 Rui Dong1,2†
Rui Dong1,2† Xuxia Wei3,2†
Xuxia Wei3,2† Kaihui Zhang1,2
Kaihui Zhang1,2 Fengling Song4,2Yuqiang Lv1,2
Fengling Song4,2Yuqiang Lv1,2 Min Gao1,2
Min Gao1,2 Dong Wang1,2
Dong Wang1,2 Jian Ma1,2
Jian Ma1,2 Zhongtao Gai1,2*
Zhongtao Gai1,2* Yi Liu1,2*
Yi Liu1,2*- 1Pediatric Research Institute, Children’s Hospital Affiliated to Shandong University (Jinan Children’s Hospital), Jinan, China
- 2Shandong Provincial Clinical Research Center for Children’s Health and Disease, Jinan, China
- 3Gastroenterology, Children’s Hospital Affiliated to Shandong University (Jinan Children’s Hospital), Jinan, China
- 4Children’s Health Department, Children’s Hospital Affiliated to Shandong University (Jinan Children’s Hospital), Jinan, China
Background: Glycogen storage diseases (GSDs) are known as a group of disorders characterized by genetic errors leading to accumulation of glycogen in various tissues. Since different types of GSD can sometimes be clinically indistinguishable, next generation sequencing is becoming a powerful tool for clinical diagnosis.
Methods: 12 patients with suspected GSDs and their parents were enrolled in this study. The clinical and laboratory data of the patients were reviewed. Causative gene variants were identified in the patients using whole exome sequencing (WES) and verified by Sanger sequencing.
Results: Genetic testing and analysis showed that 7 patients were diagnosed with GSD II (Pompe disease), 2 patients with GSD III, 1 patient with GSD VI, and 2 patients with GSD IXα. A total number of 18 variants were identified in 12 patients including 11 variants in GAA gene, 3 variants in AGL gene, 2 variants in PYGL gene and 2 variants in PHKA2 gene, of which 9 variants were reported and 9 variants were novel. SIFT, Polyphen-2, Mutation Taster, and REVEL predicted the novel variants (except GAA c.1052_1075 + 47del) to be disease-causing. The 3D structures of wild/mutant type GAA protein were predicted indicating that variants p. Trp621Gly, p. Pro541Leu, p. Ser800Ile and p. Gly293Trp might affect the proteins function via destroying hydrogen bonds or conformational constraints. Neither liver size nor laboratory findings allow for a differentiation among GSD III, GSD VI and GSD IXα.
Conclusion: Our study expanded the variation spectrum of genes associated with GSDs. WES, in combination with clinical, biochemical, and pathological hallmarks, could provide accurate results for diagnosing and sub-typing GSD and related diseases in clinical setting.
Introduction
Glycogen storage diseases (GSDs) as the innate defects of carbohydrate metabolism are caused by enzyme deficiency in the glycogenolysis or gluconeogenesis (Liang et al., 2020). There are 19 types of GSD, named in Roman numerals from 0 to XV (GSD I was divided into 4 subtypes numbered a-d), classified by enzyme deficiency and affected tissue. In aggregate, GSDs are not considered rare diseases, but every subtype of the disorder are (Molares-Vila et al., 2021). The overall estimated GSD incidence is 1 case per 10,000 live births (Beyzaei et al., 2020). Like most metabolic diseases, the vast majority of GSDs are inherited in an autosomal recessive (AR) pattern, except for X-linked type IXα (Brown et al., 2015; Szymanska et al., 2021). In GSD Ⅱ, the symptoms are caused by mutations in GAA gene and in GSD III, AGL gene. In GSD VI, the clinical manifestations are associated with mutations in PYGL gene and in GSD IXα, PHKA1 and PHKA2 gene (De Vito et al., 2019; Kim et al., 2020; Perveen et al., 2020; Grunert et al., 2021a).
GSDs are a group of clinically and genetically heterogenous diseases. The clinical manifestations of GSDs range from almost no symptoms to severe cardiac dysfunction, respiratory insufficiency, and sudden death (Molares-Vila et al., 2021). GSDs are divided into hepatic, muscle and mixed types according to glycogen accumulation in tissues (Lu et al., 2016; Zhao et al., 2019; Eghbali et al., 2020; Szymanska et al., 2021). The basic clinical symptoms of hepatic GSD are hypoglycemia and hepatomegaly. The characteristic feature of muscle GSD is progressive muscle pathology, including exercise-induced muscle weakness. In both types of GSD, there is also hypertransaminasemia. The different types of GSD are sometimes clinically indistinguishable, especially in hepatic GSDs (Choi et al., 2016). Early diagnosis and effective treatment of GSDs is necessary to prevent adverse outcomes. Clinical signs and symptoms and laboratory parameters such as hypoglycemia, hypertransaminasemia and increased triglycerides level are helpful in GSDs diagnosis. Liver biopsy is an invasive test to assess liver cell enzyme activity. Gene sequencing and genetic analysis, a technology that can provide precise diagnosis and procreation guidance and are more acceptable to patients, is increasingly used in the diagnosis of genetic diseases, including GSD (Shen and Shi, 2019; Fang et al., 2021; Huang et al., 2021; Li et al., 2021; Szymanska et al., 2021).
In this study, by clinical and next-generation sequencing analysis of 12 patients with suspected GSDs, we aimed to give them molecular diagnosis of GSD and determine the spectrum of genetic analysis specific to our cohort. As a result, we found 18 variants in GAA, AGL, PHKA2 and PYGL gene related to GSD and the 12 patients were diagnosed with GSD types II, III, VI, and IXα respectively.
Materials and methods
Study cohort
A total of 12 hospitalized children (6 female, 6 male) in unrelated families with clinically suspected GSD were recruited for this study in Children’s Hospital Affiliated to Shandong University (Jinan Children’s Hospital) from November 2018 to October 2021. All patients from the Han Chinese population in Shandong Province, China were examined and diagnosed by experienced pediatric specialists from the Neurology or Gastroenterology Department. The clinical records of 12 patients with GSD were retrospectively reviewed.
