 Ningan Xu
Ningan Xu Kangxiang Liu1†
Kangxiang Liu1† Yongjia Yang
Yongjia Yang Yan Zhong
Yan Zhong- 1Department of Child Healthcare, Hunan Children’s Hospital, University of South China, Changsha, Hunan, China
- 2The Laboratory of Developmental and Behavioral Pediatrics, Hunan Children’s Hospital Changsha, University of South China, Changsha, Hunan, China
- 3The Laboratory of Genetics and Metabolism, Hunan Children’s Research Institute (HCRI), Hunan Children’s Hospital, University of South China, Changsha, China
- 4Department of Radiology, Hunan Children’s Hospital, University of South China, Changsha, Hunan, China
Background: 3M syndrome is a rare autosomal recessive disease, characterized by intrauterine and postnatal growth retardation, facial dysmorphism, large head circumference, and skeletal changes, has rarely been reported in the Chinese population.
Methods: We describe the clinical manifestations and gene variants in four sporadic cases of 3M syndrome in Chinese individuals from different families.
Results: All cases had significant growth retardation, relative macrocephaly, and typical facial features. Exome sequencing revealed that two patients with 3M syndrome had homozygous variants of the CUL7 gene: one novel pathogenic variant and one previously reported pathogenic variant; the other two patients were heterozygous for variants in OBSL1, one of which had not been reported previously. Clinical evaluation indicated that these Chinese patients with 3M syndrome shared similar recognizable features with those reported in patients of other ethnic backgrounds, but not all patients with 3M syndrome in this study had normal development milestones. Two patients underwent recombinant human growth hormone (rhGH) therapy and showed accelerated growth in the first 2 years; however, the growth rate slowed in the third year in one case. There were no obvious adverse reactions during rhGH treatment.
Conclusion: We report one novel CUL7 and one novel OBSL1 mutation in patients with 3M syndrome. Children with short stature, specific facial features, and physical symptoms should be referred for genetic testing to obtain precise diagnosis and appropriate treatment. The effects of rhGH treatment on adult height requires long-term observation and study in a large sample.
1 Introduction
3M syndrome (OMIM #273750) is a rare autosomal recessive disease with unknown prevalence, characterized by intrauterine and postnatal growth retardation [≤4 standard deviation score (SDS)], facial dysmorphism, large head circumference, and skeletal changes, with normal intelligence and endocrine function (Al-Dosari et al., 2012; Essaddam and Saayda, 2019); the condition was first reported by Miller, McKusick, and Malvauxx in 1975 and named 3M syndrome (Miller et al., 1975). According to Nosology and Classification of Genetic Disorders of the Skeleton 2015 Revision, 3M syndrome is grouped under “Slender bone dysplasia group” (Bonafe et al., 2015). The main skeletal features are long, slender tubular bones, reduced anteroposterior diameter of the vertebral bodies (tall vertebral bodies), and delayed bone age. Previously, diagnosis of 3M syndrome depended primarily on clinical manifestations and radiographic examination. With the development of gene diagnosis technology, variants in the CUL7, OBSL1, and CCDC8 genes have been identified as responsible for 3M syndrome (Guo et al., 2020). In 2005, Huber et al. (2019) initially reported that CUL7 variants accounted for 77.5%, OBSL1 variants 16.3%, and CCDC8 variants 6%, of 3M syndrome cases; however, with continued research, changes in CUL7 were found to be responsible for the majority of cases, while OBSL1 mutations were present in more than 16.3% and those in CDCC8 less than 6%. In one study of 24 patients with 3M syndrome in Turkey, no known causative gene was detected on genetic testing in four patients who met the clinical diagnosis of 3M syndrome, suggesting that there may be other unknown pathogenic genes that contribute to 3M syndrome (Simsek-Kiper et al., 2019; Khachnaoui-Zaafrane et al., 2022).
The CUL7 gene contains 26 exons and encodes a CUL7 protein containing 16,098 amino acid residues, which has an important role in the synthesis of E3 ubiquitin ligase complexes with SKPl (S-phase kinase-associated protein 1), FBX29 (F-box protein), and ROC1(Regulator of cullins 1). Functional experiments to verify R1445X and H1464P in CUL7 mutations showed that proteins with these two mutations cannot recruit ROC1 to form a complex, indicating that loss of ubiquitination may be the main pathological mechanism underlying 3M syndrome (Flex et al., 2013). In addition, researchers have found that mitotic and cytokinesis defects caused by loss of CUL7 function may be associated with short stature in patients with 3M syndrome (Li et al., 2014).
OBSL1 consists of 23 coding exons that can generate three spliced forms, encoding proteins that including several immunoglobulin-like domains and a single fibronectin domain, including a protein of 1896 amino acids that is homologous with obscurin, interacts with proteins that anchor myosin filaments, and may be involved in stabilization of the cytoskeletal network (Geisler et al., 2007). OBSL1 missense mutations are associated with a significant increase in insulin-like growth factor binding protein 5 (IGFBP5) mRNA (Huber et al., 2010) and mouse models suggest that IGFBP overexpression inhibits the effects of insulin-like growth factor (IGF) and is associated with growth retardation. Furthermore, although the relationship between OBSL1 and CUL7 is unclear, loss of OBSL1 leads to CUL7 downregulation, implying a role for OBSL1 in maintaining CUL7 protein levels (Litterman et al., 2011).
CDCC8 is a relatively new evolutionary gene considered to be related to the occurrence and development of malignant tumors, HIV, 3M syndrome, and schizophrenia. In 2011, Hanson et al. first discovered that mutations in CCDC8 can cause 3M syndrome (Hanson et al., 2011); when the CCDC8 protein was knocked out, there was no effect on CUL7 and OBSL1 protein expression levels, but there was an influence on the centrosome localization of CUL7 in mitotic cells, indicating that CCDC8 can regulate CUL7 localization (Yan et al., 2014). In 2019, Wang et al. (2019) found that embryos from CCDC8 knockout mice also had intrauterine growth retardation; most embryos died in utero, while a few died within 7 days of birth. The cause of intrauterine growth retardation and death was related to a disorder of placental vascular development. The placental developmental disorder, intrauterine growth retardation, intrauterine death, and perinatal death in CCDC8 knockout mice is very similar to that observed in CUL7 knockout mouse embryos; however, the proliferation rate of fibroblasts in CUL7 knockout mouse embryos is low and they exhibit advanced senescence, while the proliferation state of fibroblasts in CCDC8 knockout mouse embryos is the same as that of wild-type cells. These findings suggest that CCDC8 is largely functionally similar to CUL7, but that CUL7 has other unique and independent functions.
