Klein, W. H., Tsai, S. F., D'Agati, V., et al. (1991). Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349 (6306), 257–260. doi:10.1038/349257a0
Pine, S. R., Guo, Q., Yin, C., Jayabose, S., Druschel, C. M., Sandoval, C., et al. (2007). Incidence and clinical implications of GATA1 mutations in newborns with Down syndrome. Blood 110 (6), 2128–2131. doi:10.1182/blood-2007-01-069542
Polachek, W. S., Moshrif, H. F., Franti, M., Coen, D. M., Sreenu, V. B., Strang, B. L., et al. (2016). High-throughput small interfering RNA screening identifies phosphatidylinositol 3-kinase class II alpha as important for production of human cytomegalovirus virions. J. Virol. 90 (18), 8360–8371. doi:10.1128/JVI.01134-16
Rainis, L., Toki, T., Pimanda, J. E., Rosenthal, E., Machol, K., Strehl, S., et al. (2005). The proto-oncogene ERG in megakaryoblastic leukemias. Cancer Res. 65 (17), 7596–7602. doi:10.1158/0008-5472.CAN-05-0147
Reymond, A., Friedli, M., Henrichsen, C. N., Chapot, F., Deutsch, S., Ucla, C., et al. (2001). From PREDs and open reading frames to cDNA isolation: Revisiting the human chromosome 21 transcription map. Genomics 78 (1-2), 46–54. doi:10.1006/geno.2001.6640
Rivas, M. A., Meydan, C., Chin, C. R., Challman, M. F., Kim, D., Bhinder, B., et al. (2021). Smc3 dosage regulates B cell transit through germinal centers and restricts their malignant transformation. Nat. Immunol. 22 (2), 240–253. doi:10.1038/s41590-020-00827-8
Roberts, I., Alford, K., Hall, G., Juban, G., Richmond, H., Norton, A., et al. (2013). GATA1-mutant clones are frequent and often unsuspected in babies with down syndrome: Identification of a population at risk of leukemia. Blood 122 (24), 3908–3917. doi:10.1182/blood-2013-07-515148
Saida, S., Watanabe, K., Sato-Otsubo, A., Terui, K., Yoshida, K., Okuno, Y., et al. (2013). Clonal selection in xenografted TAM recapitulates the evolutionary process of myeloid leukemia in Down syndrome. Blood 121 (21), 4377–4387. doi:10.1182/blood-2012-12-474387
Salek-Ardakani, S., Smooha, G., de Boer, J., Sebire, N. J., Morrow, M., Rainis, L., et al. (2009). ERG is a megakaryocytic oncogene. Cancer Res. 69 (11), 4665–4673. doi:10.1158/0008-5472.CAN-09-0075
Sas, V., Pasca, S., Jurj, A., Pop, L., Muramatsu, H., Ono, H., et al. (2020). MicroRNA-155-5p plays a critical role in transient leukemia of down syndrome by targeting tumor necrosis factor receptor superfamily members. Cell. Physiol. biochem. 54 (5), 994–1012. doi:10.33594/000000283
Sasine, J. P., Himburg, H. A., Termini, C. M., Roos, M., Tran, E., Zhao, L., et al. (2018). Wild-type Kras expands and exhausts hematopoietic stem cells. JCI Insight 3 (11), 98197. doi:10.1172/jci.insight.98197
Shaham, L., Vendramini, E., Ge, Y., Goren, Y., Birger, Y., Tijssen, M. R., et al. (2015). MicroRNA-486-5p is an erythroid oncomiR of the myeloid leukemias of Down syndrome. Blood 125 (8), 1292–1301. doi:10.1182/blood-2014-06-581892
Shi, X., Yang, Y., Shang, S., Wu, S., Zhang, W., Peng, L., et al. (2019). Cooperation of Dnmt3a R878H with nras G12D promotes leukemogenesis in knock-in mice: A pilot study. BMC Cancer 19 (1), 1072. doi:10.1186/s12885-019-6207-y
