 Yunting Lin1†
Yunting Lin1† Xiaohong Chen2
Xiaohong Chen2 Bobo Xie3,4
Bobo Xie3,4 Zhihong Guan1
Zhihong Guan1 Xiaodan Chen1
Xiaodan Chen1 Xiuzhen Li1†
Xiuzhen Li1† Peng Yi1
Peng Yi1 Rong Du1Huifen Mei1
Rong Du1Huifen Mei1 Li Liu1*†Wen Zhang1*
Li Liu1*†Wen Zhang1* Chunhua Zeng1*†
Chunhua Zeng1*†- 1Department of Genetics and Endocrinology, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, China
- 2Department of Endocrinology and Metabolism, Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Center for Medical Genetics and Genomics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, China
- 4The Guangxi Health Commission Key Laboratory of Medical Genetics and Genomics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, China
Objective: Wiedemann-Steiner syndrome (WSS) is a rare autosomal dominant disorder caused by deleterious heterozygous variants of the KMT2A gene. This study aims to describe the phenotypic and genotypic features of Chinese WSS patients, and assess therapeutic effects of recombinant human growth hormone (rhGH).
Methods: Eleven Chinese children with WSS were enrolled in our cohort. Their clinical, imaging, biochemical and molecular findings were analyzed retrospectively. Moreover, the phenotypic features of 41 previously reported Chinese WSS patients were reviewed and included in our analysis.
Results: In our cohort, the 11 WSS patients presented with classic clinical manifestations, but with different frequencies. The most common clinical features were short stature (90.9%) and developmental delay (90.9%), followed by intellectual disability (72.7%). The most frequent imaging features were patent ductus arteriosus (57.1%) and patent foramen ovale (42.9%) in cardiovascular system, and abnormal corpus callosum (50.0%) in the brain. In the set comprising 52 Chinese WSS patients, the most common clinical and imaging manifestations were developmental delay (84.6%), intellectual disability (84.6%), short stature (80.8%) and delayed bone age (68.0%), respectively. Eleven different variants, including three known and eight novel variants, of the KMT2A gene were identified in our 11 WSS patients without a hotspot variant. Two patients were treated with rhGH and yielded satisfactory height gains, but one developed acceleration of bone age.
Conclusion: Our study adds 11 new patients with WSS, reveals different clinical characteristics in Chinese WSS patients, and extends the mutational spectrum of the KMT2A gene. Our study also shares the therapeutic effects of rhGH in two WSS patients without GH deficiency.
1 Introduction
Wiedemann-Steiner syndrome (WSS) is a rare autosomal dominant disorder caused by deleterious heterozygous variants of the KMT2A gene located on chromosome 11q23.3. The KMT2A gene, also known as the MLL gene, is comprised of 36 exons with a range of 16.6 Kb that encodes a histone lysine methyltransferase of 3,969 amino acids (Jones et al., 2012). The KMT2A protein comprises a number of functional domains: three DNA binding AT hooks, a CXXC zinc finger domain, four PHD zinc finger domains, a BD domain, a FYRN domain, a TAD motif, a FYRC domain, a WIN motif, and a SET domain (Jones et al., 2012; Aggarwal et al., 2017; Baer et al., 2018). To date, 349 different KMT2A variants, including 273 disease-causing and 76 possible disease-causing variants, have been documented in the Human Gene Mutation Database (HGMD, Professional 2022.4, https://www.hgmd.cf.ac.uk/) to cause WSS or other related disorders or phenotypes.
As the KMT2A gene regulates the expression of multiple HOX and WNT genes and plays a critical role in early development and hematopoiesis, WSS patients present complex and variable phenotypes involving multiple systems (Aggarwal et al., 2017; Baer et al., 2018; Li et al., 2018). The classic features of WSS include developmental delay, intellectual disability, short stature, facial dysmorphism, and hypertrichosis cubiti (hairy elbows). Other additional phenotypes of feeding difficulties, epilepsy, ocular abnormalities, congenital heart disease, musculoskeletal problems, genitourinary anomalies, endocrinologic issues, immunologic dysfunction, behavioral differences, and abnormal brain magnetic resonance imaging (MRI) also have been described in WSS (Miyake et al., 2016; Baer et al., 2018; Li et al., 2018; Sheppard et al., 2021; Castiglioni et al., 2022; Durand et al., 2022).
However, the features of WSS are shared by a range of disorders, including Coffin-Siris syndrome, Nicolaides-Baraitser syndrome, Cornelia de Lange syndrome, Rubinstein-Taybi syndrome, Kabuki syndrome, Bohring-Opitz syndrome, Suleiman-El-Hattab syndrome, and Blepharophimosis-ptosis-epicanthus inversus syndrome (Bramswig et al., 2015; Miyake et al., 2016; Baer et al., 2018; Li et al., 2018; Negri et al., 2019; Jinxiu et al., 2020; Demir et al., 2021; Di Fede et al., 2021; Sheppard et al., 2021; Castiglioni et al., 2022; Riedhammer et al., 2022). The overlapping clinical signs may complicate the diagnosis of a patient. Next-generation sequencing (NGS), including whole genome sequencing (WGS), whole exome sequencing (WES) and target region sequencing (TRS), may contribute to the precise diagnosis of WSS (Bramswig et al., 2015; Negri et al., 2019; Jinxiu et al., 2020; Demir et al., 2021; Di Fede et al., 2021).
Due to highly clinical heterogeneity and phenotypic similarity with other disorders, WSS is far away from well-known. The prevalence of WSS is estimated to be less than 1/1,000,000 (Luo et al., 2021). To date, less than 400 WSS patients have been reported in the worldwide, and some affected adults are not yet recognized until their child’s diagnosis (Sheppard et al., 2021). Thus, it is probable that a number of WSS cases are underdiagnosed.
In this study, we analyzed the clinical and genetic findings of 11 Chinese children with WSS retrospectively, and reviewed the clinical presentations of 41 previously reported Chinese WSS patients, in order to better describe phenotypic characteristics and mutational spectrum of Chinese WSS patients. We also evaluated the therapeutic effects of recombinant human growth hormone (rhGH) in two patients.