Genetic analysis
Blood samples were obtained from the patients and their parents, and DNA was extracted form peripheral blood leukocytes using QIAamp DNA Blood Midi Kit (Qiagen, Shanghai, China). Mutation screening of the patients was applied by Whole exome sequencing (WES) with the Human Exome Probes P039Exome (MyGenostics, Beijing, China) on the Illumina NovaSeq5000 platform (Illumina, United States). The obtained mean exome coverage was more than 95% (>10X coverage; mean depth of over 100X). Paired-end alignment was performed with version GRCh37/hg19 of the human genome on Burrows-Wheeler Aligner software (BWA Version: 0.7.10). Variant frequencies were determined in thousands of genomes (1000 Genomes, http://www.1000genomes.org), ExAC (http://exac.broadinstitute.org/), Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS) and in-house database to remove common variants (suballelic frequency >5%). Frameshift variants, missense variants, premature stop-gain, initiation codon loss, and typical splicing site changes were prioritized. Effect on protein function of variations were predicted by SIFT, PolyPhen-2, MutationTaster, and REVEL. Descriptions of variants in Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk) and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) also were referenced. Swiss-Pdb Viewer 4.1 software was utilized to generate the 3D protein plots of wild-type and mutant type for novel missense variants. The variants identified in this study were classified according to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015).
Validation of gene mutations
The target variants screened from WES were verified by Sanger sequencing in parents and probands using ABI Prism 3700 automated sequencer (Applied Biosystems, Foster City, CA).
Statistical analysis
Statistical analysis was performed on SPSS 24.0 software (SPSS Inc. Chicago IL, USA). The Median and Interquartile range (IQR) of onset age and diagnosis age was calculated. The Shapiro–Wilk test was used to test for normality of the data. For data with non-normal distribution, Kruskal-Wallis H test was used for multiple group comparisons and Mann-Whitney U test for two group. P < 0.05 (two-tailed) were considered statistically significant.
Results
Clinical manifestations and laboratory tests
Clinical details were available for 12 patients. The age, sex, physical examination and laboratory findings at the time of diagnosis were recorded and showed in Table 1. The average age of onset was 11.5 ± 36.5 months, and the age of diagnosis was 22.5 ± 38.5 months. The initial symptoms of patients in our cohort were hypertransaminases, motor retardation due to muscle weakness, hypertrophic cardiomyopathy, and abdominal distension. Patients 1-7 had elevated liver enzymes and muscle weakness and were classified to mixed GSD, while patients 8–12 had only a liver-related phenotype without muscle involvement and were classified to hepatic GSD.
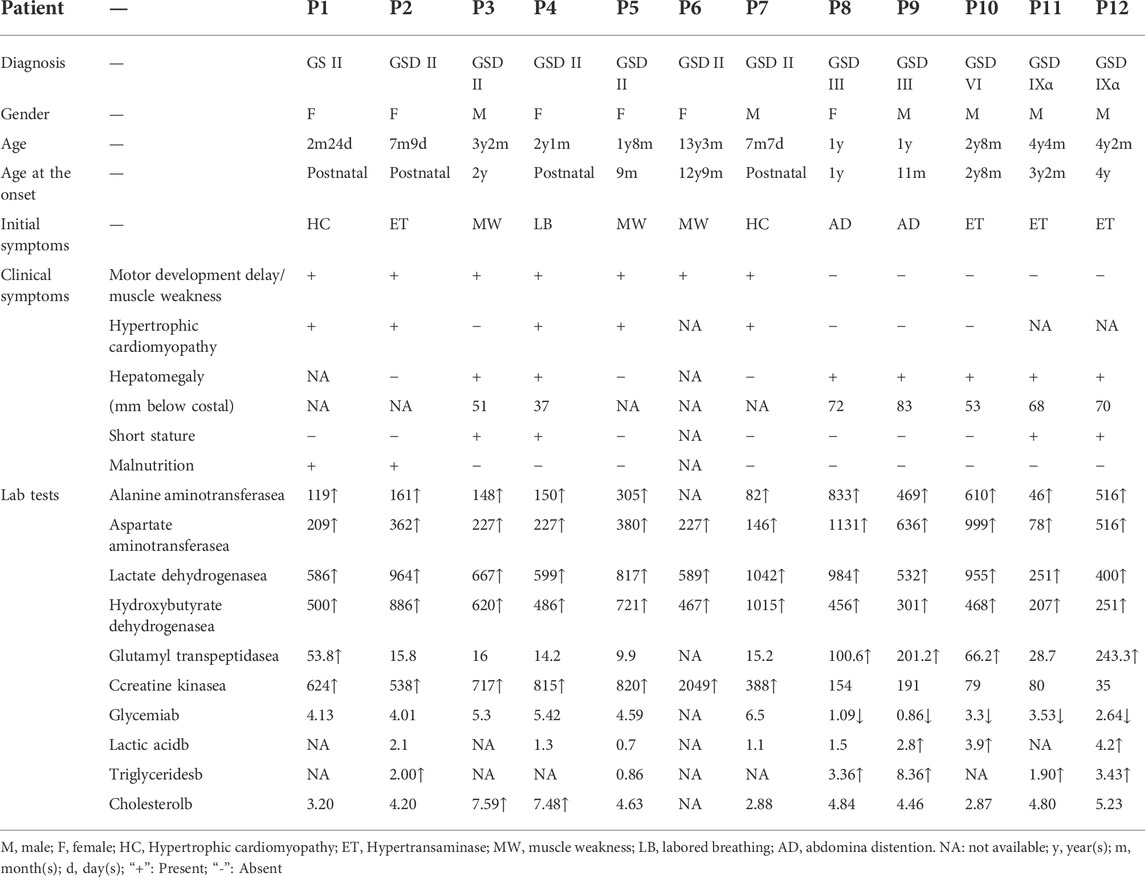
TABLE 1. | The clinical and laboratory features of the Chinese patients with GSD.
Candidate variants
Compound heterozygous/homozygous variants in GAA gene (P1-P7), AGL gene (P8, P9), or PYGL gene (P10) were detected in our patients, respectively. Patients 11–12 were detected as heterozygotes for the PHKA2 gene variant. Totally 18 different variants in four GSD related genes were found (variants c.2662G>T, c.1798C>T, and c.1622C>T of GAA gene were detected twice), of which 9 variants were reported and 9 variants were novel. The variants in the patients were inherited from their carrier parents, except for 2 male patients with PHKA2 gene variant on the X chromosome were from their mother. GSD type, variation location, variation, amino acid variation, variant type, and mutation analysis results of the variations are given in Tables 2, 3.