Management of short stature is a struggle in these patients and final height can be 5–6 SDS below the mean. GH therapy has been tried on many of these patients with variable success (Huber et al., 2011; Clayton et al., 2012; Meazza et al., 2013). Though generally thought to be ineffective, a positive response was noted in a patient with CUL7 mutation in 2015 (Deeb et al., 2015). One 3M patient was treated with rhIGF1, and the result showed this therapy with minimal height benefit but accelerated weight gain (Yang and Patni, 2020). To date, no more than 200 cases of 3M syndrome have been reported worldwide, and there have only been nine publications reporting 15 patients with 3M syndrome in China. Here, we summarize the clinical features and auxiliary examination results of four patients with 3M syndrome from four Chinese families, and report one novel variant each in CUL7 and OBSL1, expanding the molecular spectrum of the syndrome. The purpose of this study is to improve awareness of rare syndromes and to better understand specific genetic diseases in small for gestational age (SGA) children. Our findings will help to improve the accurate diagnosis and inform clinical decisions on the efficacy of growth hormone treatment.
2 Materials and methods
2.1 Subjects
The study protocol was approved by the Academic Committee of Hunan Children’s Hospital (Approval No. HCHLL-2021-18, Changsha City, Hunan Province, China). Twelve family members (Han Chinese ethnicity), including four children and their parents, provided written informed consent to participate in this study. Parental permission for publishing patient photos was obtained, except for Case 1. Clinical data related to the children were collected through the electronic medical record system, including age at first diagnosis, clinical manifestations, growth and development history, maternal history of pregnancy and childbirth, physical examination, laboratory test results, gene sequencing results, treatment, and prognosis.
2.2 Endocrine laboratory tests
Insulin-like growth factor 1 (IGF-1) and IGF-binding protein 3 (IGFBP-3) levels were tested using an IMMULITE 2000 XPi immunoassay analyzer (Siemens, Munich, Germany). Insulin, thyroid function, growth hormone stimulation, and adreno-cortico-tropic-hormone (ACTH) tests were performed using a Roche electrochemiluminescence immunoassay system (Roche, Basel, Switzerland).
2.3 Radiological examination
Bone age (BA), and pelvic and spinal images were obtained using a Kangda Direct Digital Radiography System, and magnetic resonance imaging (MRI) performed using a Siemens 1.5T system; slice thickness, 5 mm and slice spacing, 0.1 mm. Spin echo sequence was detected in the sagittal and coronal planes, and proton density weighted processing, T2WI weighted processing and T1WI weighted processing conducted. After scanning, imaging data were read and analyzed by the same two film readers. BA was measured by Tanner and Whitehouse 3 (TW3) method.
2.4 Genetic testing
DNA was isolated from peripheral blood using a DNA Isolation Kit (Blood DNA Kit V2, CW2553), in accordance with a standard method as we used before (Shen et al., 2022). Genomic DNA was evaluated and quantified using a Qubit fluorometer. DNA samples from each patient were fragmented into 180–280 bp segments using a Covaris bath sonicator. Libraries were prepared and captured using a KAPA Library Preparation Kit (Kapa Biosystems, KR0453), following the manufacturer’s instructions. Quality-passed libraries were sequenced on the Illumina Novaseq 6,000 platform (Illumina, Inc., United States). Raw BCL files were converted into FASTQ files, with 12 Gb of sequence obtained for each sample (average yield, approximately 16.3 Gb; error rate <0.1%). Variants were annotated and filtered by Ingenuity Pathway Analysis (https://variants.ingenuity.com). Common variants (frequency >0.5% in the ExAC, ESP, or 1000G databases) were excluded. Variants were classified following the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology standards and guidelines (Richards et al., 2015), the Rules for combining criteria to classify sequence variants are shown in Table 1. We can All putative pathogenic variants detected in the patients by whole exome sequencing were confirmed by Sanger sequencing. Parents of probands were also checked by segregation analysis.

TABLE 1. Rules for combining criteria to classify sequence variants.
3 Results
3.1 Clinical information
Among the four cases included in this study, two were male and two were female, and their age at diagnos is ranged from 19 months to 11 years 4 months. All four children had obvious short stature (height ≤2 SD), relatively enlarged head circumference, normal intelligence, and particular facial features, such as triangular face, wide forehead, low nose bridge, and thick lips (see Tables 2, 3; Figure 1 for details). Compared with the two children with OBSL1 gene mutations (116.1 cm, −4.3SD and 132.4 cm, −2.5SD), the two with CUL7 gene mutations had more severe short stature (90.4 cm, −5.1SD and 96.0 cm, −4.1SD).
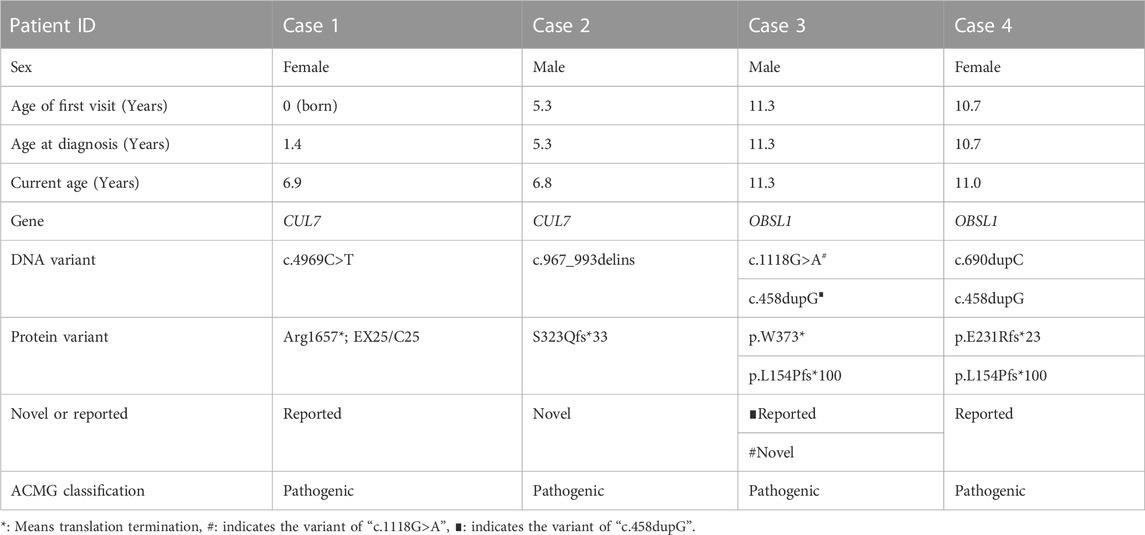
TABLE 2. Demographic characteristics and genetic variants in four patients diagnosed with 3-M syndrome.