2 Materials and methods
2.1 Patients
Eleven WSS children from 11 unrelated families were enrolled in our cohort. Among them, eight patients (P1-P8) were enrolled in Guangzhou Women and Children’s Medical Center, one patient (P9) was enrolled in the Second Affiliated Hospital of Guangxi Medical University, and two patients (P10 and P11) were enrolled in Wuhan Children’s Hospital. All subjects are Chinese. P9 is of Zhuang ethnicity, while the other 10 patients are of Han ethnicity. All parents are asymptomatic and non-consanguineous.
2.2 Clinical information
The clinical information was collected and evaluated by clinicians. The physical examinations were performed by physicians, skeletal X-ray and brain MRI or computed tomography (CT) were performed by radiologists, ultrasonography was performed by sonologists, electrocardiogram (ECG) was performed by cardiologists, and electroencephalogram (EEG) was performed by neurologists. Biochemical parameters of blood samples were detected in hospital’s clinical laboratory center, and serum hormones were detected in hospital’s endocrinology and metabolism laboratory.
2.3 WES
Eleven patients were subjected to WES with a proband-parents trio or proband-only strategy. As described previously (Lin et al., 2020), genomic DNA (gDNA) was extracted from peripheral blood samples using DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany). The workflow of WES was strictly according to the manufacturers’ protocol. gDNA was randomly interrupted to an average size of 180–280 bp by Covaris S220 ultrasonicator (Covaris, Woburn, United States). The fragmented products were end repaired and phosphorylated, followed by A-tailing and ligation at the 3’ ends with paired-end adaptors (Illumina, San Diego, United States). Subsequently, the prepared DNA library was purified using Agencourt AMPure SPRI beads (Beckman Coulter, Brea, United States) and detected by Agilent 2,100 Bioanalyzer and real-time PCR (Agilent, Santa Clara, United States). At last, the exome sequences were enriched from the qualified library using Agilent SureSelect Human All Exon V6 kit (Agilent, Santa Clara, United States) and sequenced on Illumina Novaseq 6,000 platform (Illumina, San Diego, United States) for paired-end 150 bp reads. The acquired data were processed upon an established analysis pipeline for variant calling and functional annotation to identify the disease-causing variants.
2.4 Molecular analysis of the KMT2A gene
To validate the candidate causative mutational site identified by WES, the classic Sanger sequencing was carried out using primers targeting the specific exons of the KMT2A gene (NG_027813.1, NM_001197104.2). The exon sequences together with adjoining intron boundaries were amplified by polymerase chain reaction (PCR) and sequenced using an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, United States). The sequencing chromatograms were read by Chromas software (Technelysium, South Brisbane, Australia), while the exported sequences were aligned with the reference sequence using DNAMAN software (Lynnon Corporation, Vaudreuil, Canada). In addition, for gross deletions covering several exons of the KMT2A gene, a quantitative PCR (qPCR) of gDNA was conducted using a BIO-RAD CFX96 Real-Time System (BIO-RAD, Hercules, United States).
The dbSNP database (https://www.ncbi.nlm.nih.gov/snp/) was employed to exclude the polymorphic alleles, while HGMD and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) databases were engaged for the confirmation of known pathogenic variants. For novel variants, the pathogenicity was evaluated according to the ACMG guidelines (Richards et al., 2015).
2.5 Treatment and follow-up
Five patients (P1, P4, P5, P8 and P10) in our cohort had clinic follow-ups, while three of them (P4, P5 and P10) kept regular clinic visits. Growth hormone (GH) provocation test and serum insulin-like growth factor (IGF-1) were assessed in these three patients.
Two patients (P4 and P10) were treated with rhGH (Jintropin, GeneScience Pharmaceuticals, Changchun, China). Clinical follow-up of P4 was performed with an interval of 1 month in the first 3 months after treatment and subsequently with an interval of 3 months, and her height, weight and serum IGF-1 were measured at every visit. P10 visited clinic with an interval of 3–6 months, but had a bad compliance as only her serum IGF-1 was monitored at every visit whereas her height and weight were measured irregularly.
2.6 Statistical analysis
SPSS Statistics 17.0 software (SPSS, Chicago, United States) was used to calculate means and standard deviations. Prism 6 GraphPad (GraphPad, San Diego, United States) was used to draw curve and line charts.
3 Results
3.1 Clinical characteristics
Among 11 WSS children in our cohort, eight were males and three were females, with a male-to-female ratio of 2.7:1. Most of them (7/10, 70.0%) presented prenatal growth retardation with a birth length of 45.5 ± 3.3 cm and a birth weight of 2.60 ± 0.47 kg at full-term gestation, and all of them showed postnatal growth retardation with a height standard deviation score (SDS) of −3.6 ± 1.3 and a weight SDS of −3.7 ± 1.5 at the first visit (Li et al., 2009) (Supplementary Table S1).
As shown in Supplementary Table S1, the clinical features of 11 WSS patients were highly complex and variable. The most frequent clinical features were short stature (10/11, 90.9%) and developmental delay (10/11, 90.9%), followed by intellectual disability (8/11, 72.7%). Other common clinical features presented in more than half of WSS patients included low-set ears, slim and muscular build, thick eyebrows, long eyelashes, and hypertrichosis of the back.
Skeletal X-ray, brain MRI or CT, abdominal and urinary ultrasonography, cardiac ultrasonography, ECG, and EEG were conducted in six, ten, five, seven, seven, and four patients, respectively. The most frequent imaging features were patent ductus arteriosus (4/7, 57.1%) and patent foramen ovale (3/7, 42.9%) in cardiovascular system, and abnormal corpus callosum (5/10, 50.0%) in the brain (Supplementary Table S1).
All the 11 patients showed normal liver, renal and thyroid functions, and no obvious signs of metabolic acidosis were revealed.