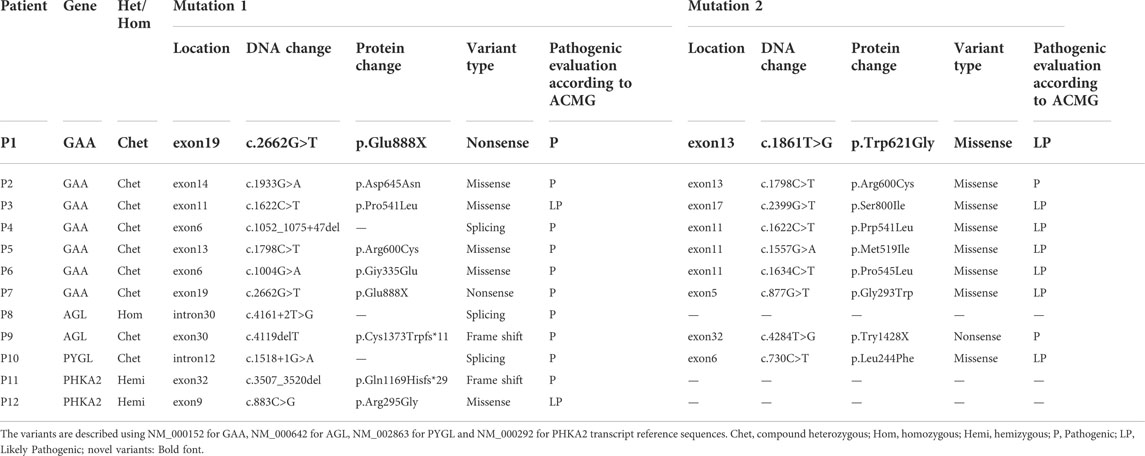
TABLE 2. | Mutations analyzed by WES and classified according to the ACMG guidelines.
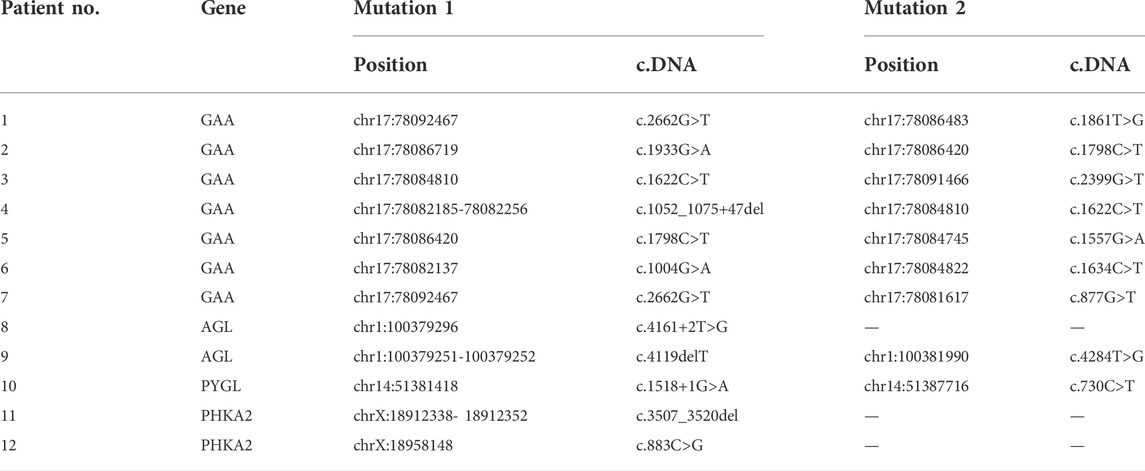
TABLE 3. | Chr positions of the variants in GAA, AGL, PHKA2 and PYGL gene.
The nine novel variants were determined after checked in all available databases and publications, including 1000 Genomes, ExAC, gnomAD, in-house databases and published articles. The scores of SIFT, Polyphen-2, Mutation Taster, and REVEL predicted that the novel variants (except c.1052_1075 + 47del) have a deleterious effect on the function of the protein (Table 4). No predictions of impact were available for variant c.1052_1075 + 47del by bioinformatics algorithms. In the novel variants, 3 splicing and 1 frameshift mutations was classified as pathogenic, 5 missense mutations as likely pathogenic according to the ACMG guidelines (Richards et al., 2015).
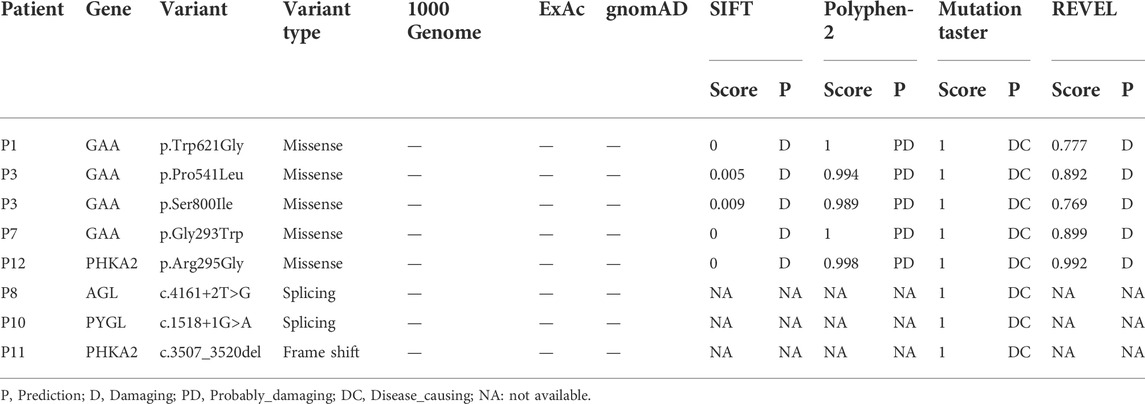
TABLE 4. | Effect on protein function of the novel variations predicted by SIFT, PolyPhen-2, MutationTaster, and REVEL.