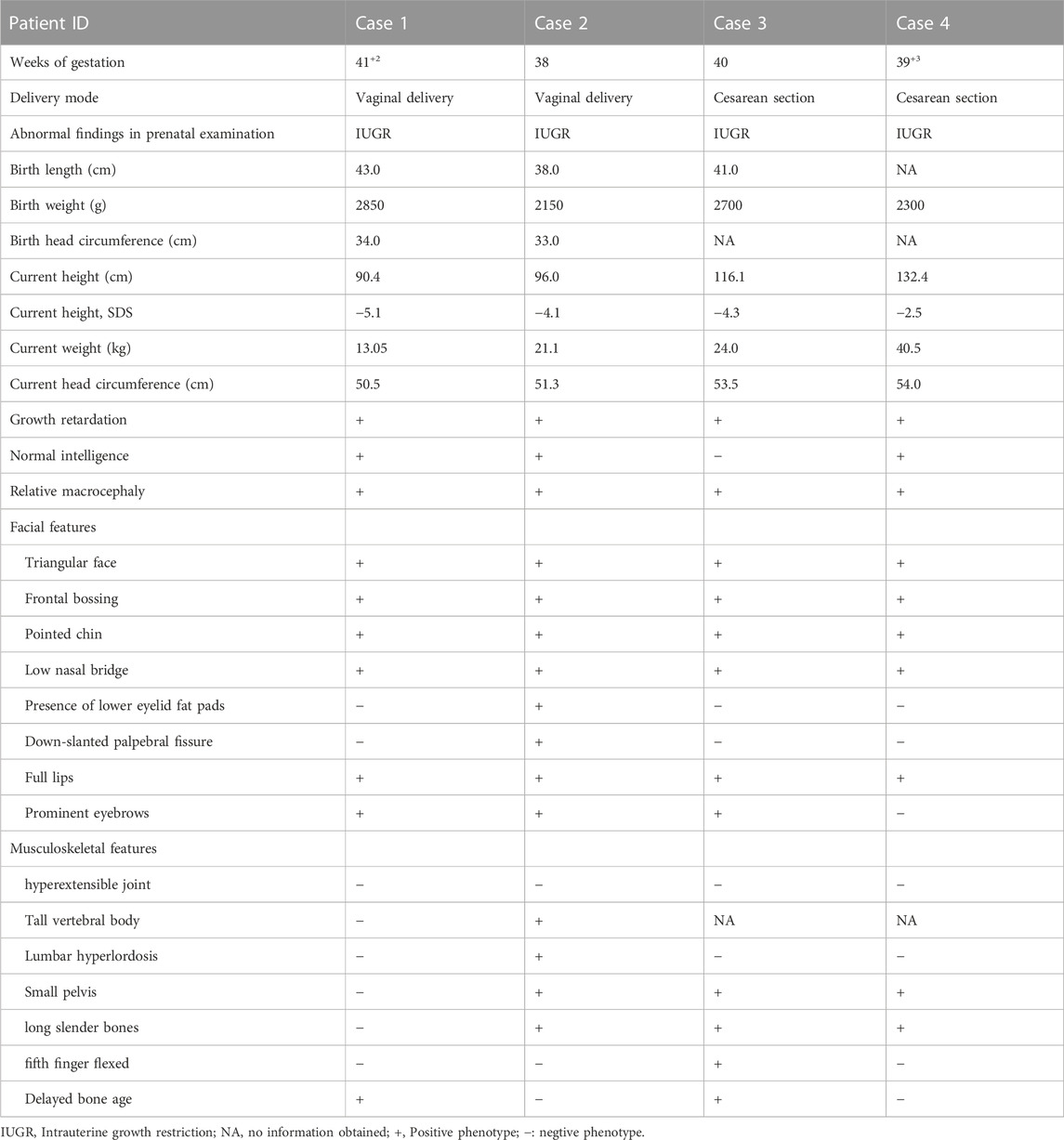
TABLE 3. Clinical features of four patients diagnosed with 3-M syndrome.
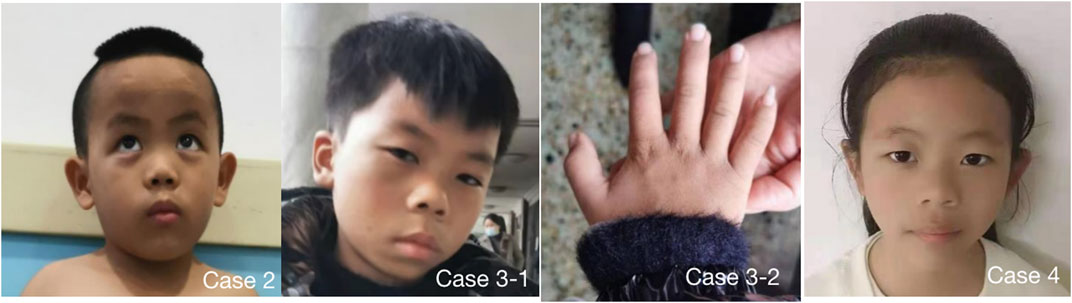
FIGURE 1. 4 cases showed obvious special facial features. Case 2 had triangular face, wide forehead, pointed chin, low bridge of nose, lower eyelid fat pad, lateral canthus oblique, thick lips and thick eyebrows; Case 3 had triangular face, wide forehead, pointed chin, low nasal bridge, thick lips, thick eyebrows and flexion of the fifth finger of right hand (Case 3-2), Case 4 had triangular face, wide forehead, pointed chin, low nasal bridge and thick lips. Case 1 refused to publish portrait photos.