In addition, to fully investigate the phenotypic spectrum of Chinese WSS patients, 41 of 43 previously reported Chinese WSS patients with available identify information were reviewed and included in our analysis [(Sun et al., 2017; Gao et al., 2018; Li et al., 2018; Chen et al., 2019; Dai et al., 2019; Shangguan et al., 2019; Jinxiu et al., 2020; Wang et al., 2021a; Wang et al., 2021b; Feng et al., 2021; Lee et al., 2021; Luo et al., 2021; Sheppard et al., 2021; Xue et al., 2021; Chen et al., 2022; Wu et al., 2022; Yu et al., 2022), and other 4 literatures in Chinese], which increases the total sample size to 52.
Phenotypic heterogeneity was also shown in this set comprising 52 Chinese WSS patients. The male-to-female ratio was 1.3:1. Over half of them (23/45, 51.1%) and most of them (48/52, 92.3%) had prenatal and postnatal growth retardation, respectively. The most frequent clinical features were developmental delay (44/52, 84.6%), intellectual disability (44/52, 84.6%), and short stature (42/52, 80.8%). Other common clinical features presented in more than half of patients included hypertelorism, flat nasal bridge, thick hairs, long eyelashes, hypertrichosis of the back, hypertrichosis of lower limbs, and low hairline. The most frequent imaging feature was delayed bone age (17/25, 68.0%) (Supplementary Table S1).
3.2 Mutational spectrum
Eleven different variants, including three known and eight novel variants, of the KMT2A gene were identified in 11 WSS patients. All of them were heterozygous with a de novo pattern. According to the ACMG guidelines, all eleven KMT2A variants (six truncating, three missense, one splicing and one gross deletion) were classified as pathogenic or likely pathogenic (Table 1).
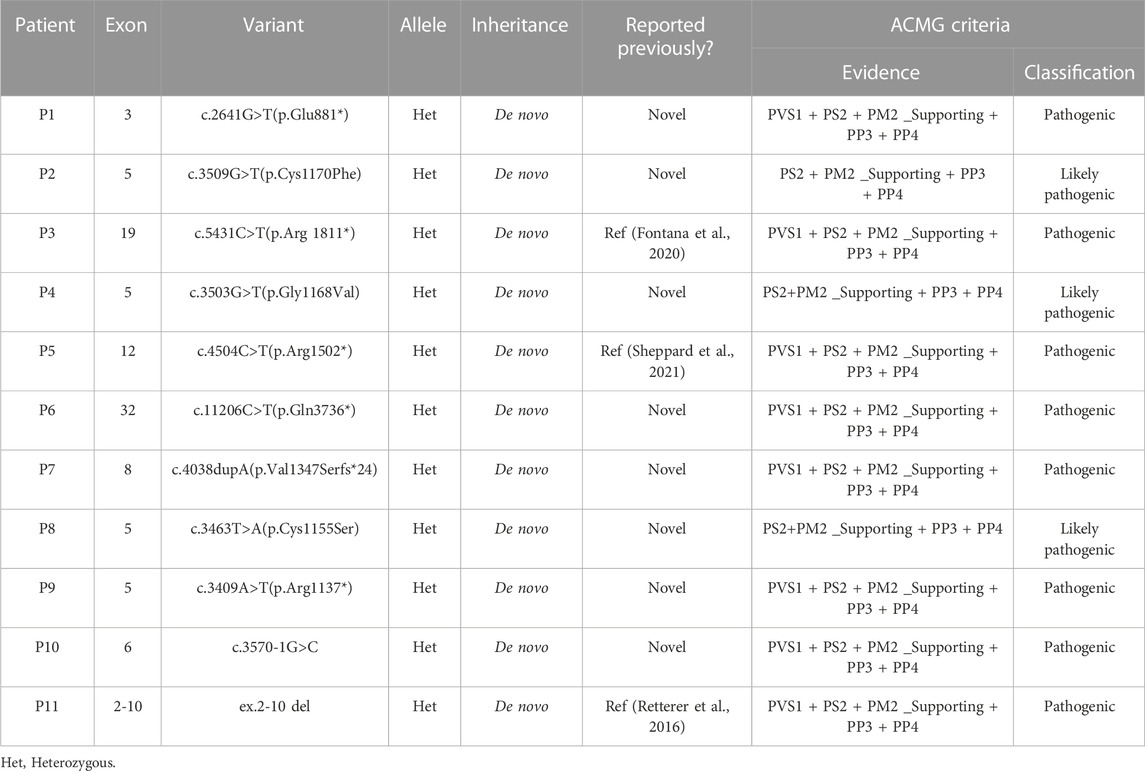
TABLE 1. Deleterious KMT2A variants identified in 11 Chinese WSS patients.
Among eleven identified KMT2A variants, six (6/11, 54.5%) were occurred in functional domains of the KMT2A protein: c.3463T>A(p.Cys1155Ser), c.3503G>T(p.Gly1168Val) and c.3509G>T(p.Cys1170Phe) missense variants were located at the CXXC domain; c.4504C>T(p.Arg1502*) nonsense variant was located at the PHD domain; c.11206C>T(p.Gln3736*) nonsense variant was located at the FYRC domain; while ex.2-10 del covered the AT hooks, CXXC domain and a part of PHD domain. Of the other five variants, four loss-of-function (LoF) variants, including c.2641G>T(p.Glu881*) and c.3409A>T(p.Arg1137*) nonsense variants, c.3570-1G>C splicing variant, and c.4038dupA(p.Val1347Serfs*24) frameshift variant, were distributed near by the CXXC domain, while c.5431C>T(p.Arg1811*) nonsense variant was close to the BD domain (Figure 1).