There were 4 mutations of GAA and 1 of PHKA2 in the novel missense variants. The 3D protein structures of GAA with novel missense variants were predicted by Swiss-Pdb Viewer 4.1 indicating the variants might affect the proteins function via destroying hydrogen bonds, or conformational constraints which resulted in the proteins into disordered extended structures (Figure 1). The protein structure of PHKA2 with p. Arg295Gly was not constructed because there was no suitable model in SWISS-MODEL database. Silico analysis showed that the p. Arg295 in PHKA2 was highly conservative in different species of human, Macaque, canine, Cattle, et al. (Figure 2).
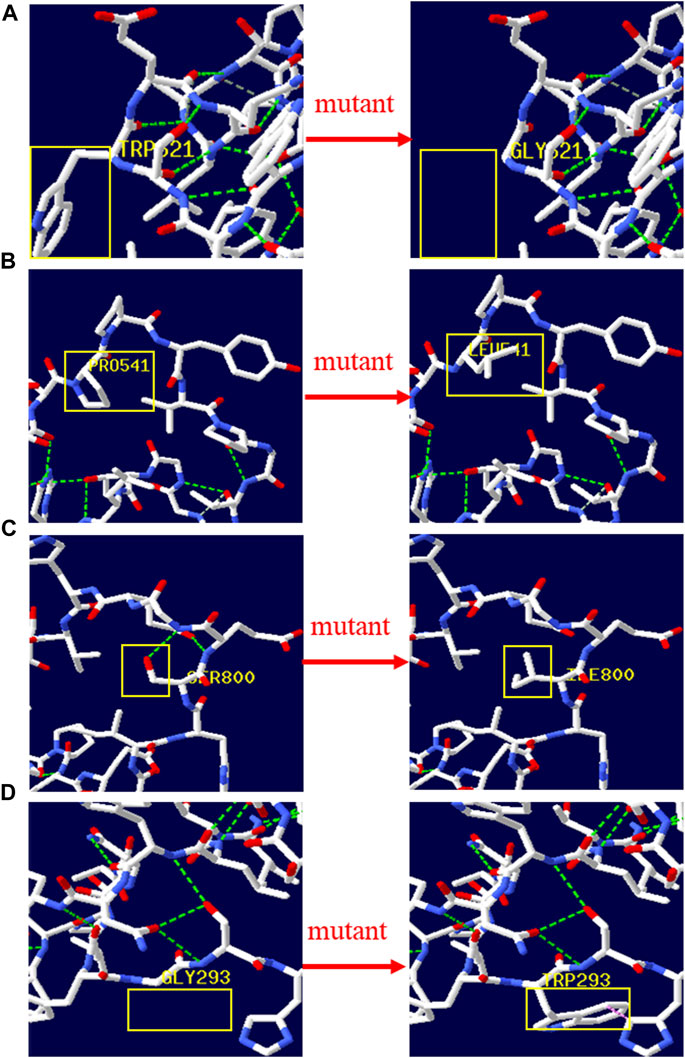
FIGURE 1. 3D structure of wild type and mutant type of novel variants in GAA. The yellow boxes marked the sites of wild‐type Gly293, Prp541, Trp621, Ser800 and mutated Gly621, Leu541, Ile800, Trp293. The 3D protein structures of novel variants in GAA gene were predicted by Swiss‐Pdb Viewer 4.1 indicating the missense mutations might affect the proteins function via destroying hydrogen bonds, or conformational constraints which resulted in the proteins into disordered extended structures.

FIGURE 2. Conservation analysis of the p.Arg295Gly mutation in PHKA2. Silico analysis of p.Arg295 in PHKA2 shows the site highly conservative in different species of human, Macaque, canine, Cattle, etc.
Genetic diagnosis and clinical analysis
Gene sequencing showed that 7 patients were diagnosed with GSD II, 2 patients with GSD III, 1 patient with GSD VI, and 2 patients with GSD IXα. In the study group, the most frequent subtype of GSD was type Ⅱ with 58.3% (n = 7), followed by GSD III (n = 2) and IXα with 16.7% (n = 2).
Seven GSD II patients were divided into infantile-onset Pompe disease (IOPD) (P1, P2, P7) group and late-onset Pompe disease (LOPD) (P3-P6) group according to appearance cardiomyopathy within/out 12 months and age of onset (Molares-Vila et al., 2021). The initial symptoms of GSD II patients were Hypertrophic cardiomyopathy (66.6%) and Hypertransaminase (33.3%) in IOPD group, while exercise-induced muscle weakness (75.0%) and labored breathing (25.0%) in LOPD group. In our study, laboratory findings of IOPD and LOPD were not significantly different except for CK values (p > 0.05).
There were 5 patients with hepatic GSD in our study, including 2 patients with GSD III, 1 patient with GSD VI, and 2 patients with GSD IXα. Hepatomegaly, hypoglycemia and hypertransaminase were observed in all patients, hypertriglyceridemia in 80% (4/5) patients and hyperlactate in 60% (3/5, 1 undetected) patients. The first and main symptom of patients (P8, P9) with GSD III was progressive abdominal distention (100%) due to enlarged liver. The main symptom observed in the patient (P10-P12) with GSD VI or IXα was hypertransaminasemia (100%) detected on laboratory examination accompanied by hepatomegaly. Neither liver size nor laboratory findings allow for a differentiation among GSD III, GSD VI and GSD IXα (p > 0.05). At diagnosis, both patients with GSD IX had a height shorter than -2 SDS, while growth retardation was not found in GSD III and GSD VI patients. Between GSD II group and the hepatic GSD group, significant differences in liver size and partial laboratory findings (glutamyl transpeptidasea, hydroxybutyrate dehydrogenasea, creatine kinasea, Glycemia, Lactic acid) were found.
Treatment and follow-up
Of seven GSD II patients, six received enzyme replacement therapy (ERT) (Myozyme®) injection, 4 of whom responded to treatment, 2 were discontinued due to death with severe myocardial hypertrophy and pulmonary hypertension and hypersensitivity, and 1 patient lost visit. Two patients with GSD III received oral raw cornstarch and increased dietary protein after diagnosis. During treatment, they occasionally experienced convulsions due to fasting hypoglycemia, and developed symptoms of short stature and delayed motor development. In patients with GSD VI and IXα, after receiving oral raw cornstarch, the clinical symptoms basically disappeared except for the hypertransaminases.