3.2 Laboratory examination
Routine blood and urine tests, liver and kidney function, electrolytes, blood glucose, and blood lipids were in the normal range in all cases, as were thyroid function, ACTH, and cortisol. IGF-1 and IGFBP-3 levels were also in the normal range. One patient had a peak value of 5–10 ng/mL in the growth hormone stimulation test, while the other three children had peak values >10 ng/mL (see Table 4 for details).
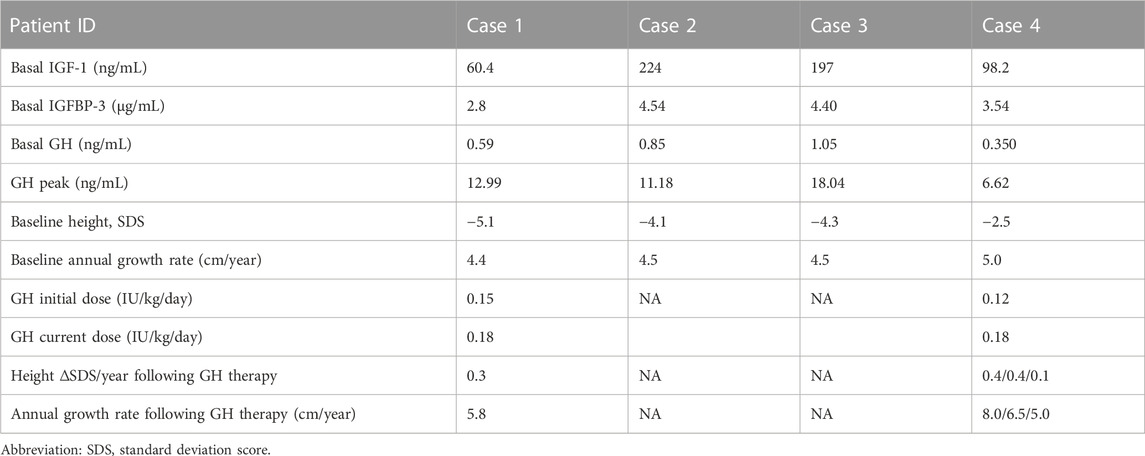
TABLE 4. Growth hormone stimulation test results and treatment information.
3.3 Imaging examination
Imaging examination showed that three patients had a small pelvis (Figure 2) and long tubular bones (Figure 3), and one patient had typical tall vertebral body of 3M syndrome (Figure 4). Two patients showed BA was significantly younger than chronological age (CA) (Case 1: BA 4.0/CA 5.3; Case 3: BA8.7/CA11.3), and one patient showed advanced bone age (Case 4: BA 11.9/CA10.7). No abnormality was found on MRI examination of the head and pituitary glands of any patient.
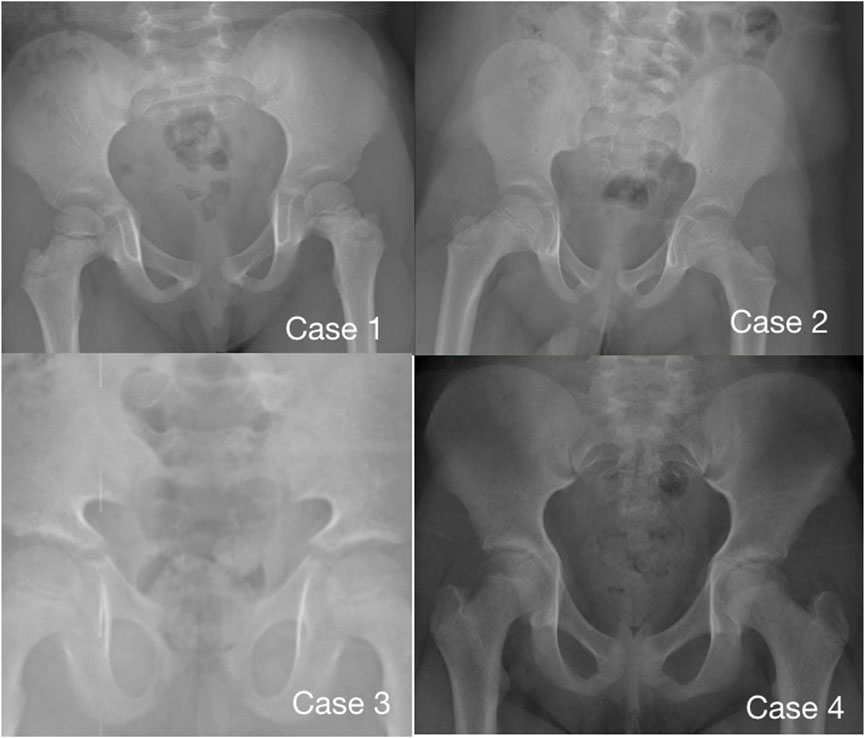
FIGURE 2. Pelvic radiographs of 4 children with 3M syndrome. No obvious abnormality was found in Case 1. The pelvic measurements of Cases 2, 3 and 4 are smaller than those of their peers. Case 3 shows a typical triangular pelvis.