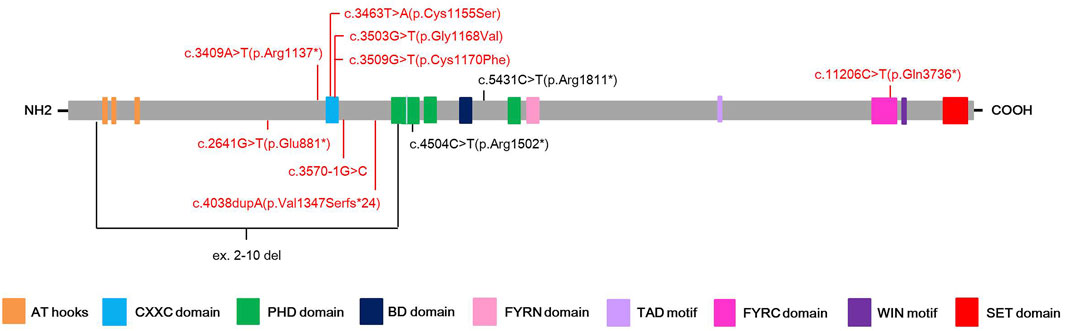
FIGURE 1. Schematic presentation of the KMT2A protein with domain structure and the localization of the eleven different variants found in this study. Red color highlights the novel variants, while black color stands for known pathogenic variants.
3.3 Treatment and follow-up
In our cohort, five patients (5/11, 45.5%) had clinic follow-ups ranging from 5 months to 2.3 years. Among them, P4, P5 and P10 had regular visits. They all showed short stature below −2 height SDS at the first visit and underwent GH provocation test to check for GH deficiency (GHD) (Supplementary Table S1). The GH peak level and IGF-1 level of P5 were 5.65 ng/mL and 36.6 ng/mL (reference range of IGF-1: 50–286 ng/mL) respectively, indicating GHD, whereas no evidence supported GHD in P4 and P10. Consistently, not P4 and P10 but P5 presented with delayed bone age at examination. However, only P4 and P10 received rhGH treatment based on low height gain and the parents’ permission.
P4 and P10 started rhGH treatment at 8.1 years old and 5.3 years old with a dose of 0.12 IU/kg/day and 0.14 IU/kg/day respectively. Comparing with P5 without a medical intervention, the growth trajectories of P4 and P10 improved significantly with a catch-up growth (Figure 2A), and their serum IGF-1 levels also markedly increased (Figure 2B). The height SDS of P10 increased from −2.6 to −0.3 after rhGH treatment of 2.3 years as well as P4 from −3.4 to −2.8 after rhGH treatment of 6 months, whereas P5 remained of a short stature with the height SDS from −4.6 to −5.0 during 1.9 years follow-up (Figure 2A; Supplementary Table S1).
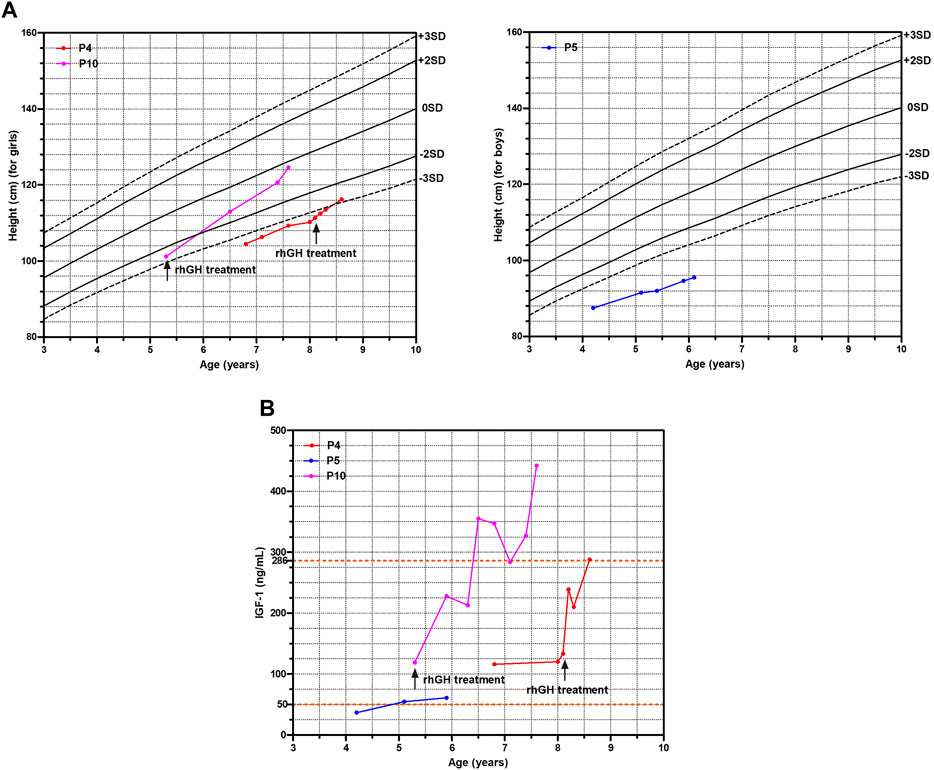
FIGURE 2. The growth curve and IGF-1 levels of three WSS children. (A) Growth curve. (B) IGF-1 levels.
However, growth acceleration of the bone age after rhGH treatment was worth nothing in P10 from approximately 5 years and 9 months at 5.3 years old to 7 years and 10 months at 6.5 years old, and 10 years at 7.6 years old according to the standards of Greulich and Pyle (Greulich and Pyle, 1959). In contrast, the bone age of P4 was consistent with her physical age at the first visit of 6.8 years old, and gradually showed a trend of growth slowdown with a bone age of approximately 7 years and 6 months at 8.1 years old when initiating rhGH treatment. After rhGH treatment for 2 months at 8.3 years old, the bone age of P4 remained the relatively delayed value of 7 years and 6 months (Figure 3).