Discussion
Diagnosis and distribution of GSDs
The molecular mechanism of GSD is variation in genes encoding key enzymes in glycogenolysis or gluconeogenesis (Ying et al., 2020). With the development of gene sequencing technology, identification of variants by next-generation sequencing (NGS) has partially replaced traditional invasive examinations, such as liver biopsy, muscle biopsy, etc. As the accurate standard for diagnosis of GSD (Sperb-Ludwig et al., 2019; Beyzaei et al., 2020; Szymanska et al., 2021). In the present study, 18 different heterozygous candidate mutations (9 novel and 9 reported) were identified in 12 Chinese GSD families: 7 families carrying GAA mutations (7/12, 58.3%), 2 carrying AGL mutations (2/12, 16.7%), 1 carrying PYGL mutation (1/12, 8.3%), and 2 carrying PHKA2 mutations (2/12, 16.7%). According to the ACMG guidelines (Richards et al., 2015), 4 of the 9 novel variants was classified as pathogenic, 5 classified as likely pathogenic, and all of them have not been reported in any human gene mutation databases or publications (Table 2). Early and accurate diagnosis of GSD is an important step in realizing personalized management, helping physicians to formulate the best treatment plan for patients and minimizing adverse outcomes (Retterer et al., 2016; Beyzaei et al., 2021; Unnisa et al., 2022). The median age of the 12 patients diagnosed in this study was 22 months (including 5 cases less than or equal to 1 year old), meaning that an earlier diagnosis and treatment is more conducive to reducing the risk of organ damage progression and death.
It has been known the geographical distribution of GSD subtypes and genotype was different. In a cohort of Iranian, GSD III (AGL), GSD IX (PHKB and PHKG2) was the major type of GSD (Beyzaei et al., 2021). In Turkish, the genetic analysis showed that the highest proportion of GSDs was type III with 39.5% (n = 15), followed by type I with 36.8% (n = 14) (Cakar et al., 2020). While in Spain, Vega et al. reported that more than three-quarters of patients with GSDs were type III or IXα (Vega et al., 2016). In our study cohort, more than half of the patients were diagnosed as GSD II which suggested that type II may be the most common subtype in the Chinese GSD population.
Patients with GSD II
GSD II (MIM# 232300), also known as Pompe disease, caused by deficiency of the enzyme acid α-glucosidase encoded by GAA, presents as a progressive myopathy due to accumulation of glycogen in lysosomes and cell destruction (van Capelle et al., 2016). GSD II presents as a broad clinical spectrum of phenotypes with considerable variation in age of onset, symptom/presenting sign, degree of severity, organ involvement, and nature history (Holzwarth et al., 2022). In our study, 7 patients were detected compound heterozygous mutations in GAA gene with an age ranging from birth to 12 years old. Three patients (P1, P2, P7) manifesting generalized myasthenia and hypertrophic cardiomyopathy within the first months of life were IOPD. Two of them also showed severe malnutrition. Four patients (P3-P6) who presented as progressive limb-girdle muscle weakness with hyperCKemia were LOPD. Noteworthily, although P4 developed dyspnea shortly after birth due to respiratory muscle involvement, but she had relatively mild myocardial damage and was classified as LOPD (Hout et al., 2003; Kishnani et al., 2006; Chawla et al., 2022).
Of the GAA variants reported worldwide, c.-32-13T>G is the most common, especially in Caucasian populations. In Asia Pacific, p. Asp645Glu, p. Trp746Cys, p. Gly576Ser, p. Glu888X were most reported (Bergsma et al., 2019; Reuser et al., 2019; Zhao et al., 2019). While in our study, p. Glu888X were detected in P1 and P7, and no other Asia-Pacific high-frequency variants were found. Previous genotype-phenotype correlation research have shown that variants p. Glu888X, p. Asp645Asn, and p. Arg600Cys were associated with IOPD (Reuser et al., 2019). The conclusion was confirmed in our study that 4 patients (P1, P2, P5, and P7) carried one or both of the above variants manifest as early-onset or severe symptom.
We identified 6 reported variants and 5 novel variants of GAA in seven patients with GSD II. Four of the new variants were missense variants and all of them located in the GAA protein domain: p. Gly293Trp maps to the N-terminal Beta Sheet domain, p. Prp541Leu and p. Trp621Gly maps to the catalytic GH31 domain, p. Ser800Ile Maps to the Proximal Beta Sheet field. The four variants were predicted to be disease-causing by SIFT, Mutation taster, REVEL and probably-damaging by Polyphen. The 3D protein structures of GAA protein were predicted and indicated that the novel missense mutations might affect the proteins function via destroying hydrogen bonds, or conformational constraints which resulted in the proteins into disordered extended structures (Figure 1). In addition, p. Gly293Arg, resulting in co-located but different amino acid changes with p. Gly293Trp, has been described in two LOPD patients and identified as pathogenic (Loscher et al., 2018). Another new variant was c.1052_1075 + 47del, a splice variant lacking 71 nucleotides spanning exon 6 and intron 6 that could theoretically affect normal mRNA splicing. However, bioinformatics algorithms were unable to predict the effect of this variant. According to ACMG criteria, four novel missense variants were classified as probable pathogenic variants and one novel splice variant was classified as pathogenic.