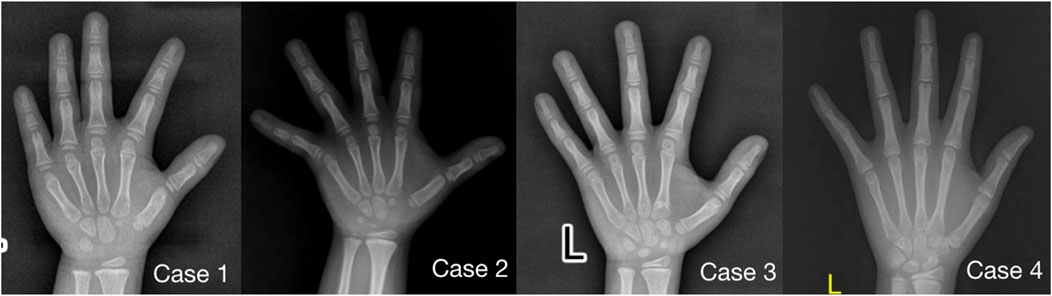
FIGURE 3. Anterior-posterior radiographs of the metacarpal and phalangeal bones of four children with 3M syndrome. The picture of Case 1 was taken at 5.3 years old, without obvious slender changes of tubular bone, and the BA was 4.0 years old; Case 2 was taken at the age of 5.3 years old, with slender tubular bone and BA basically consistent with CA; Case 3 was taken at 11.3 years old, the tubular bone was slender and the BA was 8.5 years old, significantly younger than CA; Case 4 was taken at 10.7 years old, with slender tubular bone and BA was 11.9 years old, significantly elder than CA.
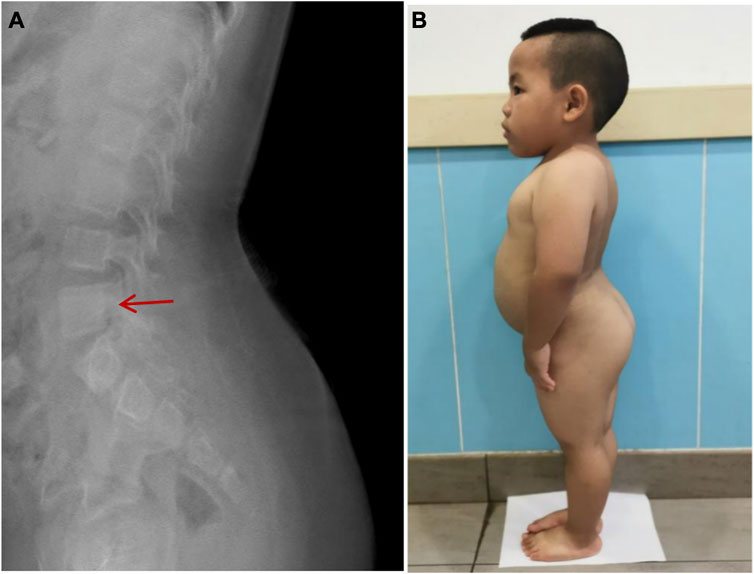
FIGURE 4. High vertebral body and lumbar hyperlordosis changes in Case 2.(A): The typical high vertebral body and lumbar hyperlordosis (red arrow). (B): Case 2 showed the special posture of waist and buttocks.
3.4 Genetic testing
All four patients were sporadic and five mutations were identified by whole exome sequencing, all of which were verified by Sanger sequencing (Figure 5). Case 1 had a homozygous variant, c.4717C>T (p.R1573*) in exon 25 of CUL7, which has been reported previously (Xuyun et al., 2017) and was adjudicated as a pathogenic variant (PVS1+PM2+PM3) according to the ACMG guidelines. In Case 2, a homozygous variant c.967_993delinsCAGCTGG was found in exon 4 of CUL7 gene. The variant causes a frameshift in the reading frame, resulting in the mutation of the 323rd amino acid of the CUL7-encoded protein from serine to L-Glutamine and an premature stop codon (p.S323Qfs * 33). Given that such variant occurred nearing to the N-terminal of the protein, it may cause nonsense-mediated decay at mRNA level. Then the variant fulfill the criteria of null variant (PVS1). The is absent in the public exome database (PM2). The patient’s clinical phenotype is in consistent to 3M syndrome, and 3M syndrome is a rare disorder (PP4). According to the ACMG genetic variation classification criteria and guidelines, it was determined that the variant c.967_993delinsCAGCTGG was pathogenic (PVS1 + PM2 + PP4). Case 3 carried compound heterozygous variants in exon 1 (c.458dupG; p. L154Pfs*100) and exon 2 (c.1118G>A; P.W373*) of OBSL1, where the c.1118G>A variant has not previously been reported in the literature. The mutation causes a change in base 1,118 from G to A, leading to premature stop codon, resulting in protein truncation (PVS1), which is very rare in population data and not a polymorphism (PM2); the patient phenotype was highly and specifically consistent with 3M disease (PP4) and therefore c.1118G>A was classified as a pathogenic variant (PVS1 + PM2 + PP4) according to the ACMG guidelines. OBSL gene encodes a protein which contains 1896 amino acids. The c.1118G>A variant was a truncation variant from the N-teminal (373*). Such novel variant was high likely resulting in a haploinsufficienty which as the consequence of non-sense mediated decay. The c.458dupG variant was previously documented (Hanson et al., 2009) and can be classified as a pathogenic variant according to the ACMG guidelines (PVS1 + PM2 + PM3 + PP4). Two compound heterozygous variants, c.690dupC (p.E231Rfs*23) and c.458dupG (p.L154Pfs*100), in exon 1 of OBSL1, were detected in Case 4. Both variants were previously reported in the literature (Zhang et al., 2019), and are classified as pathogenic variants (PVS1 + PM2 + PP4) according to the ACMG guidelines.