Shimizu, R., Kobayashi, E., Engel, J. D., and Yamamoto, M. (2009). Induction of hyperproliferative fetal megakaryopoiesis by an N-terminally truncated GATA1 mutant. Genes cells. 14 (9), 1119–1131. doi:10.1111/j.1365-2443.2009.01338.x
Shukla, S., Kavak, E., Gregory, M., Imashimizu, M., Shutinoski, B., Kashlev, M., et al. (2011). CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479 (7371), 74–79. doi:10.1038/nature10442
Si, X., Zhang, X., Hao, X., Li, Y., Chen, Z., Ding, Y., et al. (2016). Upregulation of miR-99a is associated with poor prognosis of acute myeloid leukemia and promotes myeloid leukemia cell expansion. Oncotarget 7 (47), 78095–78109. doi:10.18632/oncotarget.12947
Singh, A., Mandal, A., Guru, V., Srinivasan, S., and Seth, R. (2017). Transient abnormal myelopoiesis: A varied spectrum of clinical presentation. J. Hematol. 6 (1), 25–28. doi:10.14740/jh306w
Singh, A., Mencia-Trinchant, N., Griffiths, E. A., Altahan, A., Swaminathan, M., Gupta, M., et al. (2022). Mutant PPM1D- and TP53-driven hematopoiesis populates the hematopoietic compartment in response to peptide receptor radionuclide therapy. JCO Precis. Oncol. 6, e2100309. doi:10.1200/PO.21.00309
Song, L., Jiang, W., Liu, W., Ji, J. H., Shi, T. F., Zhang, J., et al. (2016). Protein tyrosine phosphatases receptor type D is a potential tumour suppressor gene inactivated by deoxyribonucleic acid methylation in paediatric acute myeloid leukaemia. Acta Paediatr. 105 (3), e132–141. doi:10.1111/apa.13284
Soukup, A. A., and Bresnick, E. H. (2020). GATA2 +9.5 enhancer: From principles of hematopoiesis to genetic diagnosis in precision medicine. Curr. Opin. Hematol. 27 (3), 163–171. doi:10.1097/MOH.0000000000000576
Stankiewicz, M. J., and Crispino, J. D. (2013). AKT collaborates with ERG and Gata1s to dysregulate megakaryopoiesis and promote AMKL. Leukemia 27 (6), 1339–1347. doi:10.1038/leu.2013.33
Stankiewicz, M. J., and Crispino, J. D. (2009). ETS2 and ERG promote megakaryopoiesis and synergize with alterations in GATA-1 to immortalize hematopoietic progenitor cells. Blood 113 (14), 3337–3347. doi:10.1182/blood-2008-08-174813
Stankov, K., Popovic, S., and Mikov, M. (2014). C-KIT signaling in cancer treatment. Curr. Pharm. Des. 20 (17), 2849–2880. doi:10.2174/13816128113199990593
Sureau, L., Orvain, C., Ianotto, J. C., Ugo, V., Kiladjian, J. J., Luque Paz, D., et al. (2021). Efficacy and tolerability of janus kinase inhibitors in myelofibrosis: A systematic review and network meta-analysis. Blood Cancer J. 11 (7), 135. doi:10.1038/s41408-021-00526-z
Sweeney, T. R., Dhote, V., Guca, E., Hellen, C. U. T., Hashem, Y., Pestova, T. V., et al. (2021). Functional role and ribosomal position of the unique N-terminal region of DHX29, a factor required for initiation on structured mammalian mRNAs. Nucleic Acids Res. 49 (22), 12955–12969. doi:10.1093/nar/gkab1192
Thoms, J. A. I., Truong, P., Subramanian, S., Knezevic, K., Harvey, G., Huang, Y., et al. (2021). Disruption of a GATA2-TAL1-ERG regulatory circuit promotes erythroid transition in healthy and leukemic stem cells. Blood 138 (16), 1441–1455. doi:10.1182/blood.2020009707