Patients with GSD III, VI and IXα
GSD III (AGL, MIM# 232400), VI (PYGL, MIM# 232700) and IXα (PHKA2, MIM# 306000) belong to hepatic GSD with basic features of hepatomegaly and hypoglycemia (Beyzaei et al., 2020). They share commonalities also in other clinical signs: metabolic abnormalities, short stature, and motor development delay/muscle weakness with aging, that lead to challenging to identify the subtypes of patients with hepatic GSD, especially between VI and IX (Qu et al., 2020; Szymanska et al., 2021). However, some subtypes have been found to have specific phenotypic features, for example growth delay is more common in GSD IX patients than in VI patients and hypoglycemia was more pronounced in GSD III than GSD VI and IX (Kishnani et al., 2019; Fernandes et al., 2020; Grunert et al., 2021b). In our study, there were 2 patients of GSD III, 1 patient of GSD VI and 2 patients of GSD IXα. Hepatomegaly, hypoglycemia and hypertransaminase were observed in all patients, hypertriglyceridemia in 80% patients and hyperlactate in 60% patients. Neither liver size nor laboratory findings allow for a differentiation among GSD III, GSD VI and GSD IXα in our study, but only patients with GSD IXα had a height below -2 SDS, which was consistent with the findings of Grunert et al. (Grunert et al., 2021b). None of our patients had significant motor retardation when diagnosed. Interestingly, the initial symptom of two GSD III patients was abdominal distention due to hepatomegaly, whereas GSD VI and IX patients presented with elevated transaminases inadvertently during upper respiratory tract infection.
We identified 3 variants of AGL gene in patients with GSD III: p. Cys1373Trpfs*11 and p. Try1428X were detected compound heterozygous in patient 9 and already reported in Chinese population (Lu et al., 2016); c.4161 + 2T>G as a novel splicing variation was detected to be homozygous in P8 while her parents were from two unrelated families. c.4161 + 2T>G was predicted to affect normal splicing of mRNA and evaluated as disease-causing by Mutation taster. According to the standard of ACMG, c.4161 + 2T>G was classified as pathogenic mutation. Notably, the 3 variants were concentrated in the 30th intron or exon, and the 2 patients with them have similar phenotypes and onset times.
GSD VI is rare relative to other types of GSD, with an incidence of 1/60,000 to 1/85,000 (Zhan et al., 2021). As the only causative gene of GSD VI, PYGL gene has been recorded 80 types of mutations in the HGMD database (22 April 2022), among which missense/nonsense mutations are the most common. So far, 17 Chinese patients with GSD VI have been reported, and no PYGL hotspot variant has been found in this group (Lu et al., 2020; Luo et al., 2020; Zhan et al., 2021). For P10, we identified compound heterozygous point mutations in PYGL through WES, including a recurrent missense mutation p. Leu244Phe on exon 6 and a novel splicing mutation c.1518 + 1G>A on intron 12 (Lu et al., 2020). c.1518 + 1G>A was predicted to resulting in forming incorrect mRNA molecule and evluated as disease-causing by Mutation taster. Variant frequency was 0 in the database of 1000 Genomes, gnomAD and ExAC. According to the ACMG standard, c.1518 + 1G>A was classified as a pathogenic variant.
In liver form of GSD IXα, the disease is inherited in an X-linked recessive manner associated with PHKA2 mutations (Fang et al., 2021; Mori et al., 2022). As of 22 April 2022, 166 PHKA2 variants were recorded in the HGMD database, with missense/nonsense mutations (greater than 50%) being the most common, followed by small deletion variants. In this report, we detected two novel variants in PHKA2, c.3507_3520del (p.Gln1169Hisfs*29) and p. Arg295Gly. p. Gln1169Hisfs*29 lead to fourteen-nucleotide deletion within exon 32 of PHKA2, that seemed to result in a frameshift introducing a premature stop codon after 2602 bp of amino acid residue 1196 of 1235. The variant was predicted disease-causing by Mutation taster and evaluated as a pathogenic variant refer to ACMG guidelines. Notably, the PHKA2 gene had a total of 33 exons, and one-fifth (34/166) of the reported variants were located in or spanned exons 30–33, suggesting that the C-terminus of the PHKA2 was a variant-dense region. p. Arg295Gly is an unreported missense variant, but p. Arg295His (Fernandes et al., 2020) and p. Arg295Cys (Ban et al., 2003) had been described in GSD IXα patients and identified as pathogenic. Silico analysis of p. Arg295 in PHKA2 shows the site highly conservative in different species of human, Macaque, canine, Cattle, et al. SIFT, Mutation taster and Polyphen predicted that p. Arg295Gly was damaging to protein function. Therefore, p. Arg295Gly was assessed as likely pathogenic according to ACMG criteria.
Conclusion
This study reported 12 Chinese patients with Glycogen storage disease and identified 18 gene variants of GAA, AGL, PHKA2 and PYGL (9 novel and 9 reported) by whole-exome sequencing. Our study expanded the variation spectrum of gene associated with GSDs. Next-generation sequencing was considered the preferred diagnostic method for GSD because invasive liver biopsy can be avoided.
Data availability statement
The data presented in the study are deposited in the GenBank repository, accession number OP117444-OP117449.
Ethics statement
The studies involving human participants were reviewed and approved by Medical Ethics Committee of Children’s Hospital Affiliated to Shandong University (Jinan Children’s Hospital). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
ZG and YL conceived and designed this study. Experiments were conducted by RD, KZ, YL, MG, DW, and JM. Data were analyzed by RD, KZ, and YL. Clinical diagnosis of the patients was undertaken by FS, RD, and ZG. The paper was written by RD and XW. RD and XW contributed equally to this work.
Funding
This work was supported by the National Natural Science Foundation of China (81671362 and 32000075), the financial funding from Department of Science and Technology of Shandong Province (ZR2020QC066 and ZR2020QH102), the financial funding from Jinan Science and Technology Bureau (201907007).