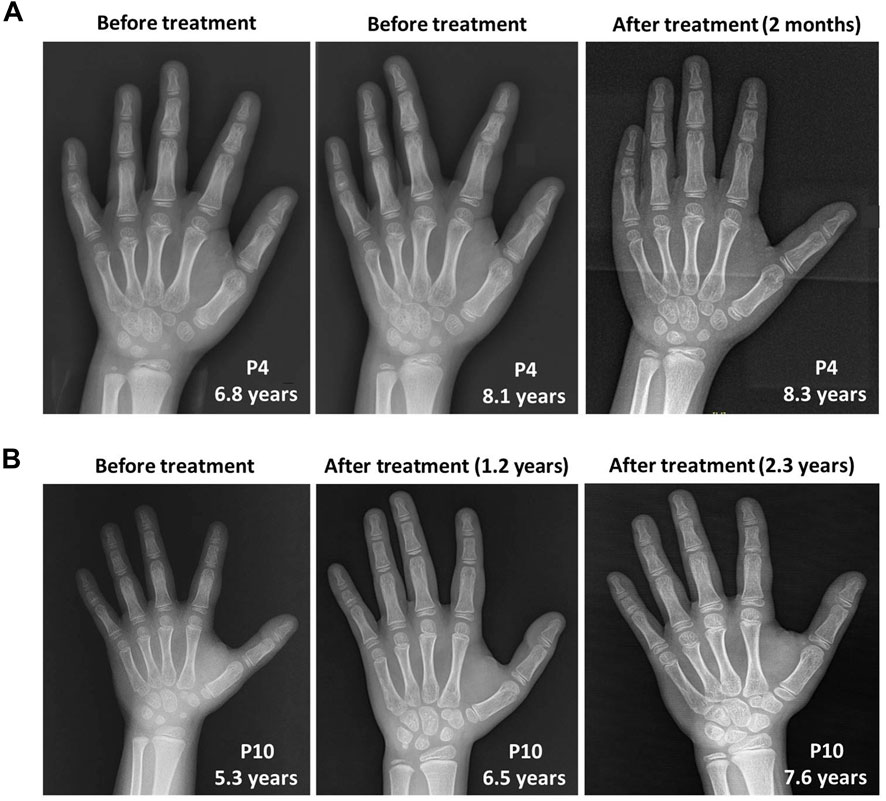
FIGURE 3. The skeletal X-ray images of two WSS patients before and after rhGH treatment. (A) P4. (B) P10.
4 Discussion
WSS is a rare disease and far away from well-known in China. To best of our knowledge, 43 Chinese WSS patients have been reported [(Sun et al., 2017; Gao et al., 2018; Li et al., 2018; Chen et al., 2019; Dai et al., 2019; Shangguan et al., 2019; Jinxiu et al., 2020; Wang et al., 2021a; Wang et al., 2021b; Feng et al., 2021; Lee et al., 2021; Luo et al., 2021; Sheppard et al., 2021; Xue et al., 2021; Chen et al., 2022; Wu et al., 2022; Yu et al., 2022), and other 4 literatures in Chinese]. Among them, only one was Chinese cohort study (Li et al., 2018). To better describe clinical and genetic features of WSS patients in China and evaluate therapeutic effects of rhGH, we enrolled this cohort of 11 WSS children.
Consistent with the prior knowledge on WSS, most patients in our cohort manifested typical clinical symptoms of short stature, developmental delay, intellectual disability, facial dysmorphism, and hypertrichosis. However, the frequencies of some features were diverse with three previous studies, the largest WSS cohort, another Chinese WSS cohort and the French WSS cohort (Baer et al., 2018; Li et al., 2018; Sheppard et al., 2021).
As shown in Table 2, short stature was the most common phenotype found in 90.9% children of our cohort, whereas had a lower incidence in other cohorts. The percentage of patients with abnormal corpus callosum was higher in our cohort, whereas strabismus, abnormal dentition, sacral dimple, hypertrichosis elbows, and hypotonia affected fewer patients than other cohorts. Noticeably, aggressive behavior appeared in the other three cohorts with different frequencies, whereas absent in our cohort. Advanced or delayed bone age was revealed in another Chinese WSS cohort (9/10, 90.0%) and the French WSS cohort (12/15, 80.0%) with high frequency, whereas with a lower than 40% frequency in our cohort (2/6, 33.3%) and the largest WSS cohort (11/29, 37.9%). Feeding difficulties were mentioned in more than 60% cases of the largest WSS cohort and the French WSS cohort, whereas approximately 30% in our cohort and another Chinese WSS cohort. The differences in phenotypic frequencies may result from the specificity of ethnicity, limitations of cohort size and whether the symptoms are recognized correctly.
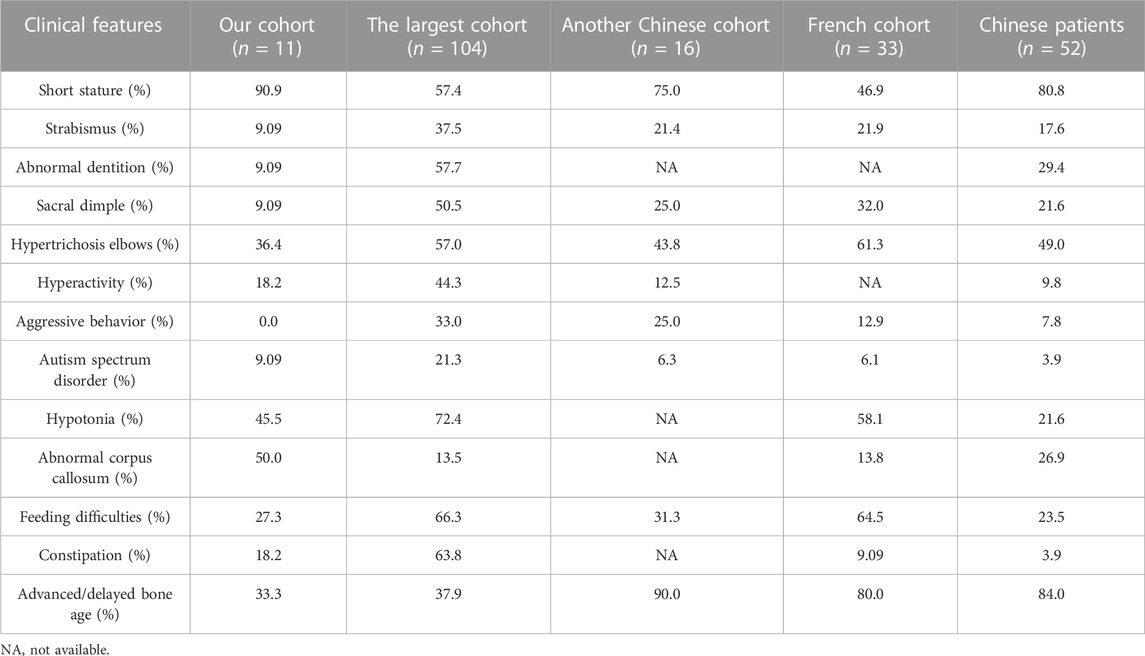
TABLE 2. Clinical features of different frequencies comparing with other WSS cohorts.