Tian, L., Chavez, M., Chang, G. S., Helton, N. M., Katerndahl, C. D. S., Miller, C. A., et al. (2021). Kdm6a deficiency restricted to mouse hematopoietic cells causes an age- and sex-dependent myelodysplastic syndrome-like phenotype. PLoS One 16 (11), e0255706. doi:10.1371/journal.pone.0255706
Todisco, G., Creignou, M., Gallì, A., Guglielmelli, P., Rumi, E., Roncador, M., et al. (2021). Co-mutation pattern, clonal hierarchy, and clone size concur to determine disease phenotype of SRSF2P95-mutated neoplasms. Leukemia 35 (8), 2371–2381. doi:10.1038/s41375-020-01106-z
Toki, T., Katsuoka, F., Kanezaki, R., Xu, G., Kurotaki, H., Sun, J., et al. (2005). Transgenic expression of BACH1 transcription factor results in megakaryocytic impairment. Blood 105 (8), 3100–3108. doi:10.1182/blood-2004-07-2826
Torrano, V., Chernukhin, I., Docquier, F., D'Arcy, V., León, J., Klenova, E., et al. (2005). CTCF regulates growth and erythroid differentiation of human myeloid leukemia cells. J. Biol. Chem. 280 (30), 28152–28161. doi:10.1074/jbc.M501481200
Tsai, M. H., Hou, J. W., Yang, C. P., Yang, P. H., Chu, S. M., Hsu, J. F., et al. (2011). Transient myeloproliferative disorder and GATA1 mutation in neonates with and without Down syndrome. Indian J. Pediatr. 78 (7), 826–832. doi:10.1007/s12098-010-0312-x
Uffmann, M., Rasche, M., Zimmermann, M., von Neuhoff, C., Creutzig, U., Dworzak, M., et al. (2017). Therapy reduction in patients with down syndrome and myeloid leukemia: The international ML-DS 2006 trial. Blood 129 (25), 3314–3321. doi:10.1182/blood-2017-01-765057
Van Der Werf, I., Wojtuszkiewicz, A., Yao, H., Sciarrillo, R., Meggendorfer, M., Hutter, S., et al. (2021). SF3B1 as therapeutic target in FLT3/ITD positive acute myeloid leukemia. Leukemia 35 (9), 2698–2702. doi:10.1038/s41375-021-01273-7
Vara, N., Liu, Y., Yan, Y., Lensing, S. Y., Colorado, N., Robinson, D., et al. (2020). Sustained fetal hematopoiesis causes juvenile death from leukemia: Evidence from a dual-age-specific mouse model. Blood Adv. 4 (15), 3728–3740. doi:10.1182/bloodadvances.2020002326
Viny, A. D., Bowman, R. L., Liu, Y., Lavallée, V. P., Eisman, S. E., Xiao, W., et al. (2019). Cohesin members Stag1 and Stag2 display distinct roles in chromatin accessibility and topological control of HSC self-renewal and differentiation. Cell Stem Cell 25 (5), 682–696. e688. doi:10.1016/j.stem.2019.08.003
Volk, A., Liang, K., Suraneni, P., Li, X., Zhao, J., Bulic, M., et al. (2018). A CHAF1B-dependent molecular switch in hematopoiesis and leukemia pathogenesis. Cancer Cell 34 (5), 707–723. e707. doi:10.1016/j.ccell.2018.10.004
Vukadin, L., Kim, J. H., Park, E. Y., Stone, J. K., Ungerleider, N., Baddoo, M. C., et al. (2021). SON inhibits megakaryocytic differentiation via repressing RUNX1 and the megakaryocytic gene expression program in acute megakaryoblastic leukemia. Cancer Gene Ther. 28 (9), 1000–1015. doi:10.1038/s41417-020-00262-9
Wagenblast, E., Araújo, J., Gan, O. I., Cutting, S. K., Murison, A., Krivdova, G., et al. (2021). Mapping the cellular origin and early evolution of leukemia in Down syndrome. Science 373 (6551), eabf6202. doi:10.1126/science.abf6202