Acknowledgments
We thank the patients and their families for their participation and contribution to the work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ban, K., Sugiyama, K., Goto, K., Mizutani, F., and Togari, H. (2003). Detection of PHKA2 gene mutation in four Japanese patients with hepatic phosphorylase kinase deficiency. Tohoku J. Exp. Med. 200, 47–53. doi:10.1620/tjem.200.47
Bergsma, A. J., In 't Groen, S. L. M., van den Dorpel, J. J. A., van den Hout, H., van der Beek, N., Schoser, B., et al. (2019). A genetic modifier of symptom onset in Pompe disease. EBioMedicine 43, 553–561. doi:10.1016/j.ebiom.2019.03.048
Beyzaei, Z., Ezgu, F., Geramizadeh, B., Imanieh, M. H., Haghighat, M., Dehghani, S. M., et al. (2021). Clinical and genetic spectrum of glycogen storage disease in Iranian population using targeted gene sequencing. Sci. Rep. 11 (1), 7040. doi:10.1038/s41598-021-86338-4
Beyzaei, Z., Geramizadeh, B., and Karimzadeh, S. (2020). Diagnosis of hepatic glycogen storage disease patients with overlapping clinical symptoms by massively parallel sequencing: a systematic review of literature. Orphanet J. Rare Dis. 15 (1), 286. doi:10.1186/s13023-020-01573-8
Brown, L. M., Corrado, M. M., van der Ende, R. M., Derks, T. G., Chen, M. A., Siegel, S., et al. (2015). Evaluation of glycogen storage disease as a cause of ketotic hypoglycemia in children. J. Inherit. Metab. Dis. 38 (3), 489–493. doi:10.1007/s10545-014-9744-1
Cakar, N. E., Gezdirici, A., Topuz, H. S., and Onal, H. (2020). Novel variants in Turkish patients with glycogen storage disease. Pediatr. Int. 62 (10), 1145–1150. doi:10.1111/ped.14286
Chawla, T., Preethish-Kumar, V., Polavarapu, K., Vengalil, S., Bardhan, M., Puri, R., et al. (2022). Late onset Pompe disease with novel mutations and atypical phenotypes. J. Neuromuscul. Dis. 9 (2), 261–273. doi:10.3233/JND-210728
Choi, R., Park, H. D., Kang, B., Choi, S. Y., Ki, C. S., Lee, S. Y., et al. (2016). PHKA2 mutation spectrum in Korean patients with glycogen storage disease type IX: Prevalence of deletion mutations. BMC Med. Genet. 17, 33. doi:10.1186/s12881-016-0295-1
De Vito, E. L., Arce, S. C., Monteiro, S. G., and Vaca Ruiz, G. A. (2019). Central drive and ventilatory failure in late-onset Pompe disease: At the gates of a new phenotype. Neuromuscul. Disord. 29 (6), 444–447. doi:10.1016/j.nmd.2019.03.008
Eghbali, M., Fatemi, K. S., Salehpour, S., Abiri, M., Saei, H., Talebi, S., et al. (2020). Whole-exome sequencing uncovers novel causative variants and additional findings in three patients affected by glycogen storage disease type VI and fanconi-bickel syndrome. Front. Genet. 11, 601566. doi:10.3389/fgene.2020.601566
Fang, Y., Yu, J., Lou, J., Peng, K., Zhao, H., and Chen, J. (2021). Clinical and genetic spectra of inherited liver disease in children in China. Front. Pediatr. 9, 631620. doi:10.3389/fped.2021.631620
Fernandes, S. A., Cooper, G. E., Gibson, R. A., and Kishnani, P. S. (2020). Benign or not benign? Deep phenotyping of liver glycogen storage disease IX. Mol. Genet. Metab. 131 (3), 299–305. doi:10.1016/j.ymgme.2020.10.004
Grunert, S. C., Hannibal, L., Schumann, A., Rosenbaum-Fabian, S., Beck-Wodl, S., Haack, T. B., et al. (2021a). Identification and characterization of a novel splice site mutation associated with glycogen storage disease type VI in two unrelated Turkish families. Diagn. (Basel) 11 (3), 500. doi:10.3390/diagnostics11030500
Grunert, S. C., Hannibal, L., and Spiekerkoetter, U. (2021b). The phenotypic and genetic spectrum of glycogen storage disease type VI. Genes 12 (8), 1205. doi:10.3390/genes12081205
Holzwarth, J., Minopoli, N., Pfrimmer, C., Smitka, M., Borrel, S., Kirschner, J., et al. (2022). Clinical and genetic aspects of juvenile onset Pompe disease. Neuropediatrics 53 (1), 39–45. doi:10.1055/s-0041-1735250
Hout, H. M. P. v. d., Hop, W., Diggelen, O. P. v., Smeitink, J. A. M., Smit, G. P. A., et al. (2003). The natural course of infantile pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 112 (2), 332–340. doi:10.1542/peds.112.2.332
Huang, W., Zhan, D., Li, Y., Zheng, N., Wei, X., Bai, B., et al. (2021). Proteomics provides individualized options of precision medicine for patients with gastric cancer. Sci. China. Life Sci. 64 (8), 1199–1211. doi:10.1007/s11427-021-1966-4
Kim, T. H., Kim, K. Y., Kim, M. J., Seong, M. W., Park, S. S., Moon, J. S., et al. (2020). Molecular diagnosis of glycogen storage disease type IX using a glycogen storage disease gene panel. Eur. J. Med. Genet. 63 (6), 103921. doi:10.1016/j.ejmg.2020.103921
Kishnani, P. S., Goldstein, J., Austin, S. L., Arn, P., Bachrach, B., Bali, D. S., et al. (2019). Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of medical genetics and Genomics (ACMG). Genet. Med. 21 (4), 772–789. doi:10.1038/s41436-018-0364-2
Kishnani, P. S., Hwu, W. L., Mandel, H., Nicolino, M., Yong, F., Corzo, D., et al. (2006). A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 148 (5), 671–676. doi:10.1016/j.jpeds.2005.11.033
Li, Y., Chen, J., Xu, Q., Han, Z., Tan, F., Shi, T., et al. (2021). Single-cell transcriptomic analysis reveals dynamic alternative splicing and gene regulatory networks among pancreatic islets. Sci. China. Life Sci. 64 (1), 174–176. doi:10.1007/s11427-020-1711-x