To eliminate the influence arisen from the limited sample size, we further covered 41 previously reported Chinese patients in our analysis. After data incorporation, short stature still was one of the most common phenotypes affecting 80.8% patients. The frequencies of strabismus, abnormal dentition, sacral dimple, hypertrichosis elbows, and aggressive behavior were increased, whereas decreased in hyperactivity, autism spectrum disorder, hypotonia, abnormal corpus callosum, and constipation. Significantly, the frequency of advanced or delayed bone age rose from 33.3% (2/6) in our cohort to 84.0% (21/25) in the set involving 52 Chinese patients (Table 2).
Moreover, our data showed highly phenotypic heterogeneity especially in craniofacial and musculoskeletal system (Supplementary Table S1), suggesting that it is difficult to diagnose WSS based on clinical symptoms alone. Due to the clinical complexity, no diagnostic criteria have been developed for WSS. It is probable that a number of WSS cases are underdiagnosed or misdiagnosed as other syndromes with overlapping phenotypes (Bramswig et al., 2015; Miyake et al., 2016; Baer et al., 2018; Li et al., 2018; Negri et al., 2019; Jinxiu et al., 2020; Demir et al., 2021; Di Fede et al., 2021; Sheppard et al., 2021; Castiglioni et al., 2022; Riedhammer et al., 2022).
In this study, eleven different variants, including three known and eight novel variants, of the KMT2A gene were identified in 11 WSS children without a hotspot variant. Of the eight novel variants, seven were point mutations and one was small insertion, resulting in three missense, three nonsense, one splicing, and one frameshift change in the amino acid sequence, which expands the mutational spectrum of this disease.
When mapping to the KMT2A protein, all three missense variants found in this study were located at the CXXC domain, which fits well with the notion that the CXXC domain is a hotspot region for missense variants (Li et al., 2018). Two of the most common phenotypes in our study, short stature and developmental delay, affected all the three patients carrying missense (non-LoF) variants and seven of eight patients carrying LoF variants (87.5%) without a significant difference. However, intellectual disability, another most common phenotype, was manifested in 62.5% patients carrying LoF variants (5/8) and all the three patients carrying missense variants, showing significant difference. Two of three patients carrying missense variants (66.7%) and three of eight patients carrying LoF variants (37.5%) showed hypotonia in our cohort, which differs from the viewpoint that participants with LoF variants are more likely to have hypotonia than those with non-LoF variants (Sheppard et al., 2021). In addition, two of three patients carrying missense variants (66.7%) whereas none of patients carrying LoF variants had seizures, which supports the issue that participants with non-LoF variants are more likely to have seizures (Sheppard et al., 2021).
In WSS patients with GHD, rhGH treatment could achieve satisfactory height gains (Stoyle et al., 2018; Jung et al., 2022). However, two patients (P4 and P10) in our cohort who were not GHD also got benefit from rhGH treatment, suggesting that short stature or failure to thrive not only GHD might be the therapeutic indicator of rhGH in WSS. As sporadic tumors are not present in the germline KMT2A variants (Castiglioni et al., 2022), the tumorigenic risk of rhGH treatment in WSS patients is the same as the general population. Furthermore, accelerated skeletal maturation is another problem of rhGH treatment, especially advanced bone age itself is a symptom of WSS (Mendelsohn et al., 2014; Baer et al., 2018; Li et al., 2018; Sheppard et al., 2021). It is difficult to determine the advanced bone age of P10 in our study is caused by rhGH treatment or the disease. In addition, a longer-term follow-up of the bone age is needed in P4, although no growth acceleration of the bone age was revealed in a very short term of 2 months after rhGH treatment. Thus, the role of rhGH treatment in WSS might be double-edged and remains to be further studied.
5 Conclusion
Our study adds 11 new patients with WSS, describes their phenotypic and genotypic characteristics, and summarizes the clinical presentations of Chinese WSS patients, which enriches the patient resources and clinical data. Our study also identifies eight novel variants in the KMT2A gene, which extends the mutational spectrum. Additionally, our study shares the therapeutic effects of rhGH in two WSS patients without GHD.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Guangzhou Women and Children’s Medical Center. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
All the listed authors were involved in drafting or editing this article, and approved its submission and publishment. CZ, WZ, and LL designed the study. CZ, WZ, LL, XHC, BX, XL, PY, and RD enrolled the family and collected the clinical data. YL, XHC, BX, and XDC performed the experiments. YL, ZG, WZ, XL, and HM analyzed the data. YL and CZ wrote the paper.
Funding
This work was supported by the Research Fund of Guangzhou Science and Technology Innovation Commission (grant numbers 202102080455 and 202102020133) and the Joint Research Fund of Guangdong Basic and Applied Basic Research Commission (grant number 2021A1515220168).