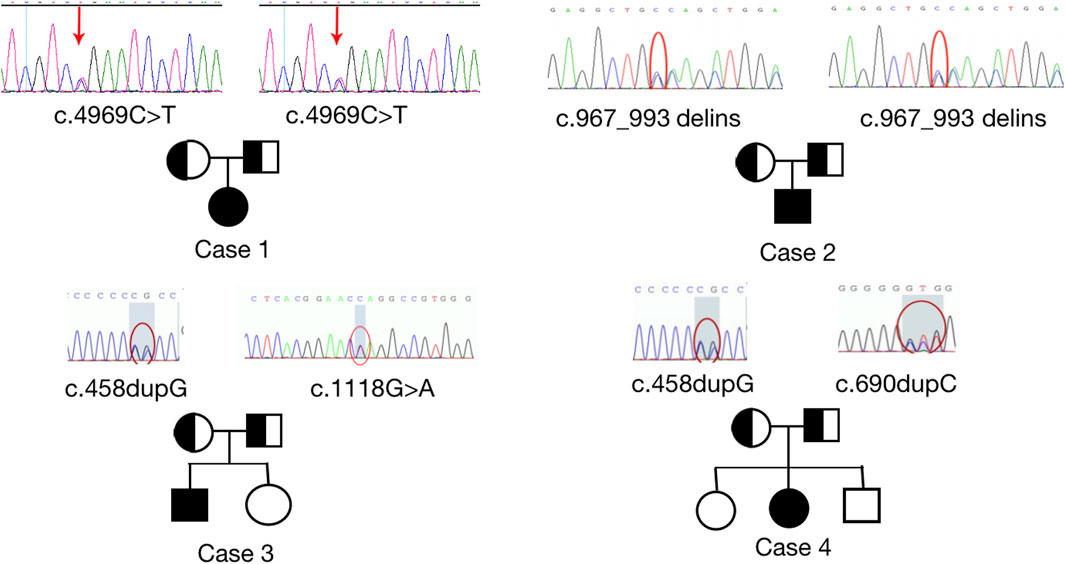
FIGURE 5. Sanger sequencing results to confirm pathogenic variants and their origin in each family. Homozygous variants, c.4969C>T; p. (Arg 1,657*) and c.967_993delinsCAGCTGG; p. (S323Qfs*33) were identified in CUL7 (upper images). Heterozygous variants, c.1118 G>A (p.W373*), c.690dupC (p.E231Rfs*23), and c.458dupG (p.L154Pfs*100) were identified in OBSL1. Sanger sequencing confirmed that the variants were inherited from the parents of the patients with 3M syndrome.
3.5 Treatment with recombinant human growth hormone (rhGH) and follow-up
Case 1 started to receive rhGH injection at 4 years old, with an initial dose of 0.15 U/(kg/day), which was gradually increased to 0.18 U/(kg/d). No discomfort occurred during the treatment. The patient was followed up for 34 months, and the height increased by 19 cm and weight increased by 6.05 kg; rhGH treatment has continued until the present time. Case 4 started to receive rhGH treatment at 8 years old, at an initial dose of 0.12 U/(kg/d) and gradually increased to 0.18 U/(kg/d). No discomfort occurred during treatment, and the height increased by 8.0 cm and 6.5 cm in the first and second years, respectively; however, the rate of increase in height slowed significantly in the third year of treatment and the parents withdrew rhGH treatment.
4 Discussion
3M syndrome is a rare hereditary disease characterized by intrauterine and postnatal growth retardation, abnormal facial features, enlarged head circumference, skeletal changes, and normal general intelligence and endocrine function, which has rarely been reported in the Chinese population, with less than 20 cases published to date (Xuyun et al., 2017; Zhang et al., 2019; Huang et al., 2021). No CDCC8 gene mutations have been found in Chinese patients. Compared with the results reported from countries other than China, the proportion of patients with 3M syndrome and mutations of the OBSL1 gene is higher in the Chinese population, while the proportion of those with CDCC8 mutations is lower (Huber et al., 2019; Simsek-Kiper et al., 2019). Short stature is the main clinical manifestation of 3M syndrome. It was reported that the height of patients with CUL7 mutations is significantly lower than that of patients with OBSL1 mutations (Hanson et al., 2009; Khachnaoui-Zaafrane et al., 2022). In this study, the SDS values of the two children with CUL7 mutations were lower than those with OBSL mutations; however, whether there is a significant difference in height between patients with different genotypes needs further investigation in a larger sample size. Notably, most of the reported 3M syndrome presented with severe dwarfism (−4∼6 SD), but the height of Case 4 remained at the level of −3 SDS before treatment and increased to −2.5 SDS after rhGH therapy. The diagnosis of 3M syndrome has been confirmed by genetic testing until rhGH treatment was unsuccessful. This gives us two hints: firstly, not all of the 3M syndrome patients are severely short, and some clinicians do not understand the clinical manifestations of this rare disease, so there may be missed or misdiagnosed patients based on China’s huge population. Secondly, when SGA patients are not responding well to rhGH therapy, or have specific clinical manifestations, such as specific facial features, skeletal changes or advanced bone age, genetic testing should be performed.
There have been no previous reports of obesity in 3M patients in other ethnicities. Ming Yang and Nivedita Patni reported that a Hispanic male 3M patient developed obesity after treatment with rhIGF1, and BMI decreased after stopping rhIGF1 (Yang and Patni, 2020). In this study, the BMI of Case2 has reached 22.89 kg/m2 (>99th percentile) and over the level of severe obesity. Whether Chinese 3M patients are more likely to develop obesity and metabolic syndrome remains to be studied in larger samples.
Patients with 3M syndrome are considered to undergo normal sexual development (Beyhan et al., 2021). We noticed that puberty initiated at the expected time in Case 4, who carried an OBSL1 gene mutation, while bone age was accelerated. Kyung Lee et al. (2020) reported a pair of sisters with 3M syndrome with central precocious puberty, who also had OBSL1 mutations. Whether precocious puberty or rapid puberty progression is a particular manifestation associated with changes in OBSL1 mutation or a phenotype more common in Asians, further clinical observation is needed. The presence of lower eyelid fat pads was previously reported as a specific phenotype associated with the OBSL1 c.458dupG variant (Huang et al., 2021); however, we found that a patient with CUL7 mutation also had changes in the lower eyelid fat pad. Overall, no specific genotype-phenotype associations have been found in patients with 3M syndrome.
To date, 69 CUL7 mutations associated with type 1 3M syndrome have been reported in the HGMD database. These mutations span almost the entire CUL7 coding sequence and inactivate CUL7 through mRNA decay or protein truncation, resulting in the inability of CUL7 protein to recruit ROC1, thus preventing substrate ubiquitination and causing cyclin D1, insulin receptor substrate-1 and other substrates to accumulate, eventually leading to disordered cell proliferation and differentiation in the quiescent zone of the growth plate, resulting in short stature (Isik et al., 2021). Further, CUL7 deletion leads to dysregulation of cell microtubule dynamics, thus affecting the binding of microtubules to the centromere, leading to defects in mitosis and cell division, indicating that CUL7 protein has a potential function in cell division regulation (Fu et al., 2022). The mitosis and cytokinesis defects caused by loss of CUL7 function may be related to the growth retardation observed in patients with 3M syndrome.