Liang, Y., Du, C., Wei, H., Zhang, C., Zhang, M., Hu, M., et al. (2020). Genotypic and clinical analysis of 49 Chinese children with hepatic glycogen storage diseases. Mol. Genet. Genomic Med. 8 (10), e1444. doi:10.1002/mgg3.1444
Loscher, W. N., Huemer, M., Stulnig, T. M., Simschitz, P., Iglseder, S., Eggers, C., et al. (2018). Pompe disease in Austria: clinical, genetic and epidemiological aspects. J. Neurol. 265 (1), 159–164. doi:10.1007/s00415-017-8686-6
Lu, C., Qiu, Z., Sun, M., Wang, W., Wei, M., and Zhang, X. (2016). Spectrum of AGL mutations in Chinese patients with glycogen storage disease type III: identification of 31 novel mutations. J. Hum. Genet. 61 (7), 641–645. doi:10.1038/jhg.2016.24
Lu, S. Q., Feng, J. Y., Liu, J., Xie, X. B., Lu, Y., and Abuduxikuer, K. (2020). Glycogen storage disease type VI can progress to cirrhosis: ten Chinese patients with GSD VI and a literature review. J. Pediatr. Endocrinol. Metab. 33 (10), 1321–1333. doi:10.1515/jpem-2020-0173
Luo, X., Hu, J., Gao, X., Fan, Y., Sun, Y., Gu, X., et al. (2020). Novel PYGL mutations in Chinese children leading to glycogen storage disease type VI: two case reports. BMC Med. Genet. 21 (1), 74. doi:10.1186/s12881-020-01010-4
Molares-Vila, A., Corbalan-Rivas, A., Carnero-Gregorio, M., Gonzalez-Cespon, J. L., and Rodriguez-Cerdeira, C. (2021). Biomarkers in glycogen storage diseases: An update. Int. J. Mol. Sci. 22 (9), 4381. doi:10.3390/ijms22094381
Mori, T., Ishikawa, A., Shigetomi, H., Fukuda, T., and Sugie, H. (2022). A novel PHKA2 variant in a Japanese boy with glycogen storage diseases type IXa. Pediatr. Int. 64 (1), e14839. doi:10.1111/ped.14839
Perveen, S., Gupta, N., Kumar, M., Kaur, P., Chowdhury, M. R., and Kabra, M. (2020). Spectrum of amyloglucosidase mutations in Asian Indian patients with Glycogen storage disease type III. Am. J. Med. Genet. A 182 (5), 1190–1200. doi:10.1002/ajmg.a.61547
Qu, Q., Qian, Q., Shi, J., Liu, H., Zhang, Y., Cui, W., et al. (2020). The novel compound heterozygous mutations in the AGL gene in a Chinese family with adult late-onset glycogen storage disease type IIIa. Front. Neurol. 11, 554012. doi:10.3389/fneur.2020.554012
Retterer, K., Juusola, J., Cho, M. T., Vitazka, P., Millan, F., Gibellini, F., et al. (2016). Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18 (7), 696–704. doi:10.1038/gim.2015.148
Reuser, A. J. J., van der Ploeg, A. T., Chien, Y. H., Llerena, J., Abbott, M. A., Clemens, P. R., et al. (2019). GAA variants and phenotypes among 1, 079 patients with Pompe disease: Data from the Pompe Registry. Hum. Mutat. 40 (11), 2146–2164. doi:10.1002/humu.23878
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Shen, Y., and Shi, T. (2019). Innovation for better health of children. Sci. China. Life Sci. 62 (12), 1561–1562. doi:10.1007/s11427-019-1589-2
Sperb-Ludwig, F., Pinheiro, F. C., Bettio Soares, M., Nalin, T., Ribeiro, E. M., Steiner, C. E., et al. (2019). Glycogen storage diseases: Twenty-seven new variants in a cohort of 125 patients. Mol. Genet. Genomic Med. 7 (11), e877. doi:10.1002/mgg3.877
Szymanska, E., Jozwiak-Dziecielewska, D. A., Gronek, J., Niewczas, M., Czarny, W., Rokicki, D., et al. (2021). Hepatic glycogen storage diseases: Pathogenesis, clinical symptoms and therapeutic management. Arch. Med. Sci. 17 (2), 304–313. doi:10.5114/aoms.2019.83063
Unnisa, Z., Yoon, J. K., Schindler, J. W., Mason, C., and van Til, N. P. (2022). Gene therapy developments for Pompe disease. Biomedicines 10 (2), 302. doi:10.3390/biomedicines10020302
van Capelle, C. I., van der Meijden, J. C., van den Hout, J. M., Jaeken, J., Baethmann, M., Voit, T., et al. (2016). Childhood Pompe disease: clinical spectrum and genotype in 31 patients. Orphanet J. Rare Dis. 11 (1), 65. doi:10.1186/s13023-016-0442-y
Vega, A. I., Medrano, C., Navarrete, R., Desviat, L. R., Merinero, B., Rodriguez-Pombo, P., et al. (2016). Molecular diagnosis of glycogen storage disease and disorders with overlapping clinical symptoms by massive parallel sequencing. Genet. Med. 18 (10), 1037–1043. doi:10.1038/gim.2015.217
Ying, S., Zhihua, Z., Yucan, Z., Yu, J., Qian, L., Bixia, Z., et al. (2020). Molecular diagnosis of panel-based next-generation sequencing approach and clinical symptoms in patients with glycogen storage disease: A single center retrospective study. Front. Pediatr. 8, 600446. doi:10.3389/fped.2020.600446
Zhan, Q., Lv, Z., Tang, Q., Huang, L., Chen, X., Yang, M., et al. (2021). Glycogen storage disease type VI with a novel PYGL mutation: Two case reports and literature review. Med. Baltim. 100 (16), e25520. doi:10.1097/MD.0000000000025520
Keywords: glycogen storage diseases, mutation, whole exome sequencing, GAA gene, AGL gene, PHKA2 gene, PYGL gene
Citation: Dong R, Wei X, Zhang K, Song F, Lv Y, Gao M, Wang D, Ma J, Gai Z and Liu Y (2022) Genotypic and phenotypic characteristics of 12 chinese children with glycogen storage diseases. Front. Genet. 13:932760. doi: 10.3389/fgene.2022.932760
Received: 05 May 2022; Accepted: 25 July 2022;
Published: 29 August 2022.
Edited by:
Tieliu Shi, Hunan University of Arts and Science, ChinaReviewed by:
Fengying Sun, Second People’s Hospital of Wuhu, ChinaZahra Beyzaei, Shiraz University of Medical Sciences, Iran
Copyright © 2022 Dong, Wei, Zhang, Song, Lv, Gao, Wang, Ma, Gai and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongtao Gai, Z2Fpemhvbmd0YW9Ac2luYS5jb20=; Yi Liu, eV9saXU5OUBzaW5hLmNvbQ==
†These authors have contributed equally to this work