Acknowledgments
The authors would like to thank the enrolled family for participation in this study. We thank the hospital’s medical detection departments for assistance with imaging and biochemical examinations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1085210/full#supplementary-material
References
Aggarwal, A., Rodriguez-Buritica, D. F., and Northrup, H. (2017). Wiedemann-Steiner syndrome: Novel pathogenic variant and review of literature. Eur. J. Med. Genet. 60, 285–288. doi:10.1016/j.ejmg.2017.03.006
Baer, S., Afenjar, A., Smol, T., Piton, A., Gérard, B., Alembik, Y., et al. (2018). Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: A study of 33 French cases. Clin. Genet. 94, 141–152. doi:10.1111/cge.13254
Bramswig, N. C., Lüdecke, H. J., Alanay, Y., Albrecht, B., Barthelmie, A., Boduroglu, K., et al. (2015). Exome sequencing unravels unexpected differential diagnoses in individuals with the tentative diagnosis of Coffin-Siris and Nicolaides-Baraitser syndromes. Hum. Genet. 134, 553–568. doi:10.1007/s00439-015-1535-8
Castiglioni, S., Di Fede, E., Bernardelli, C., Lettieri, A., Parodi, C., Grazioli, P., et al. (2022). KMT2A: Umbrella gene for multiple diseases. Genes. (Basel) 13, 514. doi:10.3390/genes13030514
Chen, K., Yang, Y., Yang, F., Xiao, X., Wu, H., Huang, X. Y., et al. (2022). Analysis of gene variation and clinical characteristics of Wiedemann-Steiner syndrome. Zhonghua Er Ke Za Zhi 60, 119–123. doi:10.3760/cma.j.cn112140-20210720-00608
Chen, M., Liu, R., Wu, C., Li, X., and Wang, Y. (2019). A novel de novo mutation (p.Pro1310Glnfs*46) in KMT2A caused wiedemann-steiner syndrome in a Chinese boy with postnatal growth retardation: A case report. Mol. Biol. Rep. 46, 5555–5559. doi:10.1007/s11033-019-04936-y
Dai, L., Fang, F., Tian, X., and Ding, C. (2019). Wiedemann-Steiner syndrome caused by KMT2A gene variation in one child. Chin. J. Appl. Clin. Pediatr. 34, 1027–1029. doi:10.3760/cma.j.issn.2095-428X.2019.13.016
Demir, S., Gürkan, H., Öz, V., Yalçıntepe, S., Atlı, E. I., and Atlı, E. (2021). Wiedemann-Steiner syndrome as a differential diagnosis of Cornelia de Lange syndrome using targeted next-generation sequencing: A case report. Mol. Syndromol. 12, 46–51. doi:10.1159/000511971
Di Fede, E., Massa, V., Augello, B., Squeo, G., Scarano, E., Perri, A. M., et al. (2021). Expanding the phenotype associated to KMT2A variants: Overlapping clinical signs between wiedemann-steiner and rubinstein-taybi syndromes. Eur. J. Hum. Genet. 29, 88–98. doi:10.1038/s41431-020-0679-8
Durand, B., Schaefer, E., Burger, P., Baer, S., Schroder, C., Mandel, J. L., et al. (2022). Neurocognitive and neurobehavioral characterization of two frequent forms of neurodevelopmental disorders: The DYRK1A and the wiedemann-steiner syndromes. Clin. Genet. 102, 296–304. doi:10.1111/cge.14190
Feng, J., Yang, C., Zhu, L., Zhang, Y., Zhao, X., Chen, C., et al. (2021). Phenotype, genotype and long-term prognosis of 40 Chinese patients with isobutyryl-CoA dehydrogenase deficiency and a review of variant spectra in ACAD8. Orphanet J. Rare Dis. 16, 392. doi:10.1186/s13023-021-02018-6
Fontana, P., Passaretti, F. F., Maioli, M., Cantalupo, G., Scarano, F., and Lonardo, F. (2020). Clinical and molecular spectrum of Wiedemann-Steiner syndrome, an emerging member of the chromatinopathy family. World J. Med. Genet. 9, 1–11. doi:10.5496/wjmg.v9.i1.1
Gao, Z. J., Jiang, Q., Chen, X. L., Chen, Q., Ji, X. N., Mao, Y. Y., et al. (2018). Study of de novo point mutations in known genes among patients with unexplained intellectual disability or developmental delay. Zhonghua Yi Xue Za Zhi 98, 3426–3432. doi:10.3760/cma.j.issn.0376-2491.2018.42.010
Greulich, W. W., and Pyle, S. I. (1959). Radiographic atlas of skeletal development of the hand and wrist. 2nd edition. Stanford, CA: Stanford University Press.
Jinxiu, L., Shuimei, L., Ming, X., Jonathan, L. C., Xiangju, L., and Wenyuan, D. (2020). Wiedemann-steiner syndrome with a de novo mutation in KMT2A: A case report. Med. Baltim. 99, e19813. doi:10.1097/MD.0000000000019813
Jones, W. D., Dafou, D., McEntagart, M., Woollard, W. J., Elmslie, F. V., Holder-Espinasse, M., et al. (2012). De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet. 91, 358–364. doi:10.1016/j.ajhg.2012.06.008
Jung, M. K., Kim, M. R., Yoo, E. G., Rhie, S., and Seo, G. H. (2022). Growth hormone deficiency in a boy with wiedemann-steiner syndrome: A case report and review. Ann. Pediatr. Endocrinol. Metab. Online ahead of print. doi:10.6065/apem.2244052.026
Lee, C. L., Chuang, C. K., Chiu, H. C., Tu, R. Y., Lo, Y. T., Chang, Y. H., et al. (2021). Wiedemann-Steiner syndrome with a pathogenic variant in KMT2A from taiwan. Child. (Basel) 8, 952. doi:10.3390/children8110952
Li, H., Ji, C. Y., Zong, X. N., and Zhang, Y. Q. (2009). Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi 47, 487–492. doi:10.3760/cma.j.issn.0578-1310.2009.07.003