OBSL1 maps to human chromosome 19q13.2-q13.32 and is a member of the UNC-89/Obscurin gene family. The OBSL protein is a key component of the cytoskeletal network and functions through interactions with myosin proteins, primarily localized to the cell membrane and around the nucleus (Wang et al., 2019). To date, 39 OBSL1 mutations have been reported as pathogenic causes of type 2 3M syndrome. Further, cells with OBSL1 knocked out have a phenotype consistent with CUL7 knockout, with changes in microtubule dynamics and mitotic dysfunction, and cells dying in polyploid or tetraploid states (Zhang et al., 2019), indicating that the two genes may have important roles in the same pathway involved in growth and development regulation, which could also explain why there is no significant difference in phenotype between patients with mutations in these two genes.
Effective treatment for 3M syndrome is still lacking, and use of growth hormone treatment is controversial. Most early studies reported that rhGH was ineffective for patients with 3M syndrome, and did not recommend growth hormone treatment (Huber et al., 2011; Meazza et al., 2013; Wang et al., 2019; Khachnaoui-Zaafrane et al., 2022); however, there have been literature reports that many patients have been effectively treated with growth hormone in recent years. Kyung Lee et al. (2020) reported two cases of children with 3M syndrome and precocious puberty who were treated with gonadotrophin-releasing hormone agonists combined with growth hormone, both of whom showed clear height catch up and improved adult height. Isik et al. (20221) reported two patients with 3M syndrome accompanied by growth hormone deficiency after treatment with rhGH, both patients showed clear height catch-up. At present, there are only a few case reports on growth hormone treatment of 3M syndrome, no large-scale clinical trials and mata meta-analysis, which will be the direction of our future research. There have been no reports of serious adverse reactions in patients with 3M syndrome after treatment with growth hormone. Further, the two patients treated with rhGH in our center exhibited accelerated growth without adverse reactions. Therefore, we believe that growth hormone treatment can be attempted for patients with 3M syndrome, without contraindication; however, its long-term effectiveness requires further study in a larger sample with long-term follow-up.
5 Conclusion
In conclusion, we identified novel truncating mutations of CUL7 and OBSL1 in Chinese patients with 3M syndrome. Skeletal manifestations may not be typical in some patients with 3M syndrome, which may be accompanied by early developmental delay. Further studies combined with skeletal surveys and growth curve analysis are needed to determine whether growth hormone treatment is effective or not. Genetic testing should be carried out for children with short stature with specific facial features and bone changes, particularly those with height < –3SD, to facilitate early diagnosis.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author
Ethics statement
The studies involving humans were approved by the Academic Committee of Hunan Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
NX: clinical data collection and article writing; KL: clinical data collection and document retrieval; YY: gene detection and data analysis; XL: imaging data analysis; YZ: study concept generation, clinical data collection, article review and revision. All authors contributed to the article and approved the submitted version.
Funding
This project was supported by Hunan Provincial Natural Science Foundation of China (2021JJ30394).
Acknowledgments
We thank all patients and their families for their support and help during this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-Dosari, M. S., Al-Shammari, M., Shaheen, R., Faqeih, E., Alghofely, M. A., Boukai, A., et al. (2012). 3M syndrome: an easily recognizable yet underdiagnosed cause of proportionate short stature. J. Pediatr. 161, 139–145. doi:10.1016/j.jpeds.2011.12.051
Beyhan, T., Zeynep, A. N., Hande, T., Gezdirici, A., Uludağ Alkaya, D., Kasap, B., et al. (2021). Natural history of facial and skeletal features from neonatal period to adulthood in a 3M syndrome cohort with biallelic CUL7 or OBSL1 variants. Eur. J. Med. Genet. 64, 1043–1046. doi:10.1016/j.ejmg.2021.104346
Bonafe, L., Cormier-Daire, V., Hall, C., Lachman, R., Mortier, G., Mundlos, S., et al. (2015). Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. Part A 167 (12), 2869–2892. doi:10.1002/ajmg.a.37365
Clayton, P. E., Hanson, D., Magee, L., Murray, P. G., Saunders, E., Abu-Amero, S. N., et al. (2012). Exploring the spectrum of 3-M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clin. Endocrinol. 77, 335–342. doi:10.1111/j.1365-2265.2012.04428.x
Deeb, A., Afandi, O., Attia, S., and Fatih, A. E. (2015). 3-M syndrome: a novel CUL7 mutation associated with respiratory distress and a good response to GH therapy. Endocrinol. Diabetes Metab. Case Rep. 2015, 150012. doi:10.1530/EDM-15-0012
Essaddam, L., and Saayda, B. B. (2019). 3M syndrome: a rare cause of short stature. Indian Pediatr. 56, 799. doi:10.1007/s13312-019-1632-1
Flex, E., Ciolfi, A., Caputo, V., Fodale, V., Leoni, C., Melis, D., et al. (2013). Loss of function of the E3 ubiquitin-protein ligase UBE3B causes Kaufman oculocerebrofacial syndrome. J. Med. Genet. 50, 493–499. doi:10.1136/jmedgenet-2012-101405
Fu, P. J., Zhang, Y. N., Hu, F. R., Qinli, D., and Huifeng, Z. (2022). Research progress in pathogenesis of 3M syndrome. Int. J. Gene. 45, 220–226. doi:10.3760/cma.j.cn231536-20210926-00119