Walters, D. K., Mercher, T., Gu, T. L., O'Hare, T., Tyner, J. W., Loriaux, M., et al. (2006). Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell 10 (1), 65–75. doi:10.1016/j.ccr.2006.06.002
Wang, A. J., Han, Y., Jia, N., Chen, P., and Minden, M. D. (2020). NPM1c impedes CTCF functions through cytoplasmic mislocalization in acute myeloid leukemia. Leukemia 34 (5), 1278–1290. doi:10.1038/s41375-019-0681-8
Wang, T., Glover, B., Hadwiger, G., Miller, C. A., di Martino, O., Welch, J. S., et al. (2019). Smc3 is required for mouse embryonic and adult hematopoiesis. Exp. Hematol. 70, 70–84. e76. doi:10.1016/j.exphem.2018.11.008
Wechsler, J., Greene, M., McDevitt, M. A., Anastasi, J., Karp, J. E., Le Beau, M. M., et al. (2002). Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat. Genet. 32 (1), 148–152. doi:10.1038/ng955
Wu, Y., Zhu, H., and Wu, H. (2020). PTEN in regulating hematopoiesis and leukemogenesis. Cold Spring Harb. Perspect. Med. 10 (10), a036244. doi:10.1101/cshperspect.a036244
Xu, G., Kato, K., Toki, T., Takahashi, Y., Terui, K., Ito, E., et al. (2006). Development of acute megakaryoblastic leukemia from a minor clone in a Down syndrome patient with clinically overt transient myeloproliferative disorder. J. Pediatr. Hematol. Oncol. 28 (10), 696–698. doi:10.1097/01.mph.0000212997.02554.f6
Xu, N., Donohoe, M. E., Silva, S. S., and Lee, J. T. (2007). Evidence that homologous X-chromosome pairing requires transcription and Ctcf protein. Nat. Genet. 39 (11), 1390–1396. doi:10.1038/ng.2007.5
Yan, F., Li, J., Milosevic, J., Petroni, R., Liu, S., Shi, Z., et al. (2021). KAT6A and ENL form an epigenetic transcriptional control module to drive critical leukemogenic gene expression programs. Cancer Discov. 12, 792–811. doi:10.1158/2159-8290.CD-20-1459
Yanagida, M., Osato, M., Yamashita, N., Liqun, H., Jacob, B., Wu, F., et al. (2005). Increased dosage of Runx1/AML1 acts as a positive modulator of myeloid leukemogenesis in BXH2 mice. Oncogene 24 (28), 4477–4485. doi:10.1038/sj.onc.1208675
Yang, Z. F., Zhang, H., Ma, L., Peng, C., Chen, Y., Wang, J., et al. (2013). GABP transcription factor is required for development of chronic myelogenous leukemia via its control of PRKD2. Proc. Natl. Acad. Sci. U. S. A. 110 (6), 2312–2317. doi:10.1073/pnas.1212904110
Yoshida, K., Toki, T., Okuno, Y., Kanezaki, R., Shiraishi, Y., Sato-Otsubo, A., et al. (2013). The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. 45 (11), 1293–1299. doi:10.1038/ng.2759
Yousefelahiyeh, M., Xu, J., Alvarado, E., Yu, Y., Salven, D., Nissen, R. M., et al. (2018). DCAF7/WDR68 is required for normal levels of DYRK1A and DYRK1B. PLoS One 13 (11), e0207779. doi:10.1371/journal.pone.0207779
Yuzawa, K., Terui, K., Toki, T., Kanezaki, R., Kobayashi, A., Sato, T., et al. (2020). Clinical, cytogenetic, and molecular analyses of 17 neonates with transient abnormal myelopoiesis and nonconstitutional trisomy 21. Pediatr. Blood Cancer 67 (4), e28188. doi:10.1002/pbc.28188
Zafar, N., Ghias, K., and Fadoo, Z. (2021). Genetic aberrations involved in relapse of pediatric acute myeloid leukemia: A literature review. Asia. Pac. J. Clin. Oncol. 17 (5), e135–e141. doi:10.1111/ajco.13367