Li, N., Wang, Y., Yang, Y., Wang, P., Huang, H., Xiong, S., et al. (2018). Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients. Orphanet J. Rare Dis. 13, 178. doi:10.1186/s13023-018-0909-0
Lin, Y., Sheng, H., Ting, T. H., Xu, A., Yin, X., Cheng, J., et al. (2020). Molecular and clinical characteristics of monogenic diabetes mellitus in southern Chinese children with onset before 3 years of age. BMJ Open Diabetes Res. Care 8, e001345. doi:10.1136/bmjdrc-2020-001345
Luo, S., Bi, B., Zhang, W., Zhou, R., Chen, W., Zhao, P., et al. (2021). Three de novo variants in KMT2A (MLL) identified by whole exome sequencing in patients with Wiedemann-Steiner syndrome. Mol. Genet. Genomic Med. 9, e1798. doi:10.1002/mgg3.1798
Mendelsohn, B. A., Pronold, M., Long, R., Smaoui, N., and Slavotinek, A. M. (2014). Advanced bone age in a girl with Wiedemann-Steiner syndrome and an exonic deletion in KMT2A (MLL). Am. J. Med. Genet. A 164A (8), 2079–2083. doi:10.1002/ajmg.a.36590
Miyake, N., Tsurusaki, Y., Koshimizu, E., Okamoto, N., Kosho, T., Brown, N. J., et al. (2016). Delineation of clinical features in Wiedemann-Steiner syndrome caused by KMT2A mutations. Clin. Genet. 89, 115–119. doi:10.1111/cge.12586
Negri, G., Magini, P., Milani, D., Crippa, M., Biamino, E., Piccione, M., et al. (2019). Exploring by whole exome sequencing patients with initial diagnosis of rubinstein-taybi syndrome: The interconnections of epigenetic machinery disorders. Hum. Genet. 138, 257–269. doi:10.1007/s00439-019-01985-y
Retterer, K., Juusola, J., Cho, M. T., Vitazka, P., Millan, F., Gibellini, F., et al. (2016). Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704. doi:10.1038/gim.2015.148
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Riedhammer, K. M., Burgemeister, A. L., Cantagrel, V., Amiel, J., Siquier-Pernet, K., Boddaert, N., et al. (2022). Suleiman-el-hattab syndrome: A histone modification disorder caused by TASP1 deficiency. Hum. Mol. Genet. 31, 3083–3094. doi:10.1093/hmg/ddac098
Shangguan, H., Hu, X., Shen, Y., Yuan, X., Zhang, Y., and Chen, R. (2019). Wiedemann-Steiner syndrome caused by novel mutation of KMT2A gene: One case report and literature review. Chin. J. Endocrinol. Metab. 35, 26–31. doi:10.3760/cma.j.issn.1000-6699.2019.01.005
Sheppard, S. E., Campbell, I. M., Harr, M. H., Gold, N., Li, D., Bjornsson, H. T., et al. (2021). Expanding the genotypic and phenotypic spectrum in a diverse cohort of 104 individuals with Wiedemann-Steiner syndrome. Am. J. Med. Genet. A 185 (6), 1649–1665. doi:10.1002/ajmg.a.62124
Stoyle, G., Banka, S., Langley, C., Jones, E. A., and Banerjee, I. (2018). Growth hormone deficiency as a cause for short stature in Wiedemann-Steiner Syndrome. Endocrinol. Diabetes Metab. Case Rep. 2018, 18–85. doi:10.1530/EDM-18-0085
Sun, Y., Hu, G., Liu, H., Zhang, X., Huang, Z., Yan, H., et al. (2017). Further delineation of the phenotype of truncating KMT2A mutations: The extended Wiedemann-Steiner syndrome. Am. J. Med. Genet. A 173, 510–514. doi:10.1002/ajmg.a.38025
Wang, J. L., Huang, K., Wu, W., Zhu, M. Q., Lin, H., Wu, D. W., et al. (2021). A case of Wiedemann-Steiner syndrome caused by a novel variation of the KMT2A gene. Zhonghua Er Ke Za Zhi 59, 516–518. doi:10.3760/cma.j.cn112140-20201231-01145
Wang, X., Zhang, G., Lu, Y., Luo, X., and Wu, W. (2021). Trio-WES reveals a novel de novo missense mutation of KMT2A in a Chinese patient with wiedemann-steiner syndrome: A case report. Mol. Genet. Genomic Med. 9, e1533. doi:10.1002/mgg3.1533
Wu, R., Tang, W., Qiu, K., Zhang, X., and Meng, Z. (2022). Identification of a novel frameshift variant in the KMT2A gene of a child with Wiedemann-Steiner syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 39, 630–633. doi:10.3760/cma.j.cn511374-20210208-00122
Xue, H., Feng, Y., Zhang, C., Ma, L., Wu, J., Li, Q., et al. (2021). Wiedemann-Steiner syndrome due to novel nonsense variant of KMT2A gene in a case. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 38, 138–140. doi:10.3760/cma.j.cn511374-20200122-00046
Keywords: Wiedemann-Steiner syndrome, KMT2A gene, clinical characteristics, genetic spectrum, therapeutic effect, Chinese
Citation: Lin Y, Chen X, Xie B, Guan Z, Chen X, Li X, Yi P, Du R, Mei H, Liu L, Zhang W and Zeng C (2023) Novel variants and phenotypic heterogeneity in a cohort of 11 Chinese children with Wiedemann-Steiner syndrome. Front. Genet. 14:1085210. doi: 10.3389/fgene.2023.1085210
Received: 31 October 2022; Accepted: 09 March 2023;
Published: 21 March 2023.
Edited by:
Xiumin Wang, Shanghai Children’s Medical Center, ChinaReviewed by:
Niu Li, Shanghai Children’s Medical Center, ChinaRowena Ng, Kennedy Krieger Institute, United States
Copyright © 2023 Lin, Chen, Xie, Guan, Chen, Li, Yi, Du, Mei, Liu, Zhang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Liu, bGlsaXVjaGluYUBxcS5jb20=; Wen Zhang, emh3MjAwMXpod0AxNjMuY29t; Chunhua Zeng, Y2h1bmh1YXplbmdAaG90bWFpbC5jb20=
†ORCID: Yunting Lin, orcid.org/0000-0001-8974-357X; Xiuzhen Li, orcid.org/0000-0001-5124-3051; Li Liu, orcid.org/0000-0002-4330-5694; Chunhua Zeng, orcid.org/0000-0001-5954-1803