Geisler, S. B., Robinson, D., Hauringa, M., Raeker, M. O., Borisov, A. B., Westfall, M. V., et al. (2007). Obscurin-like 1, OBSL1, is a novel cytoskeletal protein related to obscurin. Genomics 89, 521–531. doi:10.1016/j.ygeno.2006.12.004
Guo, L., Feng, Z., Jin, X., Yin, S., Zhang, M., Gao, Y., et al. (2020). A novel mutation within intron 17 of the CUL7 gene results in appearance of premature termination codon. Clin. Chim. Acta. 507, 23–30. doi:10.1016/j.cca.2020.04.008
Hanson, D., Murray, P. G., Black, G. C., and Clayton, P. E. (2011). The genetics of 3-M syndrome: unravelling a potential new regulatory growth pathway. Horm. Res. Paediatr. 76, 369–378. doi:10.1159/000334392
Hanson, D., Murray, P. G., Sudand, A., Temtamy, S. A., Aglan, M., Superti-Furga, A., et al. (2009). The primordial growth disorder 3-M syndrome connects ubiquitination to the cytoskeletal adaptor OBSL1. Am. J. Hum. Genet. 84, 801–806. doi:10.1016/j.ajhg.2009.04.021
Huang, Y., Mei, L., Zhang, J., Chen, X., Wang, W., and Ge, Y. (2021). Clinical and molecular genetic analysis of a patient with 3-M syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 38, 1237–1240. doi:10.3760/cma.j.cn511374-20200929-00702
Huber, C., Delezoide, A., Guimiot, F., Baumann, C., Malan, V., Le Merrer, M., et al. (2019). A large-scale mutation search reveals genetic heterogeneity in 3M syndrome. Eur. J. Hum. Genet. 17, 395–400. doi:10.1038/ejhg.2008.200
Huber, C., Fradin, M., Edouard, T., Le Merrer, M., Alanay, Y., Da Silva, D. B., et al. (2010). OBSL1, mutations in 3-M syndrome are associated with a modulation of IGFBP2, and IGFBP5, expression levels. Hum. Mut. 31, 20–26. doi:10.1002/humu.21150
Huber, C., Munnich, A., and Cormier-Daire, V. (2011). The 3M syndrome. Best. Pract. Res. Clin. Endocrinol. Metabol. 25, 143–151. doi:10.1016/j.beem.2010.08.015
Isik, E., Arican, D., Atik, D., Ooi, J. E., Darcan, S., Ozen, S., et al. (2021). A rare cause of syndromic short stature: 3M syndrome in three families. Med. Genet. A 185, 461–468. doi:10.1002/ajmg.a.61989
Khachnaoui-Zaafrane, K., Ouertani, I., Zanati, A., Kandara, H., Maazoul, F., and Mrad, R. (2022). 3M syndrome: a Tunisian seven-cases series. Eur. J. Med. Genet. 65, 104448. doi:10.1016/j.ejmg.2022.104448
Lee, I. K., Han, H. L., and Kim, Y. M. (2020). The effect of combined growth hormone and a gonadotropin-releasing hormone agonist therapy on height in Korean 3-M syndrome siblings. Yonsei Med. J. 61, 981–985. doi:10.3349/ymj.2020.61.11.981
Li, Z., Pei, X., Yan, J., Yan, F., Cappell, K. M., Whitehurst, A. W., et al. (2014). CUL9 mediates the functions of the 3M complex and ubiquitylates survivin to maintain genome integrity. Mol. Cell. 54, 805–819. doi:10.1016/j.molcel.2014.03.046
Litterman, N., Ikeuchi, Y., Gallardo, G., O'Connell, B. C., Sowa, M. E., Gygi, S. P., et al. (2011). An OBSL1-Cul7 Fbxw8, ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 9, e1001060. doi:10.1371/journal.pbio.1001060
Meazza, C., Lausch, E., Pagani, S., Bozzola, E., Calcaterra, V., Superti-Furga, A., et al. (2013). 3-M syndrome associated with growth hormone deficiency: 18 year follow-up of a patient. Ital. J. Pediatr. 39, 21. doi:10.1186/1824-7288-39-21
Miller, J. D., McKusick, V. A., Malvaux, P., Temtamy, S., and Salinas, C. (1975). The 3-M syndrome: a heritable low birthweight dwarfism. Artic. Ser. 11, 39–47.
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Shen, F., Yang, Y., Zheng, Y., Li, P., Luo, Z., Fu, Y., et al. (2022). MECOM-Related disorder: radioulnar synostosis without hematological aberration due to unique variants. Genet. Med. 24, 1139–1147. doi:10.1016/j.gim.2022.01.021
Simsek-Kiper, P. O., Taskiran, E., Kosukcu, C., Arslan, U. E., Cormier-Daire, V., Gonc, N., et al. (2019). Further expanding the mutational spectrum and investigation of genotype-phenotype correlation in 3M syndrome. Am. J. Med. Genet. 179A, 1157–1172. doi:10.1002/ajmg.a.61154
Wang, P., Yan, F., Li, Z., Yu, Y., Parnell, S. E., and Xiong, Y. (2019). Impaired plasma membrane localization of ubiquitin ligase complex underlies 3-M syndrome development. J. Clin. Invest. 129, 4393–4407. doi:10.1172/JCI129107
Xuyun, B., Hu, A., Gui, B., Wang, J., Li, N., Su, J., et al. (2017). Prenatal and early diagnosis of Chinese 3-M syndrome patients with novel pathogenic variants. Clin. Chim. Acta. 474, 159–164. doi:10.1016/j.cca.2017.09.022
Yan, J., Yan, F., Li, Z., Sinnott, B., Cappell, K. M., Yu, Y., et al. (2014). The 3M complex maintains microtubule and genome integrity. Mol. Cell. 54, 791–804. doi:10.1016/j.molcel.2014.03.047
Yang, M., and Patni, N. (2020). Effect of recombinant human insulin-like growth factor 1 therapy in a child with 3-M syndrome-1 with CUL7 gene mutation. J. Pediatr. Endocrinol. Meta 33 (12), 1609–1612. doi:10.1515/jpem-2020-0278
Keywords: 3-M syndrome, growth hormone, CUL-7, OBSL1, short stature
Citation: Xu N, Liu K, Yang Y, Li X and Zhong Y (2023) Chinese patients with 3M syndrome: clinical manifestations and two novel pathogenic variants. Front. Genet. 14:1164936. doi: 10.3389/fgene.2023.1164936
Received: 13 February 2023; Accepted: 31 July 2023;
Published: 31 August 2023.
Edited by:
Feng Chen, Children’s Hospital of Nanjing Medical University, ChinaReviewed by:
Meow-Keong Thong, University of Malaya, MalaysiaTengfei Wang, Baylor Scott and White Health, United States
Copyright © 2023 Xu, Liu, Yang, Li and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Zhong, emhvbmd5YW5AMTYzLmNvbQ==
†These authors share first authorship