Zampieri, M., Guastafierro, T., Calabrese, R., Ciccarone, F., Bacalini, M. G., Reale, A., et al. (2012). ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem. J. 441 (2), 645–652. doi:10.1042/BJ20111417
Zaslavsky, A., Chou, S. T., Schadler, K., Lieberman, A., Pimkin, M., Kim, Y. J., et al. (2013). The calcineurin-NFAT pathway negatively regulates megakaryopoiesis. Blood 121 (16), 3205–3215. doi:10.1182/blood-2012-04-421172
Zeisig, B. B., and So, C. W. E. (2021). Therapeutic opportunities of targeting canonical and noncanonical PcG/TrxG functions in acute myeloid leukemia. Annu. Rev. Genomics Hum. Genet. 22, 103–125. doi:10.1146/annurev-genom-111120-102443
Zhang, L., Li, X., Ke, Z., Huang, L., Liang, Y., Wu, J., et al. (2013). MiR-99a may serve as a potential oncogene in pediatric myeloid leukemia. Cancer Cell Int. 13 (1), 110. doi:10.1186/1475-2867-13-110
Zhang, W., Berthelet, J., Michail, C., Bui, L. C., Gou, P., Liu, R., et al. (2021). Human CREBBP acetyltransferase is impaired by etoposide quinone, an oxidative and leukemogenic metabolite of the anticancer drug etoposide through modification of redox-sensitive zinc-finger cysteine residues. Free Radic. Biol. Med. 162, 27–37. doi:10.1016/j.freeradbiomed.2020.11.027
Zhang, Y., Taylor, B. R., Shannon, K., and Clapp, D. W. (2001). Quantitative effects of Nf1 inactivation on in vivo hematopoiesis. J. Clin. Invest. 108 (5), 709–715. doi:10.1172/JCI12758
Zhou, J., Wu, H., Guo, C., Li, B., Zhou, L. L., Liang, A. B., et al. (2021). A comprehensive genome-wide analysis of long non-coding RNA and mRNA expression profiles of JAK2V617F-positive classical myeloproliferative neoplasms. Bioengineered 12 (2), 10564–10586. doi:10.1080/21655979.2021.2000226
Zhuo, M. Q., Pan, Y. X., Wu, K., Xu, Y. H., and Luo, Z. (2017). Characterization and mechanism of phosphoinositide 3-kinases (PI3Ks) members in insulin-induced changes of protein metabolism in yellow catfish Pelteobagrus fulvidraco. Gen. Comp. Endocrinol. 247, 34–45. doi:10.1016/j.ygcen.2017.04.002
Zimmermannova, O., Doktorova, E., Stuchly, J., Kanderova, V., Kuzilkova, D., Strnad, H., et al. (2017). An activating mutation of GNB1 is associated with resistance to tyrosine kinase inhibitors in ETV6-ABL1-positive leukemia. Oncogene 36 (43), 5985–5994. doi:10.1038/onc.2017.210
Keywords: leukemia, acute myeloid leukemia, acute megakaryoblastic leukemia, transient abnormal myelopoiesis, Down syndrome, epigenetics, genomics, GATA1 mutations
Citation: Li J and Kalev-Zylinska ML (2022) Advances in molecular characterization of myeloid proliferations associated with Down syndrome. Front. Genet. 13:891214. doi: 10.3389/fgene.2022.891214
Received: 07 March 2022; Accepted: 11 July 2022;
Published: 10 August 2022.
Edited by:
Mee-Hyun Lee, Dongshin University, South KoreaReviewed by:
Xujie Zhao, St. Jude Children’s Research Hospital, United StatesSonali P. Barwe, Alfred I. duPont Hospital for Children, United States
Copyright © 2022 Li and Kalev-Zylinska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jixia Li, ai5saUBhdWNrbGFuZC5hYy5ueg==; Maggie L. Kalev-Zylinska, bS5rYWxldkBhdWNrbGFuZC5hYy5ueg==
†ORCID: Jixia Li, orcid.org/0000-0003-2218-5563; Maggie L. Kalev-Zylinska, orcid.org/0000-0001-8378